
Bioengineering of Antibody Fragments: Challenges and Opportunities

Abstract
:1. Introduction
2. Technologies to Bioengineer the Antibody Fragments (Fabs and scFvs)
2.1. Hybridoma Technology
2.2. Phage Display
2.3. Transgenic Animals
2.4. Single B-Cell Technology
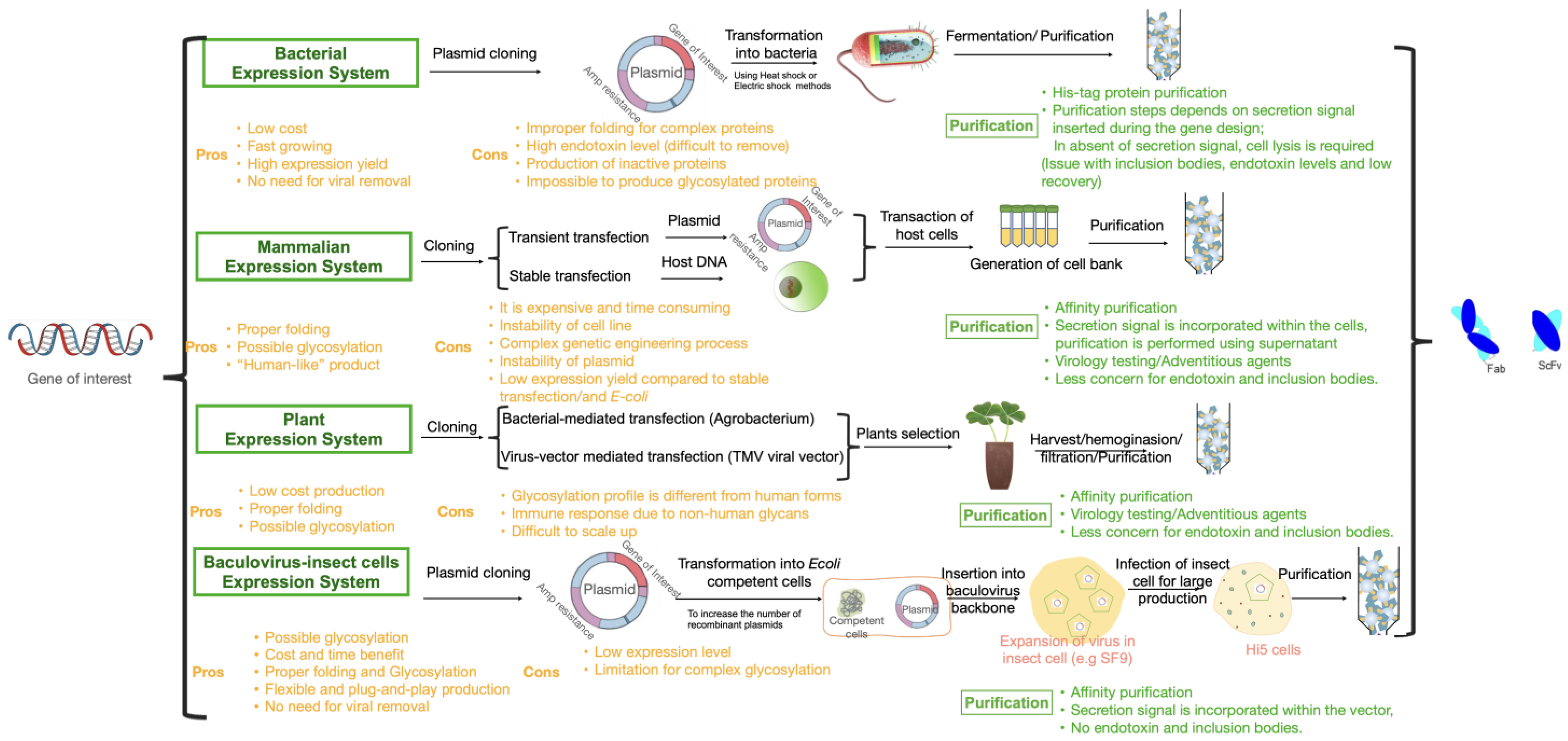
3. Heterologous Protein Expression Platforms for Antibody and Antibody Fragment Production
3.1. Bacterial Expression (E. coli)
3.2. Mammalian Cell Lines
3.3. Plant-Based Expression Systems
3.4. Insect Cell Expression System
4. Challenges and Opportunities of Different Expression Systems to Produce Antibody Fragments/Future Perspective
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- NCI Dictionary of Cancer Terms. National Cancer Institute. 2011. Available online: https://www.cancer.gov/publications/dictionaries/cancer-terms/def/biological-drug (accessed on 9 January 2023).
