
GABARAPL1 Inhibits EMT Signaling through SMAD-Tageted Negative Feedback

 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Transcriptome Analysis
2.2. Reagents and Antibodies
2.3. Tissue Samples and Immunohistochemistry (IHC)
2.4. Cell Culture and Treatments
2.5. Generation of KO Cell Lines Using the CRISPR/Cas9 Gene Editing Technology
2.6. Cells Transfection
2.7. Cell Proliferation
2.8. Cell Invasion Assays
2.9. Cell Migration Assays
2.10. Western Blotting
2.11. Quantitative RT-PCR
2.12. Confocal Microscopy
2.13. GST-Pull down Assay
2.14. Statistical Analysis
3. Results
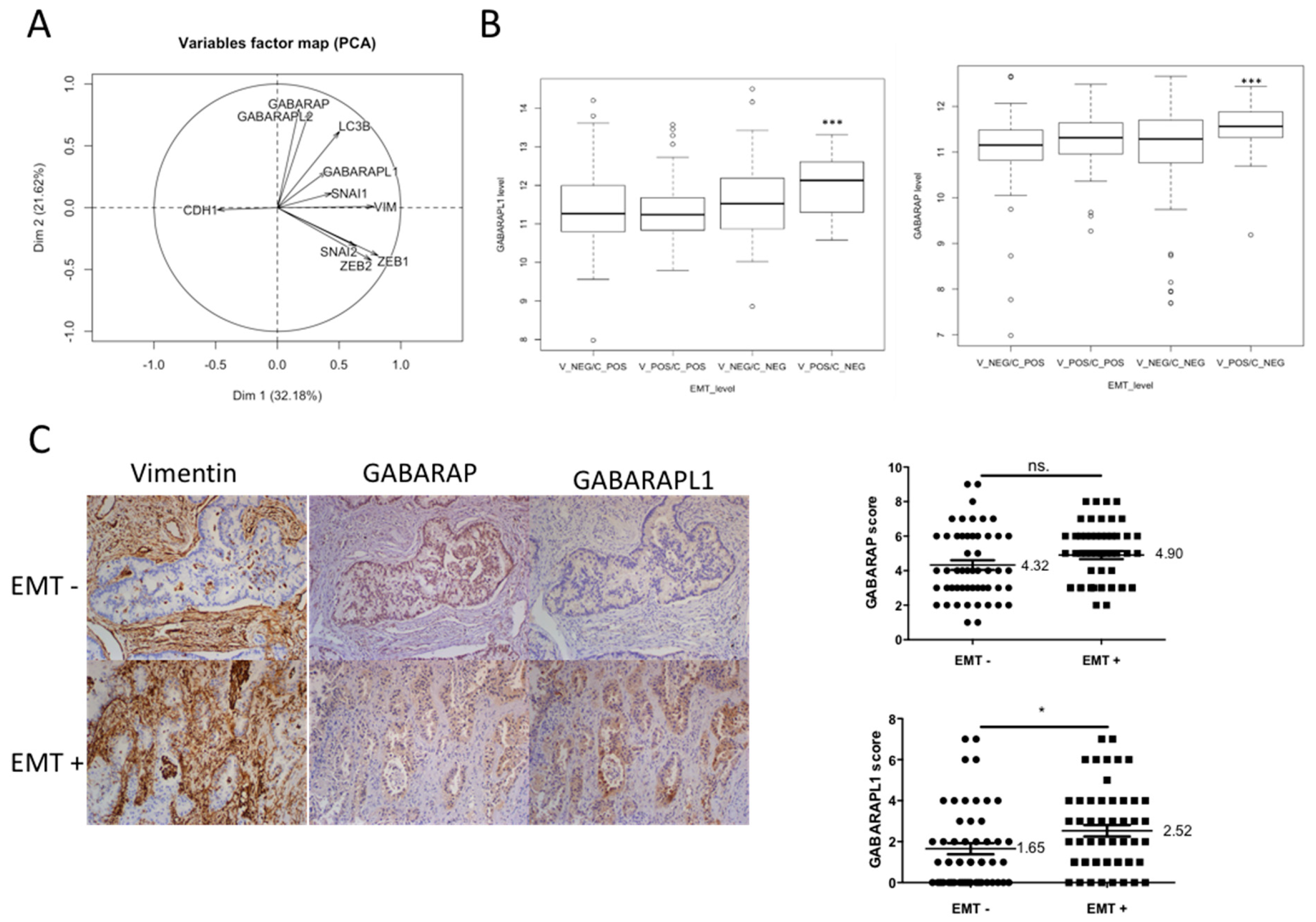
3.1. GABARAPL1 Expression Was Correlated with EMT Markers
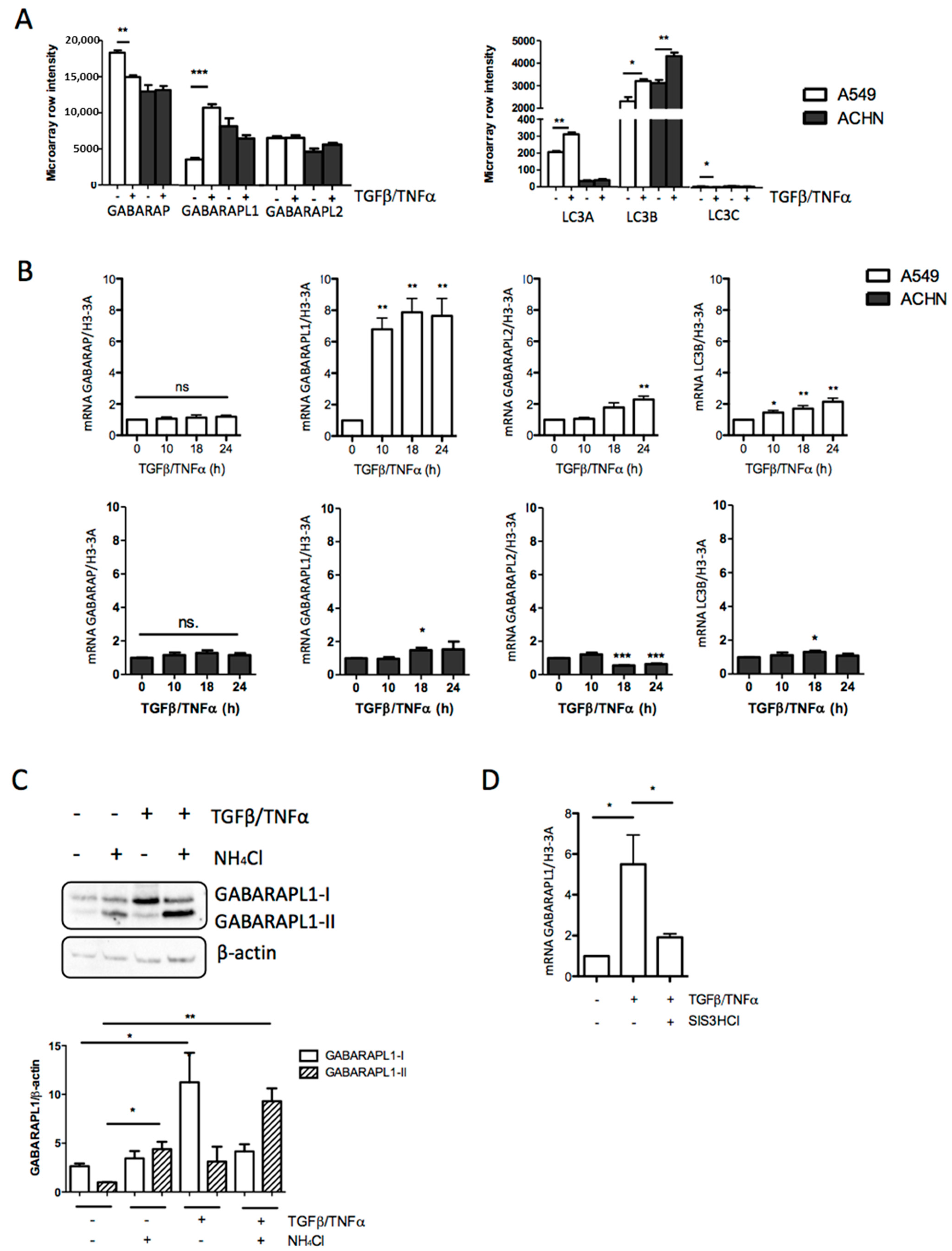
3.2. TGF-β/TNF-α-Induced EMT Was Correlated with Increased GABARAPL1 Expression
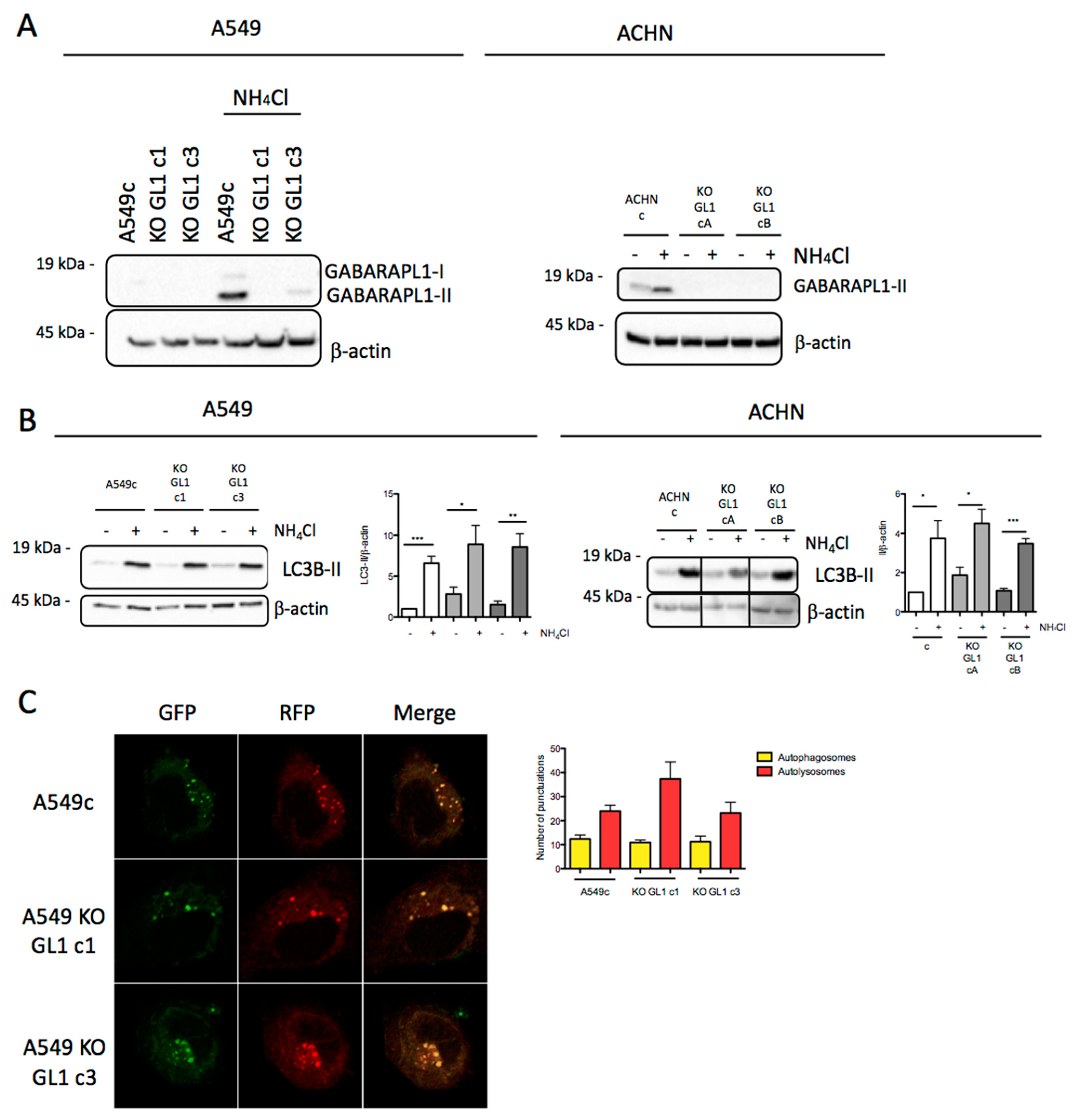
3.3. Design and Characterization of GABARAPL1 Knockout Cell Lines
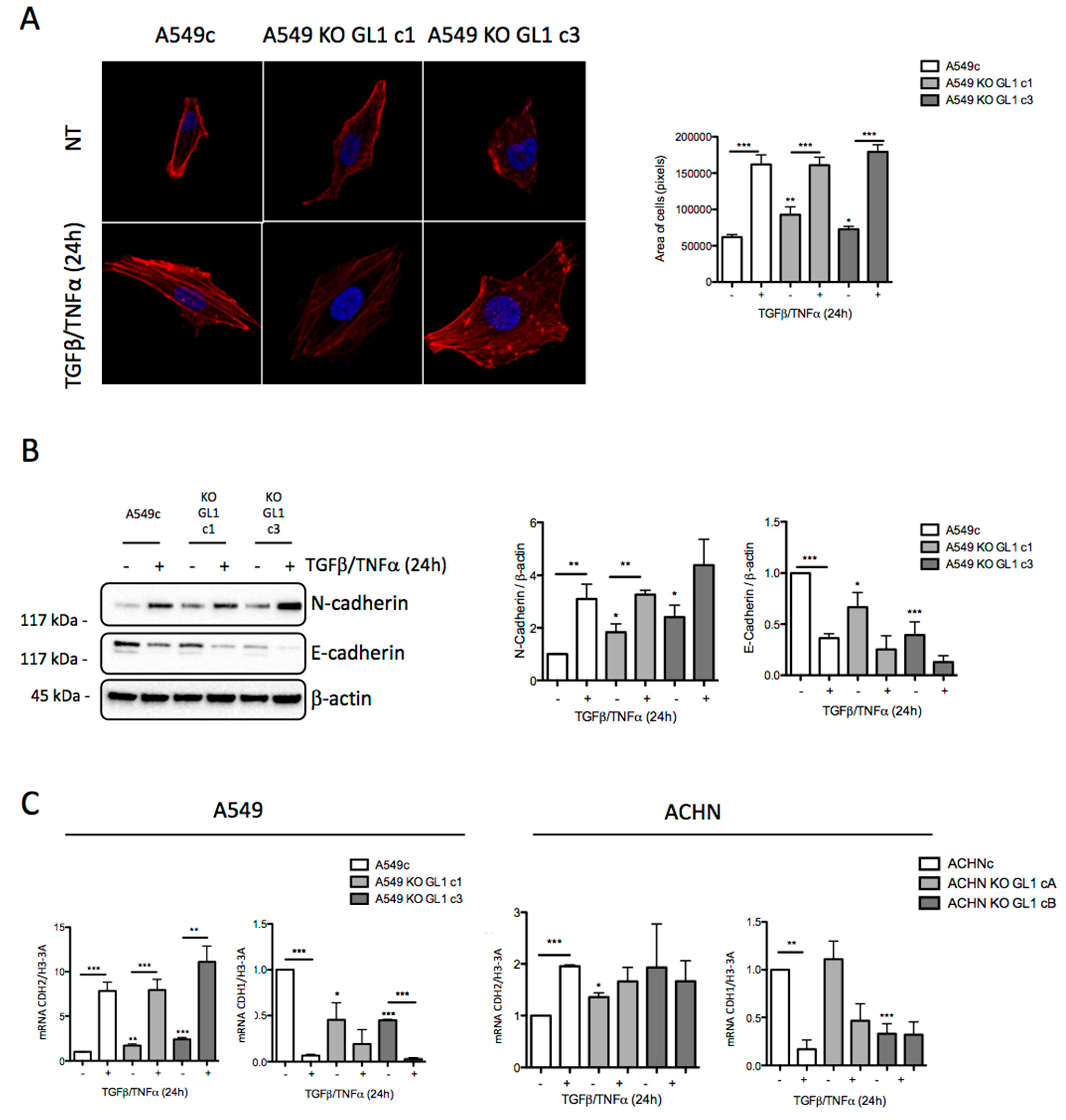
3.4. GABARAPL1 Knockout Led to the Induction of EMT
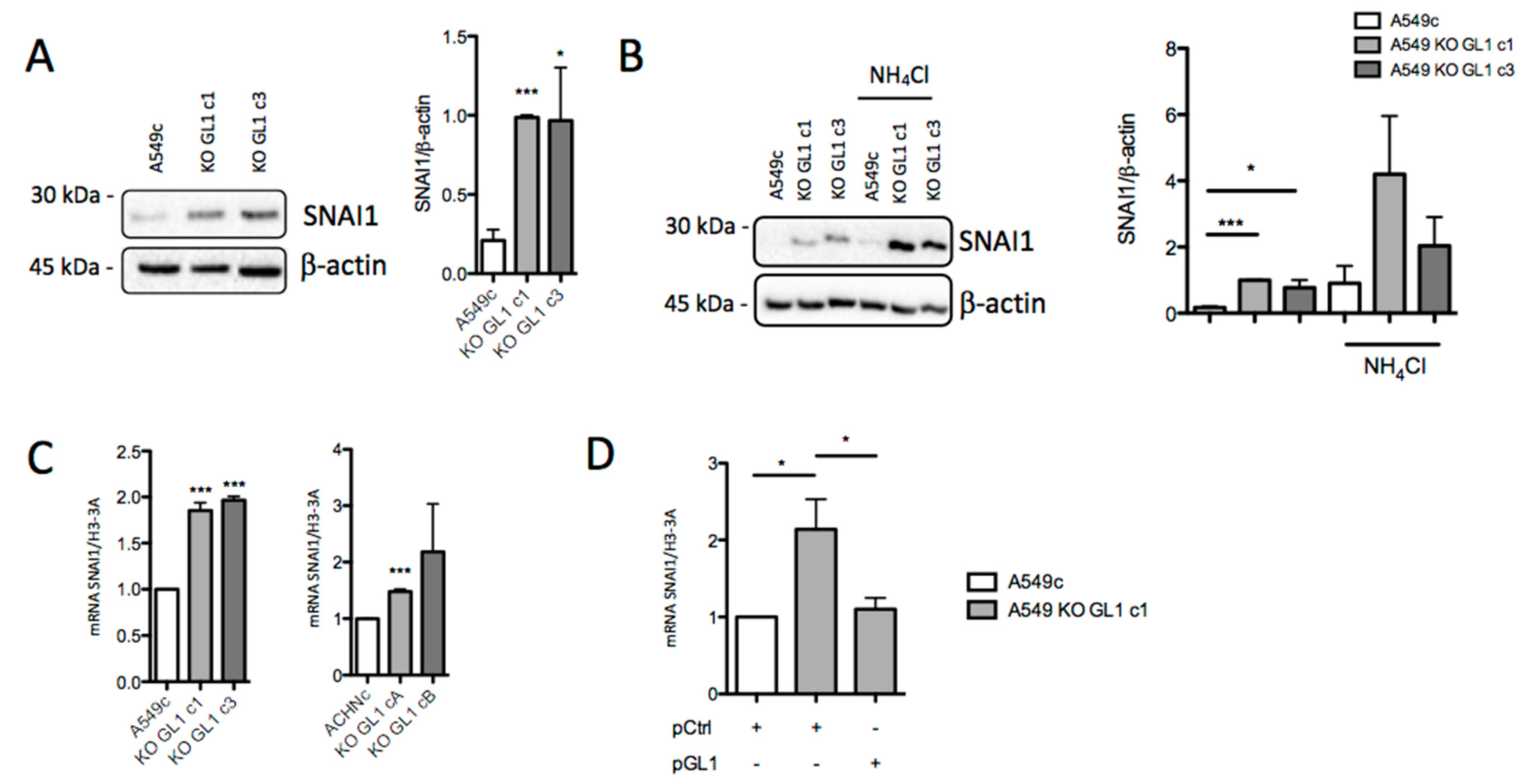
3.5. GABARAPL1 Knockout Led to an Increase in SNAI1 Levels
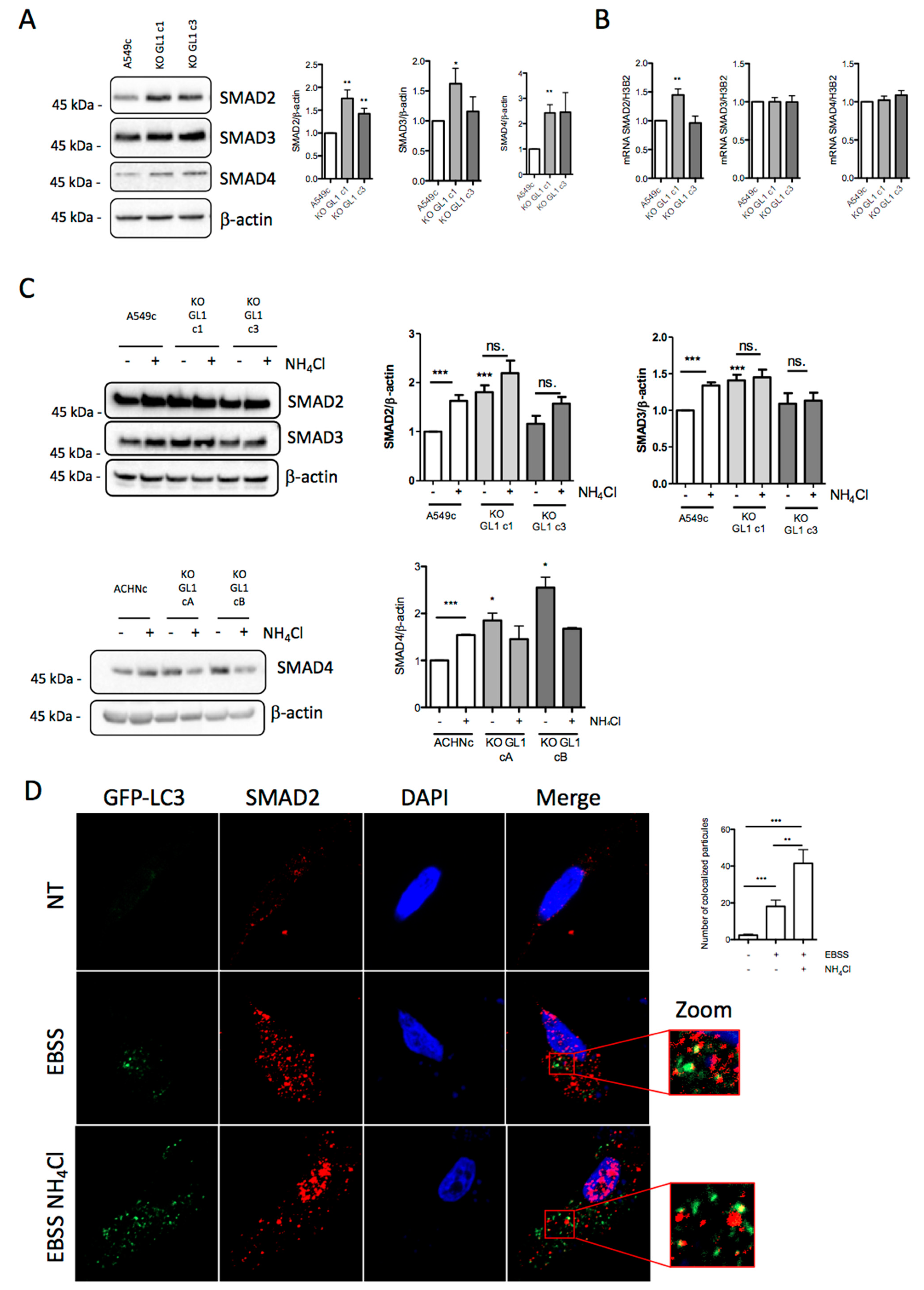
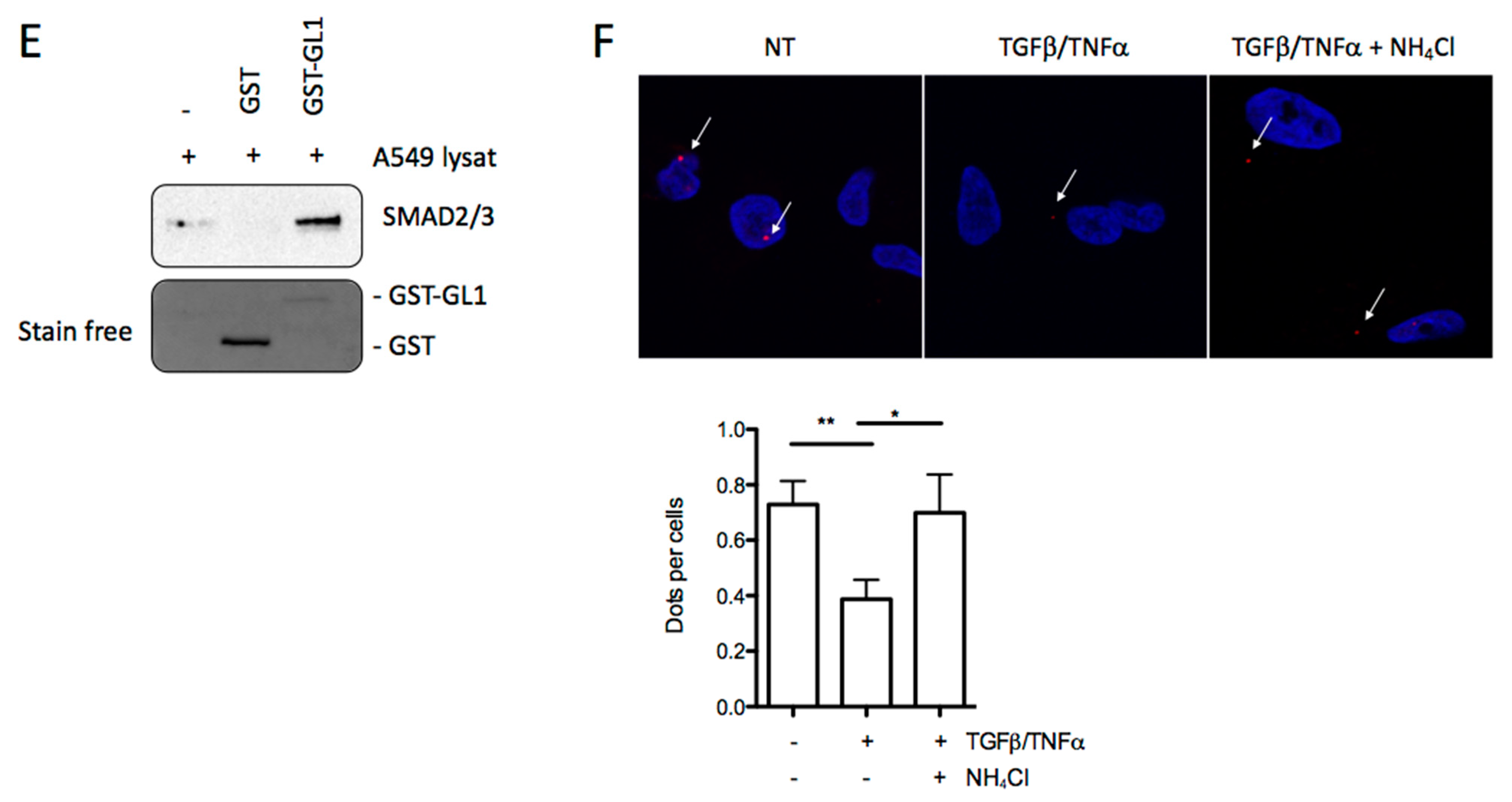
3.6. GABARAPL1 Induced the Degradation of SMAD by Autophagy
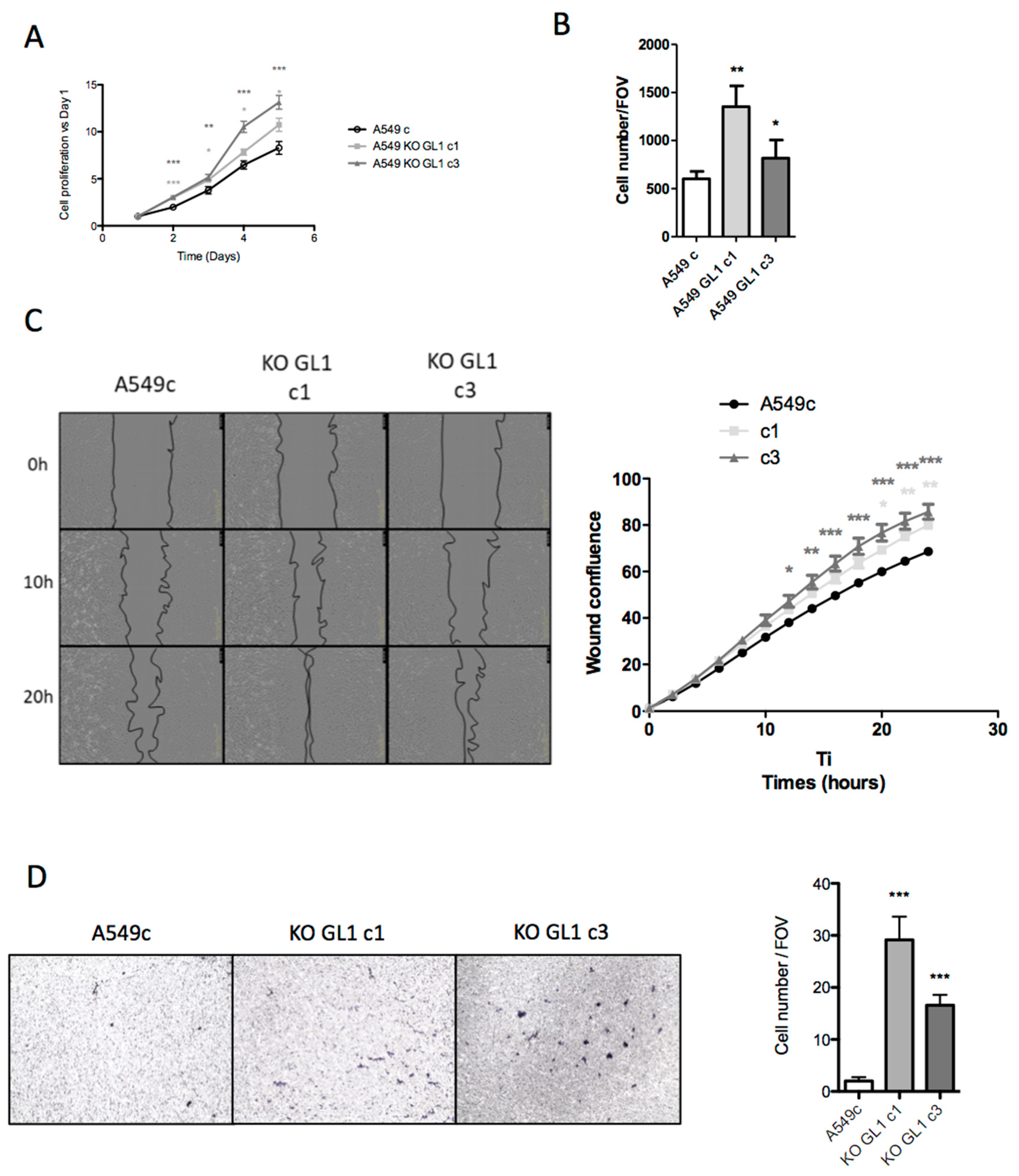
3.7. GABARAPL1 Inhibited Aggressive Cancer Phenotypes
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Brabletz, T.; Kalluri, R.; Nieto, M.A.; Weinberg, R.A. EMT in cancer. Nat. Rev. Cancer 2018, 18, 128–134. [Google Scholar] [CrossRef]
- Massagué, J. TGFβ in Cancer. Cell 2008, 134, 215–230. [Google Scholar] [CrossRef] [Green Version]
- Nieto, M.A.; Huang, R.Y.-J.; Jackson, R.A.; Thiery, J.P. EMT: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmadi, A.; Najafi, M.; Farhood, B.; Mortezaee, K. Transforming growth factor-β signaling: Tumorigenesis and targeting for cancer therapy. J. Cell. Physiol. 2018, 234, 12173–12187. [Google Scholar] [CrossRef] [PubMed]
