Mitochondrial Functions, Energy Metabolism and Protein Glycosylation are Interconnected Processes Mediating Resistance to Bortezomib in Multiple Myeloma Cells

 , ,
, ,  , ,
, ,  , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. 20S Proteasome Activity Assay
2.3. Sample Preparation and Chromatographic Conditions of the HPLC Analysis of Metabolites
2.4. ROS Evaluation
2.5. Measurement of Mitochondrial Membrane Potential
2.6. Mitochondrial Mass Evaluation
2.7. RT-qPCR
2.8. ICAM1 Expression
2.9. Transmission Electron Microscopy
2.10. Statistical Analysis
3. Results
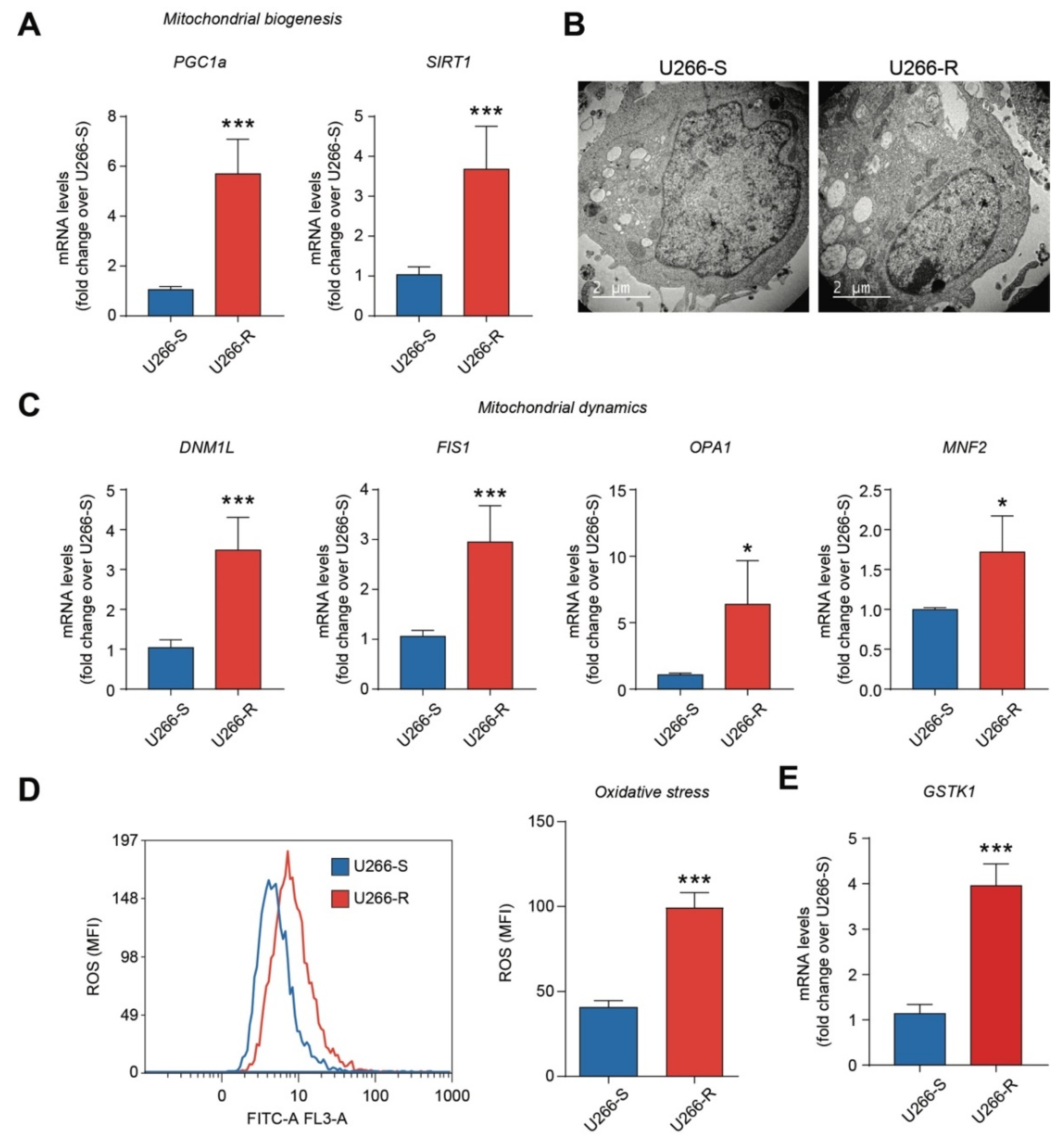
3.1. Mitochondrial Biogenesis, Mitochondrial Dynamics and the Antioxidant System are Increased in U266-R
3.2. U266-R Cells Exhibit Increased Concentrations of GTP, UTP and CTP
3.3. Redox State of Nicotinic Coenzymes in Bortezomib Sensitive and Resistant Cells
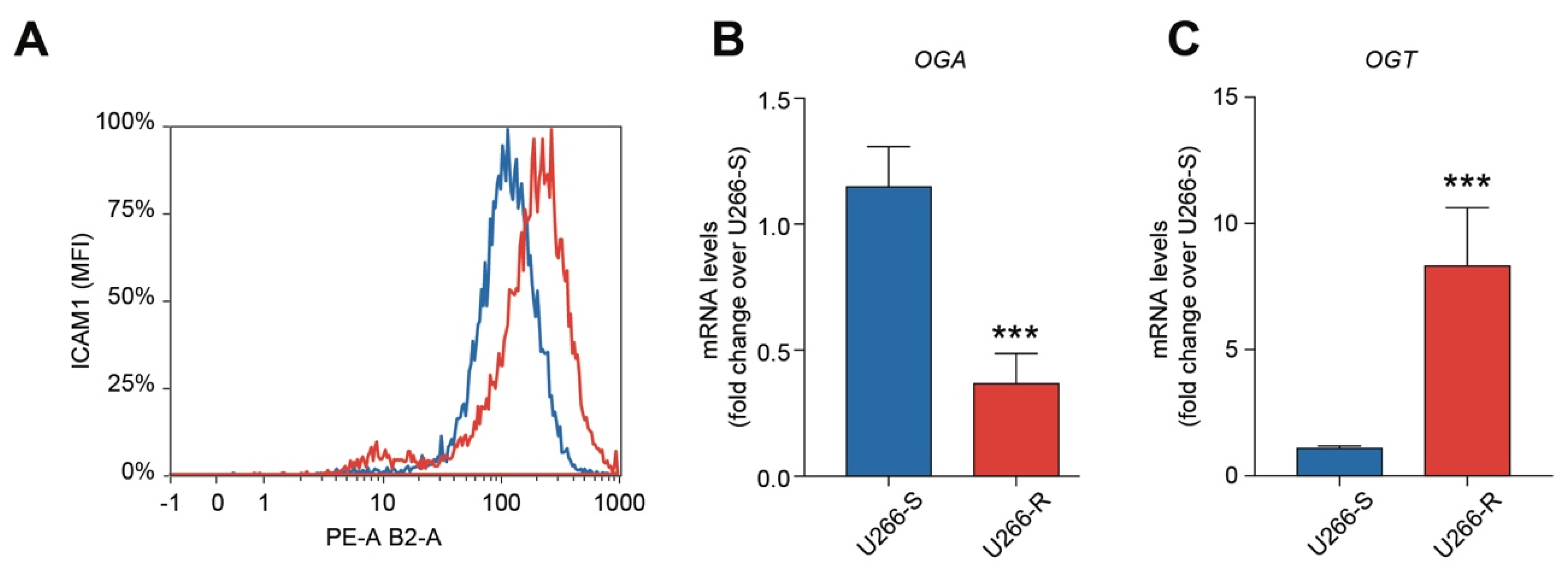
3.4. Bortezomib Resistance is Associated with Increased Glycosylation
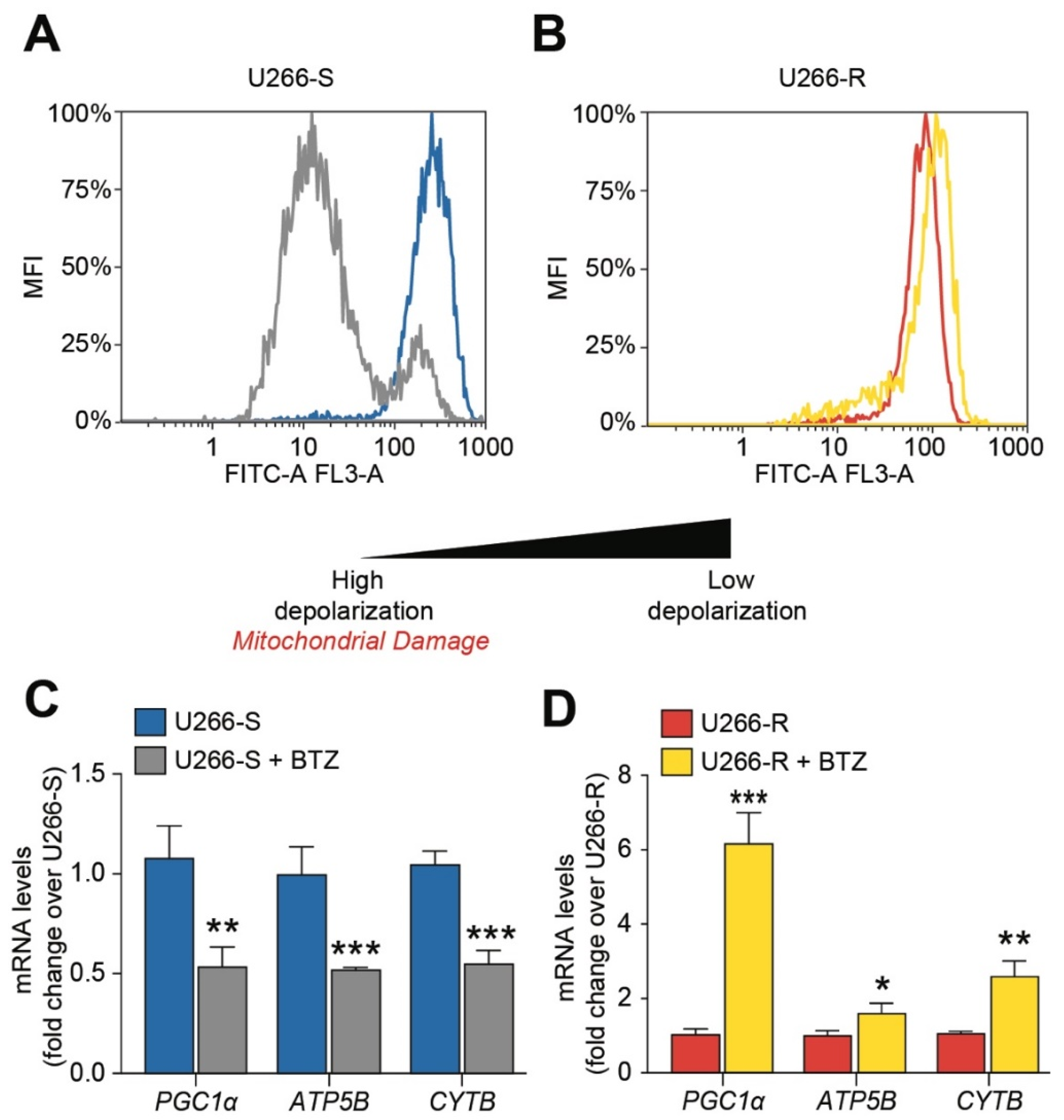
3.5. Mitochondrial Fitness after BTZ Challenge Contributes to Drug Resistance
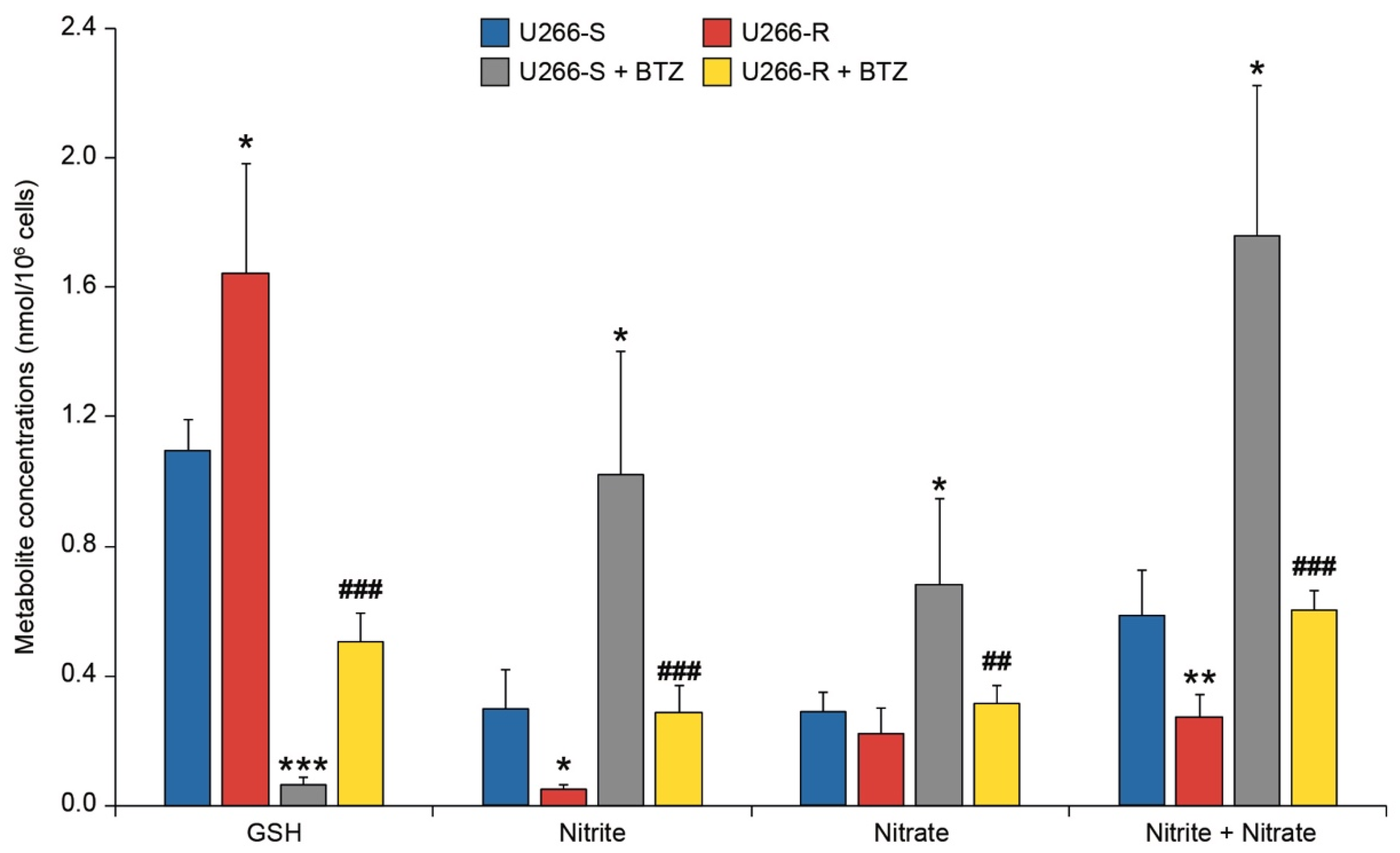
3.6. Antioxidant Status and NO Metabolism in U266-S and U266-R after Exposure to BTZ
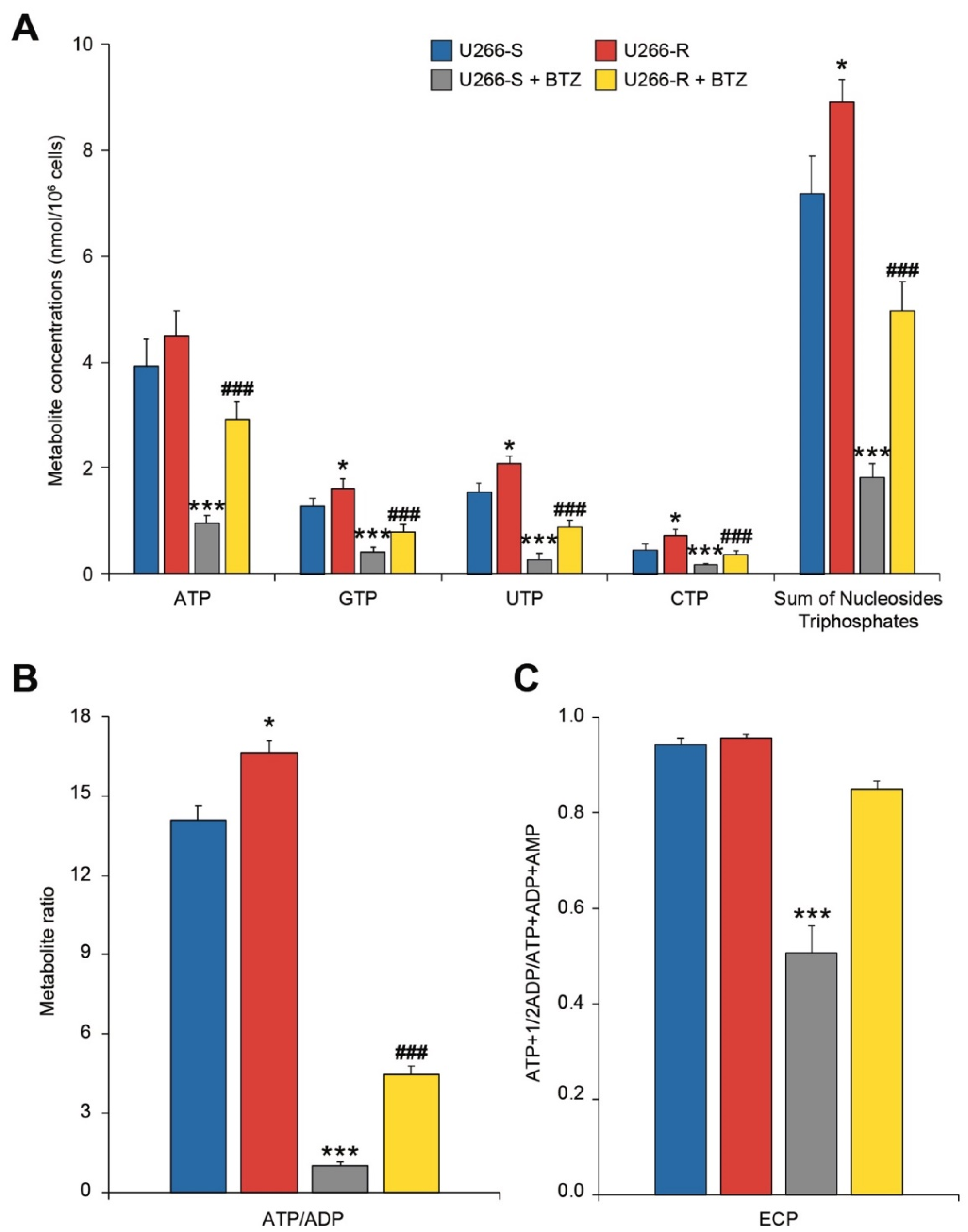
3.7. Cell Energy State in U266-S and U266-R after Exposure to BTZ
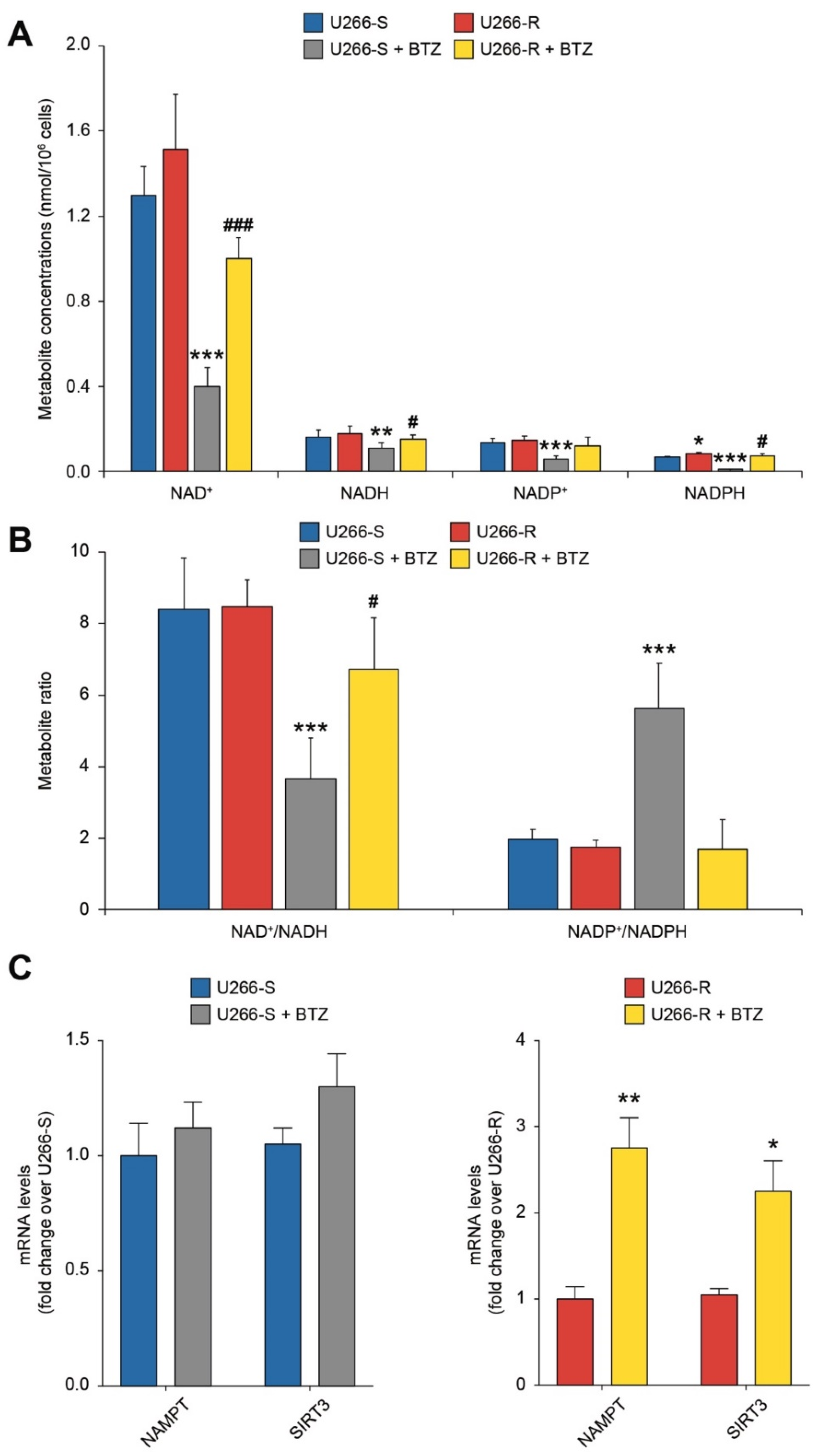
3.8. Redox State of Nicotinic Coenzymes in U266-S and U266-R after Exposure to BTZ
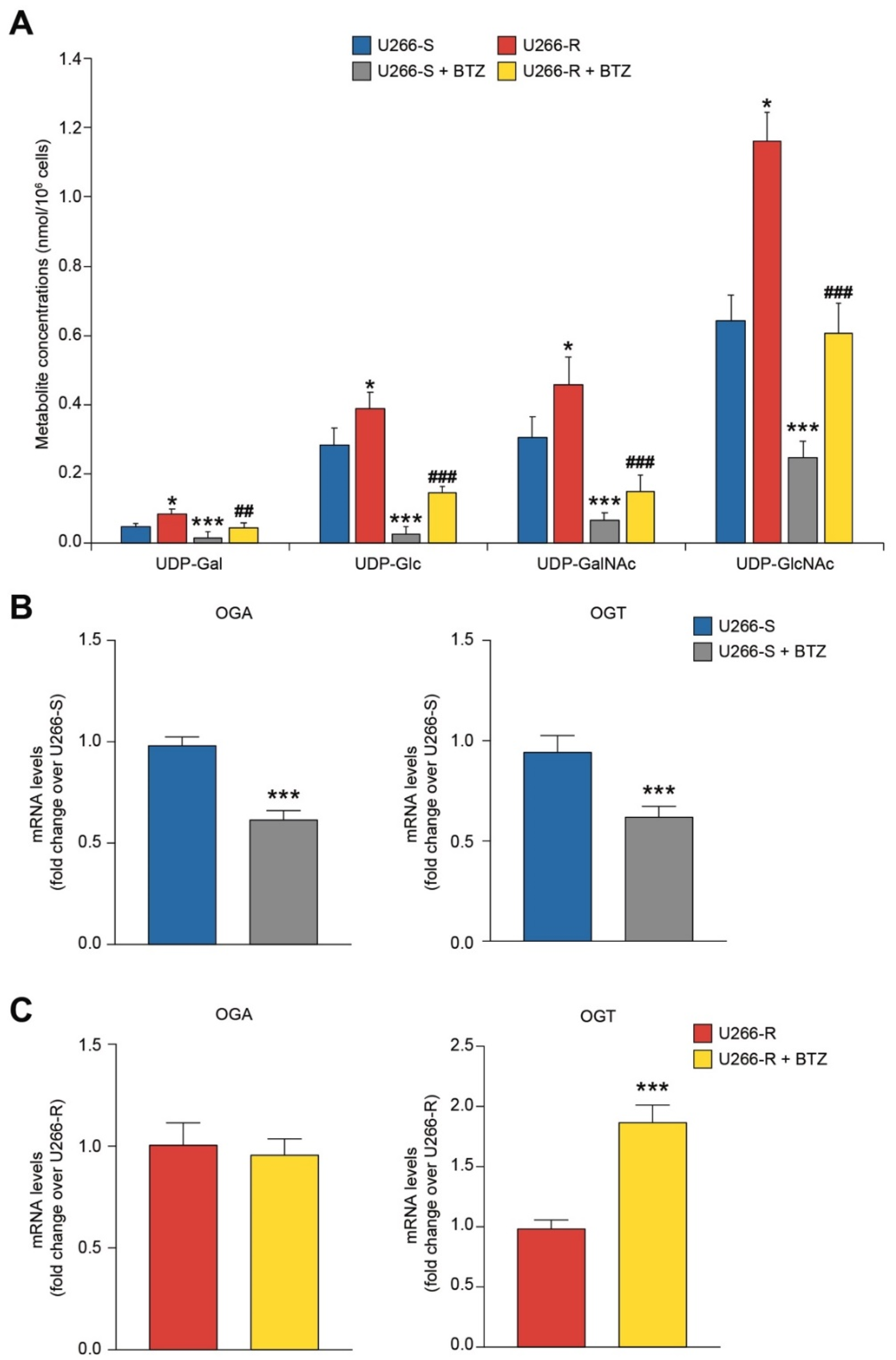
3.9. Glycosylated UDP-Derivatives in U266-S and U266-R after Exposure to BTZ
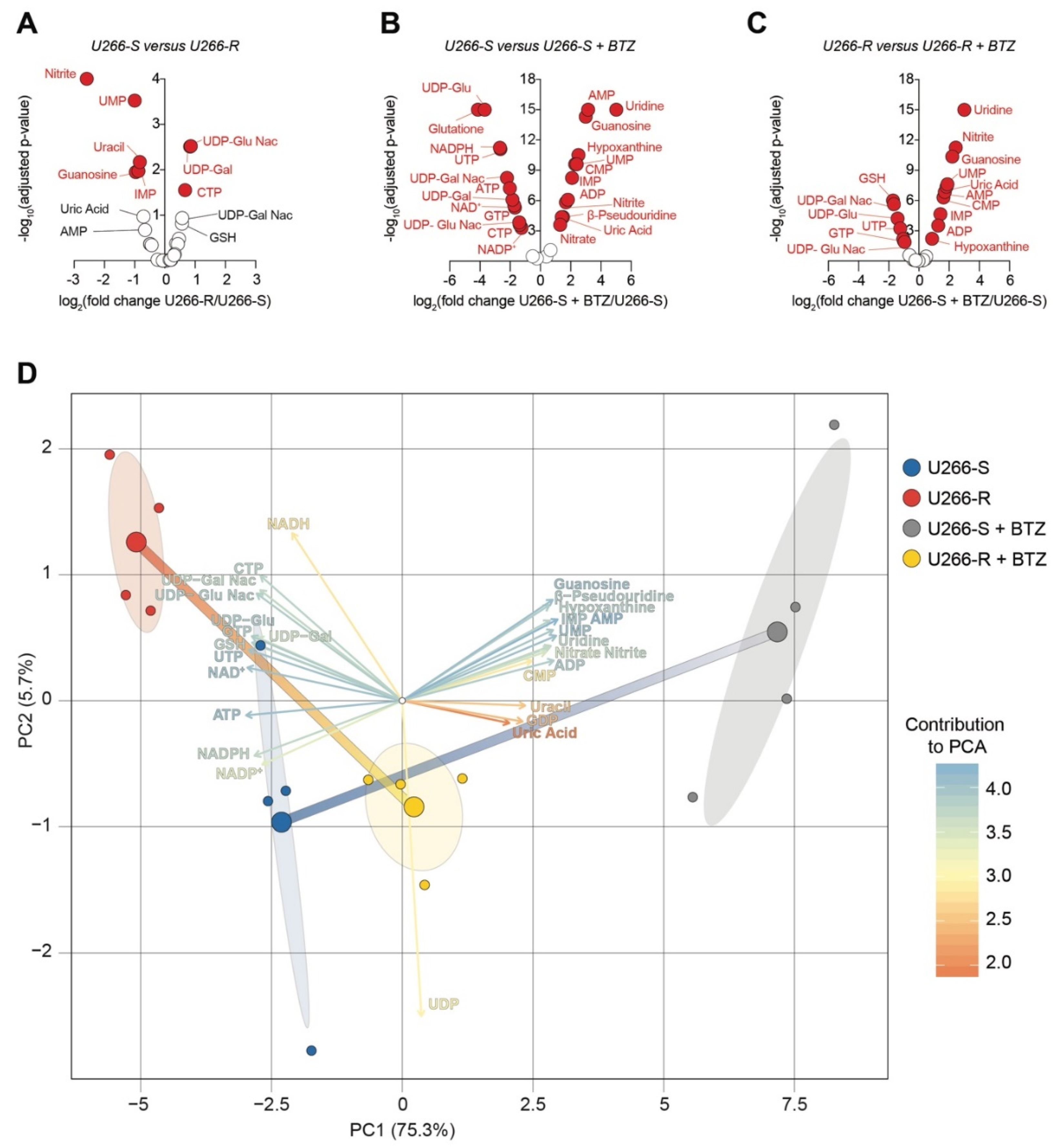
3.10. Purine and Pyrimidine Compounds in U266-S and U266-R before and after Exposure to BTZ
3.11. Principal Component Analysis of BTZ’s Effects on Metabolite Levels in U266-S Versus U266-R Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Glickman, M.H.; Maytal, V. Regulating the 26s proteasome. Curr. Top. Microbiol. Immunol. 2002, 268, 43–72. [Google Scholar] [CrossRef] [PubMed]
- Bergaggio, E.; Piva, R. Wild-type idh enzymes as actionable targets for cancer therapy. Cancers (Basel) 2019, 11, 563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tibullo, D.; Barbagallo, I.; Giallongo, C.; Vanella, L.; Conticello, C.; Romano, A.; Saccone, S.; Godos, J.; Di Raimondo, F.; Li Volti, G. Heme oxygenase-1 nuclear translocation regulates bortezomibinduced cytotoxicity and mediates genomic instability in myeloma cells. Oncotarget 2016, 7, 28868–28880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landowski, T.H.; Megli, C.J.; Nullmeyer, K.D.; Lynch, R.M.; Dorr, R.T. Mitochondrial-mediated disregulation of Ca2+ is a critical determinant of velcade (ps-341/bortezomib) cytotoxicity in myeloma cell lines. Cancer Res. 2005, 65, 3828–3836. [Google Scholar] [CrossRef] [Green Version]
