DLBCL Cells with Acquired Resistance to Venetoclax Are Not Sensitized to BIRD-2 But Can Be Resensitized to Venetoclax through Bcl-XL Inhibition

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Chemicals
2.2. Flow Cytometry: Apoptosis Assay
2.3. Western Blot
2.4. Intracellular Ca2+ Measurement in Intact Cells
2.5. siRNA Transfection of Riva VR Cells
3. Results
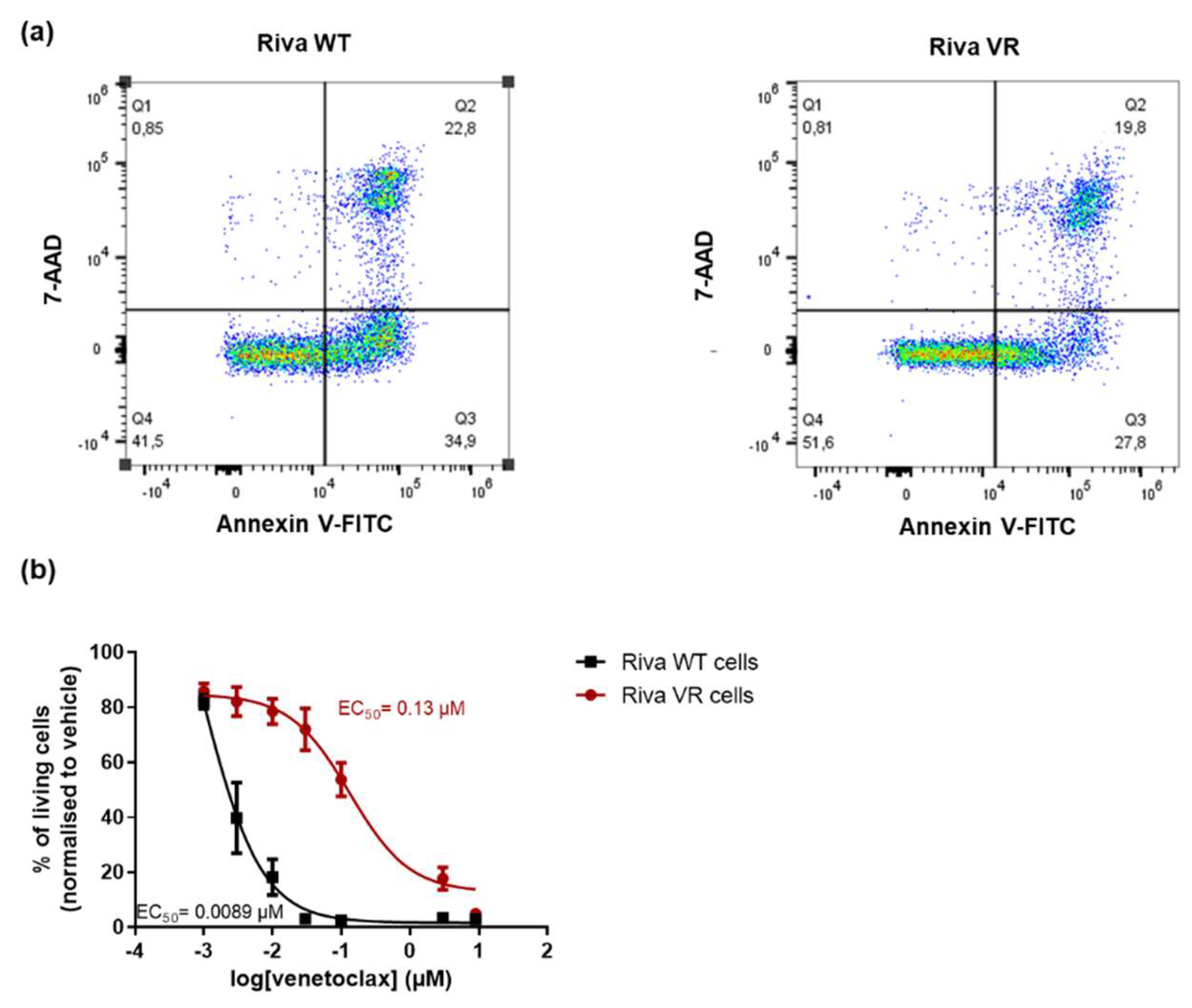
3.1. Characterization of the Venetoclax-Sensitive and -Resistant Riva Cells
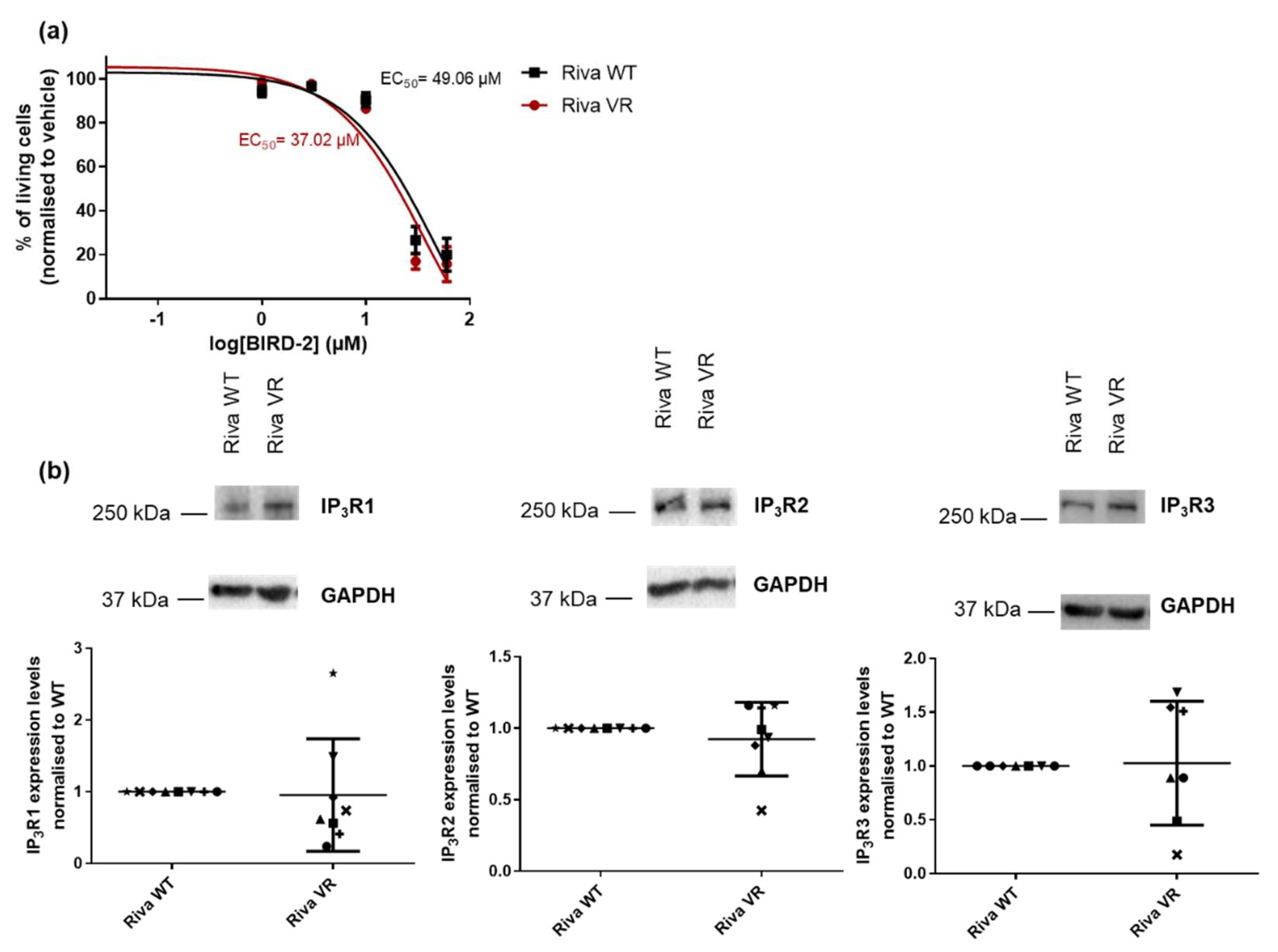
3.2. Acquired Venetoclax Resistance does not Induce Increased Sensitivity Towards BIRD-2
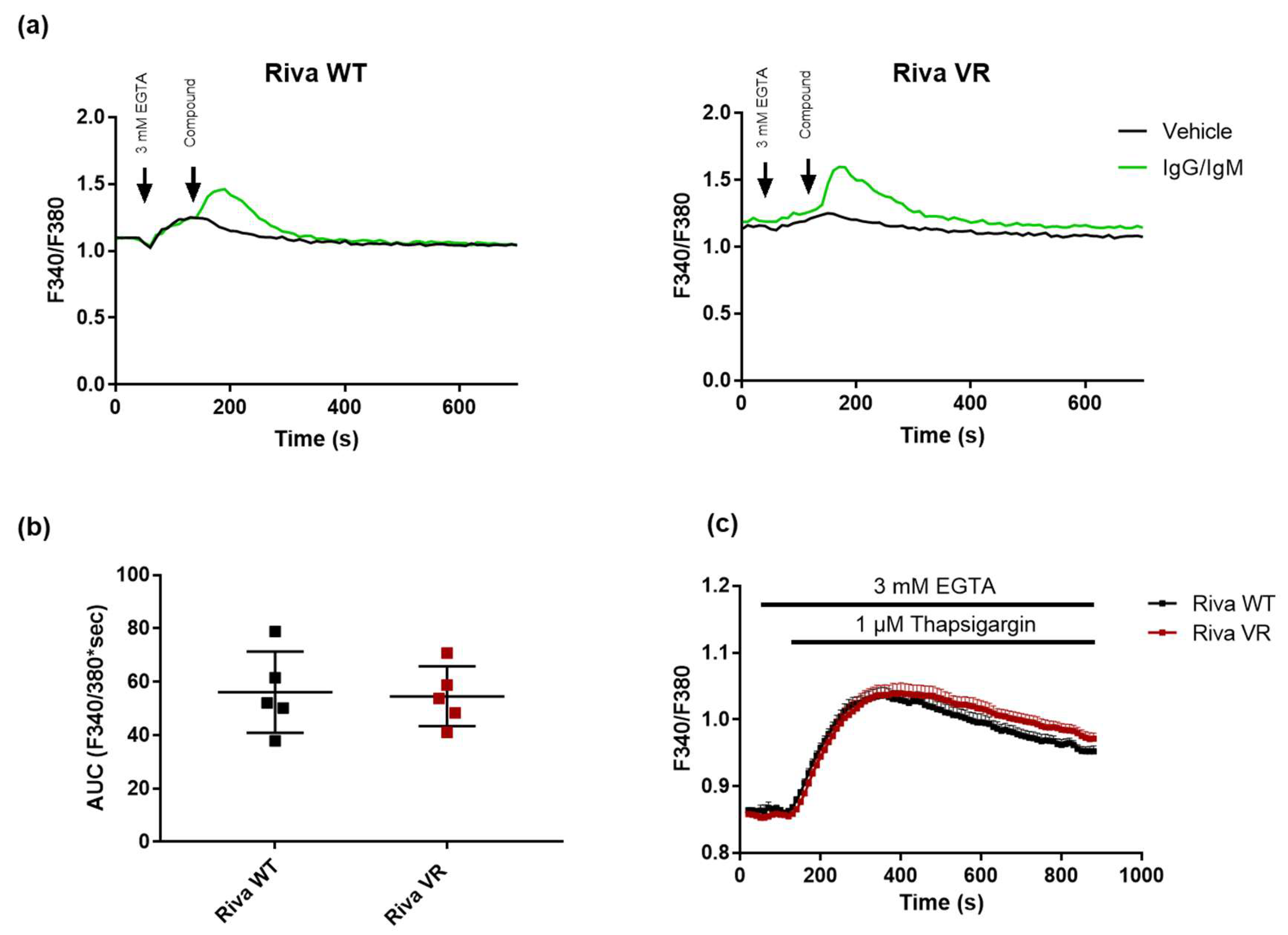
3.3. IgG/IgM-Induced Cytosolic Ca2+ Signaling and ER Store Content Do not Differ between Riva WT and Riva VR
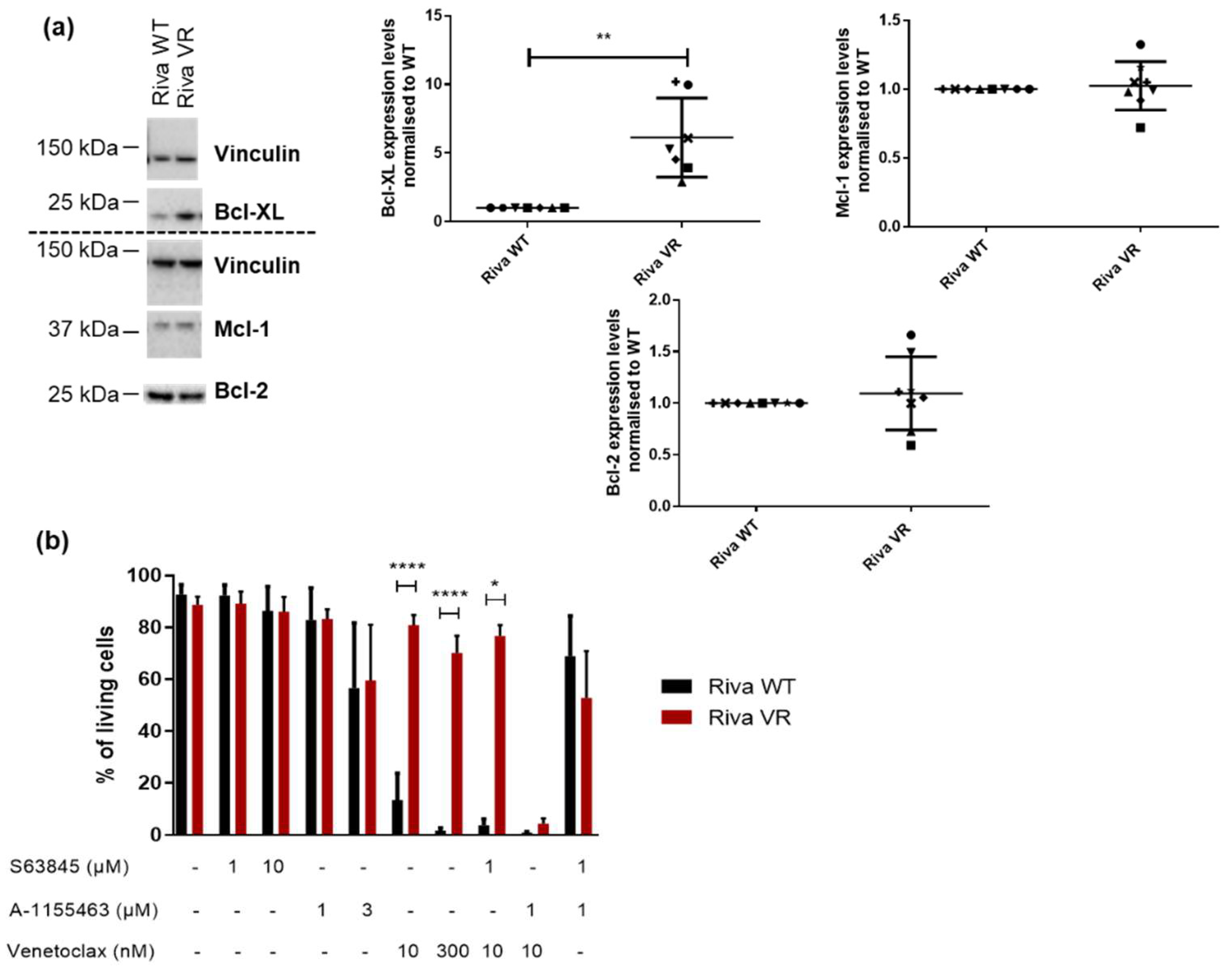
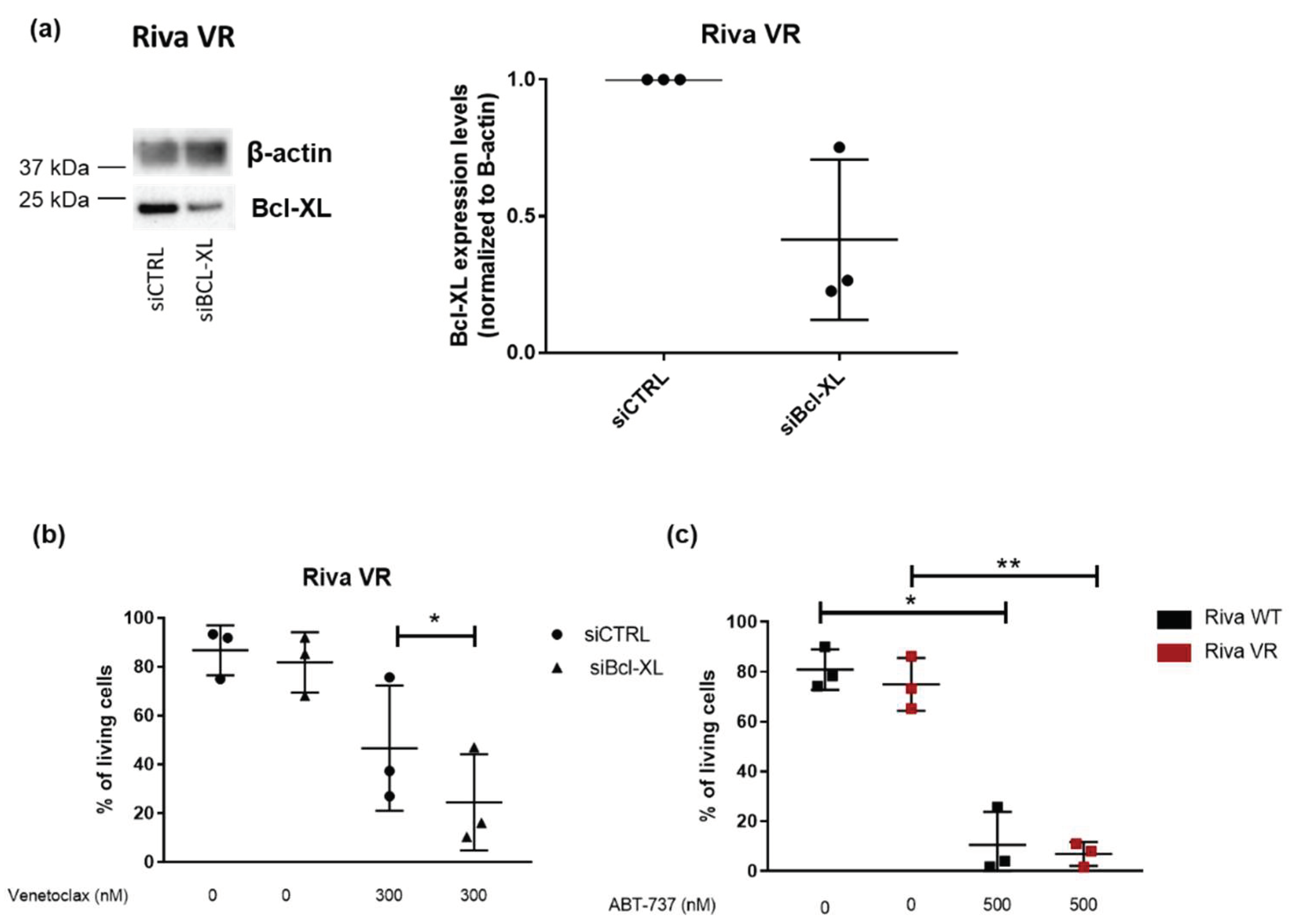
3.4. Acquired Venetoclax Resistance Is Associated with Bcl-XL Upregulation and Bcl-XL Dependence
3.5. Knockdown of Bcl-XL Resensitizes Riva VR Cells to Venetoclax
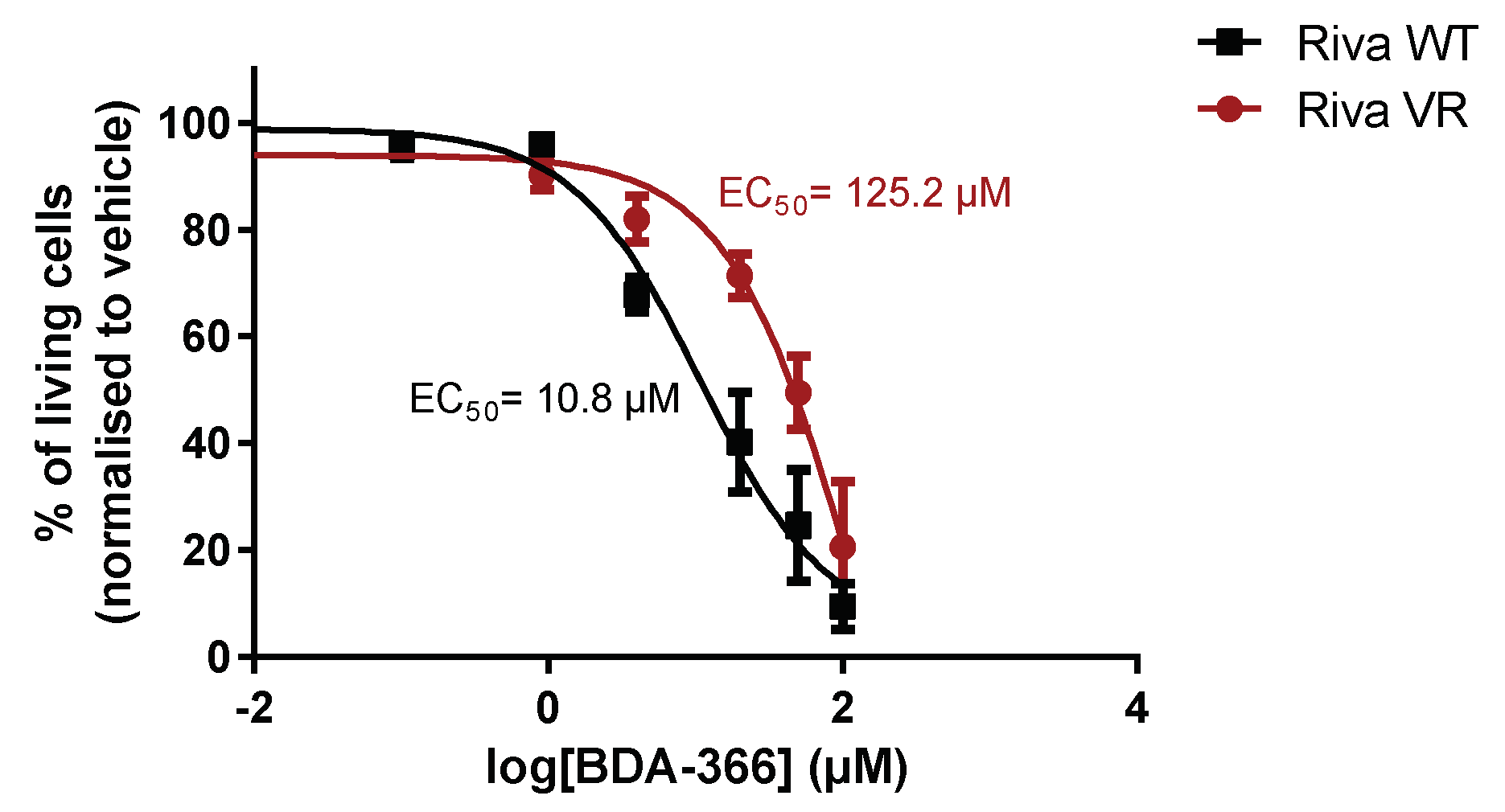
3.6. Acquired Venetoclax Resistance in Riva Cells Generates Cross-Resistance for BDA-366
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Delbridge, A.R.; Strasser, A. The BCL-2 protein family, BH3-mimetics and cancer therapy. Cell Death Differ. 2015, 22, 1071–1080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalkavan, H.; Green, D.R. MOMP, cell suicide as a BCL-2 family business. Cell Death Differ. 2018, 25, 46–55. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Letai, A.; Sarosiek, K. Regulation of apoptosis in health and disease: The balancing act of BCL-2 family proteins. Nat. Rev. Mol. Cell Biol. 2019, 20, 175–193. [Google Scholar] [CrossRef] [PubMed]
- Perini, G.F.; Ribeiro, G.N.; Pinto Neto, J.V.; Campos, L.T.; Hamerschlak, N. BCL-2 as therapeutic target for hematological malignancies. J. Hematol. Oncol. 2018, 11, 65. [Google Scholar] [CrossRef]
