
Phylogenetic and Haplotype Network Analyses of Diaporthe eres Species in China Based on Sequences of Multiple Loci
by
 , ,
, ,
Chingchai Chaisiri
1,2,3 ,
,
Xiangyu Liu
1,2,3, 
Yang Lin
2,3 ,
,
Yanping Fu
2,3,
Fuxing Zhu
3 and
Chaoxi Luo
1,2,3,* 1
Key Lab of Horticultural Plant Biology, Ministry of Education, Huazhong Agricultural University, Wuhan 430070, China
2
Key Lab of Crop Disease Monitoring and Safety Control in Hubei Province, Huazhong Agricultural University, Wuhan 430070, China
3
College of Plant Science and Technology, Huazhong Agricultural University, Wuhan 430070, China
*
Author to whom correspondence should be addressed.
Biology 2021, 10(3), 179; https://doi.org/10.3390/biology10030179
Submission received: 17 January 2021
/
Revised: 22 February 2021
/
Accepted: 25 February 2021
/
Published: 1 March 2021
(This article belongs to the Section Genetics and Genomics)
Abstract
:Simple Summary
Diaporthe eres is one of the most serious plant pathogenic fungi that affect many economically important plants. It can cause rootstock death, stem canker, stem necrosis, dead branch, shoot blight, fruit rot, leaf spot, leaf necrosis, and umbel browning. In general, morphological and molecular characterization using multiple loci sequences were performed for the identification of Diaporthe species. However, there are morphological differences due to culture conditions, and the taxonomy of species of Diaporthe is unclear because the phylogeny based on different genes gives different tree topologies. In this study, we evaluate the phylogenetic relationships and population diversity among D. eres and other Diaporthe species. Our results showed that phylogenetic analyses from concatenated multi-locus DNA sequence data could resolve the D. eres species. Furthermore, haplotype network analysis showed that no correlation existed between population diversity and distribution or hosts across China. These results could improve our understanding of the epidemiology of D. eres and provide useful information for effective disease management.
Abstract
Diaporthe eres is considered one of the most important causal agents of many plant diseases, with a broad host range worldwide. In this study, multiple sequences of ribosomal internal transcribed spacer region (ITS), translation elongation factor 1-α gene (EF1-α), beta-tubulin gene (TUB2), calmodulin gene (CAL), and histone-3 gene (HIS) were used for multi-locus phylogenetic analysis. For phylogenetic analysis, maximum likelihood (ML), maximum parsimony (MP), and Bayesian inferred (BI) approaches were performed to investigate relationships of D. eres with closely related species. The results strongly support that the D. eres species falls into a monophyletic lineage, with the characteristics of a species complex. Phylogenetic informativeness (PI) analysis showed that clear boundaries could be proposed by using EF1-α, whereas ITS showed an ineffective reconstruction and, thus, was unsuitable for speciating boundaries for Diaporthe species. A combined dataset of EF1-α, CAL, TUB2, and HIS showed strong resolution for Diaporthe species, providing insights for the D. eres complex. Accordingly, besides D. biguttusis, D. camptothecicola, D. castaneae-mollissimae, D. cotoneastri, D. ellipicola, D. longicicola, D. mahothocarpus, D. momicola, D. nobilis, and Phomopsis fukushii, which have already been previously considered the synonymous species of D. eres, another three species, D. henanensis, D. lonicerae and D. rosicola, were further revealed to be synonyms of D. eres in this study. In order to demonstrate the genetic diversity of D. eres species in China, 138 D. eres isolates were randomly selected from previous studies in 16 provinces. These isolates were obtained from different major plant species from 2006 to 2020. The genetic distance was estimated with phylogenetic analysis and haplotype networks, and it was revealed that two major haplotypes existed in the Chinese populations of D. eres. The haplotype networks were widely dispersed and not uniquely correlated to specific populations. Overall, our analyses evaluated the phylogenetic identification for D. eres species and demonstrated the population diversity of D. eres in China.
1. Introduction
The genus Diaporthe, belonging to the class of Sordariomycetes, the order of Diaporthales, and the family of Diaporthaceae, was originally established with Diaporthe eres Nitschke as the typified species [1,2]. The genus Diaporthe (asexual morph, Phomopsis) represents a group of cosmopolitan species, including saprophytic, endophytic, and pathogenic ones on different plants [3,4,5,6,7]. Furthermore, Diaporthe species were also reported as the causal agents of many important diseases in humans, mammals, and insects [8,9,10,11]. To date, over 1020 names of “Diaporthe” and around 950 names of the asexual morph “Phomopsis” have been recorded in MycoBank lists (http://www.mycobank.org (accessed on 15 July 2020), of which more than 100 Diaporthe and/or Phomopsis species have been reported in China [12,13,14,15,16,17,18,19,20,21,22].
Diaporthe eres was firstly collected with a type specimen from Ulmus sp. in Germany. It was reported that D. eres could cause shoot blight on Acer pseudoplatanus [23] and Juglans cinerea [24]. It is also responsible for umbel browning and stem necrosis on Daucus carota [25], leaf necrosis on Hedera helix [26], fruit rot on Vitis sp. [27], and stem canker and rootstock death on Malus spp. [28]. According to recent studies in China, it is responsible for branch canker, leaf blight, and root rot on Cinnamomum camphora [29], Acanthopanax senticosus, Castanea mollissima, Melia azedarace, Rhododendron simsii, Sorbus sp. [20], Juglans regia [14,20], Polygonatum sibiricum [30], Photinia fraseri cv. Red Robin [31], Coptis chinensis [32], Acer palmatum [33], Pyrus sp. [16], Vitis sp. [19], Prunus persica [13], and Pinus albicaulis [34]. It often associates with many important economic trees, e.g., camellia [35,36], camptotheca [37], citrus [18], grapevine [19,38], Japanese oak [39,40], kiwifruit [41], peach [13], pear [16], walnut [14], and so on.
Genealogical Concordance Phylogenetic Species Recognition (GCPSR) [42] represents an enhanced tool for species delimitation in the Diaporthe genus compared to morphological and biological identification [14,43]. The species concept in D. eres has greatly progressed since the molecular approach of concatenated multigene genealogies under GCPSR started to be conducted. However, the other processes, e.g., incomplete lineage sorting, recombination, and horizontal gene transfer, can cause discordances between gene trees and species trees and mask the true evolutionary relationship among closely related taxa [44]. Furthermore, the regular approach of concatenating sequence data from multiple loci under GCPSR can lead to inconsistency and poor species discrimination [45].
In recent years, insights into the species boundaries of the Diaporthe genus have been resolved based on morphological characterization combined with multi-locus phylogenetic analyses [3,4,7,46,47]. Effective multi-locus phylogenetic analyses were employed to identify Diaporthe species with ribosomal internal transcribed spacer (ITS), translation elongation factor 1-α gene (EF1-α), beta-tubulin gene (TUB2), calmodulin gene (CAL), and histone-3 gene (HIS) [3,15,47,48]. As a result, several Diaporthe species with close phylogenetic relevance were successfully demonstrated as synonyms of D. eres, including D. castaneae-mollissimae, D. cotoneastri, D. nobilis, Phomopsis fukushii [43], D. biguttusis, D. ellipicola, D. longicicola, D. mahothocarpus [14], D. camptothecicola, and D. momicola [20].
Therefore, the objectives of the study are to (i) employ different delimitation methods based on a genomic DNA sequence database to interpret species boundaries and to facilitate further species identification for D. eres, (ii) investigate Chinese populations of the D. eres species and characterize the relationship between the populations and their distributions based on sequences of multiple loci, and (iii) reconstruct phylogeny and explore the evolution of D. eres with the newly updated Chinese population.
2. Materials and Methods
2.1. D. eres and Related Species Isolates Used
Thirty-seven species, including the D. eres species complex and closely related species, were used in phylogenetic analyses. These species were originally collected in Australia, Canada, China, France, Germany, Italy, Japan, Korea, Netherlands, South Africa, Suriname, Thailand, UK, USA, and Yugoslavia, and their corresponding DNA sequences were downloaded from NCBI’s GenBank nucleotide database (www.ncbi.nlm.nih.gov (accessed on 7 April 2020)) (Table 1).
A diversity analysis of D. eres populations in China was carried out using 138 isolates that were selected based on different hosts (data from previously published literature), including Actinidia chinensis [41], Camellia sp. [35,36], Camptotheca acuminata [37], Citrus spp. [18], Juglans regia [14], Lithocarpus glabra [39,40], Prunus persica [13], Pyrus spp. [16], and Vitis spp. [19,38]. These isolates were originally collected from not only different hosts but also different areas, including 16 provinces, i.e., Beijing (BJ), Chongqing (CQ), Fujian (FJ), Gansu (GS), Hebei (HEB), Henan (HN), Hubei (HUB), Jiangsu (JS), Jiangxi (JX), Jilin (JL), Liaoning (LN), Ningxia (NX), Shandong (SD), Sichuan (SC), Yunnan (YN), and Zhejiang (ZJ).
2.2. Selection of Suitable Markers for Genetic Diversity Analysis
Based on previous studies, ITS, EF1-α, TUB2, CAL, and HIS were selected for the evaluation of species diversity of the Diaporthe genus in phylogenetic analysis. In brief, the ITS sequence was amplified with the primer set of ITS1/ITS4 [49], EF1-α with EF1-728F/EF1-986R [50], TUB2 with Bt-2a/Bt-2b [51], CAL with CAL-228F/CAL-737R [50], and HIS with CYLH3F/H3-1b [51,52]. PCR amplification protocols of the five loci are the same as those described previously [53].
2.3. Sequence Alignment and Phylogenetic Analyses
Multi-locus phylogenetic analyses were conducted to identify isolates to species level using assembled DNA sequences of five loci. DNA sequences were used for consensus analysis with minor manual editions in the DNASTAR Lasergene Core Suite software program (SaqMan v.7.1.0; DNASTAR Inc., Madison, WI, USA). Sequence alignments and comparisons of assembled sequences were performed using the L-INS-i algorithm on the MAFFT alignment online server v.7.467 [54]. The aligned sequences were checked and manually adjusted in BioEdit v.7.2.5 [55] and converted to suitable formats (PHYLIP and NEXUS) using the Alignment Transformation Environment (ALTER) website online server [56]. The resulting DNA sequences, containing all five loci, were deposited at TreeBASE (submission number: 26697). Maximum likelihood (ML) phylogenetic trees were constructed using RAxML-HPC BlackBox v.8.2.10 [57], available in the CIPRES Science Gateway v.3.3 Web Portal [58], with 1000 bootstrap replications. The general time-reversible model of evolution, including the estimation of invariable sites (GTRGAMMA + I), was performed in ML analysis. Maximum parsimony (MP) analysis was performed with 1000 replicates using Phylogenetic Analyses Using Parsimony (PAUP*) v.4.0b10 [59]. Goodness fit and bootstrap values were calculated and harvested from tree length (TL), the consistency index (CI), the retention index (RI), the rescaled consistency index (RC), and the homoplasy index (HI). A heuristic search was carried out with 1000 random stepwise addition replicates using the tree bisection-reconnection (TBR) branch-swapping algorithm on “best trees”. Gaps were treated as missing data, and all characters were weighted equally. The bootstrap support values (BS) were determined by the software to assess the robustness of MLBS and MPBS analyses; only branches with MLBS and MPBS over 70% were considered for ML/MP phylogenetic inference. Posterior probabilities values (PP) were calculated by Markov Chain Monte Carlo (MCMC) sampling in MrBayes v.3.2.2 [60]. The best-fit model of nucleotide substitution was determined with corrected Akaike information criterion (AIC) in MrModeltest v.2.3 [61] (Table S2). For BI analysis, four MCMC chains were run simultaneously, starting from random trees, for 105 generations, and trees were sampled every 100th generation. The calculation of BI analysis was stopped when the average standard deviation of split frequencies fell below 0.01. The first 10% of resulting BI trees, which represent the burn-in phase of the analysis by inspecting likelihoods and parameters in Tracer v.1.7.1 [62], were discarded, and the remaining 9000 trees were used to calculate the posterior probabilities (PP) in the majority rule consensus tree. Bayesian posterior probability values (BIPP) over 0.95 were considered for BI trees, and all trees were rooted with D. citri (CBS 135422).
