
Insights into the Binding of Receptor-Binding Domain (RBD) of SARS-CoV-2 Wild Type and B.1.620 Variant with hACE2 Using Molecular Docking and Simulation Approaches

 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Material and Methods
2.1. Structure Retrieval and Mutants’ Modeling
2.2. Modeling the RBD and ACE2 Complexes through Docking
2.3. Dynamics of the Wild-Type and B.1.620 Complexes
2.4. Post-Simulation Trajectory Analysis
2.5. Binding Energy Differences Estimation
3. Results and Discussion
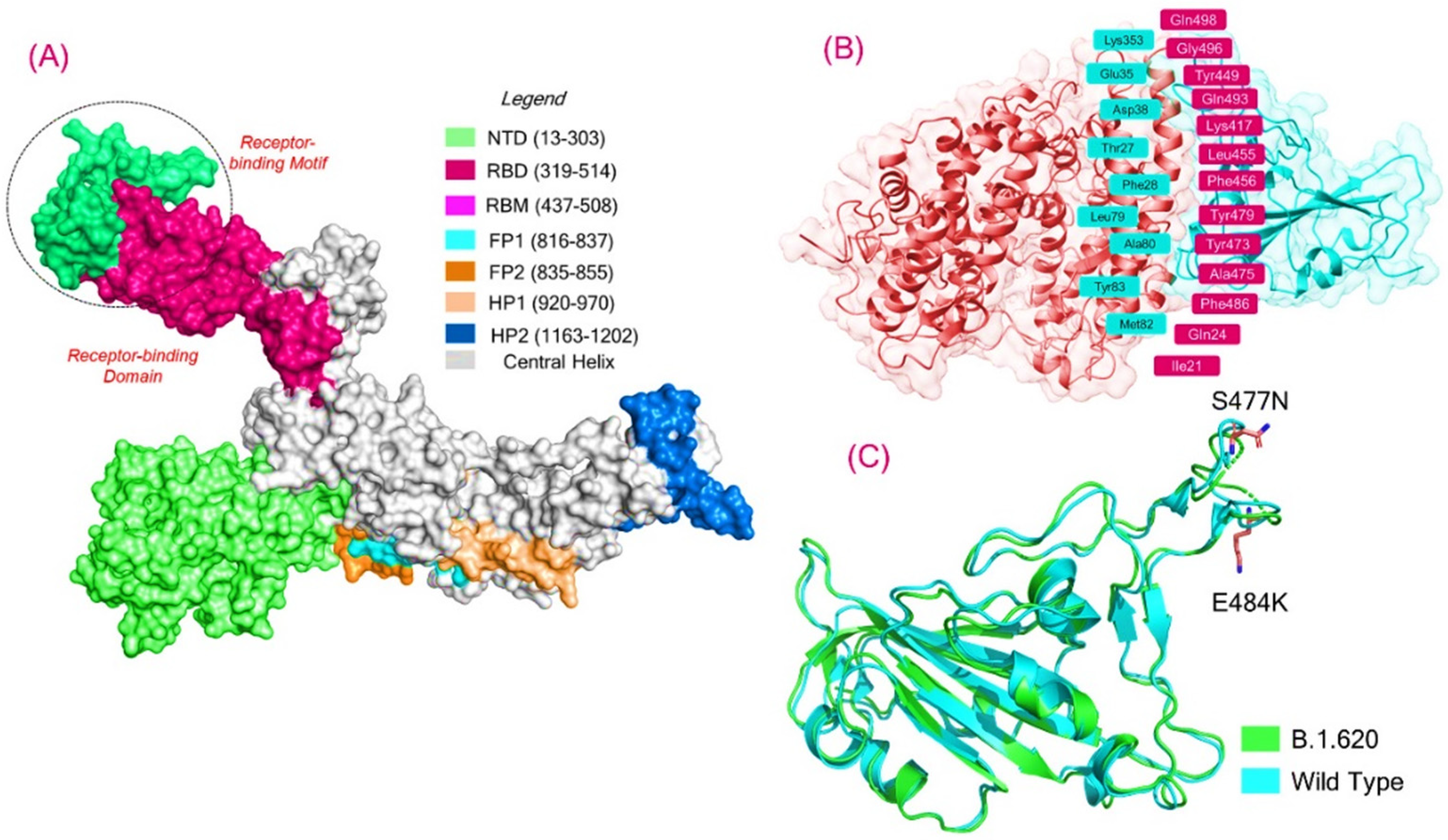
3.1. Structural Modeling and Analysis
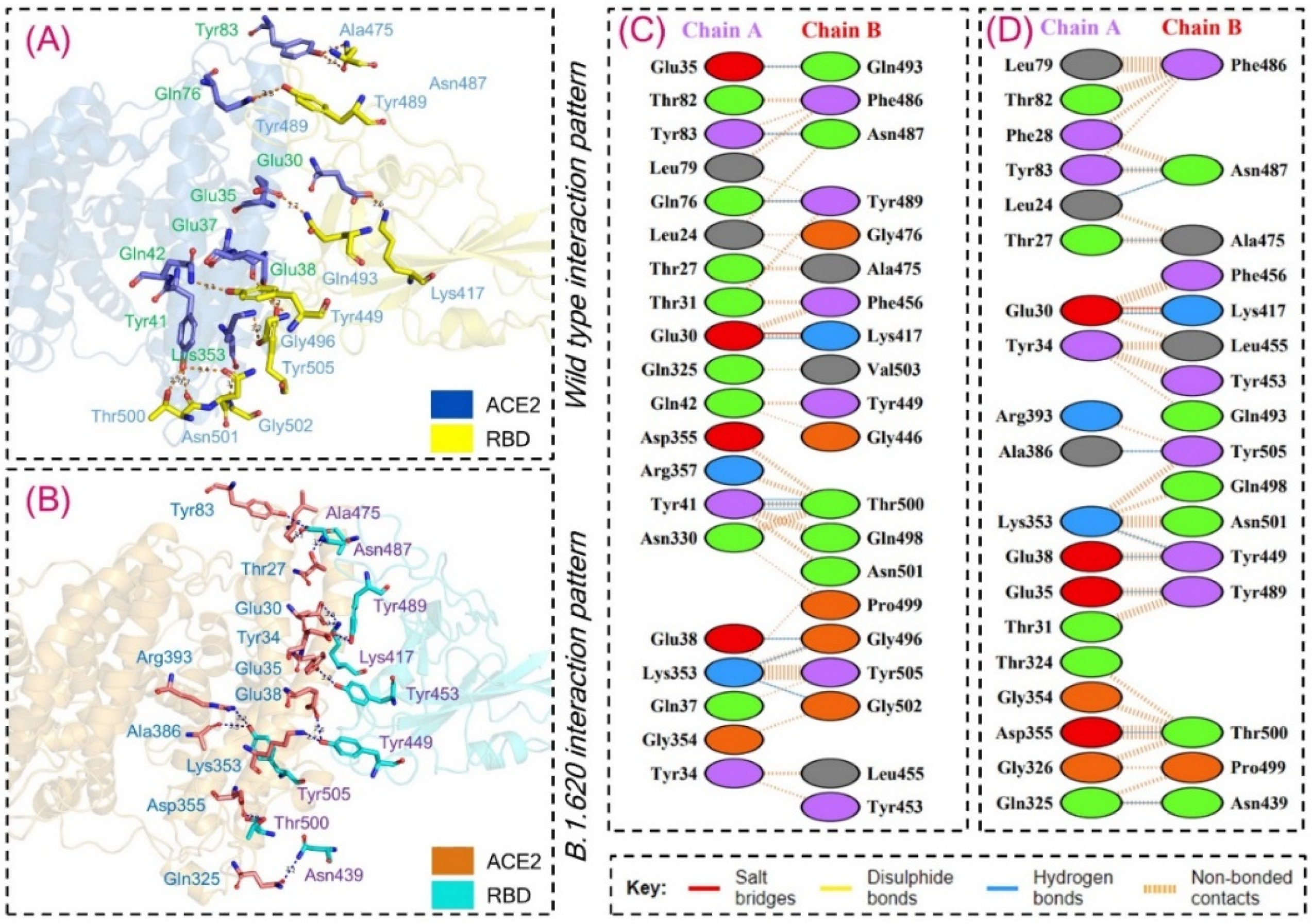
3.2. Interaction Energy and Hydrogen Bonding Network Analysis
3.3. Structural/Dynamic Features Investigation
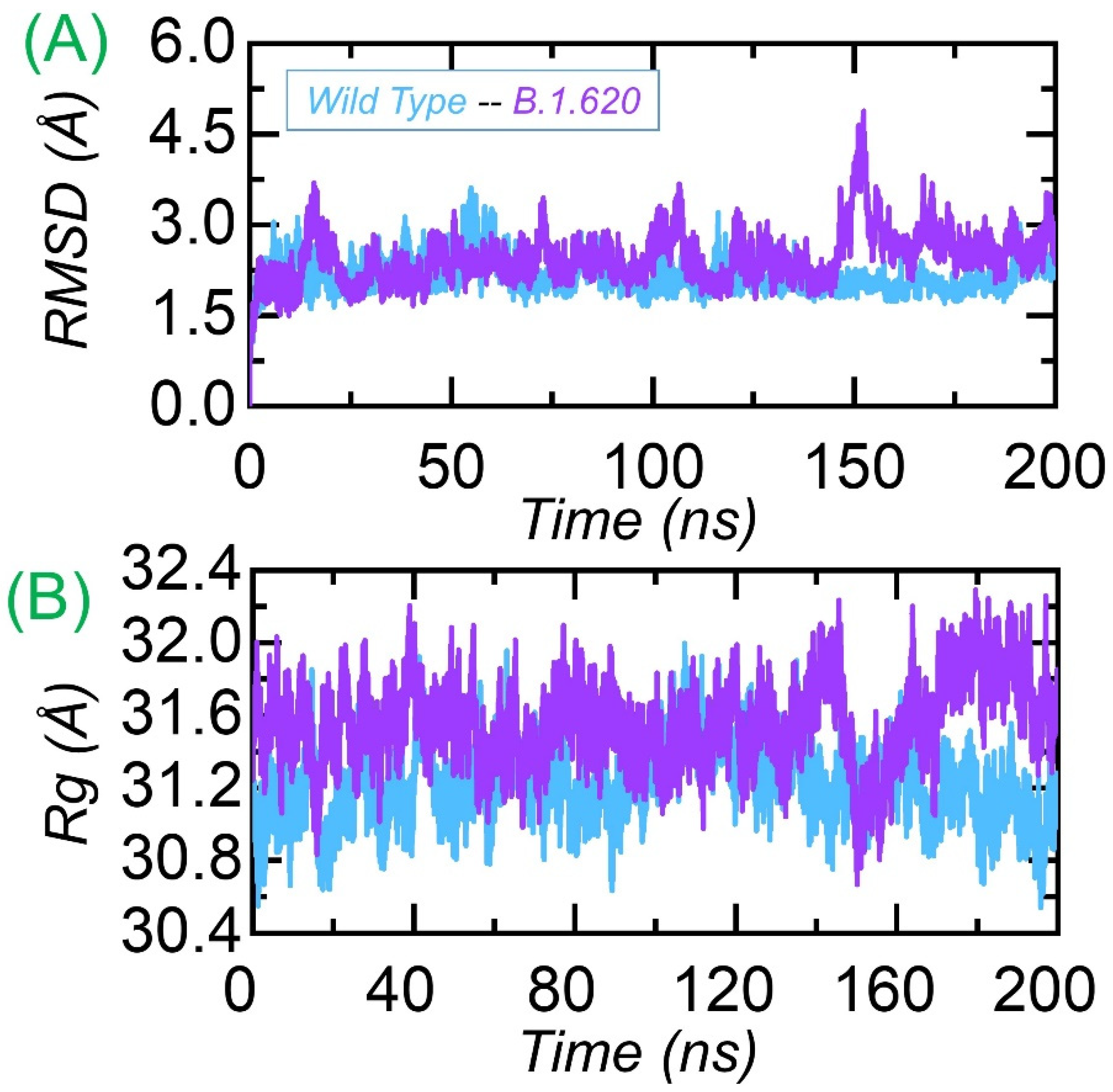
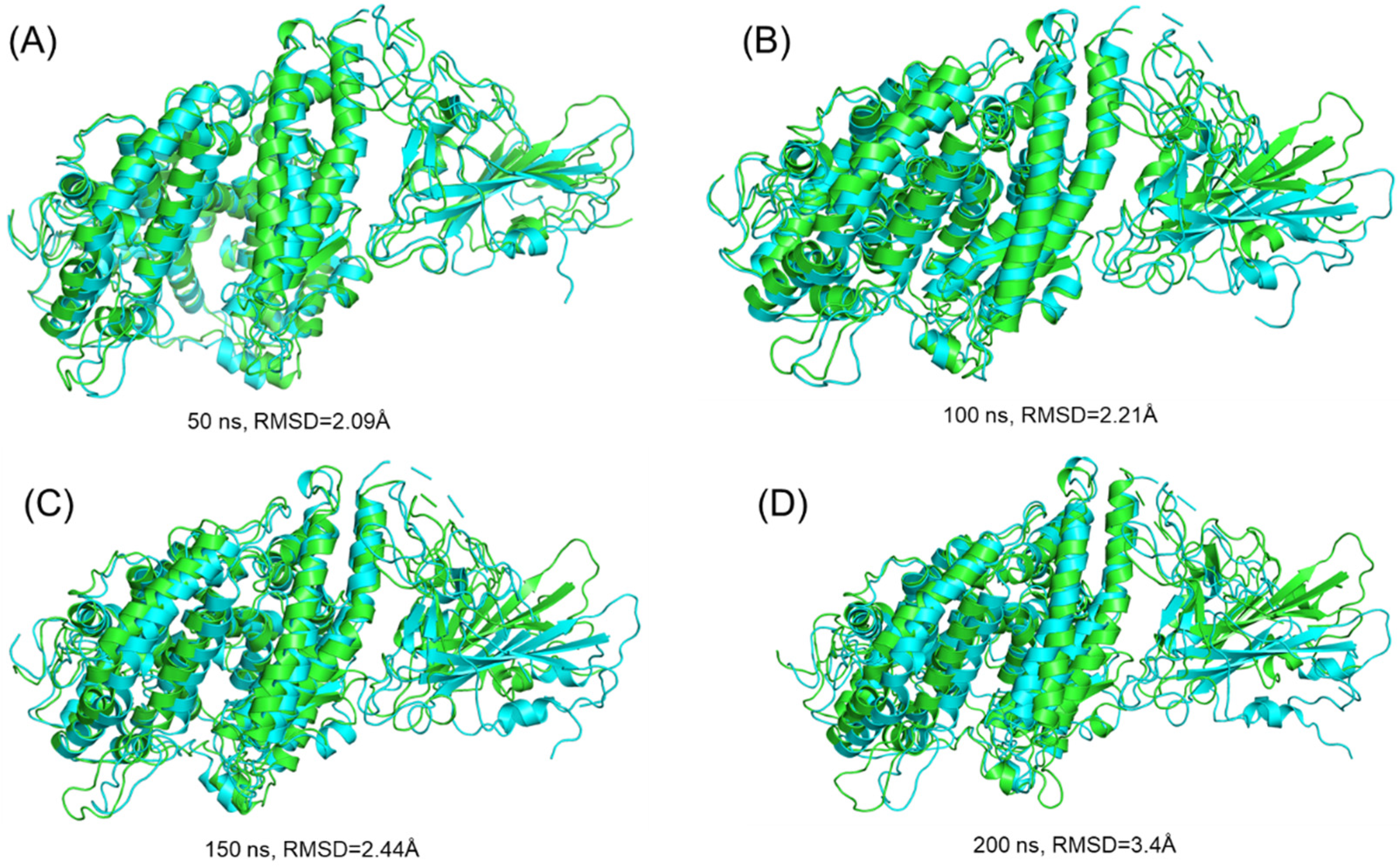
3.3.1. Dynamic Stability Calculation
3.3.2. Structural Compactness Analysis
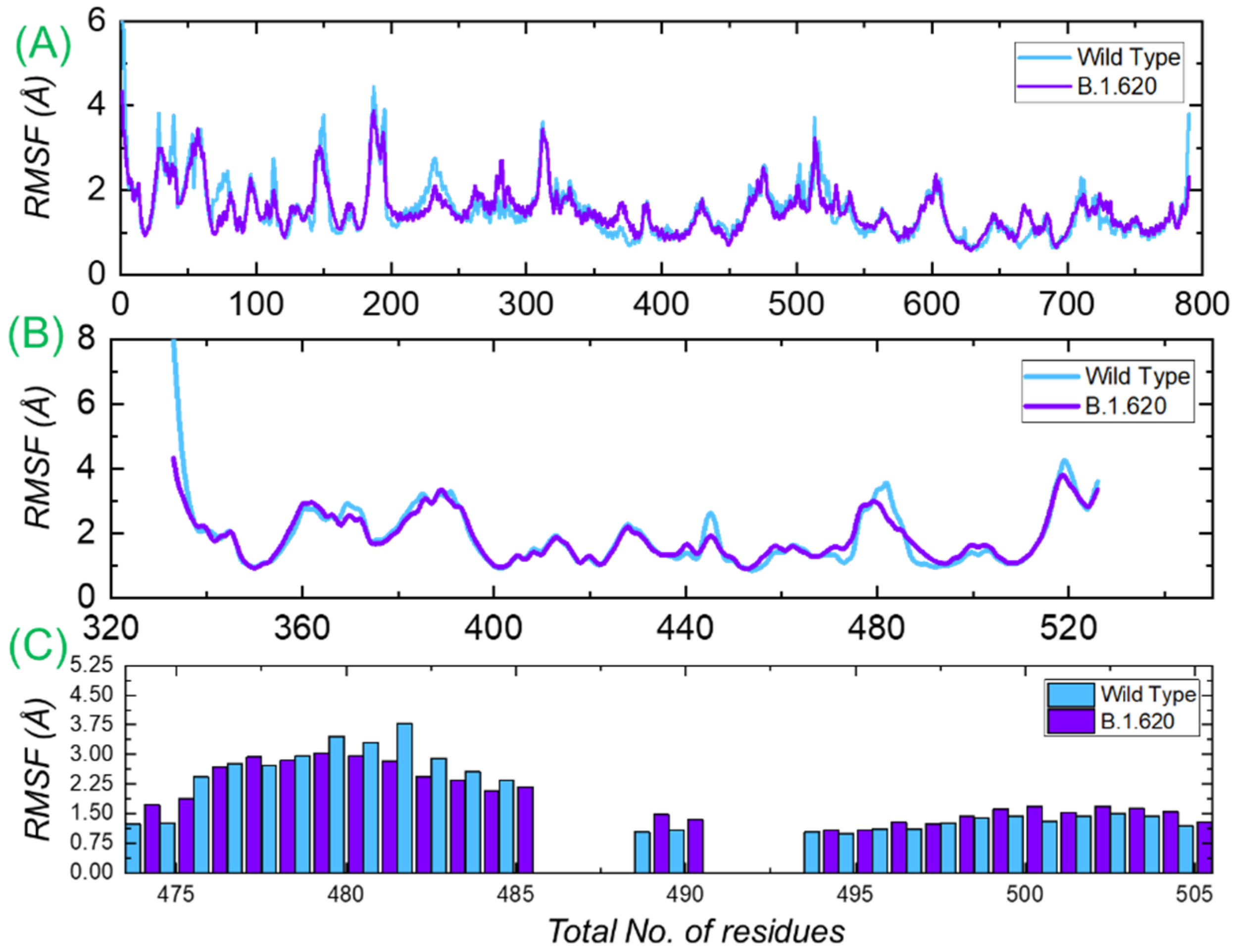
3.3.3. Residual Flexibility Analysis
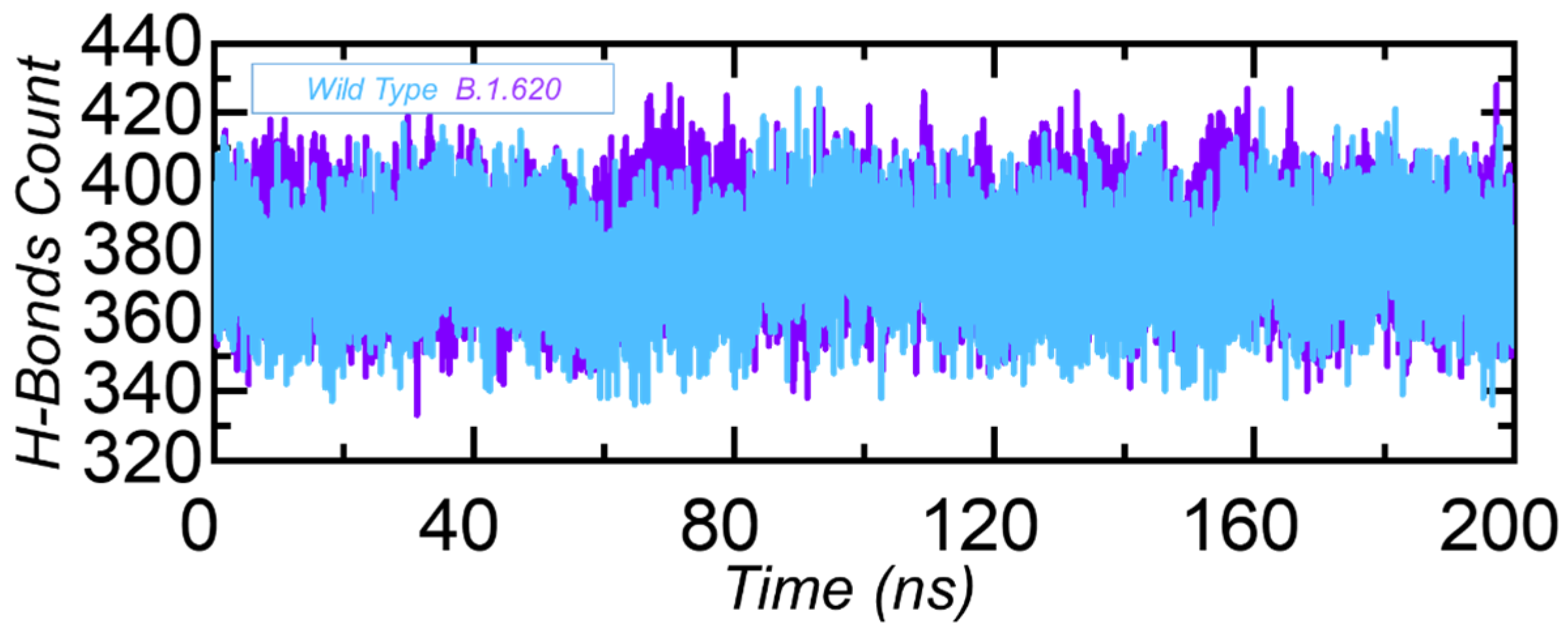
3.3.4. Hydrogen Bonding Analysis
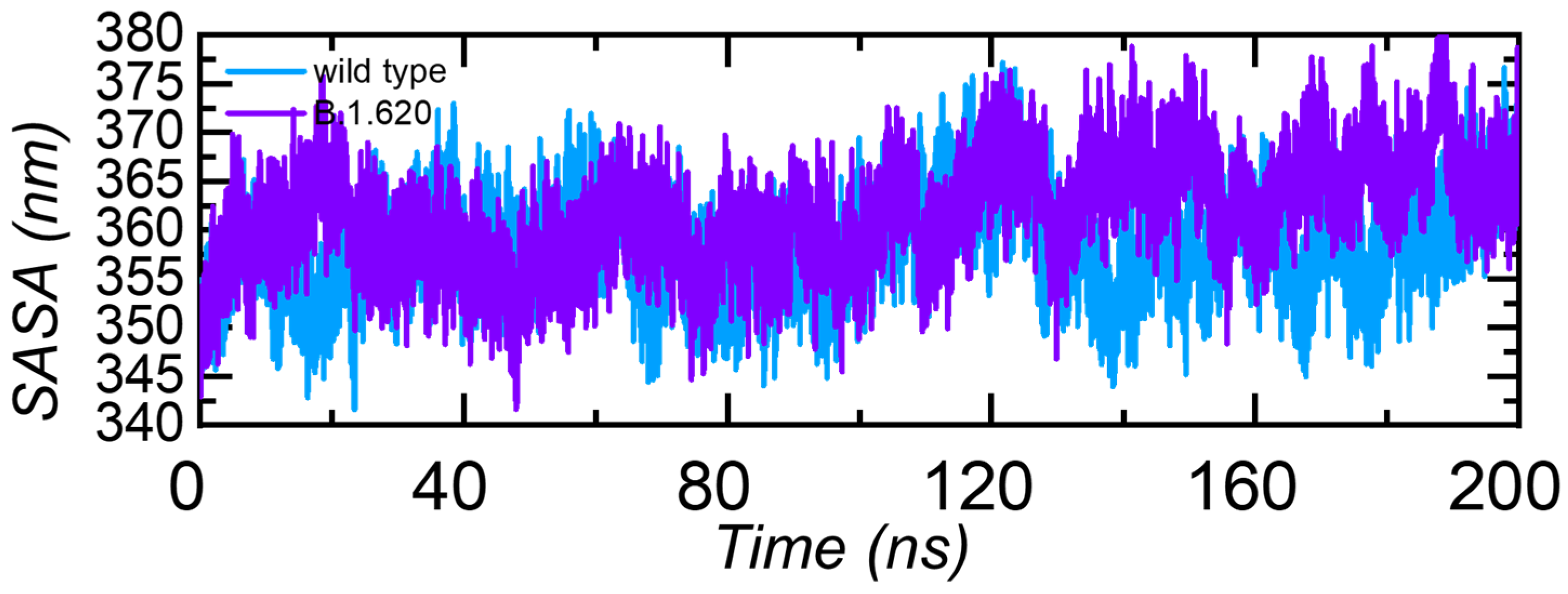
3.3.5. Solvent Accessible Surface Area (SASA)
3.4. Binding Free Energy Calculation
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.-M.; Wang, W.; Song, Z.-G.; Hu, Y.; Tao, Z.-W.; Tian, J.-H.; Pei, Y.-Y. A new coronavirus associated with human respiratory disease in china. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R. A novel coronavirus from patients with pneumonia in china, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef] [PubMed]
- ul Qamar, M.T.; Alqahtani, S.M.; Alamri, M.A.; Chen, L.-L. Structural basis of SARS-CoV-2 3clpro and anti-COVID-19 drug discovery from medicinal plants. J. Pharm. Anal. 2020, 10, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Guan, W.-j.; Ni, Z.-y.; Hu, Y.; Liang, W.-h.; Ou, C.-q.; He, J.-x.; Liu, L.; Shan, H.; Lei, C.-l.; Hui, D.S. Clinical characteristics of 2019 novel coronavirus infection in china. MedRxiv 2020, 382, 1708–1720. [Google Scholar]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X. Clinical features of patients infected with 2019 novel coronavirus in wuhan, china. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Haque, S.M.; Ashwaq, O.; Sarief, A.; Azad John Mohamed, A.K. A comprehensive review about SARS-CoV-2. Future Virol. 2020, 15, 625–648. [Google Scholar] [CrossRef]
