Cadmium-Induced Oxidative Stress: Focus on the Central Nervous System

 ,
,  ,
,  and
and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
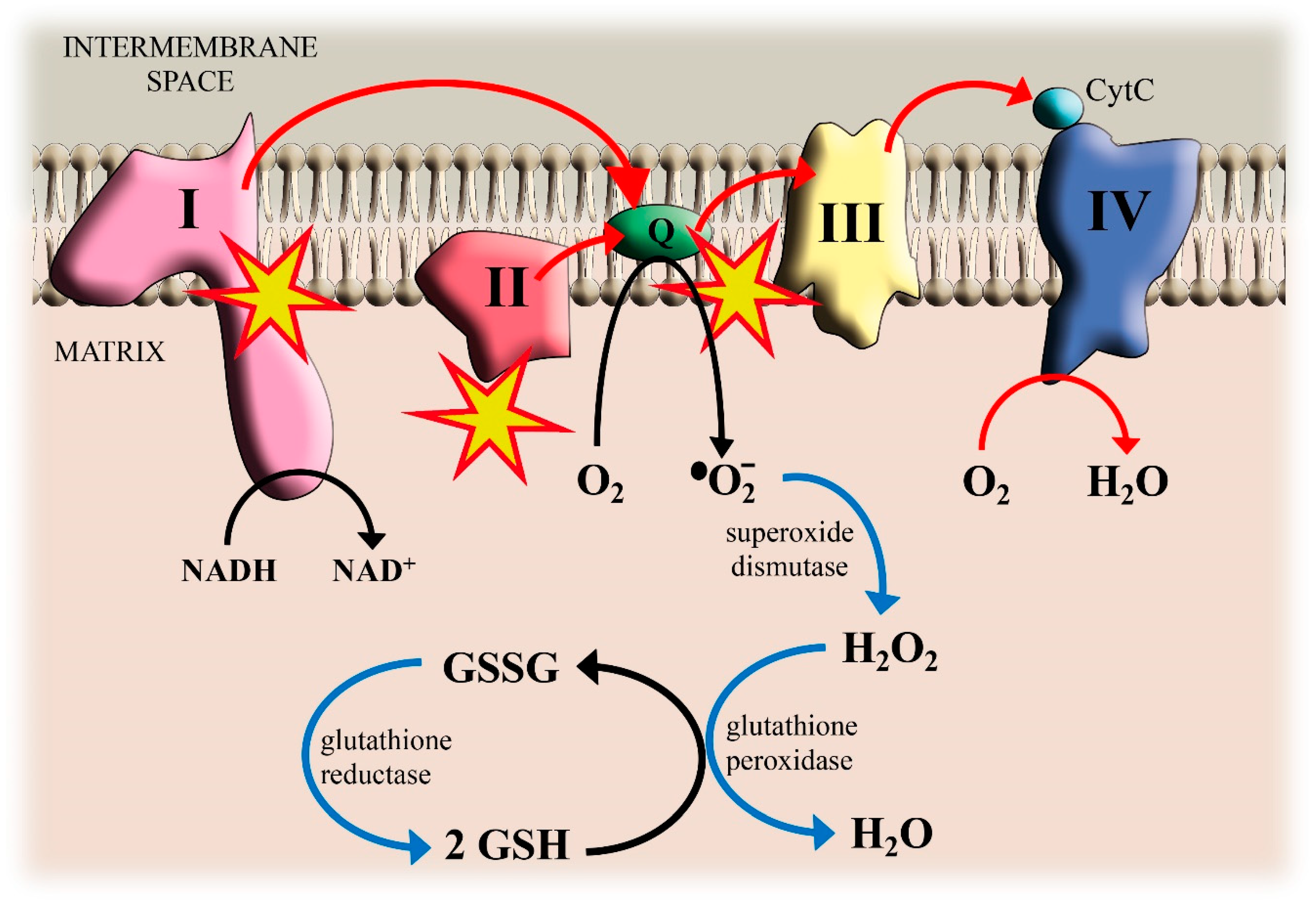
2. Cd, Mitochondrial Electron Transport Chain and ROS Production
3. Cd and Glutathione Depletion
4. Cd and Lipid Peroxidation
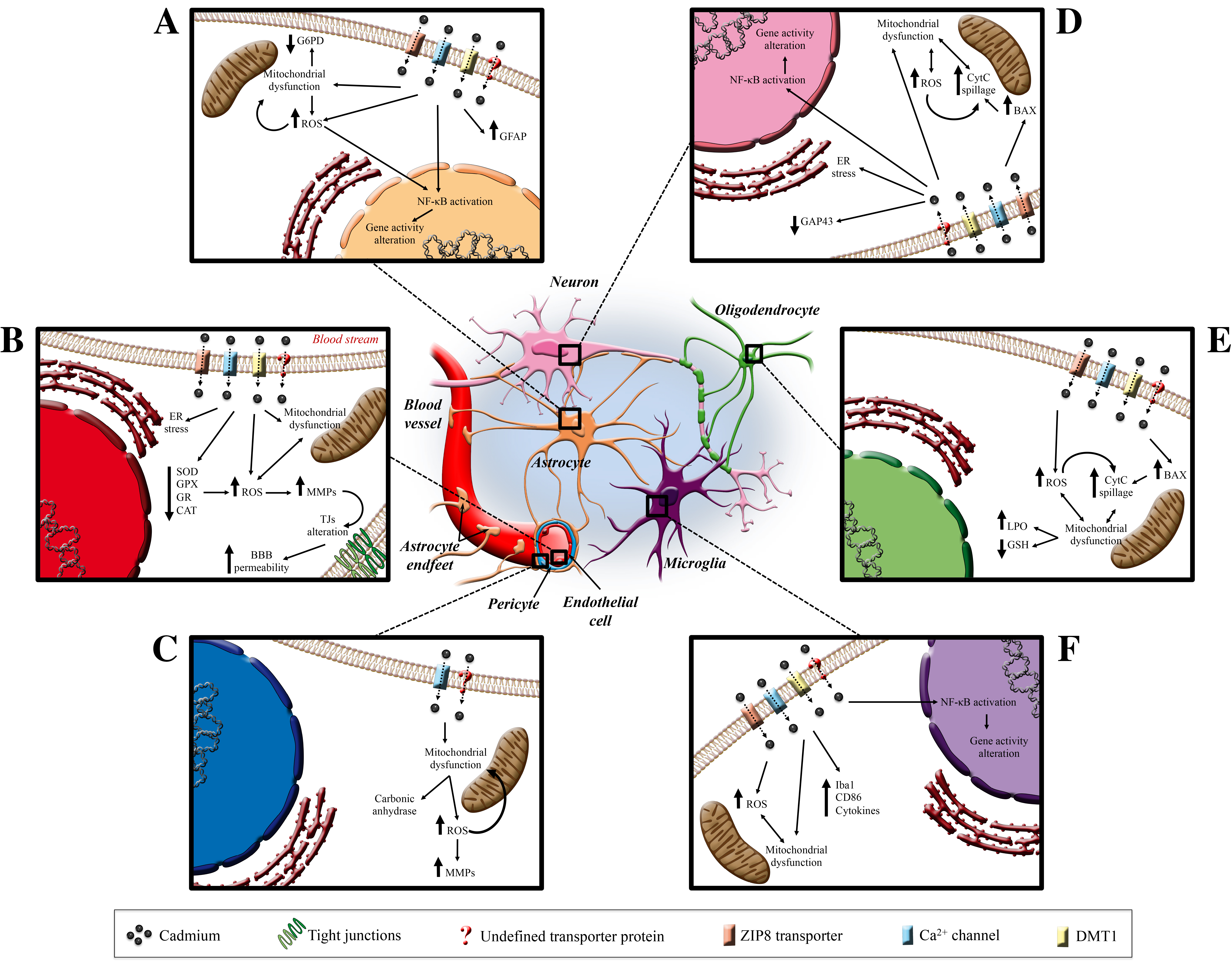
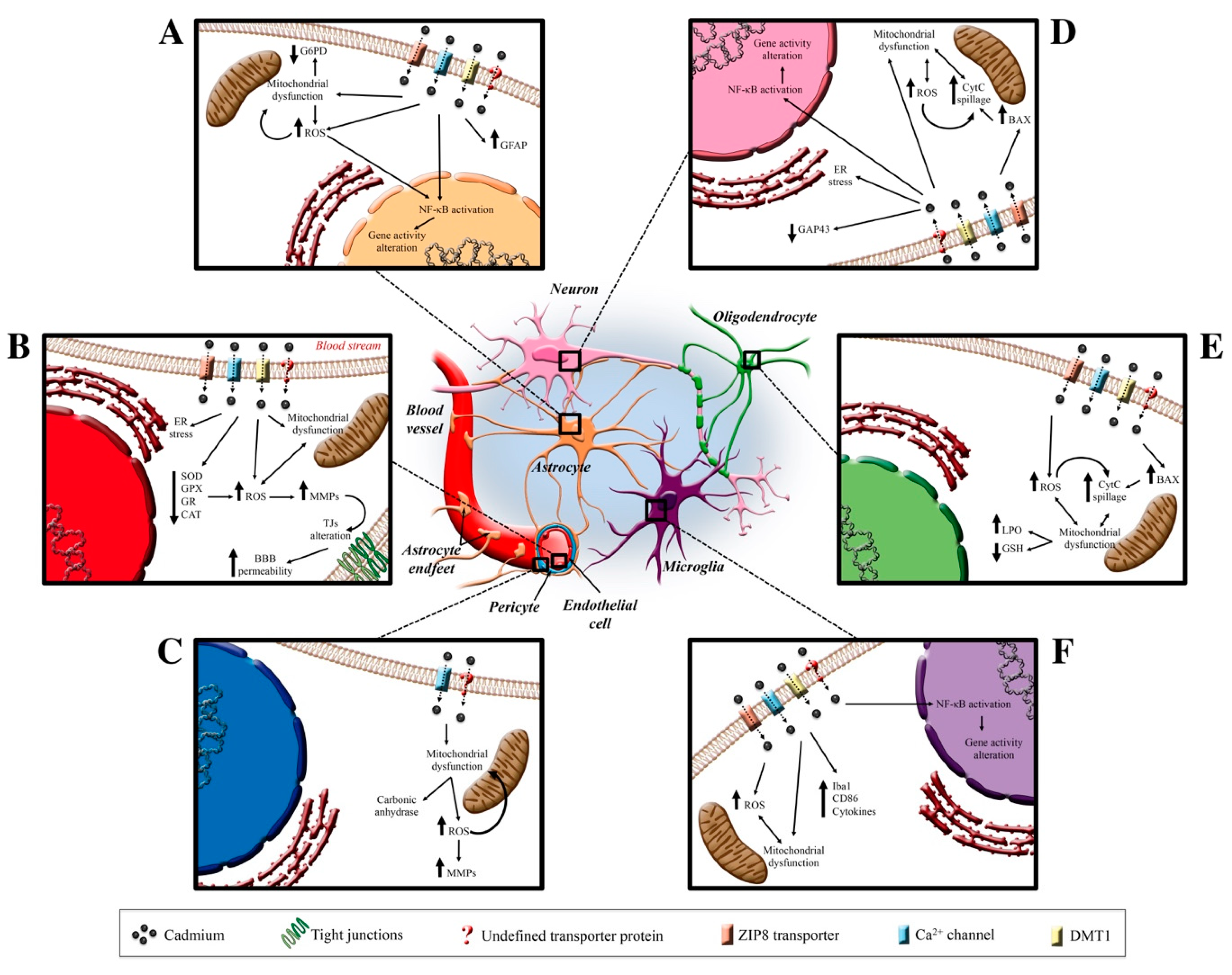
5. Cadmium and the Central Nervous System
5.1. Cadmium and Blood–Brain Barrier
5.2. Cadmium and Pericytes
5.3. Cadmium and Astrocytes
5.4. Cadmium and Microglia
5.5. Cadmium and Neurons
5.6. Cadmium and Oligodendrocytes
6. Conclusions
Funding
Conflicts of Interest
References
- Satarug, S.; Garrett, S.H.; Sens, M.A.; Sens, D.A. Cadmium, Environmental Exposure, and Health Outcomes. Environ. Health Perspect. 2010, 118, 182–190. [Google Scholar] [CrossRef]
- Anetor, J.I. Rising environmental cadmium levels in developing countries: Threat to genome stability and health. Niger J. Physiol. Sci. 2012, 27, 103–115. [Google Scholar] [CrossRef] [PubMed]
- IARC Group. Cadmium and Cadmium Compounds; Arsenic, Metals, Fibres, and Dust; International Agency for Research on Cancer: Lyon, France, 2012; pp. 121–145. [Google Scholar]
- Cuypers, A.; Plusquin, M.; Remans, T.; Jozefczak, M.; Keunen, E.; Gielen, H.; Opdenakker, K.; Nair, A.R.; Munters, E.; Artois, T.J.; et al. Cadmium stress: An oxidative challenge. Biometals 2010, 23, 927–940. [Google Scholar] [CrossRef] [PubMed]
- Ikediobi, C.; Badisa, V.; Ayuk-Takem, L.; Latinwo, L.; West, J. Response of antioxidant enzymes and redox metabolites to cadmium-induced oxidative stress in CRL-1439 normal rat liver cells. Int. J. Mol. Med. 2004. [Google Scholar] [CrossRef]
- Liu, J.; Qu, W.; Kadiiska, M.B. Role of oxidative stress in cadmium toxicity and carcinogenesis. Toxicol. Appl. Pharmacol. 2009, 238, 209–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valko, M.; Morris, H.; Cronin, M. Metals, Toxicity and Oxidative Stress. Curr. Med. Chem. 2005, 12, 1161–1208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halliwell, B.; Gutteridge, J.M. Role of free radicals and catalytic metal ions in human disease: An overview. Methods Enzym. 1990, 186, 1–85. [Google Scholar]
- Wang, X.; Hai, C. Novel insights into redox system and the mechanism of redox regulation. Mol. Biol. Rep. 2016, 43, 607–628. [Google Scholar] [CrossRef]
- Jomova, K.; Valko, M. Advances in metal-induced oxidative stress and human disease. Toxicology 2011, 283, 65–87. [Google Scholar] [CrossRef]
- Rani, A.; Kumar, A.; Lal, A.; Pant, M. Cellular mechanisms of cadmium-induced toxicity: A review. Int. J. Environ. Health Res. 2014, 24, 378–399. [Google Scholar] [CrossRef] [PubMed]
- Vara, D.; Pula, G. Reactive Oxygen Species: Physiological Roles in the Regulation of Vascular Cells. Curr. Mol. Med. 2014, 14, 1103–1125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fridovich, I. Superoxide Radical: An Endogenous Toxicant. Annu. Rev. Pharm. Toxicol. 1983, 23, 239–257. [Google Scholar] [CrossRef] [PubMed]
- Moser, C.C.; Keske, J.M.; Warncke, K.; Farid, R.S.; Dutton, P.L. Nature of biological electron transfer. Nature 1992, 355, 796–802. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nohl, H.; Jordan, W. The mitochondrial site of superoxide formation. Biochem. Biophys. Res. Commun. 1986, 138, 533–539. [Google Scholar] [CrossRef]
- Brand, M.D. Mitochondrial generation of superoxide and hydrogen peroxide as the source of mitochondrial redox signaling. Free Radic. Biol. Med. 2016, 100, 14–31. [Google Scholar] [CrossRef] [PubMed]
- Wong, H.-S.; Benoit, B.; Brand, M.D. Mitochondrial and cytosolic sources of hydrogen peroxide in resting C2C12 myoblasts. Free Radic. Biol. Med. 2019, 130, 140–150. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Ren, X.; Hu, X.; Zhou, L.; Zhang, C.; Zhang, M. Cadmium-induced apoptosis through reactive oxygen species-mediated mitochondrial oxidative stress and the JNK signaling pathway in TM3 cells, a model of mouse Leydig cells. Toxicol. Appl. Pharmacol. 2019, 368, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, C.; Ferreira de Oliveira, J.M.P.; Pinho, F.; Bastos, V.; Oliveira, H.; Peixoto, F.; Santos, C. Biochemical and transcriptional analyses of cadmium-induced mitochondrial dysfunction and oxidative stress in human osteoblasts. J. Toxicol. Environ. Health Part A 2018, 81, 705–717. [Google Scholar] [CrossRef] [PubMed]
- Alkharashi, N.A.O.; Periasamy, V.S.; Athinarayanan, J.; Alshatwi, A.A. Cadmium triggers mitochondrial oxidative stress in human peripheral blood lymphocytes and monocytes: Analysis using in vitro and system toxicology approaches. J. Trace Elem. Med. Biol. 2017, 42, 117–128. [Google Scholar] [CrossRef] [PubMed]
- Skipper, A.; Sims, J.; Yedjou, C.; Tchounwou, P. Cadmium Chloride Induces DNA Damage and Apoptosis of Human Liver Carcinoma Cells via Oxidative Stress. Int. J. Environ. Res. Public Health 2016, 13, 88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacobs, E.E.; Jacobs, M.; Sandy, D.R.; Bradley, L.B. Uncoupling of oxidative phosphorylation by cadmium ion. J. Biol. Chem. 1956, 223, 147–156. [Google Scholar]
- Müller, L.; Ohnesorge, F.K. Cadmium-induced alteration of the energy level in isolated hepatocytes. Toxicology 1984, 31, 297–306. [Google Scholar] [CrossRef]
- Cameron, I.; McNamee, P.M.; Markham, A.; Morgan, R.M.; Wood, M. The effects of cadmium on succinate and NADH-linked substrate oxidations in rat hepatic mitochondria. J. Appl. Toxicol. 1986, 6, 325–330. [Google Scholar] [CrossRef] [PubMed]
- Hirst, J.; King, M.S.; Pryde, K.R. The production of reactive oxygen species by complex I. Biochem. Soc. Trans. 2008, 36, 976–980. [Google Scholar] [CrossRef] [PubMed]