- Raffo, A.J.; Perlman, H.; Chen, M.W.; Day, M.L.; Streitman, J.S.; Buttyan, R. Overexpression of bcl-2 protects prostate cancer cells from apoptosis in vitro and confers resistance to androgen depletion in vivo. Cancer Res. 1995, 55, 4438–4445. [Google Scholar]
- Adams, J.M.; Cory, S. The BCL-2 arbiters of apoptosis and their growing role as cancer targets. Cell Death Differ 2018, 25, 27–36. [Google Scholar] [CrossRef] [Green Version]
- Montero, J.; Letai, A. Why do BCL-2 inhibitors work and where should we use them in the clinic? Cell Death Differ. 2018, 25, 56–64. [Google Scholar] [CrossRef]
- Roberts, A.W.; Davids, M.S.; Pagel, J.M.; Kahl, B.S.; Puvvada, S.D.; Gerecitano, J.F.; Kipps, T.J.; Anderson, M.A.; Brown, J.R.; Gressick, L.; et al. Targeting BCL2 with Venetoclax in Relapsed Chronic Lymphocytic Leukemia. N. Engl. J. Med. 2016, 374, 311–322. [Google Scholar] [CrossRef]
- Green, D.R. A BH3 Mimetic for Killing Cancer Cells. Cell 2016, 165, 1560. [Google Scholar] [CrossRef]
- Souers, A.J.; Leverson, J.D.; Boghaert, E.R.; Ackler, S.L.; Catron, N.D.; Chen, J.; Dayton, B.D.; Ding, H.; Enschede, S.H.; Fairbrother, W.J.; et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med. 2013, 19, 202–208. [Google Scholar] [CrossRef]
- Juarez-Salcedo, L.M.; Desai, V.; Dalia, S. Venetoclax: Evidence to date and clinical potential. Drugs Context 2019, 8, 212574. [Google Scholar] [CrossRef] [PubMed]
- Egle, A.; Harris, A.W.; Bouillet, P.; Cory, S. Bim is a suppressor of Myc-induced mouse B cell leukemia. Proc. Natl. Acad. Sci. USA 2004, 101, 6164–6169. [Google Scholar] [CrossRef] [Green Version]