- Rubinsztein, D.C.; Shpilka, T.; Elazar, Z. Mechanisms of Autophagosome Biogenesis. Curr. Biol. 2012, 22, R29–R34. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Klionsky, D.J. Mammalian autophagy: Core molecular machinery and signaling regulation. Curr. Opin. Cell Biol. 2010, 22, 124–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashford, T.P.; Porter, K.R. Cytoplasmic Components in Hepatic Cell Lysosomes. J. Cell Biol. 1962, 12, 198–202. [Google Scholar] [CrossRef]
- Fortun, J.; Dunn, W.A.; Joy, S.; Li, J.; Notterpek, L. Emerging Role for Autophagy in the Removal of Aggresomes in Schwann Cells. J. Neurosci. 2003, 23, 10672–10680. [Google Scholar] [CrossRef] [Green Version]
- Lamark, T.; Johansen, T. Aggrephagy: Selective Disposal of Protein Aggregates by Macroautophagy. Int. J. Cell Biol. 2012, 2012, 736905. [Google Scholar] [CrossRef] [Green Version]
- He, H.; Dang, Y.; Dai, F.; Guo, Z.; Wu, J.; She, X.; Pei, Y.; Chen, Y.; Ling, W.; Wu, C.; et al. Post-translational Modifications of Three Members of the Human MAP1LC3 Family and Detection of a Novel Type of Modification for MAP1LC3B. J. Biol. Chem. 2003, 278, 29278–29287. [Google Scholar] [CrossRef] [Green Version]
- Legesse-Miller, A.; Sagiv, Y.; Porat, A.; Elazar, Z. Isolation and Characterization of a Novel Low Molecular Weight Protein Involved in Intra-Golgi Traffic. J. Biol. Chem. 1998, 273, 3105–3109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mann, S.; Hammarback, J. Molecular characterization of light chain 3. A microtubule binding subunit of MAP1A and MAP1B. J. Biol. Chem. 1994, 269, 11492–11497. [Google Scholar] [CrossRef]
- Mansuy, V.; Boireau, W.; Fraichard, A.; Schlick, J.-L.; Jouvenot, M.; Delage-Mourroux, R. GEC1, a protein related to GABARAP, interacts with tubulin and GABA(A) receptor. Biochem. Biophys. Res. Commun. 2004, 325, 639–648. [Google Scholar] [CrossRef] [PubMed]
- Pellerin, I.; Vuillermoz, C.; Jouvenot, M.; Ordener, C.; Royez, M.; Adessi, G.L. Identification and characterization of an early estrogen-regulated RNA in cultured guinea-pig endometrial cells. Mol. Cell. Endocrinol. 1993, 90, R17–R21. [Google Scholar] [CrossRef]
