

Hesperidin Protects SH−SY5Y Neuronal Cells against High Glucose−Induced Apoptosis via Regulation of MAPK Signaling
 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents and Chemicals
2.2. Cell Culture
2.3. Cell Viability
2.4. Analysis of Superoxide and Hydroxyl Radicals
2.5. Intracellular Reactive Oxygen Species (ROS) Measurement
2.6. 8−Oxoguanine (8−oxoG) Detection
2.7. Western Blot Analysis
2.8. Cellular and Mitochondrial Calcium Level Detection
2.9. Hoechst 33342 Staining
2.10. Statistical Analysis
3. Results
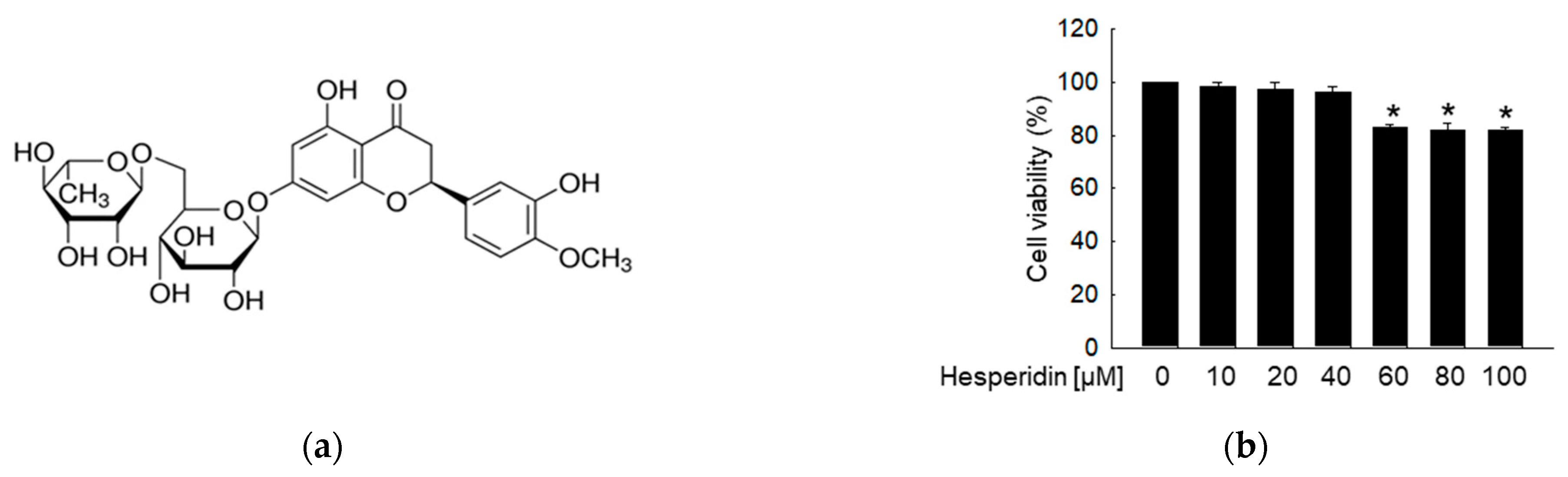
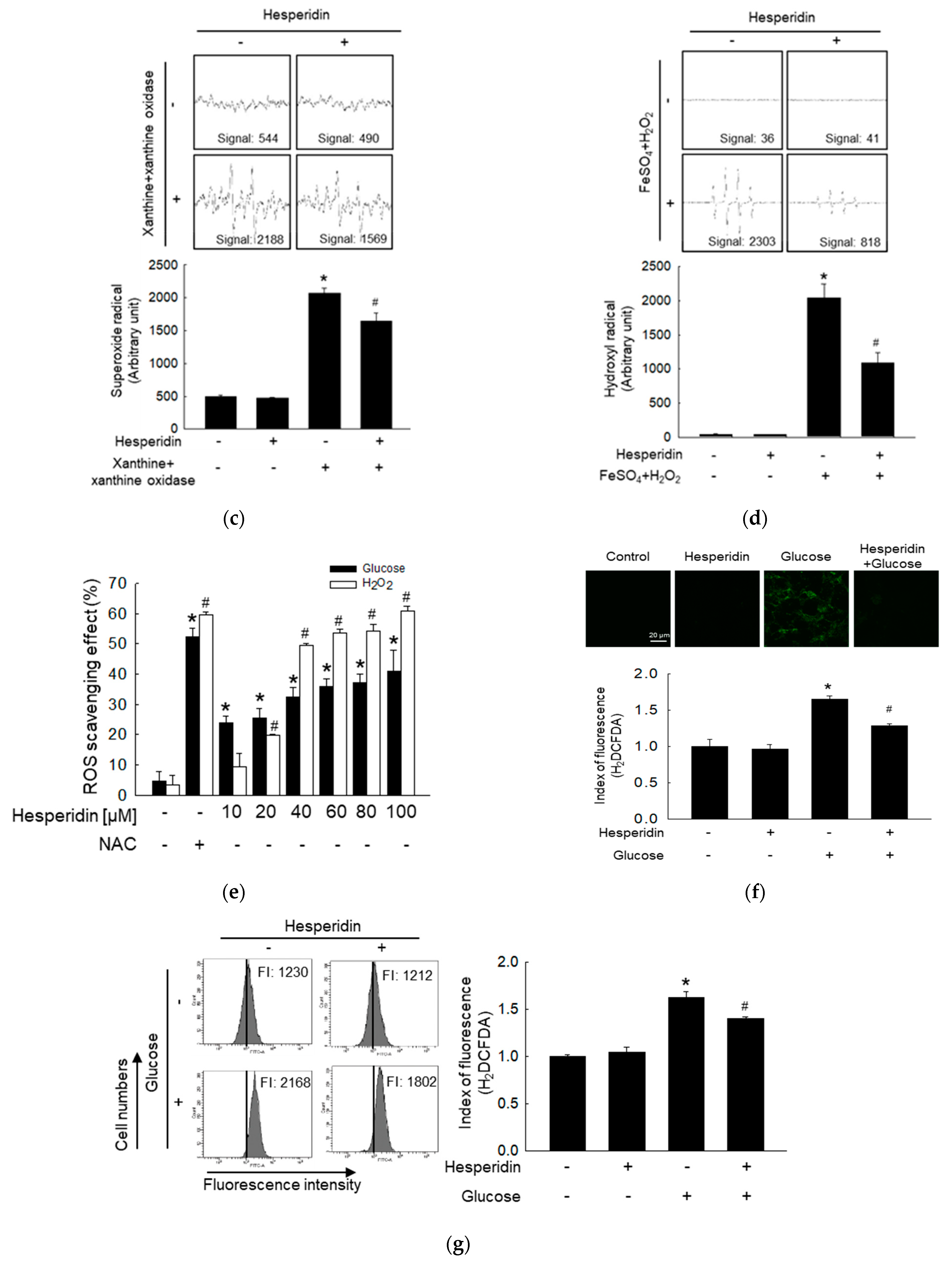
3.1. Hesperidin Protects Neuronal Cells from High Glucose−Induced Oxidative Stress
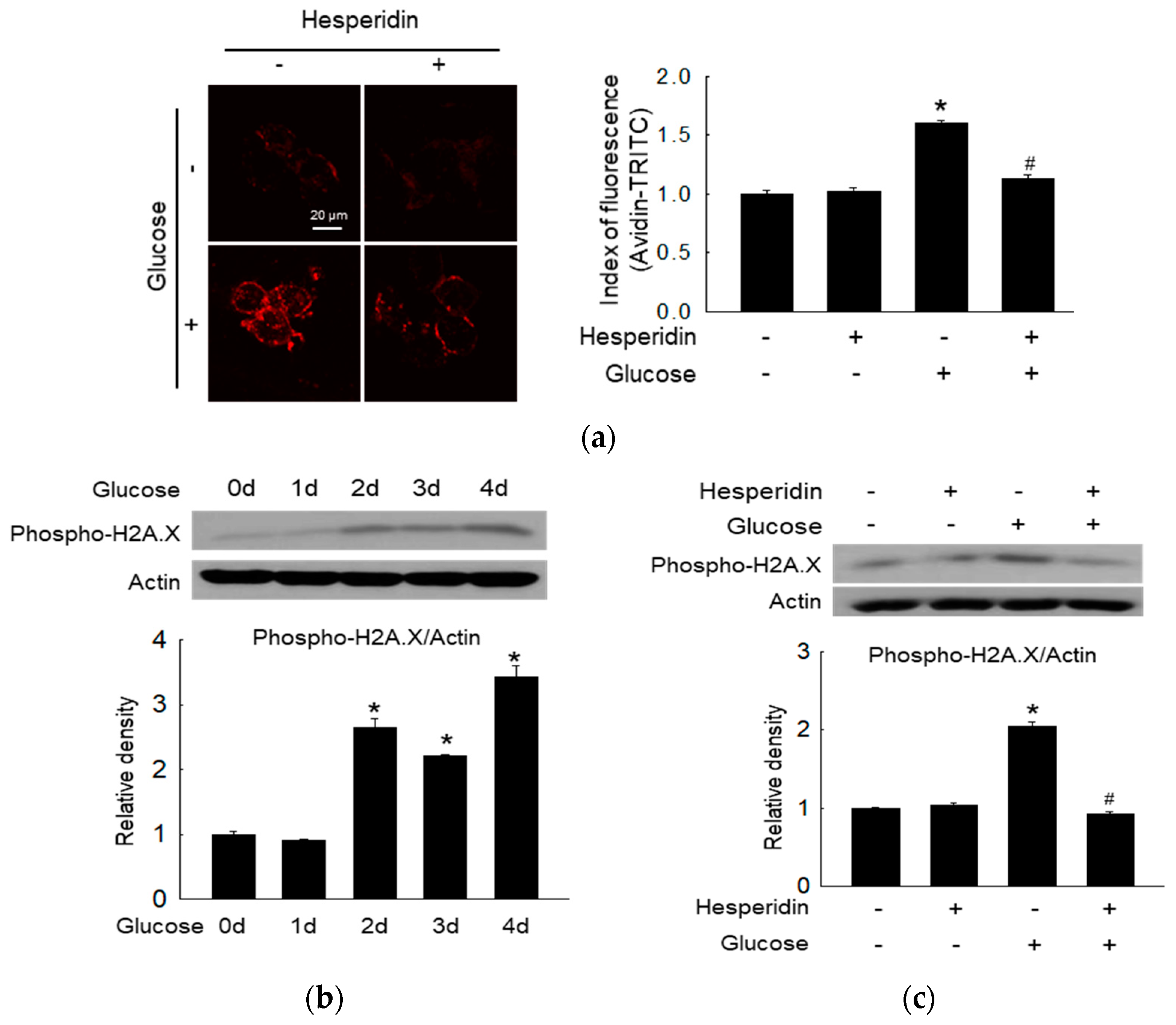
3.2. Hesperidin Ameliorated Glucose−Induced DNA Damage
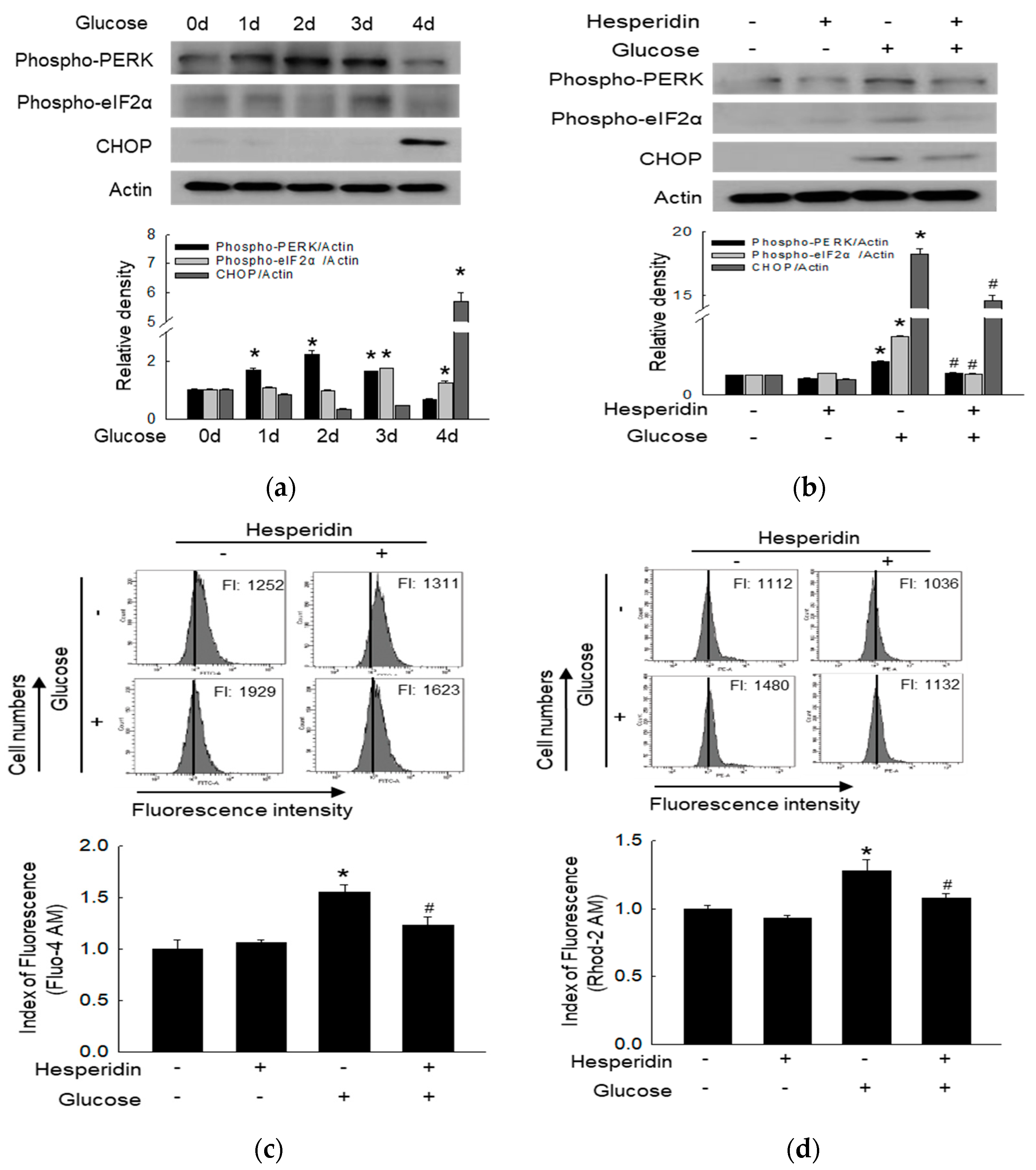
3.3. Hesperidin Suppressed Glucose−Induced ER Stress Response
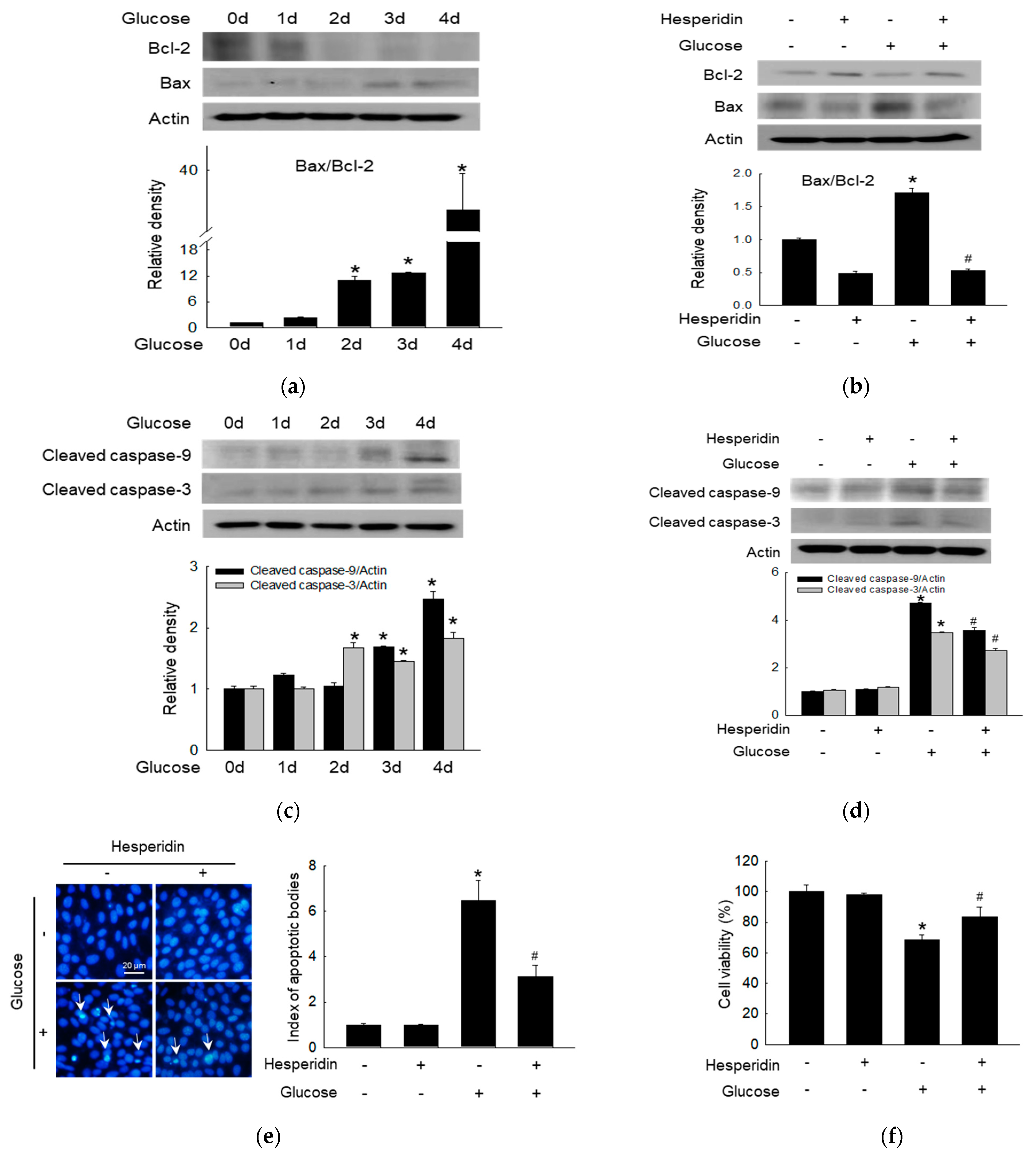
3.4. Hesperidin Protected Neuronal Cells against Glucose−Induced Apoptosis
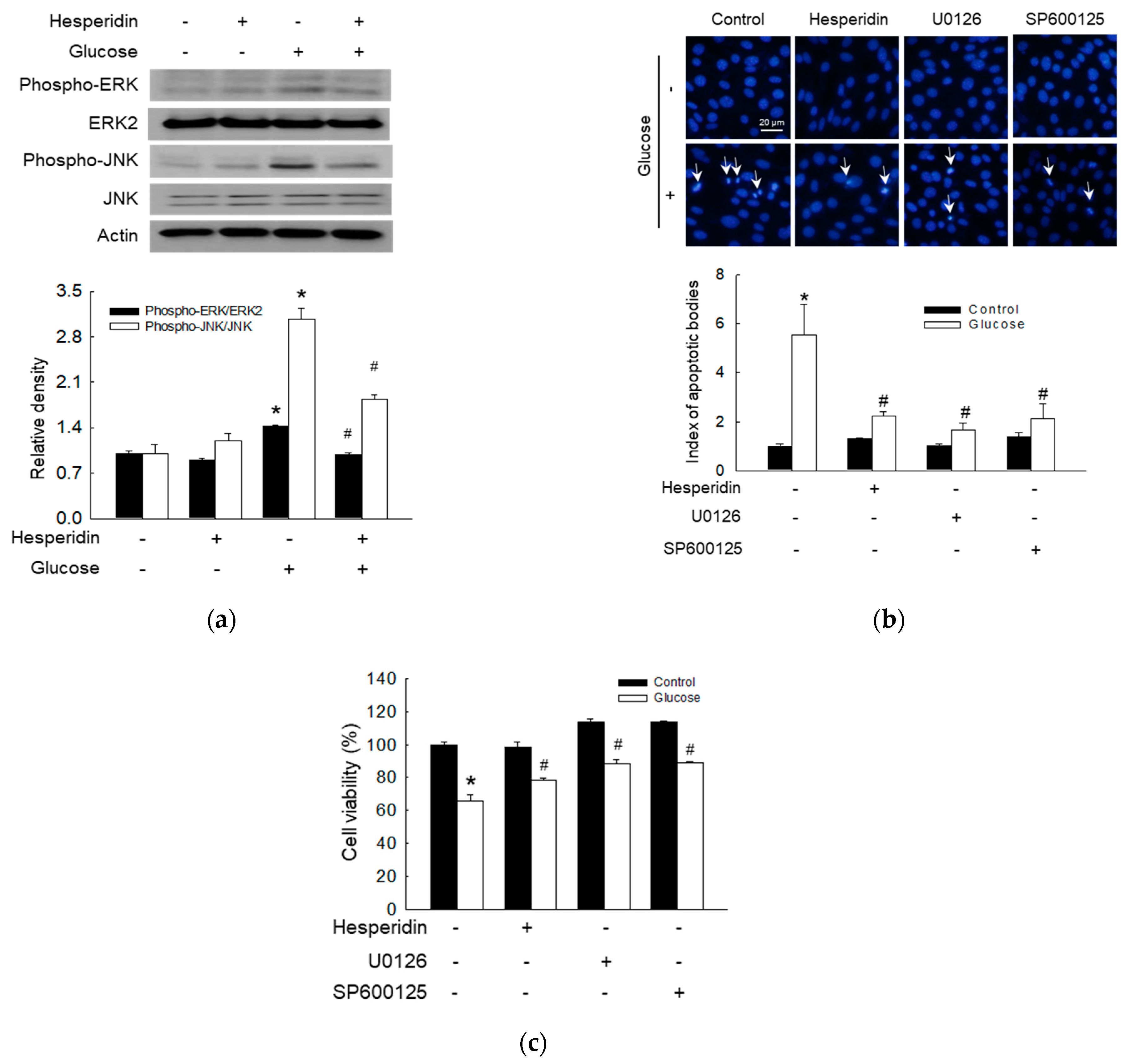
3.5. Hesperidin Protected Neuronal Cells via Attenuation of ERK/JNK Signaling Pathway
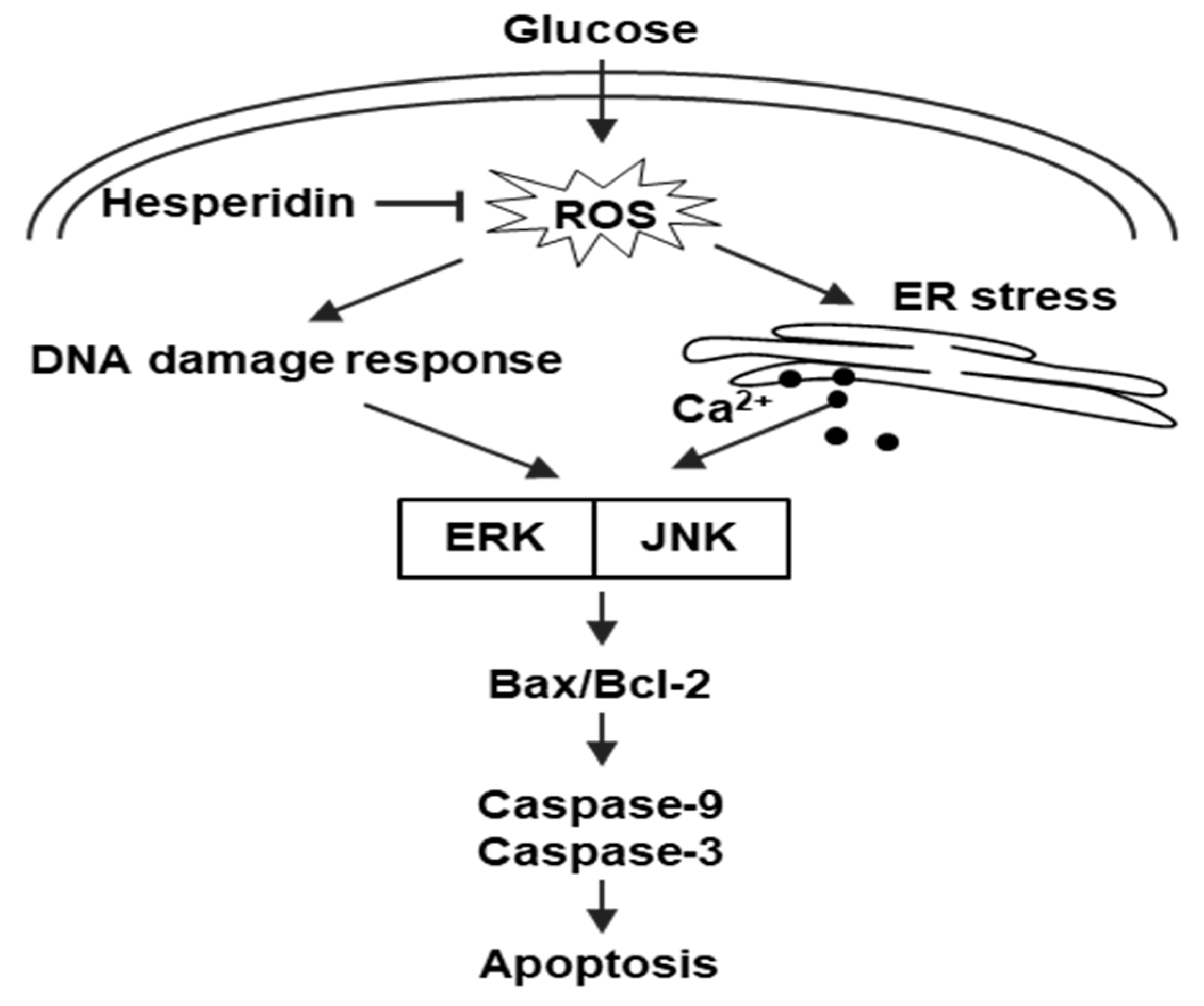
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Armstrong, D.G.; Swerdlow, M.A.; Armstrong, A.A.; Conte, M.S.; Padula, W.V.; Bus, S.A. Five year mortality and direct costs of care for people with diabetic foot complications are comparable to cancer. J. Foot Ankle Res. 2020, 13, 16. [Google Scholar] [CrossRef] [PubMed]
- Koepsell, H. Glucose transporters in brain in health and disease. Pflug. Arch. Eur. J. Physiol. 2020, 472, 1299–1343. [Google Scholar]
