Hydrogen Peroxide Mediates Artemisinin-Derived C-16 Carba-Dimer-Induced Toxicity of Human Cancer Cells

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Analytical Chemistry Approach
2.2. Cell Culture and Reagents
2.3. Toxicity Assay
2.4. Flow Cytometry Assays
2.4.1. Cell Cycle Phase Analysis
2.4.2. Reactive Oxygen Species
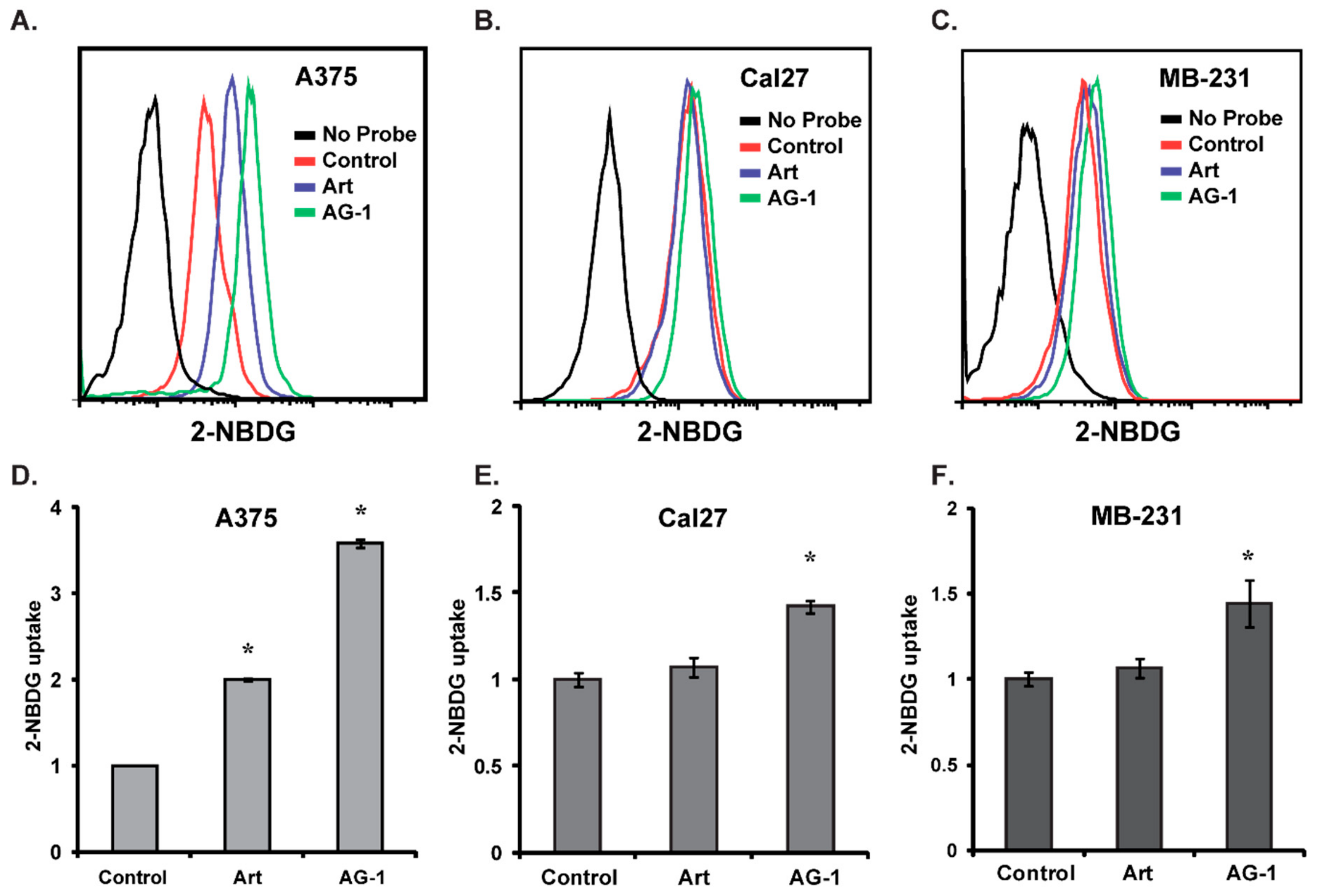
2.4.3. Glucose Uptake
2.5. Electron Paramagnetic Resonance (EPR) Spectroscopy
2.6. DNA Damage Assay
2.7. Immunoblotting Assay
2.8. Statistical Analysis
3. Results
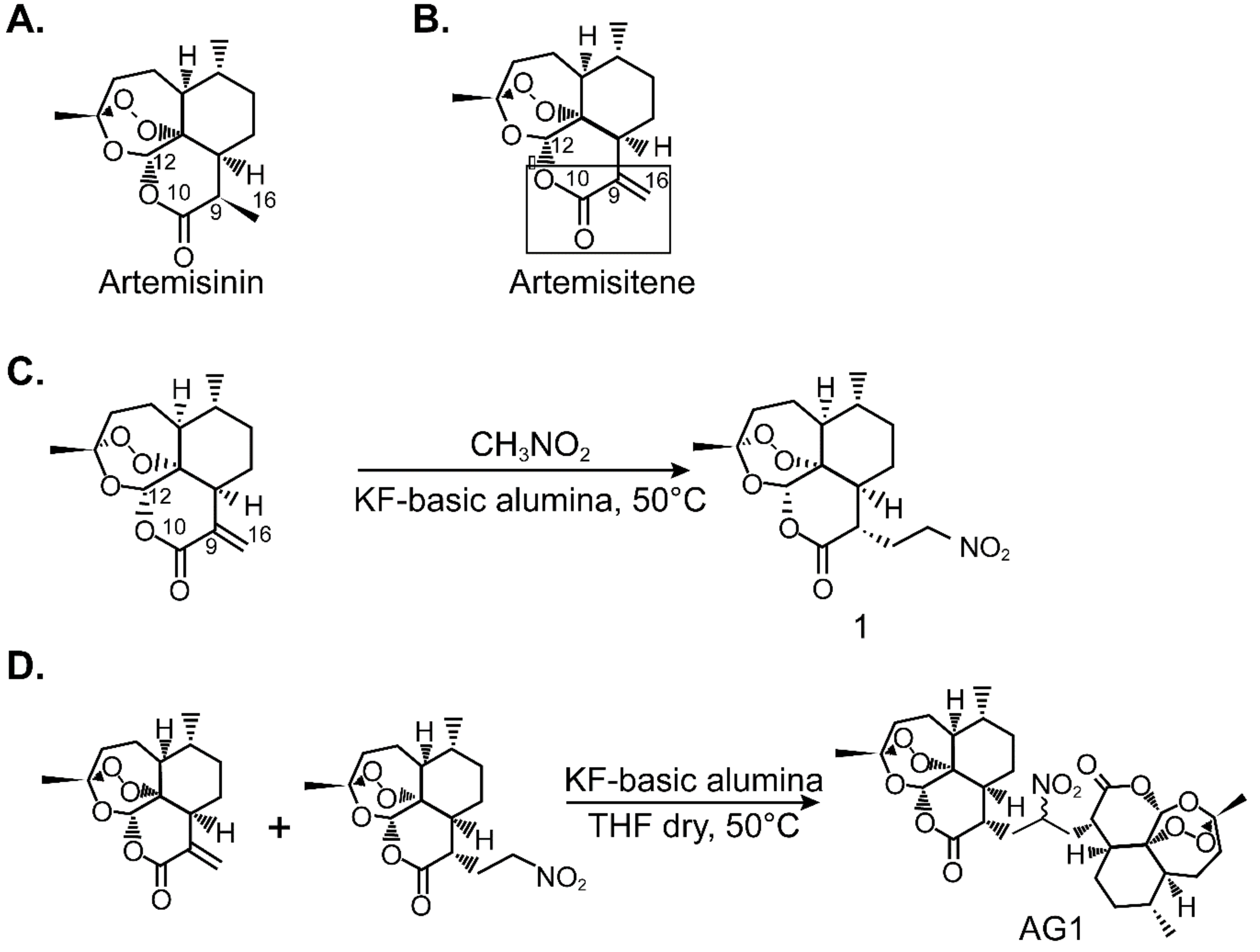
3.1. Synthesis of AG1
3.1.1. Synthesis of Artemisinin-Derived Michael Adduct
3.1.2. Synthesis of Artemisinin Dimer, AG-1
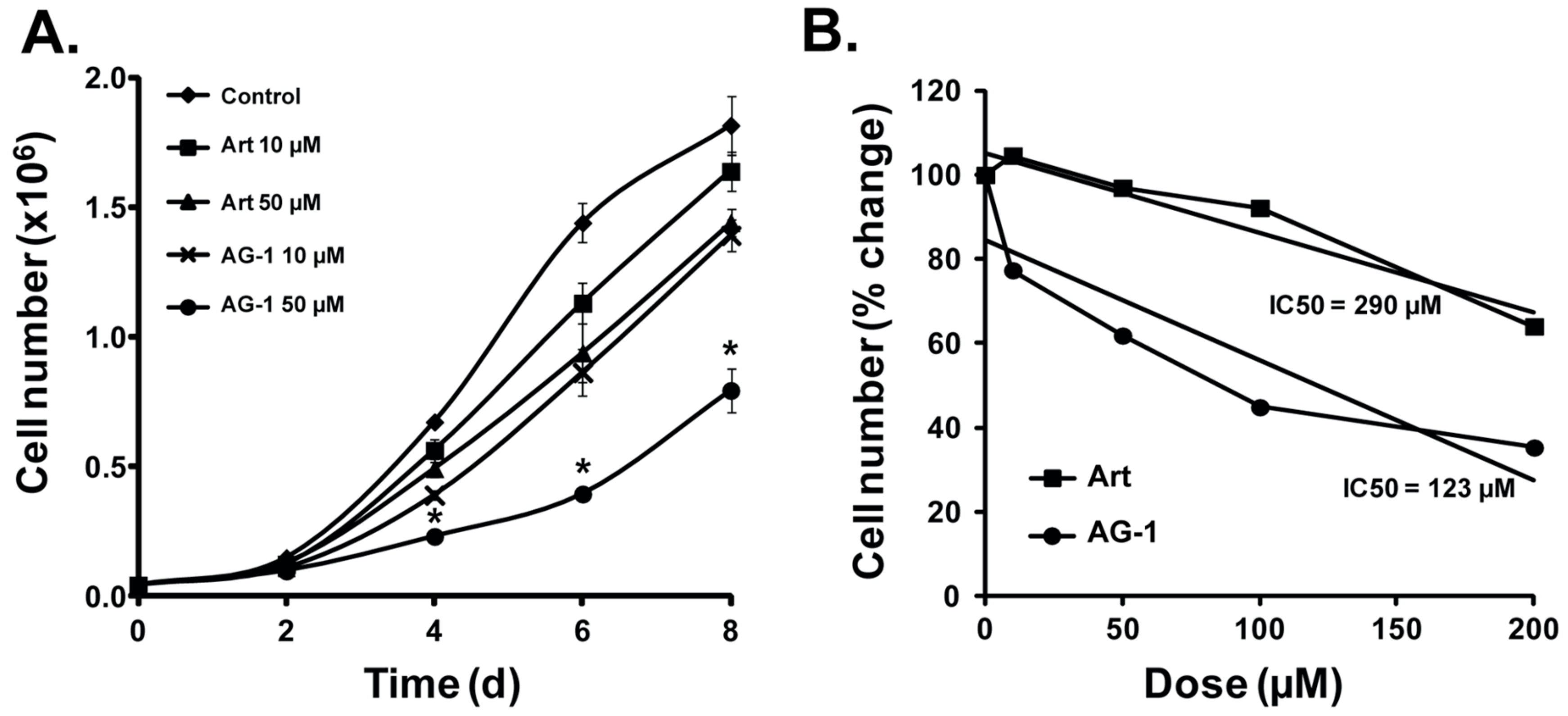
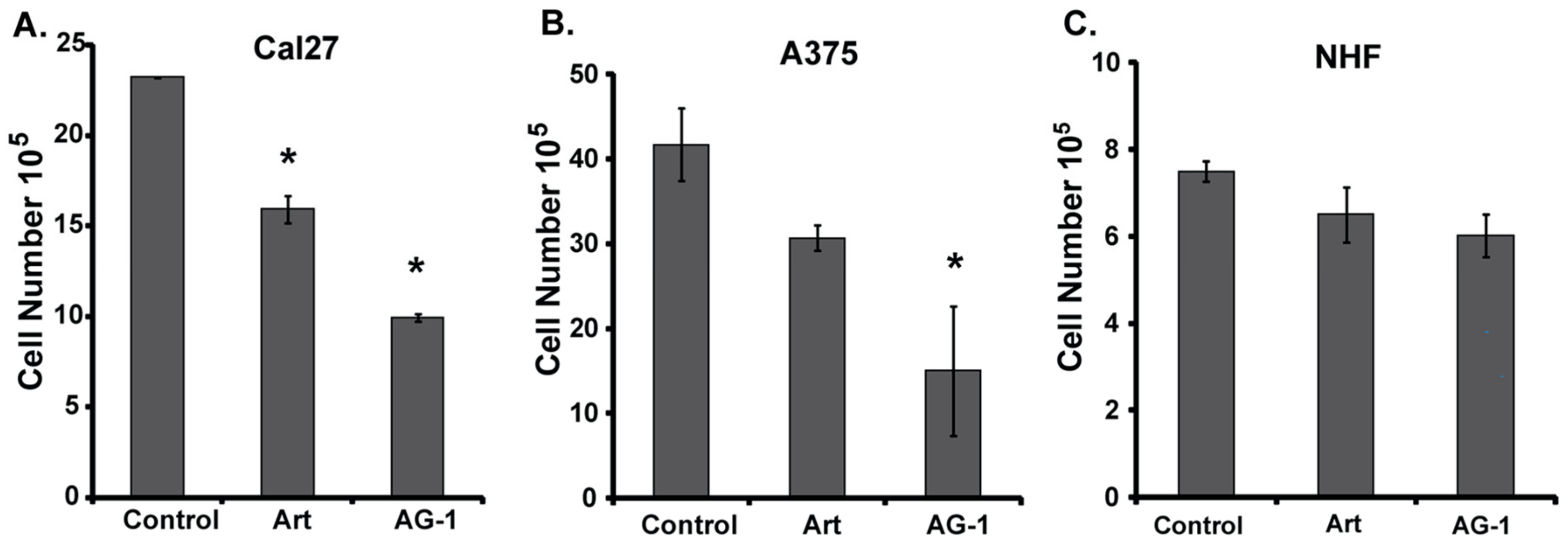
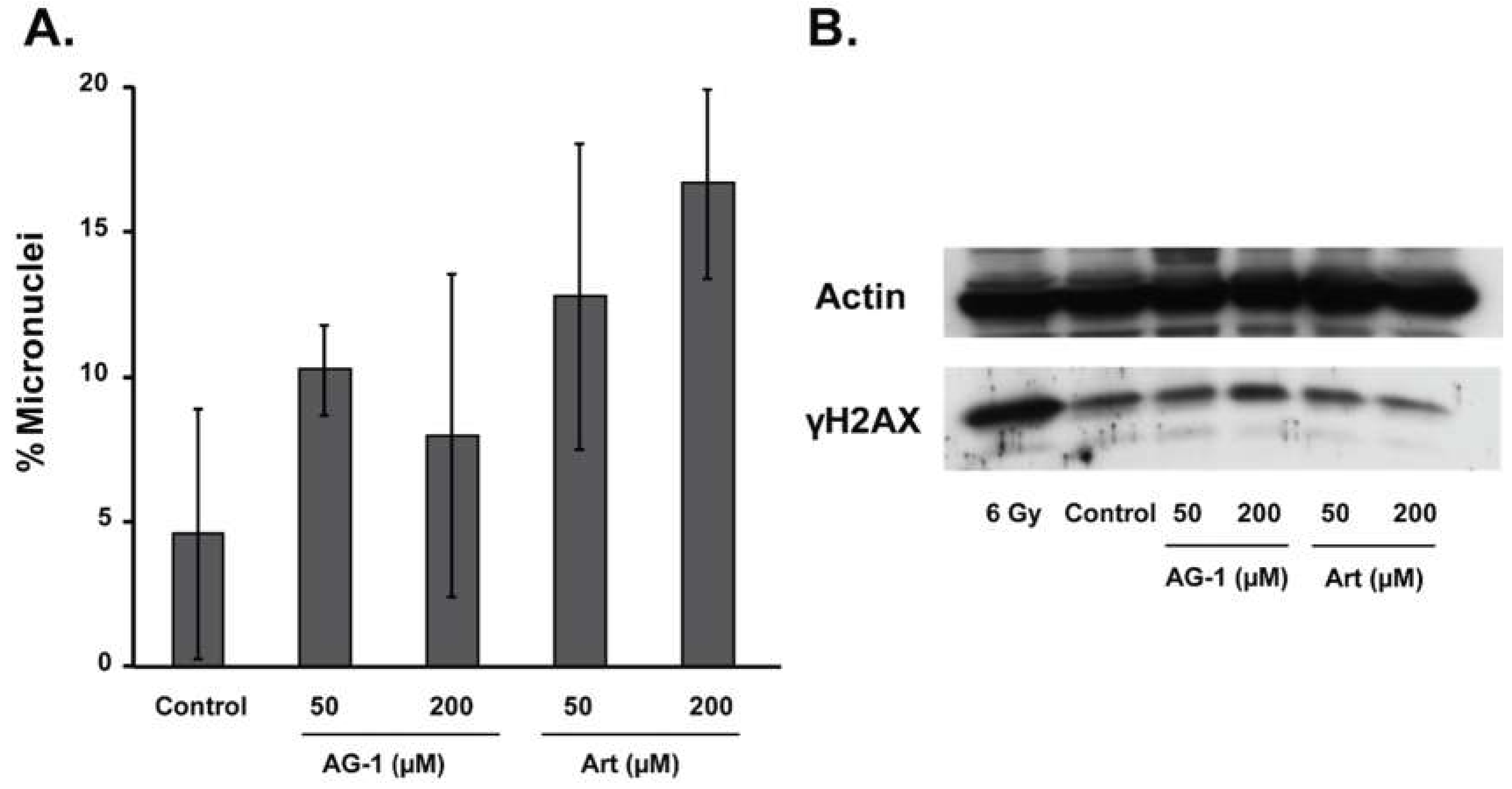
3.2. AG-1 is More Cytotoxic Than Artemisinin (Art)
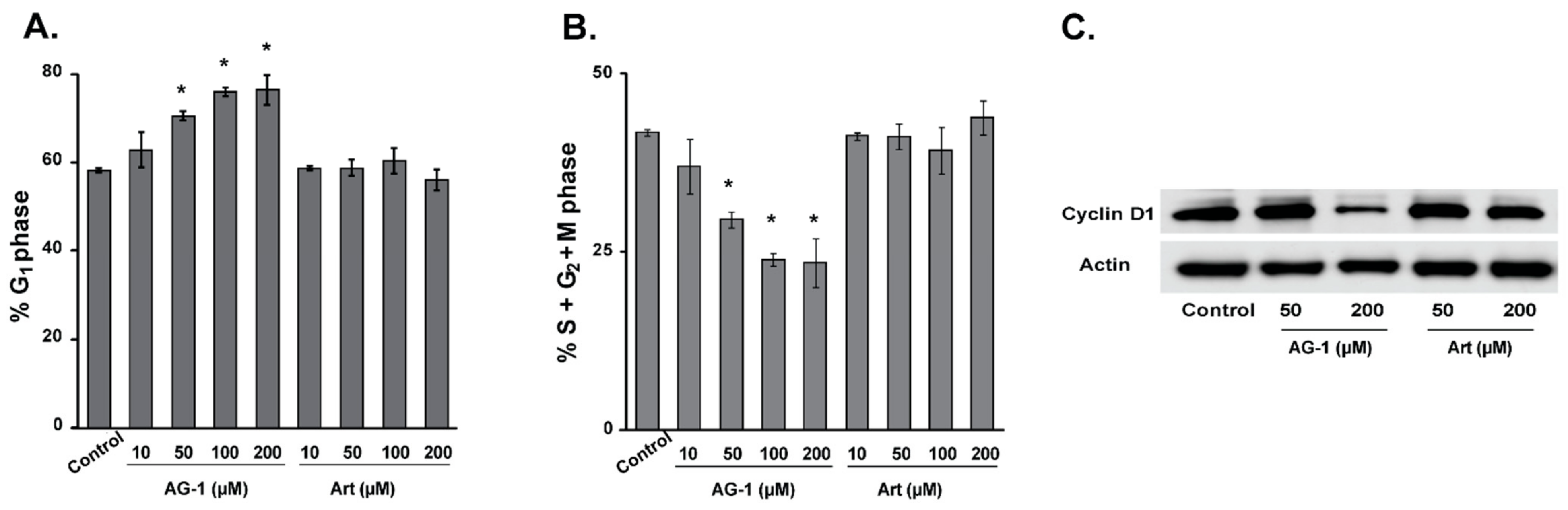
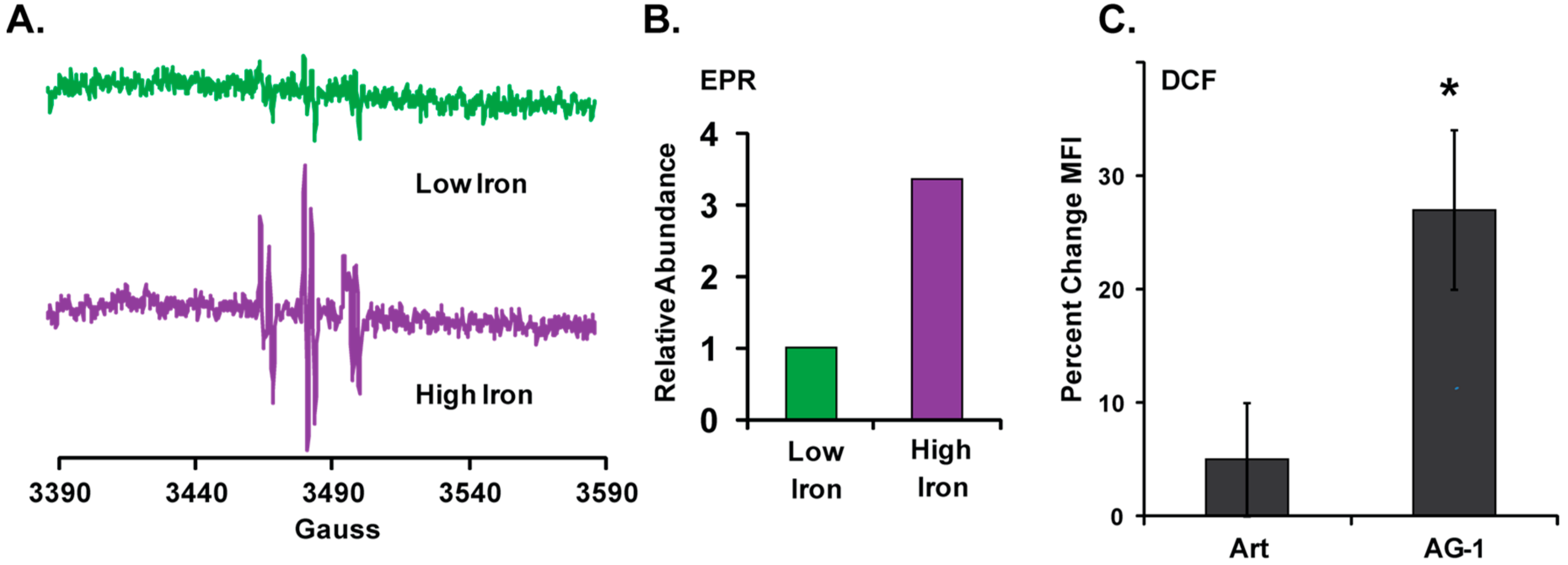
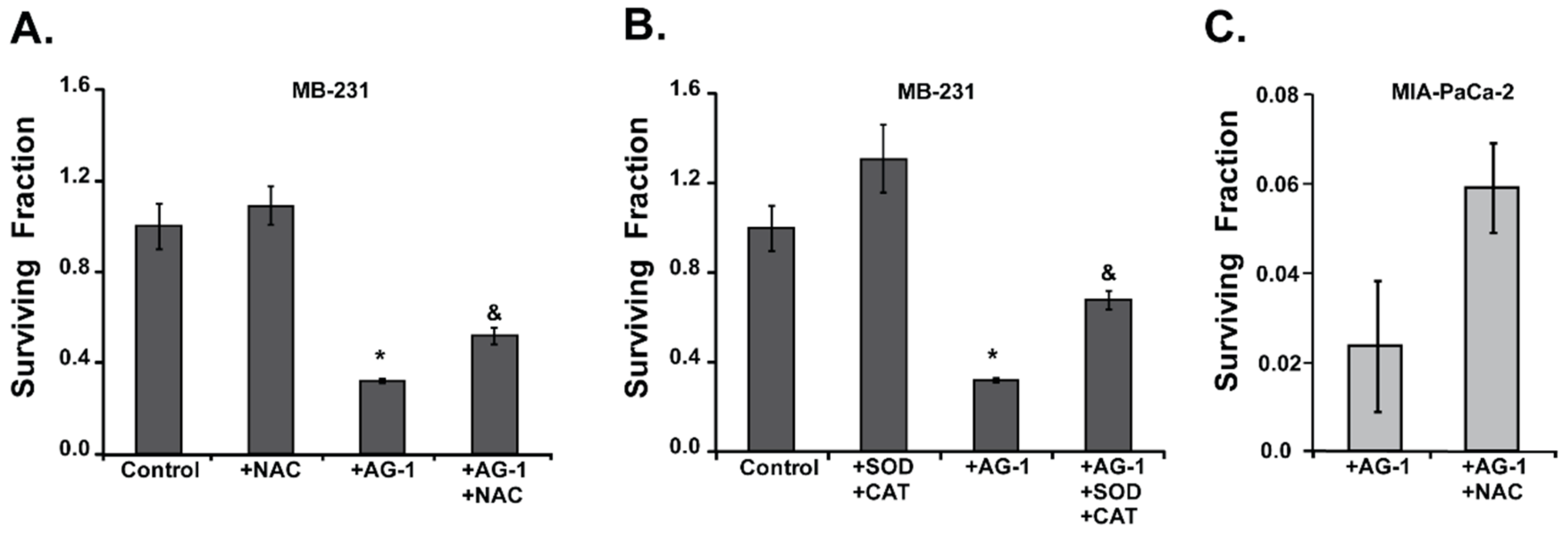
3.3. Reactive Oxygen Species (ROS: Superoxide and Hydrogen Peroxide) Regulate AG-1-induced Toxicity
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Tu, Y. The discovery of artemisinin (qinghaosu) and gifts from chinese medicine. Nat. Med. 2011, 17, 1217–1220. [Google Scholar] [CrossRef]
- Chaturvedi, D.; Goswami, A.; Saikia, P.P.; Barua, N.C.; Rao, P.G. Artemisinin and its derivatives: A novel class of anti-malarial and anti-cancer agents. Chem. Soc. Rev. 2010, 39, 435–454. [Google Scholar] [CrossRef]