- Sagiv, Y.; Legesse-Miller, A.; Porat, A.; Elazar, Z. GATE-16, a membrane transport modulator, interacts with NSF and the Golgi v-SNARE GOS-28. EMBO J. 2000, 19, 1494–1504. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Bedford, F.K.; Brandon, N.J.; Moss, S.J.; Olsen, R.W. GABA(A)-receptor-associated protein links GABA(A) receptors and the cytoskeleton. Nature 1999, 397, 69–72. [Google Scholar] [CrossRef]
- Xin, Y.; Yu, L.; Chen, Z.; Zheng, L.; Fu, Q.; Jiang, J.; Zhang, P.; Gong, R.; Zhao, S. Cloning, Expression Patterns, and Chromosome Localization of Three Human and Two Mouse Homologues of GABAA Receptor-Associated Protein. Genomics 2001, 74, 408–413. [Google Scholar] [CrossRef]
- Alemu, E.A.; Lamark, T.; Torgersen, K.M.; Birgisdottir, A.B.; Larsen, K.B.; Jain, A.; Olsvik, H.; Øvervatn, A.; Kirkin, V.; Johansen, T. ATG8 Family Proteins Act as Scaffolds for Assembly of the ULK Complex: Sequence requirements for LC3-interacting region (LIR) motifs. J. Biol. Chem. 2012, 287, 39275–39290. [Google Scholar] [CrossRef] [Green Version]
- Rogov, V.V.; Stolz, A.; Ravichandran, A.C.; Rios-Szwed, D.; Suzuki, H.; Kniss, A.; Löhr, F.; Wakatsuki, S.; Dötsch, V.; Dikic, I.; et al. Structural and functional analysis of the GABARAP interaction motif (GIM). EMBO Rep. 2017, 18, 1382–1396. [Google Scholar] [CrossRef]
- Jacquet, M.; Guittaut, M.; Fraichard, A.; Despouy, G. The function of ATG8 proteins in autophagy and cancer: Linked or unrelated? Autophagy 2020, 17, 599–611. [Google Scholar] [CrossRef] [PubMed]
- Murray, D.; Mirzayans, R.; McBride, W.H. Defenses against Pro-oxidant Forces—Maintenance of Cellular and Genomic Integrity and Longevity. Radiat. Res. 2018, 190, 331–349. [Google Scholar] [CrossRef] [PubMed]
- Goussetis, D.J.; Gounaris, E.; Wu, E.J.; Vakana, E.; Sharma, B.; Bogyo, M.; Altman, J.K.; Platanias, L.C. Autophagic degradation of the BCR-ABL oncoprotein and generation of antileukemic responses by arsenic trioxide. Blood 2012, 120, 3555–3562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belaid, A.; Cerezo, M.; Chargui, A.; Corcelle-Termeau, E.; Pedeutour, F.; Giuliano, S.; Ilie, M.; Rubera, I.; Tauc, M.; Barale, S.; et al. Autophagy Plays