- Certo, M.; Del Gaizo Moore, V.; Nishino, M.; Wei, G.; Korsmeyer, S.; Armstrong, S.A.; Letai, A. Mitochondria primed by death signals determine cellular addiction to antiapoptotic BCL-2 family members. Cancer Cell 2006, 9, 351–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jelinek, T.; Popkova, T.; Duras, J.; Mihalyova, J.; Kascak, M.; Benkova, K.; Plonkova, H.; Cerna, L.; Koristek, Z.; Simicek, M.; et al. Venetoclax plus bortezomib and dexamethasone in heavily pretreated end-stage myeloma patients without t(11;14): A real-world cohort. Hematol. Oncol. 2020. [Google Scholar] [CrossRef]
- Choi, J.H.; Bogenberger, J.M.; Tibes, R. Targeting Apoptosis in Acute Myeloid Leukemia: Current Status and Future Directions of BCL-2 Inhibition with Venetoclax and Beyond. Target Oncol. 2020, 15, 147–162. [Google Scholar] [CrossRef] [PubMed]
- Zhong, F.; Davis, M.C.; McColl, K.S.; Distelhorst, C.W. Bcl-2 differentially regulates Ca2+ signals according to the strength of T cell receptor activation. J. Cell Biol. 2006, 172, 127–137. [Google Scholar] [CrossRef]
- Monaco, G.; Beckers, M.; Ivanova, H.; Missiaen, L.; Parys, J.B.; De Smedt, H.; Bultynck, G. Profiling of the Bcl-2/Bcl-XL-binding sites on type 1 IP3 receptor. Biochem. Biophys. Res. Commun. 2012, 428, 31–35. [Google Scholar] [CrossRef] [PubMed]
- Bootman, M.D.; Bultynck, G. Fundamentals of Cellular Calcium Signaling: A Primer. Cold Spring Harb. Perspect. Biol. 2020, 12, a038802. [Google Scholar] [CrossRef] [Green Version]
- Vervliet, T.; Parys, J.B.; Bultynck, G. Bcl-2 proteins and calcium signaling: Complexity beneath the surface. Oncogene 2016, 35, 5079–5092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivanova, H.; Vervliet, T.; Monaco, G.; Terry, L.E.; Rosa, N.; Baker, M.R.; Parys, J.B.; Serysheva, I.I.; Yule, D.I.; Bultynck, G. Bcl-2-Protein Family as Modulators of IP3 Receptors and Other Organellar Ca2+ Channels. Cold Spring Harb. Perspect. Biol. 2020, 12, a035089. [Google Scholar] [CrossRef]