- Lai, H.; Singh, N.P. Oral artemisinin prevents and delays the development of 7,12-dimethylbenz[a]anthracene (dmba)-induced breast cancer in the rat. Cancer Lett. 2006, 231, 43–48. [Google Scholar] [CrossRef]
- Nam, W.; Tak, J.; Ryu, J.K.; Jung, M.; Yook, J.I.; Kim, H.J.; Cha, I.H. Effects of artemisinin and its derivatives on growth inhibition and apoptosis of oral cancer cells. Head Neck 2007, 29, 335–340. [Google Scholar] [CrossRef]
- Paik, I.H.; Xie, S.; Shapiro, T.A.; Labonte, T.; Narducci Sarjeant, A.A.; Baege, A.C.; Posner, G.H. Second generation, orally active, antimalarial, artemisinin-derived trioxane dimers with high stability, efficacy, and anticancer activity. J. Med. Chem. 2006, 49, 2731–2734. [Google Scholar] [CrossRef]
- Sundar, S.N.; Marconett, C.N.; Doan, V.B.; Willoughby, J.A., Sr.; Firestone, G.L. Artemisinin selectively decreases functional levels of estrogen receptor-alpha and ablates estrogen-induced proliferation in human breast cancer cells. Carcinogenesis 2008, 29, 2252–2258. [Google Scholar] [CrossRef] [Green Version]
- Efferth, T.; Dunstan, H.; Sauerbrey, A.; Miyachi, H.; Chitambar, C.R. The anti-malarial artesunate is also active against cancer. Int. J. Oncol. 2001, 18, 767–773. [Google Scholar] [CrossRef] [PubMed]
