Inhibition on JNK Mimics Silencing of Wnt-11 Mediated Cellular Response in Androgen-Independent Prostate Cancer Cells

 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Material and Methods
2.1. Cell Culture and Pharmacological Treatments
2.2. Cell Cycle and Apoptosis Assays
2.3. Mitochondrial Membrane Potential Loss by DiOC6 Staining and Mitotracker Red Staining
2.4. LysoTracker Red Uptake and Reactive Oxygen Species Detection
2.5. Acridine Orange Staining and GFP Transfection
2.6. Western Blotting
2.7. Real-Time PCR
2.8. siRNA Transfection
2.9. Immunostaining
2.10. Hanging Drop Assay
2.11. Migration, Invasion, and Proliferation Assays
2.12. Data Analysis
3. Results
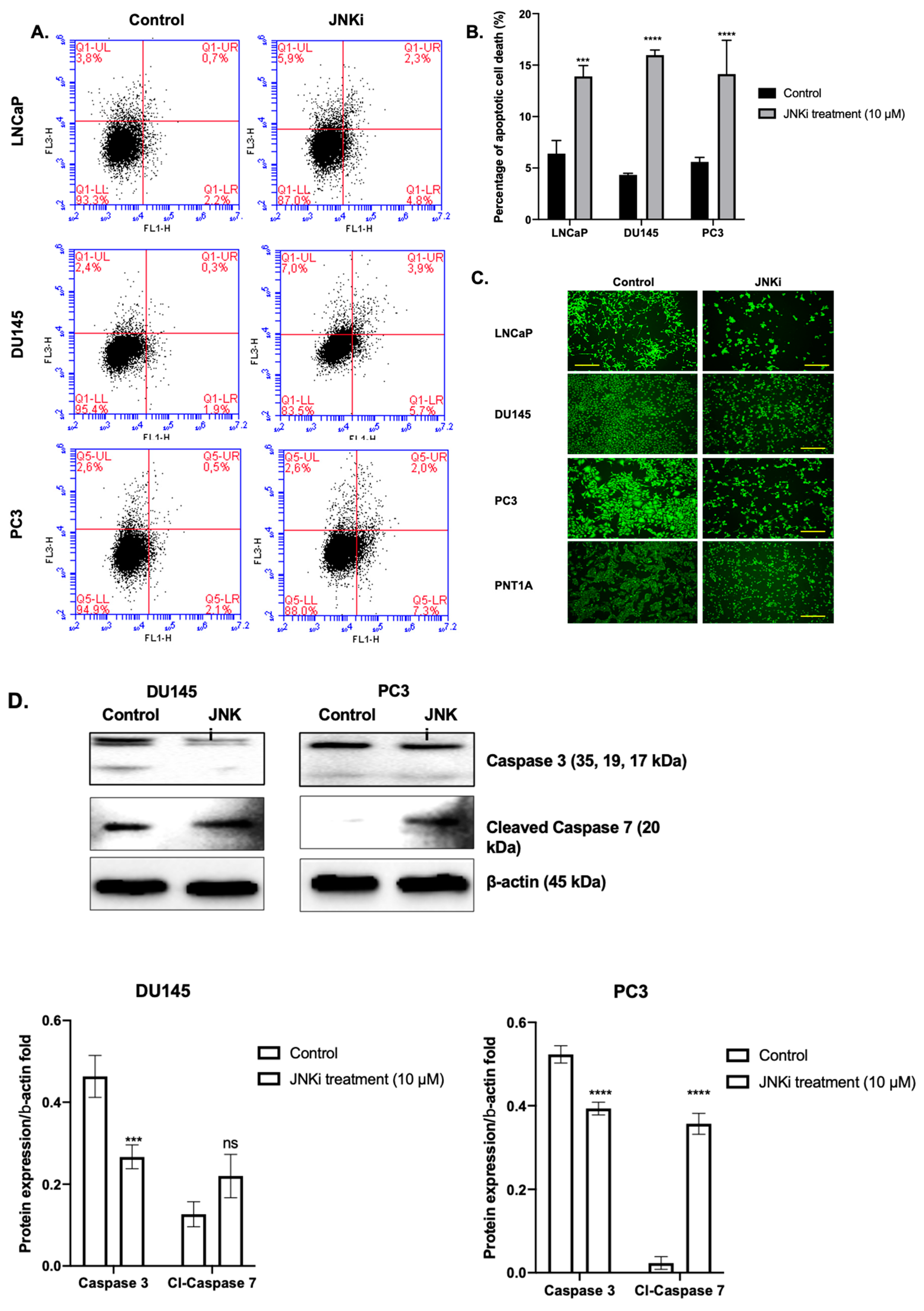
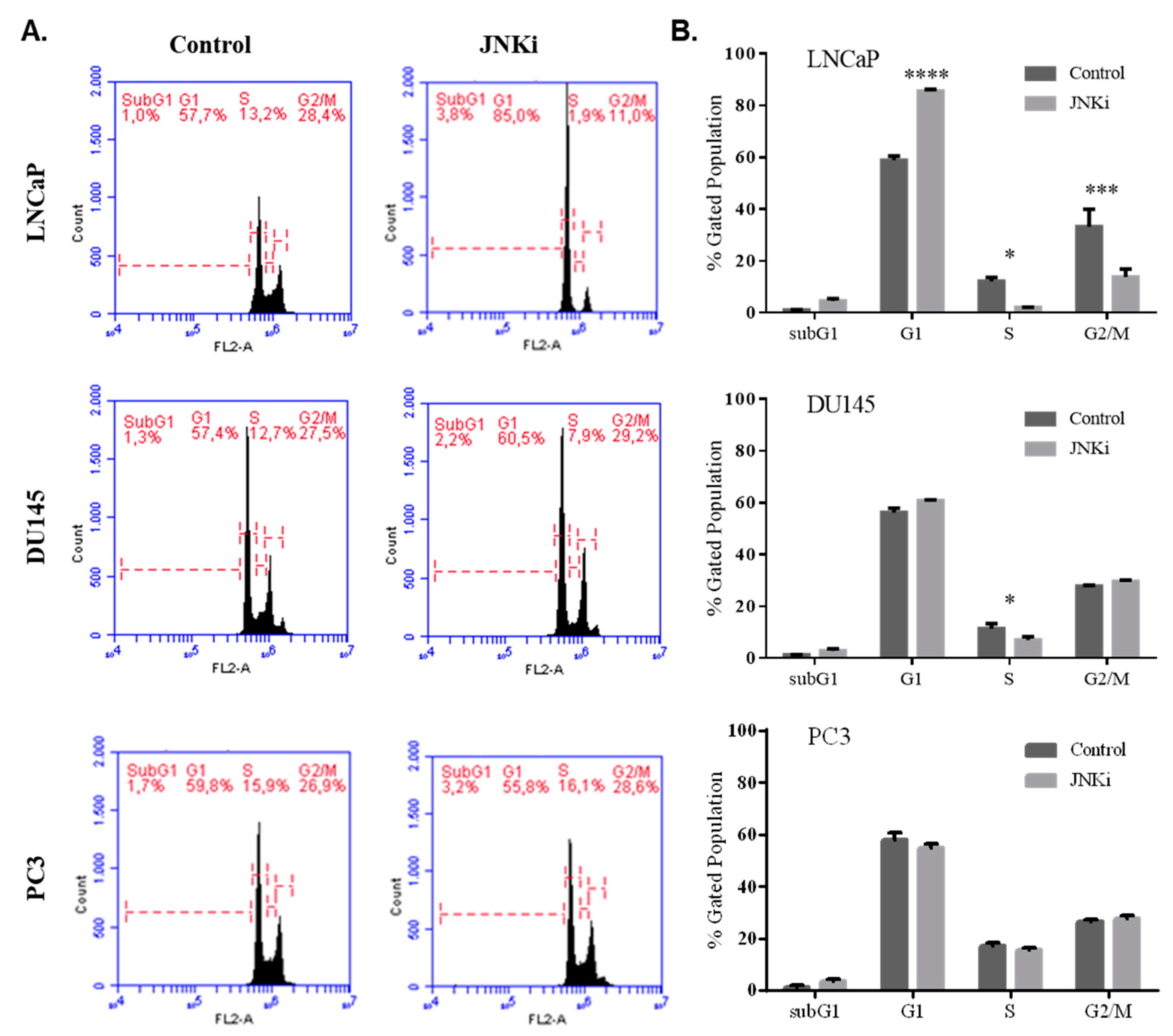
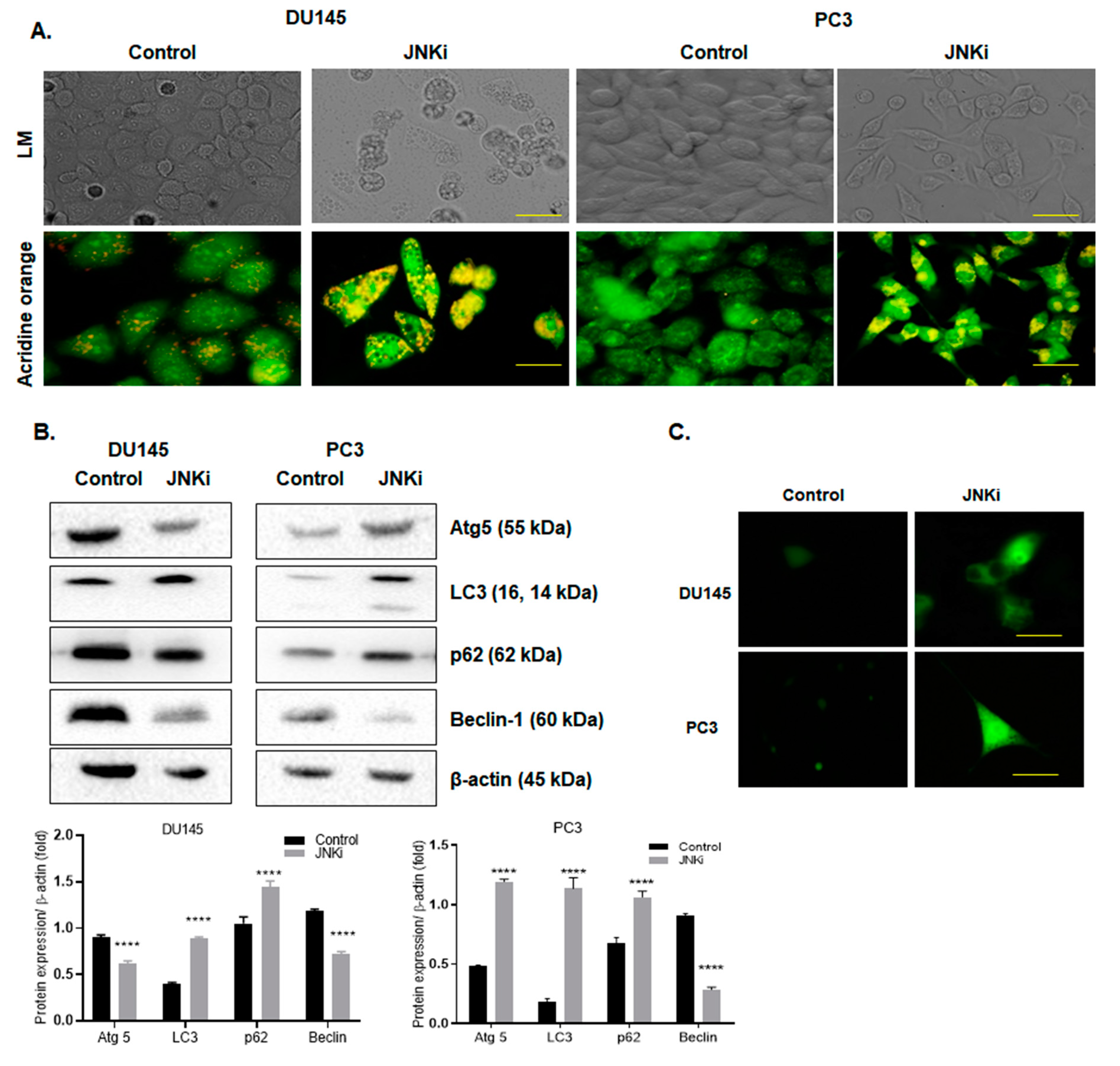
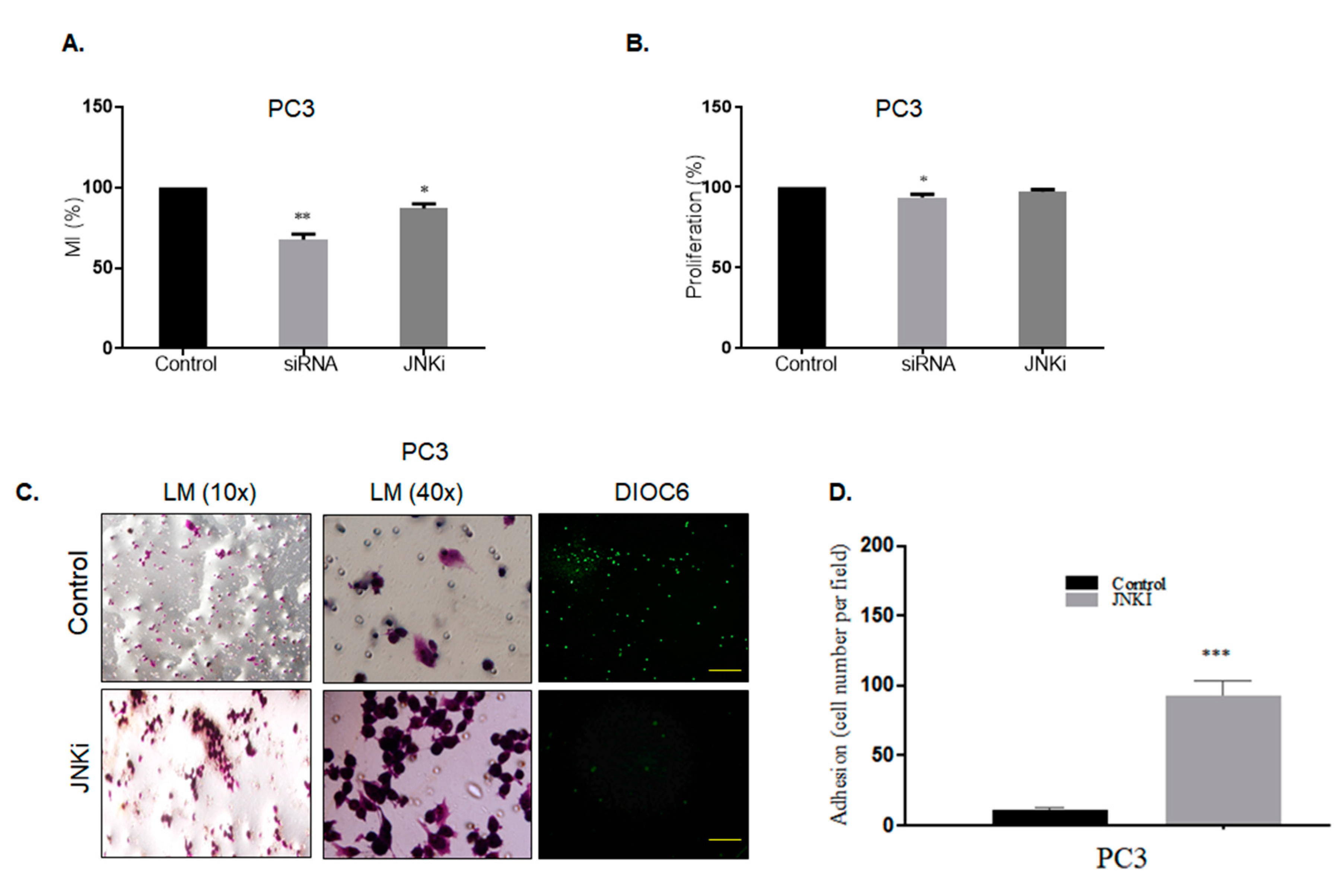
3.1. Blocking JNK Pathway Alters Cellular Fate in a Cell Type-Dependent Manner
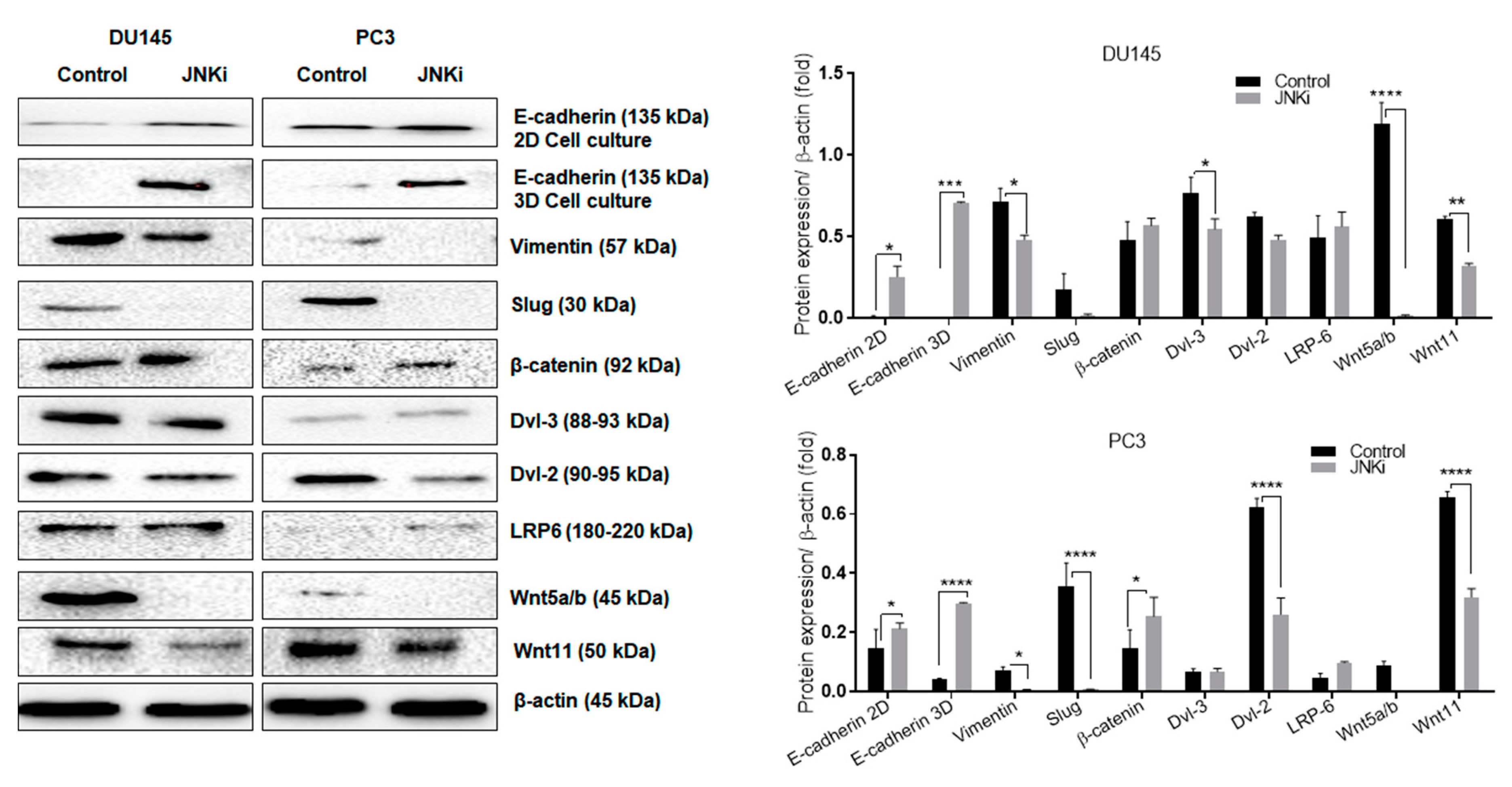
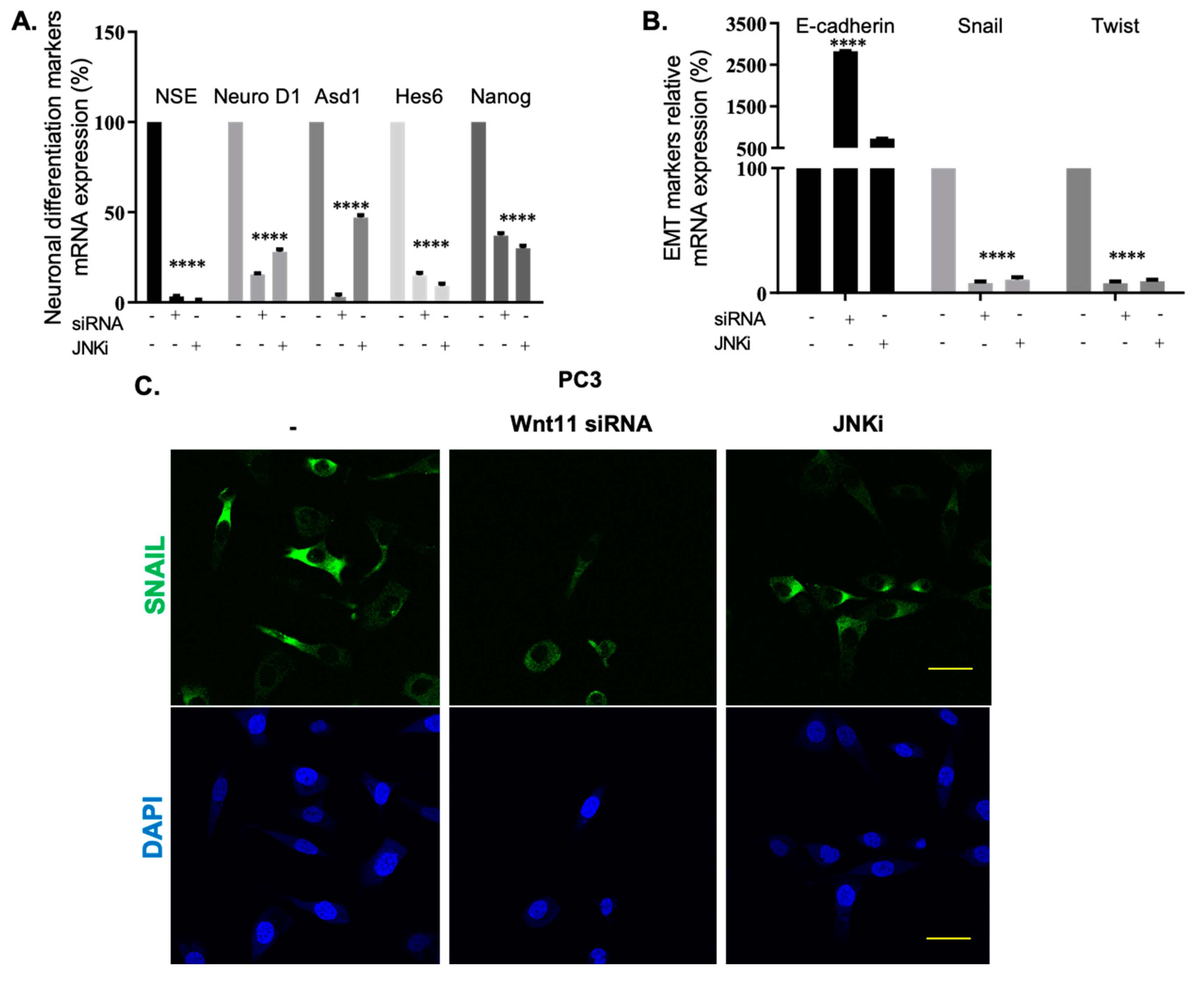
3.2. Wnt-11 Promotes EMT in PCa Cells via JNK Signalling
3.3. Effect of Silencing Wnt-11 on Cellular Migration
4. Discussion
4.1. JNKi Modulates the Survival and Death Axis in a Cell-Type Dependent Manner
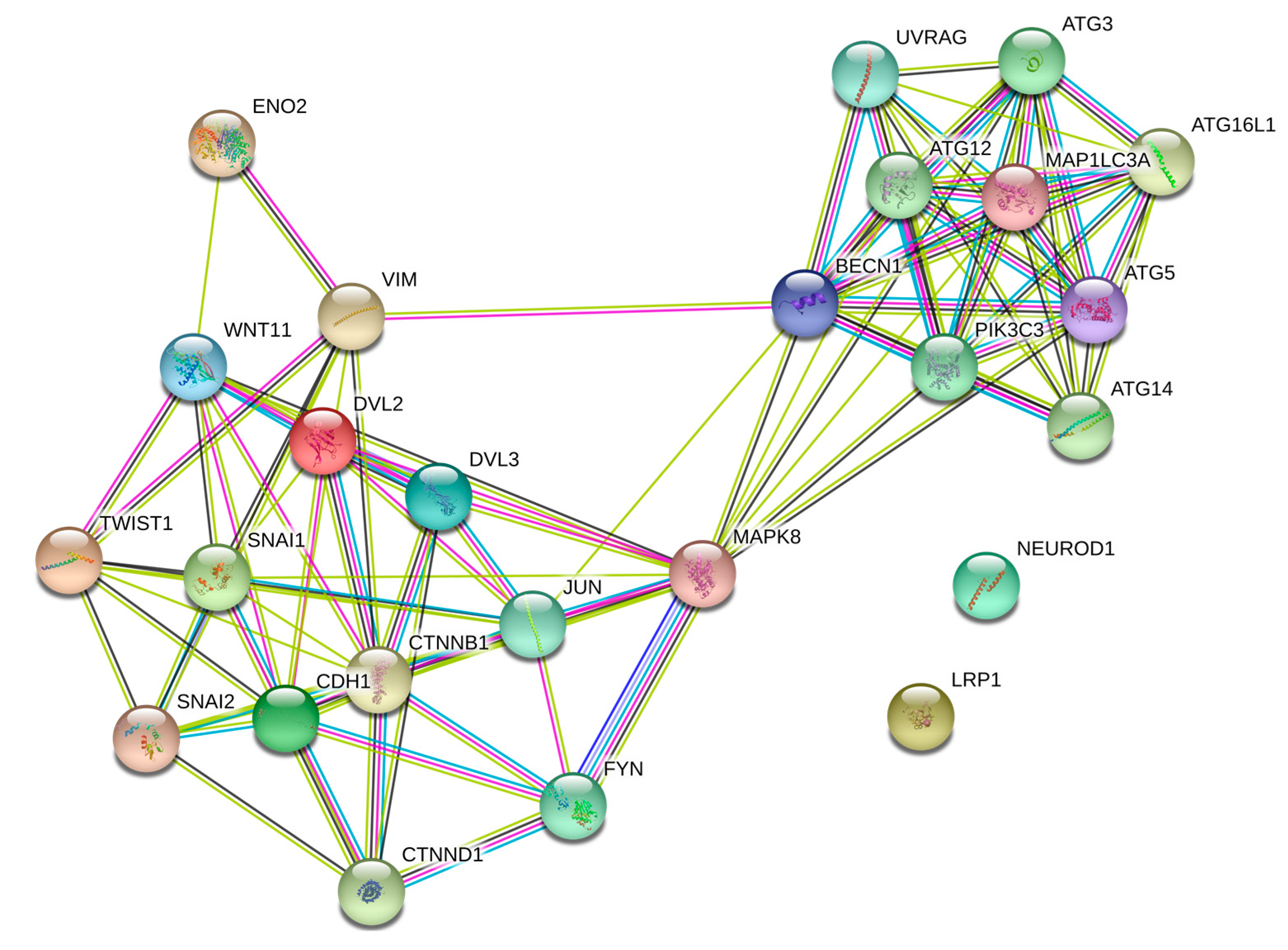
4.2. JNK and Wnt-11 Presented Similar Functional Properties in the Control of Metastasis-Associated Biomarkers
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Schatten, H. Brief overview of prostate cancer statistics, grading, diagnosis and treatment strategies. Adv. Exp. Med. Biol. 2018, 1095, 1–14. [Google Scholar] [PubMed]
- Jemal, A.; Siegel, R.; Xu, J.; Ward, E. Cancer Statistics, 2010. CA. Cancer J. Clin. 2010. [Google Scholar] [CrossRef] [PubMed]
- Ruijter, E.; Van De Kaa, C.; Miller, G.; Ruiter, D.; Debruyne, F.; Schalken, J. Molecular genetics and epidemiology of prostate carcinoma. Endocr. Rev. 1999, 20, 22–45. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, A.O.; Kuiper, G.G.J.M.; Ris-Stalpers, C.; van Rooij, H.C.J.; Romalo, G.; Trifiro, M.; Mulder, E.; Pinsky, L.; Schweikert, H.U.; Trapman, J. Androgen receptor abnormalities. J. Steroid Biochem. Mol. Biol. 1991. [Google Scholar] [CrossRef] [Green Version]
- Kopper, L.; Tímár, J. Genomics of prostate cancer: Is there anything to “translate”? Pathol. Oncol. Res. 2005, 11, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Wright, W.S.; Longo, K.A.; Dolinsky, V.W.; Gerin, I.; Kang, S.; Bennett, C.N.; Chiang, S.H.; Prestwich, T.C.; Gress, C.; Burant, C.F.; et al. Wnt10b inhibits obesity in ob/ob and agouti mice. Diabetes 2007. [Google Scholar] [CrossRef] [Green Version]
- Koushyar, S.; Grant, G.H.; Uysal-Onganer, P. The interaction of Wnt-11 and signalling cascades in prostate cancer. Tumor Biol. 2016, 37, 13049–13057. [Google Scholar] [CrossRef] [Green Version]
- Brown, M.; Strudwick, N.; Suwara, M.; Sutcliffe, L.K.; Mihai, A.D.; Ali, A.A.; Watson, J.N.; Schröder, M. An initial phase of JNK activation inhibits cell death early in the endoplasmic reticulum stress response. J. Cell Sci. 2016. [Google Scholar] [CrossRef] [Green Version]
- Saadeddin, A.; Babaei-Jadidi, R.; Spencer-Dene, B.; Nateri, A.S. The links between transcription, β-catenin/JNK signaling, and carcinogenesis. Mol. Cancer Res. 2009, 7, 1189–1196. [Google Scholar] [CrossRef] [Green Version]
- Gozdecka, M.; Lyons, S.; Kondo, S.; Taylor, J.; Li, Y.; Walczynski, J.; Thiel, G.; Breitwieser, W.; Jones, N. JNK Suppresses Tumor Formation via a Gene-Expression Program Mediated by ATF2. Cell Rep. 2014. [Google Scholar] [CrossRef]
- Lee, J.; Sohn, E.J.; Yoon, S.; Won, G.; Kim, C.G.; Jung, J.H.; Kim, S.H. Activation of JNK and IRE1 is critically involved in tanshinone I-induced p62 dependent autophagy in malignant pleural mesothelioma cells: Implication of p62 UBA domain. Oncotarget 2017. [Google Scholar] [CrossRef] [PubMed]
- Tai, T.Y.; Warner, L.N.; Jones, T.D.; Jung, S.; Concepcion, F.A.; Skyrud, D.W.; Fender, J.; Liu, Y.; Williams, A.D.; Neumaier, J.F.; et al. Antiepileptic action of c-Jun N-terminal kinase (JNK) inhibition in an animal model of temporal lobe epilepsy. Neuroscience 2017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, H.; Zhang, Y.; Chen, Q.; Lin, Y. AKT and JNK Signaling Pathways Increase the Metastatic Potential of Colorectal Cancer Cells by Altering Transgelin Expression. Dig. Dis. Sci. 2016. [Google Scholar] [CrossRef]
- Davis, R.J. Signal transduction by the JNK group of MAP kinases. Cell 2000, 103, 239–252. [Google Scholar] [CrossRef] [Green Version]
- Hübner, A.; Mulholland, D.J.; Standen, C.L.; Karasarides, M.; Cavanagh-Kyros, J.; Barrett, T.; Chi, H.; Greiner, D.L.; Tournier, C.; Sawyers, C.L.; et al. JNK and PTEN cooperatively control the development of invasive adenocarcinoma of the prostate. Proc. Natl. Acad. Sci. USA 2012, 109, 12046–12051. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Sun, C.; Tao, S.; Osunkoya, A.O.; Arnold, R.S.; Petros, J.A.; Zu, X.; Moreno, C.S. The JNK inhibitor AS602801 Synergizes with Enzalutamide to Kill Prostate Cancer Cells In Vitro and In Vivo and Inhibit Androgen Receptor Expression. Transl. Oncol. 2020. [Google Scholar] [CrossRef]
- Pakula, H.; Xiang, D.; Li, Z. A tale of two signals: AR and WNT in development and tumorigenesis of prostate and mammary gland. Cancers 2017, 9, 14. [Google Scholar] [CrossRef]
- Zhu, H.; Mazor, M.; Kawano, Y.; Walker, M.M.; Leung, H.Y.; Armstrong, K.; Waxman, J.; Kypta, R.M. Analysis of Wnt gene expression in prostate cancer: Mutual inhibition by WNT11 and the androgen receptor. Cancer Res. 2004. [Google Scholar] [CrossRef] [Green Version]
- Wei, H.; Wang, N.; Zhang, Y.; Wang, S.; Pang, X.; Zhang, J.; Luo, Q.; Su, Y.; Zhang, S. Clinical significance of Wnt-11 and squamous cell carcinoma antigen expression in cervical cancer. Med. Oncol. 2014. [Google Scholar] [CrossRef]
- Uysal-Onganer, P.; Kawano, Y.; Caro, M.; Walker, M.M.; Diez, S.; Darrington, R.S.; Waxman, J.; Kypta, R.M. Wnt-11 promotes neuroendocrine-like differentiation, survival and migration of prostate cancer cells. Mol. Cancer 2010, 9. [Google Scholar] [CrossRef] [Green Version]
- Nishioka, M.; Ueno, K.; Hazama, S.; Okada, T.; Sakai, K.; Suehiro, Y.; Okayama, N.; Hirata, H.; Oka, M.; Imai, K.; et al. Possible involvement of Wnt11 in colorectal cancer progression. Mol. Carcinog. 2013. [Google Scholar] [CrossRef]
- Uysal-Onganer, P.; Kypta, R.M. Wnt11 in 2011 - the regulation and function of a non-canonical Wnt. Acta Physiol. 2012, 204, 52–64. [Google Scholar] [CrossRef] [PubMed]
- Amorino, G.P.; Parsons, S.J. Neuroendocrine cells in prostate cancer. Crit. Rev. Eukaryot. Gene Expr. 2004, 14, 287–300. [Google Scholar] [CrossRef] [PubMed]
- Amorino, G.P.; Deeble, P.D.; Parsons, S.J. Neurotensin stimulates mitogenesis of prostate cancer cells through a novel c-Src/Stat5b pathway. Oncogene 2007. [Google Scholar] [CrossRef] [Green Version]
- Yuan, X.J.; He, Y.Q.; Liu, J.L.; Luo, H.S.; Zhang, J.H.; Cui, S. Expression of androgen receptor and its co-localization with estrogen receptor-alpha in the developing pituitary gland of sheep fetus. Histochem. Cell Biol. 2007. [Google Scholar] [CrossRef] [PubMed]
- van Amerongen, R.; Nusse, R. Towards an integrated view of Wnt signaling in development. Development 2009. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.; Zhang, B.; Liang, H.; Lu, Y.; Ai, X.; Zhang, B.; Chen, X. JNK inhibitor SP600125 enhances TGF-β-induced apoptosis of RBE human cholangiocarcinoma cells in a Smad-dependent manner. Mol. Med. Rep. 2013. [Google Scholar] [CrossRef]
- Liu, Q.; Zhang, Y.; Mao, H.; Chen, W.; Luo, N.; Zhou, Q.; Chen, W.; Yu, X. A crosstalk between the Smad and JNK signaling in the TGF-β-induced epithelial-mesenchymal transition in rat peritoneal mesothelial cells. PLoS ONE 2012. [Google Scholar] [CrossRef] [Green Version]
- Murillo-Garzón, V.; Gorroño-Etxebarria, I.; Åkerfelt, M.; Puustinen, M.C.; Sistonen, L.; Nees, M.; Carton, J.; Waxman, J.; Kypta, R.M. Frizzled-8 integrates Wnt-11 and transforming growth factor-β signaling in prostate cancer. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Kumawat, K.; Gosens, R. WNT-5A: Signaling and functions in health and disease. Cell. Mol. Life Sci. 2016, 73, 567–587. [Google Scholar] [CrossRef] [Green Version]
- Niess, H.; Camaj, P.; Renner, A.; Ischenko, I.; Zhao, Y.; Krebs, S.; Mysliwietz, J.; Jäckel, C.; Nelson, P.J.; Blum, H.; et al. Side population cells of pancreatic cancer show characteristics of cancer stem cells responsible for resistance and metastasis. Target. Oncol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Van Den Broeck, A.; Vankelecom, H.; Van Eijsden, R.; Govaere, O.; Topal, B. Molecular markers associated with outcome and metastasis in human pancreatic cancer. J. Exp. Clin. Cancer Res. 2012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dart, D.A.; Arisan, D.E.; Owen, S.; Hao, C.; Jiang, W.G.; Uysal-Onganer, P. Wnt-11 expression promotes invasiveness and correlates with survival in human pancreatic ductal adeno carcinoma. Genes 2019, 10, 921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Zhang, P.; Cai, Y.; Soofi, A.; Dressler, G.R. Activation of Wnt11 by transforming growth factor-β drives mesenchymal gene expression through non-canonical Wnt protein signaling in renal epithelial cells. J. Biol. Chem. 2012. [Google Scholar] [CrossRef] [Green Version]
- Arisan, E.D.; Rencuzogullari, O.; Freitas, I.L.; Radzali, S.; Keskin, B.; Kothari, A.; Warford, A.; Uysal-Onganer, P. Upregulated wnt-11 and mir-21 expression trigger epithelial mesenchymal transition in aggressive prostate cancer cells. Biology 2020, 9, 52. [Google Scholar] [CrossRef] [Green Version]
- STRING: Functional Protein Association Networks. Available online: https://string-db.org/cgi/about.pl?sessionId=CvE3IDazHaCE&footer_active_subpage=content (accessed on 26 June 2020).
- Li, D.D.; Wang, L.L.; Deng, R.; Tang, J.; Shen, Y.; Guo, J.F.; Wang, Y.; Xia, L.P.; Feng, G.K.; Liu, Q.Q.; et al. The pivotal role of c-Jun NH2-terminal kinase-mediated Beclin 1 expression during anticancer agents-induced autophagy in cancer cells. Oncogene 2009. [Google Scholar] [CrossRef] [Green Version]
- Shen, B.; Chao, L.; Chao, J. Pivotal role of JNK-dependent FOXO1 activation in downregulation of kallistatin expression by oxidative stress. Am. J. Physiol. Hear. Circ. Physiol. 2010. [Google Scholar] [CrossRef] [Green Version]