- Doughan, A.K.; Harrison, D.G.; Dikalov, S.I. Molecular Mechanisms of Angiotensin II–Mediated Mitochondrial Dysfunction: Linking Mitochondrial Oxidative Damage and Vascular Endothelial Dysfunction. Circ. Res. 2008, 102, 488–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dorta, D.J.; Leite, S.; DeMarco, K.C.; Prado, I.M.R.; Rodrigues, T.; Mingatto, F.E.; Uyemura, S.A.; Santos, A.C.; Curti, C. A proposed sequence of events for cadmium-induced mitochondrial impairment. J. Inorg. Biochem. 2003, 97, 251–257. [Google Scholar] [CrossRef]
- Kurochkin, I.O.; Etzkorn, M.; Buchwalter, D.; Leamy, L.; Sokolova, I.M. Top-down control analysis of the cadmium effects on molluscan mitochondria and the mechanisms of cadmium-induced mitochondrial dysfunction. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 2011, 300, R21–R31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Fiskum, G.; Schubert, D. Generation of reactive oxygen species by the mitochondrial electron transport chain. J. Neurochem. 2002, 80, 780–787. [Google Scholar] [CrossRef]
- Belyaeva, E.A. Respiratory complex II in mitochondrial dysfunction-mediated cytotoxicity: Insight from cadmium. J. Trace Elem. Med. Biol. 2018, 50, 80–92. [Google Scholar] [CrossRef]
- Andreyev, A.Y.; Kushnareva, Y.E.; Starkov, A.A. Mitochondrial metabolism of reactive oxygen species. Biochemistry 2005, 70, 15. [Google Scholar] [CrossRef] [PubMed]
- Starkov, A.A.; Fiskum, G. Regulation of brain mitochondrial H2O2 production by membrane potential and NAD(P)H redox state: ROS production by brain mitochondria. J. Neurochem. 2003, 86, 1101–1107. [Google Scholar] [CrossRef] [PubMed]
- Grivennikova, V.G.; Vinogradov, A.D. Generation of superoxide by the mitochondrial Complex I. Biochim. Biophys. Acta (BBA) Bioenergy 2006, 1757, 553–561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miccadei, S.; Floridi, A. Sites of inhibition of mitochondrial electron transport by cadmium. Chem.-Biol. Interact. 1993, 89, 159–167. [Google Scholar] [CrossRef]
- Wang, Y.; Fang, J.; Leonard, S.S.; Krishna Rao, K.M. Cadmium inhibits the electron transfer chain and induces Reactive Oxygen Species. Free Radic. Biol. Med. 2004, 36, 1434–1443. [Google Scholar] [CrossRef] [PubMed]
- Belyaeva, E.A.; Sokolova, T.V.; Emelyanova, L.V.; Zakharova, I.O. Mitochondrial Electron Transport Chain in Heavy Metal-Induced Neurotoxicity: Effects of Cadmium, Mercury, and Copper. Sci. World J. 2012, 2012, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belyaeva, E.A.; Dymkowska, D.; Więckowski, M.R.; Wojtczak, L. Reactive oxygen species produced by the mitochondrial respiratory chain are involved in Cd2+-induced injury of rat ascites hepatoma AS-30D cells. Biochim. Biophys. Acta (BBA) Bioenergy 2006, 1757, 1568–1574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Messner, K.R.; Imlay, J.A. Mechanism of Superoxide and Hydrogen Peroxide Formation by Fumarate Reductase, Succinate Dehydrogenase, and Aspartate Oxidase. J. Biol. Chem. 2002, 277, 42563–42571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinlan, C.L.; Orr, A.L.; Perevoshchikova, I.V.; Treberg, J.R.; Ackrell, B.A.; Brand, M.D. Mitochondrial Complex II Can Generate Reactive Oxygen Species at High Rates in Both the Forward and Reverse Reactions. J. Biol. Chem. 2012, 287, 27255–27264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grivennikova, V.G.; Kozlovsky, V.S.; Vinogradov, A.D. Respiratory complex II: ROS production and the kinetics of ubiquinone reduction. Biochim. Biophys. Acta (BBA) Bioenergy 2017, 1858, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Votyakova, T.V.; Reynolds, I.J. ΔΨm-Dependent and -independent production of reactive oxygen species by rat brain mitochondria: ROS and rat brain mitochondria. J. Neurochem. 2008, 79, 266–277. [Google Scholar] [CrossRef] [PubMed]
- Lambert, A.J.; Brand, M.D. Superoxide production by NADH:ubiquinone oxidoreductase (complex I) depends on the pH gradient across the mitochondrial inner membrane. Biochem. J. 2004, 382, 511–517. [Google Scholar] [CrossRef] [PubMed]
- Muller, F.L.; Liu, Y.; Abdul-Ghani, M.A.; Lustgarten, M.S.; Bhattacharya, A.; Jang, Y.C.; Van Remmen, H. High rates of superoxide production in skeletal-muscle mitochondria respiring on both complex I- and complex II-linked substrates. Biochem. J. 2008, 409, 491–499. [Google Scholar] [CrossRef] [PubMed]
- Dröse, S.; Hanley, P.J.; Brandt, U. Ambivalent effects of diazoxide on mitochondrial ROS production at respiratory chain complexes I and III. Biochim. Biophys. Acta (BBA) Gen. Subj. 2009, 1790, 558–565. [Google Scholar] [CrossRef]
- Dröse, S.; Bleier, L.; Brandt, U. A Common Mechanism Links Differently Acting Complex II Inhibitors to Cardioprotection: Modulation of Mitochondrial Reactive Oxygen Species Production. Mol. Pharm. 2011, 79, 814–822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belyaeva, E.A.; Korotkov, S.M. Mechanism of primary Cd2+-induced rat liver mitochondria dysfunction: Discrete modes of Cd2+ action on calcium and thiol-dependent domains. Toxicol. Appl. Pharmacol. 2003, 192, 56–68. [Google Scholar] [CrossRef]
- Cannino, G.; Ferruggia, E.; Luparello, C.; Rinaldi, A.M. Effects of cadmium chloride on some mitochondria-related activity and gene expression of human MDA-MB231 breast tumor cells. J. Inorg. Biochem. 2008, 102, 1668–1676. [Google Scholar] [CrossRef]
- Cannino, G.; Ferruggia, E.; Luparello, C.; Rinaldi, A.M. Mitochondrial compartment: A possible target of cadmium effects on breast epithelial cells. Mol. Cell. Biochem. 2009, 328, 75–84. [Google Scholar] [CrossRef]
