
Mechanisms and Therapeutic Effects of Benzoquinone Ring Analogs in Primary CoQ Deficiencies


Department of Neurology, Columbia University Medical Center, New York, NY 10032, USA
*
Author to whom correspondence should be addressed.
Antioxidants 2022, 11(4), 665; https://doi.org/10.3390/antiox11040665
Submission received: 8 March 2022
/
Revised: 26 March 2022
/
Accepted: 28 March 2022
/
Published: 30 March 2022
(This article belongs to the Special Issue Challenges of CoQ10 Deficiency in Diagnostic, New Models of Disease and Therapy)
Abstract
:Coenzyme Q (CoQ) is a conserved polyprenylated lipid composed of a redox-active benzoquinone ring and a long polyisoprenyl tail that serves as a membrane anchor. CoQ biosynthesis involves multiple steps, including multiple modifications of the precursor ring 4-hydroxybenzoic acid. Mutations in the enzymes involved in CoQ biosynthesis pathway result in primary coenzyme Q deficiencies, mitochondrial disorders whose clinical heterogenicity reflects the multiple biological function of CoQ. Patients with these disorders do not always respond to CoQ supplementation, and CoQ analogs have not been successful as alternative approaches. Progress made in understanding the CoQ biosynthesis pathway and studies of supplementation with 4-hydroxybenzoic acid ring analogs have opened a new area in the field of primary CoQ deficiencies treatment. Here, we will review these studies, focusing on efficacy of the different 4-hydroxybenzoic acid ring analogs, models in which they have been tested, and their mechanisms of action. Understanding how these compounds ameliorate biochemical, molecular, and/or clinical phenotypes of CoQ deficiencies is important to develop the most rational treatment for CoQ deficient patients, depending on their molecular defects.
1. Introduction
Coenzyme Q (CoQ) is a lipid with antioxidant and electron carrier capacities [1]. It was isolated and characterized by Festenstein et al. in 1955 [2] and soon after established as a compound involved in the mitochondrial respiratory chain by Crane et al. [1,3]. Since then, CoQ has been a widely studied molecule, and numerous studies have shown its roles in a variety of cellular processes [1]. For example, it functions as a cofactor for dihydroorotate dehydrogenase (DHODH) in pyrimidine biosynthesis; sulfide-quinone oxidoreductase (SQOR) in sulfide oxidation; mitochondrial glycerol-3-phosphate dehydrogenase (G3PDH), that links glycolysis, oxidative phosphorylation, and fatty acid metabolism; electron transport flavoprotein dehydrogenase (ETFDH), involved in fatty acid β-oxidation, catabolism of several amino acids, and sarcosine metabolism; proline dehydrogenase (PRODH) and proline dehydrogenase 2 (PRODH2) required for proline, glyoxylate, and arginine metabolism; and choline dehydrogenase (CHDH) that is related to glycine metabolism [4,5]. CoQ is also required for the maintenance of the lysosomal lumen acidity, necessary for the correct functioning of this organelle [6]. Thus, not surprisingly, defects in CoQ biosynthesis cause CoQ deficiency associated with a variety of human diseases [7].
Although CoQ deficiencies are potentially treatable, not all patients respond to CoQ supplementation [8]. The low bioavailability of CoQ and its inability to cross the blood–brain barrier (BBB) indicates the need for alternative therapeutic approaches [9], especially in patients with neurological involvement [10]. Some of these alternatives include supplementation with (1) CoQ reduced form (ubiquinol), instead of the oxidized one (ubiquinone) [11]; (2) water-soluble CoQ formulation, which has already shown an increase in the bioavailability [12,13]; and (3) CoQ analogs as Idebenone, mitoquinone, and vatiquinone, which have been only partially successful [10].
More recently, 4-HB analogs have emerged as potential therapeutic agents. These compounds are hypothesized to rescue CoQ production, bypassing molecular defects in different steps of the CoQ synthesis pathway. However, in vitro and in vivo studies in a variety of models have revealed that their mechanisms are more complex and require further elucidation before they can be implemented in the clinical practice.
2. CoQ Biosynthesis
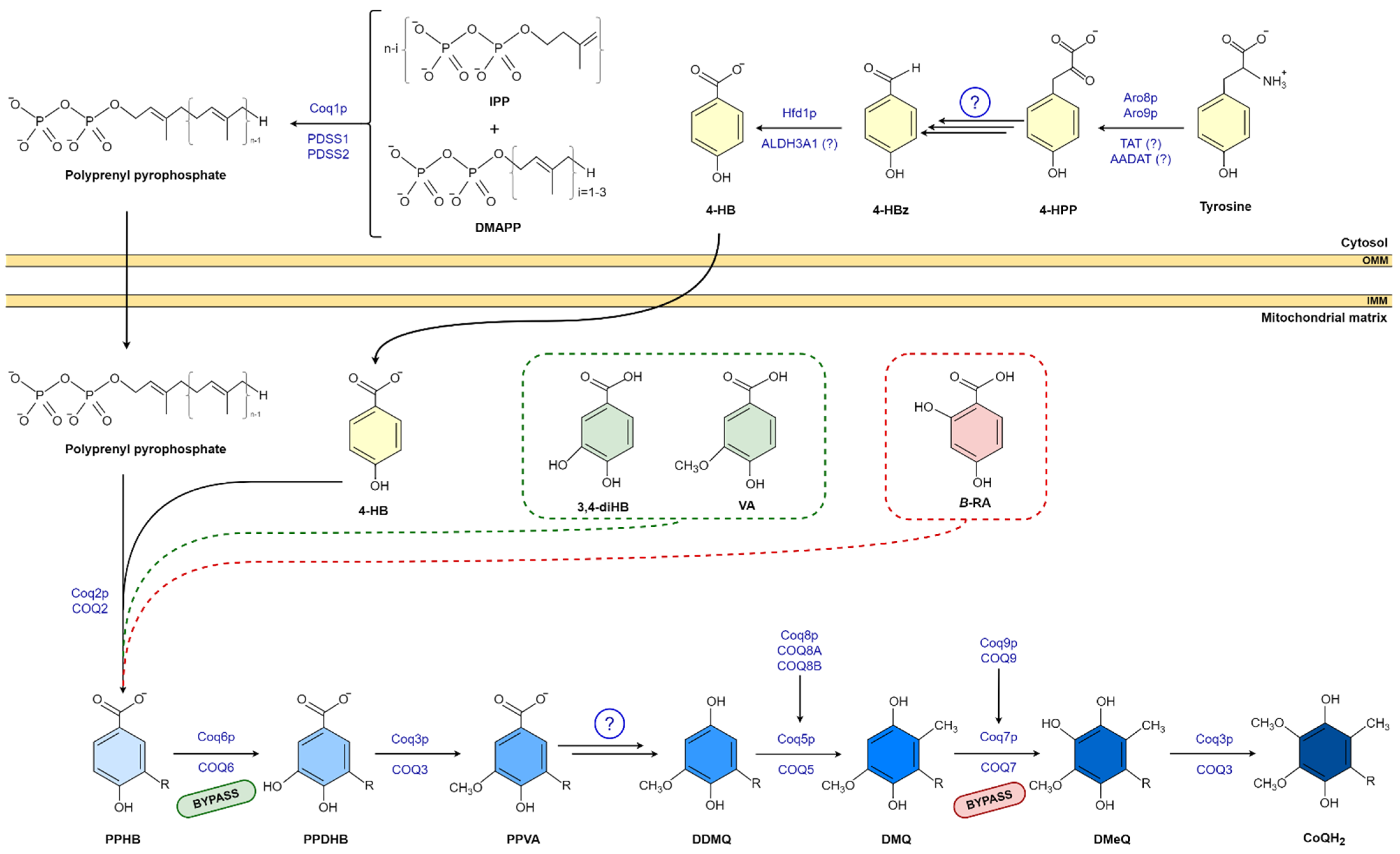
CoQ biosynthesis is a complex mechanism that has been investigated in different species, mostly in bacteria and yeast [14,15]. To date, up to 14 genes that encode proteins involved in the CoQ synthesis have been identified in yeast [14]. CoQ synthesis mainly takes place at the mitochondrial inner membrane. However, some studies have shown CoQ synthesis outside the mitochondria, specifically in the Golgi apparatus and the endoplasmatic reticulum (ER) [16,17]. In addition, recent studies in yeast have pointed out the role of the ER-mitochondria structure (ERMES) in CoQ biosynthesis, underlying the relevance of the communication between these two organelles [18,19]. The synthesis of CoQ is similar in prokaryotes and eukaryotes: a long polyisoprenoid lipid tail is initially coupled to a benzenoid precursor, and the benzenoid ring is further modified through successive steps to yield the final product [20,21,22]. The universal benzenoid precursor ring is 4-hydroxybenzoic acid (4-HB), although other alternative ring precursors have been proposed [20].
