Cellular Aging Characteristics and Their Association with Age-Related Disorders




Abstract
:1. Introduction
2. Characteristics of the Senescent Phenotype
2.1. Genome Maintenance and Epigenetic Mechanisms
2.2. RNA Maintenance and Protein Synthesis
3. Mitochondria
4. Metabolic Activity
5. Interorganellar Communication
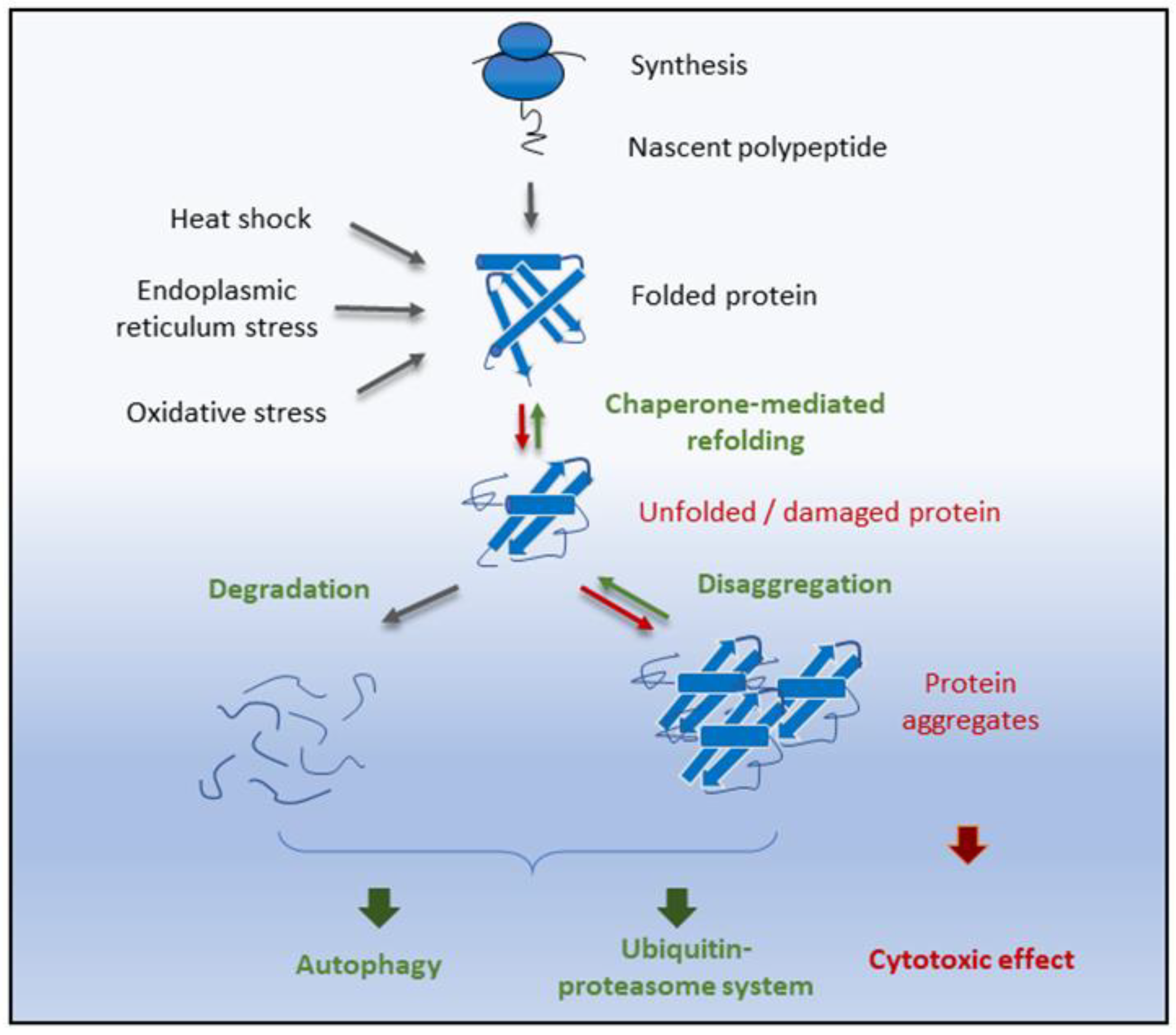
6. Proteostasis
7. Protein Carbonylation as an Aging Biomarker
8. Conclusions and Further Perspectives
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ROS | reactive oxygen species |
| DSB | DNA double-strand break |
| HR | homologous recombination |
| NHEJ | nonhomologous end-joining |
| EDARADD | Edar-associated death domain |
| TOM1L1 | Target of Myb1-like 1 membrane |
| NPTX2 | Neuronal pentraxin II |
| HAT/KAT | histone/lysine acetyltransferase |
| HDAC | histone deacetylase |
| mTOR | target of rapamycin kinase |
| PI3K | phosphoinositide 3-kinase |
| AKT | protein kinase B |
| UPS | ubiquitin–proteasome system |
| CMA | chaperone-mediated autophagy |
| DNP | 2,4-dinitrophenyl |
| DNPH | 2,4-dinitrophenylhydrazine |
References
- Raimundo, N.; Krisko, A. Cross-organelle communication at the core of longevity. Aging 2018, 10, 15–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheibye-Knudsen, M.; Fang, E.F.; Croteau, D.L.; Wilson, D.M., 3rd; Bohr, V.A. Protecting the mitochondrial powerhouse. Trends Cell Biol. 2015, 25, 158–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Díaz-Villanueva, J.F.; Díaz-Molina, R.; García-González, V. Protein Folding and Mechanisms of Proteostasis. Int. J. Mol. Sci. 2015, 16, 17193–17230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fedarko, N.S. The Biology of Aging and Frailty. Clin. Geriatr. Med. 2011, 27, 27–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, Y.; Brack, A.S. Cellular Mechanisms of Somatic Stem Cell Aging. Curr. Top. Dev. Biol. 2014, 107, 405–438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belikov, A.V. Age-related diseases as vicious cycles. Ageing Res. Rev. 2019, 49, 11–26. [Google Scholar] [CrossRef]
- Lopez-Otin, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [Green Version]
- Moeller, M.; Hirose, M.; Mueller, S.; Roolf, C.; Baltrusch, S.; Ibrahim, S.; Junghanss, C.; Wolkenhauer, O.; Jaster, R.; Köhling, R.; et al. Inbred mouse strains reveal biomarkers that are pro-longevity, antilongevity or role switching. Aging Cell 2014, 13, 729–738. [Google Scholar] [CrossRef]
- Pincus, Z.; Slack, F.J. Developmental biomarkers of aging in C. elegans. Dev. Dyn. 2010, 239, 1306–1314. [Google Scholar] [CrossRef] [Green Version]
- Gire, V.; Dulic, V. Senescence from G2 arrest, revisited. Cell Cycle 2015, 14, 297–304. [Google Scholar] [CrossRef] [Green Version]
- Kaeberlein, M.; Powers, R.W.; Steffen, K.K.; Westman, E.A.; Hu, D.; Dang, N.; Kerr, E.O.; Kirkland, K.T.; Fields, S.; Kennedy, B.K. Regulation of Yeast Replicative Life Span by TOR and Sch9 in Response to Nutrients. Science 2005, 310, 1193–1196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, C.; Zhou, C.; Kennedy, B.K. The yeast replicative aging model. Biochim. Biophys. Acta 2018, 1864, 2690–2696. [Google Scholar] [CrossRef] [PubMed]
- Kaeberlein, M. Lessons on longevity from budding yeast. Nature 2010, 464, 513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katajisto, P.; Dohla, J.; Chaffer, C.L.; Pentinmikko, N.; Marjanovic, N.; Iqbal, S.; Zoncu, R.; Chen, W.; Weinberg, R.A.; Sabatini, D.M. Stem cells. Asymmetric apportioning of aged mitochondria between daughter cells is required for stemness. Science 2015, 348, 340–343. [Google Scholar] [CrossRef] [Green Version]
- Van Deursen, J.M. The role of senescent cells in ageing. Nature 2014, 509, 439. [Google Scholar] [CrossRef] [Green Version]
- Kato, D.; Miyazawa, K.; Ruas, M.; Starborg, M.; Wada, I.; Oka, T.; Sakai, T.; Peters, G.; Hara, E. Features of replicative senescence induced by direct addition of antennapedia-p16INK4A fusion protein to human diploid fibroblasts. FEBS Lett. 1998, 427, 203–208. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.; Adams, W.J.; Alford, P.W.; McCain, M.L.; Feinberg, A.W.; Sheehy, S.P.; Goss, J.A.; Parker, K.K. Cytoskeletal prestress regulates nuclear shape and stiffness in cardiac myocytes. Exp. Biol. Med. 2015, 240, 1543–1554. [Google Scholar] [CrossRef]
- Baird, N.A.; Douglas, P.M.; Simic, M.S.; Grant, A.R.; Moresco, J.J.; Wolff, S.C.; Yates, J.R., 3rd; Manning, G.; Dillin, A. HSF-1-mediated cytoskeletal integrity determines thermotolerance and life span. Science 2014, 346, 360–363. [Google Scholar] [CrossRef] [Green Version]
- Perkins, A.D.; Lee, M.J.J.; Tanentzapf, G. The systematic identification of cytoskeletal genes required for. Sci. Data 2014, 1, 140002. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues, A.J.; do Carmo Costa, M.; Silva, T.L.; Ferreira, D.; Bajanca, F.; Logarinho, E.; Maciel, P. Absence of ataxin-3 leads to cytoskeletal disorganization and increased cell death. Biochim. Biophys. Acta 2010, 1803, 1154–1163. [Google Scholar] [CrossRef] [Green Version]