- Megallaa, M.H.; Ismail, A.A.; Zeitoun, M.H.; Khalifa, M.S. Association of diabetic foot ulcers with chronic vascular diabetic complications in patients with type 2 diabetes. Diabetes Metab. Syndr. 2019, 13, 1287–1292. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; Du, C.; Song, P.; Chen, T.; Rui, S.; Armstrong, D.G.; Deng, W. The role of oxidative stress and antioxidants in diabetic wound healing. Oxid. Med. Cell. Longev. 2021, 2021, 8852759. [Google Scholar] [CrossRef]
- Bahar, E.; Kim, H.; Yoon, H. ER stress−mediated signaling: Action potential and Ca2+ as key players. Int. J. Mol. Sci. 2016, 17, 1558. [Google Scholar] [CrossRef]
- Rosenberger, D.C.; Blechschmidt, V.; Timmerman, H.; Wolff, A.; Treede, R.D. Challenges of neuropathic pain: Focus on diabetic neuropathy. J. Neural. Transm. 2020, 127, 589–624. [Google Scholar]
- Federico, A.; Cardaioli, E.; Da Pozzo, P.; Formichi, P.; Gallus, G.N.; Radi, E. Mitochondria, oxidative stress and neurodegeneration. J. Neurol. Sci. 2012, 322, 254–262. [Google Scholar]
- Radi, E.; Formichi, P.; Battisti, C.; Federico, A. Apoptosis and oxidative stress in neurodegenerative diseases. J. Alzheimers Dis. 2014, 42 (Suppl. 3), S125–S152. [Google Scholar] [CrossRef]
- Fernyhough, P.; McGavock, J. Mechanisms of disease: Mitochondrial dysfunction in sensory neuropathy and other complications in diabetes. Handb. Clin. Neurol. 2014, 126, 353–377. [Google Scholar]
- Drake, J.; Sultana, R.; Aksenova, M.; Calabrese, V.; Butterfield, D.A. Elevation of mitochondrial glutathione by gamma−glutamylcysteine ethyl ester protects mitochondria against peroxynitrite−induced oxidative stress. J. Neurosci. Res. 2003, 74, 917–927. [Google Scholar]
- Ren, J.; Lu, Y.; Qian, Y.; Chen, B.; Wu, T.; Ji, G. Recent progress regarding kaempferol for the treatment of various diseases. Exp. Ther. Med. 2019, 18, 2759–2776. [Google Scholar] [PubMed]