The isoprene carbon units for the synthesis of CoQ side chain are derived from the mevalonate pathway in eukaryotes and some prokaryotes [20,23]. The number of isoprene units of the polyisoprenoid chain is determined by a species-specific polyprenyl diphosphate synthase (IspB in Escherichia coli (E.coli) Coq1 in Saccharomyces cerevisiae (S.cerevisiae), and PDSS1-PDSS2 in mammals). Each species has a major CoQ form: 6 isoprenyl units in S. cerevisiae, 8 units in E. coli, 9 units in mice, and 10 units in humans. This polyisoprenoid tail is attached to the position C-3 of the ring by UbiA/Coq2/COQ2 (E. coli, S. cerevisiae, and mammals, respectively) (Figure 1) [14,20,24,25,26]. Then, the ring (3-hexaprenyl-4HB; HBB) is modified to produce the final benzoquinone ring of CoQ; specifically, Coq6/COQ6 catalyzes the C-5 hydroxylation; UbiG/Coq3/COQ3 catalyzes the C-5 O-methylation; C1-decarboxylation and C1-hydroxylation enzyme(s) have not been identified; UbiE/Coq5/COQ5 catalyzes the subsequent C-2 methylation; UbiF/Coq7/COQ7 the C-6 hydroxylation; and in the final step, UbiG/Coq3/COQ3 catalyzes the C-6 O-methylation to produce CoQ [20].
Other regulatory proteins are involved in CoQ biosynthesis [20,23]. In eukaryotes, they include Coq4/COQ4, which has been hypothesized to act as a scaffolding protein that binds proteins and lipids [27]; Coq8/COQ8A-B, which are kinases that phosphorylate other COQ proteins to stabilize the Q protein complex [28,29]; Coq9/COQ9, which interacts with COQ7 and is essential for its stability and activity [30,31]; and Coq10/COQ10 and Coq11/COQ11 that have been shown to be involved in CoQ biosynthesis and transport to the mitochondrial respiratory chain, but whose exact functions remain unknown [32,33]. Some of these proteins are organized to form a multiprotein complex called the Q complex that appears to be essential for CoQ synthesis [20,23]. To date, defects in PDSS1, PDSS2, COQ2, COQ4, COQ5, COQ6, COQ7, COQ8A, COQ8B, and COQ9 have been found to cause human CoQ deficiency and disease [34].
3. 4-Hydroxybenzoic Acid Analogs
4-HB is the universal benzoquinone ring precursor for CoQ. Its synthesis varies depending on the specie. In E. coli, 4-HB is synthesized by UbiC that catalyzes a 3horismite pyruvate-lyase reaction [35,36]. It uses chorismic acid as a substrate, an intermediate of the shikimate pathway involved in the biosynthesis of aromatic amino acids [37]. In S. cerevisiae, more than one step is needed. 4-hydroxyphenyl pyruvate (4-HPP) is obtained from the shikimate pathway or from exogenous tyrosine. 4-HPP is further converted to 4-hydroxybenzaldehyde (4-Hbz) via uncharacterized steps [15], and as a final reaction, 4-Hbz is oxidized to 4-HB by the aldehyde dehydrogenase Hfd1 [15,23]. Mammals do not possess the shikimate pathway. 4-HB derives from tyrosine and phenylalanine [38,39], via a pathway that is still not well characterized [40]. The last reaction, the oxidation of 4-Hbz to 4-HB, was recently discovered in S. cerevisiae [15,23,40].
Supplementation with 4-HB has been shown to rescue CoQ levels in CoQ-deficient bacteria (Xanthomona Oryzae), plants (Arabidopsis), and yeast (S. cerevisae) with defects in XanB2, C4H, and Hfd1, respectively, that are involved in the 4-HB ring synthesis [15,23,40,41,42]. Furthermore, 4-HB supplementation in CoQ-deficient fibroblasts from three different patients carrying COQ2 mutations was able to normalize CoQ levels [43]. The normalization of CoQ levels due to the administration of COQ2 substrate suggested that bypassing a step of the CoQ biosynthesis pathway could be an effective therapy in CoQ deficiency.
3.1. 3,4-Dihydroxybenzoic Acid and Vanillic Acid
In 1977, Nambudiri et al. studied the hydroxylation of 4-HB in mammals [44]. They showed that 4-HB was hydroxylated to polyprenyl-hydroxybenzoate (PPHB) but also to a PPHB methylated derivate, vanillic acid (VA). The study showed also that rat mitochondria were able to prenylate VA and 3,4-dihydroxybenzoic acid (3,4-diHB) and introduced them in the CoQ synthesis pathway. A year later, in 1978, Goewert et al. showed that prenylated VA accumulated in a S. cerevisiae model unable to synthesize CoQ6 [45]. 3,4-diHB is a benzoquinone ring already hydroxylated in the position C3, a reaction that in CoQ biosynthesis is catalyzed by COQ6, while VA possess the O-methylation in the position C3, in CoQ biosynthesis catalyzed by COQ3. Thus, these molecules not only already carry the chemical modification necessary for the benzoquinone ring to produce the final product of CoQ biosynthesis, but they can also enter in the CoQ biosynthetic pathway through COQ2 and bypass defective modification steps.
Since the publication of the Namburidi and Goewert work, VA and 3,4-diHB, have been used in different CoQ deficiency models. Both analogs were able to rescue CoQ6 biosynthesis and respiration in a S. cerevisiae Coq6 mutant; however, the effects of the 4-HB analogs were dependent upon overexpression of Coq8, which was previously shown to stabilize the Q complex [29,46,47]. Both compounds, especially VA, were also able to restore growth in another S. cerevisiae model carrying a Coq6 human mutation [48]. In addition, surprisingly, VA supplementation partly restored CoQ biosynthesis in Drosophila garland cell nephrocytes (GCN) a model of human COQ2 nephropathy [49], indicating that VA efficacy in CoQ deficiency was not due entirely to its ability to bypass a dysfunctional step of CoQ biosynthesis, as further discussed in the next section.
VA stimulated CoQ10 biosynthesis and improved cell viability in patient-derived fibroblasts with a genetic defect in COQ9 [43]. Furthermore, VA treatment restored CoQ10 biosynthesis and, consequently, ROS levels, cellular respiration, and ATP production in a COQ6-depleted HeLa cells, where the supplementation with CoQ10 was only partially effective [50]. Additionally, VA but not 3,4-diHB, improved the pathological podocytes migration pattern in COQ6-depleted human podocytes [51].
Overall, VA seems to be more effective than 3,4-diHB in CoQ deficiency. Moreover, VA has several advantages as a potential therapeutic approach in patients: (1) In vivo, it can be produced in the liver through oxidation of vanillin [52,53]; (2) it is considered non-toxic and safe for human use by the FDA, as it is commonly used as a flavoring agent [54]; and (3) it has good bioavailability and can cross the blood–brain barrier efficiently [54], overcoming one of the major limitations of oral CoQ supplementation [50].
3.2. β-Resorcylic Acid
The β-resorcylic acid (β-RA), also named 2,4-dihydroxybenzoic acid, is a benzoquinone ring that was already hydroxylated in C2 position. This hydroxylation is naturally catalyzed by COQ7 protein in one of the last steps of CoQ biosynthesis [55].