- Monaco, G.; Decrock, E.; Akl, H.; Ponsaerts, R.; Vervliet, T.; Luyten, T.; De Maeyer, M.; Missiaen, L.; Distelhorst, C.W.; De Smedt, H.; et al. Selective regulation of IP3-receptor-mediated Ca2+ signaling and apoptosis by the BH4 domain of Bcl-2 versus Bcl-Xl. Cell Death Differ. 2012, 19, 295–309. [Google Scholar] [CrossRef] [PubMed]
- Ivanova, H.; Wagner, L.E., 2nd; Tanimura, A.; Vandermarliere, E.; Luyten, T.; Welkenhuyzen, K.; Alzayady, K.J.; Wang, L.; Hamada, K.; Mikoshiba, K.; et al. Bcl-2 and IP3 compete for the ligand-binding domain of IP3Rs modulating Ca2+ signaling output. Cell Mol. Life Sci. 2019, 76, 3843–3859. [Google Scholar] [CrossRef] [PubMed]
- Rong, Y.P.; Bultynck, G.; Aromolaran, A.S.; Zhong, F.; Parys, J.B.; De Smedt, H.; Mignery, G.A.; Roderick, H.L.; Bootman, M.D.; Distelhorst, C.W. The BH4 domain of Bcl-2 inhibits ER calcium release and apoptosis by binding the regulatory and coupling domain of the IP3 receptor. Proc. Natl. Acad. Sci. USA 2009, 106, 14397–14402. [Google Scholar] [CrossRef] [Green Version]
- Distelhorst, C.W. Targeting Bcl-2-IP3 receptor interaction to treat cancer: A novel approach inspired by nearly a century treating cancer with adrenal corticosteroid hormones. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 1795–1804. [Google Scholar] [CrossRef] [PubMed]
- Kerkhofs, M.; Vervloessem, T.; Bittremieux, M.; Bultynck, G. Recent advances in uncovering the mechanisms contributing to BIRD-2-induced cell death in B-cell cancer cells. Cell Death Dis. 2019, 10, 42. [Google Scholar] [CrossRef] [Green Version]