- Amić, D.; Davidović−Amić, D.; Bešlo, D.; Trinajstić, N. Structure−radical scavenging activity relationships of flavonoids. Croat. Chem. Acta 2003, 76, 55–61. [Google Scholar]
- Soobrattee, M.A.; Neergheen, V.S.; Luximon−Ramma, A.; Aruoma, O.I.; Bahorun, T. Phenolics as potential antioxidant therapeutic agents: Mechanism and actions. Mutat. Res. 2005, 579, 200–213. [Google Scholar] [CrossRef]
- Nicolle, E.; Souard, F.; Faure, P.; Boumendjel, A. Flavonoids as promising lead compounds in type 2 diabetes mellitus: Molecules of interest and structure−activity relationship. Curr. Med. Chem. 2011, 18, 2661–2672. [Google Scholar] [CrossRef] [PubMed]
- Ola, M.S.; Al−Dosari, D.; Alhomida, A.S. Role of oxidative stress in diabetic retinopathy and the beneficial effects of flavonoids. Curr. Pharm. Des. 2018, 24, 2180–2187. [Google Scholar] [CrossRef] [PubMed]
- Thenmozhi, A.J.; Raja, T.R.; Janakiraman, U.; Manivasagam, T. Neuroprotective effect of hesperidin on aluminium chloride induced Alzheimer’s disease in Wistar rats. Neurochem. Res. 2015, 40, 767–776. [Google Scholar] [CrossRef]
- Tamilselvam, K.; Braidy, N.; Manivasagam, T.; Essa, M.M.; Prasad, N.R.; Karthikeyan, S.; Thenmozhi, A.J.; Selvaraju, S.; Guillemin, G.J. Neuroprotective effects of hesperidin, a plant flavanone, on rotenone−induced oxidative stress and apoptosis in a cellular model for Parkinson’s disease. Oxid. Med. Cell. Longev. 2013, 2013, 102741. [Google Scholar] [CrossRef]
- Hajialyani, M.; Hosein Farzaei, M.; Echeverría, J.; Nabavi, S.M.; Uriarte, E.; Sobarzo−Sánchez, E. Hesperidin as a neuroprotective agent: A review of animal and clinical evidence. Molecules 2019, 24, 648. [Google Scholar] [CrossRef]
