
Pain in Hemophilia: Unexplored Role of Oxidative Stress


1
Division of Hematology/Oncology, Department of Medicine, University of California, Irvine, CA 92697, USA
2
VA Medical Center, Southern California Institute for Research and Education, Long Beach, CA 90822, USA
3
Division of Hematology, Oncology, and Transplantation, Department of Medicine, University of Minnesota, Minneapolis, MN 55455, USA
*
Author to whom correspondence should be addressed.
Antioxidants 2022, 11(6), 1113; https://doi.org/10.3390/antiox11061113
Submission received: 29 April 2022
/
Revised: 26 May 2022
/
Accepted: 29 May 2022
/
Published: 3 June 2022
(This article belongs to the Special Issue Reactive Oxygen Species and Oxidative Damage Mediate Neuropathic Pain)
{kind=link}
Abstract
:Hemophilia is the most common X-linked bleeding diathesis caused by the genetic deficiency of coagulation factors VIII or IX. Despite treatment advances and improvements in clinical management to prevent bleeding, management of acute and chronic pain remains to be established. Repeated bleeding of the joints leads to arthropathy, causing pain in hemophilia. However, mechanisms underlying the pathogenesis of pain in hemophilia remain underexamined. Herein, we describe the novel perspectives on the role for oxidative stress in the periphery and the central nervous system that may contribute to pain in hemophilia. Specifically, we cross examine preclinical and clinical studies that address the contribution of oxidative stress in hemophilia and related diseases that affect synovial tissue to induce acute and potentially chronic pain. This understanding would help provide potential treatable targets using antioxidants to ameliorate pain in hemophilia.
1. Introduction
Hemophilia is the most common X-linked chronic bleeding diathesis, caused by the deficiency of coagulation factor VIII (FVIII) or factor IX (FIX) in hemophilia A and hemophilia B, respectively [1]. Despite being a genetic disorder, almost one-third of patients with hemophilia (PWH) do not have a family history of the disease [2]. According to coagulation factor activity level, severity is classified as mild, moderate, or severe. The frequency of bleeding and severity are usually correlated; severe deficiency (plasma FVIII or FIX < 1 U/dL) manifests as prolonged and excessive spontaneous bleeding or secondarily to trauma [3,4,5]. Hemophilia is associated with several clinical manifestations due to bleeding-related long-term complications of which hemarthrosis is the most common and severe [6,7,8]. The early onset and recurrent bleeding result in significant structural joint deformities triggered by the release and accumulation of hemoglobin, iron, and oxygen free radicals in the joint. However, the sequence of events and the molecular mechanisms resulting in joint deterioration are incompletely understood. The morbid effects of hemarthrosis lead to joint damage, physical deformity, chronic pain, and negative effects on health-related quality of life (HRQoL) [9,10].
Hemophilia treatment is mainly replacement therapy using clotting factor concentrates, such as prevention or intervention for acute bleeding. Factor VIII replacement and other therapies reduce the severity and incidence of bleeding, but pain may persist [11]. Pain affects physical and mental health and impairs HRQoL [12,13,14,15]. The lack of evidence-based guidelines for hemophilia-specific pain is recognized globally [12,16,17,18]. Pain is one of the compelling concerns associated with joint damage from the patient’s perspective. Improvements in hemophilia treatment call for a better understanding of the pathobiological mechanisms that lead to disabling arthropathy and associated chronic pain. This review outlines our current knowledge of the pathobiology of hemophilic arthropathy, highlighting the role of oxidative stress as a leading contributor to pain.
2. Pain in Hemophilia
Pain is a debilitating consequence of hemophilia. Pain can start early in infancy and continue throughout life with limited treatment options [19,20,21]. The most commonly affected joints are the knees (45%), followed by the elbows (30%), ankles (15%), shoulders (3%), and wrists (2%) [22]. Approximately 70% of patients with severe hemophilia suffer from pain daily, which can be recurrent, acute, and/or chronic [19,23]. Acute pain occurs during joint bleeds, and their repeated recurrence causes bone/cartilage damage leading to arthropathy resulting in chronic pain [3,11,15,24]. Chronic pain is complex and is associated with neurobiological, psychological, and social changes that may maintain the pain [11].
Clinical characteristics of hemophilia pain include ‘throbbing,’ ‘sharp,’ ‘tender,’ ‘aching,’ and ‘nagging’ [23,24]. Female carriers of hemophilia have reduced joint range of motion compared to non-carrier subjects [25]. Joint pain has a negative impact even on simple activities of daily living such as walking. Changes in gait patterns, including walking speed, load bearing asymmetry, swing phase, and stance phase have been observed in patients with severe and moderate hemophilia when compared to healthy subjects [26].
In addition to physical impairment (pain, hemophilic arthropathy, organ dysfunction), bleeding episodes in hemophilia often result in mental impairments leading to restriction in activities including school and social participation that affect education and QoL [27]. Joint pain in PWH is associated with reported dependency on pain-relieving drugs and depressive episodes [16]. In children, evidence exists that musculoskeletal dysfunction and associated pain affect the HRQoL, performance, and regular activities of daily living. This can lead to avoidance of many social and physical activities, including sports. Often, there is a fear of pain due to physical activity. For example, a study on children aged 4–12 years found that although motor performance and disability did not differ significantly between children with hemophilia and their healthy peers, most patients perceived that presence of pain had an impact on their performance [28]. In addition, children with hemophilia who experience more bleeding episodes are likely to have poorer academic performance than those with fewer bleeding episodes [29].
The basis of chronic pain caused by recurrent joint bleedings, whether nociceptive or neuropathic, has not been investigated. To address this, the painDETECT-questionnaire (pDq) was used on PWH and control subjects. The pDq identifies neuropathic components in a person’s pain profile. Interestingly, this study showed a positive neuropathic pain component in nine PWH but not in controls and concluded that there is likely a potential risk to misunderstand underlying pain mechanisms in PWH [30].
3. Hemarthropathy and Oxidative Stress, Two Faces of the Same Coin
Articular cartilage is designed to bear and distribute the mechanical loads across the joints [31]. The majority of the articular cartilage’s viscoelastic and smooth structure is maintained by chondrocytes, which regulate the production of extracellular cartilage matrix (e.g., type II collagen and aggrecan) and maintain tissue homeostasis [32]. The synovial membrane is a thin specialized soft connective tissue membrane that lines the inner surface of synovial joint capsules. Unlike the avascular articular cartilage, the synovium is well innervated and vascularized, playing an essential role in transporting nutrients, debris and waste removal, immune modulation, and inflammation in the joint [33]. These specialized functions are achieved through specific cell types in the synovial membrane; macrophage-like synovial cells (type A) maintain the synovial fluid by removing wear and tear debris, and fibroblast-like synoviocytes (FLS, type B) are responsible for synthesizing and secreting major extracellular matrix proteins in synovial fluid, such as hyaluronic acid and lubricin [34].
The underlying pathogenesis of hemophilic arthropathy is not fully explored. However, evidence shows that recurrent joint bleeding creates a cascade of intra-articular inflammation, angiogenesis, and joint destruction [35]. The development of hemophilic arthritis involves three main stages: acute hemarthrosis, chronic synovitis, and degenerative arthritis [36]. The intra-articular blood effusion favors iron accumulation, released from hemoglobin, and hemosiderin deposition, both of which are believed to be pivotal for the early phase of hemophilic arthropathy, inducing cytokines and pro-angiogenic factors to progressive synovial proliferative growth and ultimately resulting in articular cartilage destruction [37,38,39]. Thus, there is a growing interest in the destructive role of reactive oxygen metabolites as an early pathogenic step resulting in chondrocyte apoptosis, with synovial inflammatory changes being secondary or parallel to the process of cartilage damage [35] (Figure 1).
3.1. Cell-Free Heme
Following a joint bleed, the synovium is overloaded with clearing the products of hemolysis. Iron, an essential constituent of hemoglobin found in erythrocytes, is a crucial breakdown product and is thought to play a significant part in inflaming the synovium [40]. The contribution of iron to the hemophilic arthropathy is multifactorial. The iron-rich breakdown product hemosiderin induces the expression of several pro-inflammatory cytokines such as interleukin (IL)-1α, IL-6, and tumor necrosis factor-alpha (TNF)-α [35,41]. Moreover, it appears that iron is involved in the initiation of synovial pannus growth by dysregulating the expression of critical genes such as c-myc and mdm2, which are responsible for synoviocyte proliferation [42,43,44]. Thus, the synovium becomes increasingly vascular and hypertrophic, and inflammatory cells are recruited to the area in greater numbers in hemophilia [40]. The activated synovium affects cartilage by producing cartilage-destructive pro-inflammatory cytokines and matrix-degrading proteinases [45], resulting in further hemarthrosis and creating a vicious cycle of bleeding and inflammation [46]. Von Drygalski et al. showed that iron deposition can occur in cartilage (not only in synovium) and established the relationship between cartilage hemosiderin in hemophilic joints and joint deterioration [47].
Additionally, iron contributes to direct cartilage damage induced by oxidative stress [48,49]. Iron plays important roles in normal cellular processes including oxygen storage and transport, and energy metabolism. Delicate regulation of iron homeostasis is required for maintaining normal cellular function, while excessive iron would damage cells by increasing the production of reactive oxygen species (ROS). Oxidants, such as hydrogen peroxide (H2O2), are produced by activated mononuclear cells and chondrocytes [40]. In the presence of erythrocyte-derived iron, H2O2 generates highly toxic hydroxyl radicals through the Fenton reaction, resulting in the peroxidation of membrane lipids, mitochondrial dysfunction, cellular protein and nucleic acid damage, and ultimately ferroptosis [50], leading to permanent cartilage damage [48,51]. Recent findings indicate that perturbation in iron homeostasis is implicated in the progression of many diseases such as osteoarthritis (OA), osteoporosis, Parkinson’s disease, and Alzheimer’s disease [52]. The association between iron overload and OA pathogenesis is broadly appreciated regarding its effect on ROS production and oxidative stress. OA is characterized by synovial tissue inflammation and cartilage degeneration [53]. Oxidative stress and mitochondrial dysfunction have been demonstrated to be an important contributor to iron overload-induced cartilage degeneration [54,55,56]. Jing et al. further confirmed these observations by showing that iron chelator or antioxidant drugs could inhibit iron overload-induced OA-related catabolic markers and mitochondrial dysfunction [53].
Interestingly, a closely related pathogenesis has been observed in sickle cell disease (SCD)-associated OA, with joint space narrowing, synovial inflammation and reduced periarticular bone mass [57]. Retrospective clinical studies indicate that iron overload may have an adverse effect on osteochondral homeostasis in SCD patients [58], as approximately 70% of patients suffering from SCD and exhibiting high serum iron levels had relatively low bone mass. In agreement with these findings, humanized sickle mice showed significant stance instability, perhaps due to pressure pain; reduced walking speed; increased stance duration, with hesitancy perhaps due to anticipation of pain; and reduced diagonal swing, which may indicate poor coordination [59]. These findings further support the contribution of free iron in osteopathic changes leading to altered gait in hemophilia.