- Distelhorst, C.W.; Bootman, M.D. Creating a New Cancer Therapeutic Agent by Targeting the Interaction between Bcl-2 and IP3 Receptors. Cold Spring Harb. Perspect. Biol. 2019, 11, a035196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, F.; Harr, M.W.; Bultynck, G.; Monaco, G.; Parys, J.B.; De Smedt, H.; Rong, Y.P.; Molitoris, J.K.; Lam, M.; Ryder, C.; et al. Induction of Ca2+-driven apoptosis in chronic lymphocytic leukemia cells by peptide-mediated disruption of Bcl-2-IP3 receptor interaction. Blood 2011, 117, 2924–2934. [Google Scholar] [CrossRef] [Green Version]
- Akl, H.; Monaco, G.; La Rovere, R.; Welkenhuyzen, K.; Kiviluoto, S.; Vervliet, T.; Molgo, J.; Distelhorst, C.W.; Missiaen, L.; Mikoshiba, K.; et al. IP3R2 levels dictate the apoptotic sensitivity of diffuse large B-cell lymphoma cells to an IP3R-derived peptide targeting the BH4 domain of Bcl-2. Cell Death Dis. 2013, 4, e632. [Google Scholar] [CrossRef]
- Bittremieux, M.; La Rovere, R.M.; Akl, H.; Martines, C.; Welkenhuyzen, K.; Dubron, K.; Baes, M.; Janssens, A.; Vandenberghe, P.; Laurenti, L.; et al. Constitutive IP3 signaling underlies the sensitivity of B-cell cancers to the Bcl-2/IP3 receptor disruptor BIRD-2. Cell Death Differ. 2019, 26, 531–547. [Google Scholar] [CrossRef]
- Vervloessem, T.; Akl, H.; Tousseyn, T.; De Smedt, H.; Parys, J.B.; Bultynck, G. Reciprocal sensitivity of diffuse large B-cell lymphoma cells to Bcl-2 inhibitors BIRD-2 versus venetoclax. Oncotarget 2017, 8, 111656–111671. [Google Scholar] [CrossRef]
- Deng, J.; Park, D.; Wang, M.; Nooka, A.; Deng, Q.; Matulis, S.; Kaufman, J.; Lonial, S.; Boise, L.H.; Galipeau, J.; et al. BCL2-BH4 antagonist BDA-366 suppresses human myeloma growth. Oncotarget 2016, 7, 27753–27763. [Google Scholar] [CrossRef] [PubMed]
- Blombery, P.; Birkinshaw, R.W.; Nguyen, T.; Gong, J.N.; Thompson, E.R.; Xu, Z.; Westerman, D.A.; Czabotar, P.E.; Dickinson, M.; Huang, D.C.S.; et al. Characterization of a novel venetoclax resistance mutation (BCL2 Phe104Ile) observed in follicular lymphoma. Br. J. Haematol. 