- Gravett, A.M.; Liu, W.M.; Krishna, S.; Chan, W.C.; Haynes, R.K.; Wilson, N.L.; Dalgleish, A.G. In vitro study of the anti-cancer effects of artemisone alone or in combination with other chemotherapeutic agents. Cancer Chemother. Pharmacol. 2011, 67, 569–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Y.Y.; Chen, T.S.; Qu, J.L.; Pan, W.L.; Sun, L.; Wei, X.B. Dihydroartemisinin (DHA) induces caspase-3-dependent apoptosis in human lung adenocarcinoma ASTC-a-1 cells. J. Biomed. Sci. 2009, 16, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Posner, G.H.; Oh, C.H.; Gerena, L.; Milhous, W.K. Extraordinarily potent antimalarial compounds: New, structurally simple, easily synthesized, tricyclic 1,2,4-trioxanes. J. Med. Chem. 1992, 35, 2459–2467. [Google Scholar] [CrossRef]
- Kalen, A.L.; Sarsour, E.H.; Venkataraman, S.; Goswami, P.C. Mn-superoxide dismutase overexpression enhances G2 accumulation and radioresistance in human oral squamous carcinoma cells. Antiox. Redox Signal. 2006, 8, 1273–1281. [Google Scholar] [CrossRef]
- Xiao, W.; Sarsour, E.H.; Wagner, B.A.; Doskey, C.M.; Buettner, G.R.; Domann, F.E.; Goswami, P.C. Succinate dehydrogenase activity regulates PCB3-quinone-induced metabolic oxidative stress and toxicity in HaCaT human keratinocytes. Arch. Toxicol. 2016, 90, 319–332. [Google Scholar] [CrossRef]
- Buettner, G.R. Spin trapping: Esr parameters of spin adducts. Free Radic. Biol. Med. 1987, 3, 259–303. [Google Scholar] [CrossRef]
- Duling, D.R. Simulation of multiple isotropic spin-trap EPR spectra. J. Magn. Reson. Ser. B 1994, 104, 105–110. [Google Scholar] [CrossRef]
