Cleavage of the APE1 N-Terminal Domain in Acute Myeloid Leukemia Cells Is Associated with Proteasomal Activity

 , , , , , ,
, , , , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Materials
2.2. Preparation of the Cell Extracts and Anti-FLAG Co-Immunoprecipitation
2.3. In Vitro Proteolysis
2.4. Western Blotting Analysis
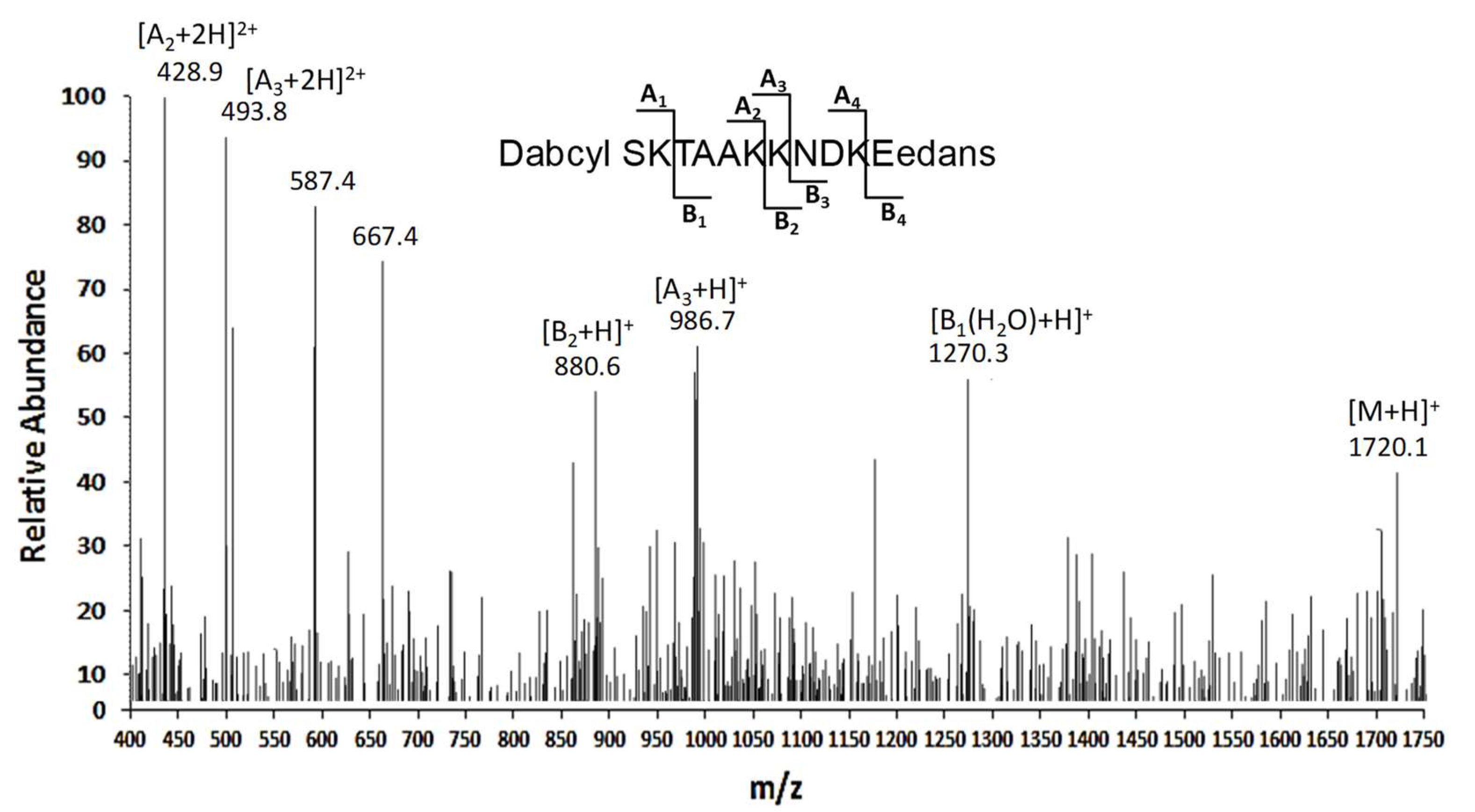
2.5. Peptide Synthesis
2.6. Proteolytic Assay
2.7. RNA Isolation, cDNA Synthesis and PCR
2.8. Recombinant Protein
2.9. Proteomic Analyses
2.9.1. Purification of Proteases
2.9.2. Protein Identification
2.9.3. Functional Enrichment Analysis
2.10. In Vitro Cleavage Assay with Purified Proteasome
3. Results
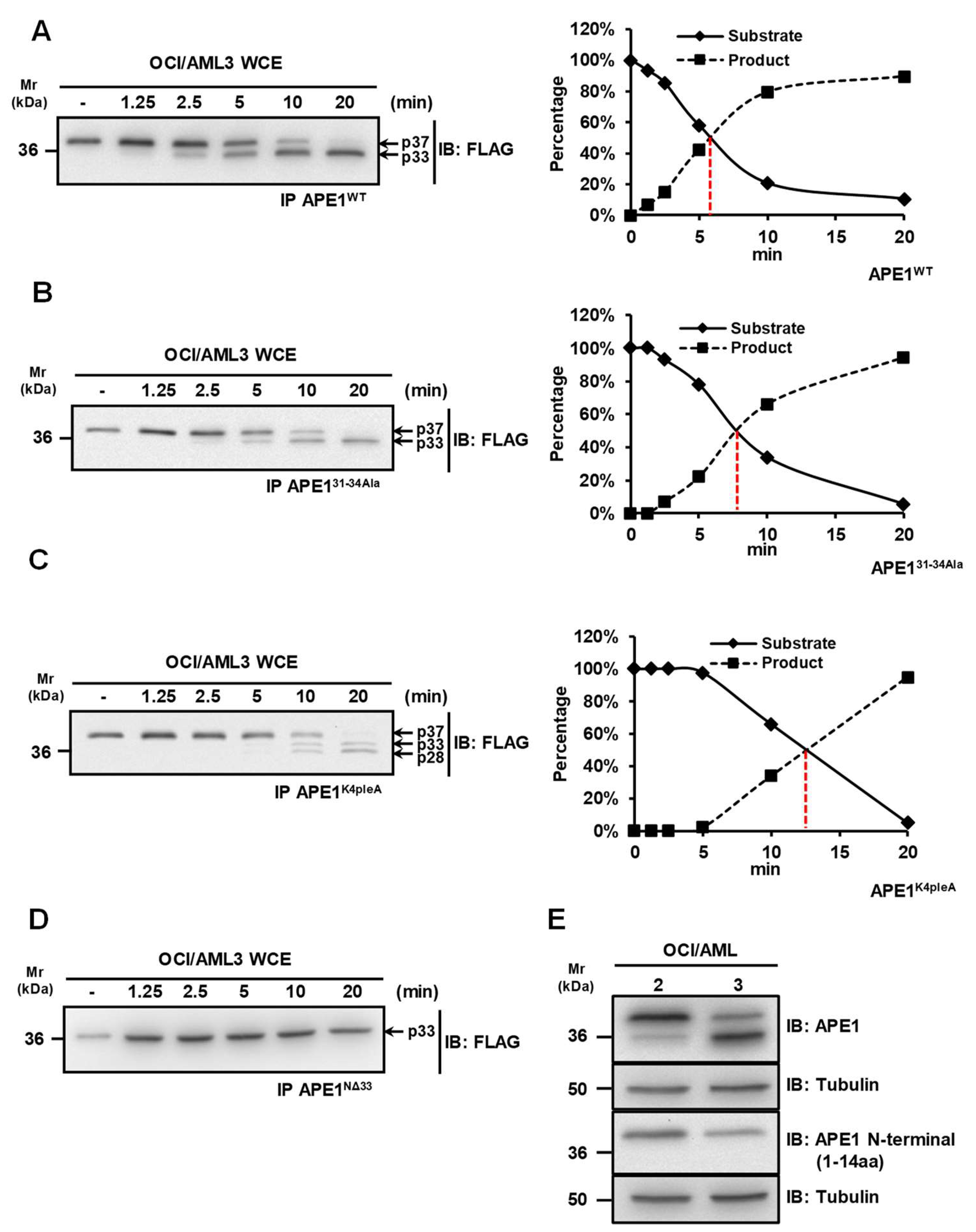
3.1. APE1 Is Cleaved at the N-Terminal Region in OCI/AML3 Cells
3.2. Characterization of the Proteolytic Sites for APE1 Cleavage
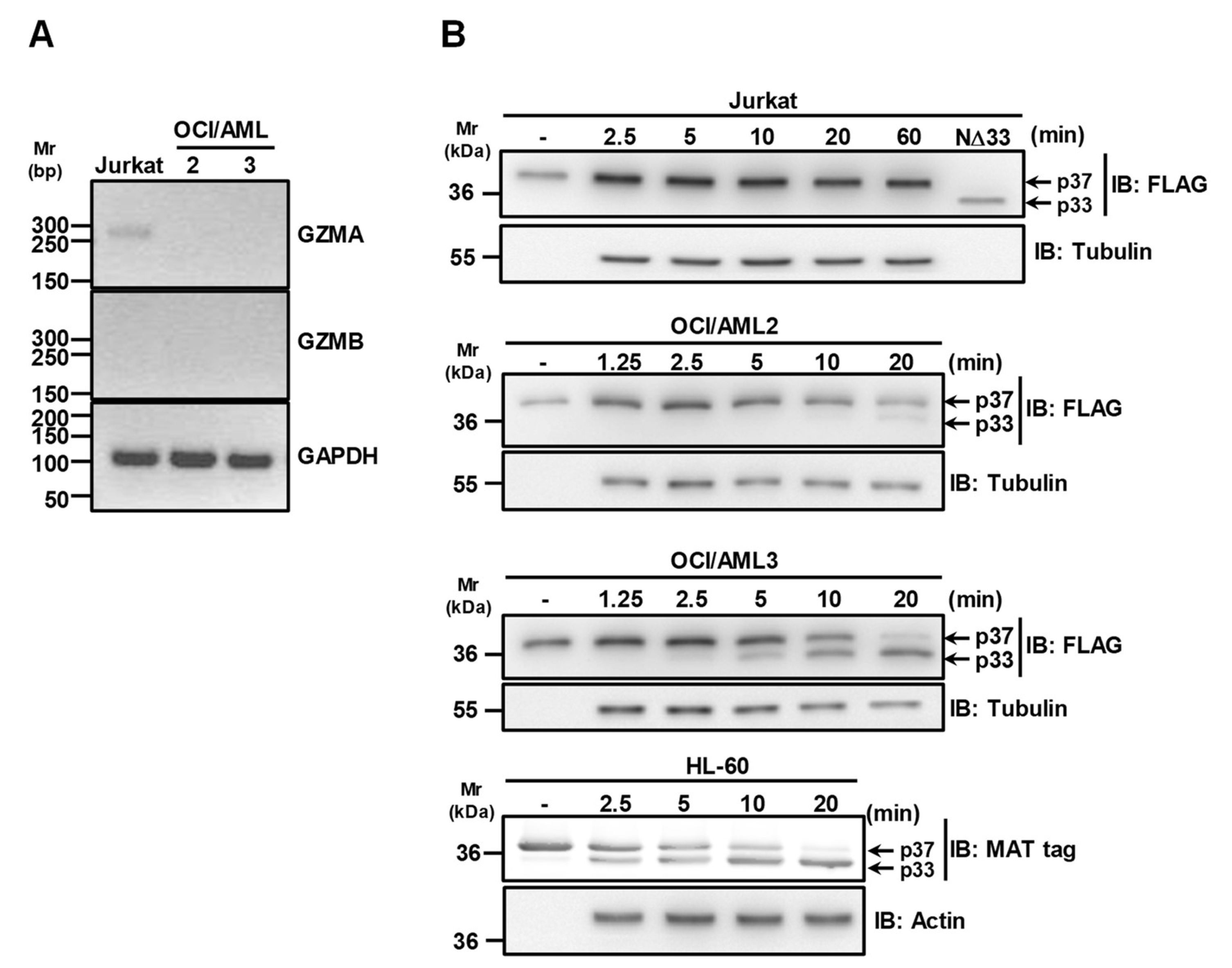
3.3. Granzymes Are not Involved in APE1 Proteolysis Observed in OCI/AML3 Cells
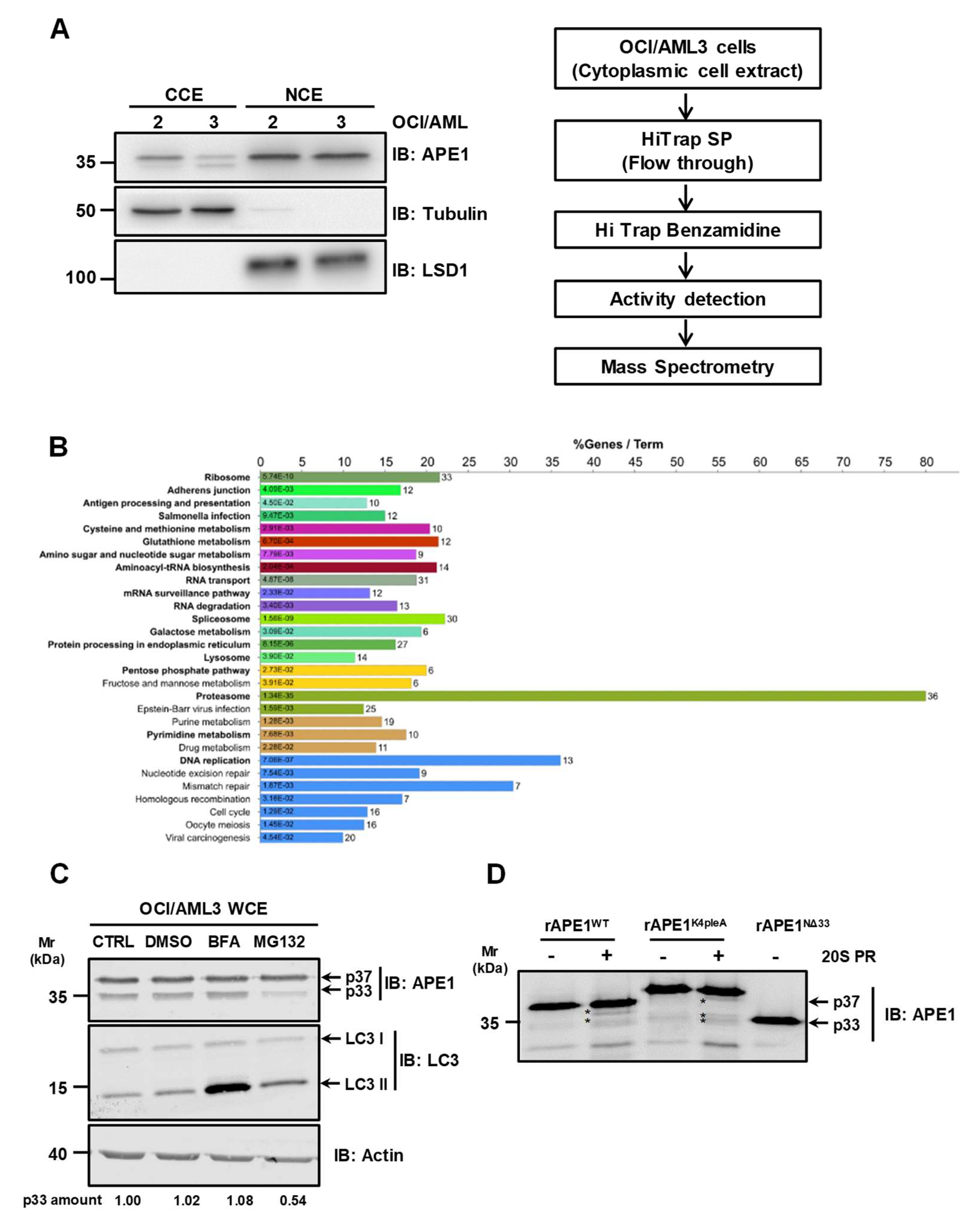
3.4. The Proteasome Machinery Is Involved in APE1 Cleavage
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sung, J.S.; Demple, B. Roles of base excision repair subpathways in correcting oxidized abasic sites in DNA. Febs J. 2006, 273, 1620–1629. [Google Scholar] [CrossRef] [PubMed]
- Mitra, S.; Izumi, T.; Boldogh, I.; Bhakat, K.; Chattopadhyay, R.; Szczesny, B. Intracellular trafficking and regulation of mammalian AP-endonuclease 1 (APE1), an essential DNA repair protein. DNA Repair 2007, 6, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Vascotto, C.; Fantini, D.; Romanello, M.; Cesaratto, L.; Deganuto, M.; Leonardi, A.; Radicella, J.P.; Kelley, M.R.; D’Ambrosio, C.; Scaloni, A.; et