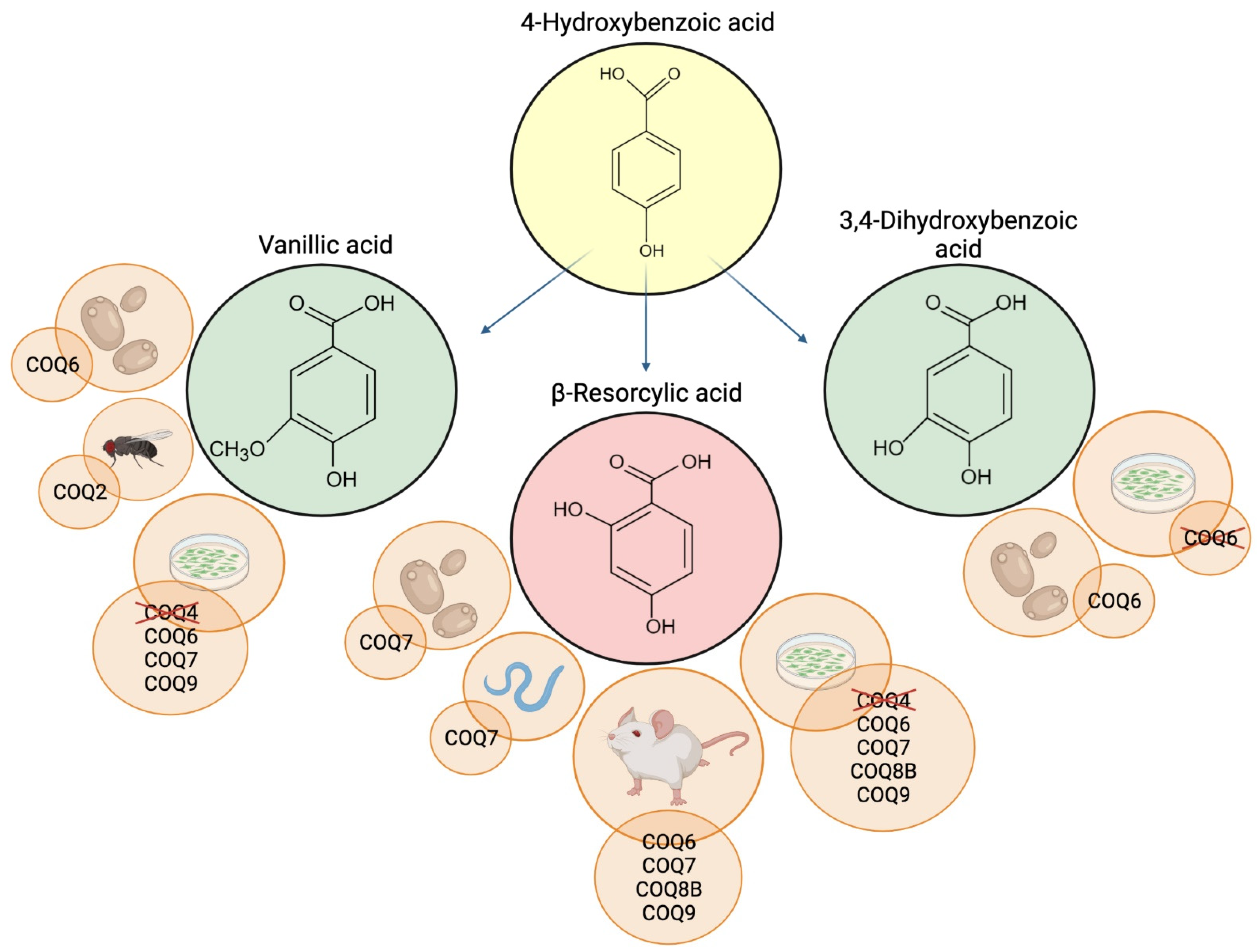
Like VA, β-RA has been widely used in the food industry; because of its ability to enhance the sweetness of other artificial sweeteners, it is added as a food flavor modifier in beverages, fish products, snack foods, and chewing gums [56]. Along with VA and 3,4-diHB, β-RA was tested in several models of CoQ deficiency (Figure 2).
In a Coq7 knockout S. cerevisiae model, β-RA supplementation has been shown to restore CoQ6 biosynthesis, after Coq8 overexpression [47,57]. By comparing β-RA and 4-HB supplementation, Xie et al. showed that 4-HB was unable to rescue CoQ6 biosynthesis and to decrease the accumulation of the toxic metabolite DMQ6 [47]. DMQ is the intermediate used by COQ7 throughout the biosynthesis of CoQ and has been shown to inhibit complexes I+III activities of the mitochondrial respiratory chain in Caenorhabditis elegans (C. elegans) [58].
β-RA supplementation was shown to rescue CoQ10 biosynthesis and mitochondrial respiration in CoQ10-deficient fibroblasts from patients with COQ7 mutations [59]. Interestingly, β-RA supplementation produced different effects in human fibroblast cell lines with two different COQ7 mutations, V141E and L111P. The V141E mutation causes severe CoQ deficiency associated with severe multisystemic disease [59], while the L111P mutation causes a milder decrease in CoQ and increase in DMQ levels, and less severe phenotype than the V141E mutation [21]. β-RA administration decreased DMQ accumulation in both cell lines, but significantly increased CoQ10 levels only in the line carrying the V141E mutation, indicating that β-RA affects CoQ10 synthesis but has different effects, depending on the extent of the loss in enzyme activity [21]. These beneficial effects of β-RA were reproduced in other COQ7 and COQ9 mutant fibroblasts from different patients [43,60].
β-RA supplementation increased CoQ9 levels also in mouse Coq7-depleted fibroblasts [56] and, when added to drinking water, was able to partially rescue the phenotype in Coq7 KO mice, increasing their weight and life span [21,56]. Amelioration of the clinical phenotype was associated with partial rescue of the levels of mitochondrial respiration defect in the kidney, lactate, and triglycerides levels in blood [56]. CoQ9 levels were increased, while accumulation of DMQ9 was decreased in heart, kidneys, and skeletal muscle [56]. This decrease in DMQ9 indicates that β-RA supplementation causes impairment of the endogenous CoQ biosynthetic pathway by competing with 4-HB, which normally acts as precursor of the CoQ benzoquinone ring [40,56,61].
Abnormalities of CoQ and DMQ levels similar to what was observed in Coq7 mutant mice were reported also in C. elegans with mutations in clk-1 (COQ7 in humans) [62], human COQ9 mutant fibroblast [60], and Coq9R239X mice [60,61].
β-RA supplementation restored survival and phenotype of Coq9R239X mutant mice, improved mitochondrial bioenergetics, and increased CoQ9 levels in kidneys, and decreased DMQ9 levels in kidney, liver, skeletal muscle, and heart [5,60,61]. Interestingly, β-RA supplementation decreased molecular, histopathological, and clinical signs of mitochondrial encephalopathy associated with CoQ9 deficiency in these Coq9R239X mice, without improving CoQ9 and DMQ9 levels, and mitochondrial bioenergetics in their brain [61]. Although the mechanisms by which β-RA supplementation rescues the phenotype of Coq9 mutant mice remain to be elucidated, all these data together support the critical role of DMQ/CoQ ratio in CoQ biosynthesis [5,61]. Furthermore, studies of β-RA supplementation in Coq9 mutant mice confirm the different effects of this compound on CoQ9 biosynthesis observed in the experiments in COQ7 mutant cells with different molecular defects. In fact, contrary to what was shown in Coq9R239X, β-RA supplementation decreased CoQ9 levels in kidney of Coq9Q95X mice, another COQ9 mutant mouse, which manifested with mild late-onset mitochondrial myopathy, associated with moderate CoQ deficiency [60]. Coq9Q95X and Coq9R239X show different levels of COQ9, as well as other COQ biosynthetic proteins [60].
β-RA administration also inhibited CoQ9 synthesis in wild-type mice and human fibroblasts [60]. Interestingly, dysregulation of CoQ9 levels in wild-type mice treated with β-RA was not associated with impairment of mitochondrial function [56].