- Gu, H.; Chen, X.; Gao, G.; Dong, H. Caspase-2 functions upstream of mitochondria in endoplasmic reticulum stress-induced apoptosis by bortezomib in human myeloma cells. Mol. Cancer 2008, 7, 2298–2307. [Google Scholar] [CrossRef] [Green Version]
- Obeng, E.A.; Carlson, L.M.; Gutman, D.M.; Harrington, W.J., Jr.; Lee, K.P.; Boise, L.H. Proteasome inhibitors induce a terminal unfolded protein response in multiple myeloma cells. Blood 2006, 107, 4907–4916. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.M.; Kim, I.Y.; Seo, M.J.; Kwon, M.R.; Choi, K.S. Nutlin-3 enhances the bortezomib sensitivity of p53-defective cancer cells by inducing paraptosis. Exp. Mol. Med. 2017, 49, e365. [Google Scholar] [CrossRef] [Green Version]
- Maiso, P.; Huynh, D.; Moschetta, M.; Sacco, A.; Aljawai, Y.; Mishima, Y.; Asara, J.M.; Roccaro, A.M.; Kimmelman, A.C.; Ghobrial, I.M. Metabolic signature identifies novel targets for drug resistance in multiple myeloma. Cancer Res. 2015, 75, 2071–2082. [Google Scholar] [CrossRef] [Green Version]
- Niewerth, D.; Kaspers, G.J.; Jansen, G.; van Meerloo, J.; Zweegman, S.; Jenkins, G.; Whitlock, J.A.; Hunger, S.P.; Lu, X.; Alonzo, T.A.; et al. Proteasome subunit expression analysis and chemosensitivity in relapsed paediatric acute leukaemia patients receiving bortezomib-containing chemotherapy. J. Hematol. Oncol. 2016, 9, 82. [Google Scholar] [CrossRef] [Green Version]
- Franke, N.E.; Niewerth, D.; Assaraf, Y.G.; van Meerloo, J.; Vojtekova, K.; van Zantwijk, C.H.; Zweegman, S.; Chan, E.T.; Kirk, C.J.; Geerke, D.P.; et al. Impaired bortezomib binding to mutant beta5 subunit of the proteasome is the underlying basis for bortezomib resistance in leukemia cells. Leukemia 2012, 26, 757–768. [Google Scholar] [CrossRef] [Green Version]
- Ruckrich, T.; Kraus, M.; Gogel, J.; Beck, A.; Ovaa, H.; Verdoes, M.; Overkleeft, H.S.; Kalbacher, H.; Driessen, C. Characterization of the ubiquitin-proteasome system in bortezomib-adapted cells. Leukemia 2009, 23, 1098–1105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gogvadze, V.; Orrenius, S.; Zhivotovsky, B. Mitochondria in cancer cells: What is so special about them? Trends Cell Biol. 2008, 18, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Vyas, S.; Zaganjor, E.; Haigis, M.C. Mitochondria and cancer. Cell 2016, 166, 555–566. [Google Scholar] [CrossRef] [PubMed]
- Zaal, E.A.; Wu, W.; Jansen, G.; Zweegman, S.; Cloos, J.; Berkers, C.R. Bortezomib resistance in multiple myeloma is associated with increased serine synthesis. Cancer Metab. 2017, 5, 7. [Google Scholar] [CrossRef] [PubMed]
- Thompson, R.M.; Dytfeld, D.; Reyes, L.; Robinson, R.M.; Smith, B.; Manevich, Y.; Jakubowiak, A.; Komarnicki, M.; Przybylowicz-Chalecka, A.; Szczepaniak, T.; et al. Glutaminase inhibitor cb-839 synergizes with carfilzomib in resistant multiple myeloma cells. Oncotarget 2017, 8, 35863–35876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soriano, G.P.; Besse, L.; Li, N.; Kraus, M.; Besse, A.; Meeuwenoord, N.; Bader, J.; Everts, B.; den Dulk, H.; Overkleeft, H.S.; et al. Proteasome inhibitor-adapted myeloma cells are largely independent from proteasome activity and show complex proteomic changes, in particular in redox and energy metabolism. Leukemia 2016, 30, 2198–2207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chauhan, D.; Li, G.; Podar, K.; Hideshima, T.; Mitsiades, C.; Schlossman, R.; Munshi, N.; Richardson, P.; Cotter, F.E.; Anderson, K.C. Targeting mitochondria to overcome conventional and bortezomib/proteasome inhibitor ps-341 resistance in multiple myeloma (mm) cells. Blood 2004, 104, 2458–2466. [Google Scholar] [CrossRef] [Green Version]
- Lazzarino, G.; Amorini, A.M.; Fazzina, G.; Vagnozzi, R.; Signoretti, S.; Donzelli, S.; Di Stasio, E.; Giardina, B.; Tavazzi, B. Single-sample preparation for simultaneous cellular redox and energy state determination. Anal. Biochem. 2003, 322, 51–59. [Google Scholar] [CrossRef]
- Tavazzi, B.; Lazzarino, G.; Leone, P.; Amorini, A.M.; Bellia, F.; Janson, C.G.; Di Pietro, V.; Ceccarelli, L.; Donzelli, S.; Francis, J.S.; et al. Simultaneous high performance liquid chromatographic separation of purines, pyrimidines, n-acetylated amino acids, and dicarboxylic acids for the chemical diagnosis of inborn errors of metabolism. Clin. Biochem. 2005, 38, 997–1008. [Google Scholar] [CrossRef]
- Romitelli, F.; Santini, S.A.; Chierici, E.; Pitocco, D.; Tavazzi, B.; Amorini, A.M.; Lazzarino, G.; Di Stasio, E. Comparison of nitrite/nitrate concentration in human plasma and serum samples measured by the enzymatic batch griess assay, ion-pairing hplc and ion-trap gc-ms: The importance of a correct removal of proteins in the griess assay. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2007, 851, 257–267. [Google Scholar] [CrossRef]