al. APE1/Ref-1 Interacts with NPM1 within Nucleoli and Plays a Role in the rRNA Quality Control Process. Mol. Cell. Biol. 2009, 29, 1834–1854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antoniali, G.; Serra, F.; Lirussi, L.; Tanaka, M.; D’Ambrosio, C.; Zhang, S.; Radovic, S.; Dalla, E.; Ciani, Y.; Scaloni, A.; et al. Mammalian APE1 controls miRNA processing and its interactome is linked to cancer RNA metabolism. Nat. Commun. 2017, 8, 797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fantini, D.; Vascotto, C.; Marasco, D.; D’Ambrosio, C.; Romanello, M.; Vitagliano, L.; Pedone, C.; Poletto, M.; Cesaratto, L.; Quadrifoglio, F.; et al. Critical lysine residues within the overlooked N-terminal domain of human APE1 regulate its biological functions. Nucleic Acids Res. 2010, 38, 8239–8256. [Google Scholar] [CrossRef] [Green Version]
- Bhakat, K.K.; Sengupta, S.; Adeniyi, V.F.; Roychoudhury, S.; Nath, S.; Bellot, L.J.; Feng, D.; Mantha, A.K.; Sinha, M.; Qiu, S.; et al. Regulation of limited N-terminal proteolysis of APE1 in tumor via acetylation and its role in cell proliferation. Oncotarget 2016, 7, 22590–22604. [Google Scholar] [CrossRef] [Green Version]
- Lirussi, L.; Antoniali, G.; Vascotto, C.; D’Ambrosio, C.; Poletto, M.; Romanello, M.; Marasco, D.; Leone, M.; Quadrifoglio, F.; Bhakat, K.K.; et al. Nucleolar accumulation of APE1 depends on charged lysine residues that undergo acetylation upon genotoxic stress and modulate its BER activity in cells. Mol. Biol. Cell 2012, 23, 4079–4096. [Google Scholar] [CrossRef]
- Busso, C.S.; Iwakuma, T.; Izumi, T. Ubiquitination of mammalian AP endonuclease (APE1) regulated by the p53-MDM2 signaling pathway. Oncogene 2009, 28, 1616–1625. [Google Scholar] [CrossRef] [Green Version]