β-RA treatment also ameliorated the phenotype, restoring the survival rate and improving renal histology in Coq8bΔPodocyte and Coq6podKO mice, two different models of CoQ-deficient nephropathy [51,63], and in COQ8B- and COQ6-depleted human podocytes [51,63]. Supplementation of β-RA prevented development of the renal dysfunction [51,63] and rescued mitochondrial dysfunction in cultured podocytes by increasing complex II+III activity [63]. We do not know the effect of β-RA treatment on CoQ biosynthesis in these models, as CoQ levels were not included in the studies; however, COQ8B and COQ6 should not be bypassed by β-RA, further indicating that the therapeutic effect of 4-HB analogs is not entirely through the restoration of CoQ synthesis.
4. Molecular Mechanisms of 4-HB Analogs
As mentioned above, the therapeutic effect of VA, β-RA, and 3,4-diHB supplementation have been demonstrated in a variety of in vitro and in vivo models of CoQ deficiency, but the mechanisms of action of these compounds are not completely understood (Table 1). The rationale behind the use of these agents is that administration of benzoquinone ring analogs, which already carry the chemical modification catalyzed by the affected protein, would bypass different steps of the CoQ biosynthetic pathway, restoring CoQ production. However, several lines of data indicate that this bypass does not always occur or is incomplete (as CoQ levels are not increased by these analogs) [5,43,61]. Furthermore, these analogs ameliorate the detrimental effects of CoQ deficiency due to defects in proteins that are not bypassed by these molecules. VA restored CoQ levels in COQ6 dysfunction models [45,46,48,50], but, surprisingly, was also beneficial in CoQ deficiencies due to defects up and downstream of the C5-hydroxylation reaction, rescuing CoQ levels in a Coq2 deficient GCN model [49] and in human COQ9 deficient fibroblasts [43]. In contrast, VA supplementation had no effect in COQ4 and COQ7 deficient cells [43]. It is possible that VA administration affects DMQ levels due to the similarities in VA and DMQ structure; alternatively, the COQ9 protein might have an additional unknown function that affects COQ6, whose defects are restored by VA supplementation [43].
β-RA has the hydroxyl group that is incorporated to the benzoquinone ring by the hydroxylase COQ7 [59]. Although COQ9 function remains unknown, its presence is needed for the stability and functioning of COQ7 [64]. As expected, β-RA supplementation was successful in COQ7- and COQ9-deficient models. However, surprisingly, β-RA is also effective in models of dysfunction of COQ6, which catalyzes a step upstream the entry of the β-RA ring, and in models of dysfunction of Coq8b/COQ8B, which is involved in the Q complex stability [47,51,63]. In fact, the rescue of the phenotype in Coq6 podocyte-conditional knockout mice does not seem to be mediated by increase in CoQ9 levels [51,63].
In COQ7 and COQ9 deficiencies, β-RA supplementation has been associated with modifications of the DMQ/CoQ ratio [5,21,43,47,56,61,62]. The rescue of the phenotypes observed in some models treated with β-RA supplementation was not associated with increases in CoQ9 levels, but rather with decreases in DMQ9 accumulation and increases in complexes I+III activities [5,43,61]. Moreover, decreases in DMQ were dose dependent [5,21,61,62]. The competition of low-affinity β-RA with the natural substrate 4-HB in entering the CoQ biosynthetic pathway and consequent reduced levels of DMQ in cases of defects in Coq9 or Coq7 have been proposed as the therapeutic mechanism of β-RA [4,40]. This hypothesis is supported by the results of combined 4-HB and β-RA supplementation, which suppresses the effects of β-RA on DMQ levels [5]. It is noteworthy that while β-RA rescues the phenotype by decreasing DMQ9, restoring mitochondrial respiration, and improving body weight and life span in CoQ9 deficient mice, it causes opposite effects in wild-type mice, with decreased body weight and CoQ9 levels, and increased levels of DMQ9 [5,56,61], again suggesting a competitive effect between 4-HB and its analogs in entering the CoQ biosynthetic pathway in vivo [40]. The efficiency of the treatments with VA and β-RA is not increased by administration of the two compounds together in CoQ10-deficient human fibroblast [43].
In summary, these data suggest that β-RA therapeutic effects in Coq7/COQ7 and Coq9/COQ9 dysfunction are mediated by decreased DMQ levels; however, additional mechanisms must be involved, as β-RA prevents the encephalopathy in Coq9 mutant mice without affecting CoQ9 and DMQ9 levels in these mice’s brains [5,61], and it recovered the pathogenic phenotype in the Coq6 and Coq8b mouse models.
Some of the results obtained in Coq6, Coq8b, and Coq9 mouse models indicate that β-RA supplementation may have other effects unrelated to the CoQ9 biosynthetic pathway. For example, the metabolic switch, mainly in kidney, observed in mice treated with β-RA might explain the therapeutic effects of this molecule in the podocyte-specific Coq6 and Coq8b knockout models [5,51,63]. Furthermore, β-RA has a strong effect on adipogenesis [5] and improves functional and morphological alterations observed in age-related obesity, reducing white adipose tissue (WAT) and adipogenesis [5]. β-RA has also been shown to have the same anti-inflammatory effect of salicylate acid derivatives in a patient with rheumatic fever [65]. 3,4-diHB has been shown to have antioxidant, anti-coagulant, and anti-inflammatory effects in diabetes mice, and neuroprotective effects in PC12 cells, by inhibiting the oligomerization of alpha-synuclein, which impairs neuronal viability [66]. 3,4-diHB is also hepatoprotective in rat hepatocytes [67] and attenuates adipogenesis-induced inflammation and mitochondrial dysfunction in 3T3-L1 adipocytes, through regulation of the AMPK pathway [68]. VA ameliorated neurodegeneration in streptozotocin-induced mice [69] and attenuates Aβ1-42-induced oxidative stress and cognitive impairment in mice [70], among other effects.
Finally, there are many other 4-HB analogs, which have failed to rescue CoQ deficiencies, such as 2,3-dihydroxybenzoic acid, 2,3-dimethoxybenzoic acid, 2-hydroxy-3-methoxybenzoic acid or the promising precursor 2,3,4-tihydroxybenzoic acid [43,47]. On the contrary, other analogs, such as para-coumarate, have only been tested in yeast [71].
Thus, the different analogs have more than one mechanism of action associated with a plethora of effects and consequent different therapeutic implications depending on the molecular defect and mechanism of disease.
5. Conclusions Remarks
Primary CoQ deficiencies are genetically and clinically heterogeneous diseases, caused by defects in proteins involved in CoQ biosynthetic pathway, which partially respond to CoQ supplementation. There is a need to better understand the mechanisms of the individual diseases and develop therapeutic approaches, which might be more suitable alternatives than CoQ supplementation. Benzoic ring analogs, such as VA and β-RA, are promising therapeutic options for patients with CoQ deficiency, depending on the molecular defect. In vitro and in vivo studies indicate that VA activates endogenous CoQ synthesis and rescues the phenotype in COQ6 deficiency, while β-RA reduces the DMQ/COQ ratio, and thus might be a suitable approach in patients with defects of CoQ biosynthesis, which cause accumulation of DMQ, as mutations in COQ9, COQ7, or COQ4. However, these precursors have additional mechanisms unrelated to their ability to affect CoQ biosynthesis. Further studies in additional animal models of primary CoQ deficiency with these and other analogs are needed to understand how they act in CoQ biosynthetic pathway, to improve their efficacy and implement their use in the clinical practice. Furthermore, these compounds have not been tested in CoQ deficiencies secondary due to mutations in genes encoding proteins not involved in the CoQ biosynthesis pathway. Biological samples from patients with these diseases often present moderated decrease in CoQ levels and reduction in CoQ biosynthetic protein and mRNA levels, without accumulation of DMQ [72,73,74,75], thus, also investigating the effects of 4-HB analogs supplementation in secondary CoQ deficiencies would be useful to gain further insight into the mechanisms of these compounds.
Author Contributions
Writing—original draft preparation, A.P.; writing—revision and editing, A.H.-G. and C.M.Q. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Department of Defense (DOD) grant PR190511 (C.M.Q.). A.H.G. is funded by a Research grant at Foreign Universities from the Fundación Alfonso Martín Escudero.