- Al-Kafaji, G.; Sabry, M.A.; Bakhiet, M. Increased expression of mitochondrial DNA-encoded genes in human renal mesangial cells in response to high glucose-induced reactive oxygen species. Mol. Med. Rep. 2016, 13, 1774–1780. [Google Scholar] [CrossRef] [Green Version]
- Vicario, N.; Pasquinucci, L.; Spitale, F.M.; Chiechio, S.; Turnaturi, R.; Caraci, F.; Tibullo, D.; Avola, R.; Gulino, R.; Parenti, R.; et al. Simultaneous activation of mu and delta opioid receptors reduces allodynia and astrocytic connexin 43 in an animal model of neuropathic pain. Mol. Neurobiol. 2019, 56, 7338–7354. [Google Scholar] [CrossRef]
- Gulino, R.; Vicario, N.; Giunta, M.A.S.; Spoto, G.; Calabrese, G.; Vecchio, M.; Gulisano, M.; Leanza, G.; Parenti, R. Neuromuscular plasticity in a mouse neurotoxic model of spinal motoneuronal loss. Int. J. Mol. Sci. 2019, 20, 1500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giusto, E.; Codrich, M.; de Leo, G.; Francardo, V.; Coradazzi, M.; Parenti, R.; Gulisano, M.; Vicario, N.; Gulino, R.; Leanza, G. Compensatory changes in degenerating spinal motoneurons sustain functional sparing in the sod1-g93a mouse model of amyotrophic lateral sclerosis. J. Comp. Neurol. 2020, 528, 231–243. [Google Scholar] [CrossRef] [PubMed]
- Lebeau, J.; Saunders, J.M.; Moraes, V.W.R.; Madhavan, A.; Madrazo, N.; Anthony, M.C.; Wiseman, R.L. The perk arm of the unfolded protein response regulates mitochondrial morphology during acute endoplasmic reticulum stress. Cell Rep. 2018, 22, 2827–2836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, P.; Srikrishna, G.; Freeze, H.H. N-glycosylation deficiency reduces icam-1 induction and impairs inflammatory response. Glycobiology 2014, 24, 392–398. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Kohler, J.J. Glycosylation of the nuclear pore. Traffic 2014, 15, 347–361. [Google Scholar] [CrossRef] [Green Version]
- Harding, T.; Baughn, L.; Kumar, S.; Van Ness, B. The future of myeloma precision medicine: Integrating the compendium of known drug resistance mechanisms with emerging tumor profiling technologies. Leukemia 2019, 33, 863–883. [Google Scholar] [CrossRef]
- Uckun, F.M.; Qazi, S.; Demirer, T.; Champlin, R.E. Contemporary patient-tailored treatment strategies against high risk and relapsed or refractory multiple myeloma. EBioMedicine 2019, 39, 612–620. [Google Scholar] [CrossRef] [Green Version]
- Guerrero-Garcia, T.A.; Gandolfi, S.; Laubach, J.P.; Hideshima, T.; Chauhan, D.; Mitsiades, C.; Anderson, K.C.; Richardson, P.G. The power of proteasome inhibition in multiple myeloma. Expert Rev. Proteom. 2018, 15, 1033–1052. [Google Scholar] [CrossRef]
