
Computational Chemistry to Repurposing Drugs for the Control of COVID-19
 ,
, 
Abstract
:1. Introduction
2. Methodology
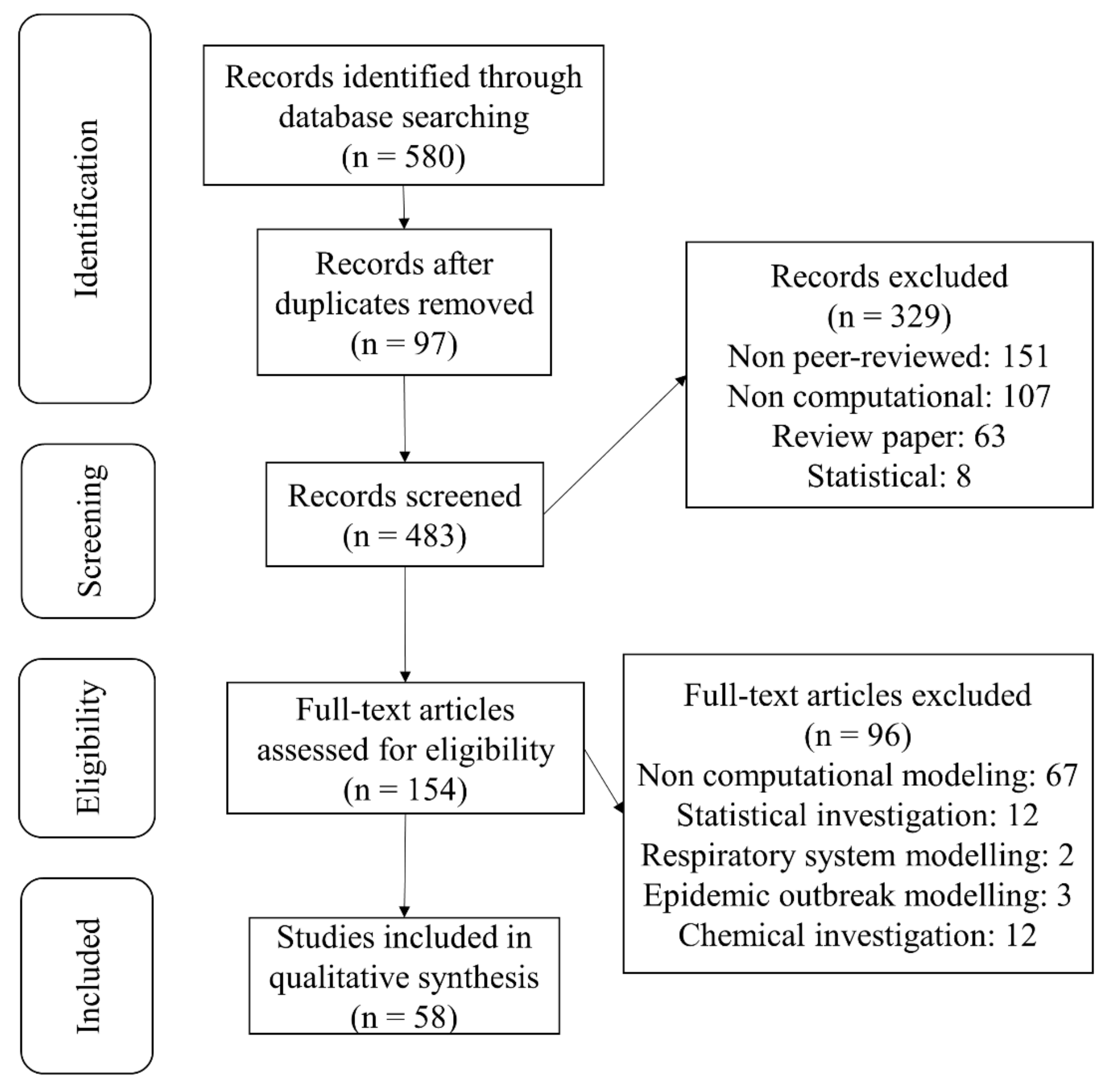
2.1. Study Selection
2.2. Data Extraction
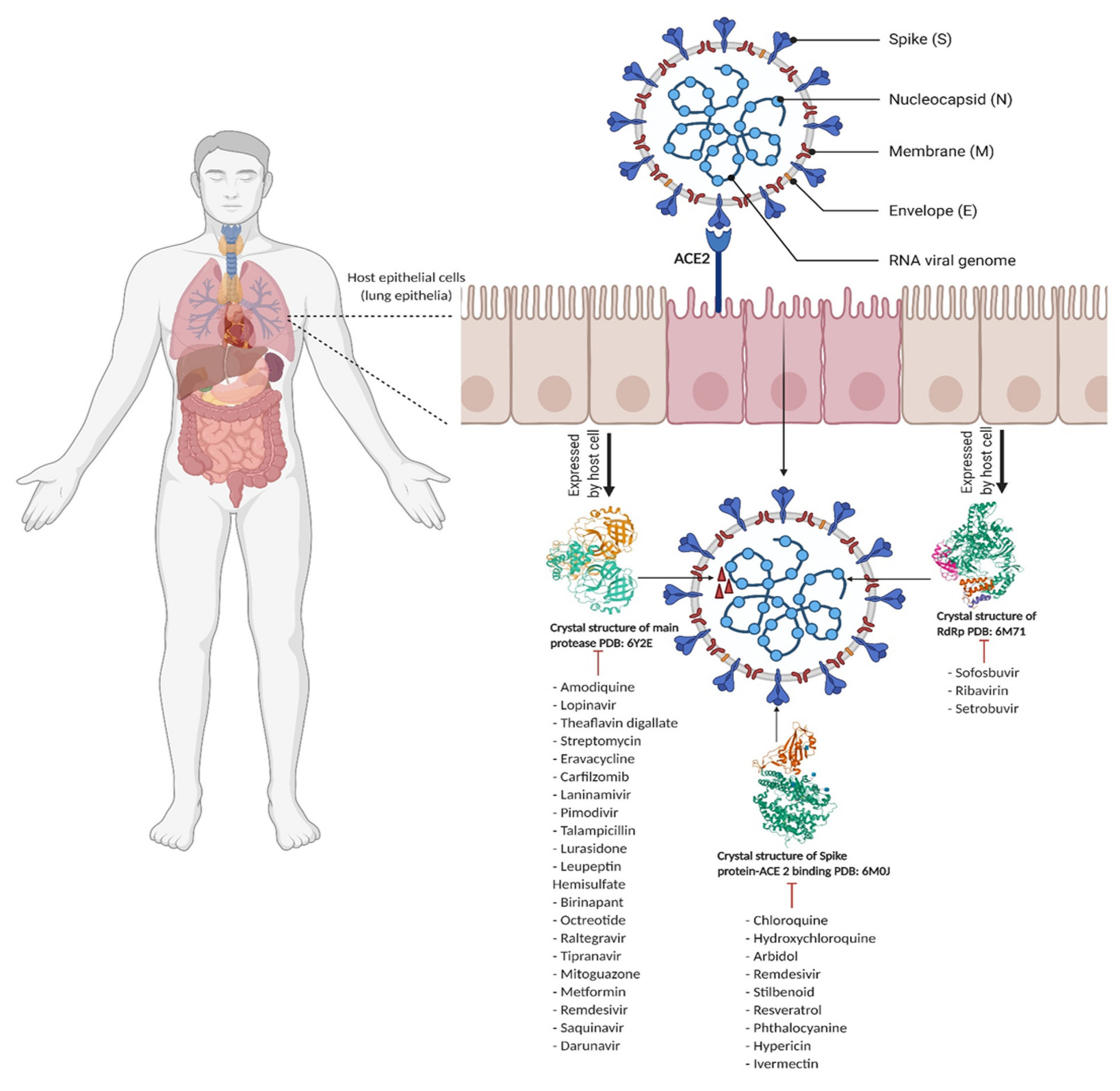
3. Computational Studies of Key SARS-CoV-2 Viral Proteins
3.1. Viral Proteases
3.2. Spike Protein-ACE2 Enzyme Target
3.3. RNA-Dependent RNA Polymerase Enzyme Target
3.4. Other Proteins and Nonstructural Proteins of SARS-CoV-2
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A novel coronavirus from patients with pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef]
- Atal, S.; Fatima, Z.; Balakrishnan, S. Approval of Itolizumab for COVID-19: A Premature Decision or Need of The Hour? BioDrugs 2020, 34, 705–711. [Google Scholar] [CrossRef] [PubMed]
- Crisafulli, S.; Isgrò, V.; La Corte, L.; Atzeni, F.; Trifirò, G. Potential role of anti-interleukin (IL)-6 drugs in the treatment of COVID-19: Rationale, clinical evidence and risks. BioDrugs 2020, 34, 415–422. [Google Scholar] [CrossRef] [PubMed]
- Ayres, J.S. A metabolic handbook for the COVID-19 pandemic. Nat. Metab. 2020, 2, 572–585. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [Green Version]
- COVID-19 CORONAVIRUS PANDEMIC. Available online: https://www.worldometers.info/coronavirus/ (accessed on 24 September 2020).