2.4. Genealogical Concordance Phylogenetic Species Recognition (GCPSR) Analysis
In this study, species boundaries were determined using genealogical concordance phylogenetic species recognition (GCPSR), as described in previous studies, in SplitsTree4 v.4.14.6 (www.splitstree.org (accessed on 26 September 2017)) [42,63,64]. Multi-locus concatenated sequence data, with EF1-α, TUB2, CAL, and HIS, were used to determine the recombination level within phylogenetically closely related species. In addition, the results of relationships between closely related species were visualized by constructing neighbor-joining (NJ) graphs.
2.5. Phylogenetic Informativeness Analysis
Phylogenetic informativeness (PI) was analyzed from taxonomically authenticated species and type-strains based on the multi-locus combined dataset of ITS, EF1-α, TUB2, CAL, and HIS. Twenty-eight representative isolates (from 23 species, including an outgroup) with a close relationship to the D. eres species complex based on phylogenetic analysis were selected to determine the profiling of phylogenetic informativeness [65]. Ultrametric trees were generated from the concatenated alignment dataset using maximum likelihood (ML) phylogenetic analysis, as described above. To estimate phylogenetic informativeness (phylogenetic informativeness per site (PI per site) and net phylogenetic informativeness (Net PI)), the corresponding partitioned alignment was harvested from the PhyDesign Web Portal at http://phydesign.townsend.yale.edu/ (accessed on 22 April 2020) [66].
2.6. Population Aggregation and Haplotype Network Analysis
To confirm the D. eres species, 138 taxa (Table S1), along with 51 taxa (Table 1), were reconstructed using multi-locus sequences of EF1-α, TUB2, CAL, and HIS (Figure S17). In order to analyze the genetic diversity for D. eres populations, isolates that have been analyzed in phylogenetic analyses were applied. In brief, an individual locus was sequenced, and the alignment and comparison of assembled sequences were performed using ClustalX v.2.0.11 [67]. Gaps were treated as the missing data of each locus, and the end of 5′- and 3′- partial sequences were trimmed in the dataset. All population genetic parameters, including the number of polymorphic (segregating) sites (S), Nei’s nucleotide diversity (π), haplotype numbers (Hap), haplotype diversity (Hd), nucleotide diversity from S (θw), and neutrality statistic information, such as Tajima’s D, Fu and Li’s D, and Fu’s Fs, were calculated using DnaSP v.6.11.01 [68] for each individual locus and combined loci. Therefore, relationships among the haplotypes were depicted with the median-joining (MJ) method in Population Analysis with Reticulate Trees (PopART: http://popart.otago.ac.nz/index.shtml (accessed on 1 June 2020)) [69].

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
List of D. eres species complex isolates used for phylogenetic analyses, with details of host, origin, and GenBank accession number.
Table 1.
List of D. eres species complex isolates used for phylogenetic analyses, with details of host, origin, and GenBank accession number.
| Diaporthe Species a | Isolate Number b | Origin | GenBank Accession Numbers c | References | ||||
|---|---|---|---|---|---|---|---|---|
| ITS | EF1-α | TUB2 | CAL | HIS | ||||
| D. acerigenaT | CFCC 52554 | China | MH121489 | MH121531 | – | MH121413 | MH121449 | [20] |
| D. alleghaniensisT | CBS 495.72 | Canada | KC343007 | KC343733 | KC343975 | KC343249 | KC343491 | [46] |
| D. alnea | CBS 146.46 | Netherlands | KC343008 | KC343734 | KC343976 | KC343250 | KC343492 | [46] |
| D. apiculatumT | CGMCC3.17533 | China | KP267896 | KP267970 | KP293476 | – | – | [15] |
| D. betulaeT | CFCC 50469 | China | KT732950 | KT733016 | KT733020 | KT732997 | KT732999 | [70] |
| D. betulinaT | CFCC 52562 | China | MH121497 | MH121539 | MH121579 | MH121421 | MH121457 | [20] |
| D. bicinctaEP | CBS 121004 | USA | KC343134 | KC343860 | KC344102 | KC343376 | KC343618 | [43,46] |
| D. celastrinaEP | CBS 139.27 | USA | KC343047 | KC343773 | KC344015 | KC343289 | KC343531 | [43,46] |
| D. celerisT | CBS 143349 | UK | MG281017 | MG281538 | MG281190 | MG281712 | MG281363 | [4] |
| D. charlesworthiiT | BRIP 54884m | Australia | KJ197288 | KJ197250 | KJ197268 | – | – | [6] |
| D. chensiensisT | CFCC 52567 | China | MH121502 | MH121544 | MH121584 | MH121426 | MH121462 | [20] |
| D. citriT | CBS 135422 | USA | KC843311 | KC843187 | KC843071 | KC843157 | MF418281 | [3,7] |
| D. citrichinensisT | CGMCC3.15225 | China | JQ954648 | JQ954666 | MF418524 | KC357494 | KJ490516 | [3,17,18] |
| D. citrichinensis | ZJUD034B | China | KJ210539 | KJ210562 | KJ420829 | KJ435042 | KJ420879 | [43] |
| D. collarianaT | MFLUCC 17-2636 | Thailand | MG806115 | MG783040 | MG783041 | MG783042 | – | [71] |
| D. conicaT | CFCC 52571 | China | MH121506 | MH121548 | MH121588 | MH121428 | MH121466 | [20] |
| D. eresEP | CBS 138594 | Germany | KJ210529 | KJ210550 | KJ420799 | KJ434999 | KJ420850 | [43] |
| D. eres (D. biguttusis) T | CGMCC3.17081 | Unknown | KF576282 | KF576257 | KF576306 | – | – | [39] |
| D. eres (D. camptothecicola) T | CFCC 51632 | China | KY203726 | KY228887 | KY228893 | KY228877 | KY228881 | [37] |
| D. eres (D. castaneae-mollissimae) T | DNP128 | China | JF957786 | KJ210561 | KJ420801 | KJ435040 | KJ420852 | [43,72] |
| D. eres (D. cotoneastri) T | CBS 439.82 | UK | FJ889450 | GQ250341 | JX275437 | JX197429 | – | [72] |
| D. eres (D. ellipicola) T | CGMCC3.17084 | China | KF576270 | KF576245 | KF576291 | – | – | [39] |
| D. eres (D. henanensis) T | CGMCC3.17639 | China | KC898258 | – | KF600608 | – | KF600609 | [73] |
| D. eres (D. longicicola) T | CGMCC3.17089 | Unknown | KF576267 | KF576242 | KF576291 | – | – | [39] |
| D. eres (D. lonicerae) T | MFLUCC 17-0963 | Italy | KY964190 | KY964146 | KY964073 | KY964116 | – | [74] |
| D. eres (D. mahothocarpus) T | CGMCC3.15181 | China | KC153096 | KC153087 | KF576312 | – | – | [39,40] |
| D. eres (D. momicola) T | CGMCC3.17466 | China | KU557563 | KU557631 | KU557587 | KU557611 | – | [13] |
| D. eres (D. nobilis) | CBS 113470 | Korea | KC343146 | KC343872 | KC344114 | KC343388 | KC343630 | [46] |
| D. eres (D. rosicola) T | MFLU 17-0646 | UK | MG828895 | MG829270 | MG843877 | – | – | [75] |
| D. eres (Phomopsis fukushii) NE | MAFF 625033 | Japan | JQ807468 | JQ807417 | KJ420814 | KJ435017 | KJ420865 | [43] |
| D. eucommiicolaH | SCHM 3607 | China | AY578071 | – | – | – | – | [76] |
| D. fraxinicolaT | CFCC 52582 | China | MH121517 | MH121559 | – | MH121435 | – | [20] |
| D. gardeniae | CBS 288.56 | Italy | KC343113 | KC343839 | KC344081 | KC343355 | KC343597 | [46] |
| D. helicisEP | CBS 138596 | France | KJ210538 | KJ210559 | KJ420828 | KJ435043 | KJ420875 | [43] |
| D. heterophyllaeT | CBS 143769 | France | MG600222 | MG600224 | MG600226 | MG600218 | MG600220 | [77] |
| D. infertilisT | CBS 230.52 | Suriname | KC343052 | KC343778 | KC344020 | KC343294 | KC343536 | [3,46] |
| D. maritimaT | DAOMC 250563 | Canada | KU552025 | KU552023 | KU574615 | – | – | [78] |
| D. neilliae | CBS 144.27 | Unknown | KC343144 | KC343870 | KC344112 | KC343386 | KC343628 | [46] |
| D. oracciniiT | CGMCC3.17531 | China | KP267863 | KP267937 | KP293443 | – | KP293517 | [15] |
| D. padinaT | CFCC 52590 | China | MH121525 | MH121567 | MH121604 | MH121443 | MH121483 | [20] |
| D. penetriteumT | CGMCC3.17532 | China | KP714505 | KP714517 | KP714529 | – | KP714493 | [15] |
| D. phragmitisT | CBS 138897 | China | KP004445 | – | KP004507 | – | KP004503 | [79] |
| D. pulla | CBS 338.89 | Yugoslavia | KC343152 | KC343878 | KC344120 | KC343394 | KC343636 | [43] |
| D. sambucusiiT | CFCC 51986 | China | KY852495 | KY852507 | KY852511 | KY852499 | KY852503 | [80] |
| D. sennicolaT | CFCC 51634 | China | KY203722 | KY228883 | KY228889 | KY228873 | – | [81] |
| D. shennongjiaensisT | CNUCC 201905 | China | MN216229 | MN224672 | MN227012 | MN224551 | MN224559 | [35] |
| D. subclavataT | CGMCC3.17257 | China | KJ490630 | KJ490509 | KJ490451 | – | KJ490572 | [18] |
| D. tibetensisT | CFCC 51999 | China | MF279843 | MF279858 | MF279873 | MF279888 | MF279828 | [14] |
| D. ukurunduensisT | CFCC 52592 | China | MH121527 | MH121569 | – | MH121445 | MH121485 | [20] |
| D. vacciniiT | CBS 160.32 | USA | KC343228 | KC343954 | KC344196 | KC343470 | KC343712 | [46] |
| D. virgiliaeT | CBS 138788 | South Africa | KP247573 | – | KP247582 | – | – | [82] |
a H (holotype), T (ex-type), EP (ex-epitype), and NE (ex-neotype) cultures are indicated with isolate numbers in bold. b BRIP: Plant Pathology Herbarium, Department of Employment, Economic, Development and Innovation, Queensland, Australia; CBS: Westerdijk Fungal Biodiversity Institute, Utrecht, The Netherlands; CFCC: China Forestry Culture Collection Center, Beijing, China; CGMCC: China General Microbiological Culture Collection, China; CNUCC: Capital Normal University Culture Collection Center, Beijing, China; DAOMC: Canadian Collection of Fungal Cultures, Agriculture, and Agri-Food Canada, Ottawa, Canada; DNP: First author’s personal collection (deposited in MFLUCC), Thailand; MAFF: NIAS GenBank Project, Ministry of Agriculture, Forestry, and Fisheries, Japan; MFLU: Herbarium of Mae Fah Luang University, Chiang Rai, Thailand; MFLUCC: Mae Fah Luang University Culture Collection, Chiang Rai, Thailand; SCHM: Mycological Herbarium of South China Agricultural University, Guangzhou, China; ZJUD: Diaporthe species culture collection at the Institute of Biotechnology, Zhejiang University, Hangzhou, China. c Ribosomal internal transcribed spacer (ITS) region of ribosomal DNA (ITS1-5.8S-ITS2), translation elongation factor 1-α (EF1-α), beta-tubulin 2 (TUB2), calmodulin (CAL), and histone-3 (HIS).
3. Results
3.1. Phylogenetic Analysis of D. eres
In this study, the concatenated DNA sequences of five loci (ITS, EF1-α, CAL, TUB2, and HIS) from 216 sequences, including 5 outgroup sequences of D. citri, were used to infer delimitation of Diaporthe species. For the reconstruction of phylogenetic trees of Diaporthe species, altogether, 51 sequences of ITS, 47 sequences of EF1-α, 36 sequences of CAL, 47 sequences of TUB2, and 35 sequences of HIS were obtained from the GenBank database. Sequences of ITS, EF1-α, CAL, TUB2, and HIS were determined as 598, 592, 542, 828, and 502 base pairs (bp), respectively. For species delimitation of the D. eres complex, 50 taxa were analyzed, with 2464 bp assembled sequences of 4 genes, including 592 bp (1–592) of EF1-α, 542 bp (593–1134) of CAL, 828 bp (1135–1962) of TUB2, and 502 bp (1963–2464) of HIS, respectively. For the 5 loci combined sequences dataset with ITS region, we filled in the end of the four-gene dataset with 598 bp (2465–3062) of ITS. ML, MP, and BI analyses were used to perform phylogenetic reconstruction for individual and combined datasets; results showed similar topology and few differences in statistical support values. A comparison of alignment properties in parsimony analyses of individual and combined loci used in phylogenetic analyses is provided in Table 2.
Phylogenetic analyses for Diaporthe species were performed using each individual locus and combined loci of DNA sequences (Figures S1–S15). Among them, phylogenetic trees, using EF1-α, EF1-α+CAL, and EF1-α+CAL+TUB2+HIS, showed clear delimitation for D. eres species, while unclear delimitation was observed using five loci sequences of EF1-α+CAL+TUB2+HIS+ITS (Figure 1). It was found that the three-loci combined dataset of EF1-α+CAL+TUB2 or EF1-α+CAL+HIS, with ML, MP, and BI analyses, was unable to separate D. bicincta (CBS 121004), D. celastrina (CBS 139.27), D. celeris (CBS 143349), D. helicis (CBS 138596), D. maritima (DAOMC 250563), D. phragmitis (CBS 138897), and D. pulla (CBS 338.89) species (Figures S12 and S13). Overall, the four-loci combined dataset of EF1-α+CAL+TUB2+HIS showed the highest reliability to identify and resolve species boundaries in the D. eres complex (Figure 1 and Figure 2).
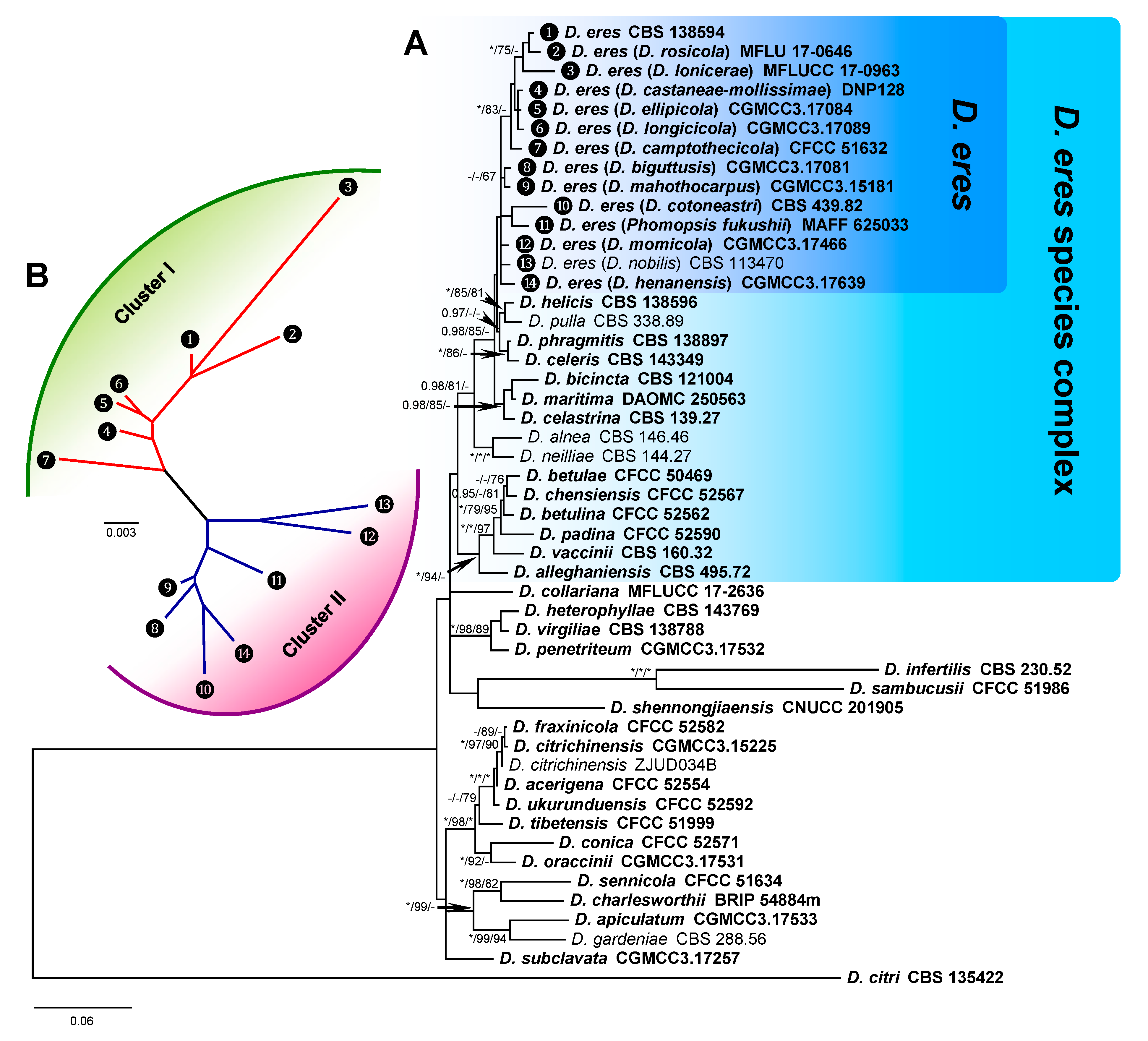
The Bayesian inference phylogenetic tree of the D. eres species complex and close species constructed with combined DNA sequences is presented in Figure 2 as an example. In the phylogenetic tree, the D. eres species complex cluster (D. eres species complex) was clearly separated from another cluster that included D. collariana, D. heterophyllae, D. virgiliae, D. penetriteum, D. infertilis, D. sambucusii, and D. shennongjiaensis. Within the D. eres species complex cluster, a subcluster that included D. eres and 13 species with other names should be the synonymous species of D. eres. The close subcluster, which included D. helicis (CBS 138596), D. pulla (CBS 338.89), D. phragmitis (CBS 138897), and D. celeris (CBS 143349), showed a relatively distinct distance with D. eres, indicating that they are different species (Figure 2A). The D. eres species was further analyzed by GCPSR analysis. The NJ tree shows the relationship between Cluster I and Cluster II (Figure 2B), indicating that genetic diversity is rich in this species.
3.2. Phylogenetic Informative Analysis
For phylogenetic informative analysis, only taxa with complete 5 loci sequences were used. As a result, the assembled DNA sequences were 3049 bp, including 544 bp of CAL (1–544), 573 bp of EF1-α (545–1117), 829 bp of TUB2 (1118–1946), 503 bp of HIS (1947–2449), and 600 bp of ITS (2450–3049). The combined dataset consisted of 28 taxa (from 23 species), including the outgroup species D. citri (CBS 135422).
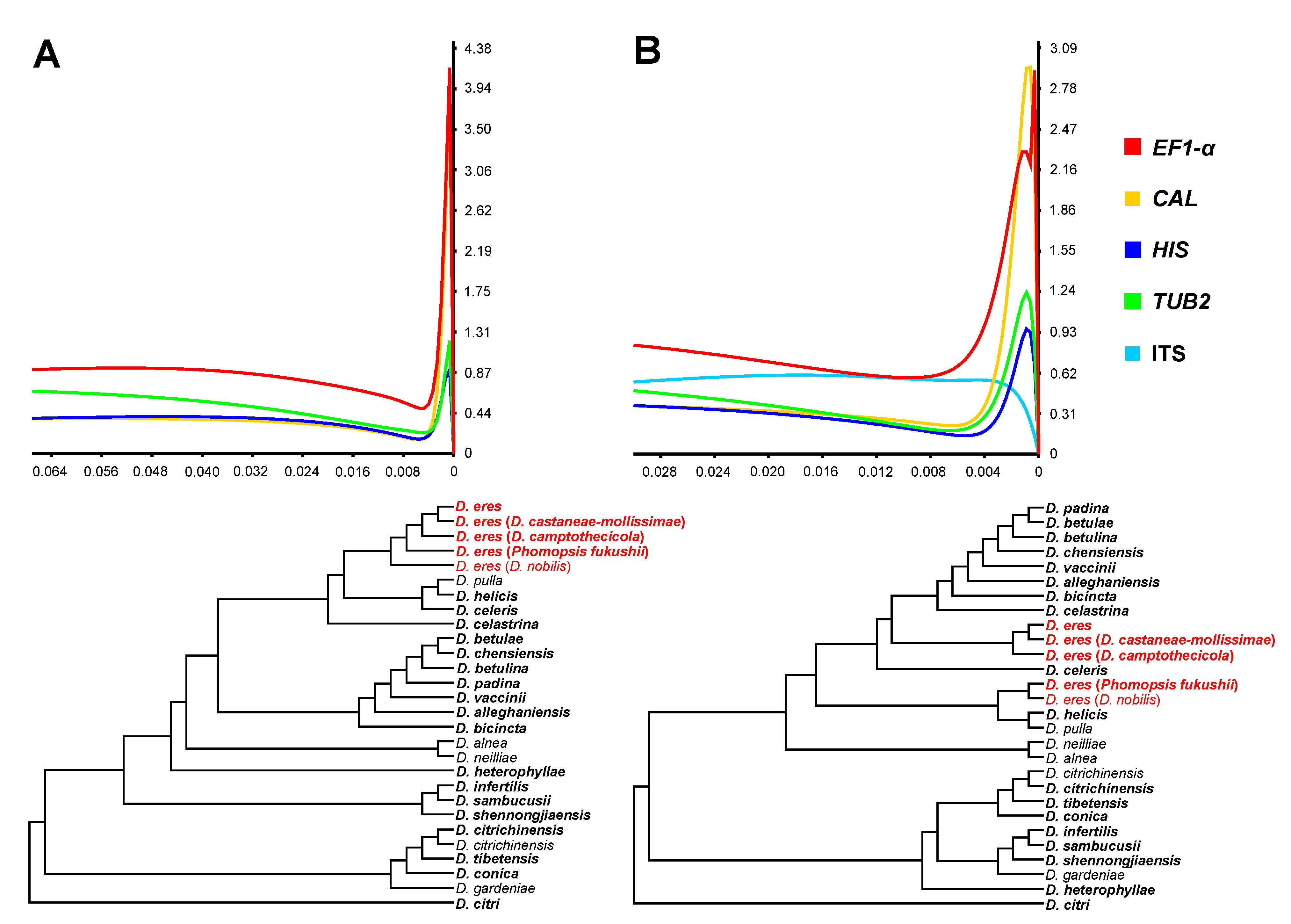
Phylogenetic informativeness (PI) profiles, both Net PI and PI per site, indicated that EF1-α and CAL displayed the highest informative sequences to resolve the phylogenetic signal at the taxonomic level. Next were HIS and TUB2, which maintained fairly high informative sequences (Figure 3 and Figure S16). ITS presented the lowest PI signal among the selected loci and was unreliable for the delimitation of the D. eres species. The combined dataset of four loci (EF1-α, TUB2, CAL, and HIS) showed better delimitation for D. eres compared to the dataset of five loci (Figure 3), further confirming that the ITS locus was lowly informative.
3.3. D. eres Species Boundaries
Based on the phylogenetic analyses using multi-locus reconstruction (EF1-α, TUB2, CAL, and HIS), the species delimitation was determined among D. eres and closely related species (Figure 1 and Figure 2). Results showed that D. eres and the other 13 species were conspecific. Among them, D. biguttusis, D. camptothecicola, D. castaneae-mollissimae, D. cotoneastri, D. ellipicola, D. longicicola, D. mahothocarpus, D. momicola, D. nobilis, and Phomopsis fukushii have already been previously considered the synonymous species of D. eres; in this study, another three species, D. henanensis, D. lonicerae, and D. rosicola, were further revealed to be synonyms of D. eres.
