
Enhanced Levels of Peroxisome-Derived H2O2 Do Not Induce Pexophagy but Impair Autophagic Flux in HEK-293 and HeLa Cells
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. DNA Manipulations and Plasmids
2.2. Cell Culture and Treatment
2.3. Antibodies
2.4. Immunoblotting
2.5. Fluorescence Microscopy
2.6. Electrophoretic Mobility Shift Assay
2.7. Flow Cytometry Analysis
2.8. Statistical Analysis
3. Results
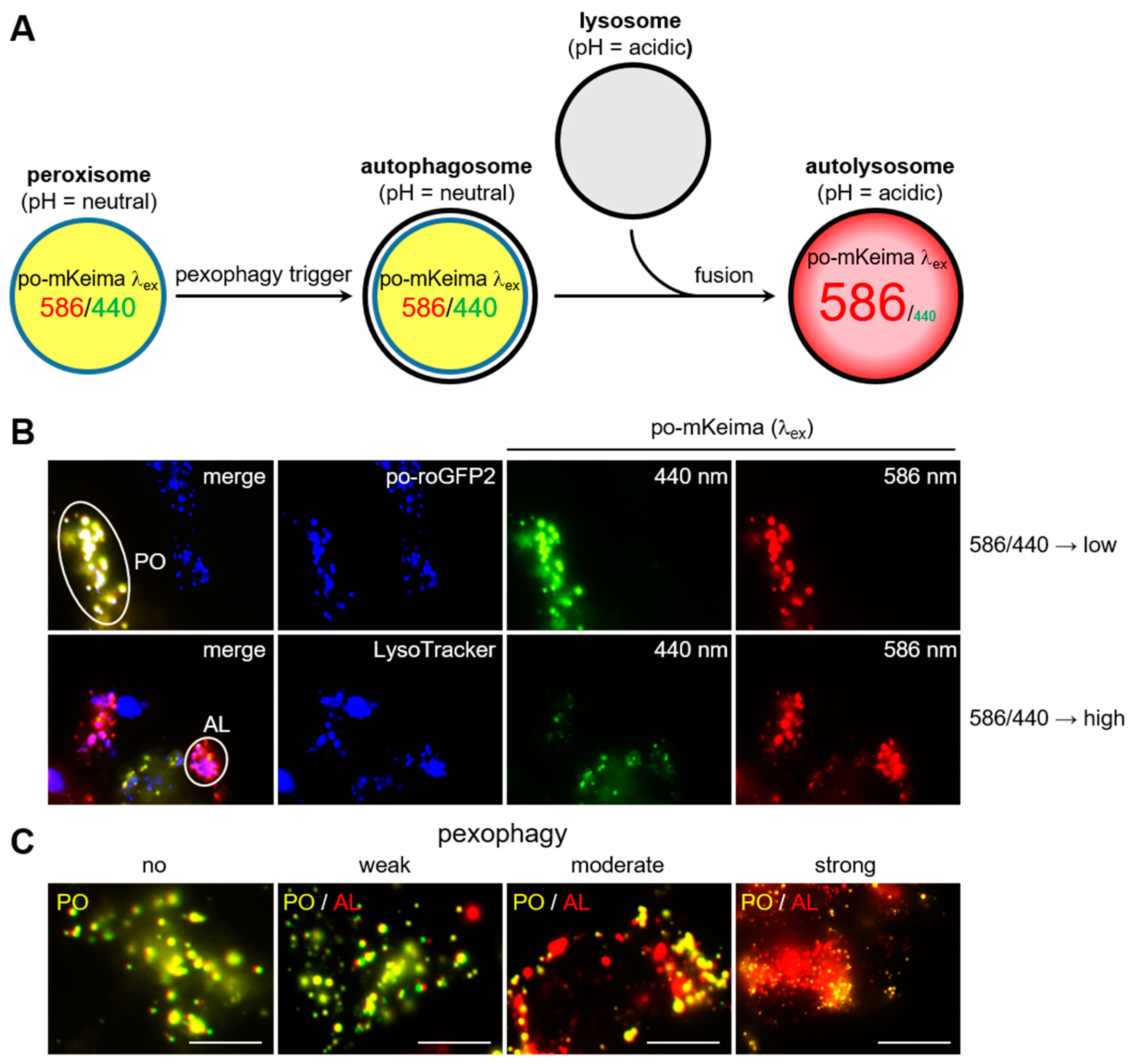
3.1. Validation of the DD-DAO/po-mKeima Flp-In T-REx Cell Lines
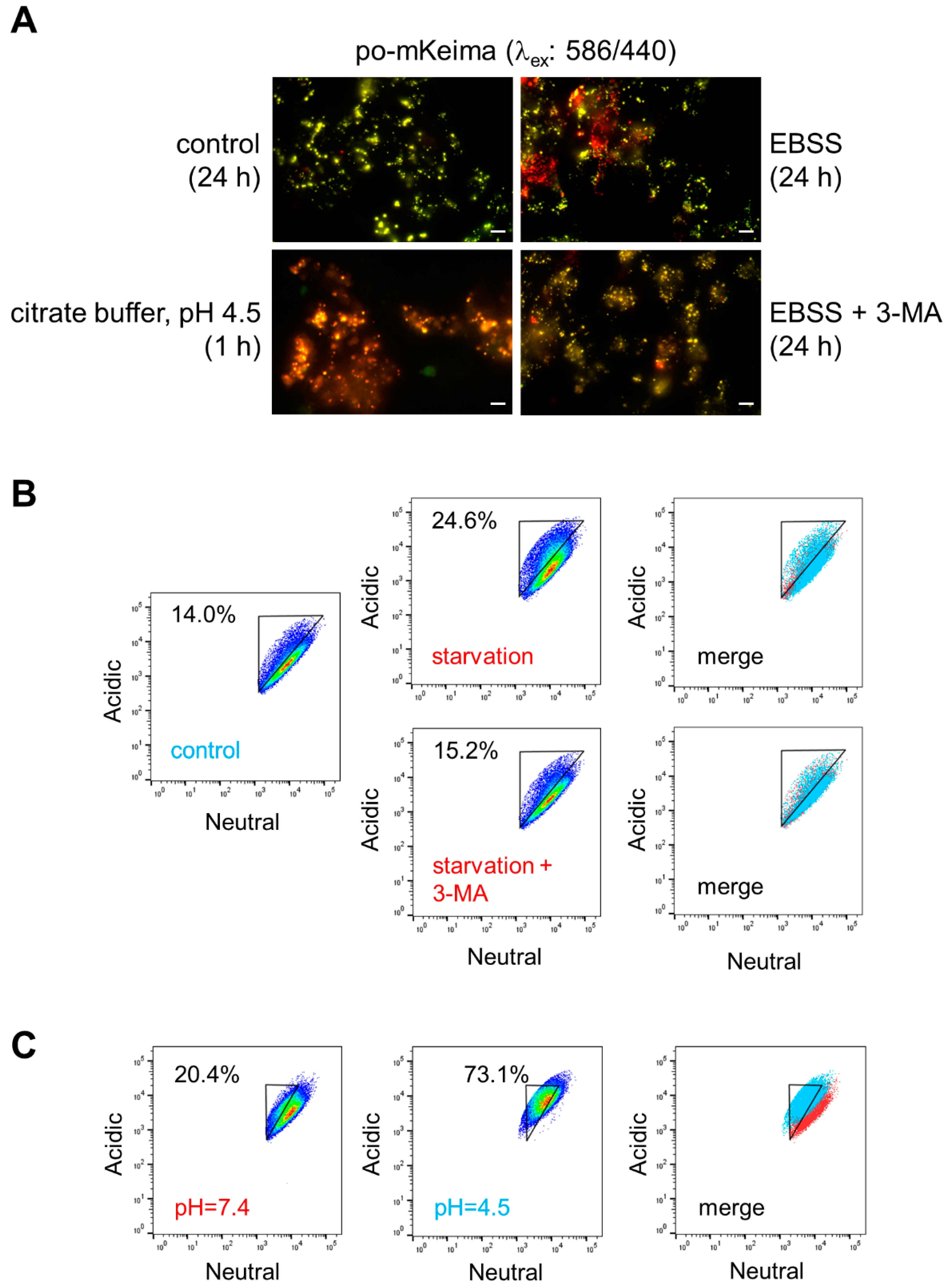
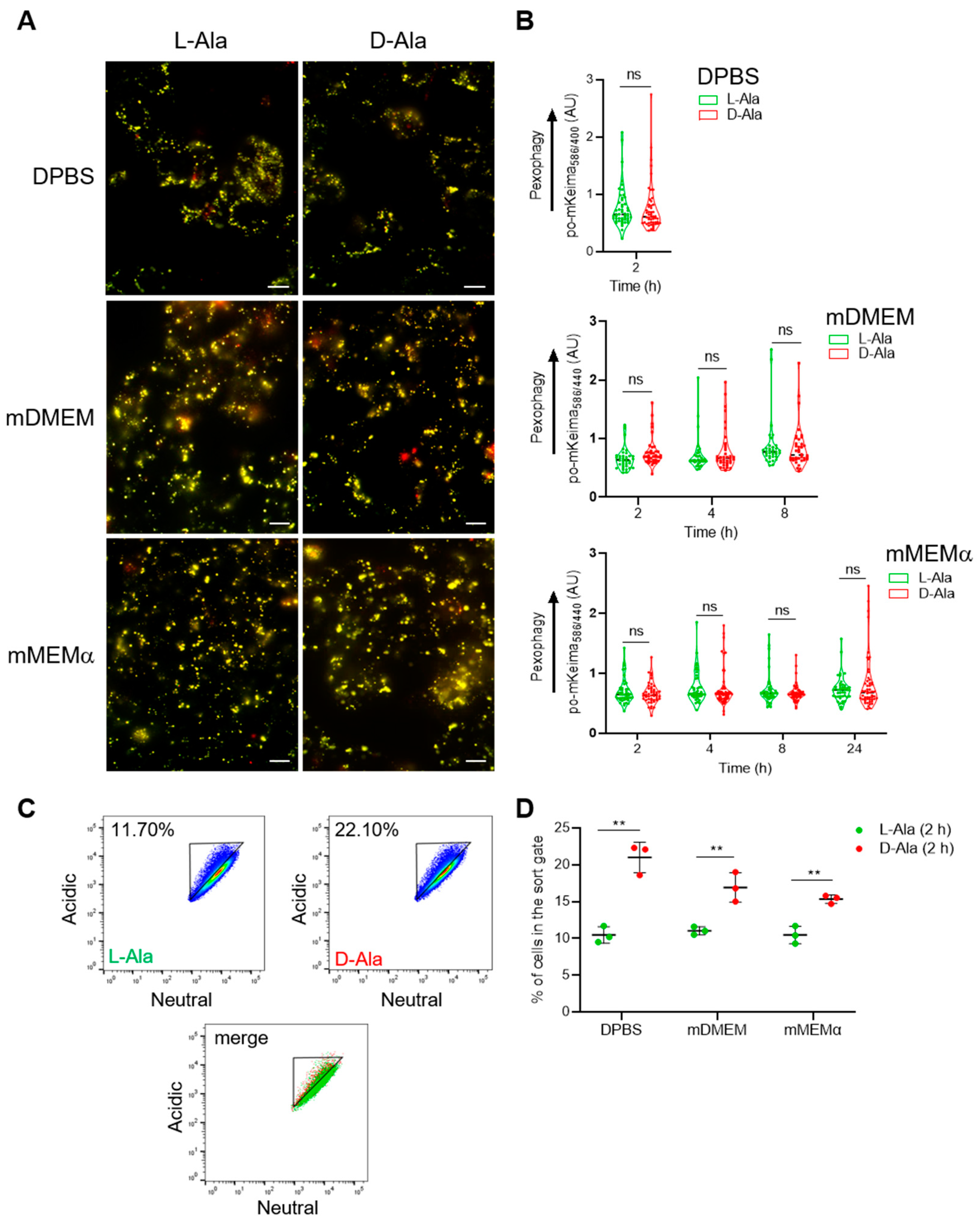
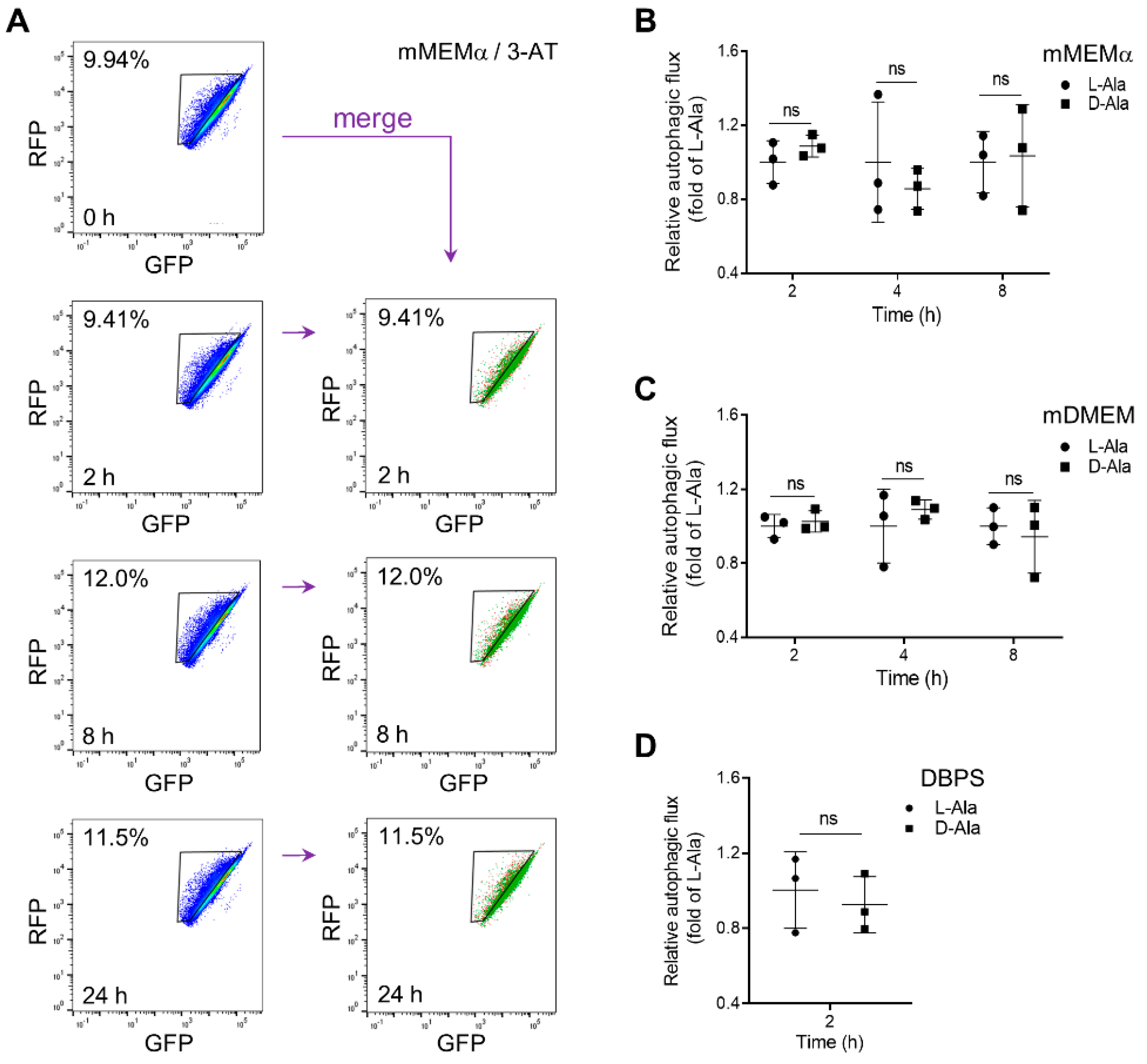
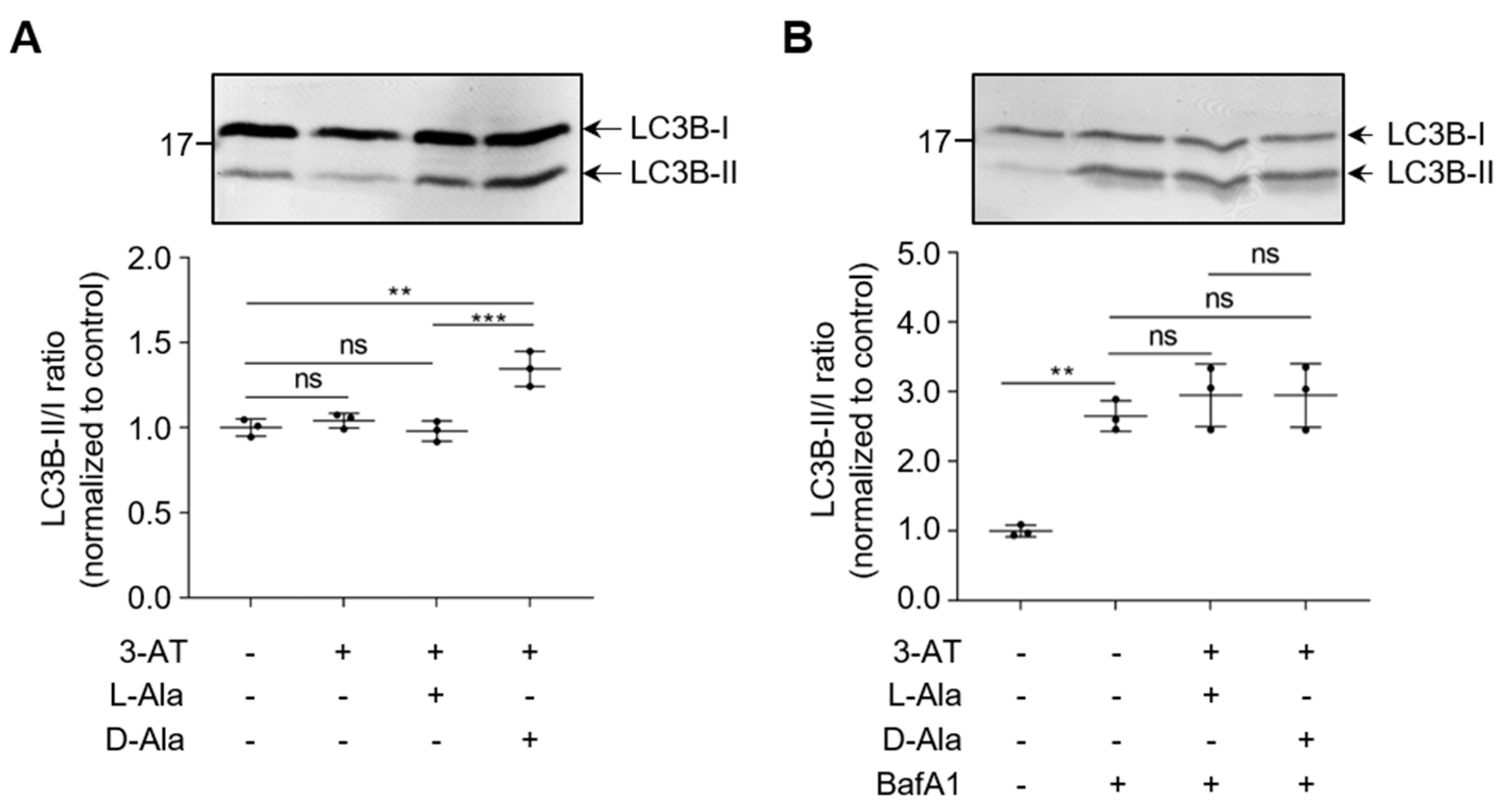
3.2. Peroxisomal H2O2 Emission Does Not Act as the Primary Trigger for Pexophagy in HEK-293 and HeLa Cells
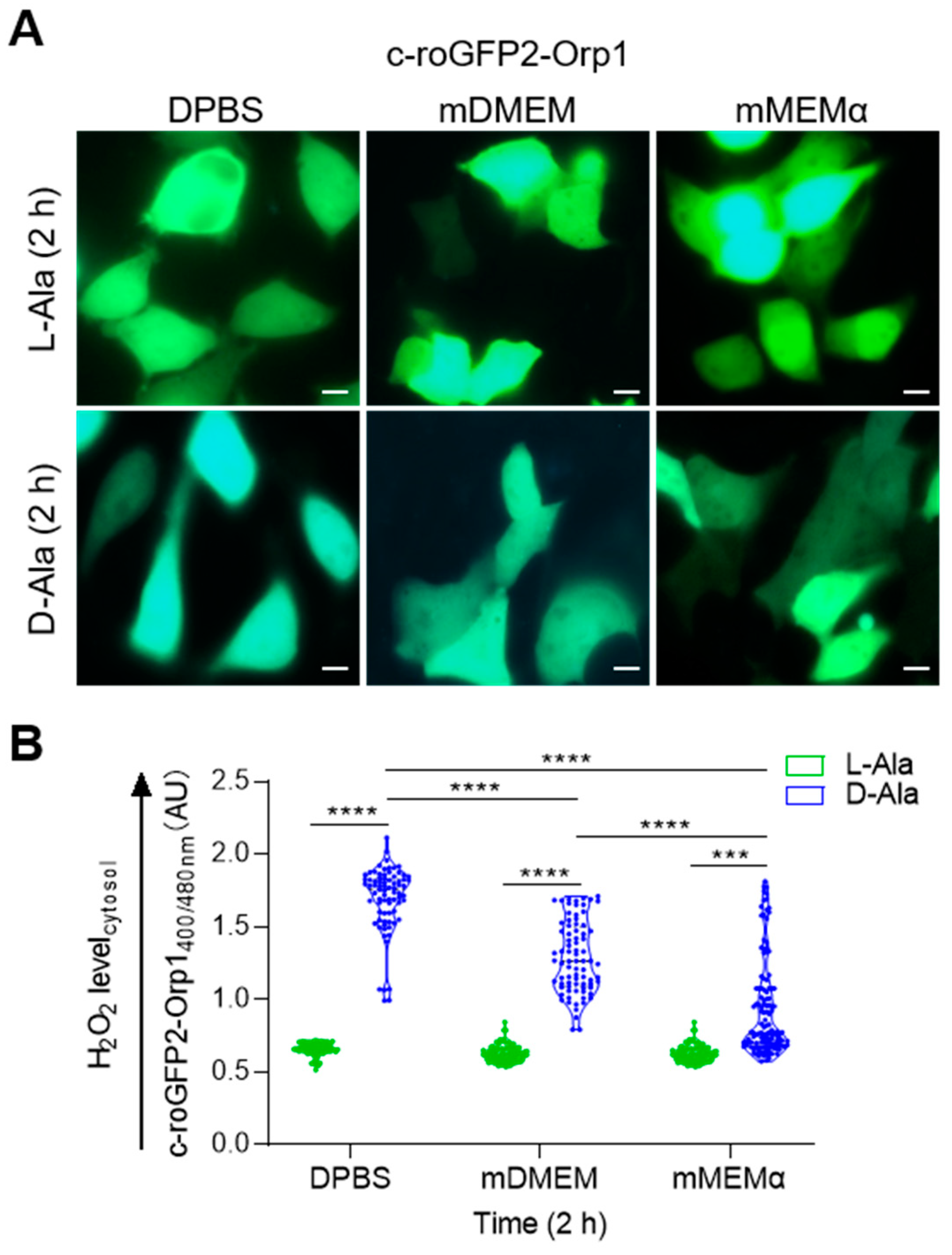
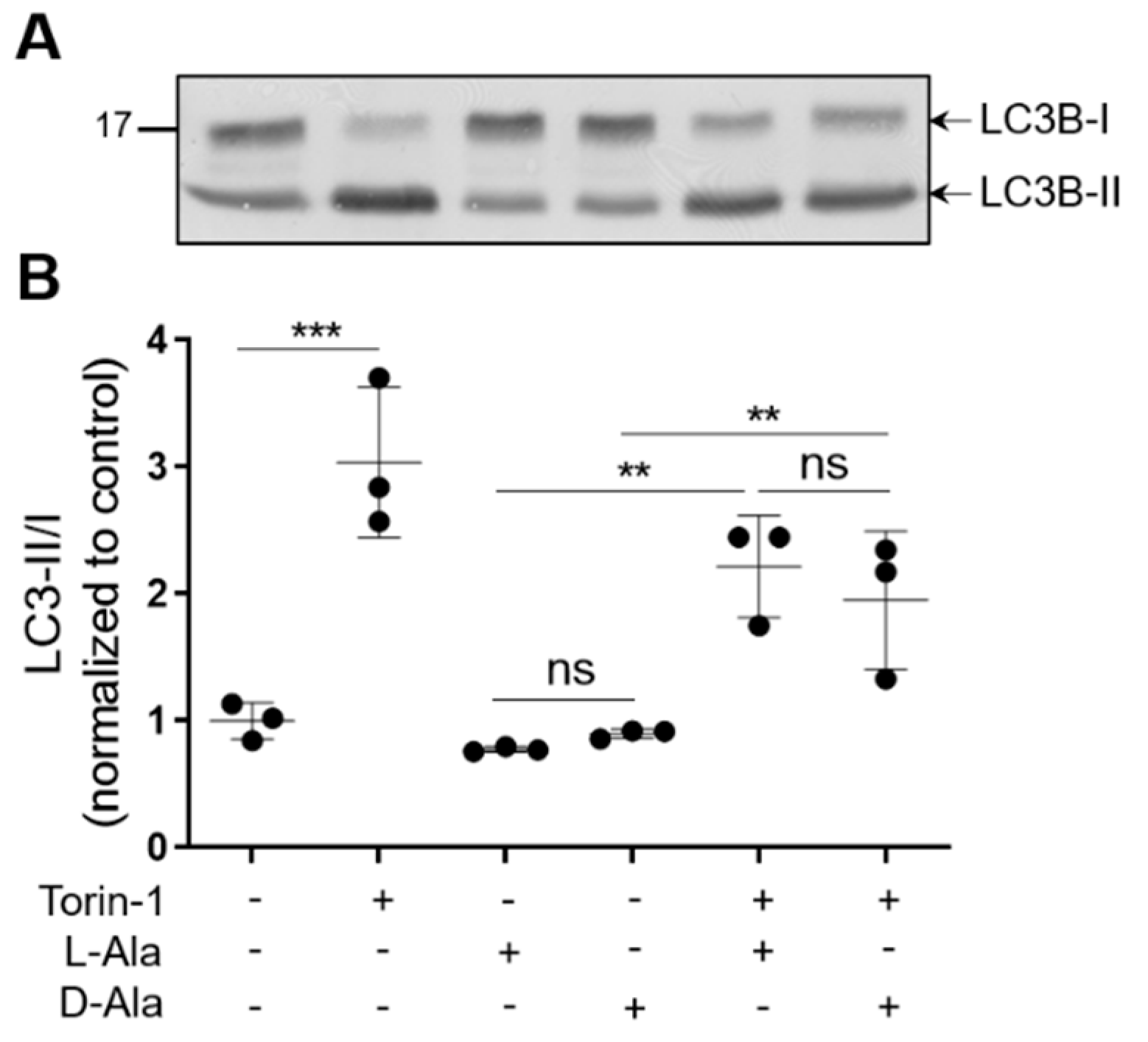
3.3. Enhanced Levels of Peroxisome-Derived H2O2 Cause Late-Stage Impairment in Autophagy
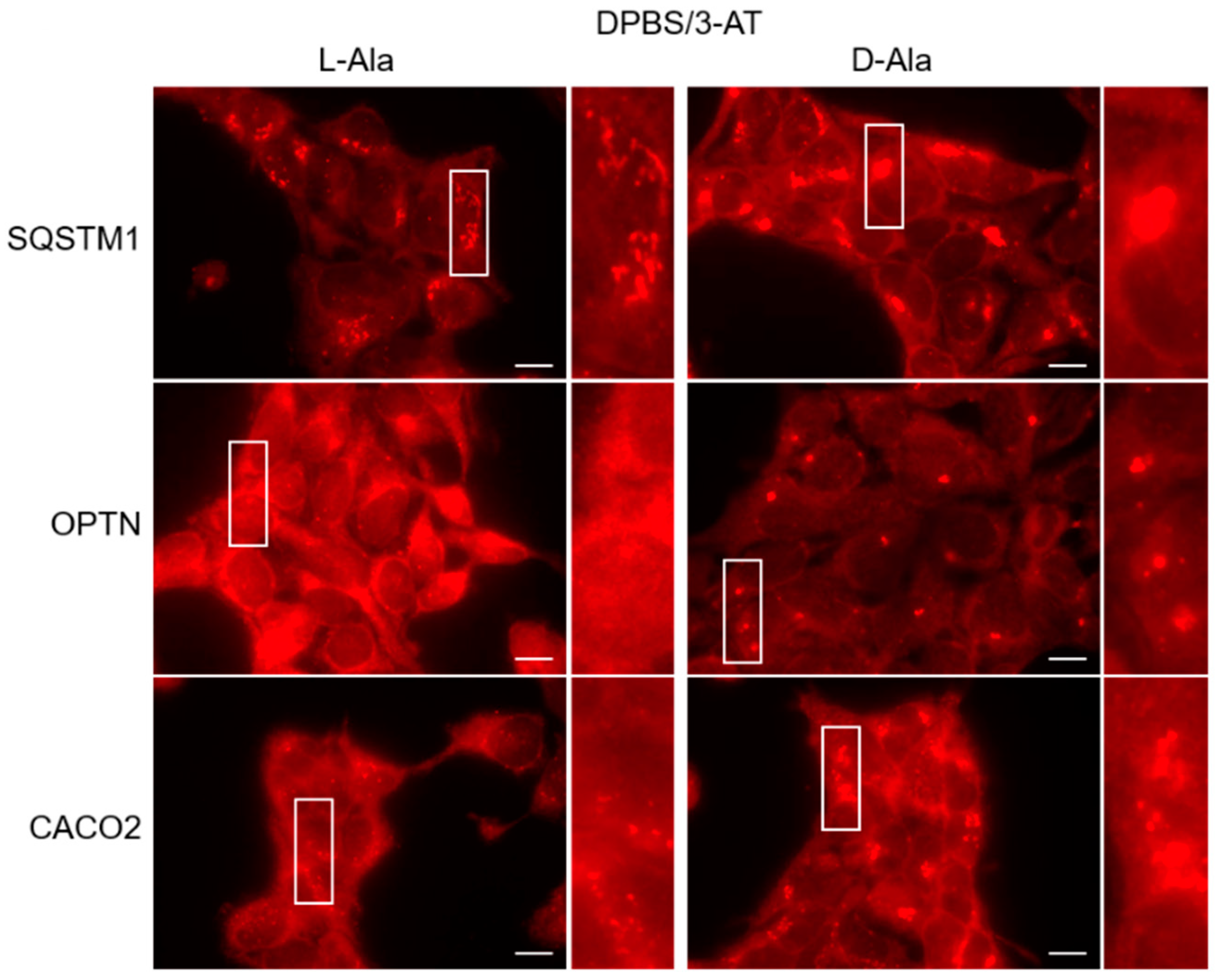
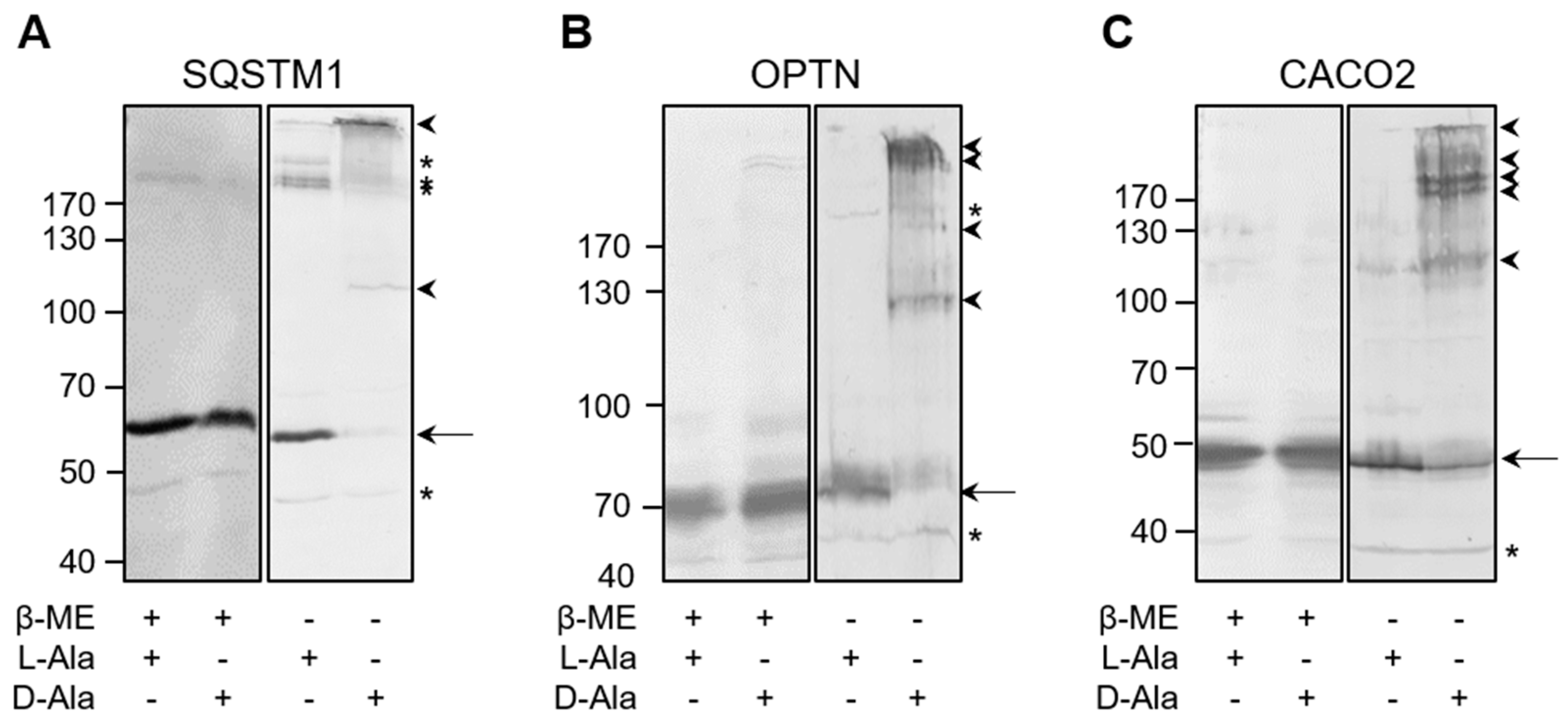
3.4. The Autophagy Receptors SQSTM1, CACO2, and OPTN Aggregate in Response to Peroxisome-Derived H2O2
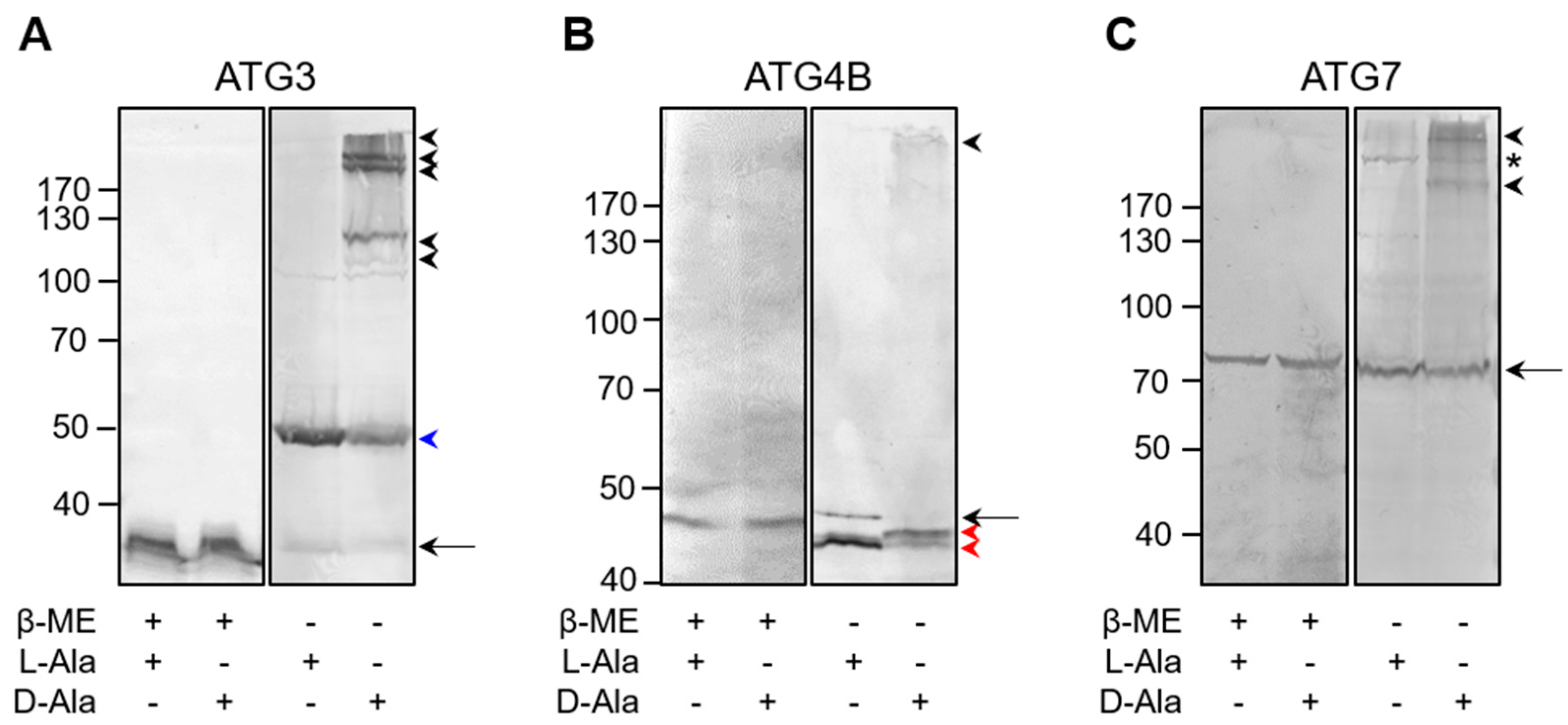
3.5. The Core Autophagy Machinery Proteins ATG3, ATG4B, and ATG7 Are Prone to Oxidation by Peroxisome-Derived H2O2
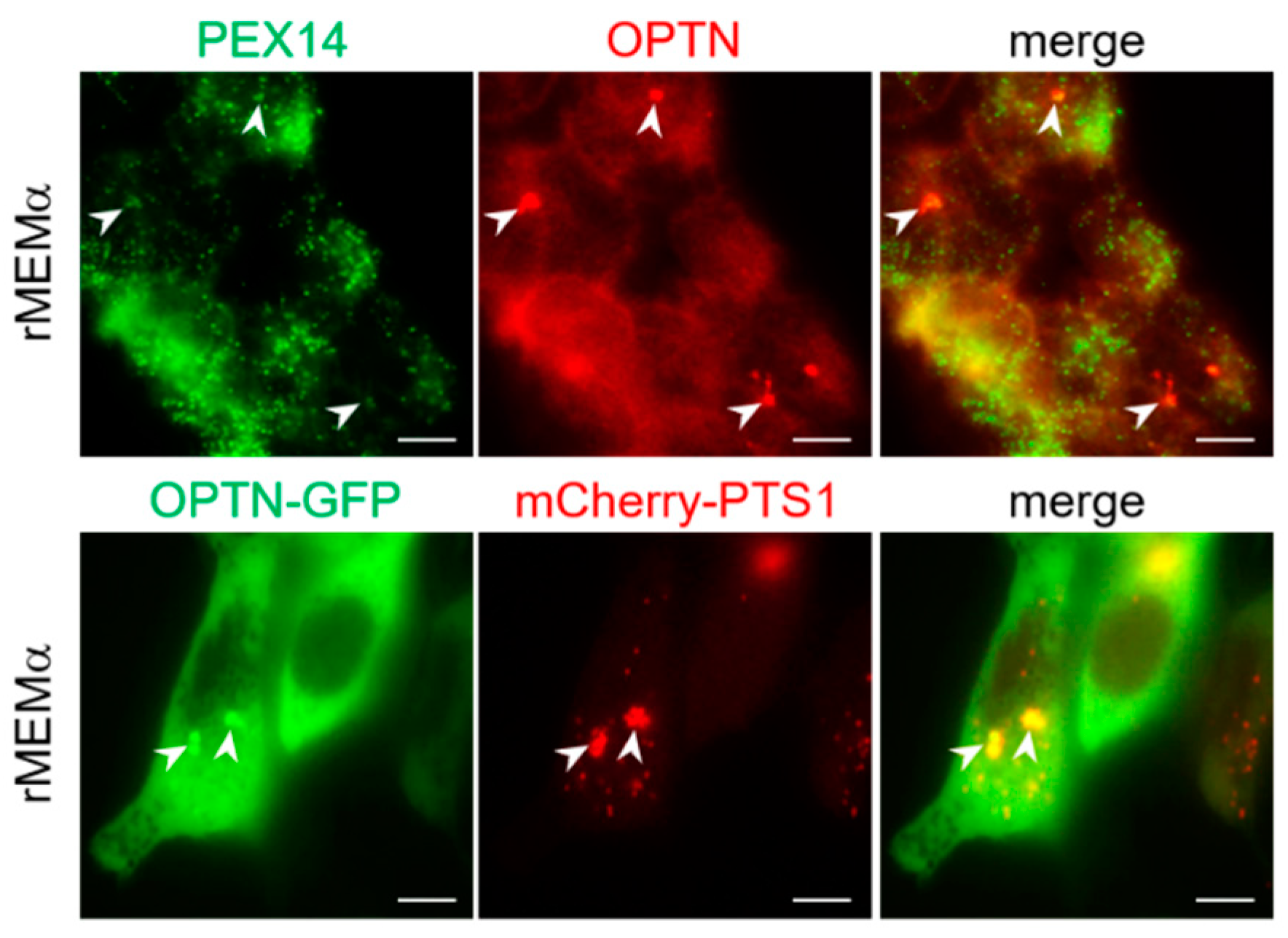
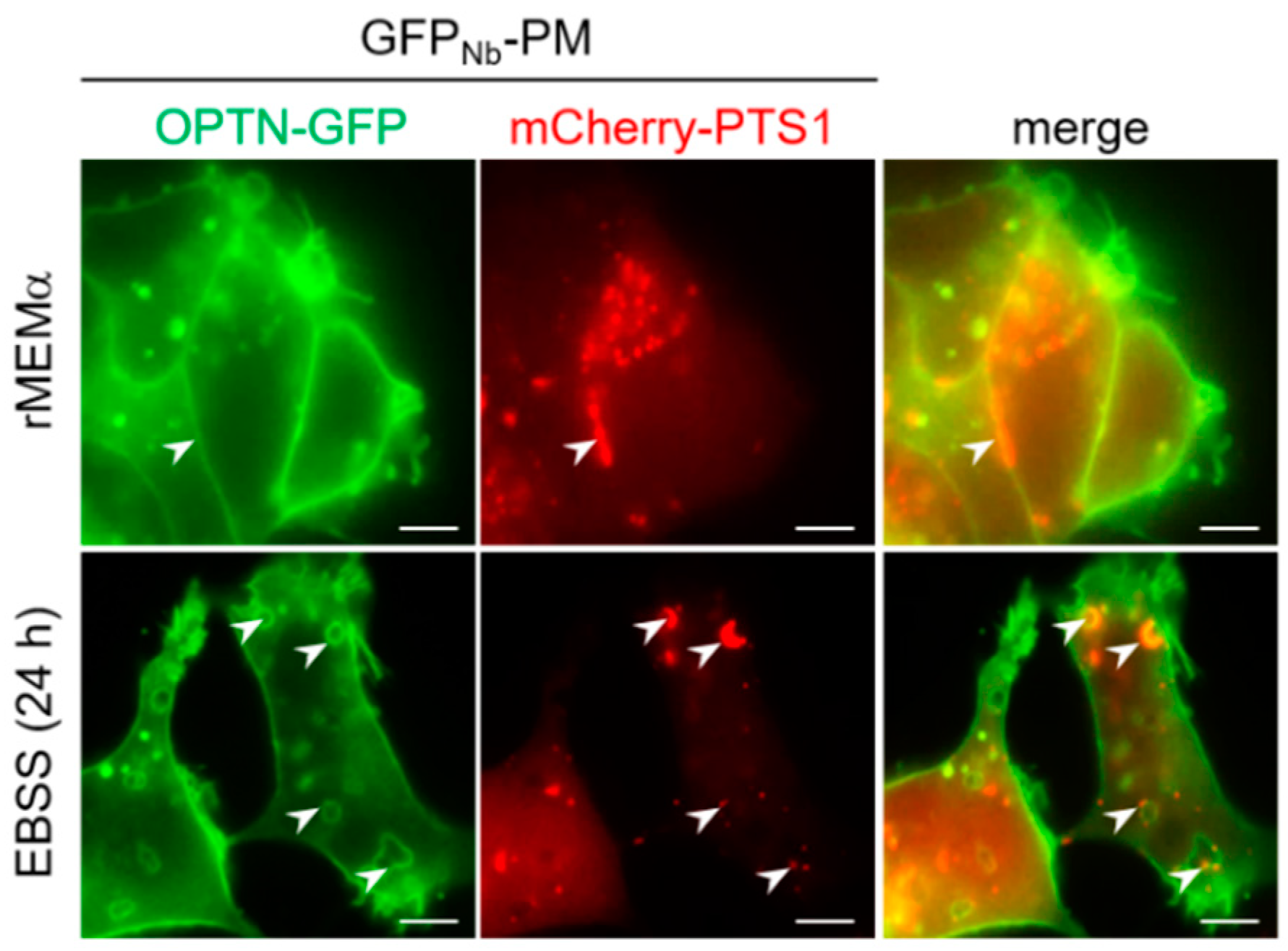
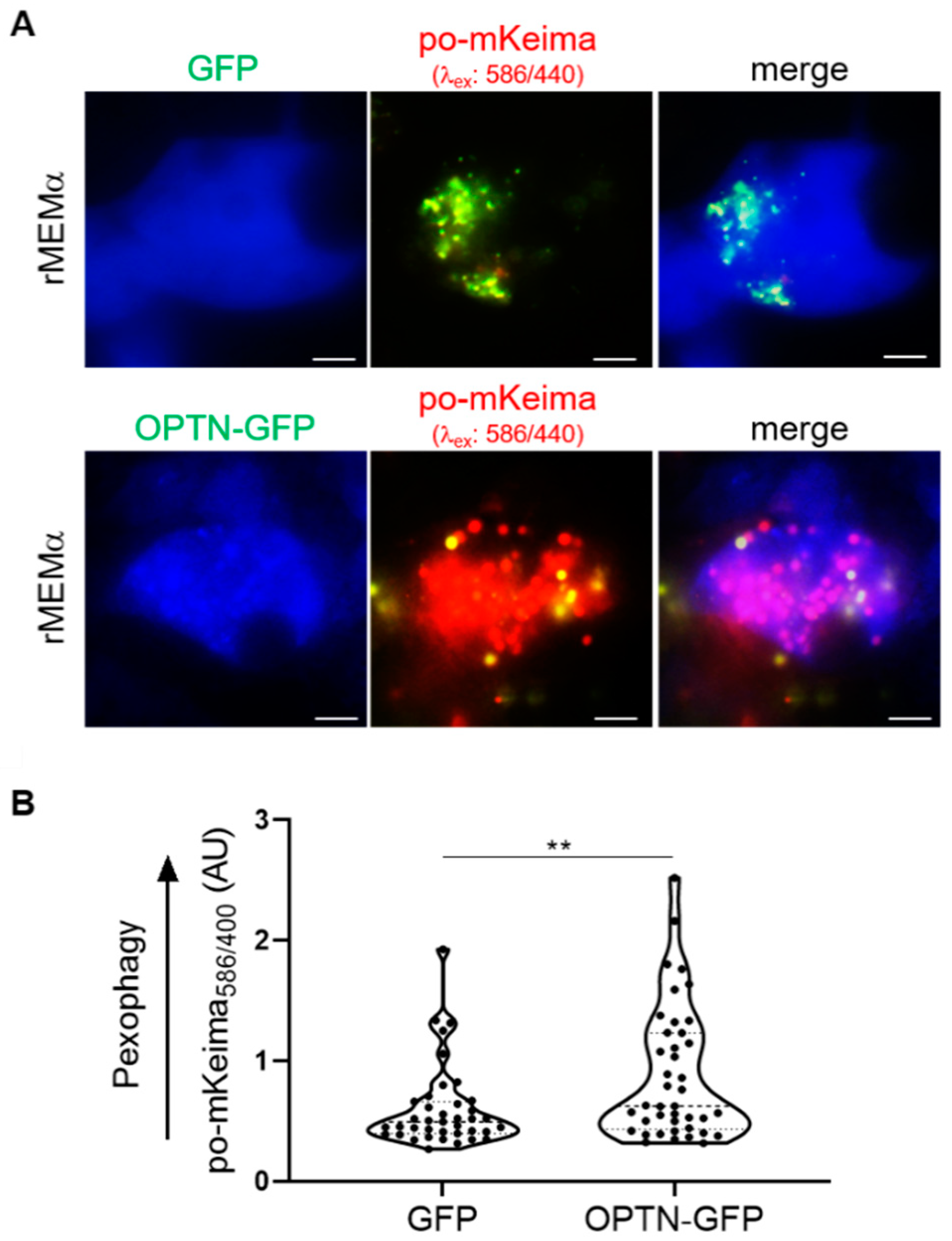
3.6. OPTN Can Be Recruited to Peroxisomes and Trigger Pexophagy
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yu, G.; Klionsky, D.J. Life and death decisions–the many faces of autophagy in cell survival and cell death. Biomolecules 2022, 12, 866. [Google Scholar] [CrossRef] [PubMed]
- Codogno, P.; Mehrpour, M.; Proikas-Cezanne, T. Canonical and non-canonical autophagy: Variations on a common theme of self-eating? Nat. Rev. Mol. Cell. Biol. 2011, 13, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Dall’Armi, C.; Devereaux, K.A.; Di Paolo, G. The role of lipids in the control of autophagy. Curr. Biol. 2013, 23, R33–R45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamark, T.; Johansen, T. Mechanisms of selective autophagy. Annu. Rev. Cell Dev. Biol. 2021, 37, 143–169. [Google Scholar] [CrossRef] [PubMed]
- Gatica, D.; Lahiri, V.; Klionsky, D.J. Cargo recognition and degradation by selective autophagy. Nat. Cell Biol. 2018, 20, 233–242. [Google Scholar] [CrossRef]