- Itakura, K.; Hirose, T.; Crea, R.; Riggs, A.D.; Heyneker, H.L.; Bolivar, F.; Boyer, H.W. Expression in Escherichia coli of a chemically synthesized gene for the hormone somatostatin. Science 1977, 198, 1056–1063. [Google Scholar] [CrossRef] [PubMed]
- Posnett, D.N.; Chiorazzi, N.; Kunkel, H.G. Monoclonal antibodies with specificity for hairy cell leukemia cells. J. Clin. Investig. 1982, 70, 254–261. [Google Scholar] [CrossRef] [PubMed]
- Tami, J.A.; Parr, M.D.; Brown, S.A.; Thompson, J.S. Monoclonal antibody technology. Am. J. Health Pharm. 1986, 43, 2816–2825. [Google Scholar] [CrossRef]
- Köhler, G.; Milstein, C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 1975, 256, 495–497. [Google Scholar] [CrossRef] [PubMed]
- Sgro, C. Side-effects of a monoclonal antibody, muromonab CD3/orthoclone OKT3: Bibliographic review. Toxicology 1995, 105, 23–29. [Google Scholar] [CrossRef]
- Balthasar, J.; Fung, H.L. Utilization of antidrug antibody fragments for the optimization of intraperitoneal drug therapy: Studies using digoxin as a model drug. J. Pharmacol. Exp. Ther. 1994, 268, 734–739. [Google Scholar]
- Nguyen, Q.D.; Das, A.; Do, D.V.; Dugel, P.U.; Gomes, A.; Holz, F.G.; Koh, A.; Pan, C.K.; Sepah, Y.J.; Patel, N.; et al. Brolucizumab: Evolution through preclinical and clinical studies and the implications for the management of neovascular age-related macular degeneration. Ophthalmology 2020, 127, 963–976. [Google Scholar] [CrossRef]
- Dugel, P.U. Hawk and harrier: Phase 3, multicenter, randomized, double-masked trials of brolucizumab for neovascular age-related macular degeneration. Ophthalmology 2020, 127, 72–84. [Google Scholar] [CrossRef]
- Jain, A.; Chea, S.; Matsumiya, W.; Halim, M.S.; Yaşar, Ç.; Kuang, G.; Sepah, Y.J.; Khanani, A.M.; Do, D.V.; Nguyen, Q.D. Severe vision loss secondary to retinal arteriolar occlusions after multiple intravitreal brolucizumab administrations. Am. J. Ophthalmol. Case Rep. 2020, 18, 100687. [Google Scholar] [CrossRef]
- Dhillon, S. Tebentafusp: First approval. Drugs 2022, 82, 703–710. [Google Scholar] [CrossRef]
- Damato, B.E.; Dukes, J.; Goodall, H.; Carvajal, R.D. Tebentafusp: T cell redirection for the treatment of metastatic uveal melanoma. Cancers 2019, 11, 971. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Kumar, N.; Parachuri, N.; Singh, S.; Bandello, F.; Kuppermann, B.D.; Loewenstein, A. Brolucizumab-related retinal vasculitis: Emerging disconnect between clinical trials and real world. Eye 2020, 35, 1292–1294. [Google Scholar] [CrossRef] [PubMed]
- Bates, A.; Power, C.A. David vs. Goliath: The structure, function, and clinical prospects of antibody fragments. Antibodies 2019, 8, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jen, E.Y.; Xu, Q.; Schetter, A.; Przepiorka, D.; Shen, Y.L.; Roscoe, D.; Sridhara, R.; Deisseroth, A.; Philip, R.; Farrell, A.T.; et al. FDA approval: Blinatumomab for patients with B-cell precursor acute lymphoblastic leukemia in morphologic remission with minimal residual disease. Clin. Cancer Res. 2019, 25, 473–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naddafi, F.; Davami, F.; Tabarzad, M.; Barkhordari, F.; Shirazi, F.H. Construction of a mammalian IRES-based expression vector to amplify a bispecific antibody; blinatumomab. Iran. J. Pharm. Res. IJPR 2019, 18, 2117–2123. [Google Scholar] [CrossRef] [PubMed]
- Dhara, V.G.; Naik, H.M.; Majewska, N.I.; Betenbaugh, M.J. Recombinant antibody production in CHO and NS0 cells: Differences and similarities. Biodrugs 2018, 32, 571–584. [Google Scholar] [CrossRef]
- Kebenko, M.; Goebeler, M.-E.; Wolf, M.; Hasenburg, A.; Seggewiss-Bernhardt, R.; Ritter, B.; Rautenberg, B.; Atanackovic, D.; Kratzer, A.; Rottman, J.B.; et al. A multicenter phase 1 study of solitomab (MT110, AMG 110), a bispecific EpCAM/CD3 T-cell engager (BiTE®) antibody construct, in patients with refractory solid tumors. Oncoimmunology 2018, 7, e1450710. [Google Scholar] [CrossRef]
- Brischwein, K.; Schlereth, B.; Guller, B.; Steiger, C.; Wolf, A.; Lutterbuese, R.; Offner, S.; Locher, M.; Urbig, T.; Raum, T.; et al. MT110: A novel bispecific single-chain antibody construct with high efficacy in eradicating established tumors. Mol. Immunol. 2006, 43, 1129–1143. [Google Scholar] [CrossRef]