- Weiss, S.R.; Leibowitz, J.L. Coronavirus pathogenesis. In Advances in Virus Research; Elsevier: Amsterdam, The Netherlands, 2011; Volume 81, pp. 85–164. [Google Scholar]
- Alamri, M.A.; ul Qamar, M.T.; Afzal, O.; Alabbas, A.B.; Riadi, Y.; Alqahtani, S.M. Discovery of anti-mers-cov small covalent inhibitors through pharmacophore modeling, covalent docking and molecular dynamics simulation. J. Mol. Liq. 2021, 330, 115699. [Google Scholar] [CrossRef]
- Alamri, M.A.; ul Qamar, M.T.; Mirza, M.U.; Alqahtani, S.M.; Froeyen, M.; Chen, L.-L. Discovery of human coronaviruses pan-papain-like protease inhibitors using computational approaches. J. Pharm. Anal. 2020, 10, 546–559. [Google Scholar] [CrossRef]
- Duchene, S.; Featherstone, L.; Haritopoulou-Sinanidou, M.; Rambaut, A.; Lemey, P.; Baele, G. Temporal signal and the phylodynamic threshold of SARS-CoV-2. Virus Evol. 2020, 6, veaa061. [Google Scholar] [CrossRef]
- Hussain, I.; Pervaiz, N.; Khan, A.; Saleem, S.; Shireen, H.; Wei, D.-Q.; Labrie, V.; Bao, Y.; Abbasi, A.A. Evolutionary and structural analysis of SARS-CoV-2 specific evasion of host immunity. Genes Immun. 2020, 21, 409–419. [Google Scholar] [CrossRef]
- Peacock, T.P.; Goldhill, D.H.; Zhou, J.; Baillon, L.; Frise, R.; Swann, O.C.; Kugathasan, R.; Penn, R.; Brown, J.C.; Sanchez-David, R.Y. The furin cleavage site in the SARS-CoV-2 spike protein is required for transmission in ferrets. Nat. Microbiol. 2021, 6, 899–909. [Google Scholar] [CrossRef]
- Mehmood, I.; Ijaz, M.; Ahmad, S.; Ahmed, T.; Bari, A.; Abro, A.; Allemailem, K.S.; Almatroudi, A.; Tahir ul Qamar, M. SARS-CoV-2: An update on genomics, risk assessment, potential therapeutics and vaccine development. Int. J. Environ. Res. Public Health 2021, 18, 1626. [Google Scholar] [CrossRef]
- Muhseen, Z.T.; Hameed, A.R.; Al-Hasani, H.M.; ul Qamar, M.T.; Li, G. Promising terpenes as SARS-CoV-2 spike receptor-binding domain (rbd) attachment inhibitors to the human ace2 receptor: Integrated computational approach. J. Mol. Liq. 2020, 320, 114493. [Google Scholar] [CrossRef] [PubMed]
- Tareq, A.M.; Emran, T.B.; Dhama, K.; Dhawan, M.; Tallei, T.E. Impact of SARS-CoV-2 delta variant (b.1.617.2) in surging second wave of COVID-19 and efficacy of vaccines in tackling the ongoing pandemic. Hum. Vaccines Immunother. 2021, 1–2. [Google Scholar] [CrossRef]
- Celik, I.; Yadav, R.; Duzgun, Z.; Albogami, S.; El-Shehawi, A.M.; Fatimawali; Idroes, R.; Tallei, T.E.; Emran, T.B. Interactions of the receptor binding domain of SARS-CoV-2 variants with hace2: Insights from molecular docking analysis and molecular dynamic simulation. Biology 2021, 10, 880. [Google Scholar] [CrossRef]
- Plante, J.A.; Mitchell, B.M.; Plante, K.S.; Debbink, K.; Weaver, S.C.; Menachery, V.D. The variant gambit: COVID’s next move. Cell Host Microbe 2021, 29, 508–515. [Google Scholar] [CrossRef] [PubMed]
- Peacock, T.P.; Penrice-Randal, R.; Hiscox, J.A.; Barclay, W.S. SARS-CoV-2 one year on: Evidence for ongoing viral adaptation. J. Gen. Virol. 2021, 102, 001584. [Google Scholar] [CrossRef]
- Zhang, L.; Jackson, C.B.; Mou, H.; Ojha, A.; Rangarajan, E.S.; Izard, T.; Farzan, M.; Choe, H. The d614g mutation in the SARS-CoV-2 spike protein reduces s1 shedding and increases infectivity. bioRxiv 2020. [Google Scholar] [CrossRef]
- Kannan, S.R.; Spratt, A.N.; Cohen, A.R.; Naqvi, S.H.; Chand, H.S.; Quinn, T.P.; Lorson, C.L.; Byrareddy, S.N.; Singh, K. Evolutionary analysis of the delta and delta plus variants of the SARS-CoV-2 viruses. J. Autoimmun. 2021, 124, 102715. [Google Scholar] [CrossRef] [PubMed]
- Messali, S.; Bertelli, A.; Campisi, G.; Zani, A.; Ciccozzi, M.; Caruso, A.; Caccuri, F. A cluster of the new SARS-CoV-2 b. 1.621 lineage in italy and sensitivity of the viral isolate to the bnt162b2 vaccine. J. Med. Virol. 2021, 93, 6468–6470. [Google Scholar] [CrossRef]
- Wink, P.L.; Volpato, F.C.Z.; Monteiro, F.L.; Willig, J.B.; Zavascki, A.P.; Barth, A.L.; Martins, A.F. First identification of SARS-CoV-2 lambda (c. 37) variant in southern brazil. Infect. Control Hosp. Epidemiol. 2021, 1–2. [Google Scholar] [CrossRef]
- Liu, Z.; VanBlargan, L.A.; Bloyet, L.-M.; Rothlauf, P.W.; Chen, R.E.; Stumpf, S.; Zhao, H.; Errico, J.M.; Theel, E.S.; Liebeskind, M.J. Identification of SARS-CoV-2 spike mutations that attenuate monoclonal and serum antibody neutralization. Cell Host Microbe 2021, 29, 477–488.e474. [Google Scholar] [CrossRef]
- Yang, T.-J.; Yu, P.-Y.; Chang, Y.-C.; Chang, N.-E.; Tsai, Y.-X.; Liang, K.-H.; Draczkowski, P.; Lin, B.; Wang, Y.-S.; Chien, Y.-C. Structure-activity relationships of b. 1.617 and other SARS-CoV-2 spike variants. bioRxiv 2021. [Google Scholar] [CrossRef]
- Annavajhala, M.K.; Mohri, H.; Wang, P.; Nair, M.; Zucker, J.E.; Sheng, Z.; Gomez-Simmonds, A.; Kelley, A.L.; Tagliavia, M.; Huang, Y. Emergence and expansion of SARS-CoV-2 b. 1.526 after identification in new york. Nature 2021, 597, 703–708. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Zia, T.; Suleman, M.; Khan, T.; Ali, S.S.; Abbasi, A.A.; Mohammad, A.; Wei, D.Q. Higher infectivity of the SARS-CoV-2 new variants is associated with k417n/t, e484k, and n501y mutants: An insight from structural data. J. Cell Physiol. 2021, 236, 7045–7057. [Google Scholar] [CrossRef]
