Restraint Stress Alters Expression of Glucocorticoid Bioavailability Mediators, Suppresses Nrf2, and Promotes Oxidative Stress in Liver Tissue

 ,
,  ,
,
Abstract
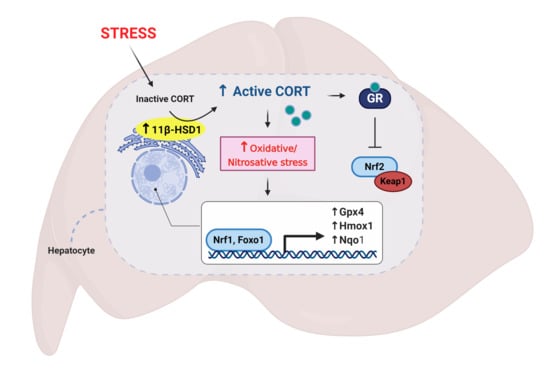
:
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Experimental Protocol
2.3. Plasma Corticosterone Measurements
2.4. mRNA Expression
2.5. Preparation of Liver Tissue for Biochemical Assays
2.6. Reduced and Oxidized Glutathione
2.7. Free Thiol Detection
2.8. General Oxidative Status
2.9. Lipid Peroxidation
2.10. Protein Carbonyl
2.11. 3-Nitrotyrosine
2.12. Immunofluorescence
2.13. Western Blotting
2.14. Protein Determination
2.15. Statistical Analyses
3. Results
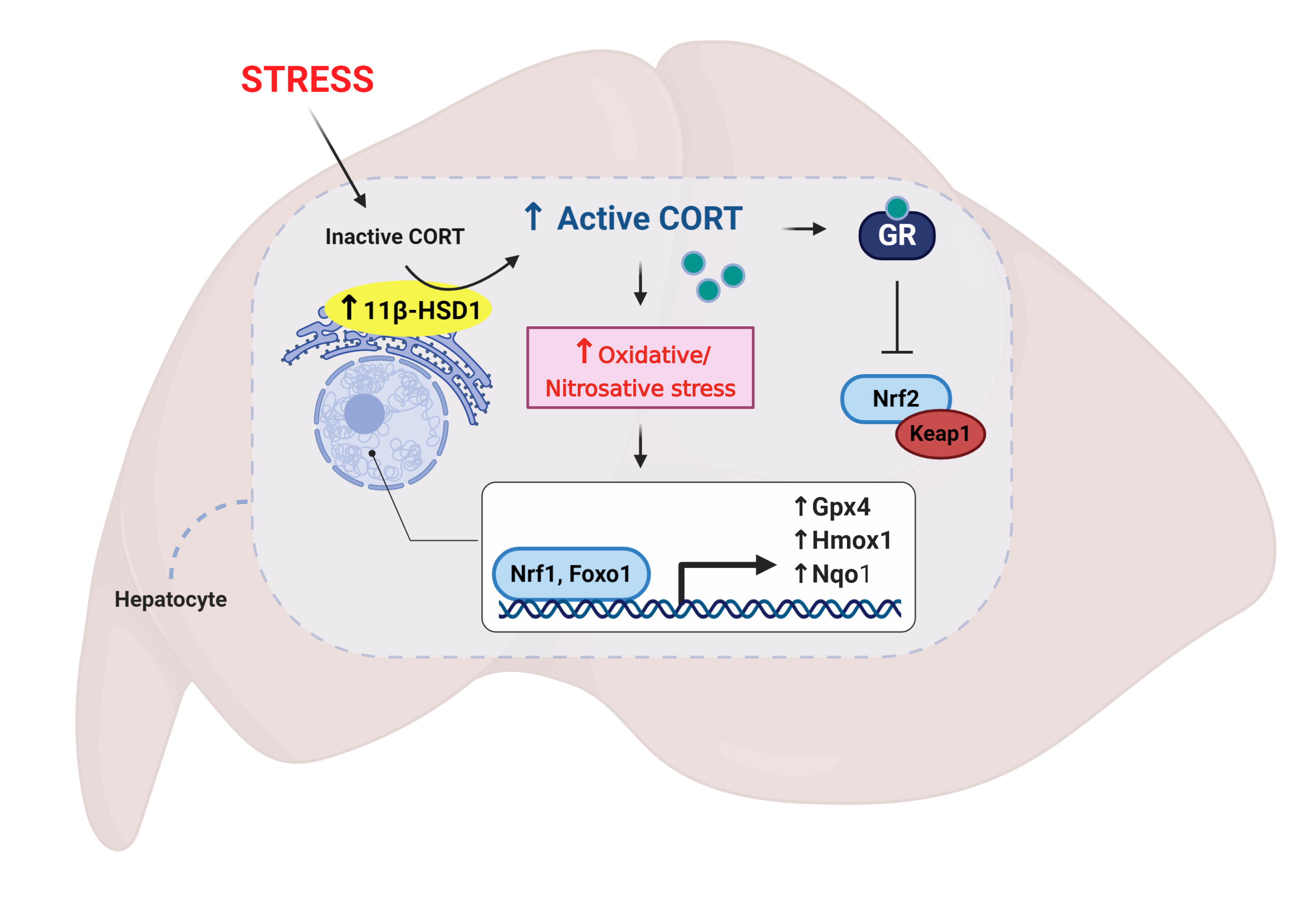
3.1. Restraint Stress-Induced Corticosterone Was Accompanied by Altered Hepatic Expression of Glucocorticoid Bioavailability Mediators
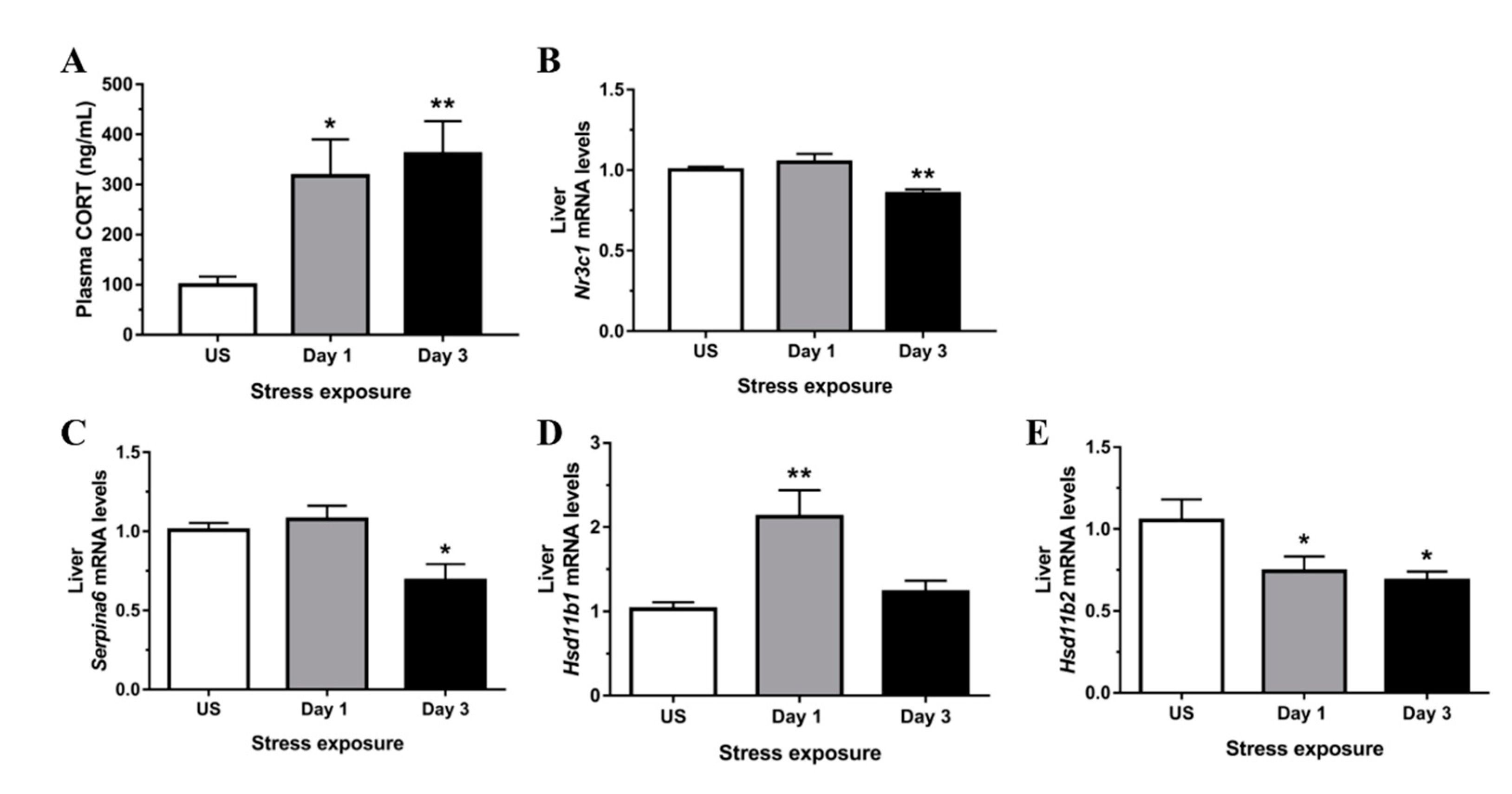
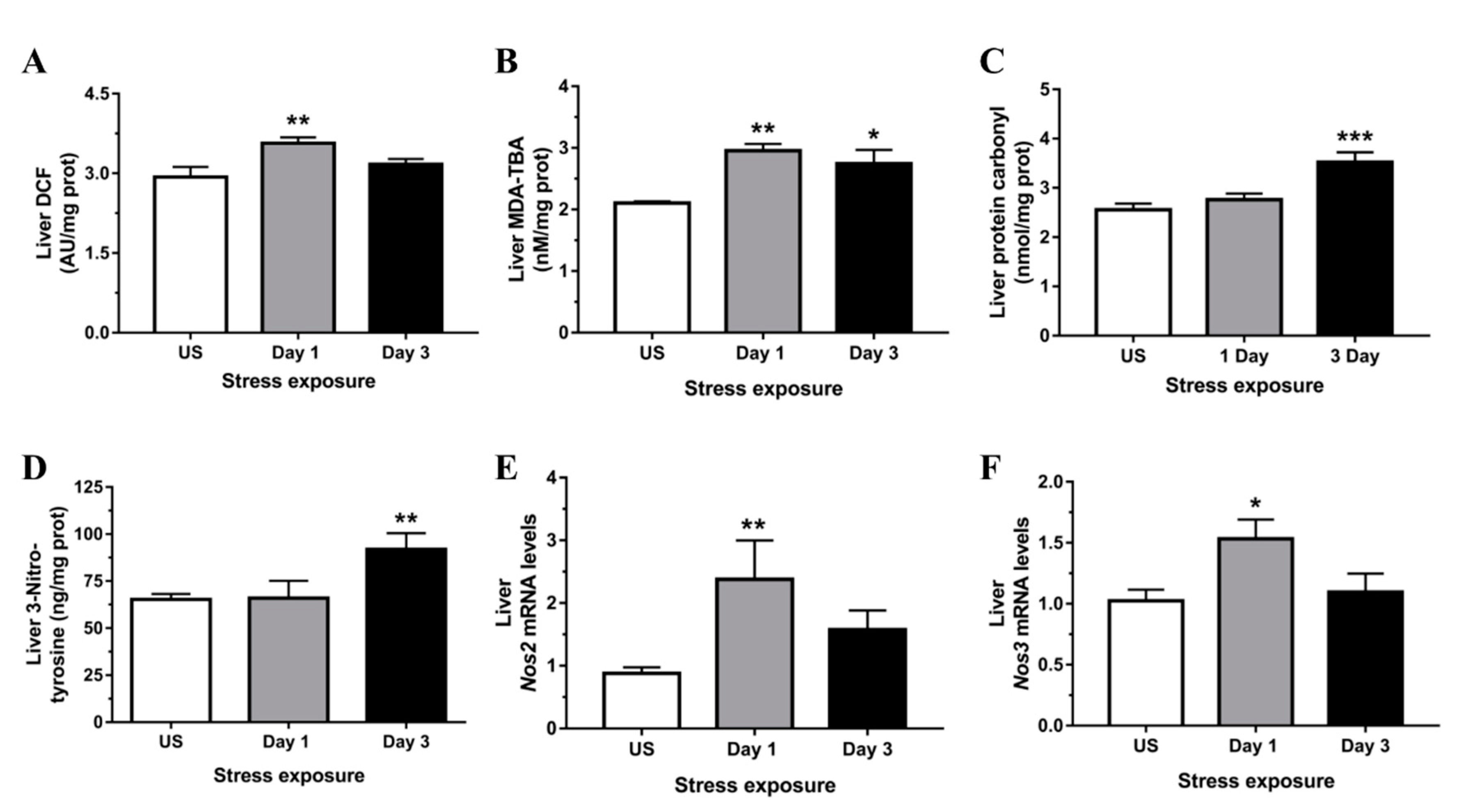
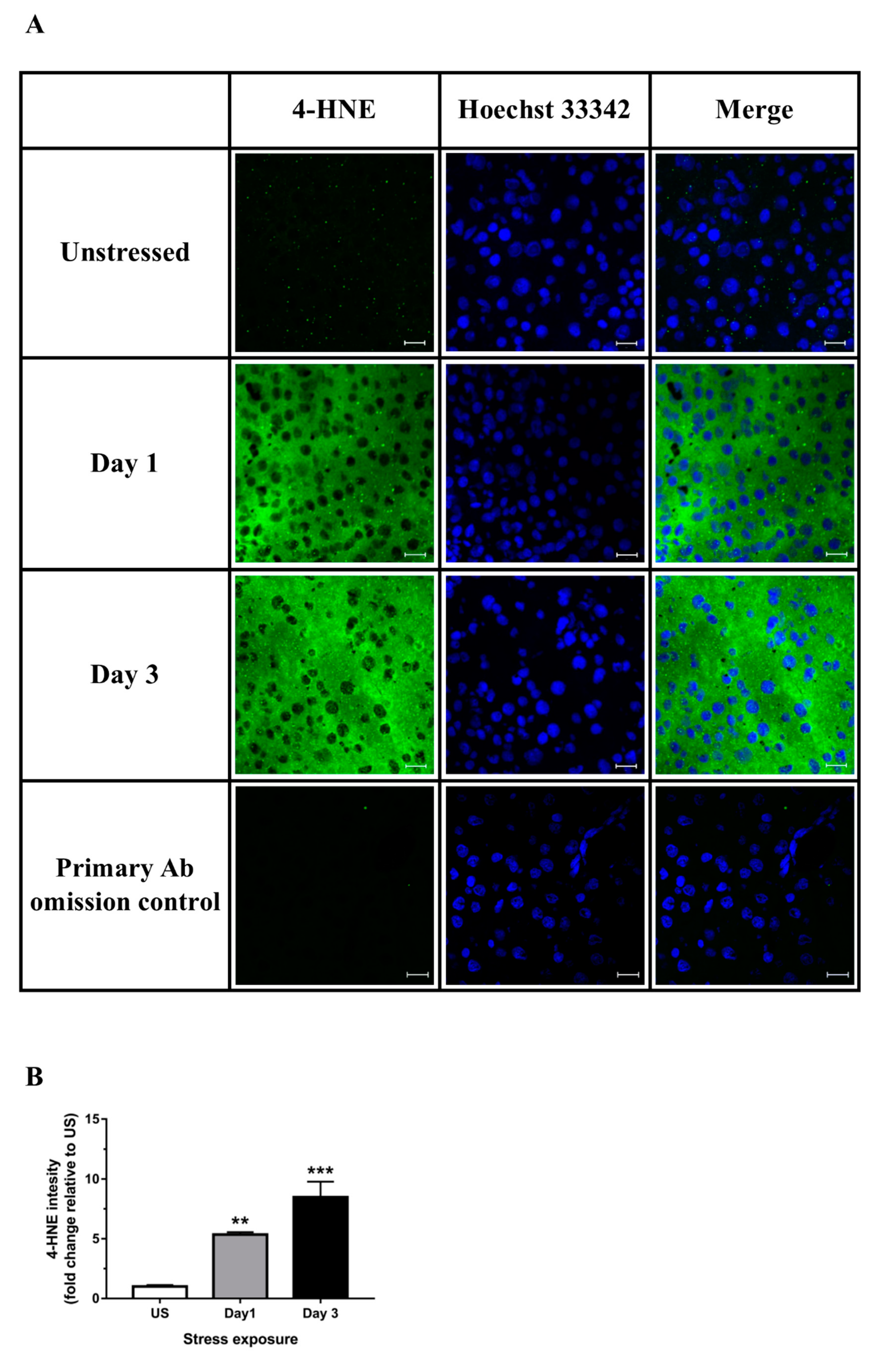
3.2. Restraint Stress-Induced Hepatic Oxidative and Nitrosative Imbalance and Oxidative Damage
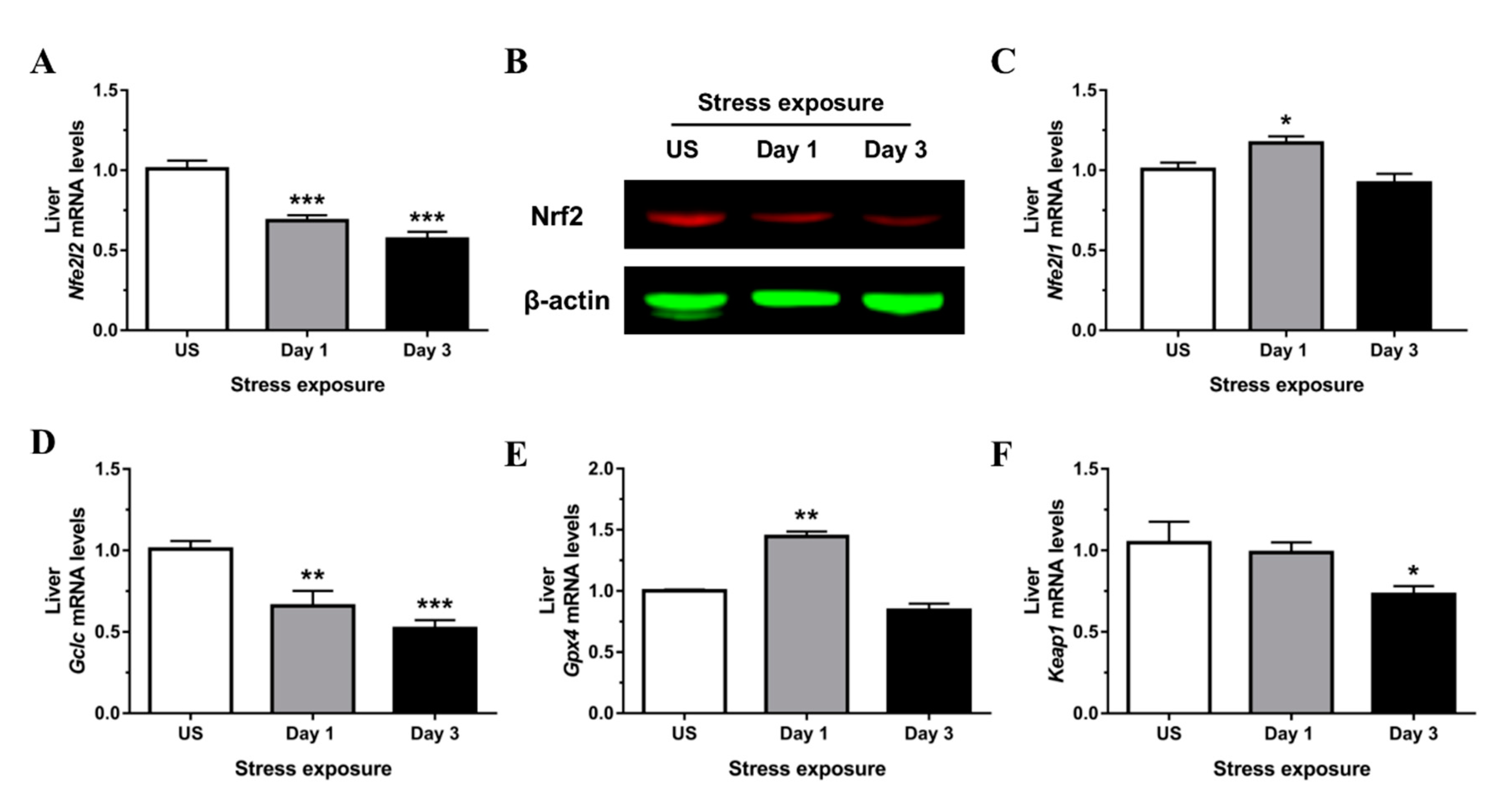
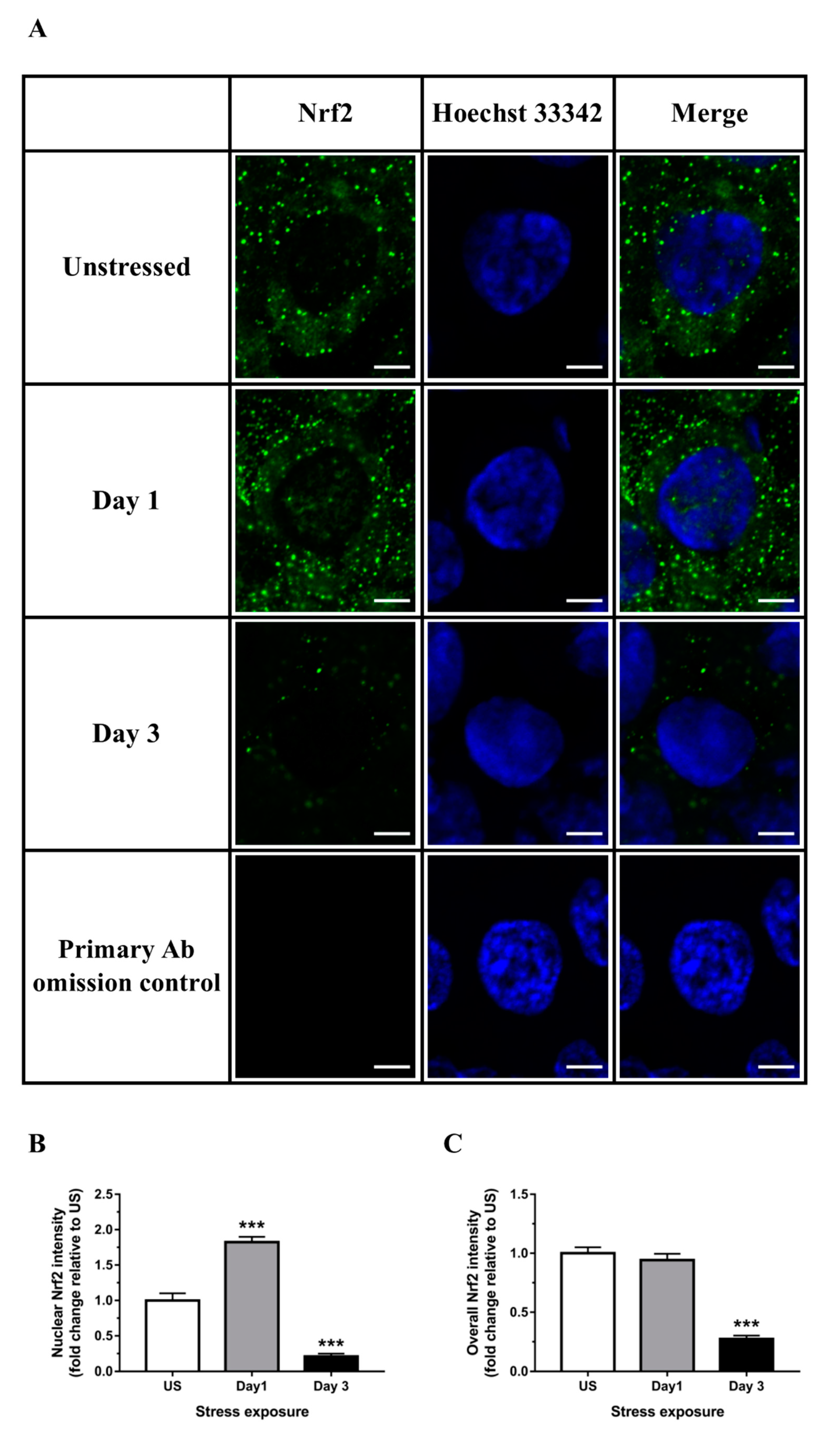
3.3. Nrf2 Was Robustly Down-regulated Following Short-Term Stress in the Liver
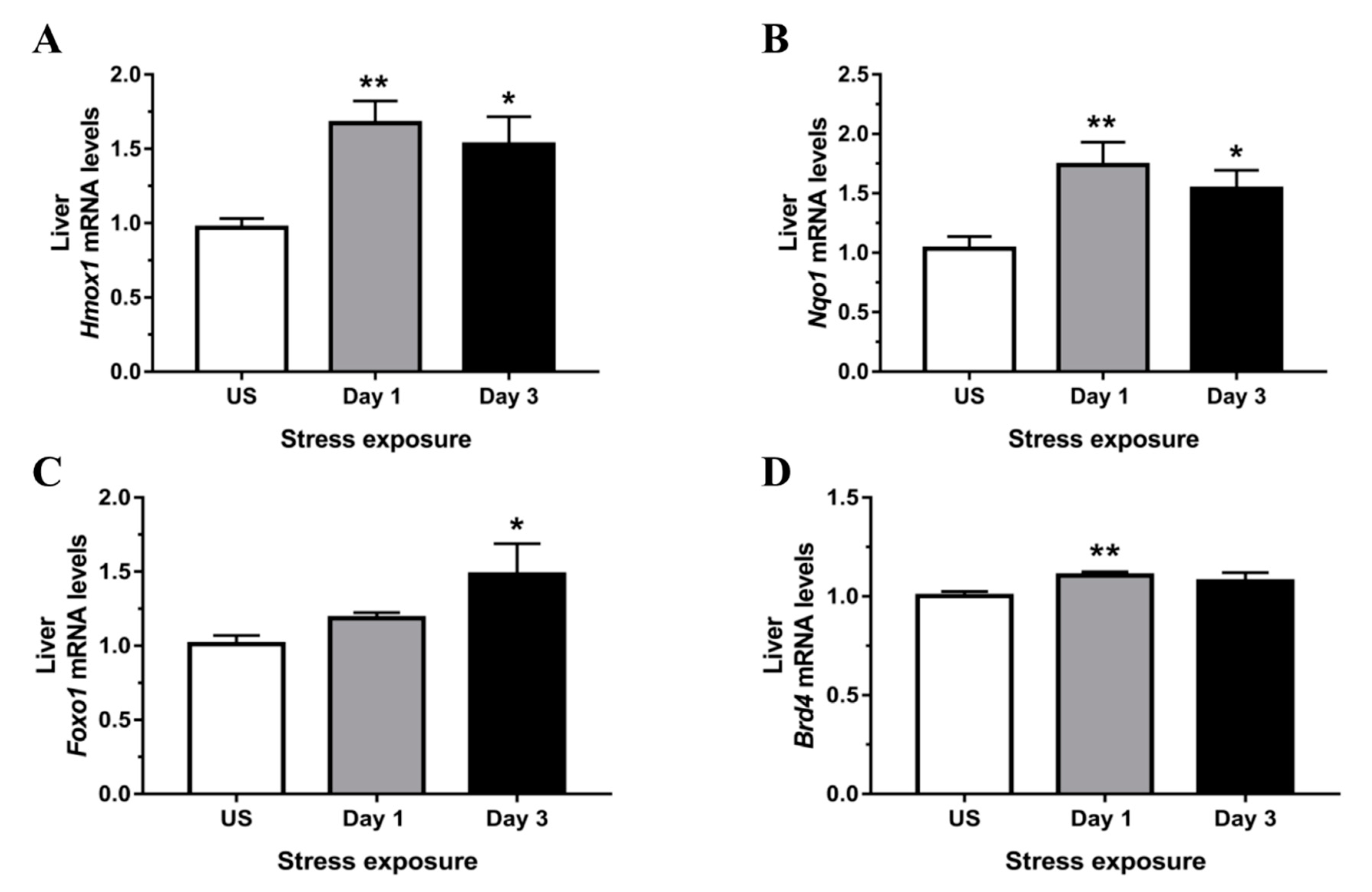
3.4. Phase II Detoxifying Enzymes Including Hmox1 and Nqo1 Were Up-regulated Following Restraint Stress
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- De Kloet, E.R.; Vreugdenhil, E.; Oitzl, M.S.; Joels, M. Brain corticosteroid receptor balance in health and disease. Endocr. Rev. 1998, 19, 269–301. [Google Scholar] [CrossRef] [PubMed]
- Gjerstad, J.K.; Lightman, S.L.; Spiga, F. Role of glucocorticoid negative feedback in the regulation of HPA axis pulsatility. Stress 2018, 21, 403–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendel, C.M. The Free Hormone Hypothesis: A Physiologically Based Mathematical Model*. Endocr. Rev. 1989, 10, 232–274. [Google Scholar] [CrossRef] [PubMed]
- Siiteri, P.K.; Murai, J.T.; Hammond, G.L.; Nisker, J.A.; Raymoure, W.J.; Kuhn, R.W. The serum transport of steroid hormones. Recent Prog. Horm. Res. 1982, 38, 457–510. [Google Scholar]
- Perogamvros, I.; Ray, D.W.; Trainer, P.J. Regulation of cortisol bioavailability—Effects on hormone measurement and action. Nat. Rev. Endocrinol. 2012, 8, 717–727. [Google Scholar] [CrossRef]
- Tinnikov, A.A. Responses of Serum Corticosterone and Corticosteroid-Binding Globulin to Acute and Prolonged Stress in the Rat. Endocrine 1999, 11, 145–150. [Google Scholar] [CrossRef]
- Fleshner, M.; Deak, T.; Spencer, R.L.; Laudenslager, M.L.; Watkins, L.R.; Maier, S.F. A long-term increase in basal levels of corticosterone and a decrease in corticosteroid-binding globulin after acute stressor exposure. Endocrinology 1995, 136, 5336–5342. [Google Scholar] [CrossRef]