2019, 186, e188–e191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guieze, R.; Liu, V.M.; Rosebrock, D.; Jourdain, A.A.; Hernandez-Sanchez, M.; Martinez Zurita, A.; Sun, J.; Ten Hacken, E.; Baranowski, K.; Thompson, P.A.; et al. Mitochondrial Reprogramming Underlies Resistance to BCL-2 Inhibition in Lymphoid Malignancies. Cancer Cell 2019, 36, 369–384. [Google Scholar] [CrossRef] [PubMed]
- Villalobos-Ortiz, M.; Ryan, J.; Mashaka, T.N.; Opferman, J.T.; Letai, A. BH3 profiling discriminates on-target small molecule BH3 mimetics from putative mimetics. Cell Death Differ. 2020, 27, 999–1007. [Google Scholar] [CrossRef]
- Oltersdorf, T.; Elmore, S.W.; Shoemaker, A.R.; Armstrong, R.C.; Augeri, D.J.; Belli, B.A.; Bruncko, M.; Deckwerth, T.L.; Dinges, J.; Hajduk, P.J.; et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumors. Nature 2005, 435, 677–681. [Google Scholar] [CrossRef]
- Vervloessem, T.; La Rovere, R.; Bultynck, G. Antagonizing Bcl-2’s BH4 domain in cancer. Aging (Albany NY) 2015, 7, 748–749. [Google Scholar] [CrossRef]
- Choudhary, G.S.; Al-Harbi, S.; Mazumder, S.; Hill, B.T.; Smith, M.R.; Bodo, J.; Hsi, E.D.; Almasan, A. MCL-1 and BCL-xL-dependent resistance to the BCL-2 inhibitor ABT-199 can be overcome by preventing PI3K/AKT/mTOR activation in lymphoid malignancies. Cell Death Dis 2015, 6, e1593. [Google Scholar] [CrossRef]
- Chen, L.; Willis, S.N.; Wei, A.; Smith, B.J.; Fletcher, J.I.; Hinds, M.G.; Colman, P.M.; Day, C.L.; Adams, J.M.; Huang, D.C. Differential targeting of prosurvival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Mol. Cell 2005, 17, 393–403. [Google Scholar] [CrossRef]
- Eichhorn, J.M.; Alford, S.E.; Sakurikar, N.; Chambers, T.C. Molecular analysis of functional redundancy among anti-apoptotic Bcl-2 proteins and its role in cancer cell survival. Exp. Cell Res. 2014, 322, 415–424. [Google Scholar] [CrossRef] [Green Version]