- Kissler, S.M.; Tedijanto, C.; Goldstein, E.; Grad, Y.H.; Lipsitch, M. Projecting the transmission dynamics of SARS-CoV-2 through the postpandemic period. Science 2020, 368, 860–868. [Google Scholar] [CrossRef] [PubMed]
- Su, S.; Wong, G.; Shi, W.; Liu, J.; Lai, A.C.; Zhou, J.; Liu, W.; Bi, Y.; Gao, G.F. Epidemiology, genetic recombination, and pathogenesis of coronaviruses. Trends Microbiol. 2016, 24, 490–502. [Google Scholar] [CrossRef] [Green Version]
- Perlman, S.; Netland, J. Coronaviruses post-SARS: Update on replication and pathogenesis. Nat. Rev. Microbiol. 2009, 7, 439–450. [Google Scholar] [CrossRef] [Green Version]
- Lu, R.; Zhao, X.; Li, J.; Niu, P.; Yang, B.; Wu, H.; Wang, W.; Song, H.; Huang, B.; Zhu, N.; et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: Implications for virus origins and receptor binding. Lancet 2020, 395, 565–574. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Geng, M.; Peng, Y.; Meng, L.; Lu, S. Molecular immune pathogenesis and diagnosis of COVID-19. J. Pharm. Anal. 2020, 10, 102–108. [Google Scholar] [CrossRef]
- Wang, N.; Shang, J.; Jiang, S.; Du, L. Subunit vaccines against emerging pathogenic human coronaviruses. Front. Microbiol. 2020, 11, 298. [Google Scholar] [CrossRef]
- Ge, X.-Y.; Li, J.-L.; Yang, X.-L.; Chmura, A.A.; Zhu, G.; Epstein, J.H.; Mazet, J.K.; Hu, B.; Zhang, W.; Peng, C.; et al. Isolation and characterization of a bat SARS-like coronavirus that uses the ACE2 receptor. Nature 2013, 503, 535–538. [Google Scholar] [CrossRef]
- Wrobel, A.G.; Benton, D.J.; Xu, P.; Roustan, C.; Martin, S.R.; Rosenthal, P.B.; Skehel, J.J.; Gamblin, S.J. SARS-CoV-2 and bat RaTG13 spike glycoprotein structures inform on virus evolution and furin-cleavage effects. Nat. Struct. Mol. Biol. 2020, 27, 763–767. [Google Scholar] [CrossRef]
- Li, W.; Moore, M.J.; Vasilieva, N.; Sui, J.; Wong, S.K.; Berne, M.A.; Somasundaran, M.; Sullivan, J.L.; Luzuriaga, K.; Greenough, T.C.; et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 2003, 426, 450–454. [Google Scholar] [CrossRef] [Green Version]
- Smith, T.R.; Patel, A.; Ramos, S.; Elwood, D.; Zhu, X.; Yan, J.; Gary, E.N.; Walker, S.N.; Schultheis, K.; Purwar, M.; et al. Immunogenicity of a DNA vaccine candidate for COVID-19. Nat. Commun. 2020, 11, 1–13. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.-H.; Nitsche, A. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 2020, 181, 271–280. [Google Scholar] [CrossRef]
- Millet, J.K.; Whittaker, G.R. Host cell proteases: Critical determinants of coronavirus tropism and pathogenesis. Virus Res. 2015, 202, 120–134. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Li, C.; Huang, A.; Xia, S.; Lu, S.; Shi, Z.; Lu, L.; Jiang, S.; Yang, Z.; Wu, Y. Potent binding of 2019 novel coronavirus spike protein by a SARS coronavirus-specific human monoclonal antibody. Emerg. Microbes Infect. 2020, 9, 382–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, L.; Yang, Y.; Zhou, Y.; Lu, L.; Li, F.; Jiang, S. MERS-CoV spike protein: A key target for antivirals. Expert Opin. Ther. Targets 2017, 21, 131–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Wong, G.; Lu, G.; Yan, J.; Gao, G.F. MERS-CoV spike protein: Targets for vaccines and therapeutics. Antivir. Res. 2016, 133, 165–177. [Google Scholar] [CrossRef]
- Kuo, L.; Hurst, K.R.; Masters, P.S. Exceptional flexibility in the sequence requirements for coronavirus small envelope protein function. J. Virol. 2007, 81, 2249–2262. [Google Scholar] [CrossRef] [Green Version]
- Schoeman, D.; Fielding, B.C. Coronavirus envelope protein: Current knowledge. Virol. J. 2019, 16, 1–22. [Google Scholar] [CrossRef] [Green Version]
- Gupta, M.K.; Vemula, S.; Donde, R.; Gouda, G.; Behera, L.; Vadde, R. In-silico approaches to detect inhibitors of the human severe acute respiratory syndrome coronavirus envelope protein ion channel. J. Biomol. Struct. Dyn. 2020, 39, 2617–2627. [Google Scholar] [CrossRef] [Green Version]
- Lin, S.-M.; Lin, S.-C.; Hsu, J.-N.; Chang, C.-K.; Chien, C.-M.; Wang, Y.-S.; Wu, H.-Y.; Jeng, U.-S.; Kehn-Hall, K.; Hou, M.-H. Structure-based stabilization of non-native protein–protein interactions of coronavirus nucleocapsid proteins in antiviral drug design. J. Med. Chem. 2020, 63, 3131–3141. [Google Scholar] [CrossRef] [PubMed]
- Zeng, W.; Liu, G.; Ma, H.; Zhao, D.; Yang, Y.; Liu, M.; Mohammed, A.; Zhao, C.; Yang, Y.; Xie, J. Biochemical characterization of SARS-CoV-2 nucleocapsid protein. Biochem. Biophys. Res. Commun. 2020, 527, 618–623. [Google Scholar] [CrossRef]
- Ahmed, S.F.; Quadeer, A.A.; McKay, M.R. Preliminary identification of potential vaccine targets for the COVID-19 coronavirus (SARS-CoV-2) based on SARS-CoV immunological studies. Viruses 2020, 12, 254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ziebuhr, J.; Snijder, E.J.; Gorbalenya, A.E. Virus-encoded proteinases and proteolytic processing in the Nidovirales. J. Gen. Virol. 2000, 81, 853–879. [Google Scholar] [CrossRef] [PubMed]
- Dong, S.; Sun, J.; Mao, Z.; Wang, L.; Lu, Y.L.; Li, J. A guideline for homology modeling of the proteins from newly discovered betacoronavirus, 2019 novel coronavirus (2019-nCoV). J. Med. Virol. 2020, 92, 1542–1548. [Google Scholar] [CrossRef]
- Rappuoli, R.; Black, S.; Bloom, D.E. Vaccines and global health: In search of a sustainable model for vaccine development and delivery. Sci. Transl. Med. 2019, 11, eaaw2888. [Google Scholar] [CrossRef]
- Ciliberto, G.; Cardone, L. Boosting the arsenal against COVID-19 through computational drug repurposing. Drug Discov. Today 2020, 25, 946–948. [Google Scholar] [CrossRef]
- Rosenberg, E.S.; Dufort, E.M.; Udo, T.; Wilberschied, L.A.; Kumar, J.; Tesoriero, J.; Weinberg, P.; Kirkwood, J.; Muse, A.; DeHovitz, J.; et al. Association of treatment with hydroxychloroquine or azithromycin with in-hospital mortality in patients with COVID-19 in New York state. JAMA 2020, 323, 2493–2502. [Google Scholar] [CrossRef]
- Molina, J.M.; Delaugerre, C.; Le Goff, J.; Mela-Lima, B.; Ponscarme, D.; Goldwirt, L.; de Castro, N. No evidence of rapid antiviral clearance or clinical benefit with the combination of hydroxychloroquine and azithromycin in patients with severe COVID-19 infection. Med. Mal. Infect. 2020, 50, 30085–30088. [Google Scholar] [CrossRef]
- Cao, B.; Wang, Y.; Wen, D.; Liu, W.; Wang, J.; Fan, G.; Ruan, L.; Song, B.; Cai, Y.; Wei, M. A trial of lopinavir–ritonavir in adults hospitalized with severe Covid-19. N. Engl. J. Med. 2020, 382, 1787–1799. [Google Scholar] [CrossRef]
