
Overview of Evidence-Based Chemotherapy for Oral Cancer: Focus on Drug Resistance Related to the Epithelial-Mesenchymal Transition



Abstract
:1. Introduction
2. Antimetabolites
2.1. Methotrexate

2.2. 5-Fluorouracil
2.3. Capecitabine

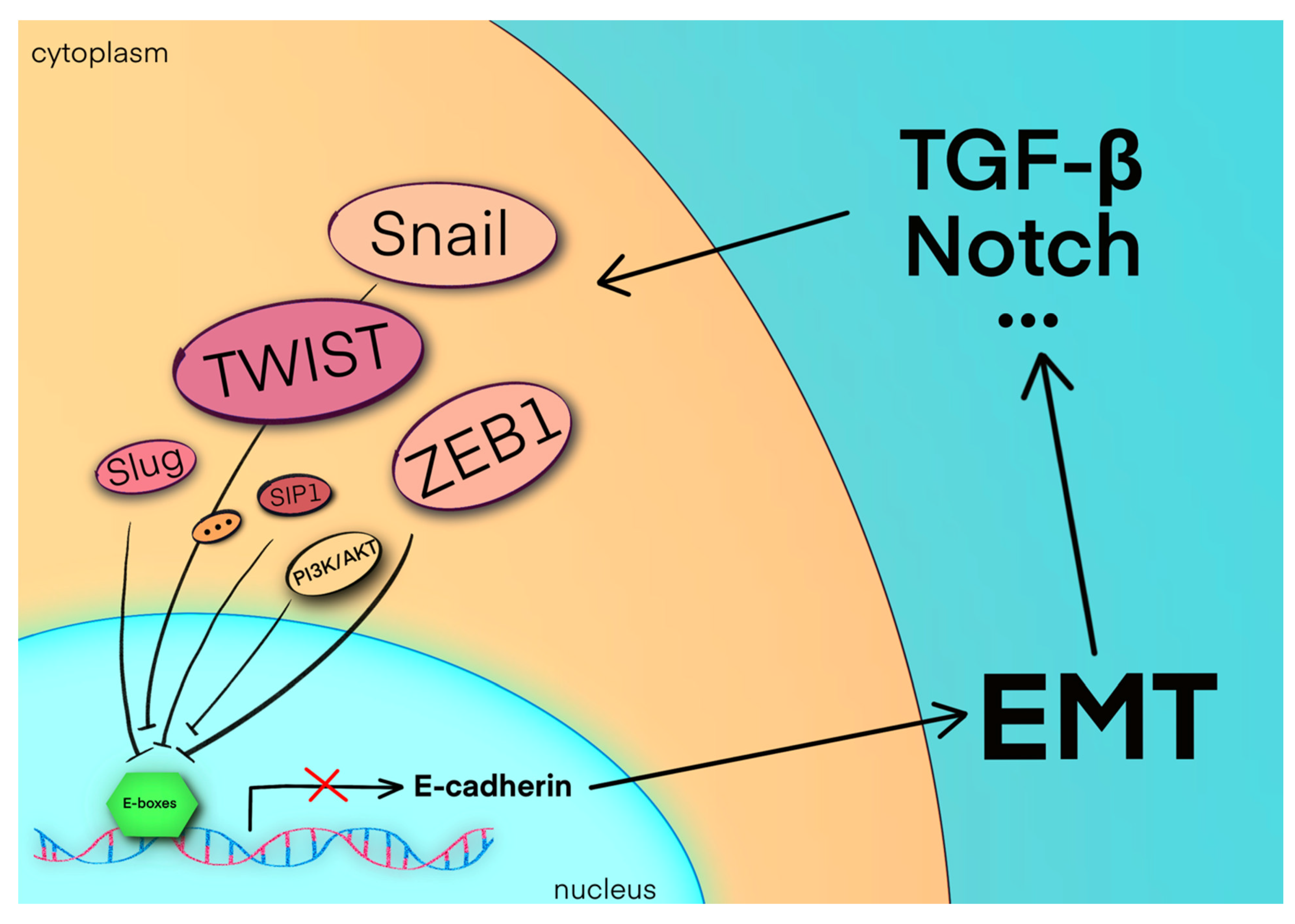{kind=link}
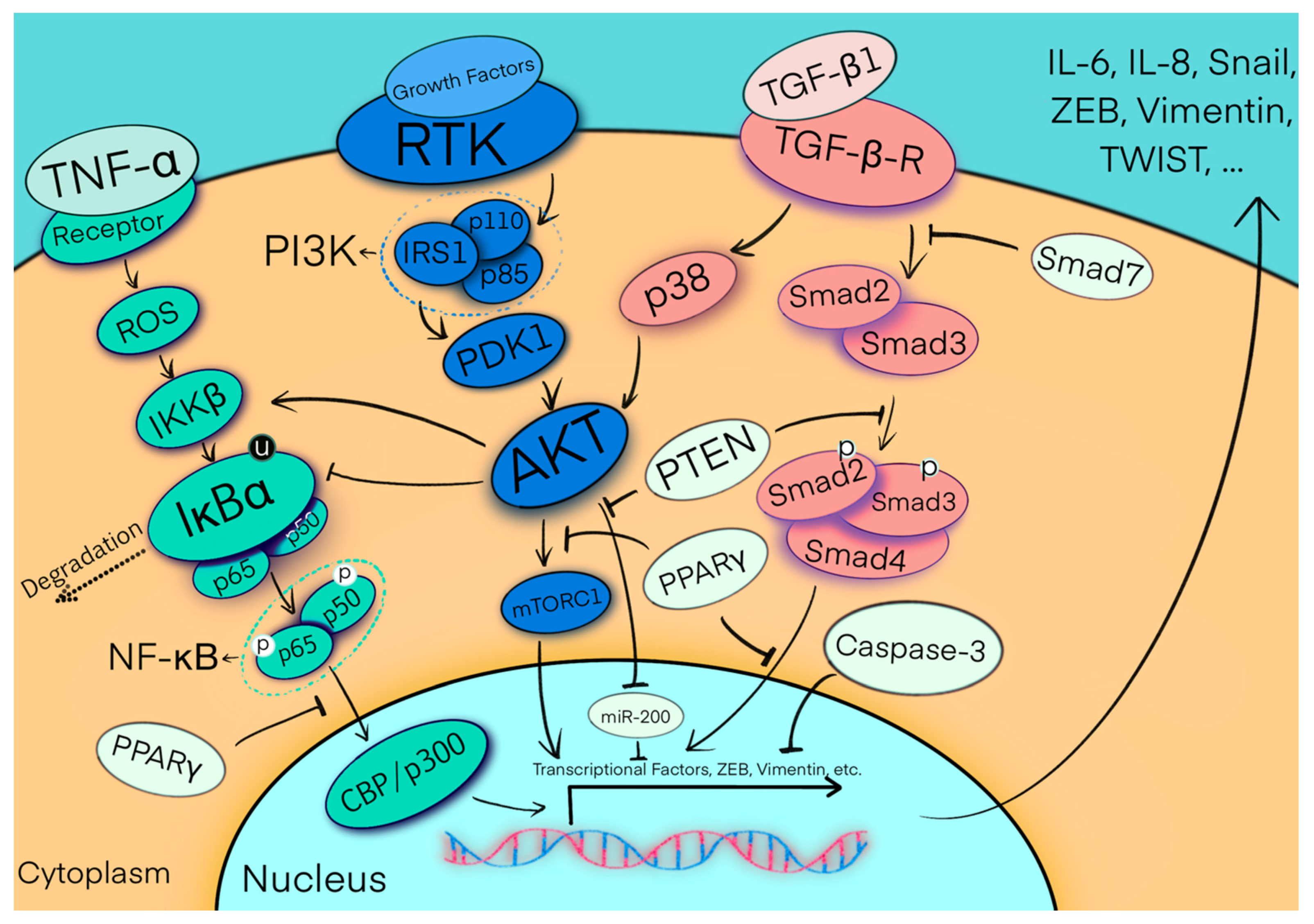{kind=link}
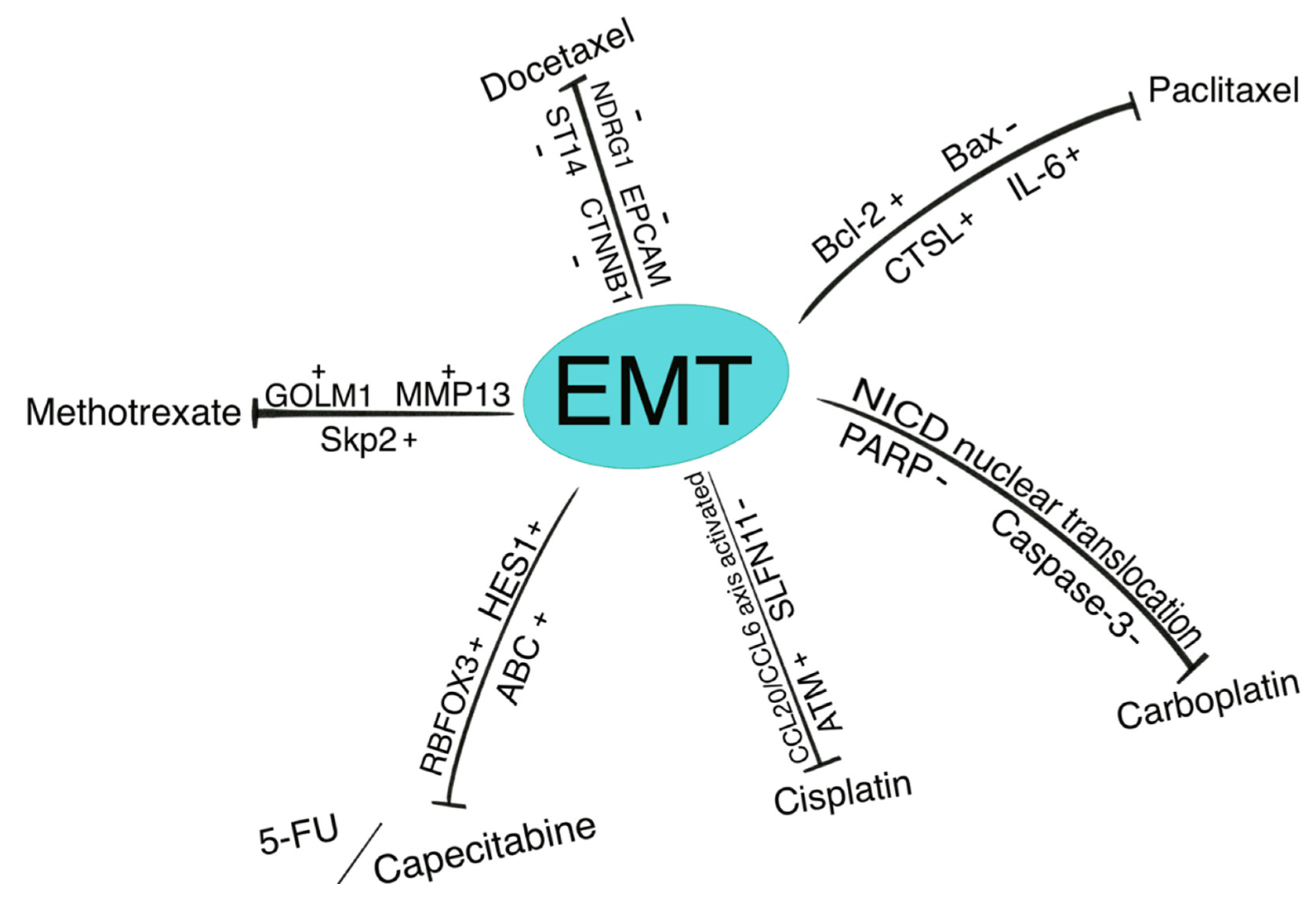{kind=link}
| Reference | Outcomes | 5-FU | Capecitabine | p-Value | |
|---|---|---|---|---|---|
| Administration | Intravenous administration | Oral administration | |||
| Toxicity (Grade III/IV) | [69] | Diarrhea | 12.7% | 16.6% | 0.001 |
| [70] | Diarrhea Stomatitis Hand-foot syndrome | 58.2% 61.6% 6.2% | 47.7% 24.3% 53.5% | <0.001 <0.001 <0.001 | |
| [71] | Acute adverse effects | 28.21% | 26.82% | >0.05 | |
| [72] | Stomatitis Hand-foot syndrome | 16% 1% | 3% 18% | <0.0001 <0.0001 | |
| Tumor Response | [73] | Overall response rate | 17% | 26% | <0.0002 |
| [71] | Complete pathological response rate | 15.48% | 19.53% | 0.04 | |
| [72] | Overall response rate | 11.6% | 25% | 0.005 | |
| Survival | [74] | Overall survival (median survival in months) | 12.1 M | 13.2 M | 0.33 |
| [75] | Overall survival (median survival in months) | 12.8 M | 12.9 M | 0.05 | |
| [71] | Three-year disease-free survival (rate) | 75.72% | 76.67% | 0.05 | |
| [72] | Overall survival (median survival in months) | 13.3 M | 12.5 M | 0.05 | |
3. Platinum-Based Agents
3.1. Cisplatin
3.2. Carboplatin
| Reference | Outcomes | Cisplatin | Carboplatin | p-Value | |
|---|---|---|---|---|---|
| Administration | Intravenous administration | Intravenous administration | |||
| Toxicity (Grade III/IV) | [103] | Neutropenia Diarrhea Stomatitis | 60.0% 1.9% 0.7% | 51.5% 1.5% 0.0% | NC NC NC |
| [104] | Nephrotoxicity Neurotoxicity | 37% 33% | 8% 4% | 0.03 0.02 | |
| [105] | Nausea/Vomiting Neutropenia | 17.7% 0.0% | 5.6% 1.1 | 0.0129 >0.05 | |
| Tumor Response | [103] | Objective response rate | 49.3% | 48.5% | >0.05 |
| [104] | Overall clinical response rate | 71% | 41% | 0.04 | |
| [106] | Objective response rate | 33.75% | 27.125% | 0.001 | |
| [107] | Objective response rate | 57.03% | 42.19% | 0.02 | |
| [105] | Objective response rate | 35.6% | 23.6% | 0.05 | |
| Survival | [103] | Overall survival (median survival in months) | 12.87 M | 10 M | 0.05 |
| [106] | Overall survival (median survival in months) | 9.2 M | 8.4 M | 0.05 | |
| [105] | Overall survival (median survival in months) | 8.75 M | 8 M | 0.05 | |
4. Plant Alkaloids
4.1. Paclitaxel

4.2. Docetaxel
5. Discussion
6. Future Perspectives
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ashrafizadeh, M.; Zarrabi, A.; Hushmandi, K.; Kalantari, M.; Mohammadinejad, R.; Javaheri, T.; Sethi, G. Association of the Epithelial–Mesenchymal Transition (EMT) with cisplatin resistance. Int. J. Mol. Sci. 2020, 21, 4002. [Google Scholar] [CrossRef]
- Warnakulasuriya, S. Living with oral cancer: Epidemiology with particular reference to prevalence and life-style changes that influence survival. Oral Oncol. 2010, 46, 407–410. [Google Scholar] [CrossRef] [PubMed]
- Muwonge, R.; Ramadas, K.; Sankila, R.; Thara, S.; Thomas, G.; Vinoda, J.; Sankaranarayanan, R. Role of tobacco smoking, chewing and alcohol drinking in the risk of oral cancer in Trivandrum, India: A nested case-control design using incident cancer cases. Oral Oncol. 2008, 44, 446–454. [Google Scholar] [CrossRef] [PubMed]
- Gupta, N.; Gupta, R.; Acharya, A.K.; Patthi, B.; Goud, V.; Reddy, S.; Garg, A.; Singla, A. Changing trends in oral cancer—A global scenario. Nepal J. Epidemiol. 2017, 6, 613–619. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Trufero, J.; Isla, D.; Adansa, J.C.; Irigoyen, A.; Hitt, R.; Gil-Arnaiz, I.; Lambea, J.; Lecumberri, M.J.; Cruz, J.J. Phase II study of capecitabine as palliative treatment for patients with recurrent and metastatic squamous head and neck cancer after previous platinum-based treatment. Br. J. Cancer 2010, 102, 1687–1691. [Google Scholar] [CrossRef] [Green Version]
- Binmadi, N.O.; Basile, J.R. Perineural invasion in oral squamous cell carcinoma: A discussion of significance and review of the literature. Oral Oncol. 2011, 47, 1005–1010. [Google Scholar] [CrossRef] [PubMed]
- Kohn, W.G.; Malvitz, D.M.; Park, B.Z. Preventing and controlling oral and pharyngeal cancer; recommendations from a National Strategic Planning Conference. MMWR Recomm. Rep. 1998, 47, 1–12. [Google Scholar]
- Vokes, E.E.; Weichselbaum, R.R.; Lippman, S.M.; Hong, W.K. Head and neck cancer. N. Engl. J. Med. 1993, 328, 184–194. [Google Scholar] [CrossRef] [PubMed]
- Pivot, X.; Chamorey, E.; Guardiola, E.; Magné, N.; Thyss, A.; Otto, J.; Giroux, B.; Mouri, Z.; Schneider, M.; Milano, G. Phase I and pharmacokinetic study of the association of capecitabine-cisplatin in head and neck cancer patients. Ann. Oncol. 2003, 14, 1578–1586. [Google Scholar] [CrossRef]
- Minami, K.; Ueda, N.; Ishimoto, K.; Tsujiuchi, T. Lysophosphatidic Acid Receptor-2 (LPA2)-mediated signaling enhances chemoresistance in melanoma cells treated with anticancer drugs. Mol. Cell. Biochem. 2020, 469, 89–95. [Google Scholar] [CrossRef]
- Zhang, Z.; Qiu, N.; Yin, J.; Zhang, J.; Liu, H.; Guo, W.; Liu, M.; Liu, T.; Chen, D.; Luo, K.; et al. SRGN crosstalks with YAP to maintain chemoresistance and stemness in breast cancer cells by modulating HDAC2 expression. Theranostics 2020, 10, 4290–4307. [Google Scholar] [CrossRef] [PubMed]
- Son, H.; Moon, A. Epithelial-Mesenchymal transition and cell invasion. Toxicol. Res. 2010, 26, 245–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haensel, D.; Dai, X. Epithelial-to-Mesenchymal transition in cutaneous wound healing: Where we are and where we are heading. Dev. Dyn. 2018, 247, 473–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loh, C.Y.; Chai, J.; Tang, T.; Wong, W.; Sethi, G.; Shanmugam, M.; Chong, P.; Looi, C. The E-Cadherin and N-Cadherin switch in epithelial-to-mesenchymal transition: Signaling, therapeutic implications, and challenges. Cells 2019, 8, 1118. [Google Scholar] [CrossRef] [Green Version]
- Garg, M. Epithelial-Mesenchymal transition-activating transcription factors-multifunctional regulators in cancer. World J. Stem Cells 2013, 5, 188. [Google Scholar] [CrossRef]
- Sun, T.; Qin, Y.; Zhong, W. Epithelial-Mesenchymal transition and its regulation in tumor metastasis. In Tumor Metastasis; Xu, K., Ed.; InTechOpen: Rijeka, Croatia, 2016; pp. 217–231. ISBN 978-953-51-2630-0. [Google Scholar]
- Fulcher, C.D.; Haigentz, M.; Ow, T.J. The education committee of the American Head and Neck Society (AHNS) AHNS Series: Do you know your guidelines? Principles of Treatment for locally advanced or unresectable head and neck squamous cell carcinoma. Head Neck 2018, 40, 676–686. [Google Scholar] [CrossRef]
- Koźmiński, P.; Halik, P.K.; Chesori, R.; Gniazdowska, E. Overview of dual-acting drug methotrexate in different neurological diseases, autoimmune pathologies and cancers. Int. J. Mol. Sci. 2020, 21, 3483. [Google Scholar] [CrossRef]