- Li, D.; Ding, Z.; Du, K.; Ye, X.; Cheng, X. Reactive oxygen species as a link between antioxidant pathways and autophagy. Oxid. Med. Cell. Longev. 2021, 2021, 5583215. [Google Scholar] [CrossRef]
- Ornatowski, W.; Lu, Q.; Yegambaram, M.; Garcia, A.E.; Zemskov, E.A.; Maltepe, E.; Fineman, J.R.; Wang, T.; Black, S.M. Complex interplay between autophagy and oxidative stress in the development of pulmonary disease. Redox Biol. 2020, 36, 101679. [Google Scholar] [CrossRef]
- Yun, H.R.; Jo, Y.H.; Kim, J.; Shin, Y.; Kim, S.S.; Choi, T.G. Roles of autophagy in oxidative stress. Int. J. Mol. Sci. 2020, 21, 3289. [Google Scholar] [CrossRef]
- Scherz-Shouval, R.; Shvets, E.; Elazar, Z. Oxidation as a post-translational modification that regulates autophagy. Autophagy 2007, 3, 371–373. [Google Scholar] [CrossRef] [Green Version]
- Redza-Dutordoir, M.; Averill-Bates, D.A. Interactions between reactive oxygen species and autophagy. Biochim. Biophys. Acta Mol. Cell. Res. 2021, 1868, 119041. [Google Scholar] [CrossRef]
- Zhou, J.; Li, X.Y.; Liu, Y.J.; Feng, J.; Wu, Y.; Shen, H.M.; Lu, G.D. Full-coverage regulations of autophagy by ROS: From induction to maturation. Autophagy 2022, 18, 1240–1255. [Google Scholar] [CrossRef] [PubMed]
- Lennicke, C.; Cochemé, H.M. Redox metabolism: ROS as specific molecular regulators of cell signaling and function. Mol. Cell 2021, 81, 3691–3707. [Google Scholar] [CrossRef] [PubMed]
- Sies, H.; Belousov, V.V.; Chandel, N.S.; Davies, M.J.; Jones, D.P.; Mann, G.E.; Murphy, M.P.; Yamamoto, M.; Winterbourn, C. Defining roles of specific reactive oxygen species (ROS) in cell biology and physiology. Nat. Rev. Mol. Cell Biol. 2022, 23, 499–515. [Google Scholar] [CrossRef]
- Sengupta, A.; Molkentin, J.D.; Yutzey, K.E. FoxO transcription factors promote autophagy in cardiomyocytes. J. Biol. Chem. 2009, 284, 28319–28331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Wang, N.; Xu, D.; Ma, Q.; Chen, Y.; Xu, S.; Xia, Q.; Zhang, Y.; Prehn, J.H.M.; Wang, G.; et al. Oxidation of multiple MiT/TFE transcription factors links oxidative stress to transcriptional control of autophagy and lysosome biogenesis. Autophagy 2020, 16, 1683–1696. [Google Scholar] [CrossRef]