Conflicts of Interest
The authors declare that they have no conflict of interest with the contents of this article.
References
- Turunen, M.; Olsson, J.; Dallner, G. Metabolism and function of coenzyme Q. Biochim. Biophys. Acta (BBA)—Biomembr. 2004, 1660, 171–199. [Google Scholar] [CrossRef] [Green Version]
- Festenstein, G.N.; Heaton, F.W.; Lowe, J.S.; Morton, R.A. A constituent of the unsaponifiable portion of animal tissue lipids. Biochem. J. 1955, 59, 558–566. [Google Scholar] [CrossRef] [PubMed]
- Crane, F.L.; Hatefi, Y.; Lester, R.L.; Widmer, C. Isolation of a quinone from beef heart mitochondria. Biochim. Biophys. Acta 1957, 25, 220–221. [Google Scholar] [CrossRef]
- González-García, P.; Barriocanal-Casado, E.; Díaz-Casado, M.E.; López-Herrador, S.; Hidalgo-Gutiérrez, A.; López, L.C. Animal Models of Coenzyme Q Deficiency: Mechanistic and Translational Learnings. Antioxidants 2021, 10, 1687. [Google Scholar] [CrossRef] [PubMed]
- Hidalgo-Gutiérrez, A.; Barriocanal-Casado, E.; Díaz-Casado, M.E.; González-García, P.; Chiozzi, R.Z.; Acuña-Castroviejo, D.; López, L.C. β-RA Targets Mitochondrial Metabolism and Adipogenesis, Leading to Therapeutic Benefits against CoQ Deficiency and Age-Related Overweight. Biomedicines 2021, 9, 1457. [Google Scholar] [CrossRef]
- Heaton, R.A.; Heales, S.; Rahman, K.; Sexton, D.W.; Hargreaves, I. The Effect of Cellular Coenzyme Q10 Deficiency on Lysosomal Acidification. J. Clin. Med. 2020, 9, 1923. [Google Scholar] [CrossRef] [PubMed]
- Alcazar-Fabra, M.; Trevisson, E.; Brea-Calvo, G. Clinical syndromes associated with Coenzyme Q10 deficiency. Essays Biochem. 2018, 62, 377–398. [Google Scholar] [CrossRef]
- Hirano, M.; Garone, C.; Quinzii, C.M. CoQ10 deficiencies and MNGIE: Two treatable mitochondrial disorders. Biochim. Biophys. Acta (BBA)—Gen. Subj. 2012, 1820, 625–631. [Google Scholar] [CrossRef] [Green Version]
- Wainwright, L.; Hargreaves, I.P.; Georgian, A.R.; Turner, C.; Dalton, R.N.; Abbott, N.J.; Heales, S.J.R.; Preston, J.E. CoQ10 Deficient Endothelial Cell Culture Model for the Investigation of CoQ10 Blood–Brain Barrier Transport. J. Clin. Med. 2020, 9, 3236. [Google Scholar] [CrossRef]
- Suárez-Rivero, J.M.; Pastor-Maldonado, C.J.; Povea-Cabello, S.; Álvarez-Córdoba, M.; Villalón-García, I.; Munuera-Cabeza, M.; Suárez-Carrillo, A.; Talaverón-Rey, M.; Sánchez-Alcázar, J.A. Coenzyme Q 10 Analogues: Benefits and Challenges for Therapeutics. Antioxidants 2021, 10, 236. [Google Scholar] [CrossRef]
- García-Corzo, L.; Luna-Sánchez, M.; Doerrier, C.; Ortiz, F.; Escames, G.; Acuña-Castroviejo, D.; López, L.C. Ubiquinol-10 ameliorates mitochondrial encephalopathy associated with CoQ deficiency. Biochim. Biophys. Acta (BBA)—Mol. Basis Dis. 2014, 1842, 893–901. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.Y.; Song, J.F. Inclusion of coenzyme Q10 with beta-cyclodextrin studied by polarography. Yao Xue Xue Bao 2006, 41, 671–674. [Google Scholar]
- Pastor-Maldonado, C.J.; Suárez-Rivero, J.M.; Povea-Cabello, S.; Álvarez-Córdoba, M.; Villalón-García, I.; Munuera-Cabeza, M.; Suárez-Carrillo, A.; Talaverón-Rey, M.; Sánchez-Alcázar, J.A. Coenzyme Q10: Novel Formulations and Medical Trends. Int. J. Mol. Sci. 2020, 21, 8432. [Google Scholar] [CrossRef]
- Awad, A.M.; Bradley, M.C.; Fernández-Del-Río, L.; Nag, A.; Tsui, H.S.; Clarke, C.F. Coenzyme Q10 deficiencies: Pathways in yeast and humans. Essays Biochem. 2018, 62, 361–376. [Google Scholar] [CrossRef] [Green Version]
- Payet, L.-A.; Leroux, M.; Willison, J.C.; Kihara, A.; Pelosi, L.; Pierrel, F. Mechanistic Details of Early Steps in Coenzyme Q Biosynthesis Pathway in Yeast. Cell Chem. Biol. 2016, 23, 1241–1250. [Google Scholar] [CrossRef] [Green Version]
- Mugoni, V.; Postel, R.; Catanzaro, V.; De Luca, E.; Turco, E.; Digilio, G.; Silengo, L.; Murphy, M.P.; Medana, C.; Stainier, D.Y.; et al. Ubiad1 Is an Antioxidant Enzyme that Regulates eNOS Activity by CoQ10 Synthesis. Cell 2013, 152, 504–518. [Google Scholar] [CrossRef] [Green Version]
- Teclebrhan, H.; Jakobsson-Borin, A.; Brunk, U.; Dallner, G. Relationship between the endoplasmic reticulum-Golgi membrane system and ubiquinone biosynthesis. Biochim. Biophys. Acta (BBA)—Lipids Lipid Metab. 1995, 1256, 157–165. [Google Scholar] [CrossRef]
- Eisenberg-Bord, M.; Tsui, H.S.; Antunes, D.; del Rio, L.F.; Bradley, M.C.; Dunn, C.; Nguyen, T.P.T.; Rapaport, D.; Clarke, C.F.; Schuldiner, M. The Endoplasmic Reticulum-Mitochondria Encounter Structure Complex Coordinates Coenzyme Q Biosynthesis. Contact 2019, 2, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Subramanian, K.; Jochem, A.; Le Vasseur, M.; Lewis, S.; Paulson, B.R.; Reddy, T.R.; Russell, J.D.; Coon, J.J.; Pagliarini, D.J.; Nunnari, J. Coenzyme Q biosynthetic proteins assemble in a substrate-dependent manner into domains at ER–mitochondria contacts. J. Cell Biol. 2019, 218, 1353–1369. [Google Scholar] [CrossRef]
- Fernández-Del-Río, L.; Clarke, C. Coenzyme Q Biosynthesis: An Update on the Origins of the Benzenoid Ring and Discovery of New Ring Precursors. Metabolites 2021, 11, 385. [Google Scholar] [CrossRef]
- Wang, Y.; Smith, C.; Parboosingh, J.S.; Khan, A.; Innes, M.; Hekimi, S. Pathogenicity of two COQ7 mutations and responses to 2,4-dihydroxybenzoate bypass treatment. J. Cell. Mol. Med. 2017, 21, 2329–2343. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Hekimi, S. The Complexity of Making Ubiquinone. Trends Endocrinol. Metab. 2019, 30, 929–943. [Google Scholar] [CrossRef] [PubMed]
- Stefely, J.A.; Pagliarini, D.J. Biochemistry of Mitochondrial Coenzyme Q Biosynthesis. Trends Biochem. Sci. 2017, 42, 824–843. [Google Scholar] [CrossRef] [PubMed]
- Abby, S.S.; Kazemzadeh, K.; Vragniau, C.; Pelosi, L.; Pierrel, F. Advances in bacterial pathways for the biosynthesis of ubiquinone. Biochim. Biophys. Acta 2020, 1861, 148259. [Google Scholar] [CrossRef]
- Hayashi, K.; Ogiyama, Y.; Yokomi, K.; Nakagawa, T.; Kaino, T.; Kawamukai, M. Functional Conservation of Coenzyme Q Biosynthetic Genes among Yeasts, Plants, and Humans. PLoS ONE 2014, 9, e99038. [Google Scholar] [CrossRef] [Green Version]