- Rooswinkel, R.W.; van de Kooij, B.; de Vries, E.; Paauwe, M.; Braster, R.; Verheij, M.; Borst, J. Antiapoptotic potency of Bcl-2 proteins primarily relies on their stability, not binding selectivity. Blood 2014, 123, 2806–2815. [Google Scholar] [CrossRef] [Green Version]
- Al-Harbi, S.; Choudhary, G.S.; Ebron, J.S.; Hill, B.T.; Vivekanathan, N.; Ting, A.H.; Radivoyevitch, T.; Smith, M.R.; Shukla, G.C.; Almasan, A. miR-377-dependent BCL-xL regulation drives chemotherapeutic resistance in B-cell lymphoid malignancies. Mol. Cancer 2015, 14, 185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tausch, E.; Close, W.; Dolnik, A.; Bloehdorn, J.; Chyla, B.; Bullinger, L.; Dohner, H.; Mertens, D.; Stilgenbauer, S. Venetoclax resistance and acquired BCL2 mutations in chronic lymphocytic leukemia. Haematologica 2019, 104, e434–e437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharon, D.; Cathelin, S.; Mirali, S.; Di Trani, J.M.; Yanofsky, D.J.; Keon, K.A.; Rubinstein, J.L.; Schimmer, A.D.; Ketela, T.; Chan, S.M. Inhibition of mitochondrial translation overcomes venetoclax resistance in AML through activation of the integrated stress response. Sci. Transl. Med. 2019, 11, eaax2863. [Google Scholar] [CrossRef] [PubMed]
- Leverson, J.D.; Cojocari, D. Hematologic Tumor Cell Resistance to the BCL-2 Inhibitor Venetoclax: A Product of Its Microenvironment? Front Oncol. 2018, 8, 458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blombery, P.; Thompson, E.R.; Nguyen, T.; Birkinshaw, R.W.; Gong, J.N.; Chen, X.; McBean, M.; Thijssen, R.; Conway, T.; Anderson, M.A.; et al. Multiple BCL2 mutations cooccurring with Gly101Val emerge in chronic lymphocytic leukemia progression on venetoclax. Blood 2020, 135, 773–777. [Google Scholar] [CrossRef] [PubMed]