Diaporthe eres Nitschke 1870 [83].
= Diaporthe henanensis Y. Yang, H.Y. Wu & M. Zhang, 2016.
= Diaporthe lonicerae A.J. Dissanayake, E. Camporesi & K.D. Hyde, 2017.
= Diaporthe rosicola D.N. Wanasinghe, E.B.G. Jones & K.D. Hyde, 2018.
3.4. Population Aggregation and Haplotype Network Analysis
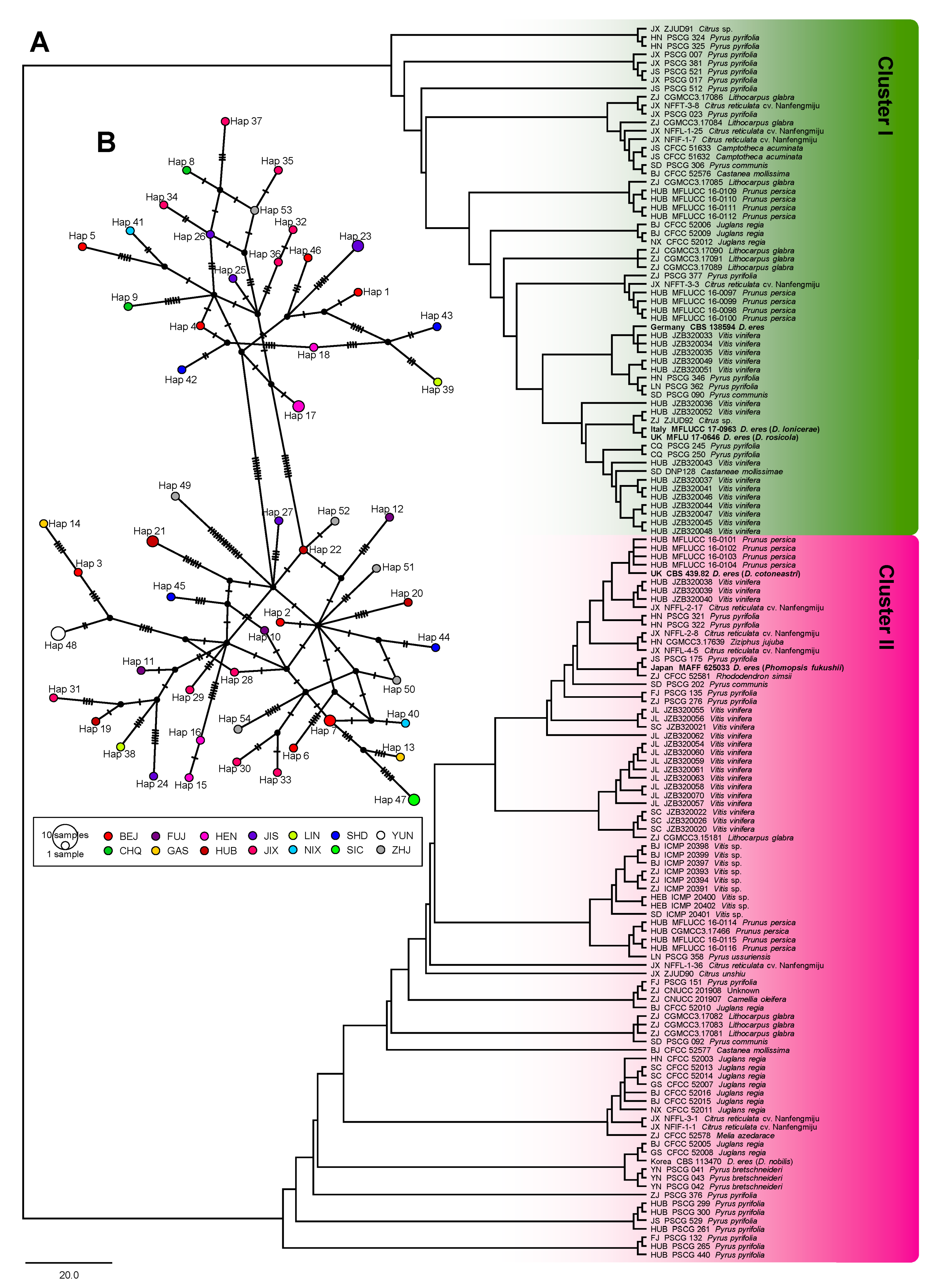
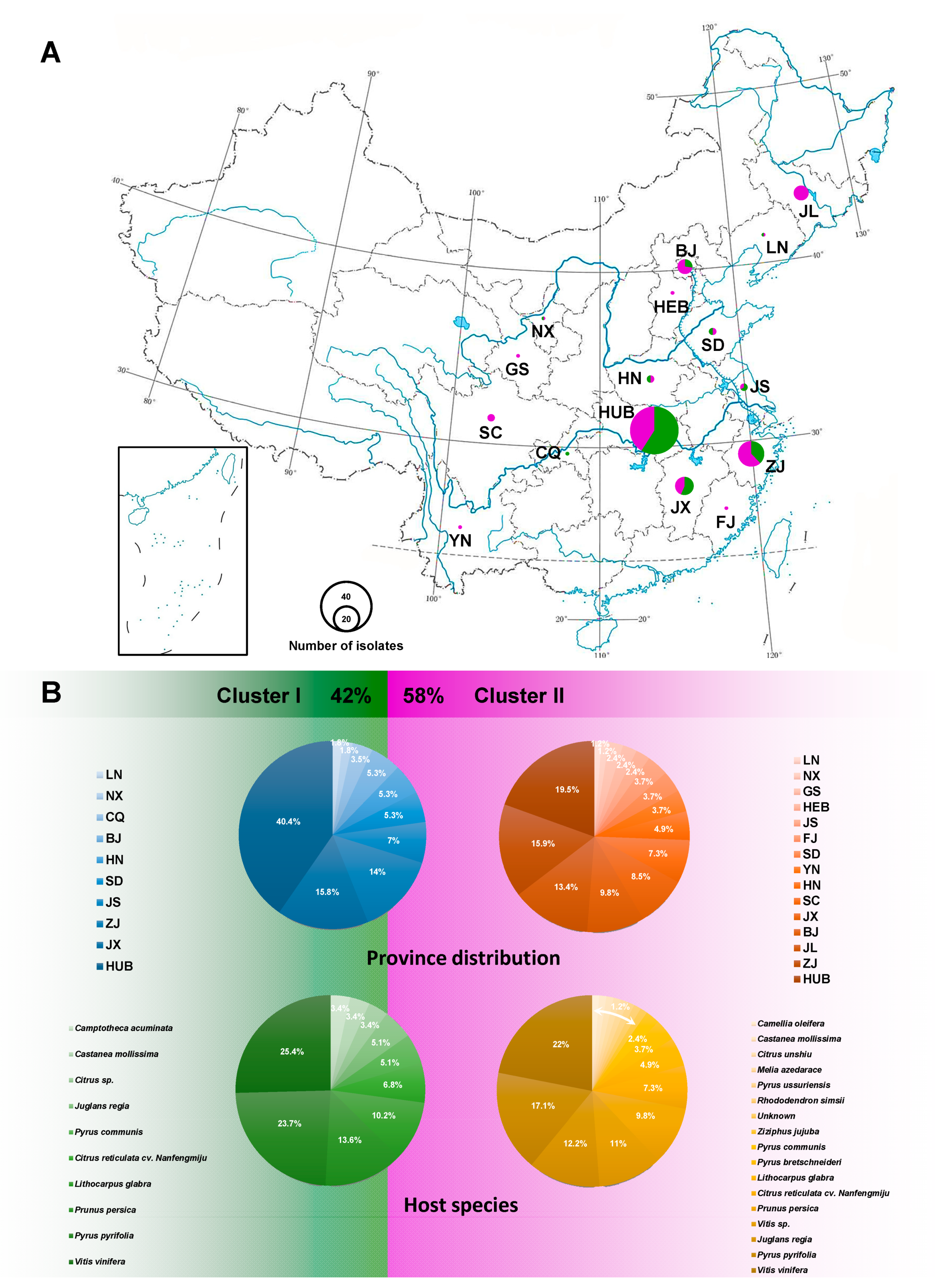
The ITS of 137 taxa, EF1-α of 132 taxa, TUB2 of 137 taxa, CAL of 118 taxa, HIS of 70 taxa, and the combined sequences of 61 taxa were 450, 278, 355, 492, 421, 1429 (four loci without ITS), and 1882 (five loci) bp in length, respectively. The analysis of genetic diversity within D. eres showed a high level of haplotype numeric (Hap) and haplotype diversity (Hd). The summary of sequence variation and indices of sequence variation within the five loci among D. eres are shown in Table 3 and Table S3. The haplotype diversity values of ITS, EF1-α, TUB2, CAL, HIS, and the combined datasets were greater than 0.5, reflecting high genetic diversity. The neutrality statistic (Tajima’s D and Fu’s Fs) results showed negative values, suggesting population expansion in D. eres isolates. We obtained similar results of population network analysis in the phylogenetic tree (Figure 4A) and the median-joining haplotype network (Figure 4B) using the combined dataset of EF1-α, TUB2, CAL, and HIS. Population connectivity was grouped into two clusters that were not correlated to specific populations of geographic distribution (Figure 5).
3.5. Phylogenetic Informative Analysis
It should be noted that median-joining haplotype network analysis was also performed based on each individual locus using DnaSP v.6.11.01. The major haplotype numbers from EF1-α, CAL, TUB2, HIS, and ITS were 29, 49, 25, 23, and 9, respectively. Analysis with the CAL locus showed two small distinct clusters: one consisted of hap 1, 18, and 21 from BJ, GS, HN, HUB, JL, LN, and SD, and another consisted of hap 5, 9, and 10 from CQ, HUB, JX, and YN. HUB isolates could be found in both clusters (Figure 5). Similarly, the analysis of the TUB2 locus also showed two small clusters. Thus, we found that haplotypes that were connected between Cluster I (hap 11) and Cluster II (hap 13, 21, and 26) were from a center part of China, i.e., HEB, HN, HUB, and JX. Analysis of EF1-α, HIS, and ITS loci showed a wide distribution and incommensurate derivative splitting by geographic distribution. These median-joining haplotype networks in each individual locus are shown in Figure S18.
4. Discussion
In this study, we used five-loci DNA sequences to understand and interpret the species boundaries of the D. eres species complex and assess the genetic diversity of D. eres populations in China. A total of 51 taxa, including 37 close species to the D. eres species complex, was applied to narrow the criteria of phylogenetic relatives using the GCPSR of phylogenetic species, while 138 D. eres isolates from various Chinese populations were examined to assess the relationship between genetic diversity and different geographic distributions.
Recently, the classification of Diaporthe species has become more dependent on a molecular approach rather than traditional morphological characterization [72,84,85]. Next-generation sequencing (NGS) technology, such as DNA barcoding, is highly efficient, more accurate, and, thus, valid for fungal identification at the species level [86,87]. The ITS sequence is commonly used for preliminary fungal identification and is recommended for identifying species boundaries in the genus Diaporthe, Diaporthaceae, and Sordariomycetes [5,77,88,89]. However, there are many intraspecific variations in the ITS locus of certain Diaporthe species. Sometimes the intraspecific variation is even greater than the interspecific variation, which makes it difficult to identify Diaporthe species using the ITS sequence alone [90,91].