- Lorenzo, P.I.; Saatcioglu, F. Inhibition of apoptosis in prostate cancer cells by androgens is mediated through downregulation of c-Jun N-terminal kinase activation. Neoplasia 2008. [Google Scholar] [CrossRef] [Green Version]
- Dou, Y.; Jiang, X.; Xie, H.; He, J.; Xiao, S. The Jun N-terminal kinases signaling pathway plays a “seesaw” role in ovarian carcinoma: A molecular aspect. J. Ovarian Res. 2019, 12, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Park, W.H. MAPK inhibitors, particularly the JNK inhibitor, increase cell death effects in H2O2-treated lung cancer cells via increased superoxide anion and glutathione depletion. Oncol. Rep. 2018. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Zhou, M.I.; Liu, G.; Huang, X.; He, W.; Gou, X.; Jiang, T. Autophagy activated by the c-Jun N-terminal kinase-mediated pathway protects human prostate cancer PC3 cells from celecoxib-induced apoptosis. Exp. Ther. Med. 2017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, H.O.; Hong, S.E.; Park, J.A.; Chang, Y.H.; Hong, Y.J.; Park, I.C.; Lee, J.K. Inhibition of JNK-mediated autophagy enhances NSCLC cell sensitivity to mTORC1/2 inhibitors. Sci. Rep. 2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.T.; Liu, H.; Mao, M.J.; Tan, Y.; Mo, X.Q.; Meng, X.J.; Cao, M.T.; Zhong, C.Y.; Liu, Y.; Shan, H.; et al. Crosstalk between autophagy and epithelial-mesenchymal transition and its application in cancer therapy. Mol. Cancer 2019, 18, 1–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.; Wang, J.; Sun, W.; Archibong, E.; Kahkoska, A.R.; Zhang, X.; Lu, Y.; Ligler, F.S.; Buse, J.B.; Gu, Z. Synthetic beta cells for fusion-mediated dynamic insulin secretion. Nat. Chem. Biol. 2018. [Google Scholar] [CrossRef]
- Lee, M.-H.; Koria, P.; Qu, J.; Andreadis, S.T. JNK phosphorylates β-catenin and regulates adherens junctions. FASEB J. 2009. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, H.; Oue, N.; Sato, A.; Hasegawa, Y.; Yamamoto, H.; Matsubara, A.; Yasui, W.; Kikuchi, A. Wnt5a signaling is involved in the aggressiveness of prostate cancer and expression of metalloproteinase. Oncogene 2010. [Google Scholar] [CrossRef] [Green Version]