- Bodo, J.; Zhao, X.; Durkin, L.; Souers, A.J.; Phillips, D.C.; Smith, M.R.; Hsi, E.D. Acquired resistance to venetoclax (ABT-199) in t(14;18) positive lymphoma cells. Oncotarget 2016, 7, 70000–70010. [Google Scholar] [CrossRef] [Green Version]
- Oppermann, S.; Ylanko, J.; Shi, Y.; Hariharan, S.; Oakes, C.C.; Brauer, P.M.; Zuniga-Pflucker, J.C.; Leber, B.; Spaner, D.E.; Andrews, D.W. High-content screening identifies kinase inhibitors that overcome venetoclax resistance in activated CLL cells. Blood 2016, 128, 934–947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pham, L.V.; Huang, S.; Zhang, H.; Zhang, J.; Bell, T.; Zhou, S.; Pogue, E.; Ding, Z.; Lam, L.; Westin, J.; et al. Strategic Therapeutic Targeting to Overcome Venetoclax Resistance in Aggressive B-cell Lymphomas. Clin. Cancer Res. 2018, 24, 3967–3980. [Google Scholar] [CrossRef] [PubMed] [Green Version]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kerkhofs, M.; Vervloessem, T.; Stopa, K.B.; Smith, V.M.; Vogler, M.; Bultynck, G. DLBCL Cells with Acquired Resistance to Venetoclax Are Not Sensitized to BIRD-2 But Can Be Resensitized to Venetoclax through Bcl-XL Inhibition. Biomolecules 2020, 10, 1081. https://doi.org/10.3390/biom10071081
Kerkhofs M, Vervloessem T, Stopa KB, Smith VM, Vogler M, Bultynck G. DLBCL Cells with Acquired Resistance to Venetoclax Are Not Sensitized to BIRD-2 But Can Be Resensitized to Venetoclax through Bcl-XL Inhibition. Biomolecules. 2020; 10(7):1081. https://doi.org/10.3390/biom10071081
Chicago/Turabian StyleKerkhofs, Martijn, Tamara Vervloessem, Kinga B. Stopa, Victoria M. Smith, Meike Vogler, and Geert Bultynck. 2020. "DLBCL Cells with Acquired Resistance to Venetoclax Are Not Sensitized to BIRD-2 But Can Be Resensitized to Venetoclax through Bcl-XL Inhibition" Biomolecules 10, no. 7: 1081. https://doi.org/10.3390/biom10071081