- Xicoy, H.; Wieringa, B.; Martens, G.J. The SH−SY5Y cell line in Parkinson’s disease research: A systematic review. Mol. Neurodegener. 2017, 12, 10. [Google Scholar]
- Kim, H.; Choi, J.; Lee, H.; Park, J.; Yoon, B.I.; Jin, S.M.; Park, K. Skin corrosion and irritation test of nanoparticles using reconstructed three−dimensional human skin model, EpiDermTM. Toxicol. Res. 2016, 32, 311–316. [Google Scholar]
- Cha, J.W.; Piao, M.J.; Kim, K.C.; Yao, C.W.; Zheng, J.; Kim, S.M.; Hyun, C.L.; Ahn, Y.S.; Hyun, J.W. The polyphenol chlorogenic acid attenuates UVB−mediated oxidative stress in human HaCaT keratinocytes. Biomol. Ther. 2014, 22, 136–142. [Google Scholar] [CrossRef] [PubMed]
- Zhen, A.X.; Piao, M.J.; Hyun, Y.J.; Kang, K.A.; Ryu, Y.S.; Cho, S.J.; Kang, H.K.; Koh, Y.S.; Ahn, M.J.; Kim, T.H.; et al. Purpurogallin protects keratinocytes from damage and apoptosis induced by ultraviolet B radiation and particulate matter 2.5. Biomol. Ther. 2019, 27, 395–403. [Google Scholar] [CrossRef] [PubMed]
- Piao, M.J.; Ahn, M.J.; Kang, K.A.; Ryu, Y.S.; Hyun, Y.J.; Shilnikova, K.; Zhen, A.X.; Jeong, J.W.; Choi, Y.H.; Kang, H.K.; et al. Particulate matter 2.5 damages skin cells by inducing oxidative stress, subcellular organelle dysfunction, and apoptosis. Arch. Toxicol. 2018, 92, 2077–2091. [Google Scholar] [PubMed]
- Wang, H.; Zhang, J.; Hu, S.H.; Tan, S.Z.; Zhang, B.; Zhou, H.M.; Peng, R.Y. Real−time microwave exposure induces calcium efflux in primary hippocampal neurons and primary cardiomyocytes. Biomed. Environ. Sci. 2018, 31, 561–571. [Google Scholar] [PubMed]