a Critical Role in the Degradation of Active RHOA, the Control of Cell Cytokinesis, and Genomic Stability. Cancer Res. 2013, 73, 4311–4322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elgendy, M.; Sheridan, C.; Brumatti, G.; Martin, S.J. Oncogenic Ras-induced expression of Noxa and Beclin-1 promotes autophagic cell death and limits clonogenic survival. Mol. Cell. 2011, 42, 23–35. [Google Scholar] [CrossRef] [Green Version]
- Yu, L.; Wan, F.; Dutta, S.; Welsh, S.; Liu, Z.; Freundt, E.; Baehrecke, E.H.; Lenardo, M. Autophagic programmed cell death by selective catalase degradation. Proc. Natl. Acad. Sci. USA 2006, 103, 4952–4957. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; He, Z.; von Rütte, T.; Yousefi, S.; Hunger, R.E.; Simon, H.-U. Down-Regulation of Autophagy-Related Protein 5 (ATG5) Contributes to the Pathogenesis of Early-Stage Cutaneous Melanoma. Sci. Transl. Med. 2013, 5, 202ra123. [Google Scholar] [CrossRef] [PubMed]
- Folkerts, H.; Hilgendorf, S.; Vellenga, E.; Bremer, E.; Wiersma, V.R. The multifaceted role of autophagy in cancer and the microenvironment. Med. Res. Rev. 2018, 39, 517–560. [Google Scholar] [CrossRef]
- Chen, Y.; Sun, H.-Q.; Eichorst, J.P.; Albanesi, J.P.; Yin, H.; Mueller, J.D. Co-mobility of GABARAP and Phosphatidylinositol 4-kinase 2A on cytoplasmic vesicles. Biochemistry 2018, 57, 3556–3559. [Google Scholar] [CrossRef] [PubMed]
- Sharif, T.; Martell, E.; Dai, C.; Kennedy, B.E.; Murphy, P.; Clements, D.R.; Kim, Y.; Lee, P.W.K.; Gujar, S.A. Autophagic homeostasis is required for the pluripotency of cancer stem cells. Autophagy 2016, 13, 264–284. [Google Scholar] [CrossRef] [Green Version]
- Catalano, M.; D’Alessandro, G.; Lepore, F.; Corazzari, M.; Caldarola, S.; Valacca, C.; Faienza, F.; Esposito, V.; Limatola, C.; Cecconi, F.; et al. Autophagy induction impairs migration and invasion by reversing EMT in glioblastoma cells. Mol. Oncol. 2015, 9, 1612–1625. [Google Scholar] [CrossRef] [Green Version]