- Busso, C.S.; Wedgeworth, C.M.; Izumi, T. Ubiquitination of human AP-endonuclease 1 (APE1) enhanced by T233E substitution and by CDK5. Nucleic Acids Res. 2011, 39, 8017–8028. [Google Scholar] [CrossRef] [Green Version]
- Chattopadhyay, R.; Wiederhold, L.; Szczesny, B.; Boldogh, I.; Hazra, T.K.; Izumi, T.; Mitra, S. Identification and characterization of mitochondrial abasic (AP)-endonuclease in mammalian cells. Nucleic Acids Res. 2006, 34, 2067–2076. [Google Scholar] [CrossRef] [Green Version]
- Fan, Z.; Beresford, P.J.; Zhang, D.; Xu, Z.; Novina, C.D.; Yoshida, A.; Pommier, Y.; Lieberman, J. Cleaving the oxidative repair protein Ape1 enhances cell death mediated by granzyme A. Nat. Immunol. 2003, 4, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Chen, J.; Zhao, T.; Fan, Z. Granzyme K degrades the redox/DNA repair enzyme Ape1 to trigger oxidative stress of target cells leading to cytotoxicity. Mol. Immunol. 2008, 45, 2225–2235. [Google Scholar] [CrossRef] [PubMed]
- Vascotto, C.; Lirussi, L.; Poletto, M.; Tiribelli, M.; Damiani, D.; Fabbro, D.; Damante, G.; Demple, B.; Colombo, E.; Tell, G. Functional regulation of the apurinic/apyrimidinic endonuclease 1 by nucleophosmin: Impact on tumor biology. Oncogene 2014, 33, 2876–2887. [Google Scholar] [CrossRef] [PubMed]
- Antoniali, G.; Lirussi, L.; Poletto, M.; Tell, G. Emerging roles of the nucleolus in regulating the DNA damage response: The noncanonical DNA repair enzyme APE1/Ref-1 as a paradigmatical example. Antioxid. Redox Signal. 