- Robak, P.; Drozdz, I.; Szemraj, J.; Robak, T. Drug resistance in multiple myeloma. Cancer Treat. Rev. 2018, 70, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Chanukuppa, V.; Taware, R.; Chatterjee, T.; Sharma, S.; More, T.H.; Taunk, K.; Kumar, S.; Santra, M.K.; Rapole, S. Current understanding of the potential of proteomics and metabolomics approaches in cancer chemoresistance: A focus on multiple myeloma. Curr. Top. Med. Chem. 2018, 18, 2584–2598. [Google Scholar] [CrossRef] [PubMed]
- Lackner, L.L. The expanding and unexpected functions of mitochondria contact sites. Trends Cell Biol. 2019, 29, 580–590. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Cuffe, L.; Szegezdi, E.; Logue, S.E.; Neary, C.; Healy, S.; Samali, A. Mechanisms of er stress-mediated mitochondrial membrane permeabilization. Int. J. Cell Biol. 2010, 2010, 170215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryoo, I.G.; Kwak, M.K. Regulatory crosstalk between the oxidative stress-related transcription factor nfe2l2/nrf2 and mitochondria. Toxicol. Appl. Pharm. 2018, 359, 24–33. [Google Scholar] [CrossRef]
- Ventura-Clapier, R.; Garnier, A.; Veksler, V. Transcriptional control of mitochondrial biogenesis: The central role of pgc-1alpha. Cardiovasc. Res. 2008, 79, 208–217. [Google Scholar] [CrossRef] [Green Version]
- Gureev, A.P.; Shaforostova, E.A.; Popov, V.N. Regulation of mitochondrial biogenesis as a way for active longevity: Interaction between the nrf2 and pgc-1alpha signaling pathways. Front. Genet. 2019, 10, 435. [Google Scholar] [CrossRef] [Green Version]
- Li, P.; Liu, Y.; Burns, N.; Zhao, K.S.; Song, R. Sirt1 is required for mitochondrial biogenesis reprogramming in hypoxic human pulmonary arteriolar smooth muscle cells. Int. J. Mol. Med. 2017, 39, 1127–1136. [Google Scholar] [CrossRef] [Green Version]
- Menzies, K.J.; Hood, D.A. The role of sirt1 in muscle mitochondrial turnover. Mitochondrion 2012, 12, 5–13. [Google Scholar] [CrossRef]
- Kong, X.; Wang, R.; Xue, Y.; Liu, X.; Zhang, H.; Chen, Y.; Fang, F.; Chang, Y. Sirtuin 3, a new target of pgc-1alpha, plays an important role in the suppression of ros and mitochondrial biogenesis. PLoS ONE 2010, 5, e11707. [Google Scholar] [CrossRef] [Green Version]
- Tilokani, L.; Nagashima, S.; Paupe, V.; Prudent, J. Mitochondrial dynamics: Overview of molecular mechanisms. Essays Biochem. 2018, 62, 341–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouchez, C.; Devin, A. Mitochondrial biogenesis and mitochondrial reactive oxygen species (ros): A complex relationship regulated by the camp/pka signaling pathway. Cells 2019, 8, 287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cork, G.K.; Thompson, J.; Slawson, C. Real talk: The inter-play between the mtor, ampk, and hexosamine biosynthetic pathways in cell signaling. Front. Endocrinol. (Lausanne) 2018, 9, 522. [Google Scholar] [CrossRef] [PubMed]