- Burness, C.B. Idarucizumab: First global approval. Drugs 2015, 75, 2155–2161. [Google Scholar] [CrossRef]
- Reilly, P.A.; Van Ryn, J.; Grottke, O.; Glund, S.; Stangier, J. Idarucizumab, a specific reversal agent for dabigatran: Mode of action, pharmacokinetics and pharmacodynamics, and safety and efficacy in phase 1 subjects. Am. J. Med. 2016, 129, S64–S72. [Google Scholar] [CrossRef] [Green Version]
- Moncalvo, F.; Espinoza, M.I.M.; Cellesi, F. Nanosized delivery systems for therapeutic proteins: Clinically validated technologies and advanced development strategies. Front. Bioeng. Biotechnol. 2020, 8, 89. [Google Scholar] [CrossRef] [PubMed]
- Goel, N.; Stephens, S. Certolizumab pegol. Mabs 2010, 2, 137–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plyukhova, A.A.; Budzinskaya, M.V.; Starostin, K.M.; Rejdak, R.; Bucolo, C.; Reibaldi, M.; Toro, M.D. Comparative safety of bevacizumab, ranibizumab, and aflibercept for treatment of neovascular age-related macular degeneration (AMD): A systematic review and network meta-analysis of direct comparative studies. J. Clin. Med. 2020, 9, 1522. [Google Scholar] [CrossRef] [PubMed]
- Usta, C.; Turgut, N.T.; Bedel, A. How abciximab might be clinically useful. Int. J. Cardiol. 2016, 222, 1074–1078. [Google Scholar] [CrossRef] [PubMed]
- Herrington-Symes, A.P.; Farys, M.; Khalili, H.; Brocchini, S. Antibody fragments: Prolonging circulation half-life special issue-antibody research. Adv. Biosci. Biotechnol. 2013, 4, 689–698. [Google Scholar] [CrossRef] [Green Version]
- Huehls, A.M.; Coupet, T.A.; Sentman, C.L. Bispecific T-cell engagers for cancer immunotherapy. Immunol. Cell Biol. 2014, 93, 290–296. [Google Scholar] [CrossRef] [Green Version]
- Ahamadi-Fesharaki, R.; Fateh, A.; Vaziri, F.; Solgi, G.; Siadat, S.D.; Mahboudi, F. Single-chain variable fragment-based bispecific antibodies: Hitting two targets with one sophisticated arrow. Mol. Ther.-Oncolytics 2019, 14, 38–56. [Google Scholar] [CrossRef] [Green Version]
- Nissim, A.; Chernajovsky, Y. Historical development of monoclonal antibody therapeutics. In Therapeutic Anti-Bodies; Springer: Berlin/Heidelberg, Germany, 2008; pp. 3–18. [Google Scholar]
- Ribatti, D. The chick embryo chorioallantoic membrane as a model for tumor biology. Exp. Cell Res. 2014, 328, 314–324. [Google Scholar] [CrossRef]
- Mitra, S.; Tomar, P.C. Hybridoma technology; advancements, clinical significance, and future aspects. J. Genet. Eng. Biotechnol. 2021, 19, 1–12. [Google Scholar] [CrossRef]
- Ganguly, S.; Wakchaure, R. Hybridoma technology: A brief review on its diagnostic and clinical significance. Pharm. Biol. Evaluations 2016, 3, 554–555. [Google Scholar]
- Luckenbach, G.-A. Some recent aspect on hybridoma technology. In Advances in Forensic Haemogenetics; Springer: Berlin/Heidelberg, Germany, 1988; p. 267. [Google Scholar]
- Parray, H.A.; Shukla, S.; Samal, S.; Shrivastava, T.; Ahmed, S.; Sharma, C. Hybridoma technology a versatile method for isolation of monoclonal antibodies, its applicability across species, limitations, advancement and future perspectives. Int. Immunopharmacol. 2020, 85, 106639. [Google Scholar] [CrossRef] [PubMed]
- Zaroff, S.; Tan, G. Hybridoma technology: The preferred method for monoclonal antibody generation for in vivo applications. Biotechniques 2019, 67, 90–92. [Google Scholar] [CrossRef] [PubMed]
- Barbas, C.F.; Burton, D.R.; Scott, J.K.; Silverman, G.J. Phage display: A laboratory manual. Q. Rev. Biol. 2001, 76, 487–488. [Google Scholar] [CrossRef]
- Pini, A.; Bracci, L. Phage display of antibody fragments. Curr. Protein Pept. Sci. 2000, 1, 155–169. [Google Scholar] [CrossRef] [PubMed]
- Hammers, C.M.; Stanley, J.R. Antibody phage display: Technique and applications. J. Investig. Dermatol. 2014, 134, 1–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Homola, J.; Yee, S.S.; Gauglitz, G. Surface plasmon resonance sensors: Review. Sens. Actuators B Chem. 1999, 54, 3–15. [Google Scholar] [CrossRef]
- Green, R.J.; Frazier, R.; Shakesheff, K.; Davies, M.; Roberts, C.; Tendler, S.J. Surface plasmon resonance analysis of dynamic biological interactions with biomaterials. Biomaterials 2000, 21, 1823–1835. [Google Scholar] [CrossRef] [PubMed]