- Leslie, N.R. The redox regulation of PI 3-kinase-dependent signaling. Antioxid. Redox Signal. 2006, 8, 1765–1774. [Google Scholar] [CrossRef] [PubMed]
- Alexander, A.; Cai, S.L.; Kim, J.; Nanez, A.; Sahin, M.; MacLean, K.H.; Inoki, K.; Guan, K.L.; Shen, J.; Person, M.D.; et al. ATM signals to TSC2 in the cytoplasm to regulate mTORC1 in response to ROS. Proc. Natl. Acad. Sci. USA 2010, 107, 4153–4158. [Google Scholar] [CrossRef] [Green Version]
- Frudd, K.; Burgoyne, T.; Burgoyne, J.R. Oxidation of Atg3 and Atg7 mediates inhibition of autophagy. Nat. Commun. 2018, 9, 95. [Google Scholar] [CrossRef] [Green Version]
- Scherz-Shouval, R.; Shvets, E.; Fass, E.; Shorer, H.; Gil, L.; Elazar, Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J. 2007, 26, 1749–1760. [Google Scholar] [CrossRef] [Green Version]
- Zheng, X.; Yang, Z.; Gu, Q.; Xia, F.; Fu, Y.; Liu, P.; Yin, X.M.; Li, M. The protease activity of human ATG4B is regulated by reversible oxidative modification. Autophagy 2020, 16, 1838–1850. [Google Scholar] [CrossRef]
- Gao, J.; Ohtsubo, M.; Hotta, Y.; Minoshima, S. Oligomerization of optineurin and its oxidative stress- or E50K mutation-driven covalent cross-linking: Possible relationship with glaucoma pathology. PLoS ONE 2014, 9, e101206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kataura, T.; Otten, E.G.; Rabanal-Ruiz, Y.; Adriaenssens, E.; Urselli, F.; Scialo, F.; Fan, L.; Smith, G.R.; Dawson, W.M.; Chen, X.; et al. NDP52 acts as a redox sensor in PINK1/Parkin-mediated mitophagy. EMBO J. 2022, e111372. [Google Scholar] [CrossRef] [PubMed]
- Cha-Molstad, H.; Lee, S.H.; Kim, J.G.; Sung, K.W.; Hwang, J.; Shim, S.M.; Ganipisetti, S.; McGuire, T.; Mook-Jung, I.; Ciechanover, A.; et al. Regulation of autophagic proteolysis by the N-recognin SQSTM1/p62 of the N-end rule pathway. Autophagy 2018, 14, 359–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carroll, B.; Otten, E.G.; Manni, D.; Stefanatos, R.; Menzies, F.M.; Smith, G.R.; Jurk, D.; Kenneth, N.; Wilkinson, S.; Passos, J.F.; et al. Oxidation of SQSTM1/p62 mediates the link between redox state and protein homeostasis. Nat. Commun. 2018, 9, 256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagakannan, P.; Eftekharpour, E. Differential redox sensitivity of cathepsin B and L holds the key to autophagy-apoptosis interplay after Thioredoxin reductase inhibition in nutritionally stressed SH-SY5Y cells. Free Radic. Biol. Med. 2017, 108, 819–831. [Google Scholar] [CrossRef]