- Bellera, C.L.; Llanos, M.; Gantner, M.E.; Rodriguez, S.; Gavernet, L.; Comini, M.; Talevi, A. Can drug repurposing strategies be the solution to the COVID-19 crisis? Expert Opin. Drug Discov. 2021, 16, 605–612. [Google Scholar] [CrossRef]
- Tabari, M.A.K.; Khoshhal, H.; Tafazoli, A.; Khandan, M.; Bagheri, A. Applying Computer Simulations in Battling with COVID-19, using pre-analyzed molecular and chemical data to face the pandemic. Inform. Med. Unlocked 2020, 21, 100458. [Google Scholar] [CrossRef] [PubMed]
- Hiranuma, N.; Park, H.; Baek, M.; Anishchenko, I.; Dauparas, J.; Baker, D. Improved protein structure refinement guided by deep learning based accuracy estimation. Nat. Commun. 2021, 12, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Venkatesan, A.; Gopal, J.; Candavelou, M.; Gollapalli, S.; Karthikeyan, K. Computational approach for protein structure prediction. Healthc. Inform. Res. 2013, 19, 137. [Google Scholar] [CrossRef] [PubMed]
- Ou-Yang, S.-S.; Lu, J.-Y.; Kong, X.-Q.; Liang, Z.-J.; Luo, C.; Jiang, H. Computational drug discovery. Acta Pharmacol. Sin. 2012, 33, 1131–1140. [Google Scholar] [CrossRef] [Green Version]
- Moitessier, N.; Englebienne, P.; Lee, D.; Lawandi, J.; Corbeil, C.R. Towards the development of universal, fast and highly accurate docking/scoring methods: A long way to go. Br. J. Pharmacol. 2008, 153, S7–S26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, X.-Y.; Zhang, H.-X.; Mezei, M.; Cui, M. Molecular docking: A powerful approach for structure-based drug discovery. Curr. Comput. Aided Drug Des. 2011, 7, 146–157. [Google Scholar] [CrossRef] [PubMed]
- Cavasotto, C.N.; Phatak, S.S. Homology modeling in drug discovery: Current trends and applications. Drug Discov. Today 2009, 14, 676–683. [Google Scholar] [CrossRef] [PubMed]
- Pagadala, N.S.; Syed, K.; Tuszynski, J. Software for molecular docking: A review. Biophys. Rev. 2017, 9, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Salmaso, V.; Moro, S. Bridging molecular docking to molecular dynamics in exploring ligand-protein recognition process: An overview. Front. Pharmacol. 2018, 9, 923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Lin, D.; Sun, X.; Curth, U.; Drosten, C.; Sauerhering, L.; Becker, S.; Rox, K.; Hilgenfeld, R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science 2020, 368, 409–412. [Google Scholar] [CrossRef] [Green Version]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Y.; Yan, L.; Huang, Y.; Liu, F.; Zhao, Y.; Cao, L.; Wang, T.; Sun, Q.; Ming, Z.; Zhang, L.; et al. Structure of the RNA-dependent RNA polymerase from COVID-19 virus. Science 2020, 368, 779–782. [Google Scholar] [CrossRef] [Green Version]
- Fischer, D.; Norel, R.; Wolfson, H.; Nussinov, R. Surface motifs by a computer vision technique: Searches, detection, and implications for protein–ligand recognition. Proteins Struct. Funct. Bioinform. 1993, 16, 278–292. [Google Scholar] [CrossRef]
- Rarey, M.; Kramer, B.; Lengauer, T.; Klebe, G. A fast flexible docking method using an incremental construction algorithm. J. Mol. Biol. 1996, 261, 470–489. [Google Scholar] [CrossRef] [Green Version]
- Miranker, A.; Karplus, M. Functionality maps of binding sites: A multiple copy simultaneous search method. Proteins Struct. Funct. Bioinform. 1991, 11, 29–34. [Google Scholar] [CrossRef]
- Hart, T.N.; Read, R.J. A multiple-start Monte Carlo docking method. Proteins Struct. Funct. Bioinform. 1992, 13, 206–222. [Google Scholar] [CrossRef]
- Oshiro, C.M.; Kuntz, I.D.; Dixon, J.S. Flexible ligand docking using a genetic algorithm. J. Comput. Aided Mol. Des. 1995, 9, 113–130. [Google Scholar] [CrossRef]
- Naqvi, A.A.; Mohammad, T.; Hasan, G.M.; Hassan, M. Advancements in docking and molecular dynamics simulations towards ligand-receptor interactions and structure-function relationships. Curr. Top. Med. Chem. 2018, 18, 1755–1768. [Google Scholar] [CrossRef]
- Tanchuk, V.Y.; Tanin, V.O.; Vovk, A.I.; Poda, G. A new, improved hybrid scoring function for molecular docking and scoring based on AutoDock and AutoDock Vina. Chem. Biol. Drug Des. 2016, 87, 618–625. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Fu, A.; Zhang, L. Progress in molecular docking. Quant. Biol. 2019, 7, 83–89. [Google Scholar] [CrossRef] [Green Version]
- Hodos, R.A.; Kidd, B.A.; Shameer, K.; Readhead, B.P.; Dudley, J.T. In silico methods for drug repurposing and pharmacology. Wiley Interdiscip. Rev. Syst. Biol. Med. 2016, 8, 186–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohamed, K.; Yazdanpanah, N.; Saghazadeh, A.; Rezaei, N. Computational drug discovery and repurposing for the treatment of Covid-19: A systematic review. Bioorganic Chem. 2021, 106, 104490. [Google Scholar] [CrossRef] [PubMed]
- Stewart, L.A.; Clarke, M.; Rovers, M.; Riley, R.D.; Simmonds, M.; Stewart, G.; Tierney, J.F. Preferred reporting items for a systematic review and meta-analysis of individual participant data: The PRISMA-IPD statement. JAMA 2015, 313, 1657–1665. [Google Scholar] [CrossRef]
- Schoggins, J.W. Interferon-stimulated genes: Roles in viral pathogenesis. Curr. Opin. Virol. 2014, 6, 40–46. [Google Scholar] [CrossRef]
- Mengist, H.M.; Fan, X.; Jin, T. Designing of improved drugs for COVID-19: Crystal structure of SARS-CoV-2 main protease M pro. Signal Transduct. Target. Ther. 2020, 5, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Lin, D.; Kusov, Y.; Nian, Y.; Ma, Q.; Wang, J.; Von Brunn, A.; Leyssen, P.; Lanko, K.; Neyts, J.; et al. α-Ketoamides as broad-spectrum inhibitors of coronavirus and enterovirus replication: Structure-based design, synthesis, and activity assessment. J. Med. Chem. 2020, 63, 4562–4578. [Google Scholar] [CrossRef]
- Hatada, R.; Okuwaki, K.; Mochizuki, Y.; Handa, Y.; Fukuzawa, K.; Komeiji, Y.; Okiyama, Y.; Tanaka, S. Fragment molecular orbital based interaction analyses on COVID-19 main protease-inhibitor N3 complex (PDB ID: 6LU7). J. Chem. Inf. Model. 2020, 60, 3593–3602. [Google Scholar] [CrossRef]
- Deng, X.; Hackbart, M.; Mettelman, R.C.; O’Brien, A.; Mielech, A.M.; Yi, G.; Kao, C.C.; Baker, S.C. Coronavirus nonstructural protein 15 mediates evasion of dsRNA sensors and limits apoptosis in macrophages. Proc. Natl. Acad. Sci. USA 2017, 114, E4251–E4260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gosert, R.; Kanjanahaluethai, A.; Egger, D.; Bienz, K.; Baker, S.C. RNA replication of mouse hepatitis virus takes place at double-membrane vesicles. J. Virol. 2002, 76, 3697–3708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ulferts, R.; Ziebuhr, J. Nidovirus ribonucleases: Structures and functions in viral replication. RNA Biol. 2011, 8, 295–304. [Google Scholar] [CrossRef] [Green Version]