- Venkatesha, V.A.; Venkataraman, S.; Sarsour, E.H.; Kalen, A.L.; Buettner, G.R.; Robertson, L.W.; Lehmler, H.J.; Goswami, P.C. Catalase ameliorates polychlorinated biphenyl-induced cytotoxicity in nonmalignant human breast epithelial cells. Free Radic. Biol. Med. 2008, 45, 1094–1102. [Google Scholar] [CrossRef] [Green Version]
- Goswami, A.; Saikia, P.P.; Saikia, B.; Barua, N.C. Dinitroaliphatics as linkers: Application in the synthesis of novel artemisinin carba-dimer. Mol. Divers. 2011, 15, 707–712. [Google Scholar] [CrossRef]
- Sarsour, E.H.; Agarwal, M.; Pandita, T.K.; Oberley, L.W.; Goswami, P.C. Manganese superoxide dismutase protects the proliferative capacity of confluent normal human fibroblasts. J. Biol. Chem. 2005, 280, 18033–18041. [Google Scholar] [CrossRef] [Green Version]
- Du, C.; Gao, Z.; Venkatesha, V.A.; Kalen, A.L.; Chaudhuri, L.; Spitz, D.R.; Cullen, J.J.; Oberley, L.W.; Goswami, P.C. Mitochondrial ROS and radiation induced transformation in mouse embryonic fibroblasts. Cancer Biol. Ther. 2009, 8, 1962–1971. [Google Scholar] [CrossRef] [Green Version]
- Wagner, B.A.; Buettner, G.R.; Burns, C.P. Free radical-mediated lipid peroxidation in cells: Oxidizability is a function of cell lipid bis-allylic hydrogen content. Biochemistry 1994, 33, 4449–4453. [Google Scholar] [CrossRef]
- Von Sonntag, C.; Schuchmann, H.P. The elucidation of peroxyl radical reactions in aqueous solution with the help of radiation-chemical methods. Angew. Chem. Int. Ed. Engl. 1991, 30, 1229–1253. [Google Scholar] [CrossRef]