- Wible, D.J.; Bratton, B. Reciprocity in ROS and autophagic signaling. Curr. Opin. Toxicol. 2018, 7, 28–36. [Google Scholar] [CrossRef]
- Schrader, M.; Kamoshita, M.; Islinger, M. Organelle interplay—Peroxisome interactions in health and disease. J. Inherit. Metab. Dis. 2020, 43, 71–89. [Google Scholar] [CrossRef]
- Wanders, R.J.; Baes, M.; Ribeiro, D.; Ferdinandusse, S.; Waterham, H.R. The physiological functions of human peroxisomes. Physiol. Rev. 2023, 103, 957–1024. [Google Scholar] [CrossRef]
- Fransen, M.; Lismont, C. Redox signaling from and to peroxisomes: Progress, challenges, and prospects. Antioxid. Redox Signal. 2019, 30, 95–112. [Google Scholar] [CrossRef]
- Kim, P.K.; Hailey, D.W.; Mullen, R.T.; Lippincott-Schwartz, J. Ubiquitin signals autophagic degradation of cytosolic proteins and peroxisomes. Proc. Natl. Acad. Sci. USA 2008, 105, 20567–20574. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Lismont, C.; Revenco, I.; Hussein, M.A.; Costa, C.F.; Fransen, M. The peroxisome-autophagy redox connection: A double-edged sword? Front. Cell Dev. Biol. 2021, 9, 814047. [Google Scholar] [CrossRef]
- Li, J.; Wang, W. Mechanisms and functions of pexophagy in mammalian cells. Cells 2021, 10, 1094. [Google Scholar] [CrossRef] [PubMed]
- Lismont, C.; Nordgren, M.; Brees, C.; Knoops, B.; Van Veldhoven, P.P.; Fransen, M. Peroxisomes as modulators of cellular protein thiol oxidation: A new model system. Antioxid. Redox Signal. 2019, 30, 22–39. [Google Scholar] [CrossRef] [PubMed]
- Kimura, S.; Noda, T.; Yoshimori, T. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy 2007, 3, 452–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Violot, S.; Carpentier, P.; Blanchoin, L.; Bourgeois, D. Reverse pH-dependence of chromophore protonation explains the large Stokes shift of the red fluorescent protein mKeima. J. Am. Chem. Soc. 2009, 131, 10356–10357. [Google Scholar] [CrossRef]
- Park, B.C.; Shen, X.; Samaraweera, M.; Yue, B.Y. Studies of optineurin, a glaucoma gene: Golgi fragmentation and cell death from overexpression of wild-type and mutant optineurin in two ocular cell types. Am. J. Pathol. 2006, 169, 1976–1989. [Google Scholar] [CrossRef] [Green Version]