- Burton, D.R. Phage display. Immunotechnology 1995, 1, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Kretzschmar, T.; Von Rüden, T. Antibody discovery: Phage display. Curr. Opin. Biotechnol. 2002, 13, 598–602. [Google Scholar] [CrossRef]
- Lee, C.M.Y.; Iorno, N.; Sierro, F.; Christ, D. Selection of human antibody fragments by phage display. Nat. Protoc. 2007, 2, 3001–3008. [Google Scholar] [CrossRef]
- Arap, M.A. Phage display technology: Applications and innovations. Genet. Mol. Biol. 2005, 28, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Sidhu, S.S. Phage display in pharmaceutical biotechnology. Curr. Opin. Biotechnol. 2000, 11, 610–616. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Wu, S.; Si, Y.; Li, H.; Yin, X.; Peng, D. Single-chain fragment variable produced by phage display technology: Construction, selection, mutation, expression, and recent applications in food safety. Compr. Rev. Food Sci. Food Saf. 2022, 21, 4354–4377. [Google Scholar] [CrossRef] [PubMed]
- Haque, A.; Tonks, N.K. The use of phage display to generate conformation-sensor recombinant antibodies. Nat. Protoc. 2012, 7, 2127–2143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brüggemann, M.; Caskey, H.M.; Teale, C.; Waldmann, H.; Williams, G.T.; Surani, M.A.; Neuberger, M.S. A repertoire of monoclonal antibodies with human heavy chains from transgenic mice. Proc. Natl. Acad. Sci. USA 1989, 86, 6709–6713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brüggemann, M.; Neuberger, M.S. Strategies for expressing human antibody repertoires in transgenic mice. Immunol. Today 1996, 17, 391–397. [Google Scholar] [CrossRef] [PubMed]
- McCafferty, J.; Griffiths, A.D.; Winter, G.; Chiswell, D.J. Phage antibodies: Filamentous phage displaying antibody variable domains. Nature 1990, 348, 552–554. [Google Scholar] [CrossRef]
- Neuberger, M.; Brüggemann, M. Monoclonal antibodies. Mice perform a human repertoire. Nature 1997, 386, 25–26. [Google Scholar] [CrossRef]
- Spiesberger, K.; Paulfranz, F.; Egger, A.; Reiser, J.; Vogl, C.; Rudolf-Scholik, J.; Mayrhofer, C.; Grosse-Hovest, L.; Brem, G. Large-scale purification of r28M: A bispecific scFv antibody targeting human melanoma produced in transgenic cattle. PLoS ONE 2015, 10, e0140471. [Google Scholar] [CrossRef]
- Steinitz, M.; Klein, G.; Koskimies, S.; Makel, O. EB virus-induced B lymphocyte cell lines producing specific antibody. Nature 1977, 269, 420–422. [Google Scholar] [CrossRef]
- Houdebine, L.-M. Use of transgenic animals to improve human health and animal production. Reprod. Domest. Anim. 2005, 40, 269–281. [Google Scholar] [CrossRef] [PubMed]
- Houdebine, L.M. Transgenic animal models and target validation. Methods Mol. Biol. 2006, 360, 163–202. [Google Scholar]
- Soler, E.; Thépot, D.; Rival-Gervier, S.; Jolivet, G.; Houdebine, L.-M. Preparation of recombinant proteins in milk to improve human and animal health. Reprod. Nutr. Dev. 2006, 46, 579–588. [Google Scholar] [CrossRef] [PubMed]
- Moreira, P.; Perez-Cerezales, S.; Laguna, R.; Fernández-González, R.; Sanjuanbenito, B.P.; Gutierrez-Adan, A. Transgenic mouse offspring generated by ROSI. J. Reprod. Dev. 2016, 62, 37–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lillico, S.G.; Sherman, A.; McGrew, M.J.; Robertson, C.D.; Smith, J.; Haslam, C.; Barnard, P.; Radcliffe, P.A.; Mitrophanous, K.A.; Elliot, E.A.; et al. Oviduct-specific expression of two therapeutic proteins in transgenic hens. Proc. Natl. Acad. Sci. USA 2007, 104, 1771–1776. [Google Scholar] [CrossRef] [Green Version]
- Pfeifer, A. Lentiviral transgenesis—A versatile tool for basic research and gene therapy. Curr. Gene Ther. 2006, 6, 535–542. [Google Scholar] [CrossRef]
- Houdebine, L.-M. Production of pharmaceutical proteins by transgenic animals. Comp. Immunol. Microbiol. Infect. Dis. 2009, 32, 107–121. [Google Scholar] [CrossRef]
- Yong, H.Y.; Hao, Y.; Lai, L.; Li, R.; Murphy, C.N.; Rieke, A.; Wax, D.; Samuel, M.; Prather, R.S. Production of a transgenic piglet by a sperm injection technique in which no chemical or physical treatments were used for oocytes or sperm. Mol. Reprod. Dev. 2006, 73, 595–599. [Google Scholar] [CrossRef]
- Morozumi, K.; Shikano, T.; Miyazaki, S.; Yanagimachi, R. Simultaneous removal of sperm plasma membrane and acrosome before intracytoplasmic sperm injection improves oocyte activation/embryonic development. Proc. Natl. Acad. Sci. USA 2006, 103, 17661–17666. [Google Scholar] [CrossRef] [Green Version]