- Eckers, J.C.; Kalen, A.L.; Sarsour, E.H.; Tompkins, V.S.; Janz, S.; Son, J.M.; Doskey, C.M.; Buettner, G.R.; Goswami, P.C. Forkhead box m1 regulates quiescence-associated radioresistance of human head and neck squamous carcinoma cells. Radiat. Res. 2014, 182, 420–429. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Sun, B.; Pan, S.; Jiang, H.; Sun, X. Dihydroartemisinin inhibits growth of pancreatic cancer cells in vitro and in vivo. Anticancer Drugs 2009, 20, 131–140. [Google Scholar] [CrossRef]
- Nakase, I.; Gallis, B.; Takatani-Nakase, T.; Oh, S.; Lacoste, E.; Singh, N.P.; Goodlett, D.R.; Tanaka, S.; Futaki, S.; Lai, H.; et al. Transferrin receptor-dependent cytotoxicity of artemisinin-transferrin conjugates on prostate cancer cells and induction of apoptosis. Cancer Lett. 2009, 274, 290–298. [Google Scholar] [CrossRef]
- Willoughby, J.A., Sr.; Sundar, S.N.; Cheung, M.; Tin, A.S.; Modiano, J.; Firestone, G.L. Artemisinin blocks prostate cancer growth and cell cycle progression by disrupting Sp1 interactions with the cyclin-dependent kinase-4 (CDK4) promoter and inhibiting cdk4 gene expression. J. Biol. Chem. 2009, 284, 2203–2213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shterman, N.; Kupfer, B.; Moroz, C. Comparison of transferrin receptors, iron content and isoferritin profile in normal and malignant human breast cell lines. Pathobiology 1991, 59, 19–25. [Google Scholar] [CrossRef]