- Ciccozzi, M.; Benvenuto, D.; Giovanetti, M.; Bianchi, M.; Pascarella, S.; Angeletti, S. Response to Ribeiro da Silva et al, “Role of Nonstructural Proteins in the Pathogenesis of SARS-CoV-2”. J. Med. Virol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, A.; Wu, X.; Yang, Z.R. Mining viral protease data to extract cleavage knowledge. Bioinformatics 2002, 18, S5–S13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hosseini, F.S.; Amanlou, M. Anti-HCV and anti-malaria agent, potential candidates to repurpose for coronavirus infection: Virtual screening, molecular docking, and molecular dynamics simulation study. Life Sci. 2020, 258, 118205. [Google Scholar] [CrossRef]
- Kumar, Y.; Singh, H.; Patel, C.N. In silico prediction of potential inhibitors for the Main protease of SARS-CoV-2 using molecular docking and dynamics simulation based drug-repurposing. J. Infect. Public Health 2020, 13, 1210–1223. [Google Scholar] [CrossRef]
- Peele, K.A.; Chandrasai, P.; Srihansa, T.; Krupanidhi, S.; Sai, A.V.; Babu, D.J.; Indira, M.; Reddy, A.R.; Venkateswarulu, T. Molecular docking and dynamic simulations for antiviral compounds against SARS-CoV-2: A computational study. Inform. Med. Unlocked 2020, 19, 100345. [Google Scholar] [CrossRef]
- Wang, J. Fast identification of possible drug treatment of coronavirus disease-19 (COVID-19) through computational drug repurposing study. J. Chem. Inf. Model. 2020, 60, 3277–3286. [Google Scholar] [CrossRef]
- Havranek, B.; Islam, S.M. An in silico approach for identification of novel inhibitors as potential therapeutics targeting COVID-19 main protease. J. Biomol. Struct. Dyn. 2020, 1–12. [Google Scholar] [CrossRef]
- Kumar, A.; Choudhir, G.; Shukla, S.K.; Sharma, M.; Tyagi, P.; Bhushan, A.; Rathore, M. Identification of phytochemical inhibitors against main protease of COVID-19 using molecular modeling approaches. J. Biomol. Struct. Dyn. 2020, 39, 3760–3770. [Google Scholar] [CrossRef] [PubMed]
- Mittal, L.; Kumari, A.; Srivastava, M.; Singh, M.; Asthana, S. Identification of potential molecules against COVID-19 main protease through structure-guided virtual screening approach. J. Biomol. Struct. Dyn. 2020, 39, 3662–3680. [Google Scholar] [CrossRef] [PubMed]
- Joshi, T.; Sharma, P.; Joshi, T.; Pundir, H.; Mathpal, S.; Chandra, S. Structure-based screening of novel lichen compounds against SARS Coronavirus main protease (Mpro) as potentials inhibitors of COVID-19. Mol. Divers. 2020, 1–13. [Google Scholar] [CrossRef]
- Ul Qamar, M.T.; Alqahtani, S.M.; Alamri, M.A.; Chen, L.-L. Structural basis of SARS-CoV-2 3CLpro and anti-COVID-19 drug discovery from medicinal plants. J. Pharm. Anal. 2020, 10, 313–319. [Google Scholar] [CrossRef]
- Naik, V.R.; Munikumar, M.; Ramakrishna, U.; Srujana, M.; Goudar, G.; Naresh, P.; Kumar, B.N.; Hemalatha, R. Remdesivir (GS-5734) as a therapeutic option of 2019-nCOV main protease–in silico approach. J. Biomol. Struct. Dyn. 2020, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.A.; Zia, K.; Ashraf, S.; Uddin, R.; Ul-Haq, Z. Identification of chymotrypsin-like protease inhibitors of SARS-CoV-2 via integrated computational approach. J. Biomol. Struct. Dyn. 2020, 39, 2607–2616. [Google Scholar] [CrossRef] [Green Version]
- Gupta, S.; Singh, A.K.; Kushwaha, P.P.; Prajapati, K.S.; Shuaib, M.; Senapati, S.; Kumar, S. Identification of potential natural inhibitors of SARS-CoV2 main protease by molecular docking and simulation studies. J. Biomol. Struct. Dyn. 2020, 1–19. [Google Scholar] [CrossRef]
- Khan, A.; Ali, S.S.; Khan, M.T.; Saleem, S.; Ali, A.; Suleman, M.; Babar, Z.; Shafiq, A.; Khan, M.; Wei, D.-Q. Combined drug repurposing and virtual screening strategies with molecular dynamics simulation identified potent inhibitors for SARS-CoV-2 main protease (3CLpro). J. Biomol. Struct. Dyn. 2020, 1–12. [Google Scholar] [CrossRef]
- Ghosh, R.; Chakraborty, A.; Biswas, A.; Chowdhuri, S. Evaluation of green tea polyphenols as novel corona virus (SARS CoV-2) main protease (Mpro) inhibitors–an in silico docking and molecular dynamics simulation study. J. Biomol. Struct. Dyn. 2020, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Alamri, M.A.; ul Qamar, M.T.; Mirza, M.U.; Bhadane, R.; Alqahtani, S.M.; Muneer, I.; Froeyen, M.; Salo-Ahen, O.M. Pharmacoinformatics and molecular dynamics simulation studies reveal potential covalent and FDA-approved inhibitors of SARS-CoV-2 main protease 3CLpro. J. Biomol. Struct. Dyn. 2020, 1–13. [Google Scholar] [CrossRef]
- Odhar, H.A.; Ahjel, S.W.; Albeer, A.A.M.A.; Hashim, A.F.; Rayshan, A.M.; Humadi, S.S. Molecular docking and dynamics simulation of FDA approved drugs with the main protease from 2019 novel coronavirus. Bioinformation 2020, 16, 236. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Sharma, P.P.; Shankar, U.; Kumar, D.; Joshi, S.K.; Pena, L.; Durvasula, R.; Kumar, A.; Kempaiah, P.; Poonam; et al. Discovery of New Hydroxyethylamine Analogs Against 3CLpro Protein Target of SARS-CoV-2: Molecular Docking, Molecular Dynamics Simulation and Structure-Activity Relationship Studies. J. Chem. Inf. Model. 2020, 60, 5754–5770. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Pitsillou, E.; Karagiannis, C.; Darmawan, K.K.; Ng, K.; Hung, A.; Karagiannis, T.C. Interaction of the prototypical α-ketoamide inhibitor with the SARS-CoV-2 main protease active site in silico: Molecular dynamic simulations highlight the stability of the ligand-protein complex. Comput. Biol. Chem. 2020, 87, 107292. [Google Scholar] [CrossRef] [PubMed]
- Al-Khafaji, K.; Al-Duhaidahawi, D.; Tok, T.T. Using integrated computational approaches to identify safe and rapid treatment for SARS-CoV-2. J. Biomol. Struct. Dyn. 2020, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Pundir, H.; Joshi, T.; Joshi, T.; Sharma, P.; Mathpal, S.; Chandra, S.; Tamta, S. Using Chou’s 5-steps rule to study pharmacophore-based virtual screening of SARS-CoV-2 Mpro inhibitors. Mol. Divers. 2020. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, M.A.; Abdelrahman, A.H.; Hussien, T.A.; Badr, E.A.; Mohamed, T.A.; El, H.R.; Pare, P.W.; Efferth, T.; Hegazy, M.E.F. In silico Drug Discovery of Major Metabolites from Spices as SARS-CoV-2 Main Protease Inhibitors. Comput. Biol. Med. 2020, 126, 104046. [Google Scholar] [CrossRef] [PubMed]