- Varki, A.; Cummings, R.D.; Esko, J.D.; Freeze, H.H.; Stanley, P.; Marth, J.D.; Bertozzi, C.R.; Hart, G.W.; Etzler, M.E. Symbol nomenclature for glycan representation. Proteomics 2009, 9, 5398–5399. [Google Scholar] [CrossRef] [Green Version]
- Jones, D.R.; Keune, W.J.; Anderson, K.E.; Stephens, L.R.; Hawkins, P.T.; Divecha, N. The hexosamine biosynthesis pathway and o-glcnacylation maintain insulin-stimulated pi3k-pkb phosphorylation and tumour cell growth after short-term glucose deprivation. FEBS J. 2014, 281, 3591–3608. [Google Scholar] [CrossRef]
- Sacoman, J.L.; Dagda, R.Y.; Burnham-Marusich, A.R.; Dagda, R.K.; Berninsone, P.M. Mitochondrial o-glcnac transferase (mogt) regulates mitochondrial structure, function, and survival in hela cells. J. Biol. Chem. 2017, 292, 4499–4518. [Google Scholar] [CrossRef] [Green Version]
- Asthana, A.; Ramakrishnan, P.; Vicioso, Y.; Zhang, K.; Parameswaran, R. Hexosamine biosynthetic pathway inhibition leads to aml cell differentiation and cell death. Mol. Cancer 2018, 17, 2226–2237. [Google Scholar] [CrossRef] [Green Version]
- Akella, N.M.; Ciraku, L.; Reginato, M.J. Fueling the fire: Emerging role of the hexosamine biosynthetic pathway in cancer. BMC Biol. 2019, 17, 52. [Google Scholar] [CrossRef] [Green Version]
- Taparra, K.; Wang, H.; Malek, R.; Lafargue, A.; Barbhuiya, M.A.; Wang, X.; Simons, B.W.; Ballew, M.; Nugent, K.; Groves, J.; et al. O-glcnacylation is required for mutant kras-induced lung tumorigenesis. J. Clin. Investig. 2018, 128, 4924–4937. [Google Scholar] [CrossRef]
- Ricciardiello, F.; Votta, G.; Palorini, R.; Raccagni, I.; Brunelli, L.; Paiotta, A.; Tinelli, F.; D’Orazio, G.; Valtorta, S.; De Gioia, L.; et al. Inhibition of the hexosamine biosynthetic pathway by targeting pgm3 causes breast cancer growth arrest and apoptosis. Cell Death Dis. 2018, 9, 377. [Google Scholar] [CrossRef]
- Liu, Y.; Cao, Y.; Pan, X.; Shi, M.; Wu, Q.; Huang, T.; Jiang, H.; Li, W.; Zhang, J. O-glcnac elevation through activation of the hexosamine biosynthetic pathway enhances cancer cell chemoresistance. Cell Death Dis. 2018, 9, 485. [Google Scholar] [CrossRef] [PubMed]