- Ivashchenko, O.; Van Veldhoven, P.P.; Brees, C.; Ho, Y.S.; Terlecky, S.R.; Fransen, M. Intraperoxisomal redox balance in mammalian cells: Oxidative stress and interorganellar cross-talk. Mol. Biol. Cell 2011, 22, 1440–1451. [Google Scholar] [CrossRef]
- Nordgren, M.; Wang, B.; Apanasets, O.; Brees, C.; Van Veldhoven, P.P.; Fransen, M. Potential limitations in the use of KillerRed for fluorescence microscopy. J. Microsc. 2012, 245, 229–235. [Google Scholar] [CrossRef]
- Walton, P.A.; Brees, C.; Lismont, C.; Apanasets, O.; Fransen, M. The peroxisomal import receptor PEX5 functions as a stress sensor, retaining catalase in the cytosol in times of oxidative stress. Biochim. Biophys. Acta Mol. Cell. Res. 2017, 1864, 1833–1843. [Google Scholar] [CrossRef]
- Van Veldhoven, P.P.; de Schryver, E.; Young, S.G.; Zwijsen, A.; Fransen, M.; Espeel, M.; Baes, M.; Van Ael, E. Slc25a17 gene trapped mice: PMP34 plays a role in the peroxisomal degradation of phytanic and pristanic acid. Front. Cell Dev. Biol. 2020, 8, 144. [Google Scholar] [CrossRef]
- Ramazani, Y.; Knops, N.; Berlingerio, S.P.; Adebayo, O.C.; Lismont, C.; Kuypers, D.J.; Levtchenko, E.; van den Heuvel, L.P.; Fransen, M. Therapeutic concentrations of calcineurin inhibitors do not deregulate glutathione redox balance in human renal proximal tubule cells. PLoS ONE 2021, 16, e0250996. [Google Scholar] [CrossRef]
- Huybrechts, S.J.; Van Veldhoven, P.P.; Brees, C.; Mannaerts, G.P.; Los, G.V.; Fransen, M. Peroxisome dynamics in cultured mammalian cells. Traffic 2009, 10, 1722–1733. [Google Scholar] [CrossRef]
- Nadal-Ferret, M.; Gelabert, R.; Moreno, M.; Lluch, J.M. How does the environment affect the absorption spectrum of the fluorescent protein mKeima? J. Chem. Theory Comput. 2013, 9, 1731–1742. [Google Scholar] [CrossRef]
- Engedal, N.; Sønstevold, T.; Beese, C.J.; Selladurai, S.; Melcher, T.; Simensen, J.E.; Frankel, L.B.; Urbanucci, A.; Torgersen, M.L. Measuring autophagic cargo flux with keima-based probes. Methods Mol. Biol. 2022, 2445, 99–115. [Google Scholar] [CrossRef] [PubMed]