- Grassi, G.; Di Caprio, G.; Santangelo, L.; Fimia, G.M.; Cozzolino, A.M.; Komatsu, M.; Ippolito, G.; Tripodi, M.; Alonzi, T. Autophagy regulates hepatocyte identity and epithelial-to-mesenchymal and mesenchymal-to-epithelial transitions promoting Snail degradation. Cell Death Dis. 2015, 6, e1880. [Google Scholar] [CrossRef] [Green Version]
- Lv, Q.; Wang, W.; Xue, J.; Hua, F.; Mu, R.; Lin, H.; Yan, J.; Lv, X.; Chen, X.; Hu, Z.-W. DEDD Interacts with PI3KC3 to Activate Autophagy and Attenuate Epithelial–Mesenchymal Transition in Human Breast Cancer. Cancer Res. 2012, 72, 3238–3250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.; Bian, E.; Xu, Y.; Ji, X.; Tang, F.; Ma, C.; Wang, H.; Zhao, B. Meg3 Induces EMT and Invasion of Glioma Cells via Autophagy. OncoTargets Ther. 2020, 13, 989–1000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tong, H.; Yin, H.; Hossain, M.A.; Wang, Y.; Wu, F.; Dong, X.; Gao, S.; Zhan, K.; He, W. Starvation-induced autophagy promotes the invasion and migration of human bladder cancer cells via TGF-β1/Smad3-mediated epithelial-mesenchymal transition activation. J. Cell Biochem. 2018, 120, 5118–5127. [Google Scholar] [CrossRef]
- Zou, M.; Zhu, W.; Wang, L.; Shi, L.; Gao, R.; Ou, Y.; Chen, X.; Wang, Z.; Jiang, A.; Liu, K.; et al. AEG-1/MTDH-activated autophagy enhances human malignant glioma susceptibility to TGF-β1-triggered epithelial-mesenchymal transition. Oncotarget 2016, 7, 13122–13138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herfs, M.; Longuespée, R.; Quick, C.M.; Roncarati, P.; Suarez-Carmona, M.; Hubert, P.; Lebeau, A.; Bruyere, D.; Mazzucchelli, G.; Smargiasso, N.; et al. Proteomic signatures reveal a dualistic and clinically relevant classification of anal canal carcinoma. J. Pathol. 2016, 241, 522–533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hubert, P.; Herman, L.; Roncarati, P.; Maillard, C.; Renoux, V.; Demoulin, S.; Erpicum, C.; Foidart, J.-M.; Boniver, J.; Noel, A.; et al. Altered α-defensin 5 expression in cervical squamocolumnar junction: Implication in the formation of a viral/tumour-permissive microenvironment. J. Pathol. 2014, 234, 464–477. [Google Scholar] [CrossRef]