- Gourlay, C.W.; Ayscough, K.R. The actin cytoskeleton in ageing and apoptosis. FEMS Yeast Res. 2018, 5, 1193–1198. [Google Scholar] [CrossRef] [Green Version]
- Gong, C.X.; Iqbal, K. Hyperphosphorylation of microtubule-associated protein tau: A promising therapeutic target for Alzheimer disease. Curr. Med. Chem. 2008, 15, 2321–2328. [Google Scholar] [CrossRef]
- Prachar, J. Intimate contacts of mitochondria with nuclear envelope as a potential energy gateway for nucleo-cytoplasmic mRNA transport. Gen. Physiol. Biophys. 2003, 22, 525–534. [Google Scholar]
- Rowe, L.A.; Degtyareva, N.; Doetsch, P.W. DNA Damage-induced Reactive Oxygen Species (ROS) Stress Response in Saccharomyces cerevisiae. Free Radic. Biol. Med. 2008, 45, 1167–1177. [Google Scholar] [CrossRef] [Green Version]
- Langie, S.A.; Koppen, G.; Desaulniers, D.; Al-Mulla, F.; Al-Temaimi, R.; Amedei, A.; Azqueta, A.; Bisson, W.H.; Brown, D.G.; Brunborg, G.; et al. Causes of genome instability: The effect of low dose chemical exposures in modern society. Carcinogenesis 2015, 36 (Suppl. S1), S61–S88. [Google Scholar] [CrossRef]
- Lieber, M.R. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu. Rev. Biochem. 2010, 79, 181–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- David, S.S.; O’Shea, V.L.; Kundu, S. Base Excision Repair of Oxidative DNA Damage. Nature 2007, 447, 941–950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schärer, O.D. Nucleotide Excision Repair in Eukaryotes. Cold Spring Harb. Perspect. Biol. 2013, 5, a012609. [Google Scholar] [CrossRef] [PubMed]
- Jiricny, J. Postreplicative Mismatch Repair. Cold Spring Harb. Perspect. Biol. 2013, 5, a012633. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, R.J.; Karlseder, J. Telomeres: Protecting chromosomes against genome instability. Nat. Rev. Mol. Cell Biol. 2010, 11, 171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gramatges, M.M.; Bertuch, A.A. Short telomeres: From dyskeratosis congenita to sporadic aplastic anemia and malignancy. Transl. Res. J. Lab. Clin. Med. 2013, 162, 353–363. [Google Scholar] [CrossRef] [Green Version]
- Alder, J.K.; Chen, J.J.; Lancaster, L.; Danoff, S.; Su, S.C.; Cogan, J.D.; Vulto, I.; Xie, M.; Qi, X.; Tuder, R.M.; et al. Short telomeres are a risk factor for idiopathic pulmonary fibrosis. Proc. Natl. Acad. Sci. USA 2008, 105, 13051–13056. [Google Scholar] [CrossRef] [Green Version]
- Cai, Z.; Yan, L.J.; Ratka, A. Telomere shortening and Alzheimer’s disease. Neuromol. Med. 2013, 15, 25–48. [Google Scholar] [CrossRef]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef]
- Liyanage, V.R.B.; Jarmasz, J.S.; Murugeshan, N.; Del Bigio, M.R.; Rastegar, M.; Davie, J.R. DNA Modifications: Function and Applications in Normal and Disease States. Biology 2014, 3, 670–723. [Google Scholar] [CrossRef]
- Jin, B.; Li, Y.; Robertson, K.D. DNA Methylation: Superior or Subordinate in the Epigenetic Hierarchy? Genes Cancer 2011, 2, 607–617. [Google Scholar] [CrossRef] [Green Version]
- Horvath, S. DNA methylation age of human tissues and cell types. Genome Biol. 2013, 14, R115. [Google Scholar] [CrossRef] [Green Version]
- Johnson, A.A.; Akman, K.; Calimport, S.R.; Wuttke, D.; Stolzing, A.; de Magalhães, J.P. The Role of DNA Methylation in Aging, Rejuvenation, and Age-Related Disease. Rejuvenation Res. 2012, 15, 483–494. [Google Scholar] [CrossRef] [Green Version]
- Headon, D.J.; Emmal, S.A.; Ferguson, B.M.; Tucker, A.S.; Justice, M.J.; Sharpe, P.T.; Zonana, J.; Overbeek, P.A. Gene defect in ectodermal dysplasia implicates a death domain adapter in development. Nature 2001, 414, 913–916. [Google Scholar] [CrossRef]
- Franco, M.; Furstoss, O.; Simon, V.; Benistant, C.; Hong, W.J.; Roche, S. The Adaptor Protein Tom1L1 Is a Negative Regulator of Src Mitogenic Signaling Induced by Growth Factors. Mol. Cell. Biol. 2006, 26, 1932–1947. [Google Scholar] [CrossRef] [Green Version]
- Schaukowitch, K.; Reese, A.L.; Kim, S.K.; Kilaru, G.; Joo, J.Y.; Kavalali, E.T.; Kim, T.K. An intrinsic transcriptional program underlying synaptic scaling during activity suppression. Cell Rep. 2017, 18, 1512–1526. [Google Scholar] [CrossRef]
- Seto, E.; Yoshida, M. Erasers of Histone Acetylation: The Histone Deacetylase Enzymes. Cold Spring Harb. Perspect. Biol. 2014, 6, a018713. [Google Scholar] [CrossRef] [Green Version]
- Madeo, F.; Zimmermann, A.; Maiuri, M.C.; Kroemer, G. Essential role for autophagy in life span extension. J. Clin. Investig. 2015, 125, 85–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, K.T.; Min, K.T. Regulation of lifespan by histone deacetylase. Ageing Res. Rev. 2002, 1, 313–326. [Google Scholar] [CrossRef] [Green Version]
- Bhullar, K.S.; Hubbard, B.P. Lifespan and healthspan extension by resveratrol. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2015, 1852, 1209–1218. [Google Scholar] [CrossRef] [Green Version]