- Miyamoto, D.T.; Zheng, Y.; Wittner, B.S.; Lee, R.J.; Zhu, H.; Broderick, K.T.; Desai, R.; Fox, D.B.; Brannigan, B.W.; Trautwein, J.; et al. RNA-Seq of single prostate CTCs implicates noncanonical Wnt signaling in antiandrogen resistance. Science 2015. [Google Scholar] [CrossRef] [Green Version]
- Wehrli, M.; Dougan, S.T.; Caldwell, K.; O’Keefe, L.; Schwartz, S.; Valzel-Ohayon, D.; Schejter, E.; Tomlinson, A.; DiNardo, S. Arrow encodes an LDL-receptor-related protein essential for Wingless signalling. Nature 2000. [Google Scholar] [CrossRef]
- Wharton, K.A. Runnin’ with the Dvl: Proteins that associate with Dsh/Dvl and their significance to Wnt signal transduction. Dev. Biol. 2003, 253, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Pandur, P.; Läsche, M.; Eisenberg, L.M.; Kühl, M. Wnt-11 activation of a non-canonical Wnt signalling pathway is required for cardiogenesis. Nature 2002. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Jiao, L.; Hou, J.; Xu, C.; Wang, L.; Yu, Y.; Li, Y.; Yang, C.; Wang, X.; Sun, Y. Dishevelled-2 silencing reduces androgen-dependent prostate tumor cell proliferation and migration and expression of Wnt-3a and matrix metalloproteinases. Mol. Biol. Rep. 2013. [Google Scholar] [CrossRef] [PubMed]
- Dwyer, M.A.; Joseph, J.D.; Wade, H.E.; Eaton, M.L.; Kunder, R.S.; Kazmin, D.; Chang, C.Y.; McDonnell, D.P. WNT11 expression is induced by estrogen-related receptor α and β-catenin and acts in an autocrine manner to increase cancer cell migration. Cancer Res. 2010. [Google Scholar] [CrossRef] [Green Version]
- Meng, H.; Wu, J.; Huang, Q.; Yang, X.; Yang, K.; Qiu, Y.; Ren, J.; Shen, R.; Qi, H. NEDD9 promotes invasion and migration of colorectal cancer cell line HCT116 via JNK/EMT. Oncol. Lett. 2019. [Google Scholar] [CrossRef] [PubMed]