- Scheepers, C.; Everatt, J.; Amoako, D.G.; Tegally, H.; Wibmer, C.K.; Mnguni, A.; Ismail, A.; Mahlangu, B.; Lambson, B.E.; Richardson, S.I. Emergence and phenotypic characterization of c. 1.2, a globally detected lineage that rapidly accumulated mutations of concern. medRxiv 2021. [Google Scholar] [CrossRef]
- Kang, S.; Yang, M.; Hong, Z.; Zhang, L.; Huang, Z.; Chen, X.; He, S.; Zhou, Z.; Zhou, Z.; Chen, Q. Crystal structure of SARS-CoV-2 nucleocapsid protein rna binding domain reveals potential unique drug targeting sites. Acta Pharm. Sin. B 2020, 10, 1228–1238. [Google Scholar] [CrossRef]
- Tahir ul Qamar, M.; Rehman, A.; Tusleem, K.; Ashfaq, U.A.; Qasim, M.; Zhu, X.; Fatima, I.; Shahid, F.; Chen, L.-L. Designing of a next generation multiepitope based vaccine (mev) against SARS-CoV-2: Immunoinformatics and in silico approaches. PLoS ONE 2020, 15, e0244176. [Google Scholar] [CrossRef]
- Walls, A.C.; Park, Y.-J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell 2020, 181, 281–292. e286. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Gui, J.; Ahmad, W.; Haq, I.; Shahid, M.; Khan, A.A.; Shah, A.; Khan, A.; Ali, L.; Anwar, Z. The SARS-CoV-2 b. 1.618 variant slightly alters the spike rbd–ace2 binding affinity and is an antibody escaping variant: A computational structural perspective. RSC Adv. 2021, 11, 30132–30147. [Google Scholar] [CrossRef]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Moore, M.J.; Vasilieva, N.; Sui, J.; Wong, S.K.; Berne, M.A.; Somasundaran, M.; Sullivan, J.L.; Luzuriaga, K.; Greenough, T.C. Angiotensin-converting enzyme 2 is a functional receptor for the sars coronavirus. Nature 2003, 426, 450–454. [Google Scholar] [CrossRef] [Green Version]
- Bassi, D.E.; Zhang, J.; Renner, C.; Klein-Szanto, A.J. Targeting proprotein convertases in furin-rich lung cancer cells results in decreased in vitro and in vivo growth. Mol. Carcinog. 2017, 56, 1182–1188. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Khan, T.; Ali, S.; Aftab, S.; Wang, Y.; Qiankun, W.; Khan, M.; Suleman, M.; Ali, S.; Heng, W. SARS-CoV-2 new variants: Characteristic features and impact on the efficacy of different vaccines. Biomed. Pharmacother. 2021, 143, 112176. [Google Scholar] [CrossRef]