- Baildya, N.; Khan, A.A.; Ghosh, N.N.; Dutta, T.; Chattopadhyay, A.P. Screening of potential drug from Azadirachta Indica (Neem) extracts for SARS-CoV-2: An insight from molecular docking and MD-simulation studies. J. Mol. Struct. 2021, 1227, 129390. [Google Scholar] [CrossRef]
- Kavitha, K.; Sivakumar, S.; Ramesh, B. 1, 2, 4 triazolo [1, 5-a] pyrimidin-7-ones as novel SARS-CoV-2 Main protease inhibitors: In silico screening and molecular dynamics simulation of potential COVID-19 drug candidates. Biophys. Chem. 2020, 267, 106478. [Google Scholar] [CrossRef]
- Tachoua, W.; Kabrine, M.; Mushtaq, M.; Ul-Haq, Z. An in-silico evaluation of COVID-19 main protease with clinically approved drugs. J. Mol. Graph. Model. 2020, 101, 107758. [Google Scholar] [CrossRef]
- Yepes-Pérez, A.F.; Herrera-Calderon, O.; Sánchez-Aparicio, J.-E.; Tiessler-Sala, L.; Maréchal, J.-D.; Cardona, G.-W. Investigating Potential Inhibitory Effect of Uncaria tomentosa (Cat’s Claw) against the Main Protease 3CLpro of SARS-CoV-2 by Molecular Modeling. Evid. Based Complementary Altern. Med. 2020, 2020, 4932572. [Google Scholar] [CrossRef]
- Hejazi, I.I.; Beg, M.A.; Imam, M.A.; Athar, F.; Islam, A. Glossary of phytoconstituents: Can these be repurposed against SARS CoV-2? A quick in silico screening of various phytoconstituents from plant Glycyrrhiza glabra with SARS CoV-2 main protease. Food Chem. Toxicol. 2021, 150, 112057. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, K.H.; Chowdhury, M.; Mahmud, S.; Tareq, A.M.; Hanif, N.B.; Banu, N.; Reza, A.; Emran, T.B.; Simal-Gandara, J. Drug Repurposing Approach against Novel Coronavirus Disease (COVID-19) through Virtual Screening Targeting SARS-CoV-2 Main Protease. Biology 2021, 10, 2. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, W.B.; Mendanha, S.A. Molecular dynamics simulation of docking structures of SARS-CoV-2 main protease and HIV protease inhibitors. J. Mol. Struct. 2021, 1225, 129143. [Google Scholar] [CrossRef]
- Ibrahim, M.A.; Abdelrahman, A.H.; Allemailem, K.S.; Almatroudi, A.; Moustafa, M.F.; Hegazy, M.-E.F. In silico evaluation of prospective anti-COVID-19 drug candidates as potential SARS-CoV-2 main protease inhibitors. Protein J. 2021, 40, 296–309. [Google Scholar] [CrossRef]
- Fakhar, Z.; Khan, S.; AlOmar, S.Y.; Alkhuriji, A.; Ahmad, A. ABBV-744 as a potential inhibitor of SARS-CoV-2 main protease enzyme against COVID-19. Sci. Rep. 2021, 11, 1–15. [Google Scholar] [CrossRef]
- Pushkaran, A.C.; Melge, A.R.; Puthiyedath, R.; Mohan, C.G. A phytochemical-based medication search for the SARS-CoV-2 infection by molecular docking models towards spike glycoproteins and main proteases. RSC Adv. 2021, 11, 12003–12014. [Google Scholar] [CrossRef]
- Hall, D.C., Jr.; Ji, H.-F. A search for medications to treat COVID-19 via in silico molecular docking models of the SARS-CoV-2 spike glycoprotein and 3CL protease. Travel Med. Infect. Dis. 2020, 35, 101646. [Google Scholar] [CrossRef]
- Qiao, Z.; Zhang, H.; Ji, H.-F.; Chen, Q. Computational view toward the inhibition of SARS-CoV-2 spike glycoprotein and the 3CL protease. Computation 2020, 8, 53. [Google Scholar] [CrossRef]
- Ferrario, C.M.; Trask, A.J.; Jessup, J.A. Advances in biochemical and functional roles of angiotensin-converting enzyme 2 and angiotensin-(1–7) in regulation of cardiovascular function. Am. J. Physiol. Heart Circ. Physiol. 2005, 289, H2281–H2290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donoghue, M.; Hsieh, F.; Baronas, E.; Godbout, K.; Gosselin, M.; Stagliano, N.; Donovan, M.; Woolf, B.; Robison, K.; Jeyaseelan, R.; et al. A novel angiotensin-converting enzyme–related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1–9. Circ. Res. 2000, 87, e1–e9. [Google Scholar] [CrossRef] [PubMed]
- Kuba, K.; Imai, Y.; Rao, S.; Gao, H.; Guo, F.; Guan, B.; Huan, Y.; Yang, P.; Zhang, Y.; Deng, W.; et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus–induced lung injury. Nat. Med. 2005, 11, 875–879. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, T.; Perlot, T.; Rehman, A.; Trichereau, J.; Ishiguro, H.; Paolino, M.; Sigl, V.; Hanada, T.; Hanada, R.; Lipinski, S.; et al. ACE2 links amino acid malnutrition to microbial ecology and intestinal inflammation. Nature 2012, 487, 477–481. [Google Scholar] [CrossRef] [PubMed]
- Wu, A.; Peng, Y.; Huang, B.; Ding, X.; Wang, X.; Niu, P.; Meng, J.; Zhu, Z.; Zhang, Z.; Wang, J. Genome composition and divergence of the novel coronavirus (2019-nCoV) originating in China. Cell Host Microbe 2020, 27, 325–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, F.; Li, W.; Farzan, M.; Harrison, S.C. Structure of SARS coronavirus spike receptor-binding domain complexed with receptor. Science 2005, 309, 1864–1868. [Google Scholar] [CrossRef]
- Hussain, M.; Jabeen, N.; Raza, F.; Shabbir, S.; Baig, A.A.; Amanullah, A.; Aziz, B. Structural variations in human ACE2 may influence its binding with SARS-CoV-2 spike protein. J. Med Virol. 2020, 92, 1580–1586. [Google Scholar] [CrossRef] [Green Version]
- Abdelli, I.; Hassani, F.; Brikci, S.B.; Ghalem, S. In silico study the inhibition of Angiotensin converting enzyme 2 receptor of COVID-19 by Ammoides verticillata components harvested from western Algeria. J. Biomol. Struct. Dyn. 2020, 39, 3263–3276. [Google Scholar] [CrossRef]
- Ahmad, S.; Abbasi, H.W.; Shahid, S.; Gul, S.; Abbasi, S.W. Molecular Docking, Simulation and MM-PBSA Studies of Nigella Sativa Compounds: A Computational Quest to identify Potential Natural Antiviral for COVID-19 Treatment. J. Biomol. Struct. Dyn. 2020, 1–16. [Google Scholar] [CrossRef]
- Wahedi, H.M.; Ahmad, S.; Abbasi, S.W. Stilbene-based natural compounds as promising drug candidates against COVID-19. J. Biomol. Struct. Dyn. 2020, 1–10. [Google Scholar] [CrossRef]
- Lakshmi, S.A.; Shafreen, R.M.B.; Priya, A.; Shunmugiah, K.P. Ethnomedicines of Indian origin for combating COVID-19 infection by hampering the viral replication: Using structure-based drug discovery approach. J. Biomol. Struct. Dyn. 2020, 1–16. [Google Scholar] [CrossRef]
- Romeo, A.; Iacovelli, F.; Falconi, M. Targeting the SARS-CoV-2 spike glycoprotein prefusion conformation: Virtual screening and molecular dynamics simulations applied to the identification of potential fusion inhibitors. Virus Res. 2020, 286, 198068. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira, O.V.; Rocha, G.B.; Paluch, A.S.; Costa, L.T. Repurposing approved drugs as inhibitors of SARS-CoV-2 S-protein from molecular modeling and virtual screening. J. Biomol. Struct. Dyn. 2020, 1–14. [Google Scholar] [CrossRef]