- Ryan, J.M.; Cristofalo, V.J. Histone acetylation during aging of human cells in culture. Biochem. Biophys. Res. Commun. 1972, 48, 735–742. [Google Scholar] [CrossRef]
- De Magalhães, J.P.; Curado, J.; Church, G.M. Meta-analysis of age-related gene expression profiles identifies common signatures of aging. Bioinformatics 2009, 25, 875–881. [Google Scholar] [CrossRef] [Green Version]
- Frenk, S.; Houseley, J. Gene expression hallmarks of cellular ageing. Biogerontology 2018, 19, 547–566. [Google Scholar] [CrossRef] [Green Version]
- Belfort, M. Mobile self-splicing introns and inteins as environmental sensors. Curr. Opin. Microbiol. 2017, 38, 51–58. [Google Scholar] [CrossRef]
- Rudan, M.; Bou Dib, P.; Musa, M.; Kanunnikau, M.; Sobocanec, S.; Rueda, D.; Warnecke, T.; Krisko, A. Normal mitochondrial function in Saccharomyces cerevisiae has become dependent on inefficient splicing. eLife 2018, 7, e35330. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Wang, Z.; Ma, T.; Wei, G.; Ni, T. Alternative splicing in aging and age-related diseases. Cold Spring Harb Perspect Biol. 2013, 5, a012609. [Google Scholar] [CrossRef]
- Lu, T.; Pan, Y.; Kao, S.-Y.; Li, C.; Kohane, I.; Chan, J.; Yankner, B.A. Gene regulation and DNA damage in the ageing human brain. Nature 2004, 429, 883–891. [Google Scholar] [CrossRef] [PubMed]
- Deschênes, M.; Chabot, B. The emerging role of alternative splicing in senescence and aging. Aging Cell 2017, 16, 918–933. [Google Scholar] [CrossRef] [PubMed]
- Anisimova, A.S.; Alexandrov, A.I.; Makarova, N.E.; Gladyshev, V.N.; Dmitriev, S.E. Protein synthesis and quality control in aging. Aging 2018, 10, 4269–4288. [Google Scholar] [CrossRef]
- Rogers, A.N.; Chen, D.; McColl, G.; Czerwieniec, G.; Felkey, K.; Gibson, B.W.; Hubbard, A.; Melov, S.; Lithgow, G.J.; Kapahi, P. Life span extension via eIF4G inhibition is mediated by posttranscriptional remodeling of stress response gene expression in C. elegans. Cell Metab. 2011, 14, 55–66. [Google Scholar] [CrossRef] [Green Version]
- Luce, M.C.; Bunn, C.L. Decreased accuracy of protein synthesis in extracts from aging human diploid fibroblasts. Exp. Gerontol. 1989, 24, 113–125. [Google Scholar] [CrossRef]
- Li, M.; Xiao, Z.Q.; Chen, Z.C.; Li, J.L.; Li, C.; Zhang, P.F.; Li, M.Y. Proteomic analysis of the aging-related proteins in human normal colon epithelial tissue. J. Biochem. Mol. Biol. 2007, 40, 72–81. [Google Scholar] [CrossRef]
- Yang, S.; Liu, T.; Li, S.; Zhang, X.; Ding, Q.; Que, H.; Yan, X.; Wei, K.; Liu, S. Comparative proteomic analysis of brains of naturally aging mice. Neuroscience 2008, 154, 1107–1120. [Google Scholar] [CrossRef]
- Waldera-Lupa, D.M.; Kalfalah, F.; Florea, A.M.; Sass, S.; Kruse, F.; Rieder, V.; Tigges, J.; Fritsche, E.; Krutmann, J.; Busch, H.; et al. Proteome-wide analysis reveals an age-associated cellular phenotype of in situ aged human fibroblasts. Aging 2014, 6, 856–878. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.C.; Chang, H.Y. Molecular mechanisms of long noncoding RNAs. Mol. Cell 2011, 43, 904–914. [Google Scholar] [CrossRef] [Green Version]
- Gomes, A.Q.; Nolasco, S.; Soares, H. Non-Coding RNAs: Multi-Tasking Molecules in the Cell. Int. J. Mol. Sci. 2013, 14, 16010–16039. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, J.C.; Acuña, S.M.; Aoki, J.I.; Floeter-Winter, L.M.; Muxel, S.M. Long non-coding RNAs in the regulation of gene expression: Physiology and disease. Non-Coding RNA 2019, 5, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef] [Green Version]
- Garg, D.; Cohen, S.M. miRNAs and aging: A genetic perspective. Ageing Res. Rev. 2014, 17, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sen, C.K.; Roy, S. MicroRNA 21 in tissue injury and inflammation. Cardiovasc. Res. 2012, 96, 230–233. [Google Scholar] [CrossRef]
- Hooten, N.N.; Fitzpatrick, M.; Wood, W.H.; De, S.; Ejiogu, N.; Zhang, Y.; Mattison, J.A.; Becker, K.G.; Zonderman, A.B.; Evans, M.K. Age-related changes in microRNA levels in serum. Aging 2013, 5, 725–740. [Google Scholar] [CrossRef] [Green Version]
- Fernandes, D.P.; Bitar, M.; Jacobs, F.; Barry, G. Long Non-Coding RNAs in Neuronal Aging. Non-Coding RNA 2018, 4, 12. [Google Scholar] [CrossRef] [Green Version]
- Riva, P.; Ratti, A.; Venturin, M. The Long Non-Coding RNAs in Neurodegenerative Diseases: Novel Mechanisms of Pathogenesis. Curr. Alzheimer Res. 2016, 13, 1219–1231. [Google Scholar] [CrossRef]
- Sahin, E.; DePinho, R.A. Axis of ageing: Telomeres, p53 and mitochondria. Nat. Rev. Mol. Cell Biol. 2012, 13, 397. [Google Scholar] [CrossRef] [Green Version]
- Samuels, D.C.; Li, C.; Li, B.; Song, Z.; Torstenson, E.; Boyd Clay, H.; Rokas, A.; Thornton-Wells, T.A.; Moore, J.H.; Hughes, T.M.; et al. Recurrent tissue-specific mtDNA mutations are common in humans. PLoS Genet. 2013, 9, e1003929. [Google Scholar] [CrossRef] [PubMed]