- Chi, X.; Yan, R.; Zhang, J.; Zhang, G.; Zhang, Y.; Hao, M.; Zhang, Z.; Fan, P.; Dong, Y.; Yang, Y. A neutralizing human antibody binds to the n-terminal domain of the spike protein of SARS-CoV-2. Science 2020, 369, 650–655. [Google Scholar] [CrossRef]
- Ali, M.J.; Hanif, M.; Haider, M.A.; Ahmed, M.U.; Sundas, F.; Hirani, A.; Khan, I.A.; Anis, K.; Karim, A.H. Treatment options for COVID-19: A review. Front. Med. 2020, 7, 480. [Google Scholar] [CrossRef]
- Khan, A.; Khan, S.; Saleem, S.; Nizam-Uddin, N.; Mohammad, A.; Khan, T.; Ahmad, S.; Arshad, M.; Ali, S.S.; Suleman, M. Immunogenomics guided design of immunomodulatory multi-epitope subunit vaccine against the SARS-CoV-2 new variants, and its validation through in silico cloning and immune simulation. Comput. Biol. Med. 2021, 133, 104420. [Google Scholar] [CrossRef] [PubMed]
- Buchanan, C.J.; Gaunt, B.; Harrison, P.J.; Le Bas, A.; Khan, A.; Giltrap, A.M.; Ward, P.N.; Dumoux, M.; Daga, S.; Picchiotti, N. Cryptic SARS-CoV-2-spike-with-sugar interactions revealed by ‘universal’ saturation transfer analysis. bioRxiv 2021. [Google Scholar] [CrossRef]
- Wang, P.; Casner, R.G.; Nair, M.S.; Wang, M.; Yu, J.; Cerutti, G.; Liu, L.; Kwong, P.D.; Huang, Y.; Shapiro, L. Increased resistance of SARS-CoV-2 variant p. 1 to antibody neutralization. Cell Host Microbe 2021, 29, 747–751.e744. [Google Scholar] [CrossRef] [PubMed]
- Starr, T.N.; Greaney, A.J.; Hilton, S.K.; Ellis, D.; Crawford, K.H.; Dingens, A.S.; Navarro, M.J.; Bowen, J.E.; Tortorici, M.A.; Walls, A.C. Deep mutational scanning of SARS-CoV-2 receptor binding domain reveals constraints on folding and ace2 binding. Cell 2020, 182, 1295–1310.e1220. [Google Scholar] [CrossRef]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ace2 receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef] [Green Version]
- Eswar, N.; Eramian, D.; Webb, B.; Shen, M.-Y.; Sali, A. Protein structure modeling with modeller. In Structural Proteomics; Springer: Berlin/Heidelberg, Germany, 2008; pp. 145–159. [Google Scholar]
- Dominguez, C.; Boelens, R.; Bonvin, A.M. Haddock: A protein− protein docking approach based on biochemical or biophysical information. J. Am. Chem. Soc. 2003, 125, 1731–1737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laskowski, R.A.; Jabłońska, J.; Pravda, L.; Vařeková, R.S.; Thornton, J.M. Pdbsum: Structural summaries of pdb entries. Protein Sci. 2018, 27, 129–134. [Google Scholar] [CrossRef]