- Moehler, T.M.; Seckinger, A.; Hose, D.; Andrulis, M.; Moreaux, J.; Hielscher, T.; Willhauck-Fleckenstein, M.; Merling, A.; Bertsch, U.; Jauch, A.; et al. The glycome of normal and malignant plasma cells. PLoS ONE 2013, 8, e83719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiao, Z.; Dang, C.; Zhou, B.; Li, S.; Zhang, W.; Jiang, J.; Zhang, J.; Kong, R.; Ma, Y. O-linked n-acetylglucosamine transferase (ogt) is overexpressed and promotes o-linked protein glycosylation in esophageal squamous cell carcinoma. J. Biomed. Res. 2012, 26, 268–273. [Google Scholar] [CrossRef]
- Gandini, R.; Reichenbach, T.; Tan, T.C.; Divne, C. Structural basis for dolichylphosphate mannose biosynthesis. Nat. Commun. 2017, 8, 120. [Google Scholar] [CrossRef] [Green Version]
- Davis, E.J.; Davis-Van Thienen, W.I. Rate control of phosphorylation-coupled respiration by rat liver mitochondria. Arch. Biochem. Biophys. 1984, 233, 573–581. [Google Scholar] [CrossRef]
- Yamamoto, K.; Ikenaka, Y.; Ichise, T.; Bo, T.; Ishizuka, M.; Yasui, H.; Hiraoka, W.; Yamamori, T.; Inanami, O. Evaluation of mitochondrial redox status and energy metabolism of x-irradiated hela cells by lc/uv, lc/ms/ms and esr. Free Radic Res. 2018, 52, 648–660. [Google Scholar] [CrossRef] [PubMed]
- Baranowska-Bosiacka, I.; Gutowska, I.; Marchetti, C.; Rutkowska, M.; Marchlewicz, M.; Kolasa, A.; Prokopowicz, A.; Wiernicki, I.; Piotrowska, K.; Baskiewicz, M.; et al. Altered energy status of primary cerebellar granule neuronal cultures from rats exposed to lead in the pre- and neonatal period. Toxicology 2011, 280, 24–32. [Google Scholar] [CrossRef]
- Cagnetta, A.; Cea, M.; Calimeri, T.; Acharya, C.; Fulciniti, M.; Tai, Y.T.; Hideshima, T.; Chauhan, D.; Zhong, M.Y.; Patrone, F.; et al. Intracellular nad(+) depletion enhances bortezomib-induced anti-myeloma activity. Blood 2013, 122, 1243–1255. [Google Scholar] [CrossRef] [Green Version]
- Ruiz, P.D.; Hamilton, G.A.; Park, J.W.; Gamble, M.J. Macroh2a1 regulation of poly(adp-ribose) synthesis and stability prevents necrosis and promotes DNA repair. Mol. Cell Biol. 2019, 40, e00230-19. [Google Scholar] [CrossRef] [Green Version]
- Lu, P.; Hogan-Cann, A.D.; Kamboj, A.; Roy Chowdhury, S.K.; Aghanoori, M.R.; Fernyhough, P.; Anderson, C.M. Poly(adp-ribose) polymerase-1 inhibits mitochondrial respiration by suppressing pgc-1alpha activity in neurons. Neuropharmacology 2019, 160, 107755. [Google Scholar] [CrossRef]
- Owens, K.; Park, J.H.; Schuh, R.; Kristian, T. Mitochondrial dysfunction and nad(+) metabolism alterations in the pathophysiology of acute brain injury. Transl. Stroke Res. 2013, 4, 618–634. [Google Scholar] [CrossRef] [PubMed]