- Sun, N.; Youle, R.J.; Finkel, T. The Mitochondrial Basis of Aging. Mol. Cell 2016, 61, 654–666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harman, D. The biologic clock: The mitochondria? J. Am. Geriatr. Soc. 1972, 20, 145–147. [Google Scholar] [CrossRef] [PubMed]
- Khrapko, K.; Vijg, J. Mitochondrial DNA mutations and aging: Devils in the details? Trends Genet. TIG 2009, 25, 91–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, C.B.; Larsson, N.-G. Mitochondrial DNA mutations in disease and aging. J. Cell Biol. 2011, 193, 809–818. [Google Scholar] [CrossRef] [Green Version]
- Pinto, M.; Moraes, C.T. Mechanisms linking mtDNA damage and aging. Free Radic. Biol. Med. 2015, 85, 250–258. [Google Scholar] [CrossRef] [Green Version]
- Fox, R.; Kim, H.-S.; Reddick, R.L.; Kujoth, G.C.; Prolla, T.A.; Tsutsumi, S.; Wada, Y.; Smithies, O.; Maeda, N. Mitochondrial DNA polymerase editing mutation, PolgD257A, reduces the diabetic phenotype of Akita male mice by suppressing appetite. Proc. Natl. Acad. Sci. USA 2011, 108, 8779–8784. [Google Scholar] [CrossRef] [Green Version]
- Wallace, D.C. Mitochondrial diseases in man and mouse. Science 1999, 283, 1482–1488. [Google Scholar] [CrossRef] [Green Version]
- Weir, H.J.; Murray, T.K.; Kehoe, P.G.; Love, S.; Verdin, E.M.; O’Neill, M.J.; Lane, J.D.; Balthasar, N. CNS SIRT3 expression is altered by reactive oxygen species and in Alzheimer’s disease. PLoS ONE 2012, 7, e48225. [Google Scholar] [CrossRef] [Green Version]
- Santos, J.; Leitao-Correia, F.; Sousa, M.J.; Leao, C. Dietary Restriction and Nutrient Balance in Aging. Oxidative Med. Cell. Longev. 2016, 2016, 4010357. [Google Scholar] [CrossRef] [Green Version]
- Renaville, R.; Hammadi, M.; Portetelle, D. Role of the somatotropic axis in the mammalian metabolism. Domest. Anim. Endocrinol. 2002, 23, 351–360. [Google Scholar] [CrossRef]
- Milman, S.; Huffman, D.M.; Barzilai, N. The Somatotropic Axis in Human Aging: Framework for the Current State of Knowledge and Future Research. Cell Metab. 2016, 23, 980–989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Heemst, D. Insulin, IGF-1 and longevity. Aging Dis. 2010, 1, 147–157. [Google Scholar] [PubMed]
- Kenyon, C. The first long-lived mutants: Discovery of the insulin/IGF-1 pathway for ageing. Philos. Trans. R. Soc. B Biol. Sci. 2011, 366, 9–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laplante, M.; Sabatini, D.M. mTOR signaling in growth control and disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruetenik, A.; Barrientos, A. Dietary restriction, mitochondrial function and aging: From yeast to humans. Biochim. Biophys. Acta 2015, 1847, 1434–1447. [Google Scholar] [CrossRef] [Green Version]
- Katewa, S.D.; Kapahi, P. Dietary restriction and aging, 2009. Aging Cell 2010, 9, 105–112. [Google Scholar] [CrossRef]
- Zoncu, R.; Efeyan, A.; Sabatini, D.M. mTOR: From growth signal integration to cancer, diabetes and ageing. Nat. Rev. Mol. cell Biol. 2011, 12, 21–35. [Google Scholar] [CrossRef] [Green Version]
- Mihaylova, M.M.; Shaw, R.J. The AMPK signaling pathway coordinates cell growth, autophagy and metabolism. Nat. Cell Biol. 2011, 13, 1016–1023. [Google Scholar] [CrossRef]
- Salminen, A.; Kaarniranta, K. AMP-activated protein kinase (AMPK) controls the aging process via an integrated signaling network. Ageing Res. Rev. 2012, 11, 230–241. [Google Scholar] [CrossRef]
- Kaeberlein, M.; McVey, M.; Guarente, L. The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms. Genes Dev. 1999, 13, 2570–2580. [Google Scholar] [CrossRef] [Green Version]
- Jing, H.; Lin, H. Sirtuins in epigenetic regulation. Chem. Rev. 2015, 115, 2350–2375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- North, B.J.; Verdin, E. Sirtuins: Sir2-related NAD-dependent protein deacetylases. Genome Biol. 2004, 5, 224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maynard, S.; Fang, E.F.; Scheibye-Knudsen, M.; Croteau, D.L.; Bohr, V.A. DNA damage, DNA repair, aging, and neurodegeneration. Cold Spring Harb. Perspect. Med. 2015, 5, a025130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rudolph, K.L.; Chang, S.; Lee, H.-W.; Blasco, M.; Gottlieb, G.J.; Greider, C.; DePinho, R.A. Longevity, stress response, and cancer in aging telomerase-deficient mice. Cell 1999, 96, 701–712. [Google Scholar] [CrossRef] [Green Version]
- Monaghan, R.M.; Barnes, R.G.; Fisher, K.; Andreou, T.; Rooney, N.; Poulin, G.B.; Whitmarsh, A.J. A nuclear role for the respiratory enzyme CLK-1 in regulating mitochondrial stress responses and longevity. Nat. Cell Biol. 2015, 17, 782. [Google Scholar] [CrossRef] [Green Version]
- Longo, V.D.; Kennedy, B.K. Sirtuins in aging and age-related disease. Cell 2006, 126, 257–268. [Google Scholar] [CrossRef] [Green Version]
- Carafa, V.; Altucci, L.; Nebbioso, A. Dual Tumor Suppressor and Tumor Promoter Action of Sirtuins in Determining Malignant Phenotype. Front. Pharmacol. 2019, 10, 38. [Google Scholar] [CrossRef] [Green Version]