The identification of Diaporthe species based on morphological characterization is very contradictory, and a molecular approach using DNA sequences should be combined to identify species within this genus [46,47]. To redefine the boundaries of Diaporthe species, Santos et al. [47] proposed highly effective phylogenetic reconstruction using DNA barcoding sequences of multiple loci, i.e., ITS, EF1-α, TUB2, CAL, and HIS. The taxonomy of Diaporthe is complex, and many Diaporthe spp. are classified based on different criteria, according to host associations, morphological characteristics [12,92,93,94], or sequences of the ITS region [5,92,95]. It is suggested that only the type strains whose identification has been widely recognized should be accepted as references for the taxonomy of this genus [46,96,97]. In this study, several isolates, including type strains from previous publications, were selected as references with phylogenetic analysis. However, when a MegaBlast search was performed for each locus in NCBI, generally, the Diaporthe species showing the highest similarity with the sequence of each locus of the isolates were not the type strains. Thus, the species used by us in the current study were not always the same as those recovered by the single locus MegaBlast search in NCBI. The combined multi-locus phylogenetic reconstruction shows the very strong species delimitation for the D. sojae complex [48]. Fan et al. [14] demonstrated the effectiveness of 3 loci, including EF1-α, TUB2, and CAL, for the identification of the D. eres complex in walnut trees. Similarly, Yang et al. [20] and Zhou and Hou [35] also used three-locus sequences to identify D. eres species associated with different hosts in China. These studies excluded a few closely related Diaporthe species with typical reference strains. However, our study revealed that the phylogenetic analysis from the combined dataset of EF1-α, TUB2, CAL, and HIS was highly effective and strongly supported to resolve species boundaries of the D. eres species complex. This is consistent with the results obtained by Guo et al. [16]. Phylogenetic informative (PI) profiles using multi-locus phylogenetic analysis of five loci are commonly used to identify the D. eres species complex; both Net PI and PI per site showed similar results. Among different loci, EF1-α, APN2 (DNA lyase), and HIS loci are effective for the species delimitation of the D. eres species complex. It was reported that EF1-α showed the highest effectiveness to resolve the phylogenetic signal, which is concordant with the results obtained by Udayanga et al. [43]. Similarly, the highly variable EF1-α locus showed the highest effectiveness to discriminate species in the Diaporthe genus [43,47,91,98]. Our study revealed that EF1-α was reliable, but the ITS region impeded species delimitation and relatively limited phylogenetic signals when the combined DNA sequences of five loci (ITS, EF1-α, TUB2, CAL, and HIS) were used.
In previous studies, several synonyms of the D. eres species were successfully demonstrated based on phylogenetic analyses using multi-locus sequences, i.e., D. biguttusis, D. camptothecicola, D. castaneae-mollissimae, D. cotoneastri, D. ellipicola, D. longicicola, D. mahothocarpus, D. momicola, D. nobilis, and Phomopsis fukushii [14,20,43]. In the current study, we found that D. henanensis [73], D. lonicerae [74], and D. rosicola [75] were also synonyms of D. eres because these 3 species and the previously demonstrated 10 species were grouped into a single subcluster with D. eres in phylogenetic analyses.
Using population genetic analyses, Manawasighe et al. [19] demonstrated the genetic variation of D. eres associated with grapevine dieback in China and found isolates grouped according to geographic location. However, a comparison of Chinese and European D. eres isolates, using both individual- and multi-locus DNA sequences of ITS, EF1-α, TUB2, CAL, and HIS loci, did not show significant differences between the two geographical populations. In this study, D. eres were grouped into two major populations that were not correlated with geographic distribution. Interestingly, we found that isolates from the central part of China, e.g., HEB, HN, HUB, and JX, simultaneously fell into two different clusters with a significant haplotype connection, suggesting that this region is the origin of D. eres. This is consistent with the observation that HUB isolates might be the parental population of D. eres [19].
Finally, future species identification should use a highly effective molecular approach to make it simple and easy to detect D. eres in routine plant quarantine. For genetic variation and population analyses, sample sizes should be increased and comparisons should be performed with the analyses using other molecular markers, including amplified fragment length polymorphism (AFLP), random amplified polymorphic DNA (RAPD), and inter simple sequence repeat (ISSR). Further understanding should focus on the ancestor, phylogeographic and demographic history, divergence-time estimation, and migration history of D. eres.
5. Conclusions
The current study provides an overview of D. eres on several plant varieties and some valuable knowledge to identify this fungus. Phylogenetic trees were reconstructed for the D. eres species complex with combined DNA sequences of EF1-α, CAL, TUB2, and HIS. Phylogenetic analyses and phylogenetic informativeness profiles reported in this study revealed that for D. eres species identification and delimitation, the usage of the EF1-α locus represents the optimal alternative; this proposition is also supported by previous studies [43,47,91,98]. Moreover, our analyses revealed that the usage of the ITS region hampers proper species recognition within the D. eres species complex. An expansion of population connectivity among the D. eres populations was detected. One hundred and thirty-eight D. eres isolates were divided by phylogenetic analyses, and genetic distance estimation with haplotype networks revealed two clusters with strong support from population genetic parameters and neutrality of statistic informative values, indicating a high level of haplotype diversity. However, we found that the two clusters from both methods were not separated based on geographic distribution. Overall, our analyses determined the current pattern of phylogenetic identification for the D. eres species and population diversity within the Chinese isolates of D. eres. In the future, studies on the evolution of D. eres and plant-D. eres interaction should be conducted.
Supplementary Materials
The following are available online at https://www.mdpi.com/2079-7737/10/3/179/s1. Table S1: List of D. eres isolates used for population analysis, with details of origin, host, isolate number, and GenBank accession number. Table S2: The best-fit corresponding nucleotide substitution models used in BI analysis. Table S3: Sample size and haplotypes detected by distribution locality among D. eres. Figures S1–S5: Phylogenetic analysis of the D. eres species complex based on the individual locus of EF1-α, CAL, TUB2, HIS, and ITS, respectively. Figures S6–S15: Phylogenetic analysis of the D. eres species complex based on the combined loci of EF1-α+CAL, EF1-α+TUB2, EF1-α+HIS, CAL+TUB2, CAL+HIS, TUB2+HIS, EF1-α+CAL+TUB2, EF1-α+CAL+HIS, EF1-α+CAL+TUB2+HIS, and EF1-α+CAL+TUB2+HIS+ITS, respectively. (A) RAxML phylogenetic tree, (B) Parsimonious phylogenetic tree, (C) Bayesian phylogenetic tree. The trees were rooted using D. citri (CBS 135422). Maximum likelihood and maximum parsimony bootstrap values (MLBS and MPBS) >50%, Bayesian posterior probabilities values (BIPP) >0.75 are given at the branch nodes. Holotype, ex-type, ex-epitype, and ex-neotype cultures are indicated using isolate numbers in bold. The scale bar represents the expected number of changes per site. Figure S16: Phylogenetic informativeness profiles of 28 Diaporthe species, including the D. eres species complex and close species. Phylogenetic informativeness is shown as phylogenetic informativeness per site (PI per site) based on a combined multi-locus dataset. (A) Combined dataset from four loci (EF1-α+CAL+TUB2+HIS). (B) Combined dataset from five loci (EF1-α+CAL+TUB2+HIS+ITS). Values on the X-axes correspond to the relative timescale (0–1). Values on the Y-axes represent 10–3 PI per site in arbitrary units. Figure S17: Phylogenetic tree generated from maximum likelihood analysis of all available type species of Diaporthe 51 isolates (Table 1), together with 138 isolates (Table S1), based on combined sequences of EF1-α, CAL, TUB2, and HIS. Figure S18: Median-joining (MJ) haplotype networks were analyzed to evaluate the genetic diversity in D. eres. (A) translation elongation factor 1-α gene (EF1-α), (B) calmodulin gene (CAL), (C) beta-tubulin 2 gene (TUB2), (D) histone-3 gene (HIS), and € ribosomal internal transcribed spacer regions (ITS). Each circle represents a unique haplotype, and its size reflects the number of individuals expressing that haplotype. Crosshatches are indicative of the number of nucleotide differences between haplotypes. Color codes denote geographic populations. Geographic location abbreviation, BJ: Beijing; CQ: Chongqing; FJ: Fujian; GS: Gansu; HEB: Hebei; HN: Henan; HUB: Hubei; JS: Jiangsu; JX: Jiangxi; JL: Jilin; LN: Liaoning; NX: Ningxia; SD: Shandong; SC: Sichuan; YN: Yunnan; ZJ: Zhejiang.
Author Contributions
Conceptualization, C.C.; methodology, C.C., X.L., Y.L., Y.F., and F.Z.; software, C.C.; validation, C.C., X.L., and C.L.; formal analysis, C.C. and X.L.; investigation, C.C., X.L., Y.L., Y.F., and F.Z.; data curation, C.C., X.L., Y.L., and C.L.; writing—original draft preparation, C.C., X.L., and C.L.; writing—review and editing, Y.L., Y.F., F.Z., and C.L.; visualization, C.C. and X.L.; supervision, Y.L. and C.L.; project administration and funding acquisition, Y.L., Y.F., and C.L. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Fundamental Research Funds for the Central Universities (No. 2662020ZKPY018) and the National Key Research and Development Program of China (No. 2017YFD020200103).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Alignments generated during the current study are available in TreeBASE (accession http://purl.org/phylo/treebase/phylows/study/TB2:S26697 (accessed on 2 August 2020)). All sequence data are available in the NCBI GenBank, following the accession numbers in the manuscript.
Acknowledgments
The authors sincerely thank the reviewers for their contributions during the revision process.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript or in the decision to publish the results.
References
- Senanayake, I.C.; Crous, P.W.; Groenewald, J.Z.; Maharachchikumbura, S.S.N.; Jeewon, R.; Phillips, A.J.L.; Bhat, J.D.; Perera, R.H.; Li, Q.R.; Li, W.J.; et al. Families of Diaporthales based on morphological and phylogenetic evidence. Stud. Mycol. 2017, 86, 217–296. [Google Scholar] [CrossRef]
- Wehmeyer, L.E. The genus Diaporthe Nitschke and its segregates. Univ. Mich. Stud. Sci. Ser. 1933, 9, 1–349. [Google Scholar]
- Guarnaccia, V.; Crous, P.W. Emerging citrus diseases in Europe caused by species of Diaporthe. IMA Fungus 2017, 8, 317–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guarnaccia, V.; Groenewald, J.Z.; Woodhall, J.; Armengol, J.; Cinelli, T.; Eichmeier, A.; Ezra, D.; Fontaine, F.; Gramaje, D.; Gutierrez-Aguirregabiria, A.; et al. Diaporthe diversity and pathogenicity revealed from a broad survey of grapevine diseases in Europe. Persoonia 2018, 40, 135–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos, J.M.; Phillips, A.J.L. Resolving the complex of Diaporthe (Phomopsis) species occurring on Foeniculum vulgare in Portugal. Fungal Divers. 2009, 34, 111–125. [Google Scholar]
- Thompson, S.M.; Tan, Y.P.; Shivas, R.G.; Neate, S.M.; Morin, L.; Bissett, A.; Aitken, E.A.B. Green and brown bridges between weeds and crops reveal novel Diaporthe species in Australia. Persoonia 2015, 35, 39–49. [Google Scholar] [CrossRef] [Green Version]
- Udayanga, D.; Castlebury, L.A.; Rossman, A.Y.; Hyde, K.D. Species limits in Diaporthe: Molecular re-assessment of D. citri, D. cytosporella, D. foeniculina and D. rudis. Persoonia 2014, 32, 83–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Reyne, A.; López-Medrano, F.; Morales, J.M.; Esteban, C.G.; Martín, I.; Eraña, I.; Meije, Y.; Lalueza, A.; Alastruey-Izquierdo, A.; Rodríguez-Tudela, J.L.; et al. Cutaneous infection by Phomopsis longicolla in a renal transplant recipient from guinea: First report of human infection by this fungus. Transpl. Infect. Dis. 2011, 13, 204–207. [Google Scholar] [CrossRef] [PubMed]
- Laosakul, K.; Youngchim, S.; Chuamanochan, M.; Rujiwetpongstorn, R.; Tovanabutra, N.; Chiewchanvit, S. Phaeohyphomycosis caused by Diaporthe phaseolorum in an immunocompetent patient in Thailand: A case report. Access Microbiol. 2020, 2. [Google Scholar] [CrossRef]
- Warmelo, K.T.; Marasas, W.F.O. Phomopsis leptostromiformis: The causal fungus of lupinosis, a mycotoxicosis, in sheep. Mycologia 1972, 64, 316–324. [Google Scholar] [CrossRef]
- Webber, J.F.; Gibbs, J.N. Colonization of elm bark by Phomopsis oblonga. Trans. Br. Mycol. Soc. 1984, 82, 348–352. [Google Scholar] [CrossRef]
- Chi, P.K.; Jiang, Z.D.; Xiang, M.M. Flora Fungorum Sinicorum; Science Press: Beijing, China, 2007; Volume 34. (In Chinese) [Google Scholar]
- Dissanayake, A.J.; Zhang, W.; Liu, M.; Hyde, K.D.; Zhao, W.S.; Li, X.H.; Yan, J.Y. Diaporthe species associated with peach tree dieback in Hubei, China. Mycosphere 2017, 8, 533–549. [Google Scholar] [CrossRef]
- Fan, X.L.; Yang, Q.; Bezerra, J.D.P.; Alvarez, L.V.; Tian, C.M. Diaporthe from walnut tree (Juglans regia) in China, with insight of the Diaporthe eres complex. Mycol. Progress 2018, 17, 841–853. [Google Scholar] [CrossRef]
- Gao, Y.H.; Liu, F.; Duan, W.J.; Crous, P.W.; Cai, L. Diaporthe is paraphyletic. IMA Fungus 2017, 8, 153–187. [Google Scholar] [CrossRef]
- Guo, Y.S.; Crous, P.W.; Bai, Q.; Fu, M.; Yang, M.M.; Wang, X.H.; Du, Y.M.; Hong, N.; Xu, W.X.; Wang, G.P. High diversity of Diaporthe species associated with pear shoot canker in China. Persoonia 2020, 45, 132–162. [Google Scholar] [CrossRef]
- Huang, F.; Hou, X.; Dewdney, M.M.; Fu, Y.S.; Chen, G.Q.; Hyde, K.D.; Li, H.Y. Diaporthe species occurring on citrus in China. Fungal Divers. 2013, 61, 237–250. [Google Scholar] [CrossRef]
- Huang, F.; Udayanga, D.; Wang, X.H.; Hou, X.; Mei, X.F.; Fu, Y.S.; Hyde, K.D.; Li, H.Y. Endophytic Diaporthe associated with citrus: A phylogenetic reassessment with seven new species from China. Fungal Biol. 2015, 119, 331–347. [Google Scholar] [CrossRef]
- Manawasighe, I.S.; Dissanayake, A.J.; Li, X.H.; Liu, M.; Wanasinghe, D.N.; Xu, J.P.; Zhao, W.S.; Zhang, W.; Zhou, Y.Y.; Hyde, K.D.; et al. High genetic diversity and species complexity of Diaporthe associated with grapevine dieback in China. Front. Microbiol. 2019, 10, 1936. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Fan, X.L.; Guarnaccia, V.; Tian, C.M. High diversity of Diaporthe species associated with dieback diseases in China, with twelve new species described. MycoKeys 2018, 39, 97–149. [Google Scholar] [CrossRef] [Green Version]
- Yang, Q.; Jiang, N.; Tian, C.M. Three new Diaporthe species from Shaanxi province, China. MycoKeys 2020, 67, 1–18. [Google Scholar] [CrossRef]
- Dissanayake, A.J.; Chen, Y.Y.; Liu, J.K. Unravelling Diaporthe species associated with woody hosts from Karst Formations (Guizhou) in China. J. Fungi 2020, 6, 251. [Google Scholar] [CrossRef] [PubMed]
- Anagnostakis, S.L. Diaporthe eres (Phomopsis oblonga) as a pathogen of butternut (Juglans cinerea) in connecticut. Plant Dis. 2007, 91, 1198. [Google Scholar] [CrossRef] [PubMed]
- Quaroni, S.; Sardi, P.; Locci, R. Apical blight in Acer pseudoplatanus associated with Diaporthe eres Nits. (Phomopsis acerina Pir. et Cart). Riv. Patol. Veg. 1980, 16, 109. [Google Scholar]
- Bastidea, F.; Sérandat, I.; Gombertc, J.; Laurentc, E.; Moreld, E.; Koloppe, J.; Guillerminf, P.L.; Hamona, B.; Simoneaua, P.; Berruyera, R.; et al. Characterization of fungal pathogens (Diaporthe angelicae and D. eres) responsible for umbel browning and stem necrosis on carrot in France. Plant Pathol. 2016, 66, 221–227. [Google Scholar]
- Meepagala, K.M.; Briscoe, W.E.; Techen, N.; Johnson, R.D.; Clausen, B.M.; Duke, S.O. Isolation of a phytotoxic isocoumarin from Diaporthe eres-infected Hedera helix (english ivy) and synthesis of its phytotoxic analogs. Pest Manag. Sci. 2018, 74, 37–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lorenzini, M.; Zapparoli, G. Identification of Pestalotiopsis bicilita, Diplodia seriata and Diaporthe eres causing fruit rot in withered grapes in Italy. Eur. J. Plant Pathol. 2018, 151, 1089–1093. [Google Scholar] [CrossRef]
- Ali, S.; Renderos, W.; Bevis, E.; Hebb, J.; Abbasi, P.A. Diaporthe eres causes stem cankers and death of young apple rootstocks in Canada. Can. J. Plant Pathol. 2019, 42, 218–227. [Google Scholar] [CrossRef]
- Li, D.; Zhang, H.Y.; Song, Q.N.; Liu, J.; Yang, Q.P.; Luan, F.G.; Li, D. First of Diaporthe eres causing branch canker on Cinnamomum camphora (camphor tree) in Jiangxi province, China. Plant Dis. 2021. First Look. [Google Scholar]
- Tao, H.; Wang, H.; Huang, S.X.; Zhang, Y.; Zhang, Z.H.; Liu, W.; Shi, N.X.; Zhu, F.; Ji, Z.L.; Chen, X.R. Identification and characterization of Diaporthe eres causing leaf blight disease on the medicinal herb Polygonatum sibiricum. J. Gen. Plant Pathol. 2020, 86, 468–476. [Google Scholar] [CrossRef]
- Song, L.M.; Wang, X.; Chen, T.G.; Liang, W.X.; Yang, Q.Q. First report of Diaporthe eres leaf spot on Photinia x fraseri ‘red robin’ in Qingdao, China. Plant Dis. 2019, 103, 159. [Google Scholar] [CrossRef]
- Mei, P.Y.; Song, X.H.; Zhu, Z.Y.; Li, L.Y. First report of Diaporthe eres causing root rot of Coptis chinensis Franchet. Plant Dis. 2021. First Look. [Google Scholar] [CrossRef]
- Zhao, Y.Q.; Geng, G.M.; Tian, Y.L.; Hu, B.S. First report of shoot blight of Japanese maple caused by Diaporthe eres in China. J. Plant Pathol. 2019, 101, 179. [Google Scholar] [CrossRef] [Green Version]
- Sun, Z.Q.; Xue, C.Y.; Wang, Y.F.; Xu, C.; Zang, R.; Geng, Y.H.; Miao, Y.C.; Wu, H.Y.; Zhang, M. First report of Diaporthe eres causing twig dieback of white bark pine in China. Plant Dis. 2020, 104, 1862. [Google Scholar] [CrossRef]
- Zhou, H.; Hou, C.L. Three new species of Diaporthe from China based on morphological characters and DNA sequence data analyses. Phytotaxa 2019, 422, 157–174. [Google Scholar] [CrossRef]
- Li, Y.; Tan, P.; Zhao, D.G. Diaporthe nobilis, a new record on Camellia sinensis in Guizhou province, China. Mycosphere 2017, 8, 1–8. [Google Scholar] [CrossRef]
- Yang, Q.; Fan, X.L.; Du, Z.; Liang, Y.M.; Tian, C.M. Diaporthe camptothecicola sp. nov. on Camptotheca acuminata in China. Mycotaxon 2017, 132, 591–601. [Google Scholar] [CrossRef]
- Dissanayake, A.J.; Liu, M.; Zhang, W.; Chen, Z.; Udayanga, D.; Chukeatirote, E.; Li, X.H.; Yan, J.Y.; Hyde, K.D. Morphological and molecular characterisation of Diaporthe species associated with grapevine trunk disease in China. Fungal Biol. 2015, 119, 283–294. [Google Scholar] [CrossRef]
- Gao, Y.H.; Su, Y.Y.; Sun, W.; Cai, L. Diaporthe species occurring on Lithocarpus glabra in China, with descriptions of five new species. Fungal Biol. 2015, 119, 295–309. [Google Scholar] [CrossRef]
- Gao, Y.H.; Sun, W.; Su, Y.Y.; Cai, L. Three new species of Phomopsis in Gutianshan nature reserve in China. Mycol. Progress 2014, 13, 111–121. [Google Scholar] [CrossRef]
- Li, L.; Pan, H.; Chen, M.Y.; Zhang, S.J.; Zhong, C.H. Isolation and identification of pathogenic fungi causing postharvest fruits rot kiwifruit (Actinidia chinensis) in China. J. Phytopathol. 2017, 165, 782–790. [Google Scholar] [CrossRef]
- Taylor, J.W.; Jacobson, D.J.; Kroken, S.; Kasuga, T.; Geiser, D.M.; Hibbett, D.S.; Fisher, M.C. Phylogenetic species recognition and species concepts in fungi. Fungal Genet. Biol. 2000, 31, 21–32. [Google Scholar] [CrossRef] [Green Version]
- Udayanga, D.; Castlebury, L.A.; Rossman, A.Y.; Chukeatirote, E.; Hyde, K.D. Insights into the genus Diaporthe: Phylogenetic species delimitation in the D. eres species complex. Fungal Divers. 2014, 67, 203–229. [Google Scholar] [CrossRef] [Green Version]
- Degnan, J.H.; Rosenberg, N.A. Gene tree discordance, phylogenetic inference and the multispecies coalescent. Trends Ecol. Evol. 2009, 2009, 6. [Google Scholar] [CrossRef] [PubMed]
- Kubatko, L.S.; Degnan, J.H. Inconsistency of phylogenetic estimates from concatenated data under coalescence. Syst. Biol. 2007, 56, 17–24. [Google Scholar] [CrossRef] [Green Version]
- Gomes, R.R.; Glienke, C.; Videira, S.I.R.; Lombard, L.; Groenewald, J.Z.; Crous, P.W. Diaporthe: A genus of endophytic, saprobic and plant pathogenic fungi. Persoonia 2013, 31, 1–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos, L.; Alves, A.; Alves, R. Evaluating multi-locus phylogenies for species boundaries determination in the genus Diaporthe. PeerJ 2017, 5, 1–26. [Google Scholar] [CrossRef] [Green Version]
- Udayanga, D.; Castlebury, L.A.; Rossman, A.Y.; Chukeatirote, E.; Hyde, K.D. The Diaporthe sojae species complex: Phylogenetic re-assessment of pathogens associated with soybean, cucurbits and other field crops. Fungal Biol. 2015, 119, 383–407. [Google Scholar] [CrossRef]
- White, T.J.; Bruns, T.; Lee, S.; Taylor, J. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In PCR Protocols: A Guide to Methods and Applications; Innis, M.A., Gelfand, D.H., Sninsky, J.J., White, T.J., Eds.; Academic Press: San Diego, CA, USA, 1990; pp. 315–322. [Google Scholar]