- Hyun, Y.J.; Piao, M.J.; Kang, K.A.; Zhen, A.X.; Fernando, P.D.S.M.; Kang, H.K.; Ahn, Y.S.; Hyun, J.W. Effect of fermented fish oil on fine particulate matter−induced skin aging. Mar. Drugs 2019, 17, 61. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.J.; Kang, K.A.; Piao, M.J.; Ryu, Y.S.; Fernando, P.D.S.M.; Zhen, A.X.; Hyun, Y.J.; Ahn, M.J.; Kang, H.K.; Hyun, J.W. 7,8−Dihydroxyflavone protects high glucose−damaged neuronal cells against oxidative stress. Biomol. Ther. 2019, 27, 85–91. [Google Scholar] [CrossRef]
- Nishikawa, T.; Edelstein, D.; Du, X.L.; Yamagishi, S.; Matsumura, T.; Kaneda, Y.; Yorek, M.A.; Beebe, D.; Oates, P.J.; Hammes, H.P.; et al. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature 2000, 404, 787–790. [Google Scholar] [CrossRef]
- Liu, Y.; Sun, L.; Ma, Y.; Wei, B.; Gao, M.; Shang, L. High glucose and bupivacaine-induced cytotoxicity is mediated by enhanced apoptosis and impaired autophagy via the PERK-ATF4-CHOP and IRE1-TRAF2 signaling pathways. Mol. Med. Rep. 2019, 20, 2832–2842. [Google Scholar] [CrossRef]
- Singh, A.; Kukreti, R.; Saso, L.; Kukreti, S. Oxidative stress: A key modulator in neurodegenerative diseases. Molecules 2019, 24, 1583. [Google Scholar] [CrossRef]
- Chen, T.; Zhu, J.; Wang, Y.H.; Hang, C.H. ROS−mediated mitochondrial dysfunction and ER stress contribute to compression−induced neuronal injury. Neuroscience 2019, 416, 268–280. [Google Scholar] [CrossRef]
- Sugimoto, M.; Ko, R.; Goshima, H.; Koike, A.; Shibano, M.; Fujimori, K. Formononetin attenuates H2O2−induced cell death through decreasing ROS level by PI3K/Akt−Nrf2−activated antioxidant gene expression and suppressing MAPK−regulated apoptosis in neuronal SH−SY5Y cells. Neurotoxicology 2021, 85, 186–200. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Shen, W.B.; Yang, P.; Dong, D.; Sun, W.; Yang, P. High glucose inhibits neural stem cell differentiation through oxidative stress and endoplasmic reticulum stress. Stem. Cells Dev. 2018, 27, 745–755. [Google Scholar] [CrossRef] [PubMed]