- Holzenberger, M.; Dupont, J.; Ducos, B.; Leneuve, P.; Géloën, A.; Even, P.C.; Cervera, P.; Le Bouc, Y. IGF-1 receptor regulates lifespan and resistance to oxidative stress in mice. Nature 2003, 421, 182. [Google Scholar] [CrossRef]
- Massagué, J.; Chen, Y.-G. Controlling TGF-β signaling. Genes Dev. 2000, 14, 627–644. [Google Scholar]
- Colak, S.; Ten Dijke, P. Targeting TGF-beta Signaling in Cancer. Trends Cancer 2017, 3, 56–71. [Google Scholar] [CrossRef]
- Carlson, M.E.; Conboy, I.M. Regulating the Notch pathway in embryonic, adult and old stem cells. Curr. Opin. Pharmacol. 2007, 7, 303–309. [Google Scholar] [CrossRef] [PubMed]
- Weinmaster, G. The ins and outs of notch signaling. Mol. Cell. Neurosci. 1997, 9, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Polychronidou, E.; Vlachakis, D.; Vlamos, P.; Baumann, M.; Kossida, S. Notch signaling and ageing. Adv. Exp. Med. Biol. 2015, 822, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Carlson, M.E.; Silva, H.S.; Conboy, I.M. Aging of signal transduction pathways, and pathology. Exp. Cell Res. 2008, 314, 1951–1961. [Google Scholar] [CrossRef] [Green Version]
- Moon, R. Wnt and β-catenin signaling: Diseases and therapies. Nat. Rev. Genet. 2004, 5, 689–699. [Google Scholar] [CrossRef]
- Tan, P.; Wang, Y.J.; Li, S.; Wang, Y.; He, J.Y.; Chen, Y.Y.; Deng, H.Q.; Huang, W.; Zhan, J.K.; Liu, Y.S. The PI3K/Akt/mTOR pathway regulates the replicative senescence of human VSMCs. Mol. Cell. Biochem. 2016, 422, 1–10. [Google Scholar] [CrossRef]
- Johnson, S.C.; Rabinovitch, P.S.; Kaeberlein, M. mTOR is a key modulator of ageing and age-related disease. Nature 2013, 493, 338–345. [Google Scholar] [CrossRef] [Green Version]
- Kolch, W. Coordinating ERK/MAPK signaling through scaffolds and inhibitors. Nat. Rev. Mol. Cell Biol. 2005, 6, 827–837. [Google Scholar] [CrossRef]
- Slack, C. Ras signaling in aging and metabolic regulation. Nutr. Healthy Aging 2017, 4, 195–205. [Google Scholar] [CrossRef] [Green Version]
- Zou, J.; Lei, T.; Guo, P.; Yu, J.; Xu, Q.; Luo, Y.; Ke, R.; Huang, D. Mechanisms shaping the role of ERK1/2 in cellular senescence. Mol. Med. Rep. 2019, 19, 759–770. [Google Scholar]
- Jazwinski, S.M. The retrograde response: A conserved compensatory reaction to damage from within and from without. Prog. Mol. Biol. Transl. Sci. 2014, 127, 133–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heintz, C.; Doktor, T.K.; Lanjuin, A.; Escoubas, C.; Zhang, Y.; Weir, H.J.; Dutta, S.; Silva-Garcia, C.G.; Bruun, G.H.; Morantte, I.; et al. Splicing factor 1 modulates dietary restriction and TORC1 pathway longevity in C. elegans. Nature 2017, 541, 102–106. [Google Scholar] [CrossRef] [PubMed]
- Baker, M.J.; Palmer, C.S.; Stojanovski, D. Mitochondrial protein quality control in health and disease. Br. J. Pharmacol. 2014, 171, 1870–1889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pellegrino, M.W.; Nargund, A.M.; Haynes, C.M. Signaling the mitochondrial unfolded protein response. Biochim. Biophys. Acta 2013, 1833, 410–416. [Google Scholar] [CrossRef] [Green Version]
- Perić, M.; Lovrić, A.; Šarić, A.; Musa, M.; Bou Dib, P.; Rudan, M.; Nikolić, A.; Sobočanec, S.; Mikecin, A.; Dennerlein, S.; et al. TORC1-mediated sensing of chaperone activity alters glucose metabolism and extends lifespan. Aging Cell 2017, 16, 994–1005. [Google Scholar] [CrossRef]
- Murley, A.; Nunnari, J. The emerging network of mitochondria-organelle contacts. Mol. Cell 2016, 61, 648–653. [Google Scholar] [CrossRef] [Green Version]
- Rusinol, A.E.; Cui, Z.; Chen, M.H.; Vance, J.E. A unique mitochondria-associated membrane fraction from rat liver has a high capacity for lipid synthesis and contains pre-Golgi secretory proteins including nascent lipoproteins. J. Biol. Chem. 1994, 269, 27494–27502. [Google Scholar]
- Leite, M.F.; Thrower, E.C.; Echevarria, W.; Koulen, P.; Hirata, K.; Bennett, A.M.; Ehrlich, B.E.; Nathanson, M.H. Nuclear and cytosolic calcium are regulated independently. Proc. Natl. Acad. Sci. USA 2003, 100, 2975–2980. [Google Scholar] [CrossRef] [Green Version]
- Lasorsa, F.M.; Pinton, P.; Palmieri, L.; Scarcia, P.; Rottensteiner, H.; Rizzuto, R.; Palmieri, F. Peroxisomes as novel players in cell calcium homeostasis. J. Biol. Chem. 2008, 283, 15300–15308. [Google Scholar] [CrossRef] [Green Version]
- Wong, A.K.; Capitanio, P.; Lissandron, V.; Bortolozzi, M.; Pozzan, T.; Pizzo, P. Heterogeneity of Ca2+ handling among and within Golgi compartments. J. Mol. Cell Biol. 2013, 5, 266–276. [Google Scholar] [CrossRef]
- Behringer, E.J.; Segal, S.S. Impact of aging on calcium signaling and membrane potential in endothelium of resistance arteries: A role for mitochondria. J. Gerontol. Ser. A Biomed. Sci. Med Sci. 2017, 72, 1627–1637. [Google Scholar] [CrossRef] [PubMed]
- Puzianowska-Kuznicka, M.; Kuznicki, J. The ER and ageing II: Calcium homeostasis. Ageing Res. Rev. 2009, 8, 160–172. [Google Scholar] [CrossRef] [PubMed]
- Chandhok, G.; Lazarou, M.; Neumann, B. Structure, function, and regulation of mitofusin-2 in health and disease. Biol. Rev. Camb. Philos. Soc. 2018, 93, 933–949. [Google Scholar] [CrossRef] [Green Version]
- Janikiewicz, J.; Szymański, J.; Malinska, D.; Patalas-Krawczyk, P.; Michalska, B.; Duszyński, J.; Giorgi, C.; Bonora, M.; Dobrzyn, A.; Wieckowski, M.R. Mitochondria-associated membranes in aging and senescence: Structure, function, and dynamics. Cell Death Dis. 2018, 9, 1–12. [Google Scholar] [CrossRef]
- Sebastian, D.; Hernandez-Alvarez, M.I.; Segales, J.; Sorianello, E.; Munoz, J.P.; Sala, D.; Waget, A.; Liesa, M.; Paz, J.C.; Gopalacharyulu, P.; et al. Mitofusin 2 (Mfn2) links mitochondrial and endoplasmic reticulum function with insulin signaling and is essential for normal glucose homeostasis. Proc. Natl. Acad. Sci. USA 2012, 109, 5523–5528. [Google Scholar] [CrossRef] [Green Version]
- Shankar, J.; Kojic, L.D.; St-Pierre, P.; Wang, P.T.; Fu, M.; Joshi, B.; Nabi, I.R. Raft endocytosis of AMF regulates mitochondrial dynamics through Rac1 signaling and the Gp78 ubiquitin ligase. J. Cell Sci. 2013, 126, 3295–3304. [Google Scholar] [CrossRef] [Green Version]
- Fang, L.; Moore, X.L.; Gao, X.M.; Dart, A.M.; Lim, Y.L.; Du, X.J. Down-regulation of mitofusin-2 expression in cardiac hypertrophy in vitro and in vivo. Life Sci. 2007, 80, 2154–2160. [Google Scholar] [CrossRef]
- Bach, D.; Naon, D.; Pich, S.; Soriano, F.X.; Vega, N.; Rieusset, J.; Laville, M.; Guillet, C.; Boirie, Y.; Wallberg-Henriksson, H.; et al. Expression of Mfn2, the Charcot-Marie-Tooth neuropathy type 2A gene, in human skeletal muscle: Effects of type 2 diabetes, obesity, weight loss, and the regulatory role of tumor necrosis factor alpha and interleukin-6. Diabetes 2005, 54, 2685–2693. [Google Scholar] [CrossRef] [Green Version]
- Area-Gomez, E.; del Carmen Lara Castillo, M.; Tambini, M.D.; Guardia-Laguarta, C.; de Groof, A.J.C.; Madra, M.; Ikenouchi, J.; Umeda, M.; Bird, T.D.; Sturley, S.L.; et al. Upregulated function of mitochondria-associated ER membranes in Alzheimer disease. EMBO J. 2012, 31, 4106–4123. [Google Scholar] [CrossRef] [Green Version]
- Sala, A.J.; Bott, L.C.; Morimoto, R.I. Shaping proteostasis at the cellular, tissue, and organismal level. J. Cell Biol. 2017. [Google Scholar] [CrossRef] [Green Version]
- Koga, H.; Kaushik, S.; Cuervo, A.M. Protein Homeostasis and Aging: The importance of exquisite quality control. Ageing Res. Rev. 2011, 10, 205–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sampaio-Marques, B.; Ludovico, P. Linking cellular proteostasis to yeast longevity. FEMS Yeast Res. 2018, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andréasson, C.; Ott, M.; Büttner, S. Mitochondria orchestrate proteostatic and metabolic stress responses. EMBO Rep. 2019, 20, e47865. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, S.; Cuervo, A.M. Proteostasis and aging. Nat. Med. 2015, 21, 1406. [Google Scholar] [CrossRef]
- Nedelsky, N.; Todd, P.K.; Taylor, J.P. Autophagy and the ubiquitin-proteasome system: Collaborators in neuroprotection. Biochim. Biophys. Acta 2008, 1782, 691–699. [Google Scholar] [CrossRef] [Green Version]
- Klaips, C.L.; Jayaraj, G.G.; Hartl, F.U. Pathways of cellular proteostasis in aging and disease. J. Cell Biol. 2018, 217, 51–63. [Google Scholar] [CrossRef]
- Taylor, J.P.; Hardy, J.; Fischbeck, K.H. Toxic proteins in neurodegenerative disease. Science 2002, 296, 1991–1995. [Google Scholar] [CrossRef]
- Eisele, Y.S.; Monteiro, C.; Fearns, C.; Encalada, S.E.; Wiseman, R.L.; Powers, E.T.; Kelly, J.W. Targeting Protein Aggregation for the Treatment of Degenerative Diseases. Nat. Rev. Drug Discov. 2015, 14, 759–780. [Google Scholar] [CrossRef] [Green Version]
- von Mikecz, A.; Chen, M.; Rockel, T.; Scharf, A. The nuclear ubiquitin-proteasome system: Visualization of proteasomes, protein aggregates, and proteolysis in the cell nucleus. Methods Mol. Biol. 2008, 463, 191–202. [Google Scholar] [CrossRef]
- Rao, R.V.; Bredesen, D.E. Misfolded proteins, endoplasmic reticulum stress and neurodegeneration. Curr. Opin. Cell Biol. 2004, 16, 653–662. [Google Scholar] [CrossRef] [Green Version]
- Oslowski, C.M.; Urano, F. Measuring ER stress and the unfolded protein response using mammalian tissue culture system. Methods Enzymol. 2011, 490, 71–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, J. Endoplasmic Reticulum (ER) Stress in the Pathogenesis of Type 1 Diabetes. In Type 1 Diabetes; Alan, P.E., Alice, L., Eds.; IntechOpen: London, UK, 2013. [Google Scholar] [CrossRef]
- Lindholm, D.; Wootz, H.; Korhonen, L. ER stress and neurodegenerative diseases. Cell Death Differ. 2006, 13, 385. [Google Scholar] [CrossRef] [PubMed]
- Deretic, V. A Master Conductor for Aggregate Clearance by Autophagy. Dev. Cell 2010, 18, 694–696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eskelinen, E.-L. Macroautophagy in Mammalian Cells. 2013. Available online: https://www.ncbi.nlm.nih.gov/books/NBK6211/ (accessed on 12 December 2019).
- Bandyopadhyay, U.; Kaushik, S.; Varticovski, L.; Cuervo, A.M. The chaperone-mediated autophagy receptor organizes in dynamic protein complexes at the lysosomal membrane. Mol. Cell. Biol. 2008, 28, 5747–5763. [Google Scholar] [CrossRef] [Green Version]
- Mizushima, N.; Levine, B. Autophagy in mammalian development and differentiation. Nat. Cell Biol. 2010, 12, 823–830. [Google Scholar] [CrossRef]
- Wang, C.W.; Klionsky, D.J. The Molecular Mechanism of Autophagy. Mol. Med. 2003, 9, 65–76. [Google Scholar] [CrossRef] [Green Version]
- Bejarano, E.; Cuervo, A.M. Chaperone-Mediated Autophagy. Proc. Am. Thorac. Soc. 2010, 7, 29–39. [Google Scholar] [CrossRef]
- Hipp, M.S.; Kasturi, P.; Hartl, F.U. The proteostasis network and its decline in ageing. Nat. Rev. Mol. Cell Biol. 2019, 20, 421–435. [Google Scholar] [CrossRef]
- Moreau, K.L.; King, J.A. Protein misfolding and aggregation in cataract disease and prospects for prevention. Trends Mol. Med. 2012, 18, 273–282. [Google Scholar] [CrossRef] [Green Version]
- Lashuel, H.A.; Hartley, D.; Petre, B.M.; Walz, T.; Lansbury Jr, P.T. Neurodegenerative disease: Amyloid pores from pathogenic mutations. Nature 2002, 418, 291. [Google Scholar] [CrossRef]
- Milanesi, L.; Sheynis, T.; Xue, W.-F.; Orlova, E.V.; Hellewell, A.L.; Jelinek, R.; Hewitt, E.W.; Radford, S.E.; Saibil, H.R. Direct three-dimensional visualization of membrane disruption by amyloid fibrils. Proc. Natl. Acad. Sci. USA 2012, 109, 20455–20460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, Z.; Yan, L.J. Protein Oxidative Modifications: Beneficial Roles in Disease and Health. J. Biochem. Pharmacol. Res. 2013, 1, 15–26. [Google Scholar] [PubMed]
- Fedorova, M.; Bollineni, R.C.; Hoffmann, R. Protein carbonylation as a major hallmark of oxidative damage: Update of analytical strategies. Mass Spectrom. Rev. 2014, 33, 79–97. [Google Scholar] [CrossRef]
- Semchyshyn, H.M. Reactive carbonyl species in vivo: Generation and dual biological effects. Sci. World J. 2014, 2014, 417842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riley, P.A. Free radicals in biology: Oxidative stress and the effects of ionizing radiation. Int. J. Radiat. Biol. 1994, 65, 27–33. [Google Scholar] [CrossRef]
- Negre-Salvayre, A.; Coatrieux, C.; Ingueneau, C.; Salvayre, R. Advanced lipid peroxidation end products in oxidative damage to proteins. Potential role in diseases and therapeutic prospects for the inhibitors. Br. J. Pharmacol. 2008, 153, 6–20. [Google Scholar] [CrossRef] [Green Version]
- Adams, S.; Green, P.; Claxton, R.; Simcox, S.; Williams, M.V.; Walsh, K.; Leeuwenburgh, C. Reactive carbonyl formation by oxidative and non-oxidative pathways. Front. Biosci. 2001, 6, A17–A24. [Google Scholar] [CrossRef] [Green Version]
- Reznick, A.Z.; Packer, L. [38] Oxidative damage to proteins: Spectrophotometric method for carbonyl assay. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 1994; Volume 233, pp. 357–363. [Google Scholar]
- Weber, D.; Davies, M.J.; Grune, T. Determination of protein carbonyls in plasma, cell extracts, tissue homogenates, isolated proteins: Focus on sample preparation and derivatization conditions. Redox Biol. 2015, 5, 367–380. [Google Scholar] [CrossRef] [Green Version]
- Levine, R.L.; Stadtman, E.R. Oxidative modification of proteins during aging. Exp. Gerontol. 2001, 36, 1495–1502. [Google Scholar] [CrossRef]
- Gianni, P.; Jan, K.J.; Douglas, M.J.; Stuart, P.M.; Tarnopolsky, M.A. Oxidative stress and the mitochondrial theory of aging in human skeletal muscle. Exp. Gerontol. 2004, 39, 1391–1400. [Google Scholar] [CrossRef]
- Floyd, R.A.; Hensley, K. Oxidative stress in brain aging: Implications for therapeutics of neurodegenerative diseases. Neurobiol. Aging 2002, 23, 795–807. [Google Scholar] [CrossRef]
- Mutlu-Türkoğlu, Ü.; İlhan, E.; Öztezcan, S.; Kuru, A.; Aykaç-Toker, G.; Uysal, M. Age-related increases in plasma malondialdehyde and protein carbonyl levels and lymphocyte DNA damage in elderly subjects. Clin. Biochem. 2003, 36, 397–400. [Google Scholar] [CrossRef]
- Forster, M.J.; Sohal, B.H.; Sohal, R.S. Reversible effects of long-term caloric restriction on protein oxidative damage. J. Gerontol. Ser. A Biol. Sci. Med Sci. 2000, 55, B522–B529. [Google Scholar] [CrossRef]
- Pickering, A.M.; Davies, K.J. Degradation of damaged proteins: The main function of the 20S proteasome. Prog. Mol. Biol. Transl. Sci. 2012, 109, 227–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanase, M.; Urbanska, A.M.; Zolla, V.; Clement, C.C.; Huang, L.; Morozova, K.; Follo, C.; Goldberg, M.; Roda, B.; Reschiglian, P. Role of carbonyl modifications on aging-associated protein aggregation. Sci. Rep. 2016, 6, 19311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Telci, A.; Çakatay, U.; Salman, S.; Satman, I.l.; Sivas, A. Oxidative protein damage in early stage Type 1 diabetic patients. Diabetes Res. Clin. Pract. 2000, 50, 213–223. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Lauderback, C.M. Lipid peroxidation and protein oxidation in Alzheimer’s disease brain: Potential causes and consequences involving amyloid β-peptide-associated free radical oxidative stress. Free Radic. Biol. Med. 2002, 32, 1050–1060. [Google Scholar] [CrossRef]
- Túnez, I.; Sánchez-López, F.; Agüera, E.; Fernández-Bolaños, R.; Sánchez, F.M.; Tasset-Cuevas, I. Important role of oxidative stress biomarkers in Huntington’s disease. J. Med. Chem. 2011, 54, 5602–5606. [Google Scholar] [CrossRef]
- Shaw, P.J.; Ince, P.G.; Falkous, G.; Mantle, D. Oxidative damage to protein in sporadic motor neuron disease spinal cord. Ann. Neurol. Off. J. Am. Neurol. Assoc. Child Neurol. Soc. 1995, 38, 691–695. [Google Scholar] [CrossRef]
- Katerji, M.; Filippova, M.; Duerksen-Hughes, P. Approaches and Methods to Measure Oxidative Stress in Clinical Samples: Research Applications in the Cancer Field. Oxidative Med. Cell. Longev. 2019, 2019, 1279250. [Google Scholar] [CrossRef] [Green Version]
- Aydin, B.; Yagci, R.; Yılmaz, F.M.; Erdurmus, M.; Karadağ, R.; Keskin, U.; Durmus, M.; Yigitoglu, R. Prevention of selenite-induced cataractogenesis by N-acetylcysteine in rats. Curr. Eye Res. 2009, 34, 196–201. [Google Scholar] [CrossRef] [PubMed]
- Czerwińska, J.; Poznański, J.; Dębski, J.; Bukowy, Z.; Bohr, V.A.; Tudek, B.; Speina, E. Catalytic activities of Werner protein are affected by adduction with 4-hydroxy-2-nonenal. Nucleic Acids Res. 2014, 42, 11119–11135. [Google Scholar] [CrossRef] [PubMed]
- Range, S.; Dunster, C.; Knox, A.; Kelly, F. Treatment of pulmonary exacerbations of cystic fibrosis leads to improved antioxidant status. Eur. Respir. J. 1999, 13, 560–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodrigo, R.; Libuy, M.; Feliú, F.; Hasson, D. Oxidative stress-related biomarkers in essential hypertension and ischemia-reperfusion myocardial damage. Dis. Markers 2013, 35, 773–790. [Google Scholar] [CrossRef] [Green Version]