- Morici, G.; Agnello, M.; Spagnolo, F.; Roccheri, M.C.; Liegro, C.M.D.; Rinaldi, A.M. Confocal microscopy study of the distribution, content and activity of mitochondria during Paracentrotus lividus development. J. Microsc. 2007, 228, 165–173. [Google Scholar] [CrossRef]
- Al-Ghafari, A.; Elmorsy, E.; Fikry, E.; Alrowaili, M.; Carter, W.G. The heavy metals lead and cadmium are cytotoxic to human bone osteoblasts via induction of redox stress. PLoS ONE 2019, 14, e0225341. [Google Scholar] [CrossRef] [Green Version]
- Adiele, R.C.; Stevens, D.; Kamunde, C. Differential Inhibition of Electron Transport Chain Enzyme Complexes by Cadmium and Calcium in Isolated Rainbow Trout (Oncorhynchus mykiss) Hepatic Mitochondria. Toxicol. Sci. 2012, 127, 110–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chatterjee, S.; Kundu, S.; Bhattacharyya, A. Mechanism of cadmium induced apoptosis in the immunocyte. Toxicol. Lett. 2008, 177, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.; Lim, S. A rapid and transient ROS generation by cadmium triggers apoptosis via caspase-dependent pathway in HepG2 cells and this is inhibited through N-acetylcysteine-mediated catalase upregulation. Toxicol. Appl. Pharmacol. 2006, 212, 212–223. [Google Scholar] [CrossRef] [PubMed]
- Santamaría, G.; Martínez-Diez, M.; Fabregat, I.; Cuezva, J.M. Efficient execution of cell death in non-glycolytic cells requires the generation of ROS controlled by the activity of mitochondrial H + -ATP synthase. Carcinogenesis 2006, 27, 925–935. [Google Scholar] [CrossRef] [Green Version]
- Brown, G.C.; Borutaite, V. Regulation of apoptosis by the redox state of cytochrome c. Biochim. Biophys. Acta (BBA) Bioenergy 2008, 1777, 877–881. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Lesnefsky, E.J. Depletion of cardiolipin and cytochrome c during ischemia increases hydrogen peroxide production from the electron transport chain. Free Radic. Biol. Med. 2006, 40, 976–982. [Google Scholar] [CrossRef]
- Nemmiche, S. Oxidative Signaling Response to Cadmium Exposure. Toxicol. Sci. 2017, 156, 4–10. [Google Scholar] [CrossRef] [Green Version]
- Sies, H. Glutathione and its role in cellular functions. Free Radic. Biol. Med. 1999, 27, 916–921. [Google Scholar] [CrossRef]
- Deponte, M. Glutathione catalysis and the reaction mechanisms of glutathione-dependent enzymes. Biochim. Biophys. Acta (BBA) Gen. Subj. 2013, 1830, 3217–3266. [Google Scholar] [CrossRef] [Green Version]
- Oestreicher, J.; Morgan, B. Glutathione: Subcellular distribution and membrane transport. Biochem. Cell Biol. 2019, 97, 270–289. [Google Scholar] [CrossRef] [Green Version]
- Schafer, F.Q.; Buettner, G.R. Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radic. Biol. Med. 2001, 30, 1191–1212. [Google Scholar] [CrossRef]
- Deponte, M. The Incomplete Glutathione Puzzle: Just Guessing at Numbers and Figures? Antioxid. Redox Signal. 2017, 27, 1130–1161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singhal, R.K.; Anderson, M.E.; Meister, A. Glutathione, a first line of defense against cadmium toxicity. FASEB J. 1987, 1, 220–223. [Google Scholar] [CrossRef]
- Klaassen, C.D.; Bracken, M.; Dudley, R.E.; Goering, P.L.; Hazelton, G.A.; Hjelle, J.J. Role of Sulfhydryls in the Hepatotoxicity of Organic and Metallic Compounds. Toxicol. Sci. 1985, 5, 806–815. [Google Scholar] [CrossRef]
- Chan, H.M.; Cherian, M.G. Protective roles of metallothionein and glutathione in hepatotoxicity of cadmium. Toxicology 1992, 72, 281–290. [Google Scholar] [CrossRef]
- Tandon, S.K.; Singh, S.; Prasad, S.; Khandekar, K.; Dwivedi, V.K.; Chatterjee, M.; Mathur, N. Reversal of cadmium induced oxidative stress by chelating agent, antioxidant or their combination in rat. Toxicol. Lett. 2003, 145, 211–217. [Google Scholar] [CrossRef]
- Zhang, Q.; Zhang, C.; Ge, J.; Lv, M.-W.; Talukder, M.; Guo, K.; Li, Y.; Li, J.-L. Ameliorative Effects of Resveratrol against Cadmium-induced Nephrotoxicity via Modulating Nuclear Xenobiotic Receptors Response and PINK1/Parkin-mediated Mitophagy. Food Funct. 2020, 11, 1856–1868. [Google Scholar] [CrossRef]
- Almazan, G.; Liu, H.-N.; Khorchid, A.; Sundararajan, S.; Martinez-Bermudez, A.K.; Chemtob, S. Exposure of developing oligodendrocytes to cadmium causes HSP72 induction, free radical generation, reduction in glutathione levels, and cell death. Free Radic. Biol. Med. 2000, 29, 858–869. [Google Scholar] [CrossRef]
- Figueiredo-Pereira, M.E.; Yakushin, S.; Cohen, G. Disruption of the Intracellular Sulfhydryl Homeostasis by Cadmium-induced Oxidative Stress Leads to Protein Thiolation and Ubiquitination in Neuronal Cells. J. Biol. Chem. 1998, 273, 12703–12709. [Google Scholar] [CrossRef] [Green Version]
- Im, J.-Y.; Paik, S.-G.; Han, P.-L. Cadmium-induced astroglial death proceeds via glutathione depletion. J. Neurosci. Res. 2006, 83, 301–308. [Google Scholar] [CrossRef]
- Karmakar, R.; Banik, S.; Bandyopadhyay, S.; Chatterjee, M. Cadmium-induced alterations of hepatic lipid peroxidation, glutathione S-transferase activity and reduced glutathione level and their possible correlation with chromosomal aberration in mice: A time course study. Mutat. Res./Fundam. Mol. Mech. Mutagen. 1998, 397, 183–190. [Google Scholar] [CrossRef]
- Sarkar, S.; Yadav, P.; Trivedi, R.; Bansal, A.K.; Bhatnagar, D. Cadmium-induced Lipid Peroxidation and the Status of the Antioxidant System in Rat Tissues. J. Trace Elem. Med. Biol. 1995, 9, 144–149. [Google Scholar] [CrossRef]
- López, E.; Arce, C.; Oset-Gasque, M.J.; Cañadas, S.; González, M.P. Cadmium induces reactive oxygen species generation and lipid peroxidation in cortical neurons in culture. Free Radic. Biol. Med. 2006, 40, 940–951. [Google Scholar] [CrossRef] [PubMed]