- Sargent, G.; van Zutphen, T.; Shatseva, T.; Zhang, L.; Di Giovanni, V.; Bandsma, R.; Kim, P.K. PEX2 is the E3 ubiquitin ligase required for pexophagy during starvation. J. Cell Biol. 2016, 214, 677–690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutscher, M.; Sobotta, M.C.; Wabnitz, G.H.; Ballikaya, S.; Meyer, A.J.; Samstag, Y.; Dick, T.P. Proximity-based protein thiol oxidation by H2O2-scavenging peroxidases. J. Biol. Chem. 2009, 284, 31532–31540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palikaras, K.; Lionaki, E.; Tavernarakis, N. Mechanisms of mitophagy in cellular homeostasis, physiology and pathology. Nat. Cell Biol. 2018, 20, 1013–1022. [Google Scholar] [CrossRef] [PubMed]
- Georgakopoulos, N.D.; Wells, G.; Campanella, M. The pharmacological regulation of cellular mitophagy. Nat. Chem. Biol. 2017, 13, 136–146. [Google Scholar] [CrossRef] [PubMed]
- Su, L.; Zhang, J.; Gomez, H.; Kellum, J.A.; Peng, Z. Mitochondria ROS and mitophagy in acute kidney injury. Autophagy 2023, 19, 401–414. [Google Scholar] [CrossRef] [PubMed]
- Fransen, M.; Lismont, C.; Walton, P. The peroxisome-mitochondria connection: How and why? Int. J. Mol. Sci. 2017, 18, 1126. [Google Scholar] [CrossRef] [Green Version]
- Lismont, C.; Revenco, I.; Fransen, M. Peroxisomal hydrogen peroxide metabolism and signaling in health and disease. Int. J. Mol. Sci. 2019, 20, 3673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jo, D.S.; Bae, D.J.; Park, S.J.; Seo, H.M.; Kim, H.B.; Oh, J.S.; Chang, J.W.; Kim, S.Y.; Shin, J.W.; Cho, D.H. Pexophagy is induced by increasing peroxisomal reactive oxygen species in 1′10-phenanthroline-treated cells. Biochem. Biophys. Res. Commun. 2015, 467, 354–360. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.N.; Dutta, R.K.; Maharjan, Y.; Liu, Z.Q.; Lim, J.Y.; Kim, S.J.; Cho, D.H.; So, H.S.; Choe, S.K.; Park, R. Catalase inhibition induces pexophagy through ROS accumulation. Biochem. Biophys. Res. Commun. 2018, 501, 696–702. [Google Scholar] [CrossRef] [PubMed]
- Dutta, R.K.; Maharjan, Y.; Lee, J.N.; Park, C.; Ho, Y.S.; Park, R. Catalase deficiency induces reactive oxygen species mediated pexophagy and cell death in the liver during prolonged fasting. Biofactors 2021, 47, 112–125. [Google Scholar] [CrossRef]
- Jo, D.S.; Park, S.J.; Kim, A.K.; Park, N.Y.; Kim, J.B.; Bae, J.E.; Park, H.J.; Shin, J.H.; Chang, J.W.; Kim, P.K.; et al. Loss of HSPA9 induces peroxisomal degradation by increasing pexophagy. Autophagy 2020, 16, 1989–2003. [Google Scholar] [CrossRef]
- Cunningham, C.N.; Baughman, J.M.; Phu, L.; Tea, J.S.; Yu, C.; Coons, M.; Kirkpatrick, D.S.; Bingol, B.; Corn, J.E. USP30 and parkin homeostatically regulate atypical ubiquitin chains on mitochondria. Nat. Cell Biol. 2015, 17, 160–169. [Google Scholar] [CrossRef]