- Gauthier, T.; Claude-Taupin, A.; Delage-Mourroux, R.; Boyer-Guittaut, M.; Hervouet, E. Proximity Ligation In situ Assay is a Powerful Tool to Monitor Specific ATG Protein Interactions following Autophagy Induction. PLoS ONE 2015, 10, e0128701. [Google Scholar]
- Peixoto, P.; Etcheverry, A.; Aubry, M.; Missey, A.; Lachat, C.; Perrard, J.; Hendrick, E.; Delage-Mourroux, R.; Mosser, J.; Borg, C. EMT is associated with an epigenetic signature of ECM remodeling genes. Cell Death Dis. 2019, 10, 205. [Google Scholar] [CrossRef] [Green Version]
- Poillet-Perez, L.; Jacquet, M.; Hervouet, E.; Gauthier, T.; Fraichard, A.; Borg, C.; Pallandre, J.-R.; Gonzalez, B.J.; Ramdani, Y.; Boyer-Guittaut, M.; et al. GABARAPL1 tumor suppressive function is independent of its conjugation to autophagosomes in MCF-7 breast cancer cells. Oncotarget 2017, 8, 55998–56020. [Google Scholar] [CrossRef] [Green Version]
- Boyer-Guittaut, M.; Poillet, L.; Liang, Q.; Bôle-Richard, E.; Ouyang, X.; Benavides, G.A.; Chakrama, F.Z.; Fraichard, A.; Darley-Usmar, V.M.; Despouy, G.; et al. The role of GABARAPL1/GEC1 in autophagic flux and mitochondrial quality control in MDA-MB-436 breast cancer cells. Autophagy 2014, 10, 986–1003. [Google Scholar] [CrossRef]
- Tanida, I.; Ueno, T.; Kominami, E. Human Light Chain 3/MAP1LC3B Is Cleaved at Its Carboxyl-terminal Met121 to Expose Gly120 for Lipidation and Targeting to Autophagosomal Membranes. J. Biol. Chem. 2004, 279, 47704–47710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimura, S.; Noda, T.; Yoshimori, T. Dissection of the Autophagosome Maturation Process by a Novel Reporter Protein, Tandem Fluorescent-Tagged LC3. Autophagy 2007, 3, 452–460. [Google Scholar] [CrossRef] [Green Version]
- Pankiv, S.; Clausen, T.H.; Lamark, T.; Brech, A.; Bruun, J.A.; Outzen, H.; Øvervatn, A.; Bjørkøy, G.; Johansen, T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 2007, 282, 24131–24145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacomin, A.