- Case, D.A.; Aktulga, H.M.; Belfon, K.; Ben-Shalom, I.; Brozell, S.R.; Cerutti, D.; Cheatham, T., III; Cisneros, G.; Cruzeiro, V.; Darden, T. Amber 2021; University of California Press: Berkeley, CA, USA, 2021. [Google Scholar]
- Roe, D.R.; Cheatham, T.E., III. Ptraj and cpptraj: Software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef]
- Chen, F.; Liu, H.; Sun, H.; Pan, P.; Li, Y.; Li, D.; Hou, T. Assessing the performance of the mm/pbsa and mm/gbsa methods. 6. Capability to predict protein–protein binding free energies and re-rank binding poses generated by protein–protein docking. Phys. Chem. Chem. Phys. 2016, 18, 22129–22139. [Google Scholar] [CrossRef]
- Muneer, I.; Ahmad, S.; Naz, A.; Abbasi, S.W.; Alblihy, A.; Aloliqi, A.A.; Alkhayl, F.F.; Alrumaihi, F.; Ahmad, S.; El Bakri, Y. Discovery of novel inhibitors from medicinal plants for v-domain ig suppressor of t-cell activation (vista). Front. Mol. Biosci. 2021, 8, 716735. [Google Scholar] [CrossRef]
- Tahir ul Qamar, M.; Ahmad, S.; Khan, A.; Mirza, M.U.; Ahmad, S.; Abro, A.; Chen, L.-L.; Almatroudi, A.; Wei, D.-Q. Structural probing of hapr to identify potent phytochemicals to control vibrio cholera through integrated computational approaches. Comput. Biol. Med. 2021, 138, 104929. [Google Scholar] [CrossRef]
- Arif, R.; Ahmad, S.; Mustafa, G.; Mahrosh, H.S.; Ali, M.; Tahir ul Qamar, M.; Dar, H.R. Molecular docking and simulation studies of antidiabetic agents devised from hypoglycemic polypeptide-p of momordica charantia. BioMed Res. Int. 2021, 2021, 5561129. [Google Scholar] [CrossRef] [PubMed]
- Altharawi, A.; Ahmad, S.; Alamri, M.A.; ul Qamar, M.T. Structural insight into the binding pattern and interaction mechanism of chemotherapeutic agents with sorcin by docking and molecular dynamic simulation. Colloids Surf. B Biointerfaces 2021, 208, 112098. [Google Scholar] [CrossRef] [PubMed]
- Suleman, M.; ul Qamar, M.T.; Shoaib Saleem, S.A.; Ali, S.S.; Khan, H.; Akbar, F.; Khan, W.; Alblihy, A.; Alrumaihi, F.; Waseem, M. Mutational landscape of pirin and elucidation of the impact of most detrimental missense variants that accelerate the breast cancer pathways: A computational modelling study. Front. Mol. Biosci. 2021, 8, 692835. [Google Scholar] [CrossRef]