- Nyholm, S.; Mann, G.J.; Johansson, A.G.; Bergeron, R.J.; Graslund, A.; Thelander, L. Role of ribonucleotide reductase in inhibition of mammalian cell growth by potent iron chelators. J. Biol. Chem. 1993, 268, 26200–26205. [Google Scholar]
- Yu, Y.; Kovacevic, Z.; Richardson, D.R. Tuning cell cycle regulation with an iron key. Cell Cycl. 2007, 6, 1982–1994. [Google Scholar] [CrossRef] [Green Version]
- Andrews, N.C. Iron homeostasis: Insights from genetics and animal models. Nat. Rev. Genet. 2000, 1, 208–217. [Google Scholar] [CrossRef]
- Kruszewski, M. Labile iron pool: The main determinant of cellular response to oxidative stress. Mutat. Res. 2003, 531, 81–92. [Google Scholar] [CrossRef]
- Richardson, D.R.; Ponka, P. The molecular mechanisms of the metabolism and transport of iron in normal and neoplastic cells. Biochim. Biophys. Acta 1997, 1331, 1–40. [Google Scholar] [CrossRef]
- Keyer, K.; Imlay, J.A. Superoxide accelerates DNA damage by elevating free-iron levels. Proc. Natl. Acad. Sci. USA 1996, 93, 13635–13640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarsour, E.H.; Kumar, M.G.; Chaudhuri, L.; Kalen, A.L.; Goswami, P.C. Redox control of the cell cycle in health and disease. Antiox. Redox Signal. 2009, 11, 2985–3011. [Google Scholar] [CrossRef] [PubMed]
- Folz, R.J.; Crapo, J.D. Extracellular superoxide dismutase (SOD3): Tissue-specific expression, genomic characterization, and computer-assisted sequence analysis of the human ec sod gene. Genomics 1994, 22, 162–171. [Google Scholar] [CrossRef] [PubMed]