- Marcassa, E.; Kallinos, A.; Jardine, J.; Rusilowicz-Jones, E.V.; Martinez, A.; Kuehl, S.; Islinger, M.; Clague, M.J.; Urbé, S. Dual role of USP30 in controlling basal pexophagy and mitophagy. EMBO Rep. 2018, 19, e45595. [Google Scholar] [CrossRef]
- Rusilowicz-Jones, E.V.; Barone, F.G.; Martins Lopes, F.; Stephen, E.; Mortiboys, H.; Urbé, S.; Clague, M.J. Benchmarking a highly selective USP30 inhibitor for enhancement of mitophagy and pexophagy. Life Sci. Alliance 2021, 5, e202101287. [Google Scholar] [CrossRef]
- Flachbartová, Z.; Kovacech, B. Mortalin—A multipotent chaperone regulating cellular processes ranging from viral infection to neurodegeneration. Acta Virol. 2013, 57, 3–15. [Google Scholar] [CrossRef] [Green Version]
- Chen, B.H.; Chang, Y.J.; Lin, S.; Yang, W.Y. Hsc70/Stub1 promotes the removal of individual oxidatively stressed peroxisomes. Nat. Commun. 2020, 11, 5267. [Google Scholar] [CrossRef]
- Defourny, J.; Aghaie, A.; Perfettini, I.; Avan, P.; Delmaghani, S.; Petit, C. Pejvakin-mediated pexophagy protects auditory hair cells against noise-induced damage. Proc. Natl. Acad. Sci. USA 2019, 116, 8010–8017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Tripathi, D.N.; Jing, J.; Alexander, A.; Kim, J.; Powell, R.T.; Dere, R.; Tait-Mulder, J.; Lee, J.H.; Paull, T.T.; et al. ATM functions at the peroxisome to induce pexophagy in response to ROS. Nat. Cell Biol. 2015, 17, 1259–1269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lismont, C.; Revenco, I.; Li, H.; Costa, C.F.; Lenaerts, L.; Hussein, M.A.; De Bie, J.; Knoops, B.; Van Veldhoven, P.P.; Derua, R.; et al. Peroxisome-derived hydrogen peroxide modulates the sulfenylation profiles of key redox signaling proteins in Flp-In T-REx 293 cells. Front. Cell Dev. Biol. 2022, 10, 888873. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; McMillan-Ward, E.; Kong, J.; Israels, S.J.; Gibson, B. Oxidative stress induces autophagic cell death independent of apoptosis in transformed and cancer cells. Cell Death Differ. 2008, 15, 171–182. [Google Scholar] [CrossRef]
- Legakis, J.E.; Koepke, J.I.; Jedeszko, C.; Barlaskar, F.; Terlecky, L.J.; Edwards, H.J.; Walton, P.A.; Terlecky, S.R. Peroxisome senescence in human fibroblasts. Mol. Biol. Cell 2002, 13, 4243–4255. [Google Scholar] [CrossRef]