- Kawamukai, M. Biosynthesis of coenzyme Q in eukaryotes. Biosci. Biotechnol. Biochem. 2016, 80, 23–33. [Google Scholar] [CrossRef]
- Marbois, B.; Gin, P.; Gulmezian, M.; Clarke, C.F. The yeast Coq4 polypeptide organizes a mitochondrial protein complex essential for coenzyme Q biosynthesis. Biochim. Biophys. Acta (BBA)—Mol. Cell Biol. Lipids 2009, 1791, 69–75. [Google Scholar] [CrossRef] [Green Version]
- Saiki, R.; Ogiyama, Y.; Kainou, T.; Nishi, T.; Matsuda, H.; Kawamukai, M. Pleiotropic phenotypes of fission yeast defective in ubiquinone-10 production. A study from theabc1Sp (coq8Sp)mutant. BioFactors 2003, 18, 229–235. [Google Scholar] [CrossRef]
- He, C.H.; Xie, L.X.; Allan, C.M.; Tran, U.C.; Clarke, C.F. Coenzyme Q supplementation or over-expression of the yeast Coq8 putative kinase stabilizes multi-subunit Coq polypeptide complexes in yeast coq null mutants. Biochim. Biophys. Acta (BBA)—Mol. Cell Biol. Lipids 2014, 1841, 630–644. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, E.J.; Gin, P.; Gulmezian, M.; Tran, U.P.C.; Saiki, R.; Marbois, B.N.; Clarke, C.F. Saccharomyces cerevisiae Coq9 polypeptide is a subunit of the mitochondrial coenzyme Q biosynthetic complex. Arch. Biochem. Biophys. 2007, 463, 19–26. [Google Scholar] [CrossRef] [Green Version]
- Johnson, A.; Gin, P.; Marbois, B.N.; Hsieh, E.J.; Wu, M.H.; Barros, M.; Clarke, C.F.; Tzagoloff, A. COQ9, a New Gene Required for the Biosynthesis of Coenzyme Q in Saccharomyces cerevisiae. J. Biol. Chem. 2005, 280, 31397–31404. [Google Scholar] [CrossRef] [Green Version]
- Tsui, H.S.; Pham, N.V.B.; Amer, B.R.; Bradley, M.C.; Gosschalk, J.E.; Gallagher-Jones, M.; Ibarra, H.; Clubb, R.T.; Blaby, C.; Clarke, C.F. Human COQ10A and COQ10B are distinct lipid-binding START domain proteins required for coenzyme Q function. J. Lipid Res. 2019, 60, 1293–1310. [Google Scholar] [CrossRef]
- Allan, C.M.; Hill, S.; Morvaridi, S.; Saiki, R.; Johnson, J.S.; Liau, W.-S.; Hirano, K.; Kawashima, T.; Ji, Z.; Loo, J.A.; et al. A conserved START domain coenzyme Q-binding polypeptide is required for efficient Q biosynthesis, respiratory electron transport, and antioxidant function in Saccharomyces cerevisiae. Biochim. Biophys. Acta (BBA)—Mol. Cell Biol. Lipids 2012, 1831, 776–791. [Google Scholar] [CrossRef] [Green Version]
- Acosta, M.J.; Vazquez Fonseca, L.; Desbats, M.A.; Cerqua, C.; Zordan, R.; Trevisson, E.; Salviati, L. Coenzyme Q biosynthesis in health and disease. Biochim. Biophys. Acta BBA Bioenerg. 2016, 1857, 1079–1085. [Google Scholar] [CrossRef]
- Nichols, B.P.; Green, J.M. Cloning and sequencing of Escherichia coli ubiC and purification of chorismate lyase. J. Bacteriol. 1992, 174, 5309–5316. [Google Scholar] [CrossRef] [Green Version]
- Siebert, M.; Severin, K.; Heide, L. Formation of 4-hydroxybenzoate in Escherichia coli: Characterization of the ubiC gene and its encoded enzyme chorismate pyruvate-lyase. Microbiology 1994, 140, 897–904. [Google Scholar] [CrossRef] [Green Version]
- Lawrence, J.; Cox, G.B.; Gibson, F. Biosynthesis of Ubiquinone in Escherichia coli K-12: Biochemical and Genetic Characterization of a Mutant Unable to Convert Chorismate into 4-Hydroxybenzoate. J. Bacteriol. 1974, 118, 41–45. [Google Scholar] [CrossRef] [Green Version]
- Olson, R.E.; Dialamieh, G.H.; Bentley, R.; Springer, C.M.; Ramsey, V.G. Pattern Of Labeling In Coenzyme Q9 After Administration of Isotopic Acetate And Aromatic Amino Acids To Rats. J. Biol. Chem. 1965, 240, 514–523. [Google Scholar] [CrossRef]
- Olson, R.E. Biosynthesis of Ubiquinones in Animals. Klotho 1967, 24, 551–574. [Google Scholar] [CrossRef]
- Pierrel, F. Impact of Chemical Analogs of 4-Hydroxybenzoic Acid on Coenzyme Q Biosynthesis: From Inhibition to Bypass of Coenzyme Q Deficiency. Front. Physiol. 2017, 8, 436. [Google Scholar] [CrossRef] [Green Version]
- Zhou, L.; Wang, J.-Y.; Wang, J.; Poplawsky, A.; Lin, S.; Zhu, B.; Chang, C.; Zhou, T.; Zhang, L.-H.; He, Y.-W. The diffusible factor synthase XanB2 is a bifunctional chorismatase that links the shikimate pathway to ubiquinone and xanthomonadins biosynthetic pathways. Mol. Microbiol. 2012, 87, 80–93. [Google Scholar] [CrossRef]
- Block, A.; Widhalm, J.R.; Fatihi, A.; Cahoon, R.E.; Wamboldt, Y.; Elowsky, C.; Mackenzie, S.A.; Cahoon, E.B.; Chapple, C.; Dudareva, N.; et al. The Origin and Biosynthesis of the Benzenoid Moiety of Ubiquinone (Coenzyme Q) in Arabidopsis. Plant Cell 2014, 26, 1938–1948. [Google Scholar] [CrossRef] [Green Version]
- Herebian, D.; Seibt, A.; Smits, S.H.J.; Bünning, G.; Freyer, C.; Prokisch, H.; Karall, D.; Wredenberg, A.; Wedell, A.; López, L.C.; et al. Detection of 6-demethoxyubiquinone in CoQ 10 deficiency disorders: Insights into enzyme interactions and identification of potential therapeutics. Mol. Genet. Metab. 2017, 121, 216–223. [Google Scholar] [CrossRef]
- Nambudiri, A.M.D.; Brockman, D.; Alam, S.S.; Rudney, H. Alternate routes for ubiquinone biosynthesis in rats. Biochem. Biophys. Res. Commun. 1977, 76, 282–288. [Google Scholar] [CrossRef]
- Goewert, R.R.; Sippel, C.; Grimm, M.F.; Olson, R.E. Identification of 3-methoxy-4-hydroxy-5-hexaprenylbenzoic acid as a new intermediate in ubiquinone biosynthesis by Saccharomyces cerevisiae. FEBS Lett. 1978, 87, 219–221. [Google Scholar] [CrossRef] [Green Version]
- Ozeir, M.; Mühlenhoff, U.; Webert, H.; Lill, R.; Fontecave, M.; Pierrel, F. Coenzyme Q Biosynthesis: Coq6 Is Required for the C5-Hydroxylation Reaction and Substrate Analogs Rescue Coq6 Deficiency. Chem. Biol. 2011, 18, 1134–1142. [Google Scholar] [CrossRef]
- Xie, L.X.; Ozeir, M.; Tang, J.Y.; Chen, J.Y.; Jaquinod, S.-K.; Fontecave, M.; Clarke, C.F.; Pierrel, F. Overexpression of the Coq8 Kinase in Saccharomyces cerevisiae coq Null Mutants Allows for Accumulation of Diagnostic Intermediates of the Coenzyme Q6 Biosynthetic Pathway. J. Biol. Chem. 2012, 287, 23571–23581. [Google Scholar] [CrossRef] [Green Version]