- Kawami, M.; Harabayashi, R.; Miyamoto, M.; Harada, R.; Yumoto, R.; Takano, M. Methotrexate-Induced epithelial–mesenchymal transition in the alveolar epithelial cell line A549. Lung 2016, 194, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Kushwaha, V.S.; Gupta, S.; Husain, N.; Khan, H.; Negi, M.; Jamal, N.; Ghatak, A. Gefitinib, methotrexate and methotrexate plus 5-fluorouracil as palliative treatment in recurrent head and neck squamous cell carcinoma. Cancer Biol. Ther. 2015, 16, 346–351. [Google Scholar] [CrossRef] [Green Version]
- Purcell, W.T.; Ettinger, D.S. Novel antifolate drugs. Curr. Oncol. Rep. 2003, 5, 114–125. [Google Scholar] [CrossRef] [PubMed]
- Tortochaux, J.; Tao, Y.; Tournay, E.; Lapeyre, M.; Lesaunier, F.; Bardet, E.; Janot, F.; Lusinchi, A.; Benhamou, E.; Bontemps, P.; et al. Randomized phase III trial (GORTEC 98-03) comparing re-irradiation plus chemotherapy versus methotrexate in patients with recurrent or a second primary head and neck squamous cell carcinoma, treated with a palliative intent. Radiother. Oncol. 2011, 100, 70–75. [Google Scholar] [CrossRef]
- Rajagopalan, P.T.R.; Zhang, Z.; McCourt, L.; Dwyer, M.; Benkovic, S.J.; Hammes, G.G. Interaction of dihydrofolate reductase with methotrexate: Ensemble and single-molecule kinetics. Proc. Natl. Acad. Sci. USA 2002, 99, 13481–13486. [Google Scholar] [CrossRef] [Green Version]
- Goodsell, D.S. The molecular perspective: Methotrexate. Oncologist 1999, 4, 340–341. [Google Scholar] [CrossRef] [Green Version]
- Harner-Foreman, N.; Vadakekolathu, J.; Laversin, S.A.; Mathieu, M.G.; Reeder, S.; Pockley, A.G.; Rees, R.C.; Boocock, D.J. A novel spontaneous model of Epithelial-Mesenchymal Transition (EMT) using a primary prostate cancer derived cell line demonstrating distinct stem-like characteristics. Sci. Rep. 2017, 7, 40633. [Google Scholar] [CrossRef] [Green Version]
- Kang, Y.; Massagué, J. Epithelial-Mesenchymal transitions: Twist in development and metastasis. Cell 2004, 118, 277–279. [Google Scholar] [CrossRef] [Green Version]
- Serrano-Gomez, S.J.; Maziveyi, M.; Alahari, S.K. Regulation of epithelial-mesenchymal transition through epigenetic and post-translational modifications. Mol. Cancer 2016, 15, 18. [Google Scholar] [CrossRef] [Green Version]
- Hirano, T.; Satow, R.; Kato, A.; Tamura, M.; Murayama, Y.; Saya, H.; Kojima, H.; Nagano, T.; Okabe, T.; Fukami, K. Identification of novel small compounds that restore E-Cadherin expression and inhibit tumor cell motility and invasiveness. Biochem. Pharmacol. 2013, 86, 1419–1429. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.Y.; Yang, P.M.; Chang, Y.F.; Marquez, V.E.; Chen, C.C. Methotrexate induces apoptosis through P53/P21-dependent pathway and increases E-Cadherin expression through downregulation of HDAC/EZH2. Biochem. Pharmacol. 2011, 81, 510–517. [Google Scholar] [CrossRef] [PubMed]
- Li, R.M.; Chu, R.; Yu, Y.; Song, W.Y. Down-Expression of GOLM1 enhances the chemo-sensitivity of cervical cancer to methotrexate through modulation of the MMP13/EMT axis. Am. J. Cancer Res. 2018, 8, 964. [Google Scholar]
- Vincent-Chong, V.K.; Salahshourifar, I.; Karen-Ng, L.P.; Siow, M.Y.; Kallarakkal, T.G.; Ramanathan, A.; Yang, Y.H.; Khor, G.H.; Rahman, Z.A.A.; Ismail, S.M.; et al. Overexpression of MMP13 is associated with clinical outcomes and poor prognosis in oral squamous cell carcinoma. Sci. World J. 2014, 2014, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, J.; Wang, G.; Wu, J.; Zuo, Y.; Zhang, J.; Chen, J. Arsenic trioxide inhibits Skp2 expression to increase chemosensitivity to gemcitabine in pancreatic cancer cells. Am. J. Transl. Res. 2019, 11, 991–997. [Google Scholar]
- Ding, L.; Li, R.; Han, X.; Zhou, Y.; Zhang, H.; Cui, Y.; Wang, W.; Bai, J. Inhibition of Skp2 suppresses the proliferation and invasion of osteosarcoma cells. Oncol. Rep. 2017, 38, 933–940. [Google Scholar] [CrossRef] [Green Version]
- Ding, L.; Wang, C.; Cui, Y.; Han, X.; Zhou, Y.; Bai, J.; Li, R. S-Phase kinase-associated protein 2 is involved in epithelial-mesenchymal transition in methotrexate-resistant osteosarcoma cells. Int. J. Oncol. 2018, 52, 1841–1852. [Google Scholar] [CrossRef] [Green Version]
- Yan, G.; Ru, Y.; Wu, K.; Yan, F.; Wang, Q.; Wang, J.; Pan, T.; Zhang, M.; Han, H.; Li, X.; et al. GOLM1 promotes prostate cancer progression through activating PI3K-AKT-mTOR signaling. Prostate 2018, 78, 166–177. [Google Scholar] [CrossRef]
- Wang, Z.; Gao, D.; Fukushima, H.; Inuzuka, H.; Liu, P.; Wan, L.; Sarkar, F.H.; Wei, W. Skp2: A novel potential therapeutic target for prostate cancer. Biochim. Biophys. Acta Rev. Cancer 2012, 1825, 11–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Fukushima, H.; Inuzuka, H.; Wan, L.; Liu, P.; Gao, D.; Sarkar, F.H.; Wei, W. Skp2 is a promising therapeutic target in breast cancer. Front. Oncol. 2012, 1, 57. [Google Scholar] [CrossRef] [Green Version]
- Mamillapalli, R.; Gavrilova, N.; Mihaylova, V.T.; Tsvetkov, L.M.; Wu, H.; Zhang, H.; Sun, H. PTEN regulates the ubiquitin-dependent degradation of the CDK inhibitor P27KIP1 through the ubiquitin E3 ligase SCFSKP2. Curr. Biol. 2001, 11, 263–267. [Google Scholar] [CrossRef] [Green Version]
- Sakabe, T.; Tsuchiya, H.; Kanki, K.; Azumi, J.; Gonda, K.; Mizuta, Y.; Yamada, D.; Wada, H.; Shomori, K.; Nagano, H.; et al. Identification of the genes chemosensitizing hepatocellular carcinoma cells to interferon-α/5-fluorouracil and their clinical significance. PLoS ONE 2013, 8, e56197. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Liu, W.B.; Huang, D.B.; Jia, W.; Ji, C.S.; Hu, B. Targeting PCDH20 gene by MicroRNA-122 confers 5-FU resistance in hepatic carcinoma. Am. J. Cancer Res. 2016, 6, 1681. [Google Scholar] [PubMed]
- Gao, S. Chemoembolization alone vs combined chemoembolization and hepatic arterial infusion chemotherapy in inoperable hepatocellular carcinoma patients. World J. Gastroenterol. 2015, 21, 10443. [Google Scholar] [CrossRef]
- Johnston, P.G.; Kaye, S. Capecitabine: A novel agent for the treatment of solid tumors. Anti Cancer Drugs 2001, 12, 639–646. [Google Scholar] [CrossRef]
- Duan, W.; Zhang, Y.P.; Hou, Z.; Huang, C.; Zhu, H.; Zhang, C.Q.; Yin, Q. Novel insights into NeuN: From neuronal marker to splicing regulator. Mol. Neurobiol. 2016, 53, 1637–1647. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Huang, Y.; Wang, H.; Wu, D. MiR-106a-5p promotes 5-FU resistance and the metastasis of colorectal cancer by targeting TGFβR2. Int. J. Clin. Exp. Pathol. 2018, 11, 5622–5634. [Google Scholar] [PubMed]
- Pourhanifeh, M.H.; Mahjoubin-Tehran, M.; Shafiee, A.; Hajighadimi, S.; Moradizarmehri, S.; Mirzaei, H.; Asemi, Z. MicroRNAs and exosomes: Small molecules with big actions in multiple myeloma pathogenesis. Int. Union Biochem. Mol. Biol. Life 2020, 72, 314–333. [Google Scholar] [CrossRef] [PubMed]
- Naeli, P.; Yousefi, F.; Ghasemi, Y.; Savardashtaki, A.; Mirzaei, H. The role of MicroRNAs in lung cancer: Implications for diagnosis and therapy. Curr. Mol. Med. 2020, 20, 90–101. [Google Scholar] [CrossRef]
- Creugny, A.; Fender, A.; Pfeffer, S. Regulation of primary MicroRNA processing. FEBS Lett. 2018, 592, 1980–1996. [Google Scholar] [CrossRef]
- Beezhold, K.J.; Castranova, V.; Chen, F. Microprocessor of MicroRNAs: Regulation and potential for therapeutic intervention. Mol. Cancer 2010, 9, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.; Wu, X.; Li, Y.; Lu, W.; Zheng, F.; Zhang, C.; Long, Q.; Qiu, H.; Li, Y.; Ge, Q.; et al. RBFOX3 regulates the chemosensitivity of cancer cells to 5-Fluorouracil via the PI3K/AKT, EMT and Cytochrome-C/Caspase pathways. Cell Physiol. Biochem. 2018, 46, 1365–1380. [Google Scholar] [CrossRef]