2014, 20, 621–639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burra, S.; Marasco, D.; Malfatti, M.C.; Antoniali, G.; Virgilio, A.; Esposito, V.; Demple, B.; Galeone, A.; Tell, G. Human AP-endonuclease (Ape1) activity on telomeric G4 structures is modulated by acetylatable lysine residues in the N-terminal sequence. DNA Repair 2019, 73, 129–143. [Google Scholar] [CrossRef] [PubMed]
- Ciaccio, C.; Tundo, G.R.; Grasso, G.; Spoto, G.; Marasco, D.; Ruvo, M.; Gioia, M.; Rizzarelli, E.; Coletta, M. Somatostatin: A novel substrate and a modulator of insulin-degrading enzyme activity. J. Mol. Biol. 2009, 385, 1556–1567. [Google Scholar] [CrossRef] [Green Version]
- Hochegger, K.; Eller, P.; Huber, J.M.; Bernhard, D.; Mayer, G.; Zlabinger, G.J.; Rosenkranz, A.R. Expression of granzyme A in human polymorphonuclear neutrophils. Immunology 2007, 121, 166–173. [Google Scholar] [CrossRef]
- D’Ambrosio, C.; Arena, S.; Fulcoli, G.; Scheinfeld, M.H.; Zhou, D.; D’Adamio, L.; Scaloni, A. Hyperphosphorylation of JNK-interacting protein 1, a protein associated with Alzheimer disease. Mol. Cell Proteom. 2006, 5, 97–113. [Google Scholar] [CrossRef] [Green Version]
- Bruschi, M.; Sinico, R.A.; Moroni, G.; Pratesi, F.; Migliorini, P.; Galetti, M.; Murtas, C.; Tincani, A.; Madaio, M.; Radice, A.; et al. Glomerular autoimmune multicomponents of human lupus nephritis in vivo: α-enolase and annexin AI. J. Am. Soc. Nephrol. 2014, 25, 2483–2498. [Google Scholar] [CrossRef] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Bindea, G.; Mlecnik, B.; Hackl, H.; Charoentong, P.; Tosolini, M.; Kirilovsky, A.; Fridman, W.-H.; Pagès, F.; Trajanoski, Z.; Galon, J. ClueGO: A Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 2009, 25, 1091–1093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshida, A.; Urasaki, Y.; Waltham, M.; Bergman, A.