- Wood, C.S.; Koepke, J.I.; Teng, H.; Boucher, K.K.; Katz, S.; Chang, P.; Terlecky, L.J.; Papanayotou, I.; Walton, P.A.; Terlecky, S.R. Hypocatalasemic fibroblasts accumulate hydrogen peroxide and display age-associated pathologies. Traffic 2006, 7, 97–107. [Google Scholar] [CrossRef]
- Koepke, J.I.; Wood, C.S.; Terlecky, L.J.; Walton, P.A.; Terlecky, S.R. Progeric effects of catalase inactivation in human cells. Toxicol. Appl. Pharmacol. 2008, 232, 99–108. [Google Scholar] [CrossRef]
- Wurzer, B.; Zaffagnini, G.; Fracchiolla, D.; Turco, E.; Abert, C.; Romanov, J.; Martens, S. Oligomerization of p62 allows for selection of ubiquitinated cargo and isolation membrane during selective autophagy. Elife 2015, 4, e08941. [Google Scholar] [CrossRef]
- Chen, W.; Shen, T.; Wang, L.; Lu, K. Oligomerization of selective autophagy receptors for the targeting and degradation of protein aggregates. Cells 2021, 10, 1989. [Google Scholar] [CrossRef]
- Dutta, R.K.; Lee, J.N.; Maharjan, Y.; Park, C.; Choe, S.K.; Ho, Y.S.; Kwon, H.M.; Park, R. Catalase-deficient mice induce aging faster through lysosomal dysfunction. Cell Commun. Signal. 2022, 20, 192. [Google Scholar] [CrossRef]
- Qiu, Y.; Wang, J.; Li, H.; Yang, B.; Wang, J.; He, Q.; Weng, Q. Emerging views of OPTN (optineurin) function in the autophagic process associated with disease. Autophagy 2022, 18, 73–85. [Google Scholar] [CrossRef] [PubMed]














Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, H.; Lismont, C.; Costa, C.F.; Hussein, M.A.F.; Baes, M.; Fransen, M. Enhanced Levels of Peroxisome-Derived H2O2 Do Not Induce Pexophagy but Impair Autophagic Flux in HEK-293 and HeLa Cells. Antioxidants 2023, 12, 613. https://doi.org/10.3390/antiox12030613
Li H, Lismont C, Costa CF, Hussein MAF, Baes M, Fransen M. Enhanced Levels of Peroxisome-Derived H2O2 Do Not Induce Pexophagy but Impair Autophagic Flux in HEK-293 and HeLa Cells. Antioxidants. 2023; 12(3):613. https://doi.org/10.3390/antiox12030613
Chicago/Turabian StyleLi, Hongli, Celien Lismont, Cláudio F. Costa, Mohamed A. F. Hussein, Myriam Baes, and Marc Fransen. 2023. "Enhanced Levels of Peroxisome-Derived H2O2 Do Not Induce Pexophagy but Impair Autophagic Flux in HEK-293 and HeLa Cells" Antioxidants 12, no. 3: 613. https://doi.org/10.3390/antiox12030613