- Spencer, R.L.; Miller, A.H.; Moday, H.; McEwen, B.S.; Blanchard, R.J.; Blanchard, D.C.; Sakai, R.R. Chronic social stress produces reductions in available splenic type II corticosteroid receptor binding and plasma corticosteroid binding globulin levels. Psychoneuroendocrinology 1996, 21, 95–109. [Google Scholar] [CrossRef]
- Tannenbaum, B.; Rowe, W.; Sharma, S.; Diorio, J.; Steverman, A.; Walker, M.; Meaney, M.J. Dynamic Variations in Plasma Corticosteroid-Binding Globulin and Basal HPA Activity following Acute Stress in Adult Rats. J. Neuroendocr. 1997, 9, 163–168. [Google Scholar] [CrossRef]
- Groeneweg, F.L.; Karst, H.; De Kloet, E.R.; Joëls, M. Rapid non-genomic effects of corticosteroids and their role in the central stress response. J. Endocrinol. 2011, 209, 153–167. [Google Scholar] [CrossRef] [Green Version]
- Chapman, K.E.; Coutinho, A.E.; Zhang, Z.; Kipari, T.; Savill, J.S.; Seckl, J.R. Changing glucocorticoid action: 11β-Hydroxysteroid dehydrogenase type 1 in acute and chronic inflammation. J. Steroid Biochem. Mol. Biol. 2013, 137, 82–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomlinson, J.W.; Walker, E.A.; Bujalska, I.J.; Draper, N.; Lavery, G.G.; Cooper, M.S.; Hewison, M.; Stewart, P.M. 11beta-hydroxysteroid dehydrogenase type 1: A tissue-specific regulator of glucocorticoid response. Endocr. Rev. 2004, 25, 831–866. [Google Scholar] [CrossRef] [PubMed]
- Spiers, J.G.; Chen, H.-J.C.; Cuffe, J.S.M.; Sernia, C.; Lavidis, N.A. Acute restraint stress induces rapid changes in central redox status and protective antioxidant genes in rats. Psychoneuroendocrinology 2016, 67, 104–112. [Google Scholar] [CrossRef] [PubMed]
- Seckl, J.R. 11beta-hydroxysteroid dehydrogenases: Changing glucocorticoid action. Curr. Opin. Pharmacol. 2004, 4, 597–602. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, P.; Krozowski, Z. Role of the 11beta-hydroxysteroid dehydrogenase type 2 in blood pressure regulation. Kidney Int. 2000, 57, 1374–1381. [Google Scholar] [CrossRef] [Green Version]
- Diederich, S.; Eigendorff, E.; Burkhardt, P.; Quinkler, M.; Bumke-Vogt, C.; Rochel, M.; Seidelmann, D.; Esperling, P.; Oelkers, W.; Bahr, V. 11beta-hydroxysteroid dehydrogenase types 1 and 2: An important pharmacokinetic determinant for the activity of synthetic mineralo- and glucocorticoids. J. Clin. Endocrinol. Metab. 2002, 87, 5695–5701. [Google Scholar] [CrossRef] [Green Version]
- Ali, F.F.; Rifaai, R.A. Preventive effect of omega-3 fatty acids in a rat model of stress-induced liver injury. J. Cell Physiol. 2019, 234, 11960–11968. [Google Scholar] [CrossRef]