3.2. Bone Destruction
Repeated bleeding episodes in hemophilia leave the synovial membrane hypertrophied and villous with intense neovascularization. Over time, chronic synovitis is established with pannus, thinning of the cartilage, bony changes, and, eventually, marked joint dysfunction (i.e., reduced range of movement, stiff or unstable joints, swelling, and pain with use) [36]. It is noteworthy that osteoporosis has been frequently reported among PWH and correlated with the severity of arthropathy, which might reflect an imbalance between bone resorption and bone formation [60,61]. A possible mechanism for the bone resorption might be the reduction of thrombin, which is known to mediate the proliferation of osteoblasts via protease-activated receptor-1 (PAR-1) mediated proliferation of osteoblasts [62], shifting the balance towards osteoclastic activity.
Mechanisms underlying bone turnover are regulated strictly with the molecular triad of osteoprotegerin (OPG), the receptor activator of nuclear factor κB (RANK), and the RANK ligand (RANKL) [63,64]. The osteoclast precursors express the receptor RANK on their surface; following binding of RANK to its ligand RANKL, osteoclastogenesis is induced and potentially enhanced by cytokines (e.g., TNF-α, IL-1, and IL-17) that promote both inflammation and bone resorption [65,66]. RANKL is a transmembrane ligand synthesized by synovial cells and lymphocytes that induce osteoclast differentiation and maturation resulting in bone resorption [67]. On the other hand, the RANK/RANKL function is balanced by the OPG, which competes with RANKL for binding to RANK and thus negatively regulates osteoclast differentiation [68,69]. It is suggested that an imbalance between OPG and RANKL contributes to the hemophilic bone changes evidenced by decreased OPG levels and the strong expression of RANK and RANKL [70].
Oxidative stress is involved in the pathogenesis of bone loss in many disorders by favoring osteoclastogenesis, inhibiting the mineralization, and reducing osteoblast activity [71,72,73]. The expression of RANKL and OPG is sensitive to increased oxidative status, which induces RANKL up-regulation and OPG down-regulation through the activation of protein kinases of downstream signaling including mitogen activated protein kinases (ERK1/2, JNK) [74,75,76]. Moreover, oxidative stress blocks the activation of osteoblasts and induces osteocyte apoptosis, causing an imbalance in favor of osteoclastogenesis [77].
To date, limited experimental studies have targeted the direct antioxidant effects on bone remodeling. However, limited data suggest a correlation between reduced plasma antioxidant levels and osteoporosis [78,79,80]. Furthermore, antioxidants such as vitamin C, E, and N-acetylcysteine (NAC) might positively affect bone health in individuals with osteoporosis [81,82,83]. Together, these findings suggest a role of oxidative stress in cartilage/bone damage in hemophilic arthropathy, which may underlie the pathobiology of pain in hemophilia.
3.3. Inflammation
Iron and other blood components play a role in initiating the inflammatory process in hemarthropathy. This inflammatory process involves the synovial tissue, i.e., synovitis, and is characterized by recruitment of inflammatory cells, tissue hypertrophy, and neoangiogenesis [84]. Recent studies highlight the crucial role of inflammasome in the hemophilic arthropathy-related inflammatory process, which regulates the production of pro-inflammatory cytokines, including IL-1β. Moreover, IL-1α, IL-6, IL-1β, and TNF-α activate monocytes/macrophages [35]. The activation of these phagocytic cells may contribute to tissue damage in multiple ways. They trigger the release of proteases such as matrix metalloproteinases (MMPs), the production of inducible nitric oxide (NO), and tissue plasminogen activation [85,86]. These, in turn, play a role in activating other cells, including T cells, fibroblasts, and osteoclasts, through a variety of inflammatory mediators, resulting in articular cartilage and subchondral bone destruction [35].
Activated phagocytic cells contribute to oxidative stress in hemarthropathy. As a part of their defensive role, activated phagocytic cells undergo an oxidative burst, producing ROS that target and kill pathogens [87]. The phagocytic cells undergo the oxidative burst via the NADPH oxidase (NOX) system. This results in increased oxygen consumption and superoxide (O2−) production, which is converted to H2O2 either spontaneously or catalyzed by superoxide dismutase (SOD) [88]. SOD is present in three isoforms; of which the SOD1 gene is often constitutively expressed and not as easily inducible as other SODs. At the same time, many inflammatory cytokines have been implicated in the induction of SOD2, including interferon-gamma (IFN-γ), TNF-α, IL-1β, IL-4, and IL-6, which are capable of rapidly modulating SOD2 gene transcription [89]. NF-κB was identified as the most crucial transcriptional factor regulating cytokine-mediated induction of SOD2 gene expression [90]. Moreover, the presence of excess iron contributes to a surge in hydroxide (OH−) and hydroxyl radical production via the Fenton reaction [84].
In addition to the oxidative burst role played by the phagocytic cells, neutrophils also contribute to oxidative stress. Remarkably, the neutrophil-associated enzyme myeloperoxidase can oxidize halides such as chloride (Cl−) and convert hydrogen peroxide into hypochlorous acid (HOCl). As a result, H2O2 can further interact with HOCl, producing singlet oxygen, another highly reactive and damaging radical [91].
3.4. Angiogensis
Angiogenesis is a hallmark feature of several arthritic conditions, including hemophilic arthropathy. Vascular endothelial growth factor (VEGF), a potent angiogenic factor, was elevated in patients with severe hemophilia both in circulation and synovial tissue [92,93]. Zetterberg et al. showed that angiogenesis is increased in advanced hemophilic joint disease with increased expression of VEGF and microvessel density [94]. This correlation between disease activity and high serum VEGF-A levels has also been shown in rheumatoid arthritis (RA) [37]. Interestingly, VEGF-A expression is induced essentially by hypoxia [35,92]. A study that involved collagen-induced arthritis (CIA) in mice demonstrated that tissue oxygenation is dysregulated in response to movement in arthritic mice compared to healthy non-arthritic mice [95]. Exposure to hypoxic conditions favors the increase of ROS, and despite the proliferation of new blood vessels, they are dysfunctional and fail to restore tissue-oxygen homeostasis [96].
NOX pathway is also active in nonphagocytic cells, including endothelial cells, vascular smooth muscle cells, fibroblasts, and chondrocytes [97,98,99]. It may be involved in the proliferation, differentiation, and migration of endothelial cells and thus contribute to the angiogenesis process [100,101,102], creating repetitive cycles of hypoxia and reoxygenation that favor chronic oxidative stress within the microenvironment of the affected arthritic joint [103,104]. NOX-1 silencing and antioxidants treatment were demonstrated to decrease angiogenic response of endothelial cells [105,106], suggesting a contribution of iron and oxidative stress in the promotion of dysfunctional angiogenesis. These findings implicate the active role of NOX–derived ROS in angiogenesis, presenting oxidative stress as a possible initiating factor in driving angiogenesis and thus a potential therapeutic target.
4. Oxidative Stress as a Contributor to Pain in Hemophilia
Recurrent joint bleedings result in chronic pain, which is more complicated than acute pain and adversely affects PWH. The underlying mechanism of pain in hemophilia remains to be elucidated [11,30,107,108,109,110]. Based on other chronic pain conditions with joint pain such as osteoarthritis (OA) or rheumatoid arthritis (RA), the possible mechanisms may include features of nociceptive and/or neuropathic pain [111,112]. Pain is currently categorized into nociceptive pain (often inflammatory) resulting from tissue damage; neuropathic pain involving injury to the somatosensory nervous system; and idiopathic pain, which has no identified cause [113,114,115]. These distinctions are important because of the specificity of analgesic therapy to be used. Correctly identifying when chronic pain can be classified as neuropathic is vital because common analgesics, such as non-steroidal anti-inflammatory drugs (NSAIDs), poorly control neuropathic pain [116]. Neuropathic pain is more likely to respond to nonstandard analgesia (including tricyclic antidepressants, 5-hydroxytryptamine–noradrenaline reuptake inhibitors, or gabapentinoids) than to NSAIDs. Nociception is the neural process of encoding noxious stimuli [115]. Oxidative stress contributes to the complex and profound changes that underlie clinical disorders associated with pain. In this section, we highlight the role of oxidative stress as an underlying mechanism for chronic pain in hemophilic arthropathy and a possible target for future therapeutic trials.
4.1. Nociceptive Pain
Oxidative stress refers to a condition where the production of oxidants exceeds the capacity for their neutralization, which causes damage to cell membrane, lipids, nucleic acids, and proteins [117]. For decades, the connection between oxidative stress status and nociception has been evident [118]. ROS and reactive nitrogen species (RNS) have been shown to contribute directly to the destructive and proliferative synovitis in persons with RA [119]. This pathobiology may have similarities to another inherited blood disorder, sickle cell disease (SCD), accompanied by chronic and acute pain in a microenvironment replete with cell-free heme, oxidative stress, inflammation, and pain [120]. Recent observations have also suggested that oxidative stress might be involved in bone pathogenesis, including osteoporosis, bone cancer development, diabetes-induced bone complications, and inflammatory joint diseases [121].
Marked inflammation and synovial hypertrophy noted in hemophilic arthropathy resemble the pathological mechanisms observed in RA, while the progressive degeneration of the hyaline cartilage mimics that seen in OA [84]. These processes may occur in parallel leading to degenerative arthritis that progresses until the joint is destroyed [36]. Joint inflammation earlier in life could be a key factor in the development and maintenance of chronic arthritic pain. Early joint inflammation ‘primes’ the joint by sensitizing the peripheral nerve fibers leading to acute pain [122]. This may occur through sensitization of primary afferents in the periphery as well as central sensitization of the spinal second order neurons leading to sustained chronic pain [123]. It is believed that both inflammatory and oxidative processes contribute to the pathogenesis of the acute nociceptive phase; however, oxidative stress accounts for the maintenance of the nociceptive process in the chronic phase [124].
The pathogenesis of hemarthropathy encompasses the recruitment of inflammatory cells resulting in the production of ROS (e.g., H2O2, superoxide (2O2), and peroxynitrite (ONOOH)) [125,126]. Oxidative stress degrades cellular membranes, nucleic acids, and extracellular components, leading to the accumulation of damaged proteins in the tissue [117]. Degradation products and cellular content containing oxidized molecules contribute to the exacerbation of synovial inflammation and exacerbation of pain [127]. Free radicals oxidize lipids such as linoleic acid (LA) or arachidonic acid (AA) into their active metabolites. Oxidized LA and AA activate the transient receptor potential vanilloid 1 (TRPV1), a multimodal receptor involved in the transduction of nociceptive and thermal stimuli [128]. Patwardhan et al. demonstrated that the injections of oxidized LA metabolites into rodent hind paw elicited spontaneous nocifensive behavior and thermal hyperalgesia via a TRPV1-dependent mechanism [129]. Recent studies have implicated that nerve growth factor (NGF) triggered the development of persistent thermal and mechanical hyperalgesia mediated by oxidative processes via the production of endogenous TRPV1 agonists and activation of the channel [130]. Moreover, the administration of the combined oxidative enzyme inhibitor/antioxidant significantly reduced thermal and mechanical nociception, denoting a positive role for oxidative processes in persistent nociception from NGF [131]. Both H2O2 and ONOO− are involved in pain deriving from inflammation, mainly through the cyclooxygenase-2 (COX2)/prostaglandin E2 (PGE2) pathway [127]. PGE2 contributes to the generation of both inflammatory and neuropathic pain conditions, acting on four G protein-coupled receptor’s (GPCR) E prostanoid receptor subtypes 1, 2, 3, and 4 (EP1-4). PGE2 directly excites nociceptors and potentiates the sensitizing effects of other pain mediators such as ATP, bradykinin, and capsaicin [132]. This body of literature supports that oxidative stress is associated with and contributes to maintenance of chronic pain [118]. This contribution could be nociceptive (inflammatory) as well as neuropathic.