- Sinha, S.K.; Prasad, S.K.; Islam, M.A.; Gurav, S.S.; Patil, R.B.; AlFaris, N.A.; Aldayel, T.S.; AlKehayez, N.M.; Wabaidur, S.M.; Shakya, A. Identification of bioactive compounds from Glycyrrhiza glabra as possible inhibitor of SARS-CoV-2 spike glycoprotein and non-structural protein-15: A pharmacoinformatics study. J. Biomol. Struct. Dyn. 2020, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, S.; Malik, Y.S.; Tomar, S. Identification of SARS-CoV-2 cell entry inhibitors by drug repurposing using in silico structure-based virtual screening approach. Front. Immunol. 2020, 11, 1664. [Google Scholar] [CrossRef]
- Trezza, A.; Iovinelli, D.; Santucci, A.; Prischi, F.; Spiga, O. An integrated drug repurposing strategy for the rapid identification of potential SARS-CoV-2 viral inhibitors. Sci. Rep. 2020, 10, 1–8. [Google Scholar]
- Unni, S.; Aouti, S.; Thiyagarajan, S.; Padmanabhan, B. Identification of a repurposed drug as an inhibitor of Spike protein of human coronavirus SARS-CoV-2 by computational methods. J. Biosci. 2020, 45, 1–20. [Google Scholar] [CrossRef]
- Khan, A.A.; Baildya, N.; Dutta, T.; Ghosh, N.N. Inhibitory efficiency of potential drugs against SARS-CoV-2 by blocking human angiotensin converting enzyme-2: Virtual screening and molecular dynamics study. Microb. Pathog. 2021, 152, 104762. [Google Scholar] [CrossRef]
- Hansen, J.L.; Long, A.M.; Schultz, S.C. Structure of the RNA-dependent RNA polymerase of poliovirus. Structure 1997, 5, 1109–1122. [Google Scholar] [CrossRef] [Green Version]
- Subissi, L.; Posthuma, C.C.; Collet, A.; Zevenhoven-Dobbe, J.C.; Gorbalenya, A.E.; Decroly, E.; Snijder, E.J.; Canard, B.; Imbert, I. One severe acute respiratory syndrome coronavirus protein complex integrates processive RNA polymerase and exonuclease activities. Proc. Natl. Acad. Sci. USA 2014, 111, E3900–E3909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pachetti, M.; Marini, B.; Benedetti, F.; Giudici, F.; Mauro, E.; Storici, P.; Masciovecchio, C.; Angeletti, S.; Ciccozzi, M.; Gallo, R.C. Emerging SARS-CoV-2 mutation hot spots include a novel RNA-dependent-RNA polymerase variant. J. Transl. Med. 2020, 18, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.; Song, W.; Huang, H.; Sun, Q. Pharmacological therapeutics targeting RNA-dependent RNA polymerase, proteinase and spike protein: From mechanistic studies to clinical trials for COVID-19. J. Clin. Med. 2020, 9, 1131. [Google Scholar] [CrossRef] [Green Version]
- Elfiky, A.A. Anti-HCV, nucleotide inhibitors, repurposing against COVID-19. Life Sci. 2020, 248, 117477. [Google Scholar] [CrossRef] [PubMed]
- Elfiky, A.A. Ribavirin, Remdesivir, Sofosbuvir, Galidesivir, and Tenofovir against SARS-CoV-2 RNA dependent RNA polymerase (RdRp): A molecular docking study. Life Sci. 2020, 253, 117592. [Google Scholar] [CrossRef] [PubMed]
- Elfiky, A.A. SARS-CoV-2 RNA dependent RNA polymerase (RdRp) targeting: An in silico perspective. J. Biomol. Struct. Dyn. 2020, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Aouidate, A.; Ghaleb, A.; Chtita, S.; Aarjane, M.; Ousaa, A.; Maghat, H.; Sbai, A.; Choukrad, M.B.; Bouachrine, M.; Lakhlifi, T. Identification of a novel dual-target scaffold for 3CLpro and RdRp proteins of SARS-CoV-2 using 3D-similarity search, molecular docking, molecular dynamics and ADMET evaluation. J. Biomol. Struct. Dyn. 2020, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Borquaye, L.S.; Gasu, E.N.; Ampomah, G.B.; Kyei, L.K.; Amarh, M.A.; Mensah, C.N.; Nartey, D.; Commodore, M.; Adomako, A.K.; Acheampong, P. Alkaloids from Cryptolepis sanguinolenta as Potential Inhibitors of SARS-CoV-2 Viral Proteins: An In Silico Study. BioMed Res. Int. 2020, 2020, 5324560. [Google Scholar] [CrossRef]
- Yuen, C.-K.; Lam, J.-Y.; Wong, W.-M.; Mak, L.-F.; Wang, X.; Chu, H.; Cai, J.-P.; Jin, D.-Y.; To, K.K.-W.; Chan, J.F.-W.; et al. SARS-CoV-2 nsp13, nsp14, nsp15 and orf6 function as potent interferon antagonists. Emerg. Microbes Infect. 2020, 1–29. [Google Scholar] [CrossRef]
- Kim, Y.; Jedrzejczak, R.; Maltseva, N.I.; Wilamowski, M.; Endres, M.; Godzik, A.; Michalska, K.; Joachimiak, A. Crystal structure of Nsp15 endoribonuclease NendoU from SARS-CoV-2. Protein Sci. 2020, 29, 1596–1605. [Google Scholar] [CrossRef]
- Bhardwaj, K.; Palaninathan, S.; Alcantara, J.M.O.; Yi, L.L.; Guarino, L.; Sacchettini, J.C.; Kao, C.C. Structural and functional analyses of the severe acute respiratory syndrome coronavirus endoribonuclease Nsp15. J. Biol. Chem. 2008, 283, 3655–3664. [Google Scholar] [CrossRef] [Green Version]
- Ricagno, S.; Egloff, M.-P.; Ulferts, R.; Coutard, B.; Nurizzo, D.; Campanacci, V.; Cambillau, C.; Ziebuhr, J.; Canard, B. Crystal structure and mechanistic determinants of SARS coronavirus nonstructural protein 15 define an endoribonuclease family. Proc. Natl. Acad. Sci. USA 2006, 103, 11892–11897. [Google Scholar] [CrossRef] [Green Version]
- Sharma, A.; Tiwari, V.; Sowdhamini, R. Computational search for potential COVID-19 drugs from FDA-approved drugs and small molecules of natural origin identifies several anti-virals and plant products. J. Biosci. 2020, 45, 100. [Google Scholar] [CrossRef] [PubMed]
- Yadav, R.; Imran, M.; Dhamija, P.; Suchal, K.; Handu, S. Virtual screening and dynamics of potential inhibitors targeting RNA binding domain of nucleocapsid phosphoprotein from SARS-CoV-2. J. Biomol. Struct. Dyn. 2020, 1–16. [Google Scholar] [CrossRef]
- Sarma, P.; Shekhar, N.; Prajapat, M.; Avti, P.; Kaur, H.; Kumar, S.; Singh, S.; Kumar, H.; Prakash, A.; Dhibar, D.P. In-silico homology assisted identification of inhibitor of RNA binding against 2019-nCoV N-protein (N terminal domain). J. Biomol. Struct. Dyn. 2020, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borkotoky, S.; Banerjee, M. A computational prediction of SARS-CoV-2 structural protein inhibitors from Azadirachta indica (Neem). J. Biomol. Struct. Dyn. 2020, 1–17. [Google Scholar] [CrossRef]
- Krishnan, D.A.; Sangeetha, G.; Vajravijayan, S.; Nandhagopal, N.; Gunasekaran, K. Structure-based drug designing towards the identification of potential anti-viral for COVID-19 by targeting endoribonuclease NSP15. Inform. Med. Unlocked 2020, 20, 100392. [Google Scholar] [CrossRef] [PubMed]
- Chandra, A.; Gurjar, V.; Qamar, I.; Singh, N. Identification of Potential Inhibitors of SARS-COV-2 Endoribonuclease (EndoU) from FDA Approved Drugs: A Drug Repurposing Approach to find Therapeutics for COID19. J. Biomol. Struct. Dyn. 2020, 1–16. [Google Scholar] [CrossRef]
- Chikhale, R.V.; Gurav, S.S.; Patil, R.B.; Sinha, S.K.; Prasad, S.K.; Shakya, A.; Shrivastava, S.K.; Gurav, N.S.; Prasad, R.S. Sars-cov-2 host entry and replication inhibitors from Indian ginseng: An in-silico approach. J. Biomol. Struct. Dyn. 2020, 1–12. [Google Scholar] [CrossRef]
- Tazikeh-Lemeski, E.; Moradi, S.; Raoufi, R.; Shahlaei, M.; Janlou, M.A.M.; Zolghadri, S. Targeting SARS-COV-2 non-structural protein 16: A virtual drug repurposing study. J. Biomol. Struct. Dyn. 2020, 1–14. [Google Scholar] [CrossRef]
- Jiang, Y.; Liu, L.; Manning, M.; Bonahoom, M.; Lotvola, A.; Yang, Z.; Yang, Z.-Q. Structural analysis, virtual screening and molecular simulation to identify potential inhibitors targeting 2′-O-ribose methyltransferase of SARS-CoV-2 coronavirus. J. Biomol. Struct. Dyn. 2020, 1–16. [Google Scholar] [CrossRef]
- Hossain, M.K.; Hassanzadeganroudsari, M.; Feehan, J.; Apostolopoulos, V. COVID-19 vaccines in the pipeline, are antibodies adequate? Vaccines 2021, 9, 241. [Google Scholar] [CrossRef] [PubMed]
- Hossain, M.K.; Hassanzadeganroudsari, M.; Apostolopoulos, V. Why METH users are at high risk of fatality due to COVID-19 infection? Expert Rev. Vaccines 2020. [Google Scholar] [CrossRef] [PubMed]
- Hossain, M.K.; Hassanzadeganroudsari, M.; Apostolopoulos, V. The emergence of new strains of SARS-CoV-2. What does it mean for COVID-19 vaccines? Expert Rev. Vaccines 2021, 1–4. [Google Scholar] [CrossRef]
- Taccone, F.S.; Gorham, J.; Vincent, J.-L. Hydroxychloroquine in the management of critically ill patients with COVID-19: The need for an evidence base. Lancet Respir. Med. 2020, 8, 539–541. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, D.; Du, G.; Du, R.; Zhao, J.; Jin, Y.; Fu, S.; Gao, L.; Cheng, Z.; Lu, Q.; et al. Remdesivir in adults with severe COVID-19: A randomised, double-blind, placebo-controlled, multicentre trial. Lancet 2020, 395, 1569–1578. [Google Scholar] [CrossRef]
- Sanders, J.M.; Monogue, M.L.; Jodlowski, T.Z.; Cutrell, J.B. Pharmacologic treatments for coronavirus disease 2019 (COVID-19): A review. JAMA 2020, 323, 1824–1836. [Google Scholar] [CrossRef]
- Ferner, R.E.; Aronson, J.K. Chloroquine and hydroxychloroquine in covid-19. BMJ 2020, 369, m1432. [Google Scholar] [CrossRef] [Green Version]
- Geleris, J.; Sun, Y.; Platt, J.; Zucker, J.; Baldwin, M.; Hripcsak, G.; Labella, A.; Manson, D.K.; Kubin, C.; Barr, R.G.; et al. Observational study of hydroxychloroquine in hospitalized patients with Covid-19. N. Engl. J. Med. 2020, 382, 2411–2418. [Google Scholar] [CrossRef]
- Theoharides, T.; Conti, P. Dexamethasone for COVID-19? Not so fast. J. Biol. Regul. Homeost Agents 2020, 34. [Google Scholar] [CrossRef]
- Chen, H.; Wang, F.; Zhang, P.; Zhang, Y.; Chen, Y.; Fan, X.; Cao, X.; Liu, J.; Yang, Y.; Wang, B.; et al. Management of cytokine release syndrome related to CAR-T cell therapy. Front. Med. 2019, 13, 610–617. [Google Scholar] [CrossRef]
- Giles, A.J.; Hutchinson, M.-K.N.; Sonnemann, H.M.; Jung, J.; Fecci, P.E.; Ratnam, N.M.; Zhang, W.; Song, H.; Bailey, R.; Davis, D. Dexamethasone-induced immunosuppression: Mechanisms and implications for immunotherapy. J. Immunother. Cancer 2018, 6, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Gordon, C.J.; Tchesnokov, E.P.; Feng, J.Y.; Porter, D.P.; Götte, M. The antiviral compound remdesivir potently inhibits RNA-dependent RNA polymerase from Middle East respiratory syndrome coronavirus. J. Biol. Chem. 2020, 295, 4773–4779. [Google Scholar] [CrossRef] [Green Version]
- Sadeghi, A.; Ali Asgari, A.; Norouzi, A.; Kheiri, Z.; Anushirvani, A.; Montazeri, M.; Hosamirudsai, H.; Afhami, S.; Akbarpour, E.; Aliannejad, R.; et al. Sofosbuvir and daclatasvir compared with standard of care in the treatment of patients admitted to hospital with moderate or severe coronavirus infection (COVID-19): A randomized controlled trial. J. Antimicrob. Chemother. 2020, 75, 3379–3385. [Google Scholar] [CrossRef]
- Nourian, A.; Khalili, H. Sofosbuvir as a potential option for the treatment of COVID-19. Acta Biomed. Atenei Parm. 2020, 91, 239. [Google Scholar]
- Sisay, M. 3CLpro inhibitors as a potential therapeutic option for COVID-19: Available evidence and ongoing clinical trials. Pharmacol. Res. 2020, 156, 104779. [Google Scholar] [CrossRef] [PubMed]
- Tang, B.; Li, S.; Xiong, Y.; Tian, M.; Yu, J.; Xu, L.; Zhang, L.; Li, Z.; Ma, J.; Wen, F.; et al. COVID-19 pneumonia in a hemodialysis patient. Kidney Med. 2020, 2, 354–358. [Google Scholar] [CrossRef] [PubMed]
- Javed, H.; Meeran, M.F.N.; Jha, N.K.; Ojha, S. Carvacrol, a Plant Metabolite Targeting Viral Protease (Mpro) and ACE2 in Host Cells Can Be a Possible Candidate for COVID-19. Front. Plant Sci. 2020, 11. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| No. | Ligand | Molecular Docking | Molecular Dynamics | Ref | |||||
|---|---|---|---|---|---|---|---|---|---|
| - | - | Docking score (Kcal/mol) | Software | Methodology | Binding free energy (Kcal/mol) | Force-field | Simulation time (ns) | RMSD (Å) | |
| 1 | Simeprevier | −11.33 | AutoDuck 4.2 | Not stated | −252.54 ± 85.69 | CHARMM36 | 150 | Not stated | [68] |
| 2 | Lopinavir-Ritonavir | −10.6 | AutoDock Vina | Lamarckian genetic algorithm (GA) in combination with grid-based energy estimation | Not stated | AMBER14 | 10 | 1.5–2.458 | [69] |
| 3 | lopinavir | −9.918 | Schrödinger | HTVS, SP and XP docking modes | Not stated | OPLS | 20 | 2.3 | [70] |
| 4 | carfilzomib | −8.6 | Schrödinger | Glide flexible dockin | −13.8 ± 0.2 | AMBER FF14SB | 125 | Not stated | [71] |
| 5 | PubChem ID: 118098670 | −10.0 | AutoDoc Vina | Hybris scoring function inspired by X-score | Not stated | CHARMM36 | 100 | 3 | [72] |
| 6 | Carvacrol | −4 | AutoDuck 4.2 | Not stated | −19.77 ± 2.24 | GROMOS 96 43a1 | 50 | 2.3–3 | [73] |
| 7 | Leupeptin Hemisulfate | −9.257 | Schrödinger | Not stated | −80.784 | OPLS3 | 200 | Not stated | [74] |
| 8 | Rhizocarpic acid | −9.11 | AutoDock Vina | Xscore as scoring function | −13.81 | CHARMM36 | 10 | 1.7 ± 0.2 | [75] |
| 9 | Pubchem ID: 11610052 | −16.35 | MOE | Not stated | Not stated | GROMOS 96 | 50 | 1.7 ± 0.02 | [76] |
| 10 | Remdesivir | −8.2 | AutoDock 4.2 | Not stated | Not stated | OPLS 2005 | 100 | 1.86 | [77] |