- Aziz, N.M.; Ragy, M.M.; Ahmed, S.M. Somatostatin analogue, Octreotide, improves restraint stress-induced liver injury by ameliorating oxidative stress, inflammatory response, and activation of hepatic stellate cells. Cell Stress Chaperones 2018, 23, 1237–1245. [Google Scholar] [CrossRef]
- Zhu, Q.; Gu, L.; Wang, Y.; Jia, L.; Zhao, Z.; Peng, S.; Lei, L. The role of alpha-1 and alpha-2 adrenoceptors in restraint stress-induced liver injury in mice. PLoS ONE 2014, 9, e92125. [Google Scholar] [CrossRef]
- Jia, H.M.; Li, Q.; Zhou, C.; Yu, M.; Yang, Y.; Zhang, H.W.; Ding, G.; Shang, H.; Zou, Z.M. Chronic unpredictive mild stress leads to altered hepatic metabolic profile and gene expression. Sci. Rep. 2016, 6, 23441. [Google Scholar] [CrossRef] [Green Version]
- Ohta, Y.; Chiba, S.; Tada, M.; Imai, Y.; Kitagawa, A. Development of oxidative stress and cell damage in the liver of rats with water-immersion restraint stress. Redox Rep. 2007, 12, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.G.; Lee, J.S.; Lee, J.S.; Han, J.M.; Son, C.G. Hepatoprotective and antioxidant effects of Myelophil on restraint stress-induced liver injury in BALB/c mice. J. Ethnopharmacol. 2012, 142, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Tonelli, C.; Chio, I.I.C.; Tuveson, D.A. Transcriptional Regulation by Nrf2. Antioxid. Redox Signal. 2018, 29, 1727–1745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, K.; Han, X.D.; Kan, Y.W. An important function of Nrf2 in combating oxidative stress: Detoxification of acetaminophen. Proc. Natl. Acad. Sci. USA 2001, 98, 4611–4616. [Google Scholar] [CrossRef] [Green Version]
- Klaassen, C.D.; Reisman, S.A. Nrf2 the rescue: Effects of the antioxidative/electrophilic response on the liver. Toxicol. Appl. Pharmacol. 2010, 244, 57–65. [Google Scholar] [CrossRef] [Green Version]
- Kensler, T.W.; Wakabayashi, N.; Biswal, S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 89–116. [Google Scholar] [CrossRef]
- Kratschmar, D.V.; Calabrese, D.; Walsh, J.; Lister, A.; Birk, J.; Appenzeller-Herzog, C.; Moulin, P.; Goldring, C.E.; Odermatt, A. Suppression of the Nrf2-dependent antioxidant response by glucocorticoids and 11beta-HSD1-mediated glucocorticoid activation in hepatic cells. PLoS ONE 2012, 7, e36774. [Google Scholar] [CrossRef] [Green Version]
- Spiers, J.G.; Chen, H.-J.C.; Sernia, C.; Lavidis, N.A. A combination of plant-derived odors reduces corticosterone and oxidative indicators of stress. Chem. Senses 2014, 39, 563–569. [Google Scholar] [CrossRef]
- Chen, H.-J.C.; Spiers, J.G.; Sernia, C.; Anderson, S.T.; Lavidis, N.A. Reactive nitrogen species contribute to the rapid onset of redox changes induced by acute immobilization stress in rats. Stress 2014, 17, 520–527. [Google Scholar] [CrossRef]