- Winter, G.; Milstein, C. Man-made antibodies. Nature 1991, 349, 293–299. [Google Scholar] [CrossRef]
- Tiller, T. Single B cell antibody technologies. New Biotechnol. 2011, 28, 453–457. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, V.; Manning, B.; O’Donnell, B.; O’Reilly, B.; O’Sullivan, D.; O’Kennedy, R.; Leonard, P. Exploiting highly ordered subnanoliter volume microcapillaries as microtools for the analysis of antibody producing cells. Anal. Chem. 2014, 87, 997–1003. [Google Scholar] [CrossRef] [PubMed]
- Lanier, L.L. Just the FACS. J. Immunol. 2014, 193, 2043–2044. [Google Scholar] [CrossRef]
- Jahan-Tigh, R.R.; Ryan, C.; Obermoser, G.; Schwarzenberger, K. Flow Cytometry. J. Investig. Dermatol. 2012, 132, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeKosky, B.J.; Ippolito, G.C.; Deschner, R.P.; Lavinder, J.J.; Wine, Y.; Rawlings, B.M.; Varadarajan, N.; Giesecke, C.; Dörner, T.; Andrews, S.F.; et al. High-throughput sequencing of the paired human immunoglobulin heavy and light chain repertoire. Nat. Biotechnol. 2013, 31, 166–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazutis, L.; Gilbert, J.; Ung, W.; Weitz, D.A.; Griffiths, A.D.; Heyman, J.A. Single-cell analysis and sorting using droplet-based microfluidics. Nat. Protoc. 2013, 8, 870–891. [Google Scholar] [CrossRef] [PubMed]
- Debs, B.E.; Utharala, R.; Balyasnikova, I.V.; Griffiths, A.D.; Merten, C.A. Functional single-cell hybridoma screening using droplet-based microfluidics. Proc. Natl. Acad. Sci. USA 2012, 109, 11570–11575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pedrioli, A.; Oxenius, A. Single B cell technologies for monoclonal antibody discovery. Trends Immunol. 2021, 42, 1143–1158. [Google Scholar] [CrossRef]
- Scheid, J.F.; Mouquet, H.; Feldhahn, N.; Walker, B.D.; Pereyra, F.; Cutrell, E.; Seaman, M.S.; Mascola, J.R.; Wyatt, R.T.; Wardemann, H.; et al. A method for identification of HIV gp140 binding memory B cells in human blood. J. Immunol. Methods 2009, 343, 65–67. [Google Scholar] [CrossRef] [Green Version]
- Khan, S.; Ullah, M.W.; Siddique, R.; Nabi, G.; Manan, S.; Yousaf, M.; Hou, H. Role of recombinant DNA technology to improve life. Int. J. Genom. 2016, 2016, 2405954. [Google Scholar] [CrossRef] [Green Version]
- Vendel, M.C.; Favis, M.; Snyder, W.B.; Huang, F.; Capili, A.D.; Dong, J.; Glaser, S.M.; Miller, B.R.; Demarest, S.J. Secretion from bacterial versus mammalian cells yields a recombinant scFv with variable folding properties. Arch. Biochem. Biophys. 2012, 526, 188–193. [Google Scholar] [CrossRef] [PubMed]
- Rosano, G.L.; Ceccarelli, E.A. Recombinant protein expression in Escherichia coli: Advances and challenges. Front. Microbiol. 2014, 5, 172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsirigotaki, A.; De Geyter, J.; Šoštaric, N.; Economou, A.; Karamanou, S. Protein export through the bacterial Sec pathway. Nat. Rev. Microbiol. 2017, 15, 21–36. [Google Scholar] [CrossRef] [PubMed]
- Rusch, S.L.; Kendall, D.A. Interactions that drive sec-dependent bacterial protein transport. Biochemistry 2007, 46, 9665–9673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elvekrog, M.M.; Walter, P. Dynamics of co-translational protein targeting. Curr. Opin. Chem. Biol. 2015, 29, 79–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wild, J.; Altman, E.; Yura, T.; Gross, C.A. DnaK and DnaJ heat shock proteins participate in protein export in Escherichia coli. Genes Dev. 1992, 6, 1165–1172. [Google Scholar] [CrossRef] [Green Version]
- Denks, K.; Vogt, A.; Sachelaru, I.; Petriman, N.A.; Kudva, R.; Koch, H.-G. The sec translocon mediated protein transport in prokaryotes and eukaryotes. Mol. Membr. Biol. 2014, 31, 58–84. [Google Scholar] [CrossRef]
- Tsukazaki, T.; Mori, H.; Echizen, Y.; Ishitani, R.; Fukai, S.; Tanaka, T.; Perederina, A.; Vassylyev, D.G.; Kohno, T.; Maturana, A.D.; et al. Structure and function of a membrane component SecDF that enhances protein export. Nature 2011, 474, 235–238. [Google Scholar] [CrossRef] [Green Version]