-C.; Samavedam, S.; Promponas, V.; Nezis, I.P. iLIR database: A web resource for LIR motif-containing proteins in eukaryotes. Autophagy 2016, 12, 1945–1953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berthier, A.; Seguin, S.; Sasco, A.J.; Bobin, J.Y.; De Laroche, G.; Datchary, J.; Saez, S.; Rodriguez-Lafrasse, C.; Tolle, F.; Fraichard, A.; et al. High expression of gabarapl1 is associated with a better outcome for patients with lymph node-positive breast cancer. Br. J. Cancer 2010, 102, 1024–1031. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, F.; Han, L.; Wu, Y.; Li, S.; Yang, X.; Wang, Y.; Ren, F.; Zhai, Y.; Wang, D.; et al. GABARAPL1 Negatively Regulates Wnt/β-catenin Signaling by Mediating Dvl2 Degradation through the Autophagy Pathway. Cell. Physiol. Biochem. 2011, 27, 503–512. [Google Scholar] [CrossRef]
- Anding, A.L.; Baehrecke, E.H. Cleaning House: Selective Autophagy of Organelles. Dev. Cell 2017, 41, 10–22. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.-Y.; Wu, T.; Ma, W.; Li, S.; Jing, W.-J.; Ma, J.; Chen, D.-M.; Guo, X.-J.; Nan, K.-J. Expression and clinical significance of autophagic protein LC3B and EMT markers in gastric cancer. Cancer Manag. Res. 2018, 10, 1479–1486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiyono, K.; Suzuki, H.I.; Matsuyama, H.; Morishita, Y.; Komuro, A.; Kano, M.; Sugimoto, K.; Miyazono, K. Autophagy Is Activated by TGF-β and Potentiates TGF-β–Mediated Growth Inhibition in Human Hepatocellular Carcinoma Cells. Cancer Res. 2009, 69, 8844–8852. [Google Scholar] [CrossRef] [Green Version]
- Zada, S.; Hwang, J.S.; Ahmed, M.; Lay, T.H.; Pham, T.M.; Kim, D.R. Control of the Epithelial-to-Mesenchymal Transition and Cancer Metastasis by Autophagy-Dependent SNAI1 Degradation. Cells 2019, 8, 129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marsh, T.; Debnath, J. Autophagy suppresses breast cancer metastasis by degrading NBR1. Autophagy 2020, 16, 1164–1165. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wang, D.; Lei, M.; Gao, J.; Cui, Y.; Jin, X.; Yu, Q.; Jiang, Y.; Guo, Y.; Liu, Y.; et al. GABARAP suppresses EMT and breast cancer progression via the AKT/mTOR signaling pathway. Aging 2021, 13, 5858–5874. [Google Scholar] [CrossRef]