- Verhoog, N.; Allie-Reid, F.; Berghe, W.V.; Smith, C.; Haegeman, G.; Hapgood, J.; Louw, A. Inhibition of corticosteroid-binding globulin gene expression by glucocorticoids involves C/EBPbeta. PLoS ONE 2014, 9, e110702. [Google Scholar] [CrossRef] [Green Version]
- Jamieson, P.M.; Chapman, K.E.; Edwards, C.R.; Seckl, J.R. 11 beta-hydroxysteroid dehydrogenase is an exclusive 11 beta- reductase in primary cultures of rat hepatocytes: Effect of physicochemical and hormonal manipulations. Endocrinology 1995, 136, 4754–4761. [Google Scholar] [CrossRef]
- Jamieson, P.M.; Walker, B.R.; Chapman, K.E.; Andrew, R.; Rossiter, S.; Seckl, J.R. 11 beta-hydroxysteroid dehydrogenase type 1 is a predominant 11 beta-reductase in the intact perfused rat liver. J. Endocrinol. 2000, 165, 685–692. [Google Scholar] [CrossRef]
- Stewart, P.M.; Krozowski, Z.S. 11 beta-Hydroxysteroid dehydrogenase. Vitam. Horm. 1999, 57, 249–324. [Google Scholar]
- Kuo, T.; McQueen, A.; Chen, T.C.; Wang, J.C. Regulation of glucose homeostasis by glucocorticoids. Adv. Exp. Med. Biol. 2015, 872, 99–126. [Google Scholar]
- Joung, J.Y.; Cho, J.H.; Kim, Y.H.; Choi, S.H.; Son, C.G. A literature review for the mechanisms of stress-induced liver injury. Brain Behav. 2019, 9, e01235. [Google Scholar] [CrossRef]
- Fratelli, M.; Demol, H.; Puype, M.; Casagrande, S.; Villa, P.; Eberini, I.; Vandekerckhove, J.; Gianazza, E.; Ghezzi, P. Identification of proteins undergoing glutathionylation in oxidatively stressed hepatocytes and hepatoma cells. Proteomics 2003, 3, 1154–1161. [Google Scholar] [CrossRef]
- Sadhukhan, S.; Han, Y.; Jin, Z.; Tochtrop, G.P.; Zhang, G.F. Glutathionylated 4-hydroxy-2-(E)-alkenal enantiomers in rat organs and their contributions toward the disposal of 4-hydroxy-2-(E)-nonenal in rat liver. Free Radic. Biol. Med. 2014, 70, 78–85. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.; Long, Y.C. Crosstalk between cystine and glutathione is critical for the regulation of amino acid signaling pathways and ferroptosis. Sci. Rep. 2016, 6, 30033. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of Ferroptotic Cancer Cell Death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef] [Green Version]
- Sahin, E.; Gumuslu, S. Stress-dependent induction of protein oxidation, lipid peroxidation and anti-oxidants in peripheral tissues of rats: Comparison of three stress models (immobilization, cold and immobilization-cold). Clin. Exp. Pharmacol. Physiol. 2007, 34, 425–431. [Google Scholar] [CrossRef]