4.2. Neuropathic Pain
The poor correlation between the radiologic signs of arthritis and severity of pain and the dissociation between disease progression and pain in RA and OA drew attention to a potential risk of misunderstanding the underlying pain mechanisms in PWH [30]. Neuropathic pain may persist long after the initiation of nerve injury, and/or may arise spontaneously or be evoked by innocuous stimuli [133,134,135]. A recent study detected a positive neuropathic pain component when PWH completed pDq, a questionnaire that identifies neuropathic components in an individual´s pain profile [30]. Moreover, non-bony ankylosed knees from hemophilia patients with greater pain showed increased protein gene product (PGP) 9.5 positive sensory nerve fiber sprouting and NGF concentrations when compared to bony-ankylosed knee of end-stage hemophilic arthropathy with milder pain [136]. Impaired conditioned pain modulation response in PWH is suggestive of the involvement of the descending inhibitory pain pathway [137]. This observation comes in line with pain-related chronic conditions that share many of the underlying features of joint damage as hemophilic arthropathies such as OA and RA. Initial studies in these chronic arthritic conditions showed nociceptive and neuropathic pain features. Interestingly, sprouting of aberrant nerve fibers was detected in arthritic joints in experimental animal model where arthritis was induced by injecting complete Freund’s adjuvant (CFA), which contributed to pain-related behavior [138,139]. The increased density of unmyelinated nociceptive C-fibers expressing calcitonin gene-related peptide (CGRP) and sympathetic fibers into areas of inflammation may contribute to the persistence of pain when inflammation has subsided [139]. Arthritic human joints have shown similar CGRP+ nerve fiber sprouting and were associated with neovascularization following joint injury [122,140].
Growing evidence points to endoneural oxidative stress as a leading cause of nerve dysfunction and neuropathic pain [141]. Among neuropathic pain models, experimentally induced chronic constriction injury (CCI) of the sciatic nerve has been widely used to induce neuropathic pain in animals comparable to that observed in patients with neuropathic pain [142]. Studies have shown that oxidative stress mainly acts through mitochondrial dysfunction and the accumulation of lipid peroxidation products such as 4-hydroxynonenal (4-HNE) and malondialdehyde. The reduction of antioxidant peptide glutathione and increased expression of superoxide dismutase were identified as essential determinants of neuropathological and behavioral consequences of CCI-induced neuropathy [143].
The contribution of oxidative stress to neuropathic pain in the central and peripheral systems are multifactorial [144,145,146,147]. The nervous system is particularly vulnerable to oxidative stress due to its high levels of phospholipids and axonal mitochondrion and its relatively weak antioxidant defense [148]. It is possible for ROS to directly modulate neuroexcitability in central synapses by increasing glutamate release from primary afferent terminals and inhibiting GABAergic interneurons [146]. Superoxides have been shown to upregulate kinase signaling in hippocampal neurons [149,150]. The involvement of signal transduction pathways, particularly protein kinase C activation, is central in the development of peripheral and central sensitization of several pain etiologies [151,152,153,154,155]. It has also been shown that ROS may lead to the release of apoptotic factors from mitochondria, resulting in the degeneration of primary afferents. Activation of apoptosis contributes to the development of neuropathy [147]. Thus, the oxidative stress replete microenvironment in hemophilia is a potential contributor to neuropathic characteristics of pain and thus a promising treatable target.
5. Novel Antioxidant Therapies for Chronic Pain in Hemophilia
The Sirtuin (SIRT) family of proteins is amongst the most promising antioxidant agents [156]. The SIRTs are believed to play an essential role in the cellular response to various stresses, such as oxidative stress [157]. SIRT1 agonists modulate inflammation, oxidative stress, and mitochondrial dysfunction in a manner that can reduce chronic pain [158]. Several studies have shown that SIRT3 can enhance the ability of mitochondria to reduce ROS levels and protect against oxidative stress through regulating vital antioxidant enzymes, such as manganese superoxide dismutase (MnSOD) [159,160,161]. There is compelling evidence that polyphenols, including resveratrol, bergamot, hydroxytyrosol, and oleuropein, have the ability to activate SIRTs directly or indirectly [160,162,163,164]. The resveratrol compounds bind to SIRT1 at the N-terminal and increase SIRT1 activity [160], indicating its potential for treating chronic neuropathic pain [165]. Curcumin is another natural polyphenol that exhibits a neuroprotective role mediated by SIRT1 induction [166].
Another family of antioxidants are flavonoids that have been shown to improve and control diverse pain biomarkers in animal models of different neuropathic conditions, including diabetic- and chemotherapy induced-neuropathy [167]. Flavonoids are potent allosteric modulators of GABAergic receptors controlling their actions [168]. Essentially, flavonoids block oxidative stress, activate glial cells, and prevent mitochondrial dysfunction to reduce peripheral neuropathic pain [169].
Probucol is an anti-oxidative and lipid-lowering drug shown in preclinical and clinical studies to reduce serum low-density lipoprotein-cholesterol (LDL-C) [170]. In addition, probucol has been shown to counteract CCI-induced neuropathy via modulating NF-κB/NLRP3 signaling and augmenting transcription factor nuclear related factor (Nrf-2) activity [171]. Current evidence suggests that Nrf2 signaling contributes to antinociceptive activity by reducing inflammation, oxidative stress, and mitochondrial dysfunction [172].
In a recent study, vitamin D3 showed an antinociceptive effect in rats with CCI preventing the increase in lipid hydroperoxide, superoxide anion generation, and hydrogen peroxide (H2O2) levels in the spinal cord [173]. Thus, reduction in oxidative stress may be contributing to vitamin D-induced antinociception in CCI rats.
An unmet need is to decipher the mechanisms of oxidative stress underlying hemophilic arthropathy and pain to develop treatable targets. Several antioxidants described above are nutraceuticals and may not be toxic and/or interfere with current therapies for hemophilia. Thus, mechanism-based targeting of oxidative stress with novel antioxidants to prevent and/or treat hemophilic arthropathy and pain requires attention.
6. Conclusions
Increased bleeding, release of cell free heme, and subsequent cytokine elevation in joints may contribute to tissue damage and peripheral nerve fiber activation in hemophilia. Together, these phenomena may drive pain in hemophilia via oxidative stress as one of the major contributors to tissue damage and pain. Production and accumulation of ROS would promote immune cell activation and pro-inflammatory cytokine release, which may promote nociceptor activation leading to exacerbation of pain. It is likely that targeting mechanisms of oxidative stress in hemophilia with novel antioxidants may ameliorate pain and tissue damage.
Author Contributions
Conceptualization, R.F. and K.G.; methodology, R.F., D.A.A. and K.G.; writing—original draft preparation, R.F., D.A.A. and K.G.; writing—review and editing, R.F., D.A.A. and K.G.; visualization, R.F. and K.G.; supervision, K.G.; funding acquisition, D.A.A. and K.G. All authors have read and agreed to the published version of the manuscript.
Funding
This work has been supported by NIH grants, R01 CA263806, RO1 HL147562 and U18 EB029354 and Susan Samueli Scholar Award to KG, and the University of California President’s Postdoctoral Fellowship to DAA. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data are available upon request.
Conflicts of Interest
The authors report the following: Kalpna Gupta: Honoraria: Tautona Group, Novartis and CSL Behring. Research Grants: Cyclerion, 1910 Genetics, Novartis, Grifols, UCI Foundation, and SCIRE Foundation.
Abbreviations
| PWH | Patients With Hemophilia |
| HRQoL | Health-Related Quality Of Life |
| pDq | Paindetect-Questionnaire |
| FLS | Fibroblast-Like Synoviocytes |
| IL | Interleukin |
| TNF-α | Tumor Necrosis Factor-Alpha |
| ROS | Reactive Oxygen Species |
| H2O2 | Hydrogen Peroxide |
| OA | Osteoarthritis |
| SCD | Sickle Cell Disease |
| MMPs | Matrix Metalloproteinases |
| NO | Nitric Oxide |
| NOX | Nadph Oxidase |
| O2− | Superoxide |
| SOD | Superoxide Dismutase |
| Cl− | Chloride |
| HOCl | Hypochlorous Acid |
| VEGF | Vascular Endothelial Growth Factor |