- Toden, S.; Okugawa, Y.; Jascur, T.; Wodarz, D.; Komarova, N.L.; Buhrmann, C.; Shakibaei, M.; Boland, C.R.; Goel, A. Curcumin mediates chemosensitization to 5-Fluorouracil through MiRNA-induced suppression of epithelial-to-mesenchymal transition in chemoresistant colorectal cancer. Carcinogenesis 2015, 36, 355–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, P.; Lai, Z.L.; Chen, H.F.; Zhang, M.; Wang, A.; Jia, T.; Sun, W.Q.; Zhu, X.M.; Chen, X.F.; Zhao, Z.; et al. Curcumin synergizes with 5-Fluorouracil by impairing AMPK/ULK1-dependent autophagy, AKT activity and enhancing apoptosis in colon cancer cells with tumor growth inhibition in xenograft mice. J. Exp. Clin. Cancer Res. 2017, 36, 190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, T.S.; Gao, W.; Chan, J.Y.W. Transcription regulation of E-Cadherin by zinc finger E-box binding homeobox proteins in solid tumors. Biomed. Res. Int. 2014, 2014, 1–10. [Google Scholar] [CrossRef]
- Wu, W.S.; Heinrichs, S.; Xu, D.; Garrison, S.P.; Zambetti, G.P.; Adams, J.M.; Look, A.T. Slug antagonizes P53-mediated apoptosis of hematopoietic progenitors by repressing puma. Cell 2005, 123, 641–653. [Google Scholar] [CrossRef] [Green Version]
- Mejlvang, J.; Kriajevska, M.; Vandewalle, C.; Chernova, T.; Sayan, A.E.; Berx, G.; Mellon, J.K.; Tulchinsky, E. Direct repression of cyclin D1 by SIP1 attenuates cell cycle progression in cells undergoing an epithelial mesenchymal transition. Mol. Biol. Cell 2007, 18, 4615–4624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Feng, M.; Zheng, G.; Chen, Y.; Wang, X.; Pen, B.; Yin, J.; Yu, Y.; He, Z. Chemoresistance to 5-Fluorouracil induces epithelial–mesenchymal transition via up-regulation of snail in MCF7 human breast cancer cells. Biochem. Biophys. Res. Commun. 2012, 417, 679–685. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, R.; Ishii, H.; Endo, K.; Hotta, A.; Fujii, E.; Miyazawa, K.; Saitoh, M. Reciprocal expression of slug and snail in human oral cancer cells. PLoS ONE 2018, 13, e0199442. [Google Scholar] [CrossRef] [PubMed]
- Ke, R.; Xu, Q.; Li, C.; Luo, L.; Huang, D. Mechanisms of AMPK in the maintenance of ATP balance during energy metabolism: AMPK and ATP balance. Cell Biol. Int. 2018, 42, 384–392. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.H.; Kim, J.H.; Kim, J.S.; Chang, J.W.; Kim, S.B.; Park, J.S.; Lee, S.K. AMP-Activated protein kinase inhibits TGF-β-, angiotensin II-, aldosterone-, high glucose-, and albumin-induced epithelial-mesenchymal transition. Am. J. Physiol. Renal Physiol. 2013, 304, F686–F697. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Ke, J.; He, Z.; Chen, Z.; Huang, Q.; Ai, W.; Wang, G.; Wei, Y.; Zou, X.; Zhang, S.; et al. HES1 promotes colorectal cancer cell resistance to 5-Fu by inducing of EMT and ABC transporter proteins. J. Cancer 2017, 8, 2802–2808. [Google Scholar] [CrossRef]
- Iqbal, H.; Pan, Q. Capecitabine for treating head and neck cancer. Expert Opin. Investig. Drugs 2016, 25, 851–859. [Google Scholar] [CrossRef]
- O’Shaughnessy, J.A.; Kaufmann, M.; Siedentopf, F.; Dalivoust, P.; Debled, M.; Robert, N.J.; Harbeck, N. Capecitabine monotherapy: Review of Studies in first-line HER-2-negative metastatic breast cancer. Oncologist 2012, 17, 476–484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghaemmaghami, F.; Behtash, N.; Yarandi, F.; Moosavi, A.; Modares, M.; Toogeh, G.; Khanafshar, N. First-Line chemotherapy with 5-FU and platinum for advanced and recurrent cancer of the cervix: A phase II study. J. Obstet. Gynaecol. 2003, 23, 422–425. [Google Scholar] [CrossRef] [PubMed]
- Aguado, C. Should capecitabine replace 5-Fluorouracil in the first-line treatment of metastatic colorectal cancer? World J. Gastroenterol. 2014, 20, 6092. [Google Scholar] [CrossRef]
- Schüller, J.; Cassidy, J.; Dumont, E.; Roos, B.; Banken, L.; Mori, K.; Reigner, B.; Utoh, M.; Weidekamm, E.; Durston, S. Preferential activation of capecitabine in tumor following oral administration to colorectal cancer patients. Cancer Chemother. Pharmacol. 2000, 45, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Ho, D.H.; Pazdur, R.; Covington, W.; Brown, N.; Huo, Y.Y.; Lassere, Y.; Kuritani, J. Comparison of 5-Fluorouracil pharmacokinetics in patients receiving continuous 5-Fluorouracil infusion and oral uracil plus N1-(2′-Tetrahydrofuryl)-5-Fluorouracil. Clin. Cancer Res. 1998, 4, 2085. [Google Scholar] [PubMed]
- Ishikawa, T.; Utoh, M.; Sawada, N.; Nishida, M.; Fukase, Y.; Sekiguchi, F.; Ishitsuka, H. Tumor selective delivery of 5-Fluorouracil by capecitabine, a new oral fluoropyrimidine carbamate, in human cancer xenografts. Biochem. Pharmacol. 1998, 55, 1091–1097. [Google Scholar] [CrossRef]
- Ishikawa, T.; Sekiguchi, F.; Fukase, Y.; Sawada, N.; Ishitsuka, H. Positive correlation between the efficacy of capecitabine and doxifluridine and the ratio of thymidine phosphorylase to dihydropyrimidine dehydrogenase activities in tumors in human cancer xenografts. Cancer Res. 1998, 58, 685–690. [Google Scholar]
- Díaz-Rubio, E.; Evans, T.R.J.; Tabernero, J.; Cassidy, J.; Sastre, J.; Eatock, M.; Bisset, D.; Regueiro, P.; Baselga, J. Capecitabine (Xeloda®) in combination with oxaliplatin: A phase I, dose-escalation study in patients with advanced or metastatic solid tumors. Ann. Oncol. 2002, 13, 558–565. [Google Scholar] [CrossRef]
- Iacovelli, R.; Pietrantonio, F.; Palazzo, A.; Maggi, C.; Ricchini, F.; de Braud, F.; Bartolomeo, M.D. Incidence and relative risk of grade 3 and 4 diarrhoea in patients treated with capecitabine or 5-Fluorouracil: A meta-analysis of published trials. Br. J. Clin. Pharmacol. 2014, 78, 1228–1237. [Google Scholar] [CrossRef]
- Cassidy, J.; Twelves, C.; Van Cutsem, E.; Hoff, P.; Bajetta, E.; Boyer, M.; Bugat, R.; Burger, U.; Garin, A.; Graeven, U.; et al. First-Line oral capecitabine therapy in metastatic colorectal cancer: A favorable safety profile compared with Intravenous5-fluorouracil/leucovorin. Ann. Oncol. 2002, 13, 566–575. [Google Scholar] [CrossRef]
- Zhu, J.; Zeng, W.; Ge, L.; Yang, X.; Wang, Q.; Wang, H. Capecitabine versus 5-Fluorouracil in neoadjuvant chemoradiotherapy of locally advanced rectal cancer: A meta-analysis. Medicine 2019, 98, e15241. [Google Scholar] [CrossRef]
- Hoff, P.M.; Ansari, R.; Batist, G.; Cox, J.; Kocha, W.; Kuperminc, M.; Maroun, J.; Walde, D.; Weaver, C.; Harrison, E.; et al. Comparison of oral capecitabine versus intravenous fluorouracil plus leucovorin as first-line treatment in 605 patients with metastatic colorectal cancer: Results of a randomized phase III study. J. Clin. Oncol. 2001, 19, 2282–2292. [Google Scholar] [CrossRef]
- Cutsem, E.V.; Hoff, P.M.; Harper, P.; Bukowski, R.M.; Cunningham, D.; Dufour, P.; Graeven, U.; Lokich, J.; Madajewicz, S.; Maroun, J.A.; et al. Oral capecitabine vs intravenous 5-Fluorouracil and leucovorin: Integrated efficacy data and novel analyses from two large, randomised, phase III trials. Br. J. Cancer 2004, 90, 1190–1197. [Google Scholar] [CrossRef] [PubMed]
- Cutsem, E.V.; Twelves, C.; Cassidy, J.; Allman, D.; Bajetta, E.; Boyer, M.; Bugat, R.; Findlay, M.; Frings, S.; Jahn, M.; et al. Oral capecitabine compared with intravenous fluorouracil plus leucovorin in patients with metastatic colorectal cancer: Results of a large phase III study. J. Clin. Oncol. 2001, 19, 4097–4106. [Google Scholar] [CrossRef]
- Twelves, C. Capecitabine as first-line treatment in colorectal cancer: Pooled data from two large, phase III trials. Eur. J. Cancer 2002, 38, 15–20. [Google Scholar] [CrossRef]
- Balog, J.Á.; Hackler, L., Jr.; Kovács, A.K.; Neuperger, P.; Alföldi, R.; Nagy, L.I.; Puskás, L.G.; Szebeni, G.J. Single cell mass cytometry revealed the immunomodulatory effect of cisplatin via downregulation of splenic CD44+, IL-17A+ MDSCs and promotion of circulating IFN-Γ+ myeloid cells in the 4T1 metastatic breast cancer model. Int. J. Mol. Sci. 2019, 21, 170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Z.; Gong, Q.; Yu, Y.; Zhu, J.; Li, W. Knockdown of Circ-ABCB10 promotes sensitivity of lung cancer cells to cisplatin via MiR-556-3p/AK4 axis. BMC Pulm. Med. 2020, 20, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zang, H.; Qian, G.; Arbiser, J.; Owonikoko, T.K.; Ramalingam, S.S.; Fan, S.; Sun, S. Overcoming acquired resistance of EGFR-mutant NSCLC cells to the third generation EGFR inhibitor, osimertinib, with the natural product honokiol. Mol. Oncol. 2020, 14, 882–895. [Google Scholar] [CrossRef] [Green Version]
- Bostan, M.; Petrică-Matei, G.; Ion, G.; Radu, N.; Mihăilă, M.; Hainăroşie, R.; Braşoveanu, L.; Roman, V.; Constantin, C.; Neagu, M. Cisplatin effect on head and neck squamous cell carcinoma cells is modulated by ERK1/2 protein kinases. Exp. Ther. Med. 2019, 18, 5041–5051. [Google Scholar] [CrossRef] [Green Version]
- Achkar, I.W.; Abdulrahman, N.; Al-Sulaiti, H.; Joseph, J.M.; Uddin, S.; Mraiche, F. Cisplatin based therapy: The role of the mitogen activated protein kinase signaling pathway. J. Transl. Med. 2018, 16, 96. [Google Scholar] [CrossRef]
- Fadejeva, I.; Olschewski, H.; Hrzenjak, A. MicroRNAs as regulators of cisplatin-resistance in non-small cell lung carcinomas. Oncotarget 2017, 8, 115754–115773. [Google Scholar] [CrossRef] [Green Version]
- Sun, C.Y.; Zhang, Q.Y.; Zheng, G.J.; Feng, B. Phytochemicals: Current strategy to sensitize cancer cells to cisplatin. Biomed. Pharmacother. 2019, 110, 518–527. [Google Scholar] [CrossRef]
- Zhang, Y.F.; Li, C.S.; Zhou, Y.; Lu, X.H. Propofol facilitates cisplatin sensitivity via LncRNA MALAT1/MiR-30e/ATG5 axis through suppressing autophagy in gastric cancer. Life Sci. 2020, 244, 117280. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Wang, W.; Wang, X.; Xu, C.; Zhang, N.; Di, W. Cisplatin-Stimulated macrophages promote ovarian cancer migration via the CCL20-CCR6 axis. Cancer Lett. 2020, 472, 59–69. [Google Scholar] [CrossRef]
- Chen, K.J.; Lin, S.Z.; Zhou, L.; Xie, H.Y.; Zhou, W.H.; Taki-Eldin, A.; Zheng, S.S. Selective recruitment of regulatory T cell through CCR6-CCL20 in hepatocellular carcinoma fosters tumor progression and predicts poor prognosis. PLoS ONE 2011, 6, e24671. [Google Scholar] [CrossRef] [PubMed]
- Kryczek, I.; Lin, Y.; Nagarsheth, N.; Peng, D.; Zhao, L.; Zhao, E.; Vatan, L.; Szeliga, W.; Dou, Y.; Owens, S.; et al. IL-22+CD4+ T cells promote colorectal cancer stemness via STAT3 transcription factor activation and induction of the methyltransferase DOT1L. Immunity 2014, 40, 772–784. [Google Scholar] [CrossRef] [Green Version]
- Walch-Rückheim, B.; Mavrova, R.; Henning, M.; Vicinus, B.; Kim, Y.J.; Bohle, R.M.; Juhasz-Böss, I.; Solomayer, E.F.; Smola, S. Stromal fibroblasts induce CCL20 through IL6/C/EBPβ to support the recruitment of Th17 cells during cervical cancer progression. Cancer Res. 2015, 75, 5248–5259. [Google Scholar] [CrossRef] [Green Version]
- Ranasinghe, R.; Eri, R. Modulation of the CCR6-CCL20 axis: A potential therapeutic target in inflammation and cancer. Medicina 2018, 54, 88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.Q.; Zhang, G.A.; Zhang, B.C.; Wang, Y.; Liu, Z.; Jiao, Y.L.; Liu, N.; Zhao, Y.R. Short low concentration cisplatin treatment leads to an epithelial mesenchymal transition-like response in DU145 prostate cancer cells. Asian Pac. J. Cancer Prev. 2015, 16, 1025–1028. [Google Scholar] [CrossRef]
- Bernstein, J.L.; WECARE Study Collaborative Group; Concannon, P. ATM, radiation, and the risk of second primary breast cancer. Int. J. Radiat. Biol. 2017, 93, 1121–1127. [Google Scholar] [CrossRef] [PubMed]
- Nanda, N.; Roberts, N.J. ATM serine/threonine kinase and its role in pancreatic risk. Genes 2020, 11, 108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murai, J.; Thomas, A.; Miettinen, M.; Pommier, Y. Schlafen 11 (SLFN11), a restriction factor for replicative stress induced by DNA-targeting anti-cancer therapies. Pharmacol. Ther. 2019, 201, 94–102. [Google Scholar] [CrossRef] [PubMed]
- Stewart, C.A.; Tong, P.; Cardnell, R.J.; Sen, T.; Li, L.; Gay, C.M.; Masrorpour, F.; Fan, Y.; Bara, R.O.; Feng, Y.; et al. Dynamic variations in Epithelial-to-Mesenchymal Transition (EMT), ATM, and SLFN11 govern response to PARP inhibitors and cisplatin in small cell lung cancer. Oncotarget 2017, 8, 28575–28587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miow, Q.H.; Tan, T.Z.; Ye, J.; Lau, J.A.; Yokomizo, T.; Thiery, J.P.; Mori, S. Epithelial-Mesenchymal status renders differential responses to cisplatin in ovarian cancer. Oncogene 2015, 34, 1899–1907. [Google Scholar] [CrossRef] [PubMed]
- Apps, M.G.; Choi, E.H.Y.; Wheate, N.J. The state-of-play and future of platinum drugs. Endocr. Relat. Cancer 2015, 22, R219–R233. [Google Scholar] [CrossRef] [Green Version]
- Peters, S.; Adjei, A.A.; Gridelli, C.; Reck, M.; Kerr, K.; Felip, E. Metastatic non-small-cell lung cancer (NSCLC): ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2012, 23, vii56–vii64. [Google Scholar] [CrossRef]
- Ettinger, D.S.; Akerley, W.; Borghaei, H.; Chang, A.C.; Cheney, R.T.; Chirieac, L.R.; D’Amico, T.A.; Demmy, T.L.; Govindan, R.; Grannis, F.W., Jr.; et al. Non-Small cell lung cancer, version 2.2013. J. Natl. Compr. Cancer Netw. 2013, 11, 645–653. [Google Scholar] [CrossRef] [Green Version]
- Azzoli, C.G.; Temin, S.; Aliff, T.; Baker, S.; Brahmer, J.; Johnson, D.H.; Laskin, J.L.; Masters, G.; Milton, D.; Nordquist, L.; et al. 2011 focused update of 2009 American Society of Clinical Oncology clinical practice guideline update on chemotherapy for stage IV non-small-cell lung cancer. J. Clin. Oncol. 2011, 29, 3825–3831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dasari, S.; Bernard Tchounwou, P. Cisplatin in cancer therapy: Molecular mechanisms of action. Eur. J. Pharmacol. 2014, 740, 364–378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santana-Davila, R.; Szabo, A.; Arce-Lara, C.; Williams, C.D.; Kelley, M.J.; Whittle, J. Cisplatin versus carboplatin-based regimens for the treatment of patients with metastatic lung cancer. An analysis of veterans health administration data. J. Thorac. Oncol. 2014, 9, 702–709. [Google Scholar] [CrossRef] [Green Version]
- Kowalski, S.; Wyrzykowski, D.; Inkielewicz-Stępniak, I. Molecular and cellular mechanisms of cytotoxic activity of vanadium compounds against cancer cells. Molecules 2020, 25, 1757. [Google Scholar] [CrossRef]