-C.; Pourquier, P.; Rothwell, D.G.; Inuzuka, M.; Weinstein, J.N.; Ueda, T.; Appella, E.; et al. Human Apurinic/Apyrimidinic Endonuclease (Ape1) and Its N-terminal Truncated Form (AN34) Are Involved in DNA Fragmentation during Apoptosis. J. Biol. Chem. 2003, 278, 37768–37776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vascotto, C.; Cesaratto, L.; Zeef, L.A.; Deganuto, M.; D’Ambrosio, C.; Scaloni, A.; Romanello, M.; Damante, G.; Taglialatela, G.; Delneri, D.; et al. Genome-wide analysis and proteomic studies reveal APE1/Ref-1 multifunctional role in mammalian cells. Proteomics 2009, 9, 1058–1074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kummer, J.A.; Kamp, A.M.; Citarella, F.; Horrevoets, A.J.; Hack, C.E. Expression of human recombinant granzyme A zymogen and its activation by the cysteine proteinase cathepsin C. J. Biol. Chem. 1996, 271, 9281–9286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshida, A.; Pourquier, P.; Pommier, Y. Purification and characterization of a Mg2+-dependent endonuclease (AN34) from etoposide-treated human leukemia HL-60 cells undergoing apoptosis. Cancer Res. 1998, 58, 2576–2582. [Google Scholar]
- Fu, Z.; Wang, B.; Wang, S.; Wu, W.; Wang, Q.; Chen, Y.; Kong, S.; Lu, J.; Tang, Z.; Ran, H.; et al. Integral proteomic analysis of blastocysts reveals key molecular machinery governing embryonic diapause and reactivation for implantation in mice. Biol. Reprod. 2014, 90, 52. [Google Scholar] [CrossRef]
- Moiseeva, T.N.; Bottrill, A.; Melino, G.; Barlev, N.A. DNA damage-induced ubiquitylation of proteasome controls its proteolytic activity. Oncotarget 2013, 4, 1338–1348. [Google Scholar] [CrossRef] [Green Version]
- Ding, J.; Fishel, M.L.; Reed, A.M.; McAdams, E.; Czader, M.; Cardoso, A.A.; Kelley, M.R. Ref-1/APE1 as Transcriptional Regulator and Novel Therapeutic Target in Pediatric T-cell Leukemia. Mol. Cancer 2017, 16, 1401–1411. [Google Scholar] [CrossRef] [Green Version]
- Kelley, M.R.; Georgiadis, M.M.; Fishel, M.L. APE1/Ref-1 role in redox signaling: Translational applications of targeting the redox function of the DNA repair/redox protein APE1/Ref-1. Curr. Mol. Pharm. 2012, 5, 36–53. [Google Scholar] [CrossRef]
- Fishel, M.L.; Colvin, E.S.; Luo, M.; Kelley, M.R.; Robertson, K.A. Inhibition of the Redox Function of APE1/Ref-1 in Myeloid Leukemia Cell Lines Results in a Hypersensitive Response to Retinoic Acid-induced Differentiation and Apoptosis. Exp. Hematol. 2010, 38, 1178–1188. [Google Scholar] [CrossRef] [Green Version]
- Shah, F.; Logsdon, D.; Messmann, R.A.; Fehrenbacher, J.C.; Fishel, M.L.; Kelley, M.R. Exploiting the Ref-1-APE1 node in cancer signaling and other diseases: From bench to clinic. Npj Precis Oncol. 2017, 1, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Limpose, K.L.; Corbett, A.H.; Doetsch, P.W. BERing the burden of damage: Pathway crosstalk and posttranslational modification of base excision repair proteins regulate DNA damage management. DNA Repair 2017, 56, 51–64. [Google Scholar] [CrossRef] [PubMed]
- Thakur, S.; Dhiman, M.; Tell, G.; Mantha, A.K. A review on protein–protein interaction network of APE1/Ref-1 and its associated biological functions. Cell Biochem. Funct. 2015, 33, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Busso, C.S.; Lake, M.W.; Izumi, T. Posttranslational modification of mammalian AP endonuclease (APE1). Cell Mol. Life Sci. 2010, 67, 3609–3620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poletto, M.; Vascotto, C.; Scognamiglio, P.L.; Lirussi, L.; Marasco, D.; Tell, G. Role of the unstructured N-terminal domain of the hAPE1 (human apurinic/apyrimidinic endonuclease 1) in the modulation of its interaction with nucleic acids and NPM1 (nucleophosmin). Biochem. J. 2013, 452, 545–557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antoniali, G.; Lirussi, L.; D’Ambrosio, C.; Dal Piaz, F.; Vascotto, C.; Casarano, E.; Marasco, D.; Scaloni, A.; Fogolari, F.; Tell, G. SIRT1 gene expression upon genotoxic damage is regulated by APE1 through nCaRE-promoter elements. Mol. Biol. Cell 2014, 25, 532–547. [Google Scholar] [CrossRef] [Green Version]
- Meisenberg, C.; Tait, P.S.; Dianova, I.I.; Wright, K.; Edelmann, M.J.; Ternette, N.; Tasaki, T.; Kessler, B.M.; Parsons, J.L.; Kwon, Y.T.; et al. Ubiquitin ligase UBR3 regulates cellular levels of the essential DNA repair protein APE1 and is required for genome stability. Nucleic Acids Res. 2012, 40, 701–711. [Google Scholar] [CrossRef]
- Scott, T.L.; Wicker, C.A.; Suganya, R.; Dhar, B.; Pittman, T.; Horbinski, C.; Izumi, T. Polyubiquitination of apurinic/apyrimidinic endonuclease 1 by Parkin. Mol. Carcinog. 2017, 56, 325–336. [Google Scholar] [CrossRef] [Green Version]
- Poletto, M.; Malfatti, M.C.; Dorjsuren, D.; Scognamiglio, P.L.; Marasco, D.; Vascotto, C.; Jadhav, A.; Maloney, D.J.; Wilson, D.M.; Simeonov, A.; et al. Inhibitors of the apurinic/apyrimidinic endonuclease 1 (APE1)/nucleophosmin (NPM1) interaction that display anti-tumor properties. Mol. Carcinog. 2015, 55, 688–704. [Google Scholar] [CrossRef] [Green Version]
