
Autoimmune versus Non-autoimmune Cutaneous Features in Monogenic Patients with Inborn Errors of Immunity
 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Patients and Method
2.1. Patients
2.2. Data Collection
2.3. Genetic Analysis
2.4. Statistical Analysis
3. Results
3.1. Population Characteristics
3.2. Cutaneous Involvements in the IEI Cohort
3.3. Dermatologic Characteristics of Different IEI Entities
3.4. Autoimmune Cutaneous Manifestations in the IEI Cohort
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tangye, S.G.; Al-Herz, W.; Bousfiha, A.; Cunningham-Rundles, C.; Franco, J.L.; Holland, S.M.; Klein, C.; Morio, T.; Oksenhendler, E.; Picard, C.; et al. Human Inborn Errors of Immunity: 2022 Update on the Classification from the International Union of Immunological Societies Expert Committee. J. Clin. Immunol. 2022, 42, 1–35. [Google Scholar] [CrossRef] [PubMed]
- Berron-Ruiz, A.; Berron-Perez, R.; Ruiz-Maldonado, R. Cutaneous markers of primary immunodeficiency diseases in children. Pediatr. Dermatol. 2000, 17, 91–96. [Google Scholar] [CrossRef] [PubMed]
- Dhouib, N.G.; Ben Khaled, M.; Ouederni, M.; Ben-Mustapha, I.; Kouki, R.; Besbes, H.; Barbouche, M.R.; Mellouli, F.; Bejaoui, M. Cutaneous Manifestations of Primary Immunodeficiency Diseases in Tunisian Children. Mediterr. J. Hematol. Infect. Dis. 2018, 10, e2018065. [Google Scholar] [PubMed]
- Moin, A.; Farhoudi, A.; Moein, M.; Pourpak, Z.; Bazargan, N. Cutaneous manifestations of primary immunodeficiency diseases in children. Iran J Allergy Asthma Immunol 2006, 5, 121–126. [Google Scholar]
- Thanveer, F. Cutaneous manifestations in primary immunodeficiency diseases. J. Ski. Sex. Transm. Dis. 2021, 3, 143–150. [Google Scholar] [CrossRef]
- Al-Herz, W.; Nanda, A. Skin manifestations in primary immunodeficient children. Pediatr. Dermatol. 2011, 28, 494–501. [Google Scholar] [CrossRef]
- Aghamohammadi, A.; Rezaei, N.; Yazdani, R.; Delavari, S.; Kutukculer, N.; Topyildiz, E.; Ozen, A.; Baris, S.; Karakoc-Aydiner, E.; Kilic, S.S.; et al. Consensus Middle East and North Africa Registry on Inborn Errors of Immunity. J. Clin. Immunol. 2021, 41, 1339–1351. [Google Scholar] [CrossRef]
- Abolhassani, H.; Kiaee, F.; Tavakol, M.; Chavoshzadeh, Z.; Mahdaviani, S.A.; Momen, T.; Yazdani, R.; Azizi, G.; Habibi, S.; Gharagozlou, M.; et al. Fourth Update on the Iranian National Registry of Primary Immunodeficiencies: Integration of Molecular Diagnosis. J. Clin. Immunol. 2018, 38, 816–832. [Google Scholar] [CrossRef]
- Seidel, M.G.; Kindle, G.; Gathmann, B.; Quinti, I.; Buckland, M.; van Montfrans, J.; Scheible, R.; Rusch, S.; Gasteiger, L.M.; Grimbacher, B.; et al. The European Society for Immunodeficiencies (ESID) Registry Working Definitions for the Clinical Diagnosis of Inborn Errors of Immunity. J. Allergy Clin. Immunol. Pract. 2019, 7, 1763–1770. [Google Scholar] [CrossRef]
- Baris, S.; Abolhassani, H.; Massaad, M.J.; Al-Nesf, M.; Chavoshzadeh, Z.; Keles, S.; Reisli, I.; Tahiat, A.; Shendi, H.M.; Elaziz, D.A.; et al. The Middle East and North Africa Diagnosis and Management Guidelines for Inborn Errors of Immunity. J. Allergy Clin. Immunol. Pract. 2023, 11, 158–180.e11. [Google Scholar] [CrossRef]
- Aghamohammadi, A.; Abolhassani, H.; Rezaei, N.; Yazdani, R. Inborn Errors of Immunity: A Practical Guide; Elsevier Science: Amsterdam, The Netherlands, 2021. [Google Scholar]
- Abolhassani, H.; Aghamohammadi, A.; Fang, M.; Rezaei, N.; Jiang, C.; Liu, X.; Pan-Hammarström, Q.; Hammarström, L. Clinical implications of systematic phenotyping and exome sequencing in patients with primary antibody deficiency. Genet. Med. 2019, 21, 243–251. [Google Scholar] [CrossRef] [PubMed]
- Abolhassani, H.; Chou, J.; Bainter, W.; Platt, C.D.; Tavassoli, M.; Momen, T.; Tavakol, M.; Eslamian, M.H.; Gharagozlou, M.; Movahedi, M.; et al. Clinical, immunologic, and genetic spectrum of 696 patients with combined immunodeficiency. J. Allergy Clin. Immunol. 2018, 141, 1450–1458. [Google Scholar] [CrossRef] [PubMed]
- Abolhassani, H.; Hammarstrom, L.; Cunningham-Rundles, C. Current genetic landscape in common variable immune deficiency. Blood 2020, 135, 656–667. [Google Scholar] [CrossRef] [PubMed]
- Fang, M.; Su, Z.; Abolhassani, H.; Itan, Y.; Jin, X.; Hammarstrom, L. VIPPID: A gene-specific single nucleotide variant pathogenicity prediction tool for primary immunodeficiency diseases. Brief Bioinform. 2022, 23, bbac176. [Google Scholar] [CrossRef] [PubMed]
- Fang, M.; Abolhassani, H.; Lim, C.K.; Zhang, J.; Hammarstrom, L. Next Generation Sequencing Data Analysis in Primary Immunodeficiency Disorders—Future Directions. J. Clin. Immunol. 2016, 36 (Suppl. 1), 68–75. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Bousfiha, A.; Jeddane, L.; Picard, C.; Al-Herz, W.; Ailal, F.; Chatila, T.; Cunningham-Rundles, C.; Etzioni, A.; Franco, J.L.; Holland, S.M.; et al. Human Inborn Errors of Immunity: 2019 Update of the IUIS Phenotypical Classification. J. Clin. Immunol. 2020, 40, 66–81. [Google Scholar] [CrossRef]
- De Wit, J.; Brada, R.J.K.; van Veldhuizen, J.; Dalm, V.A.S.H.; Pasmans, S.G.M.A. Skin disorders are prominent features in primary immunodeficiency diseases: A systematic overview of current data. Allergy 2019, 74, 464–482. [Google Scholar] [CrossRef]
- Ettinger, M.; Schreml, J.; Wirsching, K.; Berneburg, M.; Schreml, S. Skin signs of primary immunodeficiencies: How to find the genes to check. Br. J. Dermatol. 2018, 178, 335–349. [Google Scholar] [CrossRef]
- Al-Herz, W.; Zainal, M.; Nanda, A. A Prospective Survey of Skin Manifestations in Children With Inborn Errors of Immunity From a National Registry Over 17 Years. Front. Immunol. 2021, 12, 751469. [Google Scholar] [CrossRef]
- Fetter, T.; Niebel, D.; Braegelmann, C.; Wenzel, J. Skin-Associated B Cells in the Pathogenesis of Cutaneous Autoimmune Diseases—Implications for Therapeutic Approaches. Cells 2020, 9, 2627. [Google Scholar] [CrossRef] [PubMed]
- Yanaba, K.; Kamata, M.; Asano, Y.; Tada, Y.; Sugaya, M.; Kadono, T.; Tedder, T.F.; Sato, S. CD19 expression in B cells regulates atopic dermatitis in a mouse model. Am. J. Pathol. 2013, 182, 2214–2222. [Google Scholar] [CrossRef] [PubMed]
- López-Quintero, W.; Cleves, D.; Gomez-Vasco, J.D.; Pérez, P.; Patiño, J.; Medina-Valencia, D.; Pachajoa, H.; Torres-Canchala, L.; Vidal, A.; Olaya, M. Skin manifestations in pediatric patients with primary immunodeficiency diseases (PIDs) in a tertiary care hospital in Colombia. World Allergy Organ. J. 2021, 14, 100527. [Google Scholar] [CrossRef]
- Antachopoulos, C.; Walsh, T.J.; Roilides, E. Fungal infections in primary immunodeficiencies. Eur. J. Pediatr. 2007, 166, 1099–1117. [Google Scholar] [CrossRef] [PubMed]
- Leonardi, L.; Rivalta, B.; Leone, F.; Cancrini, C.; Caffarelli, C.; Marseglia, G.L.; Cardinale, F. Host Defenses to Viruses: Lessons from Inborn Errors of Immunity. Medicina 2022, 58, 248. [Google Scholar] [CrossRef] [PubMed]
- Alkhairy, O.K.; Rezaei, N.; Graham, R.R.; Abolhassani, H.; Borte, S.; Hultenby, K.; Wu, C.; Aghamohammadi, A.; Williams, D.A.; Behrens, T.W.; et al. RAC2 loss-of-function mutation in 2 siblings with characteristics of common variable immunodeficiency. J. Allergy Clin. Immunol. 2015, 135, 1380–1384.e5. [Google Scholar] [CrossRef]
- Gupta, R.; Debbaneh, M.G.; Liao, W. Genetic Epidemiology of Psoriasis. Curr. Dermatol. Rep. 2014, 3, 61–78. [Google Scholar] [CrossRef]
- Shaker, O.G.; Moustafa, W.; Essmat, S.; Abdel-Halim, M.; El-Komy, M. The role of interleukin-12 in the pathogenesis of psoriasis. Clin. Biochem. 2006, 39, 119–125. [Google Scholar] [CrossRef]
- Costagliola, G.; Cappelli, S.; Consolini, R. Autoimmunity in Primary Immunodeficiency Disorders: An Updated Review on Pathogenic and Clinical Implications. J. Clin. Med. 2021, 10, 4729. [Google Scholar] [CrossRef]
- Lehman, H.; Gordon, C. The Skin as a Window into Primary Immune Deficiency Diseases: Atopic Dermatitis and Chronic Mucocutaneous Candidiasis. J. Allergy Clin. Immunol. Pract. 2019, 7, 788–798. [Google Scholar] [CrossRef]
- Sędek, Ł.; Kulis, J.; Słota, Ł.; Twardoch, M.; Pierzyna-Świtała, M.; Perkowski, B.; Szczepański, T. The influence of fixation of biological samples on cell count and marker expression stability in flow cytometric analyses. Cent. Eur. J. Immunol. 2020, 45, 206–213. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
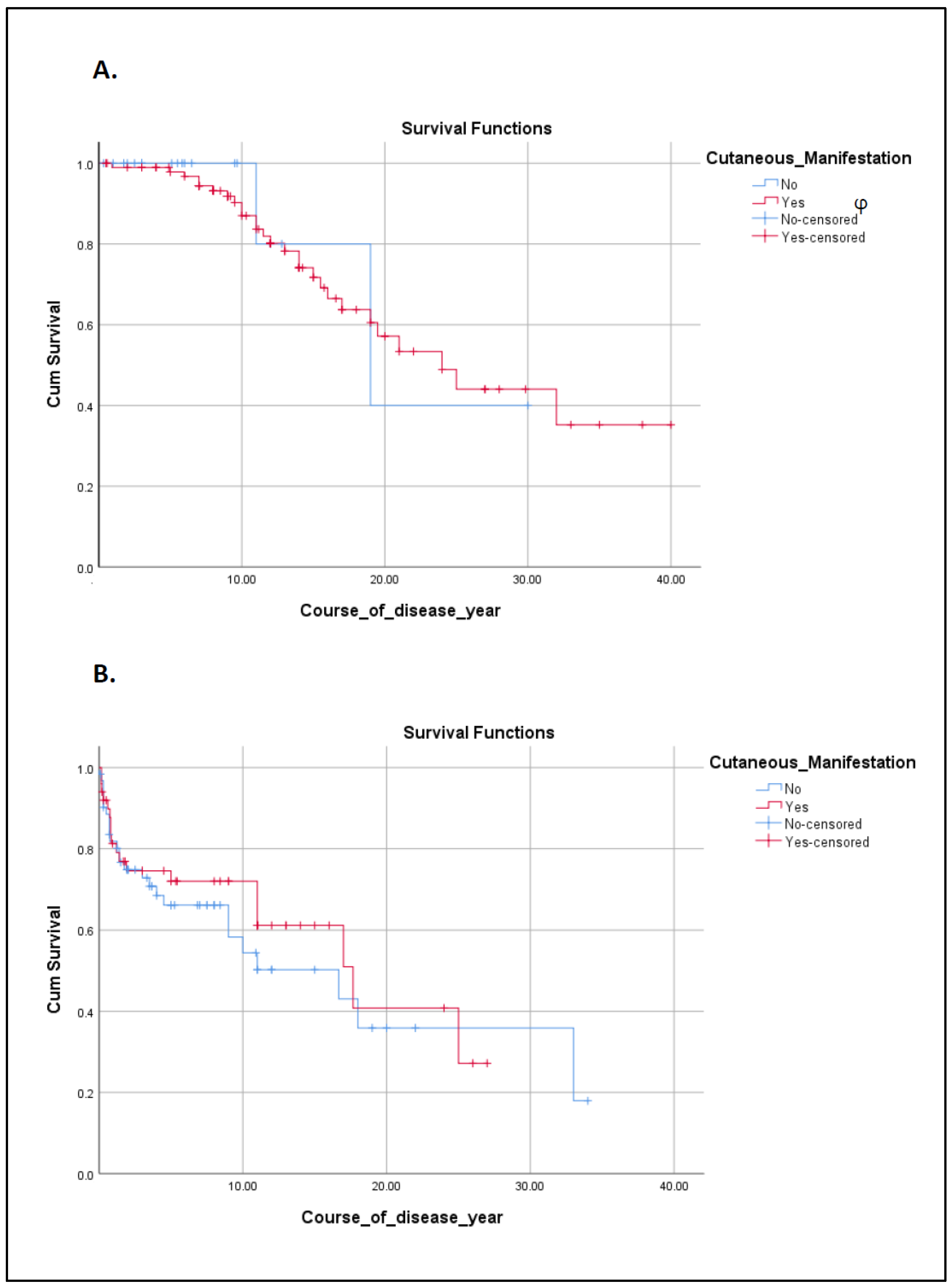{kind=link}
| Phenotypical Classifications | Presenting as a Primary Manifestation | Presenting as a Secondary Manifestation |
|---|---|---|
| Non-syndromic combined immunodeficiencies, n (%), (n = 55) | 29 (52.7%) | 26 (43.7%) |
| Syndromic combined immunodeficiencies, n (%), (n = 48) | 21 (45.7%) | 25 (54.3%) |
| Predominantly antibody deficiencies, n (%), (n = 16) | 4 (25%) | 12 (75%) |
| Diseases of immune dysregulation, n (%), (n = 33) | 5 (15.6%) | 27 (84.4%) |
| Congenital defects of phagocytes (number, function, or both), n (%), (n = 8) | 4 (50%) | 4 (50%) |
| Defects in intrinsic or innate immunity, n (%), (n = 5) | 3 (60%) | 2 (40%) |
| Cutaneous Manifestations | Total (n = 227) | Non-Syndromic Combined Immunodeficiencies (n = 55) | Syndromic Combined Immunodeficiencies (n = 106) | Predominantly Antibody Deficiencies (n = 16) | Diseases of Immune Dysregulation (n = 37) | Congenital Defects of Phagocytes (Number, Function, or Both), (n = 8) | Defects in Intrinsic or Innate Immunity (n = 5) | p-Value |
|---|---|---|---|---|---|---|---|---|
| Infectious, n (%) | 129 (56.8%) | 45 (81.8%) | 42 (39.6%) | 7 (43.8%) | 23 (62.2%) | 8 (100%) | 4 (80%) | <0.001 |
| Fungal infection, n | 35 | 19 | 4 | 1 | 8 | 1 | 2 | <0.001 |
| Viral infection, n | 16 | 3 | 4 | 0 | 7 | 2 | 0 | |
| Bacterial infection, n | 44 | 13 | 15 | 4 | 6 | 4 | 2 | |
| Non-infectious, n (%) | 188 (82.8%) | 41 (74.5%) | 101 (95.3%) | 11 (68.8%) | 30 (81.1%) | 3 (37.5%) | 2 (40%) | <0.001 |
| Eczema α, n (%) | 57 (33.5%) | 23 (41.8%) | 20 (40.8%) | 0 | 12 (32.4%) | 1 (12.5%) | 1 (20%) | 0.009 |
| Vitiligo β, n (%) | 7 (4.1%) | 2 (3.6%) | 1 (2%) | 1 (6.3%) | 3 (8.1%) | 0 | 0 | 0.686 |
| Systemic lupus erythematous, n (%) | 3 (1.8%) | 1 (1.8%) | 1 (2%) | 1 (6.3%) | 0 | 0 | 0 | 0.634 |
| Alopecia, n (%) | 10 (5.8%) | 0 | 1 (2%) | 1 (6.3%) | 8 (21.6%) | 0 | 0 | 0.001 |
| Psoriasis, n (%) | 5 (2.9%) | 0 | 1 (2%) | 2 (12.5%) | 1 (2.7%) | 0 | 1 (20%) | 0.031 |
| Ulcer, n (%) | 4 (2.4%) | 2 (3.6%) | 2 (4.1%) | 0 | 0 | 0 | 0 | 0.795 |
| Hyperpigmentation, n (%) | 7 (4.1%) | 3 (5.5%) | 4 (8.2%) | 0 | 0 | 0 | 0 | 0.504 |
| Hypopigmentation β, n (%) | 6 (3.5%) | 0 | 0 | 0 | 6 (16.2%) | 0 | 0 | 0.003 |
| Vascular-related lesion, n (%) | 77 (33.9%) | 2 (3.6%) | 74 (70.9%) | 0 | 2 (5.4%) | 0 | 0 | <0.001 |
| Atopic dermatitis α, n (%) | 13 (7.9%) | 3 (5.5%) | 5 (10.2%) | 0 | 5 (13.5%) | 0 | 0 | 0.570 |
| Blister, n (%) | 3 (1.8%) | 1 (1.8%) | 2 (4.1%) | 0 | 0 | 0 | 0 | 0.786 |
| Urticaria, n (%) | 5 (2.9%) | 0 | 1 (2%) | 1 (6.3%) | 1 (2.7%) | 2 (25%) | 0 | 0.020 |
| Undefined skin disorders ɣ | 56 (32.9%) | 14 (25.5%) | 25 (51%) | 5 (31.3%) | 9 (24.3%) | 2 (25%) | 1 (20%) | 0.068 |
| Phenotypical IEI Classifications | Total Number of Patients | Patients with Cutaneous Involvement | Skin Infections | Skin Atopic Manifestations | Skin Autoimmune Manifestations | Other Skin Manifestations |
|---|---|---|---|---|---|---|
| Non-syndromic combined immunodeficiencies | 126 | 55 (44%) | 45 (81.8%) | 25 (45.5%) | 2 (3.6%) | 17 (30.9%) |
| T-B− SCID | ||||||
| RAG1/RAG2 deficiency | 21 | 9 (42.8%) | 9 | 1 | 0 | 6 |
| ADA deficiency | 8 | 5 (62.5%) | 5 | 1 | 0 | 2 |
| Artemis deficiency | 7 | 1 (14.2%) | 1 | 0 | 0 | 1 |
| Cernnunos/XLF deficiency | 2 | 1 (50%) | 1 | 0 | 0 | 0 |
| T-B+ SCID | ||||||
| No γδ T cells [CD3E(6), CD3D(1)] | 7 | 3 (42.8%)(CD3E, CD3D) | 3 | 1 | 0 | 0 |
| JAK-3 deficiency | 6 | 3 (50%) | 3 | 3 | 0 | 0 |
| gc deficiency | 5 | 3 (60%) | 2 | 0 | 0 | 1 |
| Normal γδ T cells (PTPRC) | 1 | 1 (100%) | 1 | 0 | 0 | - |
| Others | ||||||
| CD40 ligand deficiency | 26 | 3 (11.5%) | 2 | 0 | 1 | 0 |
| DOCK8 deficiency | 25 | 21 (84%) | 14 | 17 | 1 | 5 |
| MHC class II deficiency [RFX5(2), RFXAP(1), RFXANK(4), CIITA(1)] | 8 | 3 (37.5%)(RFXANK, RFXAP, RFX5) | 3 | 1 | 0 | 1 |
| IL-21R deficiency | 2 | 0 | - | - | - | - |
| IKBKB deficiency | 2 | 1 (50%) | 0 | 1 | 0 | 0 |
| ZAP-70 deficiency | 1 | 0 | - | - | - | - |
| ICOS deficiency | 1 | 0 | - | - | - | - |
| CARD11 deficiency | 1 | 0 | - | - | - | - |
| DOCK2 deficiency | 1 | 0 | - | - | - | - |
| ITK deficiency | 1 | 0 | - | - | - | - |
| MALT1 deficiency | 1 | 1 (100%) | 1 | 0 | 0 | 1 |
| Predominantly antibody deficiencies | 108 | 16 (15%) | 7 (43.8%) | 1 (6.3%) | 4 (25%) | 5 (31.2%) |
| BTK deficiency (x-linked agammaglobulinemia) | 70 | 7 (of 69 cases with data, 10.1%) | 4 | 1 | 0 | 2 |
| IGHM deficiency (Mu heavy chain deficiency) | 13 | 3 (23.1%) | 1 | 0 | 2 | 0 |
| AID deficiency | 9 | 0 | - | - | - | - |
| PIK3R1 deficiency | 5 | 1 (20%) | 1 | 0 | 0 | 0 |
| NFKB1 deficiency | 3 | 1 (33.3%) | 0 | 0 | 0 | 1 |
| BAFF receptor deficiency | 3 | 3 (100%) | 1 | 0 | 1 | 2 |
| BLNK deficiency | 2 | 1 (50%) | 0 | 0 | 1 | 0 |
| Igα deficiency (CD79A) | 2 | 0 | - | - | - | - |
| TACI deficiency | 1 | 0 | ||||
| Syndromic combined immunodeficiencies | 131 | 106 (80.9%) | 42 (39.6%) | 22 (20.7%) | 3 (2.9%) | 86 (83.5%) |
| ATM deficiency (ataxia telangiectasia) | 78 | 74 (94.9%) | 17 | 4 | 2 | 72 |
| STAT3 deficiency (hyper IgE syndrome) | 20 | 17 (85%) | 14 | 12 | 1 | 7 |
| DNMT3B | 11 | 3 (27.2%) | 2 | 0 | 0 | 1 |
| ZBTBT24 | 6 | 3 (50%) | 1 | 0 | 0 | 3 |
| WAS deficiency (Wiskott-Aldrich syndrome) | 11 | 7 (63.6%) | 7 | 5 | 0 | 1 |
| ARPC1B deficiency | 2 | 1 (50%) | 1 | 1 | 0 | 1 |
| TTC7A deficiency (IEI with multiple intestinal atresias) | 1 | 0 | - | - | - | - |
| Purine nucleoside phosphorylase deficiency | 1 | 0 | - | - | - | - |
| IKBKG deficiency(NEMO deficiency) | 1 | 1 (100%) | 0 | 0 | 0 | 1 |
| Diseases of immune dysregulation | 107 | 38 (35.5%) | 23 (60.5%) | 14 (36.8%) | 12 (31.5%) | 13 (34.2%) |
| LRBA deficiency | 42 | 12 (28.5%) | 5 | 3 | 4 | 3 |
| IL-10RB deficiency | 13 | 3 (of 12 cases with data, 25%) | 3 | 1 | 0 | 0 |
| CD27 deficiency | 9 | 2 (22.2%) | 2 | 2 | 0 | 1 |
| RAB27A deficiency (Griscelli Sd type 2) | 6 | 3 (of 3 cases with data, 33.3%) | 1 | 1 | 0 | 3 |
| CD70 deficiency | 5 | 2 (40%) | 2 | 0 | 1 | 0 |
| AIRE deficiency (APS-1) | 4 | 4 (100%) | 3 | 0 | 2 | 0 |
| FOXP3 deficiency (immune dysregulation, polyendocrinopathy, enteropathy X-linked) | 3 | 2 (of 2 cases with data, 100%) | 2 | 2 | 1 | 1 |
| CTLA4 deficiency | 3 | 1 (33.3%) | 1 | 0 | 1 | 0 |
| XIAP deficiency (XLP2) | 3 | 2 (66.6%) | 1 | 1 | 2 | 1 |
| AP3B1 deficiency (Hermansky Pudlak syndrome type 2) | 2 | 2 | 0 | 0 | 0 | 2 |
| STAT3 gain of function mutation | 2 | 0 | 0 | 0 | 0 | 0 |
| TNFRSF6 deficiency (ALPS syndrome) | 2 | 1 (50%) | 1 | 1 | 0 | 0 |
| STXBP2 or Munc18-2 deficiency | 1 | 0 | - | - | - | - |
| SH2D1A deficiency (XLP1) | 1 | 1 (100%) | 0 | 1 | 1 | 0 |
| UNC13D or Munc13-4 deficiency | 1 | 0 | - | - | - | - |
| ITCH deficiency | 1 | 1 (100%) | 1 | 1 | 0 | 1 |
| PRF1 or perforin deficiency | 1 | 0 | - | - | - | - |
| PRKCD deficiency | 1 | 0 | - | - | - | - |
| RLTPR or CARMIL2 deficiency | 1 | 1 (100%) | 1 | 1 | 0 | 0 |
| RASGRP1 deficiency | 1 | 0 | - | - | - | - |
| RIPK1 deficiency | 1 | - | - | - | - | - |
| TPP2 or tripeptidyl-peptidase II deficiency | 2 | 0 | - | - | - | - |
| LYST deficiency (Chediak-Higashi syndrome) | 1 | 1 | 0 | 0 | 0 | 1 |
| CD25 deficiency | 1 | 0 | ||||
| Congenital defects of phagocyte number, function or both | 22 | 8 (38.1%) | 8 (100%) | 2 (25%) | 0 | 2 (25%) |
| CGD [CYBA(1), NCF1(2), NCF2(1)] | 4 | 1 (25%)(CYBA) | 1 | 0 | 0 | 0 |
| Elastase deficiency | 4 | 1 (25%) | 1 | 0 | 0 | 0 |
| Kostmann disease | 4 | 1 (25%) | 1 | 0 | 0 | 1 |
| Rac 2 deficiency | 3 | 2 (66.6%) | 2 | 2 | 0 | 1 |
| Leukocyte adhesion deficiency I | 2 | 1 (50%) | 1 | 0 | 0 | 0 |
| G-CSF receptor deficiency | 1 | 0 | - | - | - | - |
| VPS45 deficiency | 1 | 1 (100%) | 1 | 0 | 0 | 0 |
| GFI 1 deficiency | 1 | 0 | - | - | - | - |
| G6PD deficiency | 1 | 1 (100%) | 1 | 0 | 0 | 0 |
| Glycogen storage disease type 1b | 1 | 0 | - | - | - | - |
| Defects in intrinsic and innate immunity | 23 | 5 (21.7%) | 4 (80%) | 1 (20%) | 1 (20%) | 1 (20%) |
| IL-12RB1 deficiency | 19 | 3 (15.7%) | 2 | 1 | 1 | 1 |
| STAT1 deficiency | 2 | 2 (100%) | 2 | 0 | 0 | 0 |
| TYK2 deficiency | 1 | 0 | - | - | - | - |
| IL-17RA deficiency | 2 | 0 | - | - | - | - |
| Autoinflammatory disorders | 3 | 0 | - | - | - | - |
| NLRP1 deficiency | 1 | 0 | - | - | - | - |
| PLCG2 deficiency (familial cold autoinflammatory syndrome) | 1 | 0 | - | - | - | - |
| MVK or mevalonate kinase deficiency | 1 | 0 | - | - | - | - |
| Complement deficiencies | 1 | 0 | - | - | - | - |
| FCN3 or ficolin 3 deficiency | 1 | 0 | - | - | - | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sharifinejad, N.; Azizi, G.; Rasouli, S.E.; Chavoshzadeh, Z.; Mahdaviani, S.A.; Tavakol, M.; Sadri, H.; Nabavi, M.; Ebrahimi, S.S.; Shirkani, A.; et al. Autoimmune versus Non-autoimmune Cutaneous Features in Monogenic Patients with Inborn Errors of Immunity. Biology 2023, 12, 644. https://doi.org/10.3390/biology12050644
Sharifinejad N, Azizi G, Rasouli SE, Chavoshzadeh Z, Mahdaviani SA, Tavakol M, Sadri H, Nabavi M, Ebrahimi SS, Shirkani A, et al. Autoimmune versus Non-autoimmune Cutaneous Features in Monogenic Patients with Inborn Errors of Immunity. Biology. 2023; 12(5):644. https://doi.org/10.3390/biology12050644
Chicago/Turabian StyleSharifinejad, Niusha, Gholamreza Azizi, Seyed Erfan Rasouli, Zahra Chavoshzadeh, Seyed Alireza Mahdaviani, Marzieh Tavakol, Homa Sadri, Mohammad Nabavi, Sareh Sadat Ebrahimi, Afshin Shirkani, and et al. 2023. "Autoimmune versus Non-autoimmune Cutaneous Features in Monogenic Patients with Inborn Errors of Immunity" Biology 12, no. 5: 644. https://doi.org/10.3390/biology12050644