| 11 | Saquinavir | −8.5 | MOE | S-score as scoring function | −36.3026 | AMBER FF14SB | 20 | 2.72 | [78] |
| 12 | Pubchem ID: 129762283 | −9.08 | AutoDock 4.2 | Lamarckian Genetic Algorithm | Not stated | GROMOS 43a1 | 30 | 2.3–2.7 | [79] |
| 13 | Saquinavir | −9.09 | Not stated | HTVS, SP and XP docking modes | −74.4061 | Not stated | 100 | 2.5 | [80] |
| 14 | Gallocatechin-3-gallate | −9 | AutoDock Vina | Not stated | −53.5 | OPLS-AA/L | 100 | 1.45 | [81] |
| 15 | Paritaprevir | −8.8 | AutoDock Vina | MMFF94 Force-field-based | −47.15 | AMBER | 50 | 3.2 | [82] |
| 16 | Conivaptan | −8.6 | AutoDock Vina | Not stated | Not stated | Not stated | 7 | 3.25 | [83] |
| 17 | Indinavir | −8.824 | Schrödinger | XP Gscore | Not stated | Not stated | 100 | 2.771 | [84] |
| 18 | α-ketoamide 13b | −9.2 | Schrödinger | XP scoring function | −25.2 | CHARMM27 | 100 | 2.7 | [85] |
| 19 | Saquinavir | −9.856 | Schrödinger | OPLS_2005 Force-field-based | −72.17 | CHARMM27 | 50 | 0.18 | [86] |
| 20 | PubChem ID: 4167619 | −9.3 | AutoDock Vina | Not stated | −29.3 | CHARMM36 | 20 | 2.2 ± 0.3 | [87] |
| 21 | Salvianolic acid A | −9.7 | Auto Dock | genetic algorithm (GA) | −44.8 | AMBER | 40 | 2.5 | [88] |
| 22 | Desacetylgedunin | −7.3 | Autodock Vina | Not stated | Not stated | CHARMM36 | 40 | Not stated | [89] |
| 23 | ZINC ID: 000621278586 | −9.3 | AutoDock Vina | Not stated | −30.86 ± 0.57 | GROMOS 96 54a7 | 20 | 2.8 ± 0.34 | [90] |
| 24 | Lymecycline | −8.87 | Not stated | Not stated | −22.19 ± 5.23 | AMBER FF14SB | 120 | 3.10 ± 0.43 | [91] |
| 25 | Cadambine | −8.6 | AutoDock | Not stated | −51.92 ± 6.03 | Not stated | 250 | Not stated | [92] |
| 26 | Isoliquiritine apioside | −7.8 | AutoDock Vina 4.2 | Lamarkian genetic algorithm | Not stated | GROMOS 96 43a2 | 100 | 3.41 | [93] |
| 27 | Salicylamide | −7.1 | Schrödinger | HTVS, SP and XP docking modes- glide score as scoring function | −29.042 | AMBER14 | 100 | 1.11 | [94] |
| 28 | Nelfinavir | −8.3 | AutoDock Vina | Not stated | Not stated | CHARMM36 | 30 | Not stated | [95] |
| 29 | TMC-310911 | −8.3 | AutoDock Vina | Not stated | −52.8 | GAFF2 | 50 | 3.5 | [96] |
| 30 | ABBV-744 | −7.79 | Schrödinger | HTVS, SP and XP docking modes | −45.43 | Not stated | 200 | 2.45 | [97] |
| 31 | Phyllaemblicin C | −9.723 | Schrödinger | Glide molecular docking | Not Stated | GAFF | 60 | Not Stated | [98] |
| 32 | Dpnh (NADH) | −11.016 | Schrödinger | HTVS, SP and XP docking modes | Not Stated | Not Stated | Not Stated | Not Stated | [99] |
| 33 | Saquinavir | −9.5 | Autodock | MMFF94 force-field-based | Not applicable | Not applicable | Not Applicable | Not Appli-cable | [100] |
| No. | Ligand | Molecular Docking | Molecular Dynamics | Ref | |||||
|---|---|---|---|---|---|---|---|---|---|
| - | - | Docking score (Kcal/mol) | Software | Methodology | Binding free energy (Kcal/mol) | Force-field | Simulation time (ns) | RMSD (Å) | |
| 1 | Isothymol | −5.7853 | MOE | Rigid protein- flexible ligand | Not stated | Not stated | Not stated | Not stated | [108] |
| 2 | dithymoquinone | −8.6 | Autodock vina | Not stated | −26.7955 | Not stated | 100 | 2.58 | [109] |
| 3 | Resveratrol | −8 | Autodock vina | Not stated | −23.88 | AMBER AFF14SB | 50 | 1.78 | [110] |
| 4 | Orientin | −101.17 | Autodock v4.2 | Not stated | −70.6 | CHARMM | 20 | 4.6 | [111] |
| 5 | phthalocyanine | −16.3 | Autodock Vina | Lamarckian Genetic Algorithm | −66.6 | Not stated | 30 | Not stated | [112] |
| 6 | Theaflavin digallate | −8.7 | AutoDock | Not stated | –38.51 (±1.59) | GROMOS 54a7 | 18 | Not stated | [113] |
| 7 | glycyrrhizic acid | −9.2 | Autodock vina | Not stated | −79.23 | CHARMM36 | 100 | 12.3 | [114] |
| 8 | GR hydrochloride | −11.23 | Autodock vina | Lamarckian Genetic Algorithm | Not Stated | GROMOS 96 43a1 | 50 | 2.5–3.1 | [115] |
| 9 | Lumacaftor | −9.4 | AutoDock | Not stated | Not Stated | Not Stated | Not Stated | 3.2 | [116] |
| 10 | Phyllaemblicin C | −9.131 | Schrödinger | Glide molecular docking | Not Stated | GAFF | 60 | Not Stated | [98] |
| 11 | Coenzyme A | −11.555 | Schrödinger | HTVS, SP and XP docking modes | Not Stated | Not Stated | Not Stated | Not Stated | [99] |
| 12 | Bisoxatin | −7.4 | AutoDock | Not Stated | −31.94 | Not Stated | 100 | Not Stated | [117] |
| 13 | Cefpiramide | −9.1 | Autodock Vina | Not Stated | 19.09 ± 4.45 | CHARMM36 | 10 | Not Stated | [118] |
| No. | Ligand | Molecular Docking | Molecular Dynamics | Ref | |||||
|---|---|---|---|---|---|---|---|---|---|
| - | - | Docking score (Kcal/mol) | Software | Methodology | Binding free energy (Kcal/mol) | Force-field | Simulation time (ns) | RMSD (Å) | |
| 1 | IDX-184 | −9 | Autodock Vina | Not Stated | Not Stated | Not Stated | Not Stated | Not Stated | [123] |
| 2 | Sofobuvir | −9.3 | Autodock Vina | Not Stated | Not Stated | Not Stated | Not Stated | Not Stated | [124,125] |
| 3 | CAS ID: 833463-10-8 | −9.529 | Not Stated | Not Stated | Not Stated | CHARMM36 | 20 | 1.6 | [126] |
| 4 | Cryptomisrine | −9.4 | AutoDock Vina | Not Stated | −60.15 | AMBER FF14SB | 4 | 1.87 | [127] |
| Protein/NSP. | Crystal Structure | Ligand | Dock Score | Ref. |
|---|---|---|---|---|
| Nucleocapsid (N) protein |  PDB ID: 6M3M | Asinex ID: 5817 | −10.29 | [133] |
| ZINC00003118440 | −6.728 | [134] | ||
| Envelope (E) protein |  PDB ID: 5X29 | Belachinal | −11.46 | [24] |
| Nimbolin A | −11.2 | [135] | ||
| Membrane (M) protein |  PDB ID: 3I6K | Nimocin | −10.2 | [135] |
| NSP 15 |  PDB ID: 6VWW | Enamine ID: Z595015370 | −10.50 | [136] |
| Glisoxepide | −9.4 | [137] | ||
| Pubchem ID: 132519418 | −6.7 | [138] | ||
| NSP 16 |  PDB ID: 6W75 | Raltegravir | −10.3 | [139] |
| Hesperidin | −10.3 | [140] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hassanzadeganroudsari, M.; Ahmadi, A.H.; Rashidi, N.; Hossain, M.K.; Habib, A.; Apostolopoulos, V. Computational Chemistry to Repurposing Drugs for the Control of COVID-19. Biologics 2021, 1, 111-128. https://doi.org/10.3390/biologics1020007
Hassanzadeganroudsari M, Ahmadi AH, Rashidi N, Hossain MK, Habib A, Apostolopoulos V. Computational Chemistry to Repurposing Drugs for the Control of COVID-19. Biologics. 2021; 1(2):111-128. https://doi.org/10.3390/biologics1020007
Chicago/Turabian StyleHassanzadeganroudsari, Majid, Amir Hossein Ahmadi, Niloufar Rashidi, Md Kamal Hossain, Amanda Habib, and Vasso Apostolopoulos. 2021. "Computational Chemistry to Repurposing Drugs for the Control of COVID-19" Biologics 1, no. 2: 111-128. https://doi.org/10.3390/biologics1020007