- Shackelford, R.; Hirsh, S.; Henry, K.; Abdel-Mageed, A.; Kandil, E.; Coppola, D. Nicotinamide phosphoribosyltransferase and sirt3 expression are increased in well-differentiated thyroid carcinomas. Anticancer Res. 2013, 33, 3047–3052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergaggio, E.; Riganti, C.; Garaffo, G.; Vitale, N.; Mereu, E.; Bandini, C.; Pellegrino, E.; Pullano, V.; Omede, P.; Todoerti, K.; et al. Idh2 inhibition enhances proteasome inhibitor responsiveness in hematological malignancies. Blood 2019, 133, 156–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, M.J.; Han, S.J.; Kim, J.I.; Park, J.W.; Park, K.M. Mitochondrial nadp(+)-dependent isocitrate dehydrogenase deficiency increases cisplatin-induced oxidative damage in the kidney tubule cells. Cell Death Dis. 2018, 9, 488. [Google Scholar] [CrossRef]
- Diehl, F.F.; Lewis, C.A.; Fiske, B.P.; Vander Heiden, M.G. Cellular redox state constrains serine synthesis and nucleotide production to impact cell proliferation. Nat. Metab. 2019, 1, 861–867. [Google Scholar] [CrossRef]
- Seki, R.; Yamagishi, S.; Matsui, T.; Yoshida, T.; Torimura, T.; Ueno, T.; Sata, M.; Okamura, T. Pigment epithelium-derived factor (pedf) inhibits survival and proliferation of vegf-exposed multiple myeloma cells through its anti-oxidative properties. Biochem. Biophys. Res. Commun. 2013, 431, 693–697. [Google Scholar] [CrossRef]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tibullo, D.; Giallongo, C.; Romano, A.; Vicario, N.; Barbato, A.; Puglisi, F.; Parenti, R.; Amorini, A.M.; Saab, M.W.; Tavazzi, B.; et al. Mitochondrial Functions, Energy Metabolism and Protein Glycosylation are Interconnected Processes Mediating Resistance to Bortezomib in Multiple Myeloma Cells. Biomolecules 2020, 10, 696. https://doi.org/10.3390/biom10050696
Tibullo D, Giallongo C, Romano A, Vicario N, Barbato A, Puglisi F, Parenti R, Amorini AM, Saab MW, Tavazzi B, et al. Mitochondrial Functions, Energy Metabolism and Protein Glycosylation are Interconnected Processes Mediating Resistance to Bortezomib in Multiple Myeloma Cells. Biomolecules. 2020; 10(5):696. https://doi.org/10.3390/biom10050696
Chicago/Turabian StyleTibullo, Daniele, Cesarina Giallongo, Alessandra Romano, Nunzio Vicario, Alessandro Barbato, Fabrizio Puglisi, Rosalba Parenti, Angela Maria Amorini, Miriam Wissam Saab, Barbara Tavazzi, and et al. 2020. "Mitochondrial Functions, Energy Metabolism and Protein Glycosylation are Interconnected Processes Mediating Resistance to Bortezomib in Multiple Myeloma Cells" Biomolecules 10, no. 5: 696. https://doi.org/10.3390/biom10050696