- Alam, Z.I.; Daniel, S.E.; Lees, A.J.; Marsden, D.C.; Jenner, P.; Halliwell, B. A generalised increase in protein carbonyls in the brain in Parkinson’s but not incidental Lewy body disease. J. Neurochem. 1997, 69, 1326–1329. [Google Scholar] [CrossRef]
- Aksenov, M.; Aksenova, M.; Butterfield, D.; Geddes, J.; Markesbery, W. Protein oxidation in the brain in Alzheimer’s disease. Neuroscience 2001, 103, 373–383. [Google Scholar] [CrossRef]
- Ferrante, R.J.; Browne, S.E.; Shinobu, L.A.; Bowling, A.C.; Baik, M.J.; MacGarvey, U.; Kowall, N.W.; Brown, R.H., Jr.; Beal, M.F. Evidence of increased oxidative damage in both sporadic and familial amyotrophic lateral sclerosis. J. Neurochem. 1997, 69, 2064–2074. [Google Scholar] [CrossRef]
- Aryal, B.; Rao, V.A. Specific protein carbonylation in human breast cancer tissue compared to adjacent healthy epithelial tissue. PLoS ONE 2018, 13, e0194164. [Google Scholar] [CrossRef]
- Cabiscol, E.; Tamarit, J.; Ros, J. Protein carbonylation: Proteomics, specificity and relevance to aging. Mass Spectrom. Rev. 2014, 33, 21–48. [Google Scholar] [CrossRef]
- De Graff, A.M.; Hazoglou, M.J.; Dill, K.A. Highly Charged Proteins: The Achilles’ Heel of Aging Proteomes. Structure 2016, 24, 329–336. [Google Scholar] [CrossRef] [Green Version]
- Griffiths, H.R. Antioxidants and protein oxidation. Free Radic. Res. 2000, 33, S47–S58. [Google Scholar]
- Wehr, N.B.; Levine, R.L. Quantification of protein carbonylation. Methods Mol. Biol. 2013, 965, 265–281. [Google Scholar] [CrossRef] [PubMed]
- Augustyniak, E.; Adam, A.; Wojdyla, K.; Rogowska-Wrzesinska, A.; Willetts, R.; Korkmaz, A.; Atalay, M.; Weber, D.; Grune, T.; Borsa, C.; et al. Validation of protein carbonyl measurement: A multi-centre study. Redox Biol. 2015, 4, 149–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodríguez-García, A.; Morales, M.L.; Garrido-García, V.; García-Baquero, I.; Leivas, A.; Carreño-Tarragona, G.; Sánchez, R.; Arenas, A.; Cedena, T.; Ayala, R.M. Protein Carbonylation in Patients with Myelodysplastic Syndrome: An Opportunity for Deferasirox Therapy. Antioxidants 2019, 8, 508. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
| Signaling Pathway | Mechanism | Role in Senescence Context | Ref. |
|---|---|---|---|
| Genome surveillance signaling | DNA repair | Normal lifespan control Mutations in the number of DNA repair genes caused premature aging. | [93] |
| Telomere length-maintaining | Replicative lifespan control Dysfunctions were observed in telomere-mediated syndromes, such as dyskeratosis congenital, idiopathic pulmonary fibrosis, and Alzheimer’s disease. | [31,32,33,94] | |
| Tumor-suppressor expressions | Promotion of longevity through cancer prevention Splicing defects of the p53 gene occurred in progeria, vascular aging, and Alzheimer’s disease. | [53] | |
| Mitochondria and ROS signaling | Electron transport | Cellular energy control, reactive oxygen species (ROS) production/detoxification, and apoptosis A reduction of energy and ROS production could extend lifespans while reducing oxidative stress and the formation of carbonylated proteins. | [95] |
| Sirtuin deacetylase activity | Regulation of replicative lifespan In neurodegenerative diseases, sirtuin expression increased, and they acted as neuroprotective molecules in sensing and mitigating ROS. Sirtuin proteins could promote or suppress cancer development. | [78,96,97] | |
| Hormonal signaling | Insulin/IGF-1 activity | Growth, remodeling, and aging of tissues The insulin-like growth factor (IGF) system could play an important role in life processes (cell growth, division, differentiation, apoptosis, aging, and others) by binding with the receptor or activating multiple intracellular signaling cascades. Deregulation of the IGF-1 mechanism was associated with progeria, vascular aging, and Alzheimer’s disease. | [53,98] |
| Transforming growth factor (TGF)-β action | Cell growth and proliferation, migration, the regulation of the inflammatory response, wound healing, fibrosis, and cellular apoptosis Impairment of the TGF-β1 signaling pathway was demonstrated to be specific for brain cells in Alzheimer’s patients, fibrosis, and various types of human cancer, including breast, colon, and renal cancer. | [99,100] | |
| Metabolic signaling | Notch action | Embryogenesis, maintenance of tissue specific homeostasis, and stem cell differentiation The Notch pathway controlled proliferation, migration, the functions of tissue cells, as well as cross-talk between inflammatory cells and the innate immune system. Notch mutations were associated with sporadic Alzheimer’s disease and other neurodegenerative diseases such as Down syndrome, Pick’s and Prion’s disease, and CADASIL (Cerebral Autosomal Dominant Arteriopathy with Subcortical Infarcts and Leucoencephalopathy). | [101,102,103] |
| Wnt action | Embryogenesis, cell fate determination, and cell survival Increased Wnt levels inhibited myogenic differentiation in the elderly. Impaired Notch-TGF-β–Wnt balances in stem cells resulted in the loss of cellular homeostasis. | [104,105] | |
| Phosphoinositide 3-kinase (PI3K)/dependent-protein kinase B (AKT)/mTORC2 (PI3K/AKT/mTORC2) cascade | Cell proliferation, the regulation of translation, the quality control of proteins, and autophagy The regulation of pro- and antiaging signaling. The dysregulation of the PI3K–AKT–mTORC2 pathway was strongly associated with tumorigenesis. | [106,107] | |
| Serine/threonine-protein kinase (Raf)/ Mitogen-activated protein kinases (MAPK/ERK) cascade | Regulation of apoptosis, cell survival, motility, adhesion, proliferation This pathway has a role in delivering extracellular signals to the nucleus, regulating cellular behavior and longevity. | [108,109,110] | |
| Ras/cAMP-dependent protein kinase cascade | Regulation of cell survival, replicative senescence, autophagy, and cytoskeleton organization This cascade regulates caloric restriction and chronological lifespan in yeast. Mutations in Ras resulted in its activation occurring in 1/3 of human tumors (e.g., melanoma, thyroid, colon, and ovarian cancers). | [22,109] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rudzińska, M.; Parodi, A.; Balakireva, A.V.; Chepikova, O.E.; Venanzi, F.M.; Zamyatnin, A.A., Jr. Cellular Aging Characteristics and Their Association with Age-Related Disorders. Antioxidants 2020, 9, 94. https://doi.org/10.3390/antiox9020094
Rudzińska M, Parodi A, Balakireva AV, Chepikova OE, Venanzi FM, Zamyatnin AA Jr. Cellular Aging Characteristics and Their Association with Age-Related Disorders. Antioxidants. 2020; 9(2):94. https://doi.org/10.3390/antiox9020094
Chicago/Turabian StyleRudzińska, Magdalena, Alessandro Parodi, Anastasia V. Balakireva, Olga E. Chepikova, Franco M. Venanzi, and Andrey A. Zamyatnin, Jr. 2020. "Cellular Aging Characteristics and Their Association with Age-Related Disorders" Antioxidants 9, no. 2: 94. https://doi.org/10.3390/antiox9020094