- Dalle-Donne, I.; Aldini, G.; Carini, M.; Colombo, R.; Rossi, R.; Milzani, A. Protein carbonylation, cellular dysfunction, and disease progression. J. Cell. Mol. Med. 2006, 10, 389–406. [Google Scholar] [CrossRef] [PubMed]
- Spiteller, G. Linoleic acid peroxidation—The dominant lipid peroxidation process in low density lipoprotein—And its relationship to chronic diseases. Chem. Phys. Lipids 1998, 95, 105–162. [Google Scholar] [CrossRef]
- Domingues, R.M.; Domingues, P.; Melo, T.; Pérez-Sala, D.; Reis, A.; Spickett, C.M. Lipoxidation adducts with peptides and proteins: Deleterious modifications or signaling mechanisms? J. Proteom. 2013, 92, 110–131. [Google Scholar] [CrossRef]
- Fedorova, M.; Bollineni, R.C.; Hoffmann, R. Protein carbonylation as a major hallmark of oxidative damage: Update of analytical strategies: Protein carbonylation: An analytical update. Mass Spec. Rev. 2014, 33, 79–97. [Google Scholar] [CrossRef]
- Spickett, C.M. The lipid peroxidation product 4-hydroxy-2-nonenal: Advances in chemistry and analysis. Redox Biol. 2013, 1, 145–152. [Google Scholar] [CrossRef] [Green Version]
- Negre-Salvayre, A.; Coatrieux, C.; Ingueneau, C.; Salvayre, R. Advanced lipid peroxidation end products in oxidative damage to proteins. Potential role in diseases and therapeutic prospects for the inhibitors. Br. J. Pharmacol. 2008, 153, 6–20. [Google Scholar] [CrossRef] [Green Version]
- Uchida, K. Current Status of Acrolein as a Lipid Peroxidation Product. Trends Cardiovasc. Med. 1999, 9, 109–113. [Google Scholar] [CrossRef]
- Perluigi, M.; Coccia, R.; Butterfield, D.A. 4-Hydroxy-2-Nonenal, a Reactive Product of Lipid Peroxidation, and Neurodegenerative Diseases: A Toxic Combination Illuminated by Redox Proteomics Studies. Antioxid. Redox Signal. 2012, 17, 1590–1609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattson, M.P. Roles of the lipid peroxidation product 4-hydroxynonenal in obesity, the metabolic syndrome, and associated vascular and neurodegenerative disorders. Exp. Gerontol. 2009, 44, 625–633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramana, K.V.; Srivastava, S.; Singhal, S.S. Lipid Peroxidation Products in Human Health and Disease 2019. Oxidative Med. Cell. Longev. 2019, 2019, 7147235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emmi, G.; Bettiol, A.; Silvestri, E.; Di Scala, G.; Becatti, M.; Fiorillo, C.; Prisco, D. Vascular Behçet’s syndrome: An update. Intern. Emerg. Med. 2019, 14, 645–652. [Google Scholar] [CrossRef]
- Guo, L.; Chen, Z.; Cox, B.E.; Amarnath, V.; Epand, R.F.; Epand, R.M.; Davies, S.S. Phosphatidylethanolamines Modified by γ-Ketoaldehyde (γKA) Induce Endoplasmic Reticulum Stress and Endothelial Activation. J. Biol. Chem. 2011, 286, 18170–18180. [Google Scholar] [CrossRef] [Green Version]
- Reis, A.; Domingues, M.R.M.; Amado, F.M.L.; Ferrer-Correia, A.J.; Domingues, P. Radical peroxidation of palmitoyl-lineloyl-glycerophosphocholine liposomes: Identification of long-chain oxidised products by liquid chromatography–tandem mass spectrometry. J. Chromatogr. B 2007, 855, 186–199. [Google Scholar] [CrossRef]
- Pinot, F.; Kreps, S.E.; Bachelet, M.; Hainaut, P.; Bakonyi, M.; Polla, B.S. Cadmium in the Environment: Sources, Mechanisms of Biotoxicity, and Biomarkers. Rev. Environ. Health 2000, 15, 299–323. [Google Scholar] [CrossRef]
- Stohs, S. Oxidative mechanisms in the toxicity of metal ions. Free Radic. Biol. Med. 1995, 18, 321–336. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Cao, J.; Chen, D.; Liu, X.; Lu, H.; Liu, Z. Role of Oxidative Stress, Apoptosis, and Intracellular Homeostasis in Primary Cultures of Rat Proximal Tubular Cells Exposed to Cadmium. Biol. Trace Elem. Res. 2009, 127, 53–68. [Google Scholar] [CrossRef]
- Ognjanović, B.I.; Marković, S.D.; Ðorđević, N.Z.; Trbojević, I.S.; Štajn, A.Š.; Saičić, Z.S. Cadmium-induced lipid peroxidation and changes in antioxidant defense system in the rat testes: Protective role of coenzyme Q10 and Vitamin E. Reprod. Toxicol. 2010, 29, 191–197. [Google Scholar] [CrossRef]
- Patra, R.C.; Rautray, A.K.; Swarup, D. Oxidative Stress in Lead and Cadmium Toxicity and Its Amelioration. Vet. Med. Int. 2011, 2011, 457327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valko, M.; Rhodes, C.J.; Moncol, J.; Izakovic, M.; Mazur, M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem.-Biol. Interact. 2006, 160, 1–40. [Google Scholar] [CrossRef] [PubMed]
- Buha, A.; Bulat, Z.; Đukić-Ćosić, D.; Matović, V. Effects of oral and intraperitoneal magnesium treatment against cadmium-induced oxidative stress in plasma of rats. Arch. Ind. Hyg. Toxicol. 2012, 63, 247–254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elmallah, M.; Elkhadragy, M.; Al-Olayan, E.; Abdel Moneim, A. Protective Effect of Fragaria ananassa Crude Extract on Cadmium-Induced Lipid Peroxidation, Antioxidant Enzymes Suppression, and Apoptosis in Rat Testes. Int. J. Mol. Sci. 2017, 18, 957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohajeri, M.; Rezaee, M.; Sahebkar, A. Cadmium-induced toxicity is rescued by curcumin: A review. BioFactors 2017, 43, 645–661. [Google Scholar] [CrossRef] [PubMed]
- Mężyńska, M.; Brzóska, M.; Rogalska, J.; Piłat-Marcinkiewicz, B. Extract from Aronia melanocarpa L. Berries Prevents Cadmium-Induced Oxidative Stress in the Liver: A Study in a Rat Model of Low-Level and Moderate Lifetime Human Exposure to this Toxic Metal. Nutrients 2018, 11, 21. [Google Scholar] [CrossRef] [Green Version]