- Bhatti, J.S.; Sehrawat, A.; Mishra, J.; Sidhu, I.S.; Navik, U.; Khullar, N.; Kumar, S.; Bhatti, G.K.; Reddy, P.H. Oxidative stress in the pathophysiology of type 2 diabetes and related complications: Current therapeutics strategies and future perspectives. Free Radic. Biol. Med. 2022, 184, 114–134. [Google Scholar] [PubMed]
- Cheng, Y.; Liu, J.; Luan, Y.; Liu, Z.; Lai, H.; Zhong, W.; Yang, Y.; Yu, H.; Feng, N.; Wang, H.; et al. Sarm1 gene deficiency attenuates diabetic peripheral neuropathy in mice. Diabetes 2019, 68, 2120–2130. [Google Scholar] [CrossRef]
- Calabrese, V.; Cornelius, C.; Dinkova−Kostova, A.T.; Calabrese, E.J.; Mattson, M.P. Cellular stress responses, the hormesis paradigm, and vitagenes: Novel targets for therapeutic intervention in neurodegenerative disorders. Antioxid. Redox Signal. 2010, 13, 1763–1811. [Google Scholar]
- Perluigi, M.; Di Domenico, F.; Giorgi, A.; Schininà, M.E.; Coccia, R.; Cini, C.; Bellia, F.; Cambria, M.T.; Cornelius, C.; Butterfield, D.A.; et al. Redox proteomics in aging rat brain: Involvement of mitochondrial reduced glutathione status and mitochondrial protein oxidation in the aging process. J. Neurosci. Res. 2010, 88, 3498–3507. [Google Scholar] [CrossRef]
- Parhiz, H.; Roohbakhsh, A.; Soltani, F.; Rezaee, R.; Iranshahi, M. Antioxidant and anti−inflammatory properties of the citrus flavonoids hesperidin and hesperetin: An updated review of their molecular mechanisms and experimental models. Phytother. Res. 2015, 29, 323–331. [Google Scholar]
- Shi, X.; Liao, S.; Mi, H.; Guo, C.; Qi, D.; Li, F.; Zhang, C.; Yang, Z. Hesperidin prevents retinal and plasma abnormalities in streptozotocin−induced diabetic rats. Molecules 2012, 17, 12868–12881. [Google Scholar] [CrossRef]