| RA | Rheumatoid Arthritis |
| CIA | Collagen-Induced Arthritis |
| PAR-1 | Protease-Activated Receptors |
| OPG | Osteoprotegerin |
| RANKL | RANK Ligand |
| NAC | N-Acetylcysteine |
| NSAIDs | Non-Steroidal Anti-Inflammatory Drugs |
| ONOOH | Peroxynitrite |
| LA | Linoleic Acid |
| AA | Arachidonic Acid |
| TRPV1 | Transient Receptor Potential Vanilloid 1 |
| OLAMs | Oxidized LA Metabolites |
| NGF | Nerve Growth Factor |
| COX2 | Cyclooxygenase-2 |
| PGE2 | Prostaglandin E2 |
| GPCR | G Protein-Coupled Receptors |
| EP1-4 | E Prostanoid Receptor Subtypes 1, 2, 3 And 4 |
| PGP 9.5 | Protein Gene Product 9.5 |
| CFA | Complete Freund’s Adjuvant |
| CGRP | Calcitonin Gene-Related Peptide |
| CCI | Chronic Constriction Injury |
| 4-HNE | 4-Hydroxynonenal |
| MDA | Malondialdehyde |
References
- Castaman, G.; Matino, D. Hemophilia A and B: Molecular and clinical similarities and differences. Haematologica 2019, 104, 1702–1709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kulkarni, R.; Soucie, J.M.; Lusher, J.; Presley, R.; Shapiro, A.; Gill, J.; Manco-Johnson, M.; Koerper, M.; Mathew, P.; Abshire, T.; et al. Sites of initial bleeding episodes, mode of delivery and age of diagnosis in babies with haemophilia diagnosed before the age of 2 years: A report from The Centers for Disease Control and Prevention’s (CDC) Universal Data Collection (UDC) project. Haemophilia 2009, 15, 1281–1290. [Google Scholar] [CrossRef] [PubMed]
- Bolton-Maggs, P.H.; Pasi, K.J. Haemophilias A and B. Lancet 2003, 361, 1801–1809. [Google Scholar] [CrossRef]
- Pollmann, H.; Richter, H.; Ringkamp, H.; Jürgens, H. When are children diagnosed as having severe haemophilia and when do they start to bleed? A 10-year single-centre PUP study. Eur. J. Pediatrics 1999, 158, S166–S170. [Google Scholar] [CrossRef]
- Morfini, M.; Coppola, A.; Franchini, M.; Di Minno, G. Clinical use of factor VIII and factor IX concentrates. Blood Transfus. 2013, 11, s55–s63. [Google Scholar] [CrossRef]
- Rodriguez-Merchan, E.C. Musculoskeletal Complications of Hemophilia. HSS J.® 2010, 6, 37–42. [Google Scholar] [CrossRef]
- Franchini, M.; Mannucci, P.M. Co-morbidities and quality of life in elderly persons with haemophilia. Br. J. Haematol. 2010, 148, 522–533. [Google Scholar] [CrossRef]
- Philipp, C. The aging patient with hemophilia: Complications, comorbidities, and management issues. Hematol. Am. Soc. Hematol. Educ. Program 2010, 2010, 191–196. [Google Scholar] [CrossRef] [Green Version]
- Barr, R.D.; Saleh, M.; Furlong, W.; Horsman, J.; Sek, J.; Pai, M.; Walker, I. Health status and health-related quality of life associated with hemophilia. Am. J. Hematol. 2002, 71, 152–160. [Google Scholar] [CrossRef]
- van Genderen, F.R.; Westers, P.; Heijnen, L.; de Kleijn, P.; van den Berg, H.M.; Helders, P.J.; van Meeteren, N.L. Measuring patients’ perceptions on their functional abilities: Validation of the Haemophilia Activities List. Haemophilia 2006, 12, 36–46. [Google Scholar] [CrossRef]
- Auerswald, G.; Dolan, G.; Duffy, A.; Hermans, C.; Jiménez-Yuste, V.; Ljung, R.; Morfini, M.; Lambert, T.; Šalek, S.Z. Pain and pain management in haemophilia. Blood Coagul. Fibrinolysis 2016, 27, 845–854. [Google Scholar] [CrossRef] [Green Version]
- Witkop, M.; Santaella, M.; Nichols, C.D.; Lambing, A.Y.; Baumann, K.; Curtis, R.G.; Humphrey, C.; Humphries, T.J.; Newman, J.; Durben, N.; et al. Understanding the Pain Management Landscape Within the US Bleeding Disorder Community: A Multi-Center Survey. Pain Med. 2022, 23, 269–279. [Google Scholar] [CrossRef]
- Rambod, M.; Sharif, F.; Molazem, Z.; Khair, K.; von Mackensen, S. Health-Related Quality of Life and Psychological Aspects of Adults With Hemophilia in Iran. Clin. Appl. Thromb. Hemost. 2018, 24, 1073–1081. [Google Scholar] [CrossRef]
- Witkop, M.L.; Lambing, A.; Nichols, C.D.; Munn, J.E.; Anderson, T.L.; Tortella, B.J. Interrelationship between depression, anxiety, pain, and treatment adherence in hemophilia: Results from a US cross-sectional survey. Patient Prefer. Adherence 2019, 13, 1577–1587. [Google Scholar] [CrossRef] [Green Version]
- Witkop, M.; Wang, M.; Hernandez, G.; Recht, M.; Baumann, K.; Cooper, D.L. Impact of haemophilia on patients with mild-to-moderate disease: Results from the P-FiQ and B-HERO-S studies. Haemophilia 2021, 27, 8–16. [Google Scholar] [CrossRef]
- Stromer, W.; Pabinger, I.; Ay, C.; Crevenna, R.; Donnerer, J.; Feistritzer, C.; Hemberger, S.; Likar, R.; Sevelda, F.; Thom, K.; et al. Pain management in hemophilia: Expert recommendations. Wien. Klin. Wochenschr. 2021, 133, 1042–1056. [Google Scholar] [CrossRef]
- Di Minno, M.N.; Santoro, C.; Corcione, A.; Di Minno, G.; Martinelli, M.; Mancuso, M.E.; Acone, B.; Molinari, A.C.; Passeri, E.V.; Rocino, A. Pain assessment and management in Italian Haemophilia Centres. Blood Transfus. 2021, 19, 335. [Google Scholar]
- Pinto, P.R.; Paredes, A.C.; Almeida, A. Pain prevalence, characteristics, and impact among people with hemophilia: Findings from the first Portuguese survey and implications for pain management. Pain Med. 2020, 21, 458–471. [Google Scholar] [CrossRef]
- Paredes, A.C.; Teixeira, P.; Almeida, A.; Pinto, P.R. Prevalence and Interference of Chronic Pain Among People With Hemophilia: A Systematic Review and Meta-Analysis. J. Pain 2021, 22, 1134–1145. [Google Scholar] [CrossRef]
- Stromer, W.; Messerer, B.; Crevenna, R.; Hemberger, S.H.; Jauk, B.; Schwarz, R.; Streif, W.; Thom, K.; Wagner, B.; Zwiauer, K.; et al. Pain therapy for children and adolescents with hemophilia: Recommendations by an expert panel. Schmerz 2018, 32, 404–418. [Google Scholar] [CrossRef]
- Witkop, M.; Neff, A.; Buckner, T.W.; Wang, M.; Batt, K.; Kessler, C.M.; Quon, D.; Boggio, L.; Recht, M.; Baumann, K.; et al. Self-reported prevalence, description and management of pain in adults with haemophilia: Methods, demographics and results from the Pain, Functional Impairment, and Quality of life (P-FiQ) study. Haemophilia 2017, 23, 556–565. [Google Scholar] [CrossRef]
- Srivastava, A.; Brewer, A.; Mauser-Bunschoten, E.; Key, N.; Kitchen, S.; Llinas, A.; Ludlam, C.; Mahlangu, J.; Mulder, K.; Poon, M. Guidelines for the management of hemophilia. Haemophilia 2013, 19, e1–e47. [Google Scholar] [CrossRef]
- van Genderen, F.R.; Fischer, K.; Heijnen, L.; de Kleijn, P.; van den Berg, H.M.; Helders, P.J.; van Meeteren, N.L. Pain and functional limitations in patients with severe haemophilia. Haemophilia 2006, 12, 147–153. [Google Scholar] [CrossRef]
- Witkop, M.; Lambing, A.; Kachalsky, E.; Divine, G.; Rushlow, D.; Dinnen, J. Assessment of acute and persistent pain management in patients with haemophilia. Haemophilia 2011, 17, 612–619. [Google Scholar] [CrossRef]
- Sidonio, R.F.; Mili, F.D.; Li, T.; Miller, C.H.; Hooper, W.C.; DeBaun, M.R.; Soucie, M. Females with FVIII and FIX deficiency have reduced joint range of motion. Am. J. Hematol. 2014, 89, 831–836. [Google Scholar] [CrossRef] [Green Version]
- Forneris, E.; Andreacchio, A.; Pollio, B.; Mannucci, C.; Franchini, M.; Mengoli, C.; Pagliarino, M.; Messina, M. Gait analysis in children with haemophilia: First Italian experience at the Turin Haemophilia Centre. Haemophilia 2016, 22, e184–e191. [Google Scholar] [CrossRef]
- Buranahirun, C.; Walsh, K.S.; Mrakotsky, C.; Croteau, S.E.; Rajpurkar, M.; Kearney, S.; Hannemann, C.; Wilkening, G.N.; Shapiro, K.A.; Cooper, D.L. Neuropsychological function in children with hemophilia: A review of the Hemophilia Growth and Development Study and introduction of the current eTHINK study. Pediatric Blood Cancer 2020, 67, e28004. [Google Scholar] [CrossRef]
- Schoenmakers, M.A.; Gulmans, V.A.; Helders, P.J.; van den Berg, H.M. Motor performance and disability in Dutch children with haemophilia: A comparison with their healthy peers. Haemophilia 2001, 7, 293–298. [Google Scholar] [CrossRef]
- Shapiro, A.D.; Donfield, S.M.; Lynn, H.S.; Cool, V.A.; Stehbens, J.A.; Hunsberger, S.L.; Tonetta, S.; Gomperts, E.D. Defining the impact of hemophilia: The Academic Achievement in Children with Hemophilia Study. Pediatrics 2001, 108, E105. [Google Scholar] [CrossRef] [Green Version]
- Krüger, S.; Hilberg, T. Neuropathic pain in patients with haemophilia, that is the question. Hamostaseologie 2015, 35, S5–S9. [Google Scholar] [CrossRef]
- Sophia Fox, A.J.; Bedi, A.; Rodeo, S.A. The basic science of articular cartilage: Structure, composition, and function. Sports Health 2009, 1, 461–468. [Google Scholar] [CrossRef] [PubMed]
- Akkiraju, H.; Nohe, A. Role of Chondrocytes in Cartilage Formation, Progression of Osteoarthritis and Cartilage Regeneration. J. Dev. Biol. 2015, 3, 177–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, M.D. The normal synovium. Open Rheumatol. J. 2011, 5, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Di Nicola, V. Degenerative osteoarthritis a reversible chronic disease. Regen. Ther. 2020, 15, 149–160. [Google Scholar] [CrossRef]