- Doimo, M.; Trevisson, E.; Airik, R.; Bergdoll, M.; Santos-Ocaña, C.; Hildebrandt, F.; Navas, P.; Pierrel, F.; Salviati, L. Effect of vanillic acid on COQ6 mutants identified in patients with coenzyme Q10 deficiency. Biochim. Biophys. Acta (BBA)—Mol. Basis Dis. 2013, 1842, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Hermle, T.; Braun, D.A.; Helmstädter, M.; Huber, T.B.; Hildebrandt, F. Modeling Monogenic Human Nephrotic Syndrome in the Drosophila Garland Cell Nephrocyte. J. Am. Soc. Nephrol. 2016, 28, 1521–1533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez, M.J.A.; Trevisson, E.; Canton, M.; Vazquez-Fonseca, L.; Morbidoni, V.; Baschiera, E.; Frasson, C.; Pelosi, L.; Rascalou, B.; Desbats, M.A.; et al. Vanillic Acid Restores Coenzyme Q Biosynthesis and ATP Production in Human Cells LackingCOQ6. Oxidative Med. Cell. Longev. 2019, 2019, 3904905. [Google Scholar] [CrossRef] [Green Version]
- Widmeier, E.; Airik, M.; Hugo, H.; Schapiro, D.; Wedel, J.; Ghosh, C.C.; Nakayama, M.; Schneider, R.; Awad, A.M.; Nag, A.; et al. Treatment with 2,4-Dihydroxybenzoic Acid Prevents FSGS Progression and Renal Fibrosis in Podocyte-Specific Coq6 Knockout Mice. J. Am. Soc. Nephrol. 2019, 30, 393–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muskiet, F.A.; Groen, A. Urinary excretion of conjugated homovanillic acid, 3,4-dihydroxyphenylacetic acid, p-hydroxyphenylacetic acid, and vanillic acid by persons on their usual diet and patients with neuroblastoma. Clin. Chem. 1979, 25, 1281–1284. [Google Scholar] [CrossRef] [PubMed]
- Panoutsopoulos, G.; Beedham, C. Enzymatic Oxidation of Vanillin, Isovanillin and Protocatechuic Aldehyde with Freshly Prepared Guinea Pig Liver Slices. Cell. Physiol. Biochem. 2005, 15, 089–098. [Google Scholar] [CrossRef] [PubMed]
- Gitzinger, M.; Kemmer, C.; Fluri, D.A.; El-Baba, M.D.; Weber, W.; Fussenegger, M. The food additive vanillic acid controls transgene expression in mammalian cells and mice. Nucleic Acids Res. 2011, 40, e37. [Google Scholar] [CrossRef]
- Marbois, B.N.; Clarke, C.F. The COQ7 Gene Encodes a Protein in Saccharomyces cerevisiae Necessary for Ubiquinone Biosynthesis. J. Biol. Chem. 1996, 271, 2995–3004. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Oxer, D.; Hekimi, S. Mitochondrial function and lifespan of mice with controlled ubiquinone biosynthesis. Nat. Commun. 2015, 6, 6393. [Google Scholar] [CrossRef] [Green Version]
- Padilla, S.; Tran, U.C.; Jiménez-Hidalgo, M.; López-Martín, J.M.; Martín-Montalvo, A.; Clarke, C.F.; Navas, P.; Santos-Ocaña, C. Hydroxylation of demethoxy-Q6 constitutes a control point in yeast coenzyme Q6 biosynthesis. Cell. Mol. Life Sci. 2008, 66, 173–186. [Google Scholar] [CrossRef] [Green Version]
- Falk, M.J.; Kayser, E.-B.; Morgan, P.G.; Sedensky, M.M. Mitochondrial Complex I Function Modulates Volatile Anesthetic Sensitivity in C. elegans. Curr. Biol. 2006, 16, 1641–1645. [Google Scholar] [CrossRef] [Green Version]
- Freyer, C.; Stranneheim, H.; Naess, K.; Mourier, A.; Felser, A.; Maffezzini, C.; Lesko, N.; Bruhn, H.; Engvall, M.; Wibom, R.; et al. Rescue of primary ubiquinone deficiency due to a novelCOQ7defect using 2,4–dihydroxybensoic acid. J. Med. Genet. 2015, 52, 779–783. [Google Scholar] [CrossRef] [Green Version]
- Luna-Sanchez, M.; Diaz-Casado, E.; Barca, E.; Tejada, M.A.; Montilla-Garcia, A.; Cobos, E.J.; Escames, G.; Acuna-Castroviejo, D.; Quinzii, C.M.; Lopez, L.C. The clinical heterogeneity of coenzyme Q10 deficiency results from genotypic differences in the Coq9 gene. EMBO Mol. Med. 2015, 7, 670–687. [Google Scholar] [CrossRef]
- Gutiérrez, A.H.; Barriocanal-Casado, E.; Bakkali, M.; Casado, M.E.D.; Sánchez-Maldonado, L.; Romero, M.; Sayed, R.; Prehn, C.; Escames, G.; Duarte, J.; et al. β-RA reduces DMQ/CoQ ratio and rescues the encephalopathic phenotype in Coq9 R239X mice. EMBO Mol. Med. 2019, 11, e9466. [Google Scholar] [CrossRef]
- Liu, J.-L.; Yee, C.; Wang, Y.; Hekimi, S. A single biochemical activity underlies the pleiotropy of the aging-related protein CLK-1. Sci. Rep. 2017, 7, 859. [Google Scholar] [CrossRef] [Green Version]
- Widmeier, E.; Yu, S.; Nag, A.; Chung, Y.W.; Nakayama, M.; Fernández-Del-Río, L.; Hugo, H.; Schapiro, D.; Buerger, F.; Choi, W.-I.; et al. ADCK4 Deficiency Destabilizes the Coenzyme Q Complex, Which Is Rescued by 2,4-Dihydroxybenzoic Acid Treatment. J. Am. Soc. Nephrol. 2020, 31, 1191–1211. [Google Scholar] [CrossRef]
- Lohman, D.C.; Forouhar, F.; Beebe, E.T.; Stefely, M.S.; Minogue, C.E.; Ulbrich, A.; Stefely, J.A.; Sukumar, S.; Luna-Sánchez, M.; Jochem, A.; et al. Mitochondrial COQ9 is a lipid-binding protein that associates with COQ7 to enable coenzyme Q bio-synthesis. Proc. Natl. Acad. Sci. USA 2014, 111, 4697–4705. [Google Scholar] [CrossRef] [Green Version]
- Clarke, N.E.; Mosher, R.E.; Clarke, C.N. Phenolic Compounds in the Treatment of Rheumatic Fever. Circulation 1953, 7, 247–257. [Google Scholar] [CrossRef] [Green Version]
- Sahil, K.; Souravh, B. A Review on Protocatechuic Acid and Its Pharmacological Potential. ISRN Pharmacol. 2014, 2014, 952943. [Google Scholar] [CrossRef] [Green Version]
- Guan, S.; Bao, Y.-M.; Jiang, B.; An, L.-J. Protective effect of protocatechuic acid from Alpinia oxyphylla on hydrogen peroxide-induced oxidative PC12 cell death. Eur. J. Pharmacol. 2006, 538, 73–79. [Google Scholar] [CrossRef]
- Zhang, Q.; de Mejia, E.G.; Luna-Vital, D.A.; Tao, T.; Chandrasekaran, S.; Chatham, L.; Juvik, J.; Singh, V.; Kumar, D. Relationship of phenolic composition of selected purple maize (Zea mays L.) genotypes with their anti-inflammatory, anti-adipogenic and anti-diabetic potential. Food Chem. 2019, 289, 739–750. [Google Scholar] [CrossRef]
- Singh, B.; Kumar, A.; Singh, H.; Kaur, S.; Arora, S.; Singh, B. Protective effect of vanillic acid against diabetes and diabetic nephropathy by attenuating oxidative stress and upregulation of NF-κB, TN -α and COX-2 proteins in rats. Phytother. Res. 2022, 36, 1338–1352. [Google Scholar] [CrossRef]
- Amin, F.U.; Shah, S.A.; Kim, M.O. Vanillic acid attenuates Aβ1-42-induced oxidative stress and cognitive impairment in mice. Sci. Rep. 2017, 7, 40753. [Google Scholar] [CrossRef]