- Madabhushi, R.; Pan, L.; Tsai, L.H. DNA damage and its links to neurodegeneration. Neuron 2014, 83, 266–282. [Google Scholar]
- O’Brien, P.D.; Hinder, L.M.; Sakowski, S.A.; Feldman, E.L. ER stress in diabetic peripheral neuropathy: A new therapeutic target. Antioxid. Redox Signal. 2014, 21, 621–633. [Google Scholar] [CrossRef]
- Cheng, W.C.; Wen, Y.; Chiu, Y.S.; Chou, C.H.; Lim, C.J.; Lin, S.H.; Chang, J.M.; Lin, C.C. Pendulone induces apoptosis via the ROS−mediated ER−stress pathway in human non−small cell lung cancer cells. Toxicol. Vitr. 2022, 81, 105346. [Google Scholar] [CrossRef] [PubMed]
- Godoy, J.A.; Rios, J.A.; Picón−Pagès, P.; Herrera−Fernández, V.; Swaby, B.; Crepin, G.; Vicente, R.; Fernández−Fernández, J.M.; Muñoz, F.J. Mitostasis, calcium and free radicals in health, aging and neurodegeneration. Biomolecules 2021, 11, 1012. [Google Scholar] [CrossRef] [PubMed]
- Mancuso, C.; Pani, G.; Calabrese, V. Bilirubin: An endogenous scavenger of nitric oxide and reactive nitrogen species. Redox Rep. 2006, 11, 207–213. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, V.; Mancuso, C.; Calvani, M.; Rizzarelli, E.; Butterfield, D.A.; Stella, A.M. Nitric oxide in the central nervous system: Neuroprotection versus neurotoxicity. Nat. Rev. Neurosci. 2007, 8, 766–775. [Google Scholar] [CrossRef] [PubMed]
- Glory, A.; Bettaieb, A.; Averill−Bates, D.A. Mild thermotolerance induced at 40°C protects cells against hyperthermia−induced pro−apoptotic changes in Bcl−2 family proteins. Int. J. Hyperth. 2014, 30, 502–512. [Google Scholar] [CrossRef]
- Eskandari, E.; Eaves, C.J. Paradoxical roles of caspase−3 in regulating cell survival, proliferation, and tumorigenesis. J. Cell Biol. 2022, 221, e202201159. [Google Scholar]