- Tahir Ul Qamar, M.; Shahid, F.; Aslam, S.; Ashfaq, U.A.; Aslam, S.; Fatima, I.; Fareed, M.M.; Zohaib, A.; Chen, L.-L. Reverse vaccinology assisted designing of multiepitope-based subunit vaccine against SARS-CoV-2. Infect. Dis. Poverty 2020, 9, 132. [Google Scholar] [CrossRef] [PubMed]
- Vakser, I.A. Protein-protein docking: From interaction to interactome. Biophys. J. 2014, 107, 1785–1793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, A.; Wei, D.-Q.; Kousar, K.; Abubaker, J.; Ahmad, S.; Ali, J.; Al-Mulla, F.; Ali, S.S.; Nizam-Uddin, N.; Sayaf, A.M. Preliminary structural data revealed that the SARS-CoV-2 b. 1.617 variant’s rbd binds to ace2 receptor stronger than the wild type to enhance the infectivity. ChemBioChem 2021, 22, 2641–2649. [Google Scholar] [CrossRef]
- Chen, D.; Oezguen, N.; Urvil, P.; Ferguson, C.; Dann, S.; Savidge, T. Regulation of protein-ligand binding affinity by hydrogen bond pairing. Sci. Adv. 2016, 2, e1501240. [Google Scholar] [CrossRef] [Green Version]
- Chodera, J.D.; Mobley, D.L. Entropy-enthalpy compensation: Role and ramifications in biomolecular ligand recognition and design. Annu. Rev. Biophys. 2013, 42, 121–142. [Google Scholar] [CrossRef] [Green Version]
- Patil, R.; Das, S.; Stanley, A.; Yadav, L.; Sudhakar, A.; Varma, A.K. Optimized hydrophobic interactions and hydrogen bonding at the target-ligand interface leads the pathways of drug-designing. PLoS ONE 2010, 5, e12029. [Google Scholar] [CrossRef]
- Olsson, T.S.; Ladbury, J.E.; Pitt, W.R.; Williams, M.A. Extent of enthalpy–entropy compensation in protein–ligand interactions. Protein Sci. 2011, 20, 1607–1618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rehan Khalid, R.; Tahir ul Qamar, M.; Maryam, A.; Ashique, A.; Anwar, F.; Geesi, M.H.; Siddiqi, A.R. Comparative studies of the dynamics effects of bay60-2770 and bay58-2667 binding with human and bacterial h-nox domains. Molecules 2018, 23, 2141. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | Wild-Type RBD–ACE2 Complex | B.1.620 RBD–ACE2 Complex |
|---|---|---|
| HADDOCK scores | −122.6+/− 0.7 | −124.9 +/− 3.8 |
| Cluster size | 64 | 20 |
| RMSD in Å | 1.7 +/− 1.0 | 14.3 +/− 0.2 |
| vdW | −59.6 +/− 2.3 | −59.4 +/− 4.4 |
| Electrostatic energy | −181.4 +/− 15.5 | −203.5 +/− 17.1 |
| Desolvation energy | −27.1 +/− 3.4 | −25.6 +/− 2.1 |
| Restraint’s violation energy | 4.7 +/− 3.8 | 5.4 +/− 1.9 |
| Buried surface area (A2) | 1965.3 +/− 120.6 | 1906.8 +/− 52.5 |
| Z-score | −1.9 | −1.1 |
| Complex Name | ACE2 Interacting Residues | RBD Interacting Residues | Distance (Å) | Type of Bond |