- Dalbey, R.E.; Wang, P.; Van Dijl, J.M. Membrane proteases in the bacterial protein secretion and quality control pathway. Microbiol. Mol. Biol. Rev. 2012, 76, 311–330. [Google Scholar] [CrossRef] [Green Version]
- Palmer, T.; Berks, B.C. The twin-arginine translocation (Tat) protein export pathway. Nat. Rev. Genet. 2012, 10, 483–496. [Google Scholar] [CrossRef]
- Sargent, F.; Stanley, N.R.; Berks, B.C.; Palmer, T. Sec-independent protein translocation in Escherichia coli. J. Biol. Chem. 1999, 274, 36073–36082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simone, D.; Bay, D.C.; Leach, T.; Turner, R.J. Diversity and evolution of bacterial twin arginine translocase protein, TatC, reveals a protein secretion system that is evolving to fit its environmental niche. PLoS ONE 2013, 8, e78742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oertel, D.; Schmitz, S.; Freudl, R. A TatABC-type tat translocase is required for unimpaired aerobic growth of Corynebacterium glutamicum ATCC13023. PLoS ONE 2015, 10, e0123413. [Google Scholar] [CrossRef] [PubMed]
- Alami, M.; Lüke, I.; Deitermann, S.; Eisner, G.; Koch, H.-G.; Brunner, J.; Müller, M. Differential interactions between a twin-arginine signal peptide and its translocase in Escherichia coli. Mol. Cell 2003, 12, 937–946. [Google Scholar] [CrossRef]
- Lausberg, F.; Fleckenstein, S.; Kreutzenbeck, P.; Fröbel, J.; Rose, P.; Müller, M.; Freudl, R. Genetic evidence for a tight cooperation of TatB and TatC during productive recognition of twin-arginine (Tat) signal peptides in Escherichia coli. PLoS ONE 2012, 7, e39867. [Google Scholar] [CrossRef] [Green Version]
- Lüke, I.; Handford, J.I.; Palmer, T.; Sargent, F. Proteolytic processing of Escherichia coli twin-arginine signal peptides by LepB. Arch. Microbiol. 2009, 191, 919–925. [Google Scholar] [CrossRef]
- Blaudeck, N.; Kreutzenbeck, P.; Müller, M.; Sprenger, G.A.; Freudl, R. Isolation and characterization of bifunctional Escherichia coli TatA mutant proteins that allow efficient tat-dependent protein translocation in the absence of TatB. J. Biol. Chem. 2005, 280, 3426–3432. [Google Scholar] [CrossRef] [Green Version]
- Sandomenico, A.; Sivaccumar, J.P.; Ruvo, M. Evolution of Escherichia coli expression system in producing antibody recombinant fragments. Int. J. Mol. Sci. 2020, 21, 6324. [Google Scholar] [CrossRef]
- Terpe, K. Overview of bacterial expression systems for heterologous protein production: From molecular and biochemical fundamentals to commercial systems. Appl. Microbiol. Biotechnol. 2006, 72, 211–222. [Google Scholar] [CrossRef]
- Gupta, S.K.; Shukla, P. Microbial platform technology for recombinant antibody fragment production: A review. Crit. Rev. Microbiol. 2016, 43, 31–42. [Google Scholar] [CrossRef]
- Baumgarten, T.; Ytterberg, A.J.; Zubarev, R.A.; de Gier, J.-W. Optimizing recombinant protein production in the Escherichia coli periplasm alleviates stress. Appl. Environ. Microbiol. 2018, 84, e00270-18. [Google Scholar] [CrossRef] [PubMed]
- Browne, S.M.; Al-Rubeai, M. Selection methods for high-producing mammalian cell lines. Trends Biotechnol. 2007, 25, 425–432. [Google Scholar] [CrossRef] [PubMed]
- Bosques, C.J.; Collins, B.E.; Meador, J.W.; Sarvaiya, H.; Murphy, J.L.; DelloRusso, G.; Bulik, D.A.; Hsu, I.-H.; Washburn, N.; Sipsey, S.F.; et al. Chinese hamster ovary cells can produce galactose-α-1,3-galactose antigens on proteins. Nat. Biotechnol. 2010, 28, 1153–1156. [Google Scholar] [CrossRef] [PubMed]
- Chung, C.H.; Mirakhur, B.; Chan, E.; Le, Q.-T.; Berlin, J.; Morse, M.; Murphy, B.A.; Satinover, S.M.; Hosen, J.; Mauro, D.; et al. Cetuximab-induced anaphylaxis and IgE specific for galactose-α-1,3-galactose. N. Engl. J. Med. 2008, 358, 1109–1117. [Google Scholar] [CrossRef] [Green Version]
- Reusch, D.; Tejada, M.L. Fc glycans of therapeutic antibodies as critical quality attributes. Glycobiology 2015, 25, 1325–1334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chasin, L.A.; Urlaub, G. Chromosome-wide event accompanies the expression of recessive mutations in tetraploid cells. Science 1975, 187, 1091–1093. [Google Scholar] [CrossRef] [PubMed]
- Simon, A.E.; Taylor, M.W.; Bradley, W.E.; Thompson, L.H. Model involving gene inactivation in the generation of autosomal recessive mutants in mammalian cells in culture. Mol. Cell. Biol. 1982, 2, 1126–1133. [Google Scholar] [CrossRef]