- Rajaraman, G.; Wang, G.Q.; Yan, J.; Jiang, P.; Gong, Y.; Burczynski, F.J. Role of cytosolic liver fatty acid binding protein in hepatocellular oxidative stress: Effect of dexamethasone and clofibrate treatment. Mol. Cell. Biochem. 2006, 295, 27–34. [Google Scholar] [CrossRef]
- Ursini, F.; Maiorino, M. Lipid peroxidation and ferroptosis: The role of GSH and GPx4. Free Radic. Boil. Med. 2020, 152, 175–185. [Google Scholar] [CrossRef]
- Dodson, M.; Castro-Portuguez, R.; Zhang, D.D. NRF2 plays a critical role in mitigating lipid peroxidation and ferroptosis. Redox Boil. 2019, 23, 101107. [Google Scholar] [CrossRef]
- Korivi, M.; Hou, C.-W.; Huang, C.-Y.; Lee, S.-D.; Hsu, M.-F.; Yu, S.-H.; Chen, C.-Y.; Liu, Y.-Y.; Kuo, C.-H. Ginsenoside-Rg1 Protects the Liver against Exhaustive Exercise-Induced Oxidative Stress in Rats. Evid.-Based Complement. Altern. Med. 2011, 2012, 1–8. [Google Scholar] [CrossRef]
- Carfagna, S.; Napolitano, G.; Barone, D.; Pinto, G.; Pollio, A.; Venditti, P. Dietary Supplementation with the MicroalgaGaldieria sulphuraria(Rhodophyta) Reduces Prolonged Exercise-Induced Oxidative Stress in Rat Tissues. Oxidative Med. Cell. Longev. 2015, 2015, 732090. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.-H.; Zhang, X.; Roneker, C.A.; McClung, J.P.; Zhang, S.; Thannhauser, T.W.; Ripoll, D.R.; Sun, Q.; Lei, X.G. Role of copper, zinc-superoxide dismutase in catalyzing nitrotyrosine formation in murine liver. Free Radic. Boil. Med. 2008, 45, 611–618. [Google Scholar]
- Yan, M.; Huo, Y.; Yin, S.; Hu, H. Mechanisms of acetaminophen-induced liver injury and its implications for therapeutic interventions. Redox Boil. 2018, 17, 274–283. [Google Scholar] [CrossRef]
- Görg, B.; Wettstein, M.; Metzger, S.; Schliess, F.; Häussinger, D. Lipopolysaccharide-induced tyrosine nitration and inactivation of hepatic glutamine synthetase in the rat. Hepatology 2005, 41, 1065–1073. [Google Scholar] [CrossRef]
- Agarwal, R.; Hennings, L.; Rafferty, T.M.; Letzig, L.G.; McCullough, S.; James, L.P.; MacMillan-Crow, L.A.; Hinson, J.A. Acetaminophen-induced hepatotoxicity and protein nitration in neuronal nitric-oxide synthase knockout mice. J. Pharmacol. Exp. Ther. 2011, 340, 134–142. [Google Scholar] [CrossRef] [Green Version]
- Abdelmegeed, M.A.; Jang, S.; Banerjee, A.; Hardwick, J.P.; Song, B.-J. Robust protein nitration contributes to acetaminophen-induced mitochondrial dysfunction and acute liver injury. Free Radic. Boil. Med. 2013, 60, 211–222. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.-J.C.; Lee, J.K.; Yip, T.; Sernia, C.; Lavidis, N.A.; Spiers, J.G. Sub-acute restraint stress progressively increases oxidative/nitrosative stress and inflammatory markers while transiently upregulating antioxidant gene expression in the rat hippocampus. Free Radic. Boil. Med. 2019, 130, 446–457. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.-J.C.; Spiers, J.G.; Sernia, C.; Lavidis, N.A. Acute restraint stress induces specific changes in nitric oxide production and inflammatory markers in the rat hippocampus and striatum. Free Radic. Boil. Med. 2016, 90, 219–229. [Google Scholar] [CrossRef]