- Petanidis, S.; Kioseoglou, E.; Domvri, K.; Zarogoulidis, P.; Carthy, J.M.; Anestakis, D.; Moustakas, A.; Salifoglou, A. In Vitro and Ex Vivo Vanadium Antitumor Activity in (TGF-β)-Induced EMT. Synergistic Activity with Carboplatin and Correlation with Tumor Metastasis in Cancer Patients. Int. J. Biochem. Cell. Biol. 2016, 74, 121–134. [Google Scholar] [CrossRef] [PubMed]
- Lorusso, D.; Petrelli, F.; Coinu, A.; Raspagliesi, F.; Barni, S. A Systematic Review Comparing Cisplatin and Carboplatin plus Paclitaxel-Based Chemotherapy for Recurrent or Metastatic Cervical Cancer. Gynecol. Oncol. 2014, 133, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Petrioli, R.; Frediani, B.; Manganelli, A.; Barbanti, G.; Capua, B.D.; Lauretis, A.D.; Salvestrini, F.; Mondillo, S.; Francini, G. Comparison between a cisplatin-containing regimen and a carboplatin-containing regimen for recurrent or metastatic bladder cancer patients: A randomized phase II study. Cancer 1996, 77, 344–351. [Google Scholar] [CrossRef]
- Zatloukal, P.; Petruželka, L.; Zemanová, M.; Kolek, V.; Skřičková, J.; Pešek, M.; Fojtů, H.; Grygárková, I.; Sixtová, D.; Roubec, J.; et al. Gemcitabine plus cisplatin vs. gemcitabine plus carboplatin in stage IIIb and IV non-small cell lung cancer: A phase III randomized trial. Lung Cancer 2003, 41, 321–331. [Google Scholar] [CrossRef]
- Hotta, K.; Matsuo, K.; Ueoka, H.; Kiura, K.; Tabata, M.; Tanimoto, M. Meta-Analysis of randomized clinical trials comparing cisplatin to carboplatin in patients with advanced non-small-cell lung cancer. J. Clin. Oncol. 2004, 22, 3852–3859. [Google Scholar] [CrossRef]
- Galsky, M.D.; Chen, G.J.; Oh, W.K.; Bellmunt, J.; Roth, B.J.; Petrioli, R.; Dogliotti, L.; Dreicer, R.; Sonpavde, G. Comparative effectiveness of cisplatin-based and carboplatin-based chemotherapy for treatment of advanced urothelial carcinoma. Ann. Oncol. 2012, 23, 406–410. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Li, Y.; Kong, D.; Sarkar, F.H. The role of notch signaling pathway in epithelial-mesenchymal transition (EMT) during development and tumor aggressiveness. Curr. Drug Targets 2010, 11, 745–751. [Google Scholar] [CrossRef]
- Gupta, N.; Xu, Z.; El-Sehemy, A.; Steed, H.; Fu, Y. Notch3 induces epithelial-mesenchymal transition and attenuates carboplatin-induced apoptosis in ovarian cancer cells. Gynecol. Oncol. 2013, 130, 200–206. [Google Scholar] [CrossRef]
- Artavanis-Tsakonas, S. Notch signaling: Cell fate control and signal integration in development. Science 1999, 284, 770–776. [Google Scholar] [CrossRef] [Green Version]
- Iso, T.; Hamamori, Y.; Kedes, L. Notch signaling in vascular development. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 543–553. [Google Scholar] [CrossRef] [Green Version]
- Nam, Y.; Sliz, P.; Song, L.; Aster, J.C.; Blacklow, S.C. Structural basis for cooperativity in recruitment of MAML coactivators to notch transcription complexes. Cell 2006, 124, 973–983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, L.; Sun, T.; Kobayashi, K.; Gao, P.; Griffin, J.D. Identification of a family of mastermind-like transcriptional coactivators for mammalian notch receptors. Mol. Cell. Biol. 2002, 22, 7688–7700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rowinsky, E.K.; Donehower, R.C. Paclitaxel (Taxol). N. Engl. J. Med. 1995, 332, 1004–1014. [Google Scholar] [CrossRef] [PubMed]
- Serpico, A.F.; Visconti, R.; Grieco, D. Exploiting immune-dependent effects of microtubule-targeting agents to improve efficacy and tolerability of cancer treatment. Cell Death Dis. 2020, 11, 361. [Google Scholar] [CrossRef] [PubMed]
- Ramanathan, B.; Jan, K.Y.; Chen, C.H.; Hour, T.C.; Yu, H.J.; Pu, Y.S. Resistance to paclitaxel is proportional to cellular total antioxidant capacity. Cancer Res. 2005, 65, 8455–8460. [Google Scholar] [CrossRef] [Green Version]
- Kajiyama, H.; Shibata, K.; Terauchi, M.; Yamashita, M.; Ino, K.; Nawa, A.; Kikkawa, F. Chemoresistance to paclitaxel induces epithelial-mesenchymal transition and enhances metastatic potential for epithelial ovarian carcinoma cells. Int. J. Oncol. 2007, 31, 277–283. [Google Scholar] [CrossRef] [Green Version]
- Park, S.Y.; Kim, M.J.; Park, S.A.; Kim, J.S.; Min, K.N.; Kim, D.K.; Lim, W.; Nam, J.S.; Sheen, Y.Y. Combinatorial TGF-β attenuation with paclitaxel inhibits the epithelial-to-mesenchymal transition and breast cancer stem-like cells. Oncotarget 2015, 6, 37526–37543. [Google Scholar] [CrossRef] [Green Version]
- Goulet, C.R.; Champagne, A.; Bernard, G.; Vandal, D.; Chabaud, S.; Pouliot, F.; Bolduc, S. Cancer-Associated fibroblasts induce epithelial-mesenchymal transition of bladder cancer cells through paracrine IL-6 signalling. BMC Cancer 2019, 19, 137. [Google Scholar] [CrossRef] [Green Version]
- Ding, X.; Ji, J.; Jiang, J.; Cai, Q.; Wang, C.; Shi, M.; Yu, Y.; Zhu, Z.; Zhang, J. HGF-Mediated crosstalk between cancer-associated fibroblasts and MET-unamplified gastric cancer cells activates coordinated tumorigenesis and metastasis. Cell Death Dis. 2018, 9, 867. [Google Scholar] [CrossRef]
- Rincon, M.; Broadwater, G.; Harris, L.; Crocker, A.; Weaver, D.; Dressler, L.; Berry, D.; Sutton, L.; Michaelson, R.; Messino, M.; et al. Interleukin-6, multidrug resistance protein-1 expression and response to paclitaxel in women with metastatic breast cancer: Results of cancer and leukemia group B trial 159806. Breast Cancer Res. Treat. 2006, 100, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhang, F.; Cui, J.; Chen, L.; Chen, Y.; Liu, B. CAFs enhance paclitaxel resistance by inducing EMT through the IL-6/JAK2/STAT3 pathway. Oncol. Rep. 2018, 39, 2081–2090. [Google Scholar] [CrossRef] [Green Version]
- Osuala, K.O.; Sameni, M.; Shah, S.; Aggarwal, N.; Simonait, M.L.; Franco, O.E.; Hong, Y.; Hayward, S.W.; Behbod, F.; Mattingly, R.R.; et al. IL-6 signaling between ductal carcinoma in situ cells and carcinoma-associated fibroblasts mediates tumor cell growth and migration. BMC Cancer 2015, 15, 584. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Han, M.; Wang, W.; Song, Y.; Chen, G.; Wang, Z.; Liang, Z. Downregulation of Cathepsin L suppresses cancer invasion and migration by inhibiting transforming growth factor-β-mediated epithelial-mesenchymal transition. Oncol. Rep. 2015, 33, 1851–1859. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, C.; Grover, S.; Dhanjal, J.; Goyal, S.; Goyal, M.; Grover, A. Mechanistic insights into mode of action of novel natural Cathepsin L inhibitors. BMC Genom. 2013, 14, S10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tholen, M.; Wolanski, J.; Stolze, B.; Chiabudini, M.; Gajda, M.; Bronsert, P.; Stickeler, E.; Rospert, S.; Reinheckel, T. Stress-Resistant translation of Cathepsin L MRNA in breast cancer progression. J. Biol. Chem. 2015, 290, 15758–15769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, M.; Zhao, Y.; Tan, C.; Xiong, Y.; Wang, W.; Wu, F.; Fei, Y.; Wang, L.; Liang, Z. Cathepsin L upregulation-induced EMT phenotype is associated with the acquisition of cisplatin or paclitaxel resistance in A549 Cells. Acta Pharmacol. Sin. 2016, 37, 1606–1622. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Chu, F.; Chou, P.M.; Gallati, C.; Dier, U.; Mirkin, B.L.; Mousa, S.A.; Rebbaa, A. Cathepsin L inhibition suppresses drug resistance in vitro and in vivo: A putative mechanism. Am. J. Physiol. Cell Physiol. 2009, 296, C65–C74. [Google Scholar] [CrossRef]