|---|---|---|---|---|
| Wild Type | GLU30 | LYS417 | 2.56 | Hydrogen Bond |
| GLU35 | GLN493 | 2.75 | Hydrogen Bond | |
| GLU38 | GLY496 | 3.16 | Hydrogen Bond | |
| TYR41 | THR500 | 2.73 | Hydrogen Bond | |
| TYR41 | THR500 | 2.68 | Hydrogen Bond | |
| TYR41 | THR500 | 2.68 | Hydrogen Bond | |
| GLN76 | TYR489 | 3.06 | Hydrogen Bond | |
| TYR83 | ASN487 | 2.73 | Hydrogen Bond | |
| LYS353 | GLY502 | 3.15 | Hydrogen Bond | |
| LYS353 | GLY496 | 3.16 | Hydrogen Bond | |
| GLU30 | LYS417 | 2.56 | Salt Bridge | |
| B.1.620 | LEU24 | ASN487 | 3.08 | Hydrogen Bond |
| THR27 | ALA475 | 3.23 | Hydrogen Bond | |
| GLU30 | LYS417 | 2.55 | Hydrogen Bond | |
| GLU35 | TYR489 | 2.65 | Hydrogen Bond | |
| GLU38 | TYR449 | 2.61 | Hydrogen Bond | |
| TYR83 | ASN487 | 2.81 | Hydrogen Bond | |
| GLN325 | ASN439 | 3.05 | Hydrogen Bond | |
| LYS353 | TYR449 | 2.74 | Hydrogen Bond | |
| ASP355 | THR500 | 2.92 | Hydrogen Bond | |
| ALA386 | TYR505 | 3.31 | Hydrogen Bond | |
| GLU30 | LYS417 | 2.55 | Salt Bridge |
| MM/GBSA | VDW | ELE | GB | SA | Total |
|---|---|---|---|---|---|
| Wild Type | −107.63 | −592.87 | 663.18 | −13.82 | −51.14 |
| B.1.620 | −100.46 | −1129.31 | 1174.64 | −13.62 | −68.75 |
| MM/PBSA | VDW | ELE | PB | ESURF | Total |
| Wild Type | −52.21 | −76.42 | 118.74 | −6.65 | −16.54 |
| B.1.620 | −54.27 | −85.35 | 126.37 | −9.07 | −22.32 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Muhseen, Z.T.; Kadhim, S.; Yahiya, Y.I.; Alatawi, E.A.; Aba Alkhayl, F.F.; Almatroudi, A. Insights into the Binding of Receptor-Binding Domain (RBD) of SARS-CoV-2 Wild Type and B.1.620 Variant with hACE2 Using Molecular Docking and Simulation Approaches. Biology 2021, 10, 1310. https://doi.org/10.3390/biology10121310
Muhseen ZT, Kadhim S, Yahiya YI, Alatawi EA, Aba Alkhayl FF, Almatroudi A. Insights into the Binding of Receptor-Binding Domain (RBD) of SARS-CoV-2 Wild Type and B.1.620 Variant with hACE2 Using Molecular Docking and Simulation Approaches. Biology. 2021; 10(12):1310. https://doi.org/10.3390/biology10121310
Chicago/Turabian StyleMuhseen, Ziyad Tariq, Salim Kadhim, Yahiya Ibrahim Yahiya, Eid A. Alatawi, Faris F. Aba Alkhayl, and Ahmad Almatroudi. 2021. "Insights into the Binding of Receptor-Binding Domain (RBD) of SARS-CoV-2 Wild Type and B.1.620 Variant with hACE2 Using Molecular Docking and Simulation Approaches" Biology 10, no. 12: 1310. https://doi.org/10.3390/biology10121310