- McCord, J.M.; Fridovich, I. Superoxide dismutase. An enzymic function for erythrocuprein (hemocuprein). J. Biol. Chem. 1969, 244, 6049–6055. [Google Scholar]
- Oberley, T.D.; Oberley, L.W.; Slattery, A.F.; Lauchner, L.J.; Elwell, J.H. Immunohistochemical localization of antioxidant enzymes in adult syrian hamster tissues and during kidney development. Am. J. Pathol. 1990, 137, 199–214. [Google Scholar]
- Yamamoto, K.; Volkl, A.; Hashimoto, T.; Fahimi, H.D. Catalase in guinea pig hepatocytes is localized in cytoplasm, nuclear matrix and peroxisomes. Eur. J. Cell Biol. 1988, 46, 129–135. [Google Scholar]
- Halliwell, B.; Gutteridge, J.M. Biologically relevant metal ion-dependent hydroxyl radical generation. An update. FEBS Lett. 1992, 307, 108–112. [Google Scholar] [CrossRef] [Green Version]
- Sarsour, E.H.; Kalen, A.L.; Goswami, P.C. Manganese superoxide dismutase regulates a redox cycle within the cell cycle. Antiox. Redox Signal. 2014, 20, 1618–1627. [Google Scholar] [CrossRef] [Green Version]
- Laurent, A.; Nicco, C.; Chereau, C.; Goulvestre, C.; Alexandre, J.; Alves, A.; Levy, E.; Goldwasser, F.; Panis, Y.; Soubrane, O.; et al. Controlling tumor growth by modulating endogenous production of reactive oxygen species. Cancer Res. 2005, 65, 948–956. [Google Scholar]