- Yang, N.; Wang, P.; Wang, W.; Song, Y.; Liang, Z. Inhibition of Cathepsin L sensitizes human glioma cells to ionizing radiation in vitro through NF-ΚB signaling pathway. Acta Pharmacol. Sin. 2015, 36, 400–410. [Google Scholar] [CrossRef] [Green Version]
- Barberà, M.J.; Puig, I.; Domínguez, D.; Julien-Grille, S.; Guaita-Esteruelas, S.; Peiró, S.; Baulida, J.; Francí, C.; Dedhar, S.; Larue, L.; et al. Regulation of snail transcription during epithelial to mesenchymal transition of tumor cells. Oncogene 2004, 23, 7345–7354. [Google Scholar] [CrossRef] [Green Version]
- Yang, Q.; Huang, J.; Wu, Q.; Cai, Y.; Zhu, L.; Lu, X.; Chen, S.; Chen, C.; Wang, Z. Acquisition of epithelial–mesenchymal transition is associated with Skp2 expression in paclitaxel-resistant breast cancer cells. Br. J. Cancer 2014, 110, 1958–1967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inuzuka, H.; Gao, D.; Finley, L.W.S.; Yang, W.; Wan, L.; Fukushima, H.; Chin, Y.R.; Zhai, B.; Shaik, S.; Lau, A.W.; et al. Acetylation-Dependent regulation of Skp2 function. Cell 2012, 150, 179–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Einama, T.; Kagata, Y.; Tsuda, H.; Morita, D.; Ogata, S.; Ueda, S.; Takigawa, T.; Kawarabayashi, N.; Fukatsu, K.; Sugiura, Y.; et al. High-Level Skp2 expression in pancreatic ductal adenocarcinoma: Correlation with the extent of lymph node metastasis, higher histological grade, and poorer patient outcome. Pancreas 2006, 21, 542–544. [Google Scholar] [CrossRef] [Green Version]
- Zheng, W.; Zheng, J.; Ma, R.; Meng, F.; Ni, C. Relationship between levels of Skp2 and P27 in breast carcinomas and possible role of Skp2 as targeted therapy. Steroids 2005, 70, 770–774. [Google Scholar] [CrossRef]
- Dia, V.P.; Pangloli, P. Epithelial-to-Mesenchymal transition in paclitaxel-resistant ovarian cancer cells is downregulated by luteolin. J. Cell Physiol. 2017, 232, 391–401. [Google Scholar] [CrossRef]
- Zhang, D.; Sun, L.; Xian, W.; Liu, F.; Ling, G.; Xiao, L.; Liu, Y.; Peng, Y.; Haruna, Y.; Kanwar, Y.S. Low-Dose paclitaxel ameliorates renal fibrosis in rat UUO model by inhibition of TGF-β/Smad activity. Lab. Investig. 2010, 90, 436–447. [Google Scholar] [CrossRef] [Green Version]
- Adli, M.; Merkhofer, E.; Cogswell, P.; Baldwin, A.S. IKKα and IKKβ each function to regulate NF-ΚB activation in the TNF-induced/canonical pathway. PLoS ONE 2010, 5, e9428. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, S.P.; Behar, M.; Birnbaum, H.A.; Hoffmann, A.; Wright, P.E.; Ghosh, G. Analysis of the RelA: CBP/P300 interaction reveals its involvement in NF-ΚB-driven transcription. PLoS Biol. 2013, 11, e1001647. [Google Scholar] [CrossRef] [Green Version]
- Abeyrathna, P.; Su, Y. The critical role of akt in cardiovascular function. Vasc. Pharmacol. 2015, 74, 38–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horbelt, D.; Denkis, A.; Knaus, P. A portrait of transforming growth factor β superfamily signalling: Background matters. Int. J. Biochem. Cell Biol. 2012, 44, 469–474. [Google Scholar] [CrossRef]
- Verweij, J.; Clavelf, M.; Chevalier, B. Paclitaxel (Taxol™) and Docetaxel (Taxotere™): Not simply two of a kind. Ann. Oncol. 1994, 5, 495–505. [Google Scholar] [CrossRef]
- Sparreboom, A.; van Tellingen, O.; Nooijen, W.J.; Beijnen, J.H. Preclinical pharmacokinetics of paclitaxel and docetaxel. AntiCancer Drugs 1998, 9, 1–17. [Google Scholar] [CrossRef]
- Riou, J.F.; Naudin, A.; Lavelle, F. Effects of taxotere on murine and human tumor cell lines. Biochem. Biophys. Res. Commun. 1992, 187, 164–170. [Google Scholar] [CrossRef]
- Ploussard, G.; Terry, S.; Maillé, P.; Allory, Y.; Sirab, N.; Kheuang, L.; Soyeux, P.; Nicolaiew, N.; Coppolani, E.; Paule, B.; et al. Class III β-Tubulin expression predicts prostate tumor aggressiveness and patient response to docetaxel-based chemotherapy. Cancer Res. 2010, 70, 9253–9264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fojo, T.; Menefee, M. Mechanisms of multidrug resistance: The potential role of microtubule-stabilizing agents. Ann. Oncol. 2007, 18, v3–v8. [Google Scholar] [CrossRef] [PubMed]
- Darshan, M.S.; Loftus, M.S.; Thadani-Mulero, M.; Levy, B.P.; Escuin, D.; Zhou, X.K.; Gjyrezi, A.; Chanel-Vos, C.; Shen, R.; Tagawa, S.T.; et al. Taxane-Induced blockade to nuclear accumulation of the androgen receptor predicts clinical responses in metastatic prostate cancer. Cancer Res. 2011, 71, 6019–6029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guertin, D.A.; Sabatini, D.M. Defining the role of MTOR in cancer. Cancer Cell 2007, 12, 9–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marín-Aguilera, M.; Codony-Servat, J.; Reig, Ò.; Lozano, J.J.; Fernández, P.L.; Pereira, M.V.; Jiménez, N.; Donovan, M.; Puig, P.; Mengual, L.; et al. Epithelial-to-Mesenchymal transition mediates docetaxel resistance and high risk of relapse in prostate cancer. Mol. Cancer Ther. 2014, 13, 1270–1284. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; Zhang, D.; Bae, D.H.; Sahni, S.; Jansson, P.; Zheng, Y.; Zhao, Q.; Yue, F.; Zheng, M.; Kovacevic, Z.; et al. Metastasis suppressor, NDRG1, mediates its activity through signaling pathways and molecular motors. Carcinogenesis 2013, 34, 1943–1954. [Google Scholar] [CrossRef] [Green Version]
- Yae, T.; Tsuchihashi, K.; Ishimoto, T.; Motohara, T.; Yoshikawa, M.; Yoshida, G.J.; Wada, T.; Masuko, T.; Mogushi, K.; Tanaka, H.; et al. Alternative splicing of CD44 MRNA by ESRP1 enhances lung colonization of metastatic cancer cell. Nat. Commun. 2012, 3, 883. [Google Scholar] [CrossRef] [Green Version]
- Gemmill, R.M.; Roche, J.; Potiron, V.A.; Nasarre, P.; Mitas, M.; Coldren, C.D.; Helfrich, B.A.; Garrett-Mayer, E.; Bunn, P.A.; Drabkin, H.A. ZEB1-Responsive genes in non-small cell lung cancer. Cancer Lett. 2011, 300, 66–78. [Google Scholar] [CrossRef] [Green Version]
- Hao, J.; Madigan, M.C.; Khatri, A.; Power, C.A.; Hung, T.T.; Beretov, J.; Chang, L.; Xiao, W.; Cozzi, P.J.; Graham, P.H.; et al. In vitro and in vivo prostate cancer metastasis and chemoresistance can be modulated by expression of either CD44 or CD147. PLoS ONE 2012, 7, e40716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tawara, K.; Oxford, J.T.; Jorcyk, C.L. Clinical significance of interleukin (IL)-6 in cancer metastasis to bone: Potential of anti-IL-6 therapies. Cancer Manag. Res. 2011, 3, 177–189. [Google Scholar] [CrossRef] [PubMed]
- Domingo-Domenech, J.; Mellado, B.; Ferrer, B.; Truan, D.; Codony-Servat, J.; Sauleda, S.; Alcover, J.; Campo, E.; Gascon, P.; Rovira, A.; et al. Activation of nuclear factor-ΚB in human prostate carcinogenesis and association to biochemical relapse. Br. J. Cancer 2005, 93, 1285–1294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [Green Version]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.J.; Nieto, M.A. Epithelial-Mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef]
- Onder, T.T.; Gupta, P.B.; Mani, S.A.; Yang, J.; Lander, E.S.; Weinberg, R.A. Loss of E-Cadherin promotes metastasis via multiple downstream transcriptional pathways. Cancer Res. 2008, 68, 3645–3654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Opdenakker, G.; Van Damme, J. Cytokines and proteases in invasive processes: Molecular similarities between inflammation and cancer. Cytokine 1992, 4, 251–258. [Google Scholar] [CrossRef]