- Zhang, C.; Walker, L.M.; Hinson, J.A.; Mayeux, P.R. Oxidant stress in rat liver after lipopolysaccharide administration: Effect of inducible nitric-oxide synthase inhibition. J. Pharmacol. Exp. Ther. 2000, 293, 968–972. [Google Scholar]
- Palanisamy, A.P.; Cheng, G.; Sutter, A.G.; Liu, J.; Lewin, D.N.; Chao, J.; Chavin, K.D. Adenovirus-Mediated eNOS Expression Augments Liver Injury after Ischemia/Reperfusion in Mice. PLoS ONE 2014, 9, e93304. [Google Scholar] [CrossRef]
- Kawai, Y.; Garduño, L.; Theodore, M.; Yang, J.; Arinze, I.J. Acetylation-Deacetylation of the Transcription Factor Nrf2 (Nuclear Factor Erythroid 2-related Factor 2) Regulates Its Transcriptional Activity and Nucleocytoplasmic Localization. J. Boil. Chem. 2010, 286, 7629–7640. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.-C.; Nguyen, T.; Pickett, C.B.; Pacheco, R.; Ciruela, F.; Casadó, V.; Mallol, J.; Gallart, T.; Lluís, C.; Franco, R. Phosphorylation of Nrf2 at Ser-40 by Protein Kinase C Regulates Antioxidant Response Element-mediated Transcription. J. Boil. Chem. 2002, 277, 42769–42774. [Google Scholar] [CrossRef] [Green Version]
- Ki, S.H.; Cho, I.J.; Choi, D.W.; Kim, S.G. Glucocorticoid receptor (GR)-associated SMRT binding to C/EBPbeta TAD and Nrf2 Neh4/5: Role of SMRT recruited to GR in GSTA2 gene repression. Mol. Cell. Biol. 2005, 25, 4150–4165. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Liu, H.; Zhou, J.-S.; Cao, J.-F.; Zhou, X.-B.; Choi, A.M.K.; Chen, Z.-H.; Shen, H. Caveolin-1 Inhibits Expression of Antioxidant Enzymes through Direct Interaction with Nuclear Erythroid 2 p45-related Factor-2 (Nrf2). J. Boil. Chem. 2012, 287, 20922–20930. [Google Scholar] [CrossRef] [Green Version]
- Rojo, A.I.; Medina-Campos, O.N.; Rada, P.; Zúñiga-Toalá, A.; López-Gazcón, A.; Espada, S.; Pedraza-Chaverri, J.; Cuadrado, A. Signaling pathways activated by the phytochemical nordihydroguaiaretic acid contribute to a Keap1-independent regulation of Nrf2 stability: Role of glycogen synthase kinase-3. Free Radic. Boil. Med. 2012, 52, 473–487. [Google Scholar] [CrossRef]
- Kuosmanen, S.M.; Sihvola, V.; Kansanen, E.; Kaikkonen, M.U.; Levonen, A.-L. MicroRNAs mediate the senescence-associated decline of NRF2 in endothelial cells. Redox Boil. 2018, 18, 77–83. [Google Scholar] [CrossRef]
- Chan, J.Y.; Kwong, M. Impaired expression of glutathione synthetic enzyme genes in mice with targeted deletion of the Nrf2 basic-leucine zipper protein. Biochim. Biophys. Acta 2000, 1517, 19–26. [Google Scholar] [CrossRef]
- Hayes, J.D.; Dinkova-Kostova, A.T. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem. Sci. 2014, 39, 199–218. [Google Scholar] [CrossRef]
- Angeli, J.P.F.; Schneider, M.; Proneth, B.; Tyurina, Y.Y.; Tyurin, V.; Hammond, V.J.; Herbach, N.; Aichler, M.; Walch, A.; Eggenhofer, E.; et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell. Boil. 2014, 16, 1180–1191. [Google Scholar] [CrossRef] [Green Version]
- Carlson, B.A.; Tobe, R.; Yefremova, E.; Tsuji, P.A.; Hoffmann, V.J.; Schweizer, U.; Gladyshev, V.N.; Hatfield, D.L.; Conrad, M. Glutathione peroxidase 4 and vitamin E cooperatively prevent hepatocellular degeneration. Redox Boil. 2016, 9, 22–31. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Z.; Guo, S.; Copps, K.; Dong, X.; Kollipara, R.; Rodgers, J.T.; DePinho, R.A.; Puigserver, P.; White, M.F. Foxo1 integrates insulin signaling with mitochondrial function in the liver. Nat. Med. 2009, 15, 1307–1311. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Cui, Y.; Li, M.; Xu, H.; Zuo, J.; Fang, F.; Chang, Y. Cobalt Protoporphyrin Induces HO-1 Expression Mediated Partially by FOXO1 and Reduces Mitochondria-Derived Reactive Oxygen Species Production. PLoS ONE 2013, 8, e80521. [Google Scholar] [CrossRef]
- Miyauchi, T.; Uchida, Y.; Kadono, K.; Hirao, H.; Kawasoe, J.; Watanabe, T.; Ueda, S.; Okajima, H.; Terajima, H.; Uemoto, S. Up-regulation of FOXO1 and reduced inflammation by beta-hydroxybutyric acid are essential diet restriction benefits against liver injury. Proc. Natl. Acad. Sci. USA 2019, 116, 13533–13542. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Zhang, Y.; Xia, F.; Zhang, W.; Chen, P.; Yang, G. Protective effect of silencing Stat1 on high glucose-induced podocytes injury via Forkhead transcription factor O1-regulated the oxidative stress response. BMC Mol. Cell. Boil. 2019, 20, 27. [Google Scholar] [CrossRef] [Green Version]
- Hussong, M.; Borno, S.T.; Kerick, M.; Wunderlich, A.; Franz, A.; Sultmann, H.; Timmermann, B.; Lehrach, H.; Hirsch-Kauffmann, M.; Schweiger, M.R. The bromodomain protein BRD4 regulates the KEAP1/NRF2-dependent oxidative stress response. Cell Death Dis. 2014, 5, e1195. [Google Scholar] [CrossRef] [Green Version]
- Huang, M.; Zhu, L.; Garcia, J.S.; Li, M.X.; Gentles, A.J.; Mitchell, B.S. Brd4 regulates the expression of essential autophagy genes and Keap1 in AML cells. Oncotarget 2018, 9, 11665–11676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuo, H.; Wang, S.; Feng, J.; Liu, X. BRD4 contributes to high-glucose-induced podocyte injury by modulating Keap1/Nrf2/ARE signaling. Biochimie 2019, 165, 100–107. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Symbol | Gene Name | RefSeq Accession Number | Assay ID (in Applied Biosystems) |
|---|---|---|---|
| Brd4 | Bromodomain-containing protein 4 | NM_001100903.1 | Rn01535560_m1 |
| Cat | Catalase | NM_012520.2 | Rn00560930_m1 |
| Foxo1 | Forkhead box O1 | NM_001191846.2 | Rn01494868_m1 |
| Gclc | Glutamate-cysteine ligase, catalytic | NM_012815.2 | Rn00689046_m1 |
| Gpx1 | Glutathione peroxidase 1 | NM_030826.3 | Rn00577994_g1 |
| Gpx4 | Glutathione peroxidase 4 | NM_001039849.2 | Rn00820818_g1 |
| Hmox1 | Heme oxygenase 1 | NM_012580.2 | Rn00561387_m1 |
| Hsd11b1 | Hydroxysteroid 11-beta dehydrogenase 1 | NM_017080.2 | Rn00567167_m1 |
| Hsd11b2 | Hydroxysteroid 11-beta dehydrogenase 2 | NM_017081.2 | Rn04341420_g1 |
| Keap1 | Kelch-like ECH-associated protein 1 | NM_057152.2 | Rn01448220_m1 |
| Nfe2l1(Nrf1) | Nuclear factor, erythroid 2-like 1 | XM_006247236.2 | Rn01452824_m1 |
| Nfe2l2(Nrf2) | Nuclear factor, erythroid 2-like 2 | NM_031789.2 | Rn00582415_m1 |
| Nqo1 | NAD(P)H dehydrogenase, quinone 1 | NM_017000.3 | Rn00566528_m1 |
| Nos2 | Nitric oxide synthase 2, inducible | NM_012611.3 | Rn00561646_m1 |
| Nos3 | Nitric oxide synthase 3, endothelial | NM_021838.2 | Rn02132634_s1 |
| Nr3c1 | Nuclear receptor subfamily 3, group C, member 1 | NM_012576.2 | Rn00561369_m1 |
| Serpina6(Cbg) | Serpin peptidase inhibitor, clade A (alpha-1 antiproteinase, antitrypsin), member 6 | NM_001009663.1 | Rn01517119_m1 |
| Sod1 | Superoxide dismutase 1, soluble | NM_017050.1 | Rn00566938_m1 |
| Sod2 | Superoxide dismutase 2, mitochondrial | NM_017051.2 | Rn00690588_g1 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, H.-J.C.; Yip, T.; Lee, J.K.; Juliani, J.; Sernia, C.; Hill, A.F.; Lavidis, N.A.; Spiers, J.G. Restraint Stress Alters Expression of Glucocorticoid Bioavailability Mediators, Suppresses Nrf2, and Promotes Oxidative Stress in Liver Tissue. Antioxidants 2020, 9, 853. https://doi.org/10.3390/antiox9090853
Chen H-JC, Yip T, Lee JK, Juliani J, Sernia C, Hill AF, Lavidis NA, Spiers JG. Restraint Stress Alters Expression of Glucocorticoid Bioavailability Mediators, Suppresses Nrf2, and Promotes Oxidative Stress in Liver Tissue. Antioxidants. 2020; 9(9):853. https://doi.org/10.3390/antiox9090853
Chicago/Turabian StyleChen, Hsiao-Jou Cortina, Tsz Yip, Johnny K. Lee, Juliani Juliani, Conrad Sernia, Andrew F. Hill, Nickolas A. Lavidis, and Jereme G. Spiers. 2020. "Restraint Stress Alters Expression of Glucocorticoid Bioavailability Mediators, Suppresses Nrf2, and Promotes Oxidative Stress in Liver Tissue" Antioxidants 9, no. 9: 853. https://doi.org/10.3390/antiox9090853