- Menon, S.G.; Sarsour, E.H.; Kalen, A.L.; Venkataraman, S.; Hitchler, M.J.; Domann, F.E.; Oberley, L.W.; Goswami, P.C. Superoxide signaling mediates N-acetyl-l-cysteine-induced G1 arrest: Regulatory role of cyclin D1 and manganese superoxide dismutase. Cancer Res. 2007, 67, 6392–6399. [Google Scholar] [CrossRef] [Green Version]
- Menon, S.G.; Sarsour, E.H.; Spitz, D.R.; Higashikubo, R.; Sturm, M.; Zhang, H.; Goswami, P.C. Redox regulation of the G1 to s phase transition in the mouse embryo fibroblast cell cycle. Cancer Res. 2003, 63, 2109–2117. [Google Scholar] [PubMed]











© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kalen, A.L.; Wagner, B.A.; Sarsour, E.H.; Kumar, M.G.; Reedy, J.L.; Buettner, G.R.; Barua, N.C.; Goswami, P.C. Hydrogen Peroxide Mediates Artemisinin-Derived C-16 Carba-Dimer-Induced Toxicity of Human Cancer Cells. Antioxidants 2020, 9, 108. https://doi.org/10.3390/antiox9020108
Kalen AL, Wagner BA, Sarsour EH, Kumar MG, Reedy JL, Buettner GR, Barua NC, Goswami PC. Hydrogen Peroxide Mediates Artemisinin-Derived C-16 Carba-Dimer-Induced Toxicity of Human Cancer Cells. Antioxidants. 2020; 9(2):108. https://doi.org/10.3390/antiox9020108
Chicago/Turabian StyleKalen, Amanda L., Brett A. Wagner, Ehab H. Sarsour, Maneesh G. Kumar, Jessica L. Reedy, Garry R. Buettner, Nabin C. Barua, and Prabhat C. Goswami. 2020. "Hydrogen Peroxide Mediates Artemisinin-Derived C-16 Carba-Dimer-Induced Toxicity of Human Cancer Cells" Antioxidants 9, no. 2: 108. https://doi.org/10.3390/antiox9020108