
Effects of Natural Polyphenols on Oxidative Stress-Mediated Blood-Brain Barrier Dysfