
Synergistic Protective Effect of Fermented Schizandrae Fructus Pomace and Hoveniae Semen cum Fructus Extracts Mixture in the Ethanol-Induced Hepatotoxicity


Abstract
:1. Introduction
2. Materials and Methods
2.1. Preparation of Silymarin, fSFP, HSCF, and MSH
2.2. Animal Husbandry and Experiment
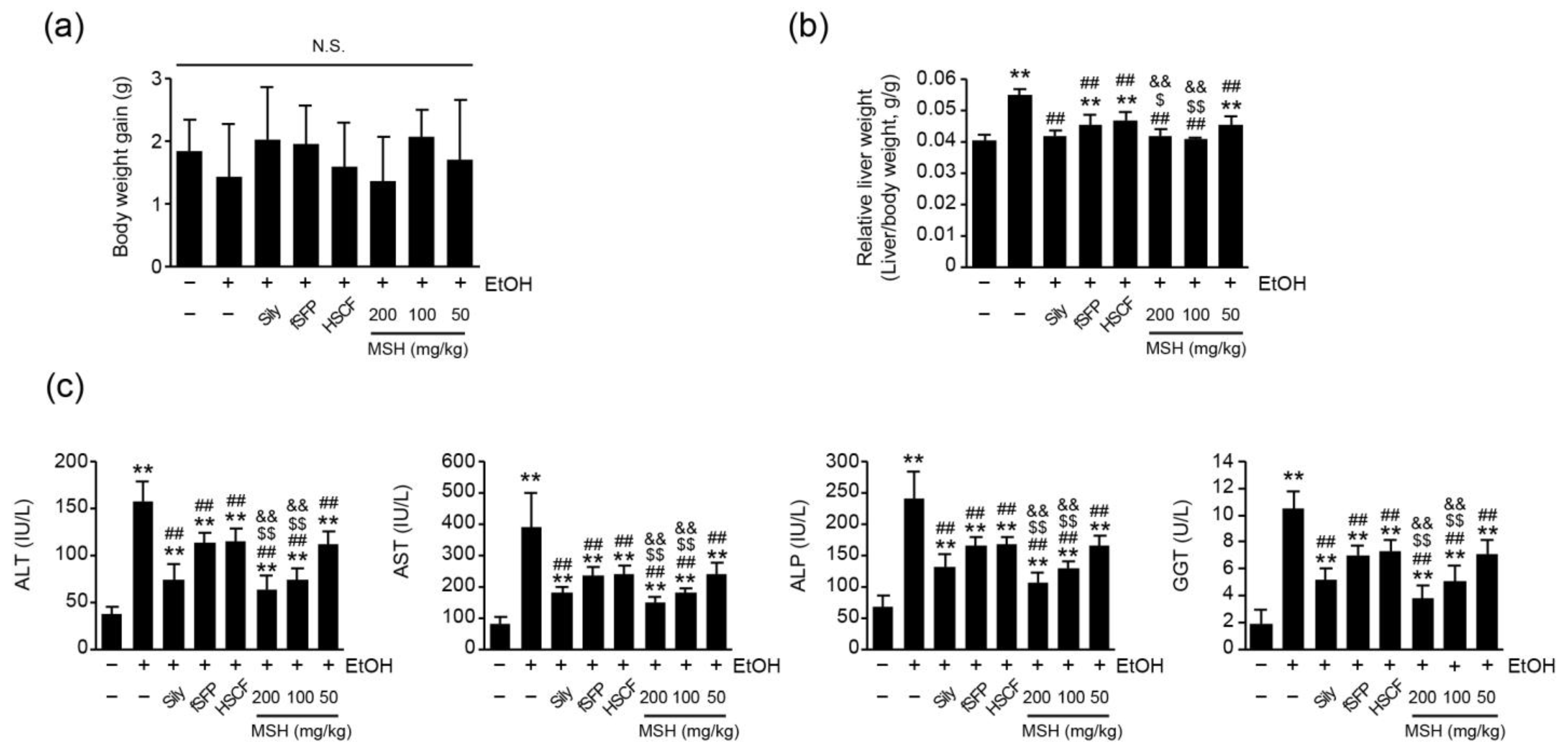
2.3. Measurements of Body and Liver Weight
2.4. Serum Biochemistry
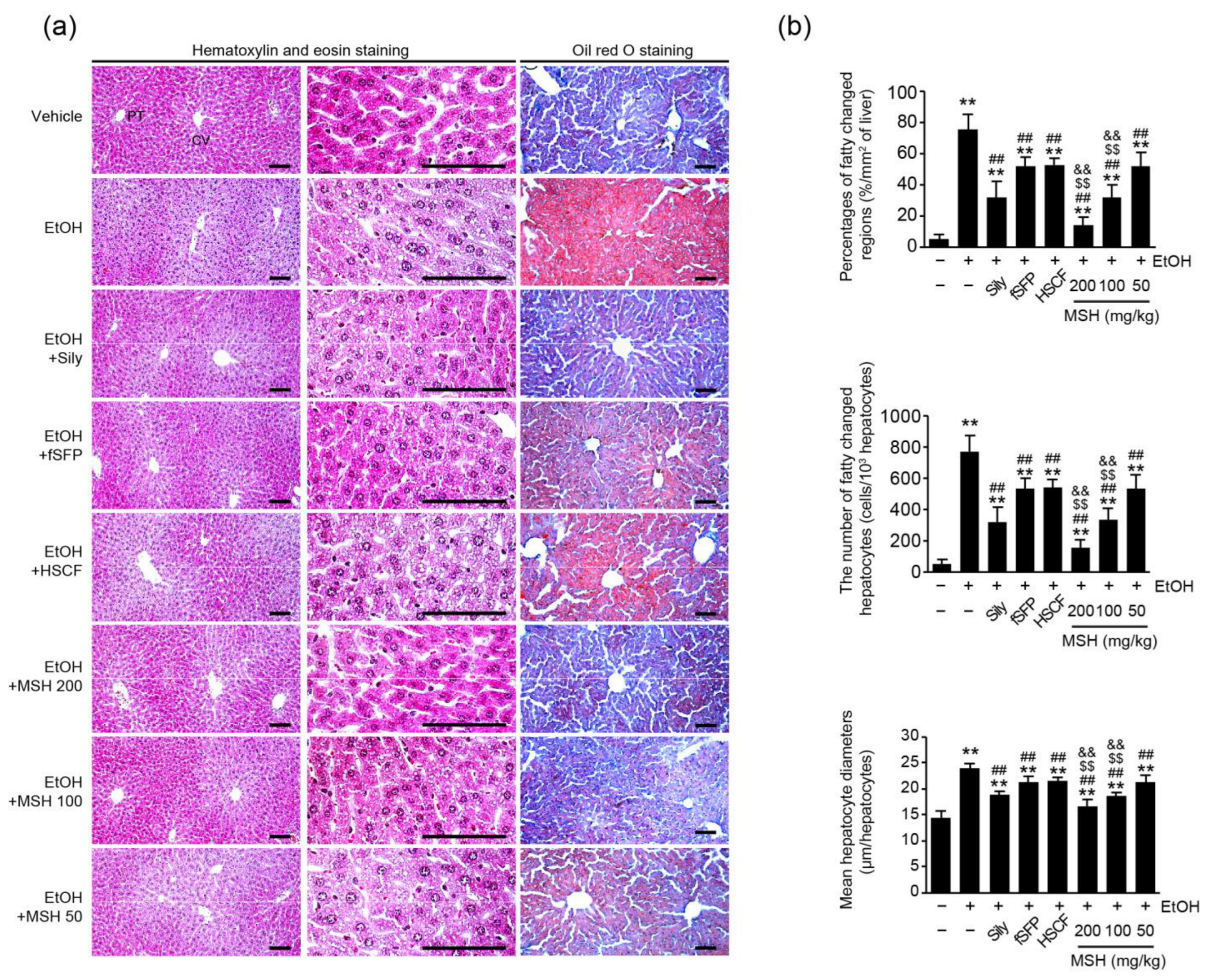
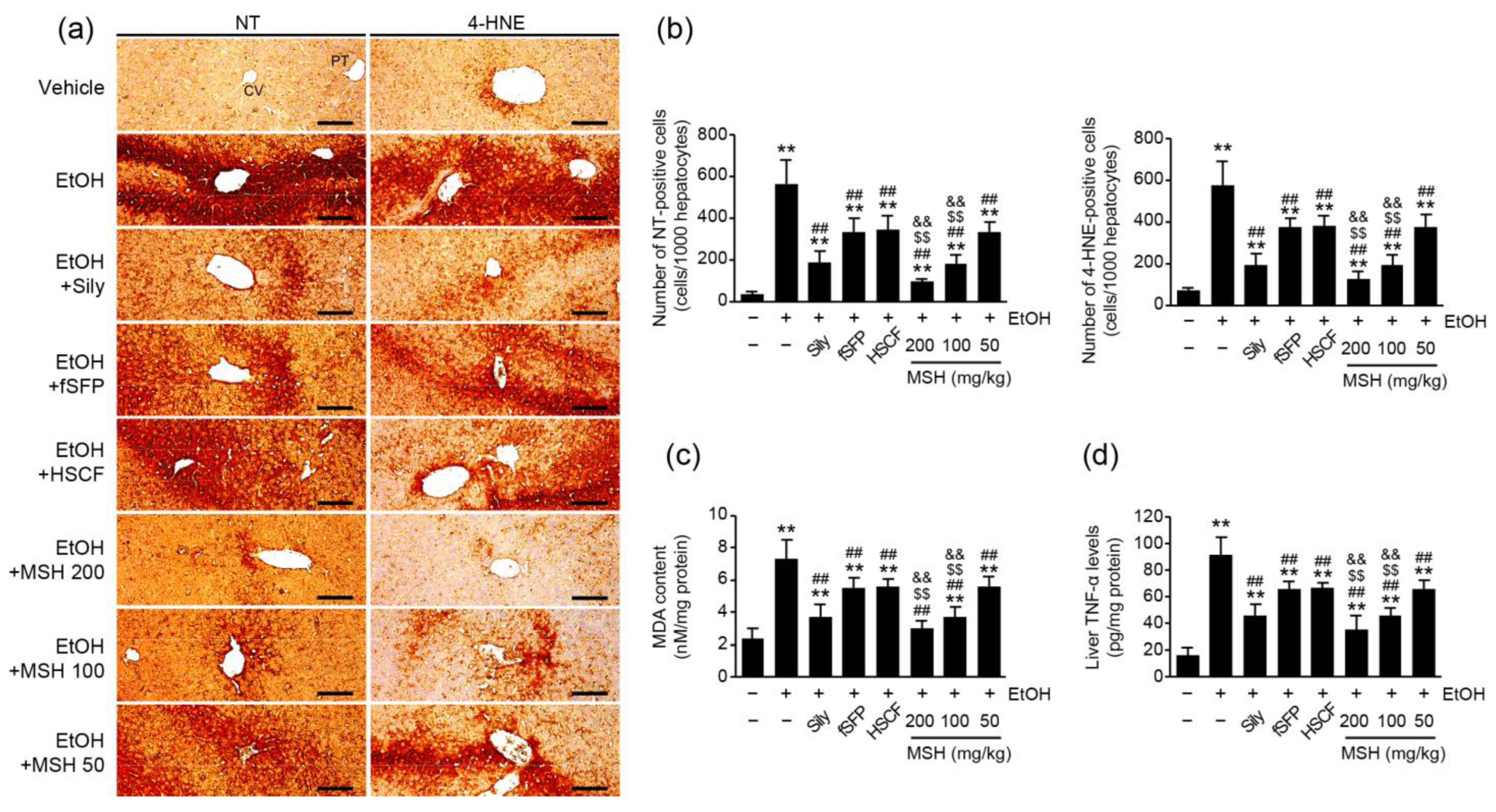
2.5. Histopathological Analysis and Immunohistochemistry
2.6. Enzyme-Linked Immunosorbent Assay (ELISA)
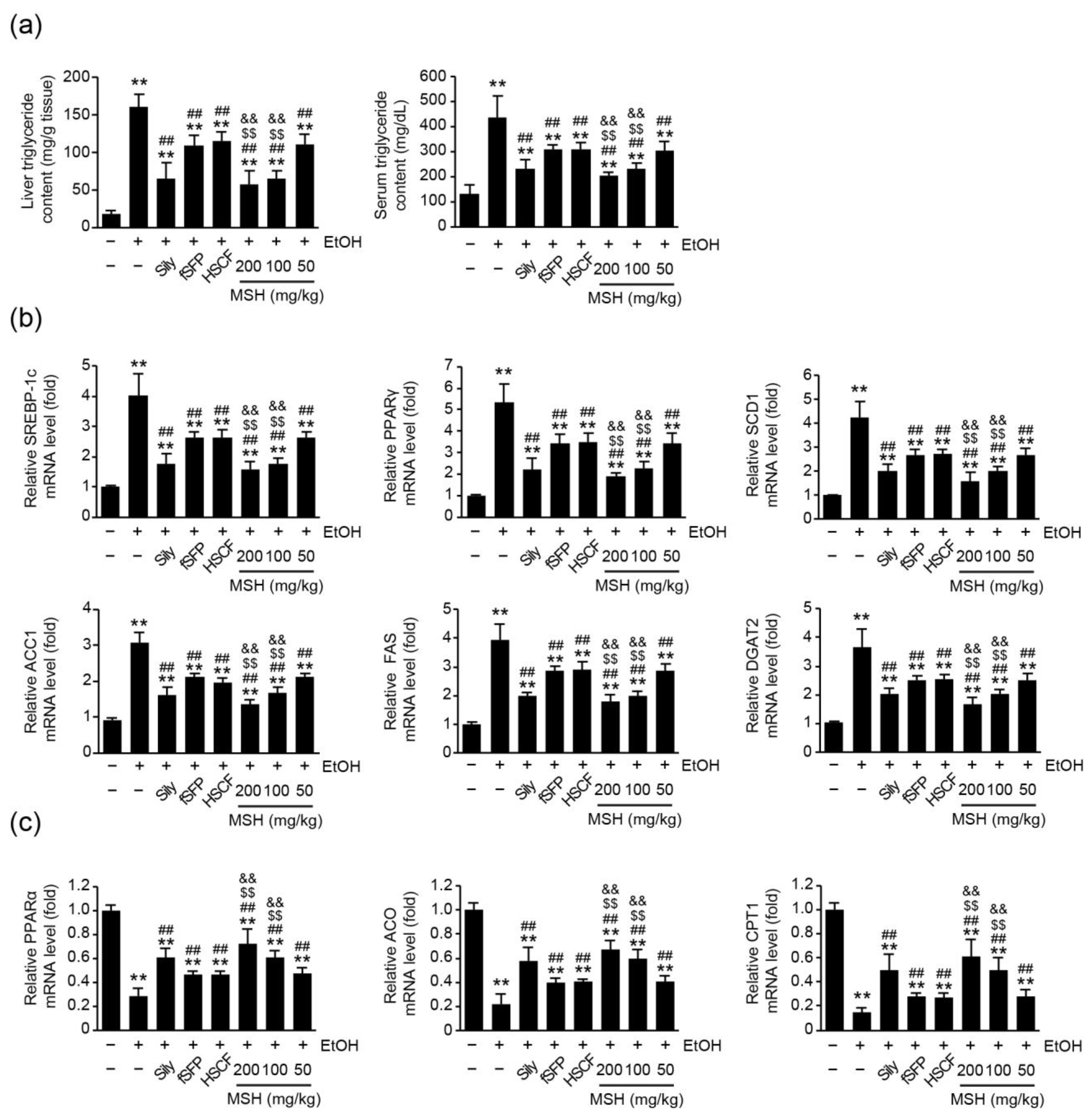
2.7. Quantitative Reverse Transcription Polymerase Chain Reaction (RT-qPCR)
2.8. Measurement of Liver Lipid Peroxidation
2.9. Measurement of Antioxidant Capacities in the Liver
2.10. Determination of Liver CYP2E1 Activity
2.11. Statistical Analysis
3. Results
3.1. MSH Synergistically Ameliorated Ethanol-Induced Liver Injury in Mice
3.2. MSH Synergistically Alleviated Ethanol-Induced Hepatic Steatosis in Mice
3.3. MSH Alleviated Ethanol-Induced Hepatic TG Accumulation via Modulating Fatty Acid Metabolism
3.4. MSH Inhibited Ethanol-Mediated Oxidative Stress and Inflammation in Mice
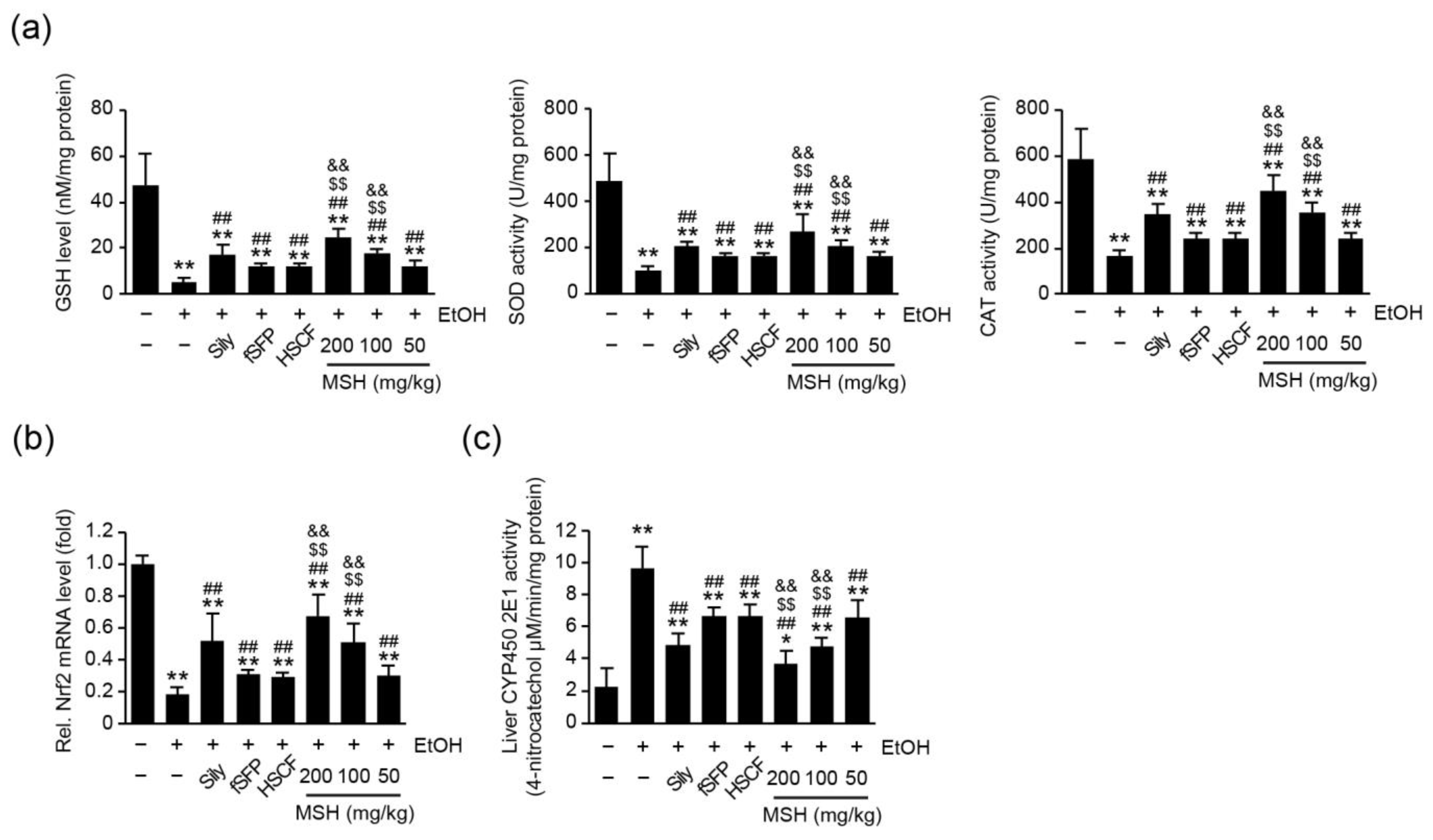
3.5. MSH Enhanced the Endogenous Antioxidant Capacity and Lowered the CYP2E1 Activity in Liver
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rehm, J.; Mathers, C.; Popova, S.; Thavorncharoensap, M.; Teerawattananon, Y.; Patra, J. Global burden of disease and injury and economic cost attributable to alcohol use and alcohol-use disorders. Lancet 2009, 373, 2223–2233. [Google Scholar] [CrossRef] [PubMed]
- Thursz, M.; Gual, A.; Lackner, C.; Mathurin, P.; Moreno, C.; Spahr, L.; Sterneck, M.; Cortez-Pinto, H. EASL Clinical Practice Guidelines: Management of alcohol-related liver disease. J. Hepatol. 2018, 69, 154–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheron, N. Alcohol and liver disease in Europe--Simple measures have the potential to prevent tens of thousands of premature deaths. J. Hepatol. 2016, 64, 957–967. [Google Scholar] [CrossRef] [PubMed]
- Gao, B.; Bataller, R. Alcoholic Liver Disease: Pathogenesis and New Therapeutic Targets. Gastroenterology 2011, 141, 1572–1585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subramaniyan, V.; Chakravarthi, S.; Jegasothy, R.; Seng, W.Y.; Fuloria, N.K.; Fuloria, S.; Hazarika, I.; Das, A. Alcohol-associated liver disease: A review on its pathophysiology, diagnosis and drug therapy. Toxicol. Rep. 2021, 8, 376–385. [Google Scholar] [CrossRef] [PubMed]
- You, M.; Fischer, M.; Deeg, M.A.; Crabb, D.W. Ethanol Induces Fatty Acid Synthesis Pathways by Activation of Sterol Regulatory Element-binding Protein (SREBP). J. Biol. Chem. 2002, 277, 29342–29347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galli, A.; Pinaire, J.; Fischer, M.; Dorris, R.; Crabb, D.W. The Transcriptional and DNA Binding Activity of Peroxisome Proliferator-activated Receptor α Is Inhibited by Ethanol Metabolism. J. Biol. Chem. 2001, 276, 68–75. [Google Scholar] [CrossRef] [Green Version]
- Cederbaum, A.I. Alcohol metabolism. Clin. Liver. Dis. 2012, 16, 667–685. [Google Scholar] [CrossRef] [Green Version]
- Cederbaum, A.I.; Lu, Y.; Wu, D. Role of oxidative stress in alcohol-induced liver injury. Arch. Toxicol. 2009, 83, 519–548. [Google Scholar] [CrossRef]
- Yuan, R.; Tao, X.; Liang, S.; Pan, Y.; He, L.; Sun, J.; Wenbo, J.; Li, X.; Chen, J.; Wang, C. Protective effect of acidic polysaccharide from Schisandra chinensis on acute ethanol-induced liver injury through reducing CYP2E1-dependent oxidative stress. Biomed. Pharmacother. 2018, 99, 537–542. [Google Scholar] [CrossRef]
- Choi, B.R.; Cho, I.J.; Jung, S.J.; Kim, J.K.; Park, S.M.; Lee, D.G.; Ku, S.K.; Park, K.M. Lemon balm and dandelion leaf extract synergistically alleviate ethanol-induced hepatotoxicity by enhancing antioxidant and anti-inflammatory activity. J. Food Biochem. 2020, 44, e13232. [Google Scholar] [CrossRef] [PubMed]
- Cho, I.; Kim, J.; Jung, J.; Sung, S.; Kim, J.; Lee, N.; Ku, S. Hepatoprotective effects of hoveniae semen cum fructus extracts in ethanol intoxicated mice. J. Exerc. Nutrition. Biochem. 2016, 20, 49–64. [Google Scholar] [CrossRef]
- Ding, R.B.; Tian, K.; Huang, L.L.; He, C.W.; Jiang, Y.; Wang, Y.T.; Wan, J.B. Herbal medicines for the prevention of alcoholic liver disease: A review. J. Ethnopharmacol. 2012, 144, 457–465. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Nie, Y.; Luo, M.; Chen, Z.; He, B. Natural Compounds: A Potential Treatment for Alcoholic Liver Disease? Front. Pharmacol. 2021, 12, 694475. [Google Scholar] [CrossRef] [PubMed]
- Sferrazza, G.; Brusotti, G.; Zonfrillo, M.; Temporini, C.; Tengattini, S.; Bononi, M.; Tateo, F.; Calleri, E.; Pierimarchi, P. Hovenia dulcis Thumberg: Phytochemistry, Pharmacology, Toxicology and Regulatory Framework for Its Use in the European Union. Molecules 2021, 26, 903. [Google Scholar] [CrossRef]
- Yang, K.; Qiu, J.; Huang, Z.; Yu, Z.; Wang, W.; Hu, H.; You, Y. A comprehensive review of ethnopharmacology, phytochemistry, pharmacology, and pharmacokinetics of Schisandra chinensis (Turcz.) Baill. and Schisandra sphenanthera Rehd. et Wils. J. Ethnopharmacol. 2022, 284, 114759. [Google Scholar] [CrossRef]
- Park, H.J.; Lee, S.J.; Song, Y.; Jang, S.H.; Ko, Y.G.; Kang, S.N.; Chung, B.Y.; Kim, H.D.; Kim, G.S.; Cho, J.H. Schisandra chinensis prevents alcohol-induced fatty liver disease in rats. J. Med. Food 2014, 17, 103–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, L.; Li, B.; Liu, X.; Meng, X. Hepatoprotective Effects of Triterpenoid Isolated from Schizandra chinensis against Acute Alcohol-Induced Liver Injury in Mice. Food Sci. Technol. Res. 2013, 19, 1003–1009. [Google Scholar] [CrossRef]
- Nagappan, A.; Jung, D.Y.; Kim, J.H.; Lee, H.; Jung, M.H. Gomisin N Alleviates Ethanol-Induced Liver Injury through Ameliorating Lipid Metabolism and Oxidative Stress. Int. J. Mol. Sci. 2018, 19, 2601. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Qu, X.-N.; Han, Y.; Zheng, S.-W.; Wang, J.; Wang, Y.-P. Ameliorative Effects of 5-Hydroxymethyl-2-furfural (5-HMF) from Schisandra chinensis on Alcoholic Liver Oxidative Injury in Mice. Int. J. Mol. Sci. 2015, 16, 2446–2457. [Google Scholar] [CrossRef]
- Park, H.; Jegal, K.H.; Choi, B.; Kim, J.K.; Ku, S.K. Hepatoprotective effect of fermented Schizandrae Fructus Pomace extract and Hoveniae Semen Cum Fructus extract combination mixtures against carbon tetrachloride-induced acute liver injured mice. Herb. Formula Sci. 2023, 31, 53–65. [Google Scholar] [CrossRef]
- Lowry, O.; Rosebrough, N.; Farr, A.L.; Randall, R. Protein Measurement with the Folin Phenol Reagent. J. Biol. Chem. 1951, 193, 265–275. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.; Deaciuc, I.; Song, M.; Lee, D.Y.; Liu, Y.; Ji, X.; McClain, C. Silymarin protects against acute ethanol-induced hepatotoxicity in mice. Alcohol. Clin. Exp. Res. 2006, 30, 407–413. [Google Scholar] [CrossRef] [PubMed]
- Tsai, J.H.; Liu, J.Y.; Wu, T.T.; Ho, P.C.; Huang, C.Y.; Shyu, J.C.; Hsieh, Y.S.; Tsai, C.C.; Liu, Y.C. Effects of silymarin on the resolution of liver fibrosis induced by carbon tetrachloride in rats. J. Viral Hepat. 2008, 15, 508–514. [Google Scholar] [CrossRef] [PubMed]
- Chen, I.S.; Chen, Y.-C.; Chou, C.-H.; Chuang, R.-F.; Sheen, L.-Y.; Chiu, C.-H. Hepatoprotection of silymarin against thioacetamide-induced chronic liver fibrosis. J. Sci. Food Agric. 2012, 92, 1441–1447. [Google Scholar] [CrossRef] [PubMed]
- Papackova, Z.; Heczkova, M.; Dankova, H.; Sticova, E.; Lodererova, A.; Bartonova, L.; Poruba, M.; Cahova, M. Silymarin prevents acetaminophen-induced hepatotoxicity in mice. PLoS ONE 2018, 13, e0191353. [Google Scholar] [CrossRef] [PubMed]
- Seitz, H.K.; Bataller, R.; Cortez-Pinto, H.; Gao, B.; Gual, A.; Lackner, C.; Mathurin, P.; Mueller, S.; Szabo, G.; Tsukamoto, H. Alcoholic liver disease. Nat. Rev. Dis. Primers. 2018, 4, 16. [Google Scholar] [CrossRef]
- Chen, L.H.; Xi, S.; Cohen, D.A. Liver antioxidant defenses in mice fed ethanol and the AIN-76A diet. Alcohol 1995, 12, 453–457. [Google Scholar] [CrossRef]
- Butura, A.; Nilsson, K.; Morgan, K.; Morgan, T.R.; French, S.W.; Johansson, I.; Schuppe-Koistinen, I.; Ingelman-Sundberg, M. The impact of CYP2E1 on the development of alcoholic liver disease as studied in a transgenic mouse model. J. Hepatol. 2009, 50, 572–583. [Google Scholar] [CrossRef]
- Morgan, K.; French, S.W.; Morgan, T.R. Production of a cytochrome P450 2E1 transgenic mouse and initial evaluation of alcoholic liver damage. Hepatology 2002, 36, 122–134. [Google Scholar] [CrossRef] [PubMed]
- Su, L.; Li, P.; Lu, T.; Mao, C.; Ji, D.; Hao, M.; Huang, Z. Protective effect of Schisandra chinensis total lignans on acute alcoholic-induced liver injury related to inhibiting CYP2E1 activation and activating the Nrf2/ARE signaling pathway. Rev. Bras. De Farmacogn. 2019, 29, 198–205. [Google Scholar] [CrossRef]
- Zhang, W.; Sun, Q.; Zhong, W.; Sun, X.; Zhou, Z. Hepatic Peroxisome Proliferator-Activated Receptor Gamma Signaling Contributes to Alcohol-Induced Hepatic Steatosis and Inflammation in Mice. Alcohol. Clin. Exp. Res. 2016, 40, 988–999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nan, Y.-M. Peroxisome proliferator-activated receptor α, a potential therapeutic target for alcoholic liver disease. World J. Gastroenterol. 2014, 20, 8055–8060. [Google Scholar] [CrossRef] [PubMed]
- Nanji, A.A.; Dannenberg, A.J.; Jokelainen, K.; Bass, N.M. Alcoholic liver injury in the rat is associated with reduced expression of peroxisome proliferator-alpha (PPARalpha)-regulated genes and is ameliorated by PPARalpha activation. J. Pharmacol. Exp. Ther. 2004, 310, 417–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, L.; Ren, W.; Li, W.; Zhao, S.; Mi, H.; Wang, R.; Zhang, Y.; Wu, W.; Nan, Y.; Yu, J. Activation of peroxisome proliferator activated receptor alpha ameliorates ethanol induced steatohepatitis in mice. Lipids Health Dis. 2011, 10, 246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischer, M.; You, M.; Matsumoto, M.; Crabb, D.W. Peroxisome Proliferator-activated Receptor α (PPARα) Agonist Treatment Reverses PPARα Dysfunction and Abnormalities in Hepatic Lipid Metabolism in Ethanol-fed Mice. J. Biol. Chem. 2003, 278, 27997–28004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diesinger, T.; Buko, V.; Lautwein, A.; Dvorsky, R.; Belonovskaya, E.; Lukivskaya, O.; Naruta, E.; Kirko, S.; Andreev, V.; Buckert, D.; et al. Drug targeting CYP2E1 for the treatment of early-stage alcoholic steatohepatitis. PLoS ONE 2020, 15, e0235990. [Google Scholar] [CrossRef]
- Lu, Y.; Wu, D.; Wang, X.; Ward, S.C.; Cederbaum, A.I. Chronic alcohol-induced liver injury and oxidant stress are decreased in cytochrome P4502E1 knockout mice and restored in humanized cytochrome P4502E1 knock-in mice. Free. Radic. Biol. Med. 2010, 49, 1406–1416. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Liu, Y.; Wang, S.; Huang, J.; Luo, Z.; Jiang, M.; Lu, Y.; Lin, Q.; Liu, H.; Cheng, N.; et al. Endoplasmic reticulum-targeted inhibition of CYP2E1 with vitamin E nanoemulsions alleviates hepatocyte oxidative stress and reverses alcoholic liver disease. Biomaterials 2022, 288, 121720. [Google Scholar] [CrossRef]
- Hartley, D.P.; Kolaja, K.L.; Reichard, J.; Petersen, D.R. 4-Hydroxynonenal and malondialdehyde hepatic protein adducts in rats treated with carbon tetrachloride: Immunochemical detection and lobular localization. Toxicol. Appl. Pharmacol. 1999, 161, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Aleynik, S.I.; Leo, M.A.; Aleynik, M.K.; Lieber, C.S. Increased Circulating Products of Lipid Peroxidation in Patients with Alcoholic Liver Disease. Alcohol. Clin. Exp. Res. 1998, 22, 192–196. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Pandey, R.; Katyal, R.; Aggarwal, H.K.; Aggarwal, R.P.; Aggarwal, S.K. Lipid peroxide levels and antioxidant status in alcoholic liver disease. Indian J. Clin. Biochem. 2005, 20, 67–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawaratani, H.; Tsujimoto, T.; Douhara, A.; Takaya, H.; Moriya, K.; Namisaki, T.; Noguchi, R.; Yoshiji, H.; Fujimoto, M.; Fukui, H. The effect of inflammatory cytokines in alcoholic liver disease. Mediat. Inflamm 2013, 2013, 495156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitazawa, T.; Nakatani, Y.; Fujimoto, M.; Tamura, N.; Uemura, M.; Fukui, H. The Production of Tumor Necrosis Factor-α by Macrophages in Rats With Acute Alcohol Loading. Alcohol. Clin. Exp. Res. 2003, 27, 72S–75S. [Google Scholar] [CrossRef]
- Iimuro, Y.; Gallucci, R.M.; Luster, M.I.; Kono, H.; Thurman, R.G. Antibodies to tumor necrosis factor alfa attenuate hepatic necrosis and inflammation caused by chronic exposure to ethanol in the rat. Hepatology 1997, 26, 1530–1537. [Google Scholar] [CrossRef] [PubMed]
- Aldred, A.; Nagy, L.E. Ethanol dissociates hormone-stimulated cAMP production from inhibition of TNF-alpha production in rat Kupffer cells. Am. J. Physiol. 1999, 276, G98–G106. [Google Scholar] [CrossRef] [PubMed]
- McClain, C.J.; Cohen, D.A. Increased tumor necrosis factor production by monocytes in alcoholic hepatitis. Hepatology 1989, 9, 349–351. [Google Scholar] [CrossRef]
- Yin, M.; Wheeler, M.D.; Kono, H.; Bradford, B.U.; Gallucci, R.M.; Luster, M.I.; Thurman, R.G. Essential role of tumor necrosis factor α in alcohol-induced liver injury in mice. Gastroenterology 1999, 117, 942–952. [Google Scholar] [CrossRef]
- Vairetti, M.; Di Pasqua, L.G.; Cagna, M.; Richelmi, P.; Ferrigno, A.; Berardo, C. Changes in Glutathione Content in Liver Diseases: An Update. Antioxidants 2021, 10, 364. [Google Scholar] [CrossRef]
- Kessova, I. Alcohol-induced liver injury in mice lacking Cu, Zn-superoxide dismutase. Hepatology 2003, 38, 1136–1145. [Google Scholar] [CrossRef] [PubMed]
- Colell, A.; Garcia-Ruiz, C.; Miranda, M.; Ardite, E.; Mari, M.; Morales, A.; Corrales, F.; Kaplowitz, N.; Fernandez-Checa, J.C. Selective glutathione depletion of mitochondria by ethanol sensitizes hepatocytes to tumor necrosis factor. Gastroenterology 1998, 115, 1541–1551. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Fu, J.; Li, L.; Chen, C.; Wang, H.; Hou, Y.; Xu, Y.; Pi, J. Nrf2 in alcoholic liver disease. Toxicol. Appl. Pharmacol. 2018, 357, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wu, K.C.; Lu, Y.F.; Ekuase, E.; Klaassen, C.D. Nrf2 protection against liver injury produced by various hepatotoxicants. Oxid. Med. Cell Longev. 2013, 2013, 305861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okawa, H.; Motohashi, H.; Kobayashi, A.; Aburatani, H.; Kensler, T.W.; Yamamoto, M. Hepatocyte-specific deletion of the keap1 gene activates Nrf2 and confers potent resistance against acute drug toxicity. Biochem. Biophys Res. Commun. 2006, 339, 79–88. [Google Scholar] [CrossRef]
- Lamle, J.; Marhenke, S.; Borlak, J.; von Wasielewski, R.; Eriksson, C.J.; Geffers, R.; Manns, M.P.; Yamamoto, M.; Vogel, A. Nuclear factor-eythroid 2-related factor 2 prevents alcohol-induced fulminant liver injury. Gastroenterology 2008, 134, 1159–1168. [Google Scholar] [CrossRef]
- He, J.L.; Zhou, Z.W.; Yin, J.J.; He, C.Q.; Zhou, S.F.; Yu, Y. Schisandra chinensis regulates drug metabolizing enzymes and drug transporters via activation of Nrf2-mediated signaling pathway. Drug. Des. Devel. Ther. 2015, 9, 127–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, I.J.; Kim, J.W.; Jung, J.J.; Sung, S.H.; Ku, S.K.; Choi, J.-S. In vitro protective effects of Hoveniae Semen cum Fructus extracts against oxidative stress. Toxicol. Environ. Health Sci. 2016, 8, 19–27. [Google Scholar] [CrossRef]
- Szopa, A.; Ekiert, R.; Ekiert, H. Current knowledge of Schisandra chinensis (Turcz.) Baill. (Chinese magnolia vine) as a medicinal plant species: A review on the bioactive components, pharmacological properties, analytical and biotechnological studies. Phytochem. Rev. 2017, 16, 195–218. [Google Scholar] [CrossRef] [Green Version]
- Guo, C.; Xue, G.; Pan, B.; Zhao, M.; Chen, S.; Gao, J.; Chen, T.; Qiu, L. Myricetin Ameliorates Ethanol-Induced Lipid Accumulation in Liver Cells by Reducing Fatty Acid Biosynthesis. Mol. Nutr. Food Res. 2019, 63, 1801393. [Google Scholar] [CrossRef]
- Wang, M.; Zhu, P.; Jiang, C.; Ma, L.; Zhang, Z.; Zeng, X. Preliminary characterization, antioxidant activity in vitro and hepatoprotective effect on acute alcohol-induced liver injury in mice of polysaccharides from the peduncles of Hovenia dulcis. Food Chem. Toxicol. 2012, 50, 2964–2970. [Google Scholar] [CrossRef]
- Buyco, D.G.; Martin, J.; Jeon, S.; Hooks, R.; Lin, C.; Carr, R. Experimental models of metabolic and alcoholic fatty liver disease. World J. Gastroenterol. 2021, 27, 1–18. [Google Scholar] [CrossRef]
- Bertola, A.; Mathews, S.; Ki, S.H.; Wang, H.; Gao, B. Mouse model of chronic and binge ethanol feeding (the NIAAA model). Nat. Protoc. 2013, 8, 627–637. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target Gene | Primer Sequence (Forward, Reverse) | Gene ID | Product Size |
|---|---|---|---|
| SREBP-1c | 5′-GATGTGCGAACTGGACACAG-3′, 5′-CATAGGGGGCGTCAAACAG-3′ | 20787 | 104 bp |
| SCD1 | 5′-CCCCTGCGGATCTTCCTTAT-3′, 5′-AGGGTCGGCGTGTGTTTCT-3′ | 20249 | 114 bp |
| ACC1 | 5′-GCCATTGGTATTGGGGCTTAC-3′, 5′-CCCGACCAAGGACTTTGTTG-3′ | 107476 | 112 bp |
| FAS | 5′-GCTGCGGAAACTTCAGGAAAT-3′, 5′-AGAGACGTGTCACTCCTGGACTT-3′ | 14104 | 84 bp |
| PPARγ | 5′-AGTGGAGACCGCCCAGG-3′, 5′-GCAGCAGGTTGTCTTGGATGT-3′ | 19016 | 64 bp |
| DGAT2 | 5′-AGTGGCAATGCTATCATCATCGT-3′, 5′-AAGGAATAAGTGGGAACCAGATCA-3′ | 67800 | 149 bp |
| PPARα | 5′-ATGCCAGTACTGCCGTTTTC-3′, 5′-GGCCTTGACCTTGTTCATGT-3′ | 19013 | 220 bp |
| ACO | 5′-GCCCAACTGTGACTTCCATT-3′, 5′-GGCATGTAACCCGTAGCACT-3′ | 74121 | 113 bp |
| CPT1 | 5′-GCACTGCAGCTCGCACATTACAA-3′, 5′-CTCAGACAGTACCTCCTTCAGGAAA-3′ | 12894 | 324 bp |
| Nrf2 | 5′-CGAGATATACGCAGGAGAGGTAAGA-3′, 5′-GCTCGACAATGTTCTCCAGCTT-3′ | 18024 | 79 bp |
| β-actin | 5′-CTGTCGAGTCGCGTCCA CCCGCGAG-3′, 5′-CTCGCGGGTGGACGCGACTCGACAG-3′ | 11461 | 516 bp |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jegal, K.-H.; Park, H.-R.; Choi, B.-R.; Kim, J.-K.; Ku, S.-K. Synergistic Protective Effect of Fermented Schizandrae Fructus Pomace and Hoveniae Semen cum Fructus Extracts Mixture in the Ethanol-Induced Hepatotoxicity. Antioxidants 2023, 12, 1602. https://doi.org/10.3390/antiox12081602
Jegal K-H, Park H-R, Choi B-R, Kim J-K, Ku S-K. Synergistic Protective Effect of Fermented Schizandrae Fructus Pomace and Hoveniae Semen cum Fructus Extracts Mixture in the Ethanol-Induced Hepatotoxicity. Antioxidants. 2023; 12(8):1602. https://doi.org/10.3390/antiox12081602
Chicago/Turabian StyleJegal, Kyung-Hwan, Hye-Rim Park, Beom-Rak Choi, Jae-Kwang Kim, and Sae-Kwang Ku. 2023. "Synergistic Protective Effect of Fermented Schizandrae Fructus Pomace and Hoveniae Semen cum Fructus Extracts Mixture in the Ethanol-Induced Hepatotoxicity" Antioxidants 12, no. 8: 1602. https://doi.org/10.3390/antiox12081602