- Ha, T.K.; Kim, D.; Kim, C.L.; Grav, L.M.; Lee, G.M. Factors affecting the quality of therapeutic proteins in recombinant Chinese hamster ovary cell culture. Biotechnol. Adv. 2021, 54, 107831. [Google Scholar] [CrossRef]
- Urlaub, G.; Chasin, L.A. Isolation of Chinese hamster cell mutants deficient in dihydrofolate reductase activity. Proc. Natl. Acad. Sci. USA 1980, 77, 4216–4220. [Google Scholar] [CrossRef] [Green Version]
- Bianchi, A.A.; McGrew, J.T. High-level expression of full-length antibodies using trans-complementing expression vectors. Biotechnol. Bioeng. 2003, 84, 439–444. [Google Scholar] [CrossRef]
- Ye, J.; Ly, J.; Watts, K.; Hsu, A.; Walker, A.; McLaughlin, K.; Berdichevsky, M.; Prinz, B.; Kersey, D.S.; D’Anjou, M.; et al. Optimization of a glycoengineered Pichia pastoris cultivation process for commercial antibody production. Biotechnol. Prog. 2011, 27, 1744–1750. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Li, F.; Button, M.; Cukan, M.; Moore, R.; Sharkey, N.; Li, H. A high-throughput purification of monoclonal antibodies from glycoengineered Pichia pastoris. Protein Expr. Purif. 2010, 74, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Schirrmann, T.; Al-Halabi, L.; Dübel, S.; Hust, M. Production systems for recombinant antibodies. Front. Biosci. 2008, 13, 4576–4594. [Google Scholar] [CrossRef] [PubMed]
- Galeffi, P.; Lombardi, A.; Di Donato, M.; Latini, A.; Sperandei, M.; Cantale, C.; Giacomini, P. Expression of single-chain antibodies in transgenic plants. Vaccine 2005, 23, 1823–1827. [Google Scholar] [CrossRef] [PubMed]
- Artsaenko, O.; Peisker, M.; Nieden, U.Z.; Fiedler, U.; Weiler, E.W.; Muntz, K.; Conrad, U. Expression of a single-chain Fv antibody against abscisic acid creates a wilty phenotype in transgenic tobacco. Plant J. 1995, 8, 745–750. [Google Scholar] [CrossRef]
- Conrad, U.; Fiedler, U. Compartment-specific accumulation of recombinant immunoglobulins in plant cells: An essential tool for antibody production and immunomodulation of physiological functions and pathogen activity. Plant Mol. Biol. 1998, 38, 101–109. [Google Scholar] [CrossRef]
- Galeffi, P.; Lombardi, A.; Pietraforte, I.; Novelli, F.; Di Donato, M.; Sperandei, M.; Tornambé, A.; Fraioli, R.; Martayan, A.; Natali, P.G.; et al. Functional expression of a single-chain antibody to ErbB-2 in plants and cell-free systems. J. Transl. Med. 2006, 4, 39. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Timko, M.P. Improving protein quantity and quality—The next level of plant molecular farming. Int. J. Mol. Sci. 2022, 23, 1326. [Google Scholar] [CrossRef]
- Felberbaum, R.S. The baculovirus expression vector system: A commercial manufacturing platform for viral vaccines and gene therapy vectors. Biotechnol. J. 2015, 10, 702–714. [Google Scholar] [CrossRef]
- Luckow, V.A.; Lee, S.C.; Barry, G.F.; Olins, P.O. Efficient generation of infectious recombinant baculoviruses by site-specific transposon-mediated insertion of foreign genes into a baculovirus genome propagated in Escherichia coli. J. Virol. 1993, 67, 4566–4579. [Google Scholar] [CrossRef] [Green Version]
- Martínez-Torrecuadrada, J.L.; Romero, S.; Núñez, A.; Alfonso, P.; Sánchez-Céspedes, M.; Casal, J.I. An efficient expression system for the production of functionally active human LKB1. J. Biotechnol. 2005, 115, 23–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Oers, M.M. Opportunities and challenges for the baculovirus expression system. J. Invertebr. Pathol. 2011, 107, S3–S15. [Google Scholar] [CrossRef] [PubMed]
- Kost, T.A.; Condreay, J.P.; Jarvis, D.L. Baculoviruses as versatile vectors for protein expression in in-sect and mammalian cells. Nat. Biotechnol. 2005, 23, 567–575. [Google Scholar] [CrossRef] [PubMed]
- Harrison, R.L.; Jarvis, D.L. Protein N-glycosylation in the baculovirus-insect cell expression system and engineering of insect cells to produce “mammalianized” recombinant glycoproteins. Adv. Virus Res. 2006, 68, 159–191. [Google Scholar] [PubMed]
- Geisler, C.; Jarvis, D.L. Innovative use of a bacterial enzyme involved in sialic acid degradation to initiate sialic acid biosynthesis in glycoengineered insect cells. Metab. Eng. 2012, 14, 642–652. [Google Scholar] [CrossRef] [Green Version]
- Shi, X.; Jarvis, D.L. Protein N-glycosylation in the baculovirus-insect cell system. Curr. Drug Targets 2007, 8, 1116–1125. [Google Scholar] [CrossRef] [Green Version]
- Kong, L.; Sheppard, N.C.; Stewart-Jones, G.B.; Robson, C.L.; Chen, H.; Xu, X.; Krashias, G.; Bonomelli, C.; Scanlan, C.N.; Kwong, P.D.; et al. Expression-system-dependent modulation of HIV-1 envelope glycoprotein antigenicity and immunogenicity. J. Mol. Biol. 2010, 403, 131–147. [Google Scholar] [CrossRef] [Green Version]
- Jarvis, D.L.; Weinkauf, C.; Guarino, L.A. Immediate early baculovirus vectors for foreign gene expression in trans-formed or infected insect cells. Protein Expr. Purif. 1996, 8, 191–203. [Google Scholar] [CrossRef]
- Hollister, J.R.; Jarvis, D.L. Engineering lepidopteran insect cells for sialoglycoprotein production by genetic transformation with mammalian 1,4-galactosyltransferase and 2,6-sialyltransferase genes. Glycobiology 2001, 11, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Aumiller, J.J.; Mabashi-Asazuma, H.; Hillar, A.; Shi, X.; Jarvis, D.L. A new glycoengineered insect cell line with an inducibly mammalianized protein N-glycosylation pathway. Glycobiology 2011, 22, 417–428. [Google Scholar] [CrossRef] [Green Version]
- Joshi, L.; Shuler, M.L.; Wood, H.A. Production of a sialylated N-linked glycoprotein in insect cells. Biotechnol. Prog. 2001, 17, 822–827. [Google Scholar] [CrossRef] [PubMed]
- Merrington, C.L.; Bailey, M.J.; Possee, R.D. Manipulation of baculovirus vectors. Mol. Biotechnol. 1997, 8, 283–297. [Google Scholar] [CrossRef] [PubMed]
- Kato, T.; Kajikawa, M.; Maenaka, K.; Park, E.Y. Silkworm expression system as a platform technology in life science. Appl. Microbiol. Biotechnol. 2009, 85, 459–470. [Google Scholar] [CrossRef] [PubMed]
- Wickham, T.J.; Nemerow, G.R. Optimization of growth methods and recombinant protein production in BTI-Tn-5B1-4 insect cells using the baculovirus expression system. Biotechnol. Prog. 1993, 9, 25–30. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Molecule Type | International Non-Proprietary Name | Target | Format | Specificity | Sequence Source | Identification | Expression System | References |
|---|---|---|---|---|---|---|---|---|
| Single Chain Fragment (scFV) | Tebentafusp | gp100, CD3 | TCR-scFv fusion protein | Bispecific | Humanized | Metastatic uveal melanoma | E. coli Bacteria | [11,12] |
| Brolucizumab | VEGF-A | scFv | Monospecific | Humanized | Necvascular age-related macular degeneration | E. coli Bacteria | [13,14] | |
| Blinatumomab | CD19, CD3 | BiTE scFv | Bispecific | Murine | Acute lymphoblastic leukemia | Chinese hamster ovary (CHO) cells | [15,16,17] | |
| Solitomab | CD3, EpCAM | BiTE scFv | Bispecific | Murine | Multiple solid tumors expressing EpCAM | Chinese hamster ovary (CHO) cells | [18,19] | |
| Fab | Idarucizumab | Dabigatran Exilate | Fab | Monospecific | Humanized | Reversal of dabigatran-induced anticoagulation | Chinese hamster ovary (CHO) cells | [17,20,21] |
| Certolizumab pegol | TNF | PEGylated Fab | Monospecific | Humanized | Crohn disease, Active Rheumatoid Arthritis, Psoriatic Arthritis | E. coli Bacteria | [22,23] | |
| Ranibizumab | VEGF | Fab | Monospecific | Humanized | Macular degeneration | E. coli Bacteria | [24] | |
| Abciximab | GPIIb/IIIa | Fab | Monospecific | Chimeric mouse/human | Prevention of blood clots in angioplasty | Murine myeloma cells (Sp2/0) | [17,25] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pirkalkhoran, S.; Grabowska, W.R.; Kashkoli, H.H.; Mirhassani, R.; Guiliano, D.; Dolphin, C.; Khalili, H. Bioengineering of Antibody Fragments: Challenges and Opportunities. Bioengineering 2023, 10, 122. https://doi.org/10.3390/bioengineering10020122
Pirkalkhoran S, Grabowska WR, Kashkoli HH, Mirhassani R, Guiliano D, Dolphin C, Khalili H. Bioengineering of Antibody Fragments: Challenges and Opportunities. Bioengineering. 2023; 10(2):122. https://doi.org/10.3390/bioengineering10020122
Chicago/Turabian StylePirkalkhoran, Sama, Wiktoria Roksana Grabowska, Hamid Heidari Kashkoli, Reihaneh Mirhassani, David Guiliano, Colin Dolphin, and Hanieh Khalili. 2023. "Bioengineering of Antibody Fragments: Challenges and Opportunities" Bioengineering 10, no. 2: 122. https://doi.org/10.3390/bioengineering10020122