- Tam, S.Y.; Wu, V.W.C.; Law, H.K.W. JNK pathway mediates low oxygen level induced epithelial–mesenchymal transition and stemness maintenance in colorectal cancer cells. Cancers 2020, 12, 224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Recio-Boiles, A.; Ilmer, M.; Rhea, P.R.; Kettlun, C.; Heinemann, M.L.; Ruetering, J.; Vykoukal, J.; Alt, E. JNK pathway inhibition selectively primes pancreatic cancer stem cells to TRAIL-induced apoptosis without affecting the physiology of normal tissue resident stem cells. Oncotarget 2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cavodeassi, F.; Carreira-Barbosa, F.; Young, R.M.; Concha, M.L.; Allende, M.L.; Houart, C.; Tada, M.; Wilson, S.W. Early stages of zebrafish eye formation require the coordinated activity of Wnt11, Fz5, and the Wnt/β-catenin pathway. Neuron 2005. [Google Scholar] [CrossRef] [Green Version]
- Westfall, T.A.; Brimeyer, R.; Twedt, J.; Gladon, J.; Olberding, A.; Furutani-Seiki, M.; Slusarski, D.C. Wnt-5/pipetail functions in vertebrate axis formation as a negative regulator of Wnt/β-catenin activity. J. Cell Biol. 2003. [Google Scholar] [CrossRef] [Green Version]
- Waxman, J.S.; Hocking, A.M.; Stoick, C.L.; Moon, R.T. Zebrafish Dapper1 and Dapper2 play distinct roles in Wnt-mediated developmental processes. Development 2004. [Google Scholar] [CrossRef] [Green Version]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arisan, E.D.; Rencuzogullari, O.; Keskin, B.; Grant, G.H.; Uysal-Onganer, P. Inhibition on JNK Mimics Silencing of Wnt-11 Mediated Cellular Response in Androgen-Independent Prostate Cancer Cells. Biology 2020, 9, 142. https://doi.org/10.3390/biology9070142
Arisan ED, Rencuzogullari O, Keskin B, Grant GH, Uysal-Onganer P. Inhibition on JNK Mimics Silencing of Wnt-11 Mediated Cellular Response in Androgen-Independent Prostate Cancer Cells. Biology. 2020; 9(7):142. https://doi.org/10.3390/biology9070142
Chicago/Turabian StyleArisan, Elif Damla, Ozge Rencuzogullari, Buse Keskin, Guy H. Grant, and Pinar Uysal-Onganer. 2020. "Inhibition on JNK Mimics Silencing of Wnt-11 Mediated Cellular Response in Androgen-Independent Prostate Cancer Cells" Biology 9, no. 7: 142. https://doi.org/10.3390/biology9070142