- Melchiorre, D.; Manetti, M.; Matucci-Cerinic, M. Pathophysiology of Hemophilic Arthropathy. J. Clin. Med. 2017, 6, 63. [Google Scholar] [CrossRef]
- Knobe, K.; Berntorp, E. Haemophilia and joint disease: Pathophysiology, evaluation, and management. J. Comorb. 2011, 1, 51–59. [Google Scholar] [CrossRef] [Green Version]
- Valentino, L.A.; Hakobyan, N.; Enockson, C. Blood-induced joint disease: The confluence of dysregulated oncogenes, inflammatory signals, and angiogenic cues. In Seminars in Hematology; Elsevier: Amsterdam, The Netherlands, 2008; Volume 45, pp. S50–S57. [Google Scholar]
- Roosendaal, G.; Lafeber, F. Pathogenesis of haemophilic arthropathy. Haemophilia 2006, 12, 117–121. [Google Scholar] [CrossRef]
- Busso, N.; Morard, C.; Salvi, R.; Péclat, V.; So, A. Role of the tissue factor pathway in synovial inflammation. Arthritis Rheum. 2003, 48, 651–659. [Google Scholar] [CrossRef]
- Pulles, A.E.; van Vulpen, L.F.D.; Coeleveld, K.; Mastbergen, S.C.; Schutgens, R.E.G.; Lafeber, F. On-demand treatment with the iron chelator deferasirox is ineffective in preventing blood-induced joint damage in haemophilic mice. Haemophilia 2021, 27, 648–656. [Google Scholar] [CrossRef]
- Aigner, T.; Soeder, S.; Haag, J. IL-1ß and BMPs-Interactive players of cartilage matrix degradation and regeneration. Eur. Cell Mater. 2006, 12, 49–56. [Google Scholar] [CrossRef]
- Mendonça, R.; Silveira, A.A.; Conran, N. Red cell DAMPs and inflammation. Inflamm. Res. 2016, 65, 665–678. [Google Scholar] [CrossRef]
- Hakobyan, N.; Kazarian, T.; Jabbar, A.A.; Jabbar, K.J.; Valentino, L.A. Pathobiology of hemophilic synovitis I: Overexpression of mdm2 oncogene. Blood 2004, 104, 2060–2064. [Google Scholar] [CrossRef] [Green Version]
- Wen, F.-Q.; Jabbar, A.A.; Chen, Y.-X.; Kazarian, T.; Patel, D.A.; Valentino, L.A. C-myc proto-oncogene expression in hemophilic synovitis: In vitro studies of the effects of iron and ceramide. Blood J. Am. Soc. Hematol. 2002, 100, 912–916. [Google Scholar] [CrossRef] [Green Version]
- Valentino, L. Blood-induced joint disease: The pathophysiology of hemophilic arthropathy. J. Thromb. Haemost. 2010, 8, 1895–1902. [Google Scholar] [CrossRef]
- Lafeber, F.; Miossec, P.; Valentino, L. Physiopathology of haemophilic arthropathy. Haemophilia 2008, 14, 3–9. [Google Scholar] [CrossRef]
- von Drygalski, A.; Barnes, R.F.; Jang, H.; Ma, Y.; Wong, J.H.; Berman, Z.; Du, J.; Chang, E.Y. Advanced magnetic resonance imaging of cartilage components in haemophilic joints reveals that cartilage hemosiderin correlates with joint deterioration. Haemophilia 2019, 25, 851–858. [Google Scholar] [CrossRef] [Green Version]
- Hooiveld, M.; Roosendaal, G.; Vianen, M.; van den Berg, M.; Bijlsma, J.; Lafeber, F. Blood-induced joint damage: Longterm effects in vitro and in vivo. J. Rheumatol. 2003, 30, 339–344. [Google Scholar]
- Hooiveld, M.J.; Roosendaal, G.; Van Den Berg, H.; Bijlsma, J.; Lafeber, F. Haemoglobin-derived iron-dependent hydroxyl radical formation in blood-induced joint damage: An in vitro study. Rheumatology 2003, 42, 784–790. [Google Scholar] [CrossRef] [Green Version]
- Xie, Y.; Hou, W.; Song, X.; Yu, Y.; Huang, J.; Sun, X.; Kang, R.; Tang, D. Ferroptosis: Process and function. Cell Death Differ. 2016, 23, 369–379. [Google Scholar] [CrossRef] [Green Version]
- Heli, H.; Mirtorabi, S.; Karimian, K. Advances in iron chelation: An update. Expert Opin. Ther. Pat. 2011, 21, 819–856. [Google Scholar] [CrossRef]
- Sousa, L.; Oliveira, M.M.; Pessôa, M.T.C.; Barbosa, L.A. Iron overload: Effects on cellular biochemistry. Clin. Chim. Acta 2020, 504, 180–189. [Google Scholar] [CrossRef]
- Jing, X.; Du, T.; Li, T.; Yang, X.; Wang, G.; Liu, X.; Jiang, Z.; Cui, X. The detrimental effect of iron on OA chondrocytes: Importance of pro-inflammatory cytokines induced iron influx and oxidative stress. J. Cell Mol. Med. 2021, 25, 5671–5680. [Google Scholar] [CrossRef]
- Wang, T.; He, C. Pro-inflammatory cytokines: The link between obesity and osteoarthritis. Cytokine Growth Factor Rev. 2018, 44, 38–50. [Google Scholar] [CrossRef]
- Cai, C.; Hu, W.; Chu, T. Interplay Between Iron Overload and Osteoarthritis: Clinical Significance and Cellular Mechanisms. Front. Cell Dev. Biol. 2022, 9, 817104. [Google Scholar] [CrossRef]
- Suantawee, T.; Tantavisut, S.; Adisakwattana, S.; Tanavalee, A.; Yuktanandana, P.; Anomasiri, W.; Deepaisarnsakul, B.; Honsawek, S. Oxidative stress, vitamin e, and antioxidant capacity in knee osteoarthritis. J. Clin. Diagn. Res. JCDR 2013, 7, 1855. [Google Scholar]
- Vanderhave, K.L.; Perkins, C.A.; Scannell, B.; Brighton, B.K. Orthopaedic manifestations of sickle cell disease. JAAOS J. Am. Acad. Orthop. Surg. 2018, 26, 94–101. [Google Scholar] [CrossRef]
- Sadat-Ali, M.; Sultan, O.; Al-Turki, H.; AlElq, A. Does high serum iron level induce low bone mass in sickle cell anemia? Biometals 2011, 24, 19–22. [Google Scholar] [CrossRef]
- Kiven, S.; Wang, Y.; Aich, A.; Argueta, D.A.; Lei, J.; Sagi, V.; Tennakoon, M.; Bedros, S.J.; Lambrecht, N.; Gupta, K. Spatiotemporal Alterations in Gait in Humanized Transgenic Sickle Mice. Front. Immunol. 2020, 11, 561947. [Google Scholar] [CrossRef]
- Katsarou, O.; Terpos, E.; Chatzismalis, P.; Provelengios, S.; Adraktas, T.; Hadjidakis, D.; Kouramba, A.; Karafoulidou, A. Increased bone resorption is implicated in the pathogenesis of bone loss in hemophiliacs: Correlations with hemophilic arthropathy and HIV infection. Ann. Hematol. 2010, 89, 67–74. [Google Scholar] [CrossRef] [Green Version]
- Christoforidis, A.; Economou, M.; Papadopoulou, E.; Kazantzidou, E.; Farmaki, E.; Tzimouli, V.; Tsatra, I.; Gompakis, N.; Athanassiou-Metaxa, M. Comparative study of dual energy X-ray absorptiometry and quantitative ultrasonography with the use of biochemical markers of bone turnover in boys with haemophilia. Haemophilia 2011, 17, e217–e222. [Google Scholar] [CrossRef]
- Pulles, A.E.; Mastbergen, S.C.; Schutgens, R.E.; Lafeber, F.P.; van Vulpen, L.F. Pathophysiology of hemophilic arthropathy and potential targets for therapy. Pharmacol. Res. 2017, 115, 192–199. [Google Scholar] [CrossRef] [PubMed]
- Boyce, B.F.; Xing, L. Biology of RANK, RANKL, and osteoprotegerin. Arthritis Res. Ther. 2007, 9, S1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vandooren, B.; Cantaert, T.; Noordenbos, T.; Tak, P.P.; Baeten, D. The abundant synovial expression of the RANK/RANKL/Osteoprotegerin system in peripheral spondylarthritis is partially disconnected from inflammation. Arthritis Rheum. 2008, 58, 718–729. [Google Scholar] [CrossRef] [PubMed]
- Saidenberg-Kermanac’h, N.; Cohen-Solal, M.; Bessis, N.; De Vernejoul, M.C.; Boissier, M.C. Role for osteoprotegerin in rheumatoid inflammation. Jt. Bone Spine 2004, 71, 9–13. [Google Scholar] [CrossRef]
- Kitaura, H.; Marahleh, A.; Ohori, F.; Noguchi, T.; Shen, W.R.; Qi, J.; Nara, Y.; Pramusita, A.; Kinjo, R.; Mizoguchi, I. Osteocyte-Related Cytokines Regulate Osteoclast Formation and Bone Resorption. Int. J. Mol. Sci. 2020, 21, 5169. [Google Scholar] [CrossRef] [PubMed]
- Ono, T.; Hayashi, M.; Sasaki, F.; Nakashima, T. RANKL biology: Bone metabolism, the immune system, and beyond. Inflamm. Regen. 2020, 40, 2. [Google Scholar] [CrossRef] [Green Version]
- Jones, D.H.; Kong, Y.Y.; Penninger, J.M. Role of RANKL and RANK in bone loss and arthritis. Ann. Rheum. Dis. 2002, 61, ii32–ii39. [Google Scholar] [CrossRef] [Green Version]
- Boyce, B.F.; Xing, L. Functions of RANKL/RANK/OPG in bone modeling and remodeling. Arch. Biochem. Biophys. 2008, 473, 139–146. [Google Scholar] [CrossRef] [Green Version]
- Melchiorre, D.; Linari, S.; Manetti, M.; Romano, E.; Sofi, F.; Matucci-Cerinic, M.; Carulli, C.; Innocenti, M.; Ibba-Manneschi, L.; Castaman, G. Clinical, instrumental, serological and histological findings suggest that hemophilia B may be less severe than hemophilia A. Haematologica 2016, 101, 219–225. [Google Scholar] [CrossRef] [Green Version]
- Lean, J.M.; Jagger, C.J.; Kirstein, B.; Fuller, K.; Chambers, T.J. Hydrogen peroxide is essential for estrogen-deficiency bone loss and osteoclast formation. Endocrinology 2005, 146, 728–735. [Google Scholar] [CrossRef]
- Manolagas, S.C. From estrogen-centric to aging and oxidative stress: A revised perspective of the pathogenesis of osteoporosis. Endocr. Rev. 2010, 31, 266–300. [Google Scholar] [CrossRef] [Green Version]
- Baek, K.H.; Oh, K.W.; Lee, W.Y.; Lee, S.S.; Kim, M.K.; Kwon, H.S.; Rhee, E.J.; Han, J.H.; Song, K.H.; Cha, B.Y.; et al. Association of oxidative stress with postmenopausal osteoporosis and the effects of hydrogen peroxide on osteoclast formation in human bone marrow cell cultures. Calcif. Tissue Int. 2010, 87, 226–235. [Google Scholar] [CrossRef]