- Xie, L.X.; Williams, K.J.; He, C.H.; Weng, E.; Khong, S.; Rose, T.E.; Kwon, O.; Bensinger, S.J.; Marbois, B.N.; Clarke, C.F. Resveratrol and para-coumarate serve as ring precursors for coenzyme Q biosynthesis. J. Lipid Res. 2015, 56, 909–919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diaz, B.G.; Barca, E.; Balreira, A.; Lopez, L.C.; Tadesse, S.; Krishna, S.; Naini, A.; Mariotti, C.; Castellotti, B.; Quinzii, C.M. Lack of aprataxin impairs mitochondrial functions via downregulation of the APE1/NRF1/NRF2 pathway. Hum. Mol. Genet. 2015, 24, 4516–4529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Compagnoni, G.M.; Kleiner, G.; Bordoni, A.; Fortunato, F.; Ronchi, D.; Salani, S.; Guida, M.; Corti, C.; Pichler, I.; Bergamini, C.; et al. Mitochondrial dysfunction in fibroblasts of Multiple System Atrophy. Biochim. Biophys. Acta (BBA)—Mol. Basis Dis. 2018, 1864, 3588–3597. [Google Scholar] [CrossRef] [PubMed]
- Barca, E.; Kleiner, G.; Tang, G.; Ziosi, M.; Tadesse, S.; Masliah, E.; Louis, E.D.; Faust, P.; Kang, U.J.; Torres, J.; et al. Decreased Coenzyme Q10 Levels in Multiple System Atrophy Cerebellum. J. Neuropathol. Exp. Neurol. 2016, 75, 663–672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yubero, D.; Montero, R.; Martín, M.Á.; Montoya, J.; Ribes, A.; Grazina, M.; Trevisson, E.; Aguilera, J.C.R.; Hargreaves, I.P.; Salviati, L.; et al. Secondary coenzyme Q 10 deficiencies in oxidative phosphorylation (OXPHOS) and non-OXPHOS disorders. Mitochondrion 2016, 30, 51–58. [Google Scholar] [CrossRef]
Figure 1.
CoQ biosynthesis pathway in eukaryotic and 4-HB analogs. Unidentified enzymes are indicated by circled question mark. R indicates the polyisproneoid tail. Abbreviations: 4-HBz, 4-hydroxybenzaldehyde; 4-HPP, 4-hydroxyphenylpyruvate; AADAT, mitochondrial alpha-aminoadipate amino-transferase; ALDH3A1, aldehyde dehydrogenase 3A1; DDMQ, 3horismit-demethyl-coenzyme Q; DMQ, 3horismit-coenzyme Q; DmeQ, 3horismit-coenzyme Q; DMAPP, dimethylallyl pyrophosphate; IPP, 3horismite3/pyrophosphate; PDSS1, prenyl (decaprenyl) diphosphate synthase subunit 1; PDSS2, prenyl (decaprenyl) diphosphate synthase subunit 2; PPDHB, polyprenyl-dihydroxybenzoate; PPHB, polyprenyl-hydroxybenzoate; PPVA, polyprenyl-vanillic acid; TAT, tyrosine aminotransferase.
Figure 1.
CoQ biosynthesis pathway in eukaryotic and 4-HB analogs. Unidentified enzymes are indicated by circled question mark. R indicates the polyisproneoid tail. Abbreviations: 4-HBz, 4-hydroxybenzaldehyde; 4-HPP, 4-hydroxyphenylpyruvate; AADAT, mitochondrial alpha-aminoadipate amino-transferase; ALDH3A1, aldehyde dehydrogenase 3A1; DDMQ, 3horismit-demethyl-coenzyme Q; DMQ, 3horismit-coenzyme Q; DmeQ, 3horismit-coenzyme Q; DMAPP, dimethylallyl pyrophosphate; IPP, 3horismite3/pyrophosphate; PDSS1, prenyl (decaprenyl) diphosphate synthase subunit 1; PDSS2, prenyl (decaprenyl) diphosphate synthase subunit 2; PPDHB, polyprenyl-dihydroxybenzoate; PPHB, polyprenyl-hydroxybenzoate; PPVA, polyprenyl-vanillic acid; TAT, tyrosine aminotransferase.

Figure 2.
Schematic representation of 4-HB analogs and the CoQ-deficient models where they were tested. Deleted proteins indicate inefficacy of the supplementation. Created with BioRender.com (accessed on 25 March 2022).
Figure 2.
Schematic representation of 4-HB analogs and the CoQ-deficient models where they were tested. Deleted proteins indicate inefficacy of the supplementation. Created with BioRender.com (accessed on 25 March 2022).


{kind=link}
{kind=link}
Table 1.
Overview of published studies on the effects of supplementation with 4-HB analogs in the different models. Rescue/improvement is indicated by + and lack of effects by -.
Table 1.
Overview of published studies on the effects of supplementation with 4-HB analogs in the different models. Rescue/improvement is indicated by + and lack of effects by -.
| Analog | Model | Molecular Defect | CoQ | DMQ | Mitochondrial Respiration | Oxidative Stress | Growth/Morphology/LIFESPAN | Other | References |
|---|---|---|---|---|---|---|---|---|---|
| 3,4-diHB | S. cerevisiae | Coq6 | + | + | [46,48] | ||||
| Human cells | COQ6 | - | [51] | ||||||
| VA | D. melanogaster | Coq2 | + | + | [49] | ||||
| Human cells | COQ4 | - | [43] | ||||||
| S. cerevisiae | Coq6 | + | + | [46,48] | |||||
| Human cells | COQ6 | + | + | + | [50] | ||||
| Human cells | COQ6 | + | [51] | ||||||
| Human cells | COQ9 | + | + | [43] | |||||
| β-RA | Human cells | COQ4 | - | [43] | |||||
| Human cells | COQ6 | + | [51] | ||||||
| Mouse | Coq6 | + | improved kidney function | [51] | |||||
| S. cerevisiae | Coq7 | + | + | [47] | |||||
| C. elegans | Coq7 | + | + | + | [63] | ||||
| Human cells | COQ7 | + | + | increased COQ proteins levels | [43] | ||||
| Human cells | COQ7 | + | + | [60] | |||||
| Human cells | COQ7 | + | + | + | [21,57] | ||||
| Mouse | Coq7 | + | + | + | + | decreased lactate and TG levels | [21] | ||
| Human cells | COQ8B | + | + | [52] | |||||
| Mouse | Coq8B | + | + | improved kidney function | [52] | ||||
| Human cells | COQ9 | + | + | increased COQ proteine levels | [43] | ||||
| Human cells | COQ9 | + | [61] | ||||||
| Mouse | Coq9 | + | + | + | + | [61,62] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Pesini, A.; Hidalgo-Gutierrez, A.; Quinzii, C.M. Mechanisms and Therapeutic Effects of Benzoquinone Ring Analogs in Primary CoQ Deficiencies. Antioxidants 2022, 11, 665. https://doi.org/10.3390/antiox11040665
AMA Style
Pesini A, Hidalgo-Gutierrez A, Quinzii CM. Mechanisms and Therapeutic Effects of Benzoquinone Ring Analogs in Primary CoQ Deficiencies. Antioxidants. 2022; 11(4):665. https://doi.org/10.3390/antiox11040665
Chicago/Turabian StylePesini, Alba, Agustin Hidalgo-Gutierrez, and Catarina M. Quinzii. 2022. "Mechanisms and Therapeutic Effects of Benzoquinone Ring Analogs in Primary CoQ Deficiencies" Antioxidants 11, no. 4: 665. https://doi.org/10.3390/antiox11040665
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.