- Carbone, I.; Kohn, L.M. A method for desianing primer sets for speciation studies in filamentous ascomycetes. Mycologia 1999, 91, 553–556. [Google Scholar] [CrossRef]
- Glass, N.L.; Donaldson, G.C. Development of primer sets designed for use with the PCR to amplify conserved genes from filamentous ascomycetes. Appl. Environ. Microb. 1995, 61, 1323–1330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crous, P.W.; Groenewald, J.Z.; Risède, J.M.; Simoneau, P.; Hywel-Jones, N.L. Calonectria species and their Cylindrocladium anamorphs: Species with clavate vesicles. Stud. Mycol. 2004, 50, 415–430. [Google Scholar] [CrossRef] [Green Version]
- Chaisiri, C.; Liu, X.Y.; Lin, Y.; Li, J.B.; Xiong, B.; Luo, C.X. Phylogenetic analysis and development of molecular tool for detection of Diaporthe citri causing melanose disease of citrus. Plants 2020, 9, 329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, K.; Rozewicki, J.; Yamada, K.D. MAFFT online service: Multiple sequence alignment, interactive sequence choice and visualization. Brief Bioinform. 2019, 20, 1160–1166. [Google Scholar] [CrossRef] [Green Version]
- Hall, A.T. BioEdit: A user-friendly biological sequence alignment editor and analysis program for windows 95/98/nt. Nucleic Acids Res 1999, 41, 95–98. [Google Scholar]
- Glez-Peña, D.; Gómez-Blanco, D.; Reboiro-Jato, M.; Fdez-Riverola, F.; Posada, D. ALTER: Program-oriented conversion of DNA and protein alignments. Nucleic Acids Res. 2010, 38, W14–W18. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.A.; Pfeiffer, W.; Schwartz, T. The CIPRES science gateway: A community resource for phylogenetic analyses. In TeraGrid Conference: Extreme Digital Discovery; San Diego Supercomputer Center: La Jolla, San Diego, CA, USA, 2011; Volume 41, pp. 1–8. [Google Scholar]
- Swofford, D.L. PAUP* Phylogenetic Analysis Using Parsimony, (*and Other Methods); Version 4.0 b10; Sinauer Associates: Sunderland, MA, USA, 2003. [Google Scholar]
- Ronquist, F.; Huelsenbeck, J.P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 2003, 19, 1572–1574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nylander, J.A.A. MrModeltest v.2. Program Distributed by the Author; Evolitionary Biology Centre, Uppsala Univeristy: Uppsala, Sweden, 2004. [Google Scholar]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior summarization in Bayesian phylogenetic using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef] [Green Version]
- Bruen, T.C.; Philippe, H.; Bryant, D. A simple and robust statistical test for detecting the presence of recombination. Genetics 2006, 172, 2665–2681. [Google Scholar] [CrossRef] [Green Version]
- Huson, D.H. SplitsTree: Analyzing and visualizing evolutionary data. Bioinformatics 1998, 14, 68–73. [Google Scholar] [CrossRef]
- Townsend, J.P. Profiling phylogenetic informativeness. Syst. Biol. 2007, 56, 222–231. [Google Scholar] [CrossRef]
- Lopez-Giraldez, F.; Townsend, J.P. PhyDesign: An online application for profiling phylogenetic informativeness. BMC Evol. Biol. 2011, 11, 152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [Green Version]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef] [PubMed]
- Leigh, J.W.; Bryant, D. PopArt: Full-feature softwere for haplotype network construction. Methods Ecol. Evol. 2015, 6, 1110–1116. [Google Scholar] [CrossRef]
- Du, Z.; Fan, X.L.; Hyde, K.D.; Yang, Q.; Liang, Y.M.; Tian, C.M. Phylogeny and morphology reveal two new species of Diaporthe from Betula spp. in China. Phytotaxa 2016, 269, 90–102. [Google Scholar] [CrossRef] [Green Version]
- Perera, R.H.; Hyde, K.D.; Dissanayake, A.J.; Jones, E.B.G.; Liu, J.K.; Wei, D.; Liu, Z.Y. Diaporthe collariana sp. nov., with prominent collarettes associated with Magnolia champaca fruits in Thailand. Stud. Fungi 2018, 3, 141–151. [Google Scholar] [CrossRef]
- Udayanga, D.; Liu, X.Z.; Crous, P.W.; McKenzie, E.H.C.; Chukeatirote, E.; Hyde, K.D. A multi-locus phylogenetic evaluation of Diaporthe (Phomopsis). Fungal Divers. 2012, 56, 157–171. [Google Scholar] [CrossRef]
- Yang, Y.; Guo, Y.X.; Zhang, Y.K.; Wu, H.Y.; Zhang, M. Diaporthe henanensis sp. nov., an endophytic fungus in Ziziphus jujuba from China. Mycotaxon 2016, 131, 645–652. [Google Scholar] [CrossRef]
- Dissanayake, A.J.; Camporesi, E.; Hyde, K.D.; Zhang, W.; Yan, J.Y.; Li, X.H. Molecular phylogenetic analysis reveals seven new Diaporthe species from Italy. Mycosphere 2017, 8, 853–877. [Google Scholar] [CrossRef]
- Wanasinghe, D.N.; Phukhamsakda, C.; Hyde, K.D.; Jeewon, R.; Lee, H.B.; Jones, E.B.G.; Tibpromma, S.; Tennakoon, D.S.; Dissanayake, A.J.; Jayasiri, S.C.; et al. Fungal diversity notes 709–839: Taxonomic and phylogenetic contributions to fungal taxa with an emphasis on fungi on Rosaceae. Fungal Divers. 2018, 89, 1–60. [Google Scholar] [CrossRef]
- Chang, C.Q.; Xi, P.G.; Xiang, M.M.; Jiang, Z.D.; Chi, P.K. New species of Phomopsis on woody plants in Hunan province. Mycosystema 2005, 24, 145–154. [Google Scholar]
- Marin-Felix, Y.; Hernández-Restrepo, M.; Wingfield, M.J.; Akulov, A.; Carnegie, A.J.; Cheewangkoon, R.; Gramaje, D.; Groenewald, J.Z.; Guarnaccia, V.; Halleen, F.; et al. Genera of phytopathogenic fungi: GOPHY 2. Stud. Mycol. 2019, 92, 47–133. [Google Scholar] [CrossRef] [PubMed]
- Tanney, J.B.; McMullin, D.R.; Green, B.D.; Miller, J.D.; Seifert, K.A. Production of antifungal and antiinsectan metabolites by the Picea endophyte Diaporthe maritima sp. nov. Fungal Biol. 2016, 120, 1448–1457. [Google Scholar] [CrossRef]
- Crous, P.W.; Wingfield, M.J.; Schumacher, R.K.; Summerell, B.A.; Giraldo, A.; Gené, J.; Guarro, J.; Wanasinghe, D.N.; Hyde, K.D.; Camporesi, E.; et al. Fungal planet description sheets: 281–319. Persoonia 2014, 33, 212–289. [Google Scholar] [CrossRef]
- Yang, Q.; Du, Z.; Tian, C.M. Phylogeny and morphology reveal two new species of Diaporthe from traditional Chinese medicine in Northeast China. Phytotaxa 2018, 336, 159–170. [Google Scholar] [CrossRef] [Green Version]
- Yang, Q.; Fan, X.L.; Du, Z.; Tian, C.M. Diaporthe species occurring on Senna bicapsularis in Southern China, with descriptions of two new species. Phytotaxa 2017, 302, 145–155. [Google Scholar] [CrossRef]
- Machingambi, N.M.; Dreyer, L.L.; Oberlander, K.C.; Roux, J.; Roets, F. Death of endemic Virgilia oroboides trees in South Africa caused by Diaporthe virgiliae sp. nov. Plant Pathol. 2015, 64, 1149–1156. [Google Scholar] [CrossRef] [Green Version]
- Nitschke, T.R.J. Pyrenomycetes germanici. In Die Kernpilze Deutschlands Bearbeitet von Dr. Th. Nitschke; Eduard Trewendt: Breslau, Germany, 1870; Volume 2, pp. 161–320. [Google Scholar]
- Castlebury, L.A.; Farr, D.F.; Rossman, A.Y.; Jaklitsch, W. Diaporthe angelicae comb. nov., a modern description and placement of Diaporthopsis in Diaporthe. Mycoscience 2003, 44, 203–208. [Google Scholar] [CrossRef]
- Crous, P.W.; Groenewald, J.Z. Hosts, species and genotypes: Opinions versus data. Australasian Plant Pathol. 2005, 34, 463–470. [Google Scholar] [CrossRef]
- Crous, P.W.; Hawksworth, D.L.; Wingfield, M.J. Identifying and naming plant-pathogenic fungi: Past, present, and future. Annu. Rev. Phytopathol. 2015, 53, 247–267. [Google Scholar] [CrossRef]
- Hibbett, D.; Abarenkov, K.; Kõljalg, U.; Öpik, M.; Chai, B.; Cole, J.; Wang, Q.; Crous, P.W.; Robert, V.; Helgason, T.; et al. Sequence-based classification and identification of fungi. Mycologia 2016, 108, 1049–1068. [Google Scholar]
- Hyde, K.D.; Nilsson, R.H.; Alias, S.A.; Ariyawansa, H.A.; Blair, J.E.; Cai, L.; de Cock, A.W.A.M.; Dissanayake, A.J.; Glockling, S.L.; Goonasekara, I.D.; et al. One stop shop: Backbones trees for important phytopathogenic genera: I (2014). Fungal Divers. 2014, 67, 21–125. [Google Scholar] [CrossRef] [Green Version]
- van Rensburg, J.C.J.; Lamprecht, S.C.; Groenewald, J.Z.; Castlebury, L.A.; Crous, P.W. Characterisation of Phomopsis spp. associated with die-back of rooibos (Aspalathus linearis) in South Africa. Stud. Mycol. 2006, 55, 65–74. [Google Scholar] [CrossRef] [Green Version]
- Farr, D.F.; Castlebury, L.A.; Rossman, A.Y.; Putnam, M.L. A new species of Phomopsis causing twig dieback of Vaccinium vitis-idaea (lingonberry). Mycol. Res. 2002, 106, 745–752. [Google Scholar] [CrossRef]
- Santos, J.M.; Correia, V.G.; Phillips, A.J.L.; Spatafora, J.W. Primers for mating-type diagnosis in Diaporthe and Phomopsis: Their use in teleomorph induction in vitro and biological species definition. Fungal Biol. 2010, 114, 255–270. [Google Scholar] [CrossRef] [PubMed]
- Mostert, L.; Crous, P.W.; Kang, J.C.; Phillips, A.J.L. Species of Phomopsis and a Libertella sp. occurring on grapevines with specific reference to South Africa: Morphological, cultural, molecular and pathological characterization. Mycologia 2001, 93, 146–167. [Google Scholar] [CrossRef]
- Uecker, F.A. A world list of Phomopsis names with notes on nomenclature, morphology and biology. Mycol. Memoirs. 1988, 13, 1–231. [Google Scholar]
- Brayford, D. Variation in Phomopsis isolates from Ulmus species in the British Isles and Italy. Mycol Res 1990, 94, 691–697. [Google Scholar] [CrossRef]
- van Niekerk, J.M.; Groenewald, J.Z.; Farr, D.F.; Fourie, P.H.; Halleen, F.; Crous, P.W. Reassessment of Phomopsis species on grapevine. Australasian Plant Pathol. 2005, 34, 27–39. [Google Scholar] [CrossRef]
- Rossman, A.Y.; Adams, G.C.; Cannon, P.F.; Castlebury, L.A.; Crous, P.W.; Gryzenhout, M.; Jaklitsch, W.M.; Mejia, L.C.; Stoykov, D.; Udayanga, D.; et al. Recommendations of generic names in Diaporthales competing for protection or use. IMA Fungus 2015, 6, 145–154. [Google Scholar] [CrossRef] [Green Version]
- Dissanayake, A.J.; Phillips, A.J.L.; Hyde, K.D.; Yan, J.Y.; Li, X.H. The current status of species in Diaporthe. Mycosphere 2017, 8, 1106–1156. [Google Scholar] [CrossRef]
- Castlebury, L.A.; Farr, D.F.; Rossman, A.Y. Phylogenetic distinction of Phomopsis isolates from cucurbits. Inoculum 2001, 52, 25. [Google Scholar]
Figure 1.
The best parsimonious trees obtained from a heuristic search for D. eres and closely related species. The tree was rooted using D. citri (CBS 135422). (A) EF1-α locus. (B) Combined dataset of 2 loci (EF1-α+CAL). (C) Combined dataset of 4 loci (EF1-α+CAL+TUB2+HIS). (D) Combined dataset of 5 loci (EF1-α+CAL+ TUB2+HIS+ITS). Taxa numbers were generated and corresponded to samples in 5 multi-locus parsimonious trees. Holotype, ex-type, ex-epitype, and ex-neotype cultures are indicated with isolate numbers in bold.
Figure 1.
The best parsimonious trees obtained from a heuristic search for D. eres and closely related species. The tree was rooted using D. citri (CBS 135422). (A) EF1-α locus. (B) Combined dataset of 2 loci (EF1-α+CAL). (C) Combined dataset of 4 loci (EF1-α+CAL+TUB2+HIS). (D) Combined dataset of 5 loci (EF1-α+CAL+ TUB2+HIS+ITS). Taxa numbers were generated and corresponded to samples in 5 multi-locus parsimonious trees. Holotype, ex-type, ex-epitype, and ex-neotype cultures are indicated with isolate numbers in bold.