- Cheng, C.; Zochodne, D.W. Sensory neurons with activated caspase−3 survive long−term experimental diabetes. Diabetes 2003, 52, 2363–2371. [Google Scholar] [CrossRef]
- Su, Z.; Han, S.; Jin, Q.; Zhou, N.; Lu, J.; Shangguan, F.; Yu, S.; Liu, Y.; Wang, L.; Lu, J.; et al. Ciclopirox and bortezomib synergistically inhibits glioblastoma multiforme growth via simultaneously enhancing JNK/p38 MAPK and NF−κB signaling. Cell Death Dis. 2021, 12, 251. [Google Scholar] [CrossRef]
- Wang, K.; Fu, X.Y.; Fu, X.T.; Hou, Y.J.; Fang, J.; Zhang, S.; Yang, M.F.; Li, D.W.; Mao, L.L.; Sun, J.Y.; et al. DSePA antagonizes high glucose−induced neurotoxicity: Evidences for DNA damage−mediated p53 phosphorylation and MAPKs and AKT pathways. Mol. Neurobiol. 2016, 53, 4363–4374. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lim, C.; Zhen, A.X.; Ok, S.; Fernando, P.D.S.M.; Herath, H.M.U.L.; Piao, M.J.; Kang, K.A.; Hyun, J.W. Hesperidin Protects SH−SY5Y Neuronal Cells against High Glucose−Induced Apoptosis via Regulation of MAPK Signaling. Antioxidants 2022, 11, 1707. https://doi.org/10.3390/antiox11091707
Lim C, Zhen AX, Ok S, Fernando PDSM, Herath HMUL, Piao MJ, Kang KA, Hyun JW. Hesperidin Protects SH−SY5Y Neuronal Cells against High Glucose−Induced Apoptosis via Regulation of MAPK Signaling. Antioxidants. 2022; 11(9):1707. https://doi.org/10.3390/antiox11091707
Chicago/Turabian StyleLim, Chaemoon, Ao Xuan Zhen, Sungwoo Ok, Pincha Devage Sameera Madushan Fernando, Herath Mudiyanselage Udari Lakmini Herath, Mei Jing Piao, Kyoung Ah Kang, and Jin Won Hyun. 2022. "Hesperidin Protects SH−SY5Y Neuronal Cells against High Glucose−Induced Apoptosis via Regulation of MAPK Signaling" Antioxidants 11, no. 9: 1707. https://doi.org/10.3390/antiox11091707