- Liu, X.; Fan, D. The Epithelial-Mesenchymal transition and cancer stem cells: Functional and mechanistic links. Curr. Pharm. Des. 2015, 21, 1279–1291. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Miyazono, K. Transforming growth factor-β signaling in epithelial-mesenchymal transition and progression of cancer. Proc. Jpn. Acad. Ser. B 2009, 85, 314–323. [Google Scholar] [CrossRef] [Green Version]
- Wendt, M.K.; Allington, T.M.; Schiemann, W.P. Mechanisms of the epithelial–mesenchymal transition by TGF-β. Future Oncol. 2009, 5, 1145–1168. [Google Scholar] [CrossRef] [Green Version]
- Winkler, J.; Abisoye-Ogunniyan, A.; Metcalf, K.J.; Werb, Z. Concepts of extracellular matrix remodelling in tumour progression and metastasis. Nat. Commun. 2020, 11, 5120. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Tilló, E.; Liu, Y.; de Barrios, O.; Siles, L.; Fanlo, L.; Cuatrecasas, M.; Darling, D.S.; Dean, D.C.; Castells, A.; Postigo, A. EMT-Activating transcription factors in cancer: Beyond EMT and tumor invasiveness. Cell. Mol. Life Sci. 2012, 69, 3429–3456. [Google Scholar] [CrossRef] [PubMed]
- Vincent, T.; Neve, E.P.A.; Johnson, J.R.; Kukalev, A.; Rojo, F.; Albanell, J.; Pietras, K.; Virtanen, I.; Philipson, L.; Leopold, P.L.; et al. A SNAIL1–SMAD3/4 transcriptional repressor complex promotes TGF-β mediated epithelial-mesenchymal transition. Nat. Cell Biol. 2009, 11, 943–950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villagrasa, P.; Díaz, V.M.; Viñas-Castells, R.; Peiró, S.; Del Valle-Pérez, B.; Dave, N.; Rodríguez-Asiain, A.; Casal, J.I.; Lizcano, J.M.; Duñach, M.; et al. Akt2 Interacts with Snail1 in the E-Cadherin promoter. Oncogene 2012, 31, 4022–4033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Tian, X.J.; Xing, J. Signal transduction pathways of EMT induced by TGF-β, SHH, and WNT and their crosstalks. J. Clin. Med. 2016, 5, 41. [Google Scholar] [CrossRef] [PubMed]
- Frías, A.; Lambies, G.; Viñas-Castells, R.; Martínez-Guillamon, C.; Dave, N.; García de Herreros, A.; Díaz, V.M. A switch in akt isoforms is required for notch-induced Snail1 expression and protection from cell death. Mol. Cell. Biol. 2016, 36, 923–940. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Wang, H.S.; Zhou, B.H.; Li, C.L.; Zhang, F.; Wang, X.F.; Zhang, G.; Bu, X.Z.; Cai, S.H.; Du, J. Epithelial–Mesenchymal transition (EMT) induced by TNF-α requires AKT/GSK-3β-mediated stabilization of snail in colorectal cancer. PLoS ONE 2013, 8, e56664. [Google Scholar] [CrossRef]
- Viñas-Castells, R.; Beltran, M.; Valls, G.; Gómez, I.; García, J.M.; Montserrat-Sentís, B.; Baulida, J.; Bonilla, F.; de Herreros, A.G.; Díaz, V.M. The hypoxia-controlled FBXL14 ubiquitin ligase targets Snail1 for proteasome degradation. J. Biol. Chem. 2010, 285, 3794–3805. [Google Scholar] [CrossRef] [Green Version]
- Bai, Y.; Sha, J.; Kanno, T. The role of carcinogenesis-related biomarkers in the Wnt pathway and their effects on epithelial–mesenchymal transition (EMT) in oral squamous cell carcinoma. Cancers 2020, 12, 555. [Google Scholar] [CrossRef] [Green Version]
- Qin, Q.; Xu, Y.; He, T.; Qin, C.; Xu, J. Normal and disease-related biological functions of Twist1 and underlying molecular mechanisms. Cell Res. 2012, 22, 90–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hui, L.; Zhang, S.; Dong, X.; Tian, D.; Cui, Z.; Qiu, X. Prognostic significance of twist and N-Cadherin expression in NSCLC. PLoS ONE 2013, 8, e62171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franco, H.L.; Casasnovas, J.; Rodriguez-Medina, J.R.; Cadilla, C.L. Redundant or separate entities?—Roles of Twist1 and Twist2 as molecular switches during gene transcription. Nucleic Acids Res. 2011, 39, 1177–1186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suarez-Carmona, M.; Lesage, J.; Cataldo, D.; Gilles, C. EMT and Inflammation: Inseparable Actors of Cancer Progression. Mol. Oncol. 2017, 11, 805–823. [Google Scholar] [CrossRef]
- Kim, M.S.; Lee, W.S.; Jeong, J.; Kim, S.J.; Jin, W. Induction of metastatic potential by TrkB via activation of IL6/JAK2/STAT3 and PI3K/AKT signaling in breast cancer. Oncotarget 2015, 6, 40158–40171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lo, H.W.; Hsu, S.C.; Xia, W.; Cao, X.; Shih, J.Y.; Wei, Y.; Abbruzzese, J.L.; Hortobagyi, G.N.; Hung, M.C. Epidermal growth factor receptor cooperates with signal transducer and activator of transcription 3 to induce epithelial-mesenchymal transition in cancer cells via up-regulation of Twist gene expression. Cancer Res. 2007, 67, 9066–9076. [Google Scholar] [CrossRef] [Green Version]
- Cheng, G.Z.; Zhang, W.; Sun, M.; Wang, Q.; Coppola, D.; Mansour, M.; Xu, L.; Costanzo, C.; Cheng, J.Q.; Wang, L.H. Twist is transcriptionally induced by activation of STAT3 and mediates STAT3 oncogenic function. J. Biol. Chem. 2008, 283, 14665–14673. [Google Scholar] [CrossRef] [Green Version]
- Gheldof, A.; Hulpiau, P.; van Roy, F.; De Craene, B.; Berx, G. Evolutionary functional analysis and molecular regulation of the ZEB transcription factors. Cell Mol. Life Sci. 2012, 69, 2527–2541. [Google Scholar] [CrossRef]
- Sánchez-Tilló, E.; Siles, L.; de Barrios, O.; Cuatrecasas, M.; Vaquero, E.C.; Castells, A.; Postigo, A. Expanding roles of ZEB factors in tumorigenesis and tumor progression. Am. J. Cancer Res. 2011, 1, 897–912. [Google Scholar]
- Postigo, A.A.; Dean, D.C. Differential expression and function of members of the Zfh-1 family of zinc finger/homeodomain repressors. Proc. Natl. Acad. Sci. USA 2000, 97, 6391–6396. [Google Scholar] [CrossRef] [Green Version]
- Baranwal, S.; Alahari, S.K. MiRNA control of tumor cell invasion and metastasis. Int. J. Cancer. 2010, 126, 1283–1290. [Google Scholar] [CrossRef] [PubMed]
- Zare, A.; Ahadi, A.; Larki, P.; Omrani, M.D.; Zali, M.R.; Alamdari, N.M.; Ghaedi, H. The clinical significance of MiR-335, MiR-124, MiR-218 and MiR-484 downregulation in gastric cancer. Mol. Biol. Rep. 2018, 45, 1587–1595. [Google Scholar] [CrossRef] [PubMed]
- Leite, K.R.M.; Sousa-Canavez, J.M.; Reis, S.T.; Tomiyama, A.H.; Camara-Lopes, L.H.; Sañudo, A.; Antunes, A.A.; Srougi, M. Change in expression of MiR-Let7c, MiR-100, and MiR-218 from high grade localized prostate cancer to metastasis. Urol. Oncol. 2011, 29, 265–269. [Google Scholar] [CrossRef]
- Wellner, U.; Schubert, J.; Burk, U.C.; Schmalhofer, O.; Zhu, F.; Sonntag, A.; Waldvogel, B.; Vannier, C.; Darling, D.; zur Hausen, A.; et al. The EMT-activator ZEB1 promotes tumorigenicity by repressing stemness-inhibiting MicroRNAs. Nat. Cell Biol. 2009, 11, 1487–1495. [Google Scholar] [CrossRef] [PubMed]
- Shimono, Y.; Zabala, M.; Cho, R.W.; Lobo, N.; Dalerba, P.; Qian, D.; Diehn, M.; Liu, H.; Panula, S.P.; Chiao, E.; et al. Downregulation of MiRNA-200c links breast cancer stem cells with normal stem cells. Cell 2009, 138, 592–603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Kumar, S.M.; Lu, H.; Liu, A.; Yang, R.; Pushparajan, A.; Guo, W.; Xu, X. MicroRNA-9 up-regulates E-Cadherin through inhibition of NF-ΚB1-Snail1 pathway in melanoma: MiR-9 inhibits melanoma progression. J. Pathol. 2012, 226, 61–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siemens, H.; Jackstadt, R.; Hünten, S.; Kaller, M.; Menssen, A.; Götz, U.; Hermeking, H. MiR-34 and Snail form a double-negative feedback loop to regulate epithelial-mesenchymal transitions. Cell Cycle 2011, 10, 4256–4271. [Google Scholar] [CrossRef] [Green Version]