- Xu, J.; Lamouille, S.; Derynck, R. TGF-β-induced epithelial to mesenchymal transition. Cell Res. 2009, 19, 156–172. [Google Scholar] [CrossRef] [PubMed]
- Birgisdottir, A.B.; Lamark, T.; Johansen, T. The LIR motif—Crucial for selective autophagy. J. Cell Sci. 2013, 126, 3237–3247. [Google Scholar] [CrossRef] [Green Version]
- Atkinson, J.M.; Ye, Y.; Gebru, M.T.; Liu, Q.; Zhou, S.; Young, M.M.; Takahashi, Y.; Lin, Q.; Tian, F.; Wang, H.-G. Time-resolved FRET and NMR analyses reveal selective binding of peptides containing the LC3-interacting region to ATG8 family proteins. J. Biol. Chem. 2019, 294, 14033–14042. [Google Scholar] [CrossRef]
- Joachim, J.; Jefferies, H.B.J.; Razi, M.; Frith, D.; Snijders, A.P.; Chakravarty, P.; Judith, D.; Tooze, S.A. Activation of ULK Kinase and Autophagy by GABARAP Trafficking from the Centrosome Is Regulated by WAC and GM130. Mol. Cell. 2015, 60, 899–913. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, T.N.; Padman, B.S.; Usher, J.; Oorschot, V.; Ramm, G.; Lazarou, M. Atg8 family LC3/GABARAP proteins are crucial for autophagosome-lysosome fusion but not autophagosome formation during PINK1/Parkin mitophagy and starvation. J. Cell Biol. 2016, 215, 857–874. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jacquet, M.; Hervouet, E.; Baudu, T.; Herfs, M.; Parratte, C.; Feugeas, J.-P.; Perez, V.; Reynders, C.; Ancion, M.; Vigneron, M.; et al. GABARAPL1 Inhibits EMT Signaling through SMAD-Tageted Negative Feedback. Biology 2021, 10, 956. https://doi.org/10.3390/biology10100956
Jacquet M, Hervouet E, Baudu T, Herfs M, Parratte C, Feugeas J-P, Perez V, Reynders C, Ancion M, Vigneron M, et al. GABARAPL1 Inhibits EMT Signaling through SMAD-Tageted Negative Feedback. Biology. 2021; 10(10):956. https://doi.org/10.3390/biology10100956
Chicago/Turabian StyleJacquet, Marine, Eric Hervouet, Timothée Baudu, Michaël Herfs, Chloé Parratte, Jean-Paul Feugeas, Valérie Perez, Célia Reynders, Marie Ancion, Marc Vigneron, and et al. 2021. "GABARAPL1 Inhibits EMT Signaling through SMAD-Tageted Negative Feedback" Biology 10, no. 10: 956. https://doi.org/10.3390/biology10100956