- Ayala, A.; Muñoz, M.F.; Argüelles, S. Lipid Peroxidation: Production, Metabolism, and Signaling Mechanisms of Malondialdehyde and 4-Hydroxy-2-Nonenal. Oxidative Med. Cell. Longev. 2014, 2014, 1–31. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Shi, L.; Li, J.; Li, L.; Wang, H.; Yang, H. Long-term cadmium exposure promoted breast cancer cell migration and invasion by up-regulating TGIF. Ecotoxicol. Environ. Saf. 2019, 175, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Darwish, W.S.; Chen, Z.; Li, Y.; Wu, Y.; Chiba, H.; Hui, S.-P. Identification of cadmium-produced lipid hydroperoxides, transcriptomic changes in antioxidant enzymes, xenobiotic transporters, and pro-inflammatory markers in human breast cancer cells (MCF7) and protection with fat-soluble vitamins. Environ. Sci. Pollut. Res. 2020, 27, 1978–1990. [Google Scholar] [CrossRef] [PubMed]
- Gaschler, M.M.; Stockwell, B.R. Lipid peroxidation in cell death. Biochem. Biophys. Res. Commun. 2017, 482, 419–425. [Google Scholar] [CrossRef] [PubMed]
- Hart, R.P.; Rose, C.S.; Hamer, R.M. Neuropsychological effects of occupational exposure to cadmium. J. Clin. Exp. Neuropsychol. 1989, 11, 933–943. [Google Scholar] [CrossRef] [PubMed]
- Viaene, M.K.; Roels, H.A.; Leenders, J.; De Groof, M.; Swerts, L.J.; Lison, D.; Masschelein, R. Cadmium: A possible etiological factor in peripheral polyneuropathy. NeuroToxicology 1999, 20, 7–16. [Google Scholar] [PubMed]
- Mèndez-Armenta, M.; Villeda-Hernàndez, J.; Barroso-Moguel, R.; Nava-Rùız, C.; Jiménez-Capdeville, M.E.; Rìos, C. Brain regional lipid peroxidation and metallothionein levels of developing rats exposed to cadmium and dexamethasone. Toxicol. Lett. 2003, 144, 151–157. [Google Scholar] [CrossRef]
- Monroe, R.; Halvorsen, S. Cadmium blocks receptor-mediated Jak/STAT signaling in neurons by oxidative stress. Free Radic. Biol. Med. 2006, 41, 493–502. [Google Scholar] [CrossRef]
- Patra, R.C.; Swarup, D.; Dwivedi, S.K. Antioxidant effects of a tocopherol, ascorbic acid and L-methionine on lead induced oxidative stress to the liver, kidney and brain in rats. Toxicology 2001, 162, 81–88. [Google Scholar] [CrossRef]
- Nazima, B.; Manoharan, V.; Miltonprabu, S. Oxidative stress induced by cadmium in the plasma, erythrocytes and lymphocytes of rats: Attenuation by grape seed proanthocyanidins. Hum. Exp. Toxicol. 2016, 35, 428–447. [Google Scholar] [CrossRef]
- Patra, R.C.; Swarup, D.; Senapati, S.K. Effects of cadmium on lipid peroxides and superoxide dismutase in hepatic, renal and testicular tissue of rats. Vet. Hum. Toxicol. 1999, 41, 65–67. [Google Scholar] [PubMed]
- Ganguly, K.; Levänen, B.; Palmberg, L.; Åkesson, A.; Lindén, A. Cadmium in tobacco smokers: A neglected link to lung disease? Eur. Respir. Rev. 2018, 27, 170122. [Google Scholar] [CrossRef] [PubMed]
- Kundu, S.; Sengupta, S.; Chatterjee, S.; Mitra, S.; Bhattacharyya, A. Cadmium induces lung inflammation independent of lung cell proliferation: A molecular approach. J. Inflamm. 2009, 6, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johri, N.; Jacquillet, G.; Unwin, R. Heavy metal poisoning: The effects of cadmium on the kidney. Biometals 2010, 23, 783–792. [Google Scholar] [CrossRef]
- Wallin, M.; Sallsten, G.; Lundh, T.; Barregard, L. Low-level cadmium exposure and effects on kidney function. Occup. Environ. Med. 2014, 71, 848–854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arroyo, V.S.; Flores, K.M.; Ortiz, L.B.; Gómez-Quiroz, L.E.; Gutiérrez-Ruiz, M.C. Liver and Cadmium Toxicity. J. Drug Metab. Toxicol. 2012, S5, 001. [Google Scholar] [CrossRef] [Green Version]
- Djordjevic, V.R.; Wallace, D.R.; Schweitzer, A.; Boricic, N.; Knezevic, D.; Matic, S.; Grubor, N.; Kerkez, M.; Radenkovic, D.; Bulat, Z.; et al. Environmental cadmium exposure and pancreatic cancer: Evidence from case control, animal and in vitro studies. Environ. Int. 2019, 128, 353–361. [Google Scholar] [CrossRef] [PubMed]
- Gluhcheva, Y.; Ivanova, J.; Ganeva, S.; Mitewa, M. Effects of Cadmium and Monensin on Spleen of Mice, Subjected to Subacute Cadmium Intoxication. J. Toxicol. Environ. Health Part A 2013, 76, 328–332. [Google Scholar] [CrossRef]
- Siu, E.R.; Mruk, D.D.; Porto, C.S.; Cheng, C.Y. Cadmium-induced testicular injury. Toxicol. Appl. Pharmacol. 2009, 238, 240–249. [Google Scholar] [CrossRef] [Green Version]
- Kazantzis, G. Cadmium, osteoporosis and calcium metabolism. Biometals 2004, 17, 493–498. [Google Scholar] [CrossRef]
- Ramírez-Bajo, M.J.; de Atauri, P.; Ortega, F.; Westerhoff, H.V.; Gelpí, J.L.; Centelles, J.J.; Cascante, M. Effects of Cadmium and Mercury on the Upper Part of Skeletal Muscle Glycolysis in Mice. PLoS ONE 2014, 9, e80018. [Google Scholar] [CrossRef] [Green Version]
- Tellez-Plaza, M.; Jones, M.R.; Dominguez-Lucas, A.; Guallar, E.; Navas-Acien, A. Cadmium Exposure and Clinical Cardiovascular Disease: A Systematic Review. Curr. Atheroscler. Rep. 2013, 15, 356. [Google Scholar] [CrossRef]
- Wang, B.; Du, Y. Cadmium and Its Neurotoxic Effects. Oxidative Med. Cell. Longev. 2013, 2013, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Abbott, N.J.; Friedman, A. Overview and introduction: The blood-brain barrier in health and disease: Blood-Brain Barrier in Health and Disease. Epilepsia 2012, 53, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Moretti, R.; Pansiot, J.; Bettati, D.; Strazielle, N.; Ghersi-Egea, J.-F.; Damante, G.; Fleiss, B.; Titomanlio, L.; Gressens, P. Blood-brain barrier dysfunction in disorders of the developing brain. Front. Neurosci. 2015, 9, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiss, N.; Miller, F.; Cazaubon, S.; Couraud, P.-O. The blood-brain barrier in brain homeostasis and neurological diseases. Biochim. Biophys. Acta (BBA) Biomembr. 2009, 1788, 842–857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thévenod, F.; Fels, J.; Lee, W.-K.; Zarbock, R. Channels, transporters and receptors for cadmium and cadmium complexes in eukaryotic cells: Myths and facts. Biometals 2019, 32, 469–489. [Google Scholar] [CrossRef] [PubMed]