- Brabletz, S.; Brabletz, T. The ZEB/MiR-200 feedback loop—A motor of cellular plasticity in development and cancer? EMBO Rep. 2010, 11, 670–677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.M.; Gaur, A.B.; Lengyel, E.; Peter, M.E. The MiR-200 family determines the epithelial phenotype of cancer cells by targeting the E-Cadherin repressors ZEB1 and ZEB2. Genes Dev. 2008, 22, 894–907. [Google Scholar] [CrossRef] [Green Version]
- Gregory, P.A.; Bert, A.G.; Paterson, E.L.; Barry, S.C.; Tsykin, A.; Farshid, G.; Vadas, M.A.; Khew-Goodall, Y.; Goodall, G.J. The MiR-200 family and MiR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat. Cell Biol. 2008, 10, 593–601. [Google Scholar] [CrossRef] [PubMed]
- Ru, P.; Steele, R.; Newhall, P.; Phillips, N.J.; Toth, K.; Ray, R.B. MiRNA-29b suppresses prostate cancer metastasis by regulating epithelial-mesenchymal transition signaling. Mol. Cancer Ther. 2012, 11, 1166–1173. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.B.; Bantounas, I.; Lee, D.Y.; Phylactou, L.; Caldwell, M.A.; Uney, J.B. Twist-1 regulates the MiR-199a/214 cluster during development. Nucleic Acids Res. 2009, 37, 123–128. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Han, Q.; Zhu, Y.; Yu, Y.; Wang, J.; Jiang, X. Down-Regulation of MiR-214 contributes to intrahepatic cholangiocarcinoma metastasis by targeting twist: MiR-214 regulates ICC metastasis. FEBS J. 2012, 279, 2393–2398. [Google Scholar] [CrossRef]
- Dong, B.; Li, S.; Zhu, S.; Yi, M.; Luo, S.; Wu, K. MiRNA-Mediated EMT and CSCs in cancer chemoresistance. Exp. Hematol. Oncol. 2021, 10, 12. [Google Scholar] [CrossRef]
- Housman, G.; Byler, S.; Heerboth, S.; Lapinska, K.; Longacre, M.; Snyder, N.; Sarkar, S. Drug resistance in cancer: An overview. Cancers 2014, 6, 1769–1792. [Google Scholar] [CrossRef] [Green Version]
- Arai, K.; Eguchi, T.; Rahman, M.M.; Sakamoto, R.; Masuda, N.; Nakatsura, T.; Calderwood, S.K.; Kozaki, K.; Itoh, M. A novel high-throughput 3D screening system for EMT inhibitors: A pilot screening discovered the EMT inhibitory activity of CDK2 inhibitor SU9516. PLoS ONE 2016, 11, e0162394. [Google Scholar] [CrossRef] [PubMed]
- Germain, A.R.; Carmody, L.C.; Morgan, B.; Fernandez, C.; Forbeck, E.; Lewis, T.A.; Nag, P.P.; Ting, A.; VerPlank, L.; Feng, Y.; et al. Identification of a selective small molecule inhibitor of breast cancer stem cells. Bioorg. Med. Chem. Lett. 2012, 22, 3571–3574. [Google Scholar] [CrossRef]
- Shah, M.Y.; Calin, G.A. MicroRNAs as therapeutic targets in human cancers. Wiley Interdiscip. Rev. RNA 2014, 5, 537–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, Z.; Chen, L.; Wang, Q.; Yin, C.; Hu, J.; Hu, X.; Fei, F.; Tang, J. MicroRNA-186 targets SKP2 to induce P27 Kip1-mediated pituitary tumor cell cycle deregulation and modulate cell proliferation. Korean J. Physiol. Pharmacol. 2019, 23, 171. [Google Scholar] [CrossRef] [Green Version]
- Wu, K.; Hu, Y.; Yan, K.; Qi, Y.; Zhang, C.; Zhu, D.; Liu, D.; Zhao, S. MicroRNA-10b confers cisplatin resistance by activating AKT/MTOR/P70S6K signaling via targeting PPARγ in esophageal cancer. J. Cell. Physiol. 2020, 235, 1247–1258. [Google Scholar] [CrossRef] [PubMed]
- Pakravan, G.; Foroughmand, A.M.; Peymani, M.; Ghaedi, K.; Hashemi, M.-S.; Hajjari, M.; Nasr-Esfahani, M.H. Downregulation of MiR-130a, antagonized doxorubicin-induced cardiotoxicity via increasing the PPAR γ expression in MESCs-derived cardiac cells. Cell Death Dis. 2018, 9, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, S.; Cui, Y.; Dong, X.; Zhang, T.; Xing, H. MicroRNA-130b attenuates dexamethasone-induced increase of lipid accumulation in porcine preadipocytes by suppressing PPAR-γ expression. Oncotarget 2017, 8, 87928–87943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, T.; Gao, F.; Zhang, X.-P. MiR-203 enhances chemosensitivity to 5-Fluorouracil by targeting thymidylate synthase in colorectal cancer. Oncol. Rep. 2015, 33, 607–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, K.; Liang, X.; Cui, D.; Wu, Y.; Shi, W.; Liu, J. MiR-1915 inhibits Bcl-2 to modulate multidrug resistance by increasing drug-sensitivity in human colorectal carcinoma cells. Mol. Carcinog. 2013, 52, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Smith, B.; Bhowmick, N. Role of EMT in metastasis and therapy resistance. J. Clin. Med. 2016, 5, 17. [Google Scholar] [CrossRef] [Green Version]
- Lawrence, M.S.; Stojanov, P.; Polak, P.; Kryukov, G.V.; Cibulskis, K.; Sivachenko, A.; Carter, S.L.; Stewart, C.; Mermel, C.H.; Roberts, S.A.; et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013, 499, 214–218. [Google Scholar] [CrossRef] [PubMed]
- Hodges, T.R.; Ott, M.; Xiu, J.; Gatalica, Z.; Swensen, J.; Zhou, S.; Huse, J.T.; de Groot, J.; Li, S.; Overwijk, W.W.; et al. Mutational burden, immune checkpoint expression, and mismatch repair in glioma: Implications for immune checkpoint immunotherapy. Neuro Oncol. 2017, 19, 1047–1057. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Saci, A.; Szabo, P.M.; Chasalow, S.D.; Castillo-Martin, M.; Domingo-Domenech, J.; Siefker-Radtke, A.; Sharma, P.; Sfakianos, J.P.; Gong, Y.; et al. EMT- and stroma-related gene expression and resistance to PD-1 blockade in urothelial cancer. Nat. Commun. 2018, 9, 3503. [Google Scholar] [CrossRef]
- Yang, B.; Bai, J.; Shi, R.; Shao, X.; Yang, Y.; Jin, Y.; Che, X.; Zhang, Y.; Qu, X.; Liu, Y.; et al. TGFB2 serves as a link between epithelial-mesenchymal transition and tumor mutation burden in gastric cancer. Int. Immunopharmacol. 2020, 84, 106532. [Google Scholar] [CrossRef]
- Ramesh, V.; Brabletz, T.; Ceppi, P. Targeting EMT in cancer with repurposed metabolic inhibitors. Trends Cancer 2020, 6, 942–950. [Google Scholar] [CrossRef]
- Terry, S.; Savagner, P.; Ortiz-Cuaran, S.; Mahjoubi, L.; Saintigny, P.; Thiery, J.-P.; Chouaib, S. New insights into the role of EMT in tumor immune escape. Mol. Oncol. 2017, 11, 824–846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Huang, H.; Remmers, N.; Hollingsworth, M.A. Loss of E-Cadherin and epithelial to mesenchymal transition is not required for cell motility in tissues or for metastasis. Tissue Barriers 2014, 2, e969112. [Google Scholar] [CrossRef] [PubMed] [Green Version]

| Categories | Agents |
|---|---|
| Antimetabolites | Methotrexate |
| 5-Fluorouracil | |
| Capecitabine | |
| Platinum-based agents | Cisplatin |
| Carboplatin | |
| Plant alkaloids | Paclitaxel |
| Docetaxel |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sha, J.; Bai, Y.; Ngo, H.X.; Okui, T.; Kanno, T. Overview of Evidence-Based Chemotherapy for Oral Cancer: Focus on Drug Resistance Related to the Epithelial-Mesenchymal Transition. Biomolecules 2021, 11, 893. https://doi.org/10.3390/biom11060893
Sha J, Bai Y, Ngo HX, Okui T, Kanno T. Overview of Evidence-Based Chemotherapy for Oral Cancer: Focus on Drug Resistance Related to the Epithelial-Mesenchymal Transition. Biomolecules. 2021; 11(6):893. https://doi.org/10.3390/biom11060893
Chicago/Turabian StyleSha, Jingjing, Yunpeng Bai, Huy Xuan Ngo, Tatsuo Okui, and Takahiro Kanno. 2021. "Overview of Evidence-Based Chemotherapy for Oral Cancer: Focus on Drug Resistance Related to the Epithelial-Mesenchymal Transition" Biomolecules 11, no. 6: 893. https://doi.org/10.3390/biom11060893