- Romagnoli, C.; Marcucci, G.; Favilli, F.; Zonefrati, R.; Mavilia, C.; Galli, G.; Tanini, A.; Iantomasi, T.; Brandi, M.L.; Vincenzini, M.T. Role of GSH/GSSG redox couple in osteogenic activity and osteoclastogenic markers of human osteoblast-like SaOS-2 cells. Febs. J. 2013, 280, 867–879. [Google Scholar] [CrossRef]
- Fontani, F.; Marcucci, G.; Iantomasi, T.; Brandi, M.L.; Vincenzini, M.T. Glutathione, N-acetylcysteine and lipoic acid down-regulate starvation-induced apoptosis, RANKL/OPG ratio and sclerostin in osteocytes: Involvement of JNK and ERK1/2 signalling. Calcif. Tissue Int. 2015, 96, 335–346. [Google Scholar] [CrossRef]
- Filaire, E.; Toumi, H. Reactive oxygen species and exercise on bone metabolism: Friend or enemy? Jt. Bone Spine 2012, 79, 341–346. [Google Scholar] [CrossRef]
- Domazetovic, V.; Marcucci, G.; Iantomasi, T.; Brandi, M.L.; Vincenzini, M.T. Oxidative stress in bone remodeling: Role of antioxidants. Clin. Cases Miner. Bone Metab. 2017, 14, 209–216. [Google Scholar] [CrossRef]
- Sendur, O.F.; Turan, Y.; Tastaban, E.; Serter, M. Antioxidant status in patients with osteoporosis: A controlled study. Jt. Bone Spine 2009, 76, 514–518. [Google Scholar] [CrossRef]
- Almeida, M.; Han, L.; Martin-Millan, M.; Plotkin, L.I.; Stewart, S.A.; Roberson, P.K.; Kousteni, S.; O’Brien, C.A.; Bellido, T.; Parfitt, A.M.; et al. Skeletal involution by age-associated oxidative stress and its acceleration by loss of sex steroids. J. Biol. Chem. 2007, 282, 27285–27297. [Google Scholar] [CrossRef] [Green Version]
- Maggio, D.; Barabani, M.; Pierandrei, M.; Polidori, M.C.; Catani, M.; Mecocci, P.; Senin, U.; Pacifici, R.; Cherubini, A. Marked decrease in plasma antioxidants in aged osteoporotic women: Results of a cross-sectional study. J. Clin. Endocrinol. Metab. 2003, 88, 1523–1527. [Google Scholar] [CrossRef] [Green Version]
- Hall, S.L.; Greendale, G.A. The relation of dietary vitamin C intake to bone mineral density: Results from the PEPI study. Calcif. Tissue Int. 1998, 63, 183–189. [Google Scholar] [CrossRef]
- Sanders, K.M.; Kotowicz, M.A.; Nicholson, G.C. Potential role of the antioxidant N-acetylcysteine in slowing bone resorption in early post-menopausal women: A pilot study. Transl. Res. 2007, 150, 215. [Google Scholar] [CrossRef]
- Morton, D.J.; Barrett-Connor, E.L.; Schneider, D.L. Vitamin C supplement use and bone mineral density in postmenopausal women. J. Bone Miner. Res. 2001, 16, 135–140. [Google Scholar] [CrossRef]
- Calcaterra, I.; Iannuzzo, G.; Dell’Aquila, F.; Di Minno, M.N.D. Pathophysiological Role of Synovitis in Hemophilic Arthropathy Development: A Two-Hit Hypothesis. Front. Physiol. 2020, 11, 541. [Google Scholar] [CrossRef]
- Øvlisen, K.; Kristensen, A.; Jensen, A.; Tranholm, M. IL-1β, IL-6, KC and MCP-1 are elevated in synovial fluid from haemophilic mice with experimentally induced haemarthrosis. Haemophilia 2009, 15, 802–810. [Google Scholar] [CrossRef]
- Rodriguez-Merchan, E. Haemophilic synovitis: Basic concepts. Haemophilia 2007, 13, 1–3. [Google Scholar] [CrossRef]
- Hitchon, C.A.; El-Gabalawy, H.S. Oxidation in rheumatoid arthritis. Arthritis Res. Ther. 2004, 6, 265–278. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, G.T.; Green, E.R.; Mecsas, J. Neutrophils to the ROScue: Mechanisms of NADPH Oxidase Activation and Bacterial Resistance. Front. Cell. Infect. Microbiol. 2017, 7, 373. [Google Scholar] [CrossRef]
- Miao, L.; St Clair, D.K. Regulation of superoxide dismutase genes: Implications in disease. Free Radic. Biol. Med. 2009, 47, 344–356. [Google Scholar] [CrossRef] [Green Version]
- Morgan, M.J.; Liu, Z.G. Crosstalk of reactive oxygen species and NF-κB signaling. Cell Res. 2011, 21, 103–115. [Google Scholar] [CrossRef] [Green Version]
- Davies, M.J.; Hawkins, C.L. The Role of Myeloperoxidase in Biomolecule Modification, Chronic Inflammation, and Disease. Antioxid. Redox Signal. 2020, 32, 957–981. [Google Scholar] [CrossRef] [Green Version]
- Acharya, S.S.; Kaplan, R.N.; Macdonald, D.; Fabiyi, O.T.; DiMichele, D.; Lyden, D. Neoangiogenesis contributes to the development of hemophilic synovitis. Blood J. Am. Soc. Hematol. 2011, 117, 2484–2493. [Google Scholar]
- Acharya, S.S. Exploration of the pathogenesis of haemophilic joint arthropathy: Understanding implications for optimal clinical management. Br. J. Haematol. 2012, 156, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Zetterberg, E.; Palmblad, J.; Wallensten, R.; Morfini, M.; Melchiorre, D.; Holmström, M. Angiogenesis is increased in advanced haemophilic joint disease and characterised by normal pericyte coverage. Eur. J. Haematol. 2014, 92, 256–262. [Google Scholar] [PubMed]
- Etherington, P.J.; Winlove, P.; Taylor, P.; Paleolog, E.; Miotla, J.M. VEGF release is associated with reduced oxygen tensions in experimental inflammatory arthritis. Clin. Exp. Rheumatol. 2002, 20, 799–805. [Google Scholar]
- Biniecka, M.; Connolly, M.; Gao, W.; Ng, C.T.; Balogh, E.; Gogarty, M.; Santos, L.; Murphy, E.; Brayden, D.; Veale, D.J.; et al. Redox-Mediated Angiogenesis in the Hypoxic Joint of Inflammatory Arthritis. Arthritis Rheumatol. 2014, 66, 3300–3310. [Google Scholar] [CrossRef] [Green Version]
- Chenevier-Gobeaux, C.; Simonneau, C.; Lemarechal, H.; Bonnefont-Rousselot, D.; Poiraudeau, S.; Rannou, F.; Anract, P.; Borderie, D. Hypoxia induces nitric oxide synthase in rheumatoid synoviocytes: Consequences on NADPH oxidase regulation. Free. Radic. Res. 2012, 46, 628–636. [Google Scholar] [CrossRef]
- Bayraktutan, U.; Blayney, L.; Shah, A.M. Molecular characterization and localization of the NAD (P) H oxidase components gp91-phox and p22-phox in endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 1903–1911. [Google Scholar] [CrossRef] [Green Version]
- Haruna, Y.; Morita, Y.; Komai, N.; Yada, T.; Sakuta, T.; Tomita, N.; Fox, D.A.; Kashihara, N. Endothelial dysfunction in rat adjuvant-induced arthritis: Vascular superoxide production by NAD(P)H oxidase and uncoupled endothelial nitric oxide synthase. Arthritis Rheum 2006, 54, 1847–1855. [Google Scholar] [CrossRef] [Green Version]
- Ray, R.; Shah, A.M. NADPH oxidase and endothelial cell function. Clin. Sci. 2005, 109, 217–226. [Google Scholar] [CrossRef] [Green Version]
- Frey, R.S.; Ushio-Fukai, M.; Malik, A.B. NADPH oxidase-dependent signaling in endothelial cells: Role in physiology and pathophysiology. Antioxid. Redox Signal. 2009, 11, 791–810. [Google Scholar] [CrossRef]
- Manea, A. NADPH oxidase-derived reactive oxygen species: Involvement in vascular physiology and pathology. Cell Tissue Res. 2010, 342, 325–339. [Google Scholar] [CrossRef]
- Simonini, G.; Matucci Cerinic, M.; Cimaz, R.; Anichini, M.; Cesaretti, S.; Zoppi, M.; Generini, S.; Falcini, F. Evidence for immune activation against oxidized lipoproteins in inactive phases of juvenile chronic arthritis. J. Rheumatol. 2001, 28, 198–203. [Google Scholar]
- Taylor, P.C.; Sivakumar, B. Hypoxia and angiogenesis in rheumatoid arthritis. Curr. Opin. Rheumatol. 2005, 17, 293–298. [Google Scholar] [CrossRef]
- Garrido-Urbani, S.; Jemelin, S.; Deffert, C.; Carnesecchi, S.; Basset, O.; Szyndralewiez, C.; Heitz, F.; Page, P.; Montet, X.; Michalik, L.; et al. Targeting vascular NADPH oxidase 1 blocks tumor angiogenesis through a PPARα mediated mechanism. PLoS ONE 2011, 6, e14665. [Google Scholar] [CrossRef]
- Colavitti, R.; Pani, G.; Bedogni, B.; Anzevino, R.; Borrello, S.; Waltenberger, J.; Galeotti, T. Reactive oxygen species as downstream mediators of angiogenic signaling by vascular endothelial growth factor receptor-2/KDR. J. Biol. Chem. 2002, 277, 3101–3108. [Google Scholar] [CrossRef] [Green Version]
- Roussel, N.A.; Chantrain, V.A.; Foubert, A.; Lambert, C.; Hermans, C.; Meeus, M.; Guillaume, S.; Lecouvet, F.; Krüger, S.; Hilberg, T.; et al. Gaining more insight into ankle pain in haemophilia: A study exploring pain, structural and functional evaluation of the ankle joint. Haemophilia 2022, 28, 480–490. [Google Scholar] [CrossRef]
- Humphries, T.J.; Kessler, C.M. Managing chronic pain in adults with haemophilia: Current status and call to action. Haemophilia 2015, 21, 41–51. [Google Scholar] [CrossRef]
- Holstein, K.; Klamroth, R.; Richards, M.; Carvalho, M.; Pérez-Garrido, R.; Gringeri, A. Pain management in patients with haemophilia: A European survey. Haemophilia 2012, 18, 743–752. [Google Scholar] [CrossRef] [Green Version]
- Humphries, T.J.; Kessler, C.M. The challenge of pain evaluation in haemophilia: Can pain evaluation and quantification be improved by using pain instruments from other clinical situations? Haemophilia 2013, 19, 181–187. [Google Scholar] [CrossRef]
- Koop, S.M.; ten Klooster, P.M.; Vonkeman, H.E.; Steunebrink, L.M.; van de Laar, M.A. Neuropathic-like pain features and cross-sectional associations in rheumatoid arthritis. Arthritis Res. Ther. 2015, 17, 237. [Google Scholar] [CrossRef] [Green Version]