Figure 2.
Phylogenetic tree of the D. eres species complex and close species inferred from a combined alignment with a multi-locus dataset (EF1-α+CAL+TUB2+HIS). (A) The majority-rule consensus tree from Bayesian inference analysis showing the phylogenetic relationships between the D. eres species complex and close species. The tree was rooted using D. citri (CBS 135422). Bayesian posterior probabilities values (BIPP) >0.95 and maximum likelihood and maximum parsimony bootstrap values (MLBS and MPBS) >70% are given at the branch nodes (BIPP/MLBS/MPBS). Fully supported branched values, with BIPP = 1.0, MLBS and MPBS = 100, are indicated with an asterisk (*). Holotype, ex-type, ex-epitype, and ex-neotype cultures are indicated with isolate numbers in bold. (B) Unrooted tree of D. eres species based on multi-locus sequences. The splits graphs were obtained using both LogDet transformation and neighbor-joining (NJ) distance transformation. Data are from the subset of 14 representative samples that were generated and corresponded to the BI phylogenetic tree.
Figure 2.
Phylogenetic tree of the D. eres species complex and close species inferred from a combined alignment with a multi-locus dataset (EF1-α+CAL+TUB2+HIS). (A) The majority-rule consensus tree from Bayesian inference analysis showing the phylogenetic relationships between the D. eres species complex and close species. The tree was rooted using D. citri (CBS 135422). Bayesian posterior probabilities values (BIPP) >0.95 and maximum likelihood and maximum parsimony bootstrap values (MLBS and MPBS) >70% are given at the branch nodes (BIPP/MLBS/MPBS). Fully supported branched values, with BIPP = 1.0, MLBS and MPBS = 100, are indicated with an asterisk (*). Holotype, ex-type, ex-epitype, and ex-neotype cultures are indicated with isolate numbers in bold. (B) Unrooted tree of D. eres species based on multi-locus sequences. The splits graphs were obtained using both LogDet transformation and neighbor-joining (NJ) distance transformation. Data are from the subset of 14 representative samples that were generated and corresponded to the BI phylogenetic tree.

Figure 3.
Phylogenetic informativeness profile and ultrametric trees of markers used for phylogenetic studies of 23 species in the D. eres species complex and close species. (A) Combined dataset from four loci (EF1-α+CAL+TUB2+HIS). (B) Combined dataset from five loci (EF1-α+CAL+TUB2+HIS+ITS). Values on the X-axes correspond to the relative timescale (0–1). Values on the Y-axes represent Net PI (103) in arbitrary units.
Figure 3.
Phylogenetic informativeness profile and ultrametric trees of markers used for phylogenetic studies of 23 species in the D. eres species complex and close species. (A) Combined dataset from four loci (EF1-α+CAL+TUB2+HIS). (B) Combined dataset from five loci (EF1-α+CAL+TUB2+HIS+ITS). Values on the X-axes correspond to the relative timescale (0–1). Values on the Y-axes represent Net PI (103) in arbitrary units.

Figure 4.
Subdivisions within the D. eres population were estimated by using genetic distance generated from the combined dataset of four loci (EF1-α+CAL+TUB2+HIS). (A) Diversification dynamics from the phylogenetic tree with the neighbor-joining (NJ) method. (B) Median-joining (MJ) haplotype network. Each circle represents a unique haplotype, and its size reflects the number of individuals expressing that haplotype. Crosshatches indicate the number of nucleotide differences between haplotypes. Color codes denote the geographic location of populations. Geographic location abbreviation, BJ: Beijing; CQ: Chongqing; FJ: Fujian; GS: Gansu; HEB: Hebei; HN: Henan; HUB: Hubei; JS: Jiangsu; JX: Jiangxi; JL: Jilin; LN: Liaoning; NX: Ningxia; SD: Shandong; SC: Sichuan; YN: Yunnan; ZJ: Zhejiang.
Figure 4.
Subdivisions within the D. eres population were estimated by using genetic distance generated from the combined dataset of four loci (EF1-α+CAL+TUB2+HIS). (A) Diversification dynamics from the phylogenetic tree with the neighbor-joining (NJ) method. (B) Median-joining (MJ) haplotype network. Each circle represents a unique haplotype, and its size reflects the number of individuals expressing that haplotype. Crosshatches indicate the number of nucleotide differences between haplotypes. Color codes denote the geographic location of populations. Geographic location abbreviation, BJ: Beijing; CQ: Chongqing; FJ: Fujian; GS: Gansu; HEB: Hebei; HN: Henan; HUB: Hubei; JS: Jiangsu; JX: Jiangxi; JL: Jilin; LN: Liaoning; NX: Ningxia; SD: Shandong; SC: Sichuan; YN: Yunnan; ZJ: Zhejiang.

Figure 5.
The population prevalence of D. eres isolates. (A) Province distribution of D. eres in China; the size of the circle indicates the number of isolates collected from that location; green and purple colors represent Clusters I and II, respectively. (B) Overall D. eres population rate (%) of two clusters displayed within province distribution and host species, respectively. Geographic location abbreviation, BJ: Beijing; CQ: Chongqing; FJ: Fujian; GS: Gansu; HEB: Hebei; HN: Henan; HUB: Hubei; JS: Jiangsu; JX: Jiangxi; JL: Jilin; LN: Liaoning; NX: Ningxia; SD: Shandong; SC: Sichuan; YN: Yunnan; ZJ: Zhejiang.
Figure 5.
The population prevalence of D. eres isolates. (A) Province distribution of D. eres in China; the size of the circle indicates the number of isolates collected from that location; green and purple colors represent Clusters I and II, respectively. (B) Overall D. eres population rate (%) of two clusters displayed within province distribution and host species, respectively. Geographic location abbreviation, BJ: Beijing; CQ: Chongqing; FJ: Fujian; GS: Gansu; HEB: Hebei; HN: Henan; HUB: Hubei; JS: Jiangsu; JX: Jiangxi; JL: Jilin; LN: Liaoning; NX: Ningxia; SD: Shandong; SC: Sichuan; YN: Yunnan; ZJ: Zhejiang.

Table 2.
Comparison of alignment properties in phylogenetic data of each individual locus and combined loci.
Table 2.
Comparison of alignment properties in phylogenetic data of each individual locus and combined loci.
| Locus a | Individual Locus | Combined Loci | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 1 + 2 | 1 + 3 | 1 + 4 | 2 + 3 | 2 + 4 | 3 + 4 | 1 + 2 + 3 | 1 + 2 + 4 | 1 + 2 + 3 + 4 | 1 + 2 + 3 + 4 + 5 | |
| No. of taxa analyzed | 47 | 36 | 47 | 35 | 51 | 47 | 50 | 49 | 50 | 41 | 49 | 50 | 49 | 50 | 51 |
| Aligned length (with gap) | 592 | 542 | 828 | 502 | 598 | 1134 | 1420 | 1094 | 1370 | 1044 | 1330 | 1962 | 1636 | 2464 | 3062 |
| Invariable characters | 289 | 319 | 449 | 355 | 466 | 608 | 738 | 644 | 768 | 674 | 804 | 1057 | 963 | 1412 | 1878 |
| Number of parsimony-informative characters | 171 | 105 | 136 | 83 | 74 | 276 | 307 | 254 | 241 | 188 | 219 | 412 | 359 | 495 | 569 |
| Number of parsimony-uninformative characters | 132 | 118 | 243 | 64 | 58 | 250 | 375 | 196 | 361 | 674 | 307 | 493 | 314 | 557 | 615 |
| Tree length (TL) | 746 | 371 | 597 | 281 | 348 | 1150 | 1379 | 1072 | 995 | 670 | 918 | 1786 | 1474 | 2124 | 2592 |
| Consistency index (CI) | 0.635 | 0.801 | 0.782 | 0.698 | 0.511 | 0.670 | 0.682 | 0.625 | 0.768 | 0.736 | 0.722 | 0.693 | 0.656 | 0.675 | 0.622 |
| Retention index (RI) | 0.698 | 0.793 | 0.712 | 0.767 | 0.739 | 0.699 | 0.676 | 0.681 | 0.714 | 0.755 | 0.688 | 0.680 | 0.688 | 0.667 | 0.640 |
| Rescaled consistency index (RC) | 0.444 | 0.635 | 0.557 | 0.535 | 0.378 | 0.468 | 0.461 | 0.426 | 0.549 | 0.555 | 0.497 | 0.471 | 0.451 | 0.451 | 0.398 |
| Homoplasy index (HI) | 0.365 | 0.199 | 0.218 | 0.302 | 0.489 | 0.330 | 0.318 | 0.375 | 0.232 | 0.264 | 0.278 | 0.307 | 0.344 | 0.325 | 0.378 |
a 1: translation elongation factor 1-α gene (EF1-α); 2: calmodulin gene (CAL); 3: beta-tubulin 2 gene (TUB2); 4: histone-3 gene (HIS); 5: ribosomal internal transcribed spacer (ITS) region of ribosomal DNA (ITS1-5.8S-ITS2).
Table 3.
Sequence variation, indices of sequence variation, and neutrality within five loci in D. eres.
Table 3.
Sequence variation, indices of sequence variation, and neutrality within five loci in D. eres.
| Gene/Locus a,b | Individual Locus | Combined Loci | ||||
|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 1 + 2 + 3 + 4 | |
| Aligned length (with gap) | 278 | 492 | 355 | 421 | 450 | 1429 |
| No. of taxa analyzed | 132 | 118 | 137 | 70 | 137 | 61 |
| No. of sites | 382 | 323 | 794 | 479 | 596 | 1464 |
| %GC | 0.548 | 0.56 | 0.567 | 0.62 | 0.53 | 0.579 |
| No. of polymorphic (segregating) sites (S) | 130 | 27 | 58 | 34 | 83 | 115 |
| Nei’s nucleotide diversity (π) | 0.026051 | 0.00558 | 0.02233 | 0.00593 | 0.33040 | 0.01056 |
| Haplotype numeric (Hap) | 43 | 24 | 36 | 19 | 59 | 54 |
| Haplotype diversity (Hd) | 0.91579 | 0.79500 | 0.92486 | 0.83520 | 0.97639 | 0.99562 |
| Nucleotide diversity from S (θw) | 0.09955 | 0.01603 | 0.03179 | 0.01676 | 0.04126 | 0.01794 |
| Tajima’s D | −2.39947 * | −1.87578 *** | −0.92870 **** | −2.07658 *** | −0.63716 **** | −1.43591 **** |
| Fu and Li’s D | −1.53703 **** | −2.58454 *** | −0.33757 **** | −3.48112 ** | −3.88613 ** | −1.67899 **** |
| Fu’s Fs | −14.6930 | −16.3734 | −6.25117 | −8.43211 | −14.1927 | −36.71492 |
a 1: translation elongation factor 1-α gene (EF1-α); 2: calmodulin gene (CAL); 3: beta-tubulin 2 gene (TUB2); 4: histone-3 gene (HIS); 5: ribosomal internal transcribed spacer (ITS) region of ribosomal DNA (ITS1-5.8S-ITS2). b * p < 0.01; ** p < 0.02; *** p < 0.05; **** p > 0.10.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Chaisiri, C.; Liu, X.; Lin, Y.; Fu, Y.; Zhu, F.; Luo, C. Phylogenetic and Haplotype Network Analyses of Diaporthe eres Species in China Based on Sequences of Multiple Loci. Biology 2021, 10, 179. https://doi.org/10.3390/biology10030179
AMA Style
Chaisiri C, Liu X, Lin Y, Fu Y, Zhu F, Luo C. Phylogenetic and Haplotype Network Analyses of Diaporthe eres Species in China Based on Sequences of Multiple Loci. Biology. 2021; 10(3):179. https://doi.org/10.3390/biology10030179
Chicago/Turabian StyleChaisiri, Chingchai, Xiangyu Liu, Yang Lin, Yanping Fu, Fuxing Zhu, and Chaoxi Luo. 2021. "Phylogenetic and Haplotype Network Analyses of Diaporthe eres Species in China Based on Sequences of Multiple Loci" Biology 10, no. 3: 179. https://doi.org/10.3390/biology10030179
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.