- Kumar Deshmukh, F.; Yaffe, D.; Olshina, M.A.; Ben-Nissan, G.; Sharon, M. The Contribution of the 20S Proteasome to Proteostasis. Biomolecules 2019, 9, 190. [Google Scholar] [CrossRef] [Green Version]
- Marasco, D.; Scognamiglio, P.L. Identification of inhibitors of biological interactions involving intrinsically disordered proteins. Int. J. Mol. Sci. 2015, 16, 7394–7412. [Google Scholar] [CrossRef] [PubMed]
- Cloos, J.; Roeten, M.S.; Franke, N.E.; van Meerloo, J.; Zweegman, S.; Kaspers, G.J.; Jansen, G. (Immuno)proteasomes as therapeutic target in acute leukemia. Cancer Metastasis Rev. 2017, 36, 599–615. [Google Scholar] [CrossRef] [PubMed]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lirussi, L.; Antoniali, G.; Scognamiglio, P.L.; Marasco, D.; Dalla, E.; D’Ambrosio, C.; Arena, S.; Scaloni, A.; Tell, G. Cleavage of the APE1 N-Terminal Domain in Acute Myeloid Leukemia Cells Is Associated with Proteasomal Activity. Biomolecules 2020, 10, 531. https://doi.org/10.3390/biom10040531
Lirussi L, Antoniali G, Scognamiglio PL, Marasco D, Dalla E, D’Ambrosio C, Arena S, Scaloni A, Tell G. Cleavage of the APE1 N-Terminal Domain in Acute Myeloid Leukemia Cells Is Associated with Proteasomal Activity. Biomolecules. 2020; 10(4):531. https://doi.org/10.3390/biom10040531
Chicago/Turabian StyleLirussi, Lisa, Giulia Antoniali, Pasqualina Liana Scognamiglio, Daniela Marasco, Emiliano Dalla, Chiara D’Ambrosio, Simona Arena, Andrea Scaloni, and Gianluca Tell. 2020. "Cleavage of the APE1 N-Terminal Domain in Acute Myeloid Leukemia Cells Is Associated with Proteasomal Activity" Biomolecules 10, no. 4: 531. https://doi.org/10.3390/biom10040531