- Eitner, A.; Hofmann, G.O.; Schaible, H.-G. Mechanisms of Osteoarthritic Pain. Studies in Humans and Experimental Models. Front. Mol. Neurosci. 2017, 10, 349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colloca, L.; Ludman, T.; Bouhassira, D.; Baron, R.; Dickenson, A.H.; Yarnitsky, D.; Freeman, R.; Truini, A.; Attal, N.; Finnerup, N.B.; et al. Neuropathic pain. Nat. Rev. Dis. Primers 2017, 3, 17002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicholson, B. Differential diagnosis: Nociceptive and neuropathic pain. Am. J. Manag. Care 2006, 12, S256–S262. [Google Scholar] [PubMed]
- St John Smith, E. Advances in understanding nociception and neuropathic pain. J. Neurol. 2018, 265, 231–238. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.H.; Seo, H.J.; Abdi, S.; Huh, B. All about pain pharmacology: What pain physicians should know. Korean J. Pain 2020, 33, 108–120. [Google Scholar] [CrossRef] [Green Version]
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative Stress: Harms and Benefits for Human Health. Oxid. Med. Cell Longev. 2017, 2017, 8416763. [Google Scholar] [CrossRef]
- Hendrix, J.; Nijs, J.; Ickmans, K.; Godderis, L.; Ghosh, M.; Polli, A. The Interplay between Oxidative Stress, Exercise, and Pain in Health and Disease: Potential Role of Autonomic Regulation and Epigenetic Mechanisms. Antioxidants 2020, 9, 1166. [Google Scholar] [CrossRef]
- Quiñonez-Flores, C.M.; González-Chávez, S.A.; Del Río Nájera, D.; Pacheco-Tena, C. Oxidative Stress Relevance in the Pathogenesis of the Rheumatoid Arthritis: A Systematic Review. Biomed. Res. Int. 2016, 2016, 6097417. [Google Scholar] [CrossRef] [Green Version]
- Telen, M.J.; Malik, P.; Vercellotti, G.M. Therapeutic strategies for sickle cell disease: Towards a multi-agent approach. Nat. Rev. Drug. Discov. 2019, 18, 139–158. [Google Scholar] [CrossRef]
- Wauquier, F.; Leotoing, L.; Coxam, V.; Guicheux, J.; Wittrant, Y. Oxidative stress in bone remodelling and disease. Trends Mol. Med. 2009, 15, 468–477. [Google Scholar] [CrossRef]
- La Hausse De Lalouviere, L.; Morice, O.; Fitzgerald, M. Altered sensory innervation and pain hypersensitivity in a model of young painful arthritic joints: Short- and long-term effects. Inflamm. Res. 2021, 70, 483–493. [Google Scholar] [CrossRef]
- Syx, D.; Tran, P.B.; Miller, R.E.; Malfait, A.M. Peripheral Mechanisms Contributing to Osteoarthritis Pain. Curr. Rheumatol. Rep. 2018, 20, 9. [Google Scholar] [CrossRef]
- Klafke, J.Z.; da Silva, M.A.; Rossato, M.F.; de Prá, S.D.; Rigo, F.K.; Walker, C.I.; Bochi, G.V.; Moresco, R.N.; Ferreira, J.; Trevisan, G. Acute and chronic nociceptive phases observed in a rat hind paw ischemia/reperfusion model depend on different mechanisms. Pflugers Arch. 2016, 468, 229–241. [Google Scholar] [CrossRef]
- Pan, L.; Yu, L.; Wang, L.; He, J.; Sun, J.; Wang, X.; Wang, H.; Bai, Z.; Feng, H.; Pei, H. Inflammatory stimuli promote oxidative stress in pancreatic acinar cells via Toll-like receptor 4/nuclear factor-κB pathway. Int. J. Mol. Med. 2018, 42, 3582–3590. [Google Scholar] [CrossRef] [Green Version]
- Mittal, M.; Siddiqui, M.R.; Tran, K.; Reddy, S.P.; Malik, A.B. Reactive oxygen species in inflammation and tissue injury. Antioxid. Redox Signal. 2014, 20, 1126–1167. [Google Scholar] [CrossRef] [Green Version]
- Zahan, O.M.; Serban, O.; Gherman, C.; Fodor, D. The evaluation of oxidative stress in osteoarthritis. Med. Pharm. Rep. 2020, 93, 12–22. [Google Scholar] [CrossRef]
- Bevan, S.; Quallo, T.; Andersson, D.A. TRPV1. In Mammalian Transient Receptor Potential (TRP) Cation Channels: Volume I.; Nilius, B., Flockerzi, V., Eds.; Springer: Berlin/Heidelberg, Germany, 2014; pp. 207–245. [Google Scholar]
- Patwardhan, A.M.; Akopian, A.N.; Ruparel, N.B.; Diogenes, A.; Weintraub, S.T.; Uhlson, C.; Murphy, R.C.; Hargreaves, K.M. Heat generates oxidized linoleic acid metabolites that activate TRPV1 and produce pain in rodents. J. Clin. Investig. 2010, 120, 1617–1626. [Google Scholar] [CrossRef]
- Mills, C.D.; Nguyen, T.; Tanga, F.Y.; Zhong, C.; Gauvin, D.M.; Mikusa, J.; Gomez, E.J.; Salyers, A.K.; Bannon, A.W. Characterization of nerve growth factor-induced mechanical and thermal hypersensitivity in rats. Eur. J. Pain 2013, 17, 469–479. [Google Scholar] [CrossRef]
- Eskander, M.A.; Ruparel, S.; Green, D.P.; Chen, P.B.; Por, E.D.; Jeske, N.A.; Gao, X.; Flores, E.R.; Hargreaves, K.M. Persistent Nociception Triggered by Nerve Growth Factor (NGF) Is Mediated by TRPV1 and Oxidative Mechanisms. J. Neurosci. 2015, 35, 8593–8603. [Google Scholar] [CrossRef] [Green Version]
- St-Jacques, B.; Ma, W. Peripheral prostaglandin E2 prolongs the sensitization of nociceptive dorsal root ganglion neurons possibly by facilitating the synthesis and anterograde axonal trafficking of EP4 receptors. Exp. Neurol. 2014, 261, 354–366. [Google Scholar] [CrossRef]
- La Hausse de Lalouvière, L.; Ioannou, Y.; Fitzgerald, M. Neural mechanisms underlying the pain of juvenile idiopathic arthritis. Nat. Rev. Rheumatol. 2014, 10, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Ji, R.R.; Chamessian, A.; Zhang, Y.Q. Pain regulation by non-neuronal cells and inflammation. Science 2016, 354, 572–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwok, C.H.T.; Learoyd, A.E.; Canet-Pons, J.; Trang, T.; Fitzgerald, M. Spinal interleukin-6 contributes to central sensitisation and persistent pain hypersensitivity in a model of juvenile idiopathic arthritis. Brain Behav. Immun. 2020, 90, 145–154. [Google Scholar] [CrossRef] [PubMed]
- Gopal, S.; Barnes, R.F.W.; Cooke, E.J.; Zhou, J.Y.; Levin, I.; Emery, P.; Hughes, T.H.; Karsdal, M.A.; Manon-Jensen, T.; von Drygalski, A. Systemic vascular basement membrane markers linked to synovial vascular remodeling are biomarkers of hemarthrosis in patients with hemophilia. J. Thromb. Haemost. 2021, 19, 1200–1211. [Google Scholar] [CrossRef]
- Krüger, S.; Hilberg, T. Understanding the pain profile in patients with haemophilia: Impaired descending pain inhibition as measured by conditioned pain modulation. Haemophilia 2020, 26, 236–242. [Google Scholar] [CrossRef] [Green Version]
- Ghilardi, J.R.; Freeman, K.T.; Jimenez-Andrade, J.M.; Coughlin, K.A.; Kaczmarska, M.J.; Castaneda-Corral, G.; Bloom, A.P.; Kuskowski, M.A.; Mantyh, P.W. Neuroplasticity of sensory and sympathetic nerve fibers in a mouse model of a painful arthritic joint. Arthritis Rheum. 2012, 64, 2223–2232. [Google Scholar] [CrossRef]
- Longo, G.; Osikowicz, M.; Ribeiro-da-Silva, A. Sympathetic fiber sprouting in inflamed joints and adjacent skin contributes to pain-related behavior in arthritis. J. Neurosci. 2013, 33, 10066–10074. [Google Scholar] [CrossRef]
- Ashraf, S.; Wibberley, H.; Mapp, P.I.; Hill, R.; Wilson, D.; Walsh, D.A. Increased vascular penetration and nerve growth in the meniscus: A potential source of pain in osteoarthritis. Ann. Rheum. Dis. 2011, 70, 523–529. [Google Scholar] [CrossRef]
- Carrasco, C.; Naziroǧlu, M.; Rodríguez, A.B.; Pariente, J.A. Neuropathic Pain: Delving into the Oxidative Origin and the Possible Implication of Transient Receptor Potential Channels. Front. Physiol. 2018, 9, 95. [Google Scholar] [CrossRef]
- Bennett, G.J.; Xie, Y.K. A peripheral mononeuropathy in rat that produces disorders of pain sensation like those seen in man. Pain 1988, 33, 87–107. [Google Scholar] [CrossRef]
- Naik, A.K.; Tandan, S.K.; Dudhgaonkar, S.P.; Jadhav, S.H.; Kataria, M.; Prakash, V.R.; Kumar, D. Role of oxidative stress in pathophysiology of peripheral neuropathy and modulation by N-acetyl-L-cysteine in rats. Eur. J. Pain 2006, 10, 573–579. [Google Scholar] [CrossRef]
- Yowtak, J.; Lee, K.Y.; Kim, H.Y.; Wang, J.; Kim, H.K.; Chung, K.; Chung, J.M. Reactive oxygen species contribute to neuropathic pain by reducing spinal GABA release. Pain 2011, 152, 844–852. [Google Scholar] [CrossRef] [Green Version]
- De Logu, F.; Nassini, R.; Materazzi, S.; Carvalho Gonçalves, M.; Nosi, D.; Rossi Degl’Innocenti, D.; Marone, I.M.; Ferreira, J.; Li Puma, S.; Benemei, S. Schwann cell TRPA1 mediates neuroinflammation that sustains macrophage-dependent neuropathic pain in mice. Nat. Commun. 2017, 8, 1–16. [Google Scholar] [CrossRef]
- Grace, P.M.; Gaudet, A.D.; Staikopoulos, V.; Maier, S.F.; Hutchinson, M.R.; Salvemini, D.; Watkins, L.R. Nitroxidative Signaling Mechanisms in Pathological Pain. Trends Neurosci. 2016, 39, 862–879. [Google Scholar] [CrossRef] [Green Version]
- Salvemini, D.; Little, J.W.; Doyle, T.; Neumann, W.L. Roles of reactive oxygen and nitrogen species in pain. Free. Radic. Biol. Med. 2011, 51, 951–966. [Google Scholar] [CrossRef] [Green Version]
- Areti, A.; Yerra, V.G.; Naidu, V.; Kumar, A. Oxidative stress and nerve damage: Role in chemotherapy induced peripheral neuropathy. Redox Biol. 2014, 2, 289–295. [Google Scholar] [CrossRef] [Green Version]
- Hongpaisan, J.; Winters, C.A.; Andrews, S.B. Strong calcium entry activates mitochondrial superoxide generation, upregulating kinase signaling in hippocampal neurons. J. Neurosci. 2004, 24, 10878–10887. [Google Scholar] [CrossRef] [Green Version]