- Shuvaev, A.N.; Kuvacheva, N.V.; Morgun, A.V.; Khilazheva, E.D.; Salmina, A.B. The Role of Ion Channels Expressed in Cerebral Endothelial Cells in the Functional Integrity of the Blood-Brain Barrier. Sovrem Teh. Med. 2016, 8, 241–250. [Google Scholar] [CrossRef]
- Shukla, A.; Shukla, G.S.; Srimal, R. Cadmium-induced alterations in blood- brain barrier permeability and its possible correlation with decreased microvessel antioxidant potential in rat. Hum. Exp. Toxicol. 1996, 15, 400–405. [Google Scholar] [CrossRef] [PubMed]
- Viaene, M.K. Neurobehavioural effects of occupational exposure to cadmium: A cross sectional epidemiological study. Occup. Environ. Med. 2000, 57, 19–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gasche, Y.; Copin, J.-C.; Sugawara, T.; Fujimura, M.; Chan, P.H. Matrix Metalloproteinase Inhibition Prevents Oxidative Stress-Associated Blood–Brain Barrier Disruption after Transient Focal Cerebral Ischemia. J. Cereb. Blood Flow Metab. 2001, 21, 1393–1400. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Estrada, E.Y.; Thompson, J.F.; Liu, W.; Rosenberg, G.A. Matrix Metalloproteinase-Mediated Disruption of Tight Junction Proteins in Cerebral Vessels is Reversed by Synthetic Matrix Metalloproteinase Inhibitor in Focal Ischemia in Rat. J. Cereb. Blood Flow Metab. 2007, 27, 697–709. [Google Scholar] [CrossRef]
- Tobwala, S.; Wang, H.-J.; Carey, J.; Banks, W.; Ercal, N. Effects of Lead and Cadmium on Brain Endothelial Cell Survival, Monolayer Permeability, and Crucial Oxidative Stress Markers in an in Vitro Model of the Blood-Brain Barrier. Toxics 2014, 2, 258–275. [Google Scholar] [CrossRef] [Green Version]
- Branca, J.J.V.; Maresca, M.; Morucci, G.; Mello, T.; Becatti, M.; Pazzagli, L.; Colzi, I.; Gonnelli, C.; Carrino, D.; Paternostro, F.; et al. Effects of Cadmium on ZO-1 Tight Junction Integrity of the Blood Brain Barrier. Int. J. Mol. Sci. 2019, 20, 6010. [Google Scholar] [CrossRef] [Green Version]
- Hellström, M.; Gerhardt, H.; Kalén, M.; Li, X.; Eriksson, U.; Wolburg, H.; Betsholtz, C. Lack of Pericytes Leads to Endothelial Hyperplasia and Abnormal Vascular Morphogenesis. J. Cell Biol. 2001, 153, 543–554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daneman, R.; Zhou, L.; Kebede, A.A.; Barres, B.A. Pericytes are required for blood–brain barrier integrity during embryogenesis. Nature 2010, 468, 562–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Q.; Yang, Y.; Fan, X. Microvascular pericytes in brain-associated vascular disease. Biomed. Pharm. 2020, 121, 109633. [Google Scholar] [CrossRef] [PubMed]
- Brown, L.S.; Foster, C.G.; Courtney, J.-M.; King, N.E.; Howells, D.W.; Sutherland, B.A. Pericytes and Neurovascular Function in the Healthy and Diseased Brain. Front. Cell. Neurosci. 2019, 13, 282. [Google Scholar] [CrossRef] [PubMed]
- Dore-Duffy, P. Pericytes: Pluripotent Cells of the Blood Brain Barrier. Curr. Pharm. Des. 2008, 14, 1581–1593. [Google Scholar] [CrossRef] [PubMed]
- Price, T.O.; Eranki, V.; Banks, W.A.; Ercal, N.; Shah, G.N. Topiramate Treatment Protects Blood-Brain Barrier Pericytes from Hyperglycemia-Induced Oxidative Damage in Diabetic Mice. Endocrinology 2012, 153, 362–372. [Google Scholar] [CrossRef] [Green Version]
- Shah, G.N.; Price, T.O.; Banks, W.A.; Morofuji, Y.; Kovac, A.; Ercal, N.; Sorenson, C.M.; Shin, E.S.; Sheibani, N. Pharmacological Inhibition of Mitochondrial Carbonic Anhydrases Protects Mouse Cerebral Pericytes from High Glucose-Induced Oxidative Stress and Apoptosis. J. Pharm. Exp. 2013, 344, 637–645. [Google Scholar] [CrossRef] [Green Version]
- May, J.M.; Jayagopal, A.; Qu, Z.; Parker, W.H. Ascorbic acid prevents high glucose-induced apoptosis in human brain pericytes. Biochem. Biophys. Res. Commun. 2014, 452, 112–117. [Google Scholar] [CrossRef] [Green Version]
- Alcazar Magana, A.; Reed, R.L.; Koluda, R.; Miranda, C.L.; Maier, C.S.; Stevens, J.F. Vitamin C Activates the Folate-Mediated One-Carbon Cycle in C2C12 Myoblasts. Antioxidants 2020, 9, 217. [Google Scholar] [CrossRef] [Green Version]
- Underly, R.G.; Levy, M.; Hartmann, D.A.; Grant, R.I.; Watson, A.N.; Shih, A.Y. Pericytes as Inducers of Rapid, Matrix Metalloproteinase-9-Dependent Capillary Damage during Ischemia. J. Neurosci. 2017, 37, 129–140. [Google Scholar] [CrossRef]
- Reynell, C. Identification and Functional Characterization of CNS Pericytes and the Role They Play in Neurovascular Coupling in Physiological and Pathological Conditions. Ph.D. Thesis, Department of Neuroscience, Physiology and Pharmacology, University College London, London, UK, 2013. [Google Scholar]
- Burdyga, T.; Borysova, L. Calcium Signalling in Pericytes. J. Vasc. Res. 2014, 51, 190–199. [Google Scholar] [CrossRef] [PubMed]
- Iadecola, C.; Nedergaard, M. Glial regulation of the cerebral microvasculature. Nat. Neurosci. 2007, 10, 1369–1376. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.-S.; Tzou, B.-C.; Liu, Y.-P.; Tsai, M.-J.; Shyue, S.-K.; Tzeng, S.-F. Inhibition of cadmium-induced oxidative injury in rat primary astrocytes by the addition of antioxidants and the reduction of intracellular calcium. J. Cell. Biochem. 2008, 103, 825–834. [Google Scholar] [CrossRef]
- Pourreza, N. Phenolic Compounds as Potential Antioxidant. Jundishapur J. Nat. Pharm. Prod. 2013, 8, 149–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, A.; Ikram, M.; Muhammad, T.; Park, J.; Kim, M.O. Caffeine Modulates Cadmium-Induced Oxidative Stress, Neuroinflammation, and Cognitive Impairments by Regulating Nrf-2/HO-1 in Vivo and in Vitro. J. Clin. Med. 2019, 8, 680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morgan, M.J.; Liu, Z. Crosstalk of reactive oxygen species and NF-κB signaling. Cell Res. 2011, 21, 103–115. [Google Scholar] [CrossRef] [Green Version]
- León-Carmona, J.R.; Galano, A. Is Caffeine a Good Scavenger of Oxygenated Free Radicals? J. Phys. Chem. B 2011, 115, 4538–4546. [Google Scholar] [CrossRef] [PubMed]
- Phuagkhaopong, S.; Ospondpant, D.; Kasemsuk, T.; Sibmooh, N.; Soodvilai, S.; Power, C.; Vivithanaporn, P. Cadmium-induced IL-6 and IL-8 expression and release from astrocytes are mediated by MAPK and NF-κB pathways. Neurotoxicology 2017, 60, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Nedzvetsky, V.S.; Sukharenko, E.V.; Kyrychenko, S.V.; Baydas, G. Soluble curcumin prevents cadmium cytotoxicity in primary rat astrocytes by improving a lack of GFAP and glucose-6-phosphate-dehydrogenase. Regul. Mech. Biosyst. 2018, 9, 501–507. [Google Scholar] [CrossRef]
- Mailloux, R.J.; Harper, M. Glucose regulates enzymatic sources of mitochondrial NADPH in skeletal muscle cells; a novel role for glucose-6-phosphate dehydrogenase. FASEB J. 2010, 24, 2495–2506. [Google Scholar] [CrossRef] [PubMed]
- Bachiller, S.; Jiménez-Ferrer, I.; Paulus, A.; Yang, Y.; Swanberg, M.; Deierborg, T.; Boza-Serrano, A. Microglia in Neurological Diseases: A Road Map to Brain-Disease Dependent-Inflammatory Response. Front. Cell. Neurosci. 2018, 12, 488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.; Yang, S.; Qian, S.Y.; Hong, J.-S.; Kadiiska, M.B.; Tennant, R.W.; Waalkes, M.P.; Liu, J. Cadmium-Induced Toxicity in Rat Primary Mid-brain Neuroglia Cultures: Role of Oxidative Stress from Microglia. Toxicol. Sci. 2007, 98, 488–494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruttkay-Nedecky, B.; Nejdl, L.; Gumulec, J.; Zitka, O.; Masarik, M.; Eckschlager, T.; Stiborova, M.; Adam, V.; Kizek, R. The Role of Metallothionein in Oxidative Stress. Int. J. Mol. Sci. 2013, 14, 6044–6066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- von Leden, R.E.; Khayrullina, G.; Moritz, K.E.; Byrnes, K.R. Age exacerbates microglial activation, oxidative stress, inflammatory and NOX2 gene expression, and delays functional recovery in a middle-aged rodent model of spinal cord injury. J. Neuroinflamm. 2017, 14, 161. [Google Scholar] [CrossRef] [PubMed]
- Liguori, I.; Russo, G.; Curcio, F.; Bulli, G.; Aran, L.; Della-Morte, D.; Gargiulo, G.; Testa, G.; Cacciatore, F.; Bonaduce, D.; et al. Oxidative stress, aging, and diseases. Clin. Interv. Aging 2018, 13, 757–772. [Google Scholar] [CrossRef] [Green Version]
- di Penta, A.; Moreno, B.; Reix, S.; Fernandez-Diez, B.; Villanueva, M.; Errea, O.; Escala, N.; Vandenbroeck, K.; Comella, J.X.; Villoslada, P. Oxidative Stress and Proinflammatory Cytokines Contribute to Demyelination and Axonal Damage in a Cerebellar Culture Model of Neuroinflammation. PLoS ONE 2013, 8, e54722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaneko, Y.S.; Ota, A.; Nakashima, A.; Mori, K.; Nagatsu, I.; Nagatsu, T. Regulation of oxidative stress in long-lived lipopolysaccharide-activated microglia. Clin. Exp. Pharm. Physiol. 2012, 39, 599–607. [Google Scholar] [CrossRef] [PubMed]
- Jha, M.K.; Jo, M.; Kim, J.-H.; Suk, K. Microglia-Astrocyte Crosstalk: An Intimate Molecular Conversation. Neuroscientist 2019, 25, 227–240. [Google Scholar] [CrossRef]
- Zuo, L.; Prather, E.R.; Stetskiv, M.; Garrison, D.E.; Meade, J.R.; Peace, T.I.; Zhou, T. Inflammaging and Oxidative Stress in Human Diseases: From Molecular Mechanisms to Novel Treatments. Int. J. Mol. Sci. 2019, 20, 4472. [Google Scholar] [CrossRef] [Green Version]
- Ehrlich, L.C.; Hu, S.; Sheng, W.S.; Sutton, R.L.; Rockswold, G.L.; Peterson, P.K.; Chao, C.C. Cytokine Regulation of Human Microglial Cell IL-8 Production. J. Immunol. 1998, 160, 1944–1948. [Google Scholar]
- Figueiredo-Pereira, M.E.; Cohen, G. The ubiquitin/proteasome pathway: Friend or foe in zinc-, cadmium-, and H2O2-induced neuronal oxidative stress. Mol. Biol. Rep. 1999, 26, 65–69. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Yuan, Y.; Zou, H.; Yang, J.; Zhao, S.; Ma, Y.; Wang, Y.; Bian, J.; Liu, X.; Gu, J.; et al. The ER stress regulator Bip mediates cadmium-induced autophagy and neuronal senescence. Sci. Rep. 2016, 6, 38091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Preston, G.M.; Brodsky, J.L. The evolving role of ubiquitin modification in endoplasmic reticulum-associated degradation. Biochem. J. 2017, 474, 445–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.; Kaufman, R.J. Protein misfolding in the endoplasmic reticulum as a conduit to human disease. Nature 2016, 529, 326–335. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y. Oxidative Stress and Apoptotic Changes of Rat Cerebral Cortical Neurons Exposed to Cadmium in Vitro. Biomed. Environ. Sci. 2012, 25, 172–181. [Google Scholar]
- Xu, M.-Y.; Wang, P.; Sun, Y.-J.; Yang, L.; Wu, Y.-J. Joint toxicity of chlorpyrifos and cadmium on the oxidative stress and mitochondrial damage in neuronal cells. Food Chem. Toxicol. 2017, 103, 246–252. [Google Scholar] [CrossRef]
- Zhao, R.; Yu, Q.; Hou, L.; Dong, X.; Zhang, H.; Chen, X.; Zhou, Z.; Ma, J.; Huang, S.; Chen, L. Cadmium induces mitochondrial ROS inactivation of XIAP pathway leading to apoptosis in neuronal cells. Int. J. Biochem. Cell Biol. 2020, 121, 105715. [Google Scholar] [CrossRef]
- Xu, B.; Chen, S.; Luo, Y.; Chen, Z.; Liu, L.; Zhou, H.; Chen, W.; Shen, T.; Han, X.; Chen, L.; et al. Calcium Signaling Is Involved in Cadmium-Induced Neuronal Apoptosis via Induction of Reactive Oxygen Species and Activation of MAPK/mTOR Network. PLoS ONE 2011, 6, e19052. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Ren, Q.; Zhang, J.; Ye, Y.; Zhang, Z.; Xu, Y.; Guo, M.; Ji, H.; Xu, C.; Gu, C.; et al. N-acetyl-L-cysteine protects against cadmium-induced neuronal apoptosis by inhibiting ROS-dependent activation of Akt/mTOR pathway in mouse brain: NAC prevents Cd neurotoxicity via targeting ROS-mTOR signalling. Neuropathol. Appl. Neurobiol. 2014, 40, 759–777. [Google Scholar] [CrossRef] [Green Version]
- Branca, J.J.V.; Morucci, G.; Maresca, M.; Tenci, B.; Cascella, R.; Paternostro, F.; Ghelardini, C.; Gulisano, M.; Di Cesare Mannelli, L.; Pacini, A. Selenium and zinc: Two key players against cadmium-induced neuronal toxicity. Toxicol. Vitr. 2018, 48, 159–169. [Google Scholar] [CrossRef] [Green Version]
- Branca, J.J.V.; Morucci, G.; Becatti, M.; Carrino, D.; Ghelardini, C.; Gulisano, M.; Di Cesare Mannelli, L.; Pacini, A. Cannabidiol Protects Dopaminergic Neuronal Cells from Cadmium. Int. J. Environ. Res. Public Health 2019, 16, 4420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taoufik, E.; Kouroupi, G.; Zygogianni, O.; Matsas, R. Synaptic dysfunction in neurodegenerative and neurodevelopmental diseases: An overview of induced pluripotent stem-cell-based disease models. Open Biol. 2018, 8, 180138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almeer, R.S.; Kassab, R.B.; AlBasher, G.I.; Alarifi, S.; Alkahtani, S.; Ali, D.; Abdel Moneim, A.E. Royal jelly mitigates cadmium-induced neuronal damage in mouse cortex. Mol. Biol. Rep. 2019, 46, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Elkhadragy, M.F.; Kassab, R.B.; Metwally, D.; Almeer, R.S.; Abdel-Gaber, R.; Al-Olayan, E.M.; Essawy, E.A.; Amin, H.K.; Abdel Moneim, A.E. Protective effects of Fragaria ananassa methanolic extract in a rat model of cadmium chloride-induced neurotoxicity. Biosci. Rep. 2018, 38, BSR20180861. [Google Scholar] [CrossRef] [Green Version]
- Al Kahtani, M. Effect of both selenium and biosynthesized nanoselenium particles on cadmium-induced neurotoxicity in albino rats. Hum. Exp. Toxicol. 2020, 39, 159–172. [Google Scholar] [CrossRef]
- Booz, G.W. Cannabidiol as an emergent therapeutic strategy for lessening the impact of inflammation on oxidative stress. Free Radic. Biol. Med. 2011, 51, 1054–1061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, Y.; Jiang, C.; Xu, H.; Sun, Y.; Hu, F.; Bian, J.; Liu, X.; Gu, J.; Liu, Z. Cadmium-Induced Apoptosis in Primary Rat Cerebral Cortical Neurons Culture Is Mediated by a Calcium Signaling Pathway. PLoS ONE 2013, 8, e64330. [Google Scholar] [CrossRef] [PubMed]
- Pabis, K.; Gundacker, C.; Giacconi, R.; Basso, A.; Costarelli, L.; Piacenza, F.; Strizzi, S.; Provinciali, M.; Malavolta, M. Zinc supplementation can reduce accumulation of cadmium in aged metallothionein transgenic mice. Chemosphere 2018, 211, 855–860. [Google Scholar] [CrossRef] [PubMed]
- Zhai, Q.; Narbad, A.; Chen, W. Dietary Strategies for the Treatment of Cadmium and Lead Toxicity. Nutrients 2015, 7, 552–571. [Google Scholar] [CrossRef] [Green Version]
- Hossain, S.; Liu, H.-N.; Nguyen, M.; Shore, G.; Almazan, G. Cadmium exposure induces mitochondria-dependent apoptosis in oligodendrocytes. Neurotoxicology 2009, 30, 544–554. [Google Scholar] [CrossRef] [PubMed]
- Lassmann, H.; van Horssen, J. Oxidative stress and its impact on neurons and glia in multiple sclerosis lesions. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2016, 1862, 506–510. [Google Scholar] [CrossRef] [PubMed]
- Peferoen, L.; Kipp, M.; van der Valk, P.; van Noort, J.M.; Amor, S. Oligodendrocyte-microglia cross-talk in the central nervous system. Immunology 2014, 141, 302–313. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Xie, D.; Fang, M.; Zhu, G.; Chen, C.; Zeng, H.; Lu, J.; Charanjit, K. Astrocyte-Derived Proinflammatory Cytokines Induce Hypomyelination in the Periventricular White Matter in the Hypoxic Neonatal Brain. PLoS ONE 2014, 9, e87420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramesh, G.; MacLean, A.G.; Philipp, M.T. Cytokines and Chemokines at the Crossroads of Neuroinflammation, Neurodegeneration, and Neuropathic Pain. Mediat. Inflamm. 2013, 2013, 1–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Domingues, H.S.; Portugal, C.C.; Socodato, R.; Relvas, J.B. Oligodendrocyte, Astrocyte, and Microglia Crosstalk in Myelin Development, Damage, and Repair. Front. Cell Dev. Biol. 2016, 4, 71. [Google Scholar] [CrossRef] [PubMed]
- Miron, V.E.; Boyd, A.; Zhao, J.-W.; Yuen, T.J.; Ruckh, J.M.; Shadrach, J.L.; van Wijngaarden, P.; Wagers, A.J.; Williams, A.; Franklin, R.J.M.; et al. M2 microglia and macrophages drive oligodendrocyte differentiation during CNS remyelination. Nat. Neurosci. 2013, 16, 1211–1218. [Google Scholar] [CrossRef] [PubMed] [Green Version]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Branca, J.J.V.; Fiorillo, C.; Carrino, D.; Paternostro, F.; Taddei, N.; Gulisano, M.; Pacini, A.; Becatti, M. Cadmium-Induced Oxidative Stress: Focus on the Central Nervous System. Antioxidants 2020, 9, 492. https://doi.org/10.3390/antiox9060492
Branca JJV, Fiorillo C, Carrino D, Paternostro F, Taddei N, Gulisano M, Pacini A, Becatti M. Cadmium-Induced Oxidative Stress: Focus on the Central Nervous System. Antioxidants. 2020; 9(6):492. https://doi.org/10.3390/antiox9060492
Chicago/Turabian StyleBranca, Jacopo J. V., Claudia Fiorillo, Donatello Carrino, Ferdinando Paternostro, Niccolò Taddei, Massimo Gulisano, Alessandra Pacini, and Matteo Becatti. 2020. "Cadmium-Induced Oxidative Stress: Focus on the Central Nervous System" Antioxidants 9, no. 6: 492. https://doi.org/10.3390/antiox9060492