- Lai, B.; Zhang, L.; Dong, L.-Y.; Zhu, Y.-H.; Sun, F.-Y.; Zheng, P. Impact of inhibition of Qo site of mitochondrial complex III with myxothiazol on persistent sodium currents via superoxide and protein kinase C in rat hippocampal CA1 cells. Neurobiol. Dis. 2006, 21, 206–216. [Google Scholar] [CrossRef]
- Fang, L.; Wu, J.; Lin, Q.; Willis, W.D. Protein kinases regulate the phosphorylation of the GluR1 subunit of AMPA receptors of spinal cord in rats following noxious stimulation. Mol. Brain Res. 2003, 118, 160–165. [Google Scholar] [CrossRef]
- Fischer, M.J.; Reeh, P.W. Sensitization to heat through G-protein-coupled receptor pathways in the isolated sciatic mouse nerve. Eur. J. Neurosci. 2007, 25, 3570–3575. [Google Scholar] [CrossRef]
- Sculptoreanu, A.; Aura Kullmann, F.; De Groat, W.C. Neurokinin 2 receptor-mediated activation of protein kinase C modulates capsaicin responses in DRG neurons from adult rats. Eur. J. Neurosci. 2008, 27, 3171–3181. [Google Scholar] [CrossRef] [Green Version]
- Kohno, T.; Wang, H.; Amaya, F.; Brenner, G.J.; Cheng, J.-K.; Ji, R.-R.; Woolf, C.J. Bradykinin enhances AMPA and NMDA receptor activity in spinal cord dorsal horn neurons by activating multiple kinases to produce pain hypersensitivity. J. Neurosci. 2008, 28, 4533–4540. [Google Scholar] [CrossRef]
- Li, K.-C.; Zheng, J.-H.; Chen, J. Involvement of spinal protein kinase C in induction and maintenance of both persistent spontaneous flinching reflex and contralateral heat hyperalgesia induced by subcutaneous bee venom in the conscious rat. Neurosci. Lett. 2000, 285, 103–106. [Google Scholar] [CrossRef]
- Grabowska, W.; Sikora, E.; Bielak-Zmijewska, A. Sirtuins, a promising target in slowing down the ageing process. Biogerontology 2017, 18, 447–476. [Google Scholar] [CrossRef] [Green Version]
- Ilari, S.; Giancotti, L.A.; Lauro, F.; Gliozzi, M.; Malafoglia, V.; Palma, E.; Tafani, M.; Russo, M.A.; Tomino, C.; Fini, M.; et al. Natural Antioxidant Control of Neuropathic Pain-Exploring the Role of Mitochondrial SIRT3 Pathway. Antioxidants 2020, 9, 1103. [Google Scholar] [CrossRef]
- Song, F.H.; Liu, D.Q.; Zhou, Y.Q.; Mei, W. SIRT1: A promising therapeutic target for chronic pain. CNS Neurosci. Ther. 2022, 28, 818–828. [Google Scholar] [CrossRef]
- Cheng, A.; Yang, Y.; Zhou, Y.; Maharana, C.; Lu, D.; Peng, W.; Liu, Y.; Wan, R.; Marosi, K.; Misiak, M. Mitochondrial SIRT3 mediates adaptive responses of neurons to exercise and metabolic and excitatory challenges. Cell Metab. 2016, 23, 128–142. [Google Scholar] [CrossRef] [Green Version]
- Ilari, S.; Giancotti, L.A.; Lauro, F.; Dagostino, C.; Gliozzi, M.; Malafoglia, V.; Sansone, L.; Palma, E.; Tafani, M.; Russo, M.A. Antioxidant modulation of sirtuin 3 during acute inflammatory pain: The ROS control. Pharmacol. Res. 2020, 157, 104851. [Google Scholar] [CrossRef]
- Gao, J.; Zheng, Z.; Gu, Q.; Chen, X.; Liu, X.; Xu, X. Deacetylation of MnSOD by PARP-regulated SIRT3 protects retinal capillary endothelial cells from hyperglycemia-induced damage. Biochem. Biophys. Res. Commun. 2016, 472, 425–431. [Google Scholar] [CrossRef]
- Muscoli, C.; Lauro, F.; Dagostino, C.; Ilari, S.; Giancotti, L.; Gliozzi, M.; Costa, N.; Carresi, C.; Musolino, V.; Casale, F. Olea Europea-derived phenolic products attenuate antinociceptive morphine tolerance: An innovative strategic approach to treat cancer pain. J. Biol. Regul. Homeost. Agents 2014, 28, 105–116. [Google Scholar]
- Lauro, F.; Giancotti, L.A.; Ilari, S.; Dagostino, C.; Gliozzi, M.; Morabito, C.; Malafoglia, V.; Raffaeli, W.; Muraca, M.; Goffredo, B.M. Inhibition of spinal oxidative stress by bergamot polyphenolic fraction attenuates the development of morphine induced tolerance and hyperalgesia in mice. PLoS ONE 2016, 11, e0156039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahman, I.; Chung, S. Dietary polyphenols, deacetylases and chromatin remodeling in inflammation. J. Nutrigenet. Nutr. 2010, 3, 220–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, Q.; Dong, W.; Zhang, L.; Yang, X. Activating Sirt1 by resveratrol suppresses Nav1.7 expression in DRG through miR-182 and alleviates neuropathic pain in rats. Channels 2020, 14, 69–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benameur, T.; Soleti, R.; Panaro, M.A.; La Torre, M.E.; Monda, V.; Messina, G.; Porro, C. Curcumin as Prospective Anti-Aging Natural Compound: Focus on Brain. Molecules 2021, 26, 4794. [Google Scholar] [CrossRef] [PubMed]
- Basu, P.; Basu, A. In Vitro and In Vivo Effects of Flavonoids on Peripheral Neuropathic Pain. Molecules 2020, 25, 1171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akbar, S.; Subhan, F.; Karim, N.; Shahid, M.; Ahmad, N.; Ali, G.; Mahmood, W.; Fawad, K. 6-Methoxyflavanone attenuates mechanical allodynia and vulvodynia in the streptozotocin-induced diabetic neuropathic pain. Biomed. Pharmacother. 2016, 84, 962–971. [Google Scholar] [CrossRef] [PubMed]
- Uddin, M.S.; Mamun, A.A.; Rahman, M.A.; Kabir, M.T.; Alkahtani, S.; Alanazi, I.S.; Perveen, A.; Ashraf, G.M.; Bin-Jumah, M.N.; Abdel-Daim, M.M. Exploring the Promise of Flavonoids to Combat Neuropathic Pain: From Molecular Mechanisms to Therapeutic Implications. Front. Neurosci. 2020, 14, 478. [Google Scholar] [CrossRef]
- Yamashita, S.; Matsuzawa, Y. Where are we with probucol: A new life for an old drug? Atherosclerosis 2009, 207, 16–23. [Google Scholar] [CrossRef]
- Derangula, K.; Javalgekar, M.; Kumar Arruri, V.; Gundu, C.; Kumar Kalvala, A.; Kumar, A. Probucol attenuates NF-κB/NLRP3 signalling and augments Nrf-2 mediated antioxidant defence in nerve injury induced neuropathic pain. Int. Immunopharmacol. 2022, 102, 108397. [Google Scholar] [CrossRef]
- Basu, P.; Averitt, D.L.; Maier, C.; Basu, A. The Effects of Nuclear Factor Erythroid 2 (NFE2)-Related Factor 2 (Nrf2) Activation in Preclinical Models of Peripheral Neuropathic Pain. Antioxidants 2022, 11, 430. [Google Scholar] [CrossRef]
- Santos, M.C.Q.; Silva, T.; Silva, F.; Siebert, C.; Kroth, A.; Silveira, E.M.S.; Wyse, A.T.S.; Partata, W.A. Effects of vitamin D administration on nociception and spinal cord pro-oxidant and antioxidant markers in a rat model of neuropathic pain. Braz. J. Med. Biol. Res. 2021, 54, e11207. [Google Scholar] [CrossRef]
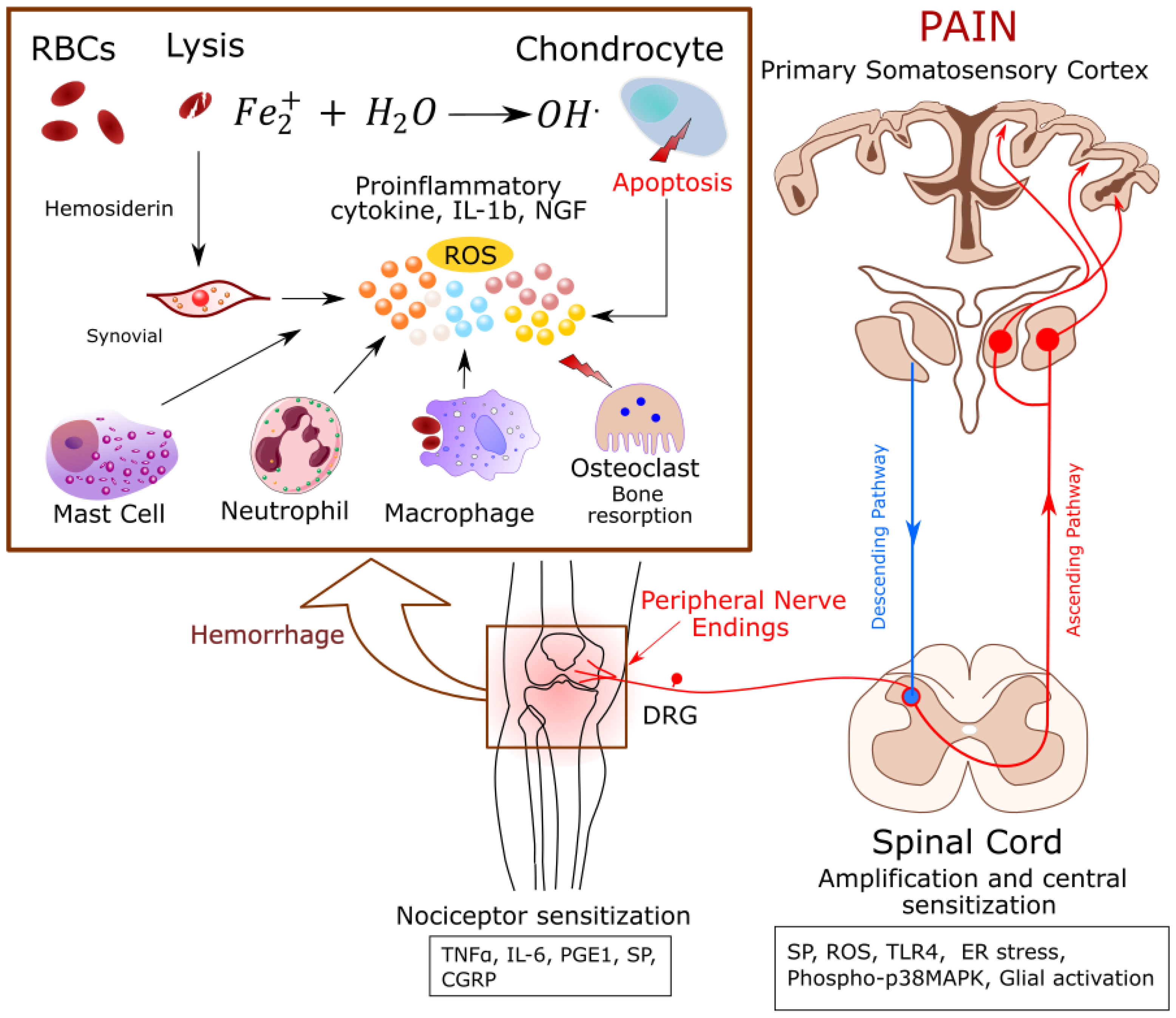
Figure 1.
Proposed mechanism(s) of pain in Hemophilia: Joint bleeding releases cell free heme and proinflammatory cytokines leading to synovitis. Red cell derived iron (Fe2+) causes oxidative stress and chondrocyte apoptosis. Inflammation activates Receptor Activator of NF-κB Ligand (RANK-L)-RANK-Osteoprotegin (OPG)-pathway, resulting in bone resorption by osteoclasts. This microenvironment of inflammation, cell damage, and oxidative stress activates nociceptors on the nerve fibers, which release neuropeptides and transmit the action potentials to the central nervous system (CNS) leading to the perception of pain. Abbreviations: CGRP: Calcitonin gene-related peptide, DRG: Dorsal root ganglion, ER stress: Endoplasmic reticulum stress, NGF: Nerve growth factor, PGE1: Prostaglandin E1, RBCs: Red blood cells, ROS: Reactive oxygen species, SP: Substance P, TLR4, Toll like receptor 4, TNF-α: Tumor necrosis factor.
Figure 1.
Proposed mechanism(s) of pain in Hemophilia: Joint bleeding releases cell free heme and proinflammatory cytokines leading to synovitis. Red cell derived iron (Fe2+) causes oxidative stress and chondrocyte apoptosis. Inflammation activates Receptor Activator of NF-κB Ligand (RANK-L)-RANK-Osteoprotegin (OPG)-pathway, resulting in bone resorption by osteoclasts. This microenvironment of inflammation, cell damage, and oxidative stress activates nociceptors on the nerve fibers, which release neuropeptides and transmit the action potentials to the central nervous system (CNS) leading to the perception of pain. Abbreviations: CGRP: Calcitonin gene-related peptide, DRG: Dorsal root ganglion, ER stress: Endoplasmic reticulum stress, NGF: Nerve growth factor, PGE1: Prostaglandin E1, RBCs: Red blood cells, ROS: Reactive oxygen species, SP: Substance P, TLR4, Toll like receptor 4, TNF-α: Tumor necrosis factor.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Fouda, R.; Argueta, D.A.; Gupta, K. Pain in Hemophilia: Unexplored Role of Oxidative Stress. Antioxidants 2022, 11, 1113. https://doi.org/10.3390/antiox11061113
AMA Style
Fouda R, Argueta DA, Gupta K. Pain in Hemophilia: Unexplored Role of Oxidative Stress. Antioxidants. 2022; 11(6):1113. https://doi.org/10.3390/antiox11061113
Chicago/Turabian StyleFouda, Raghda, Donovan A. Argueta, and Kalpna Gupta. 2022. "Pain in Hemophilia: Unexplored Role of Oxidative Stress" Antioxidants 11, no. 6: 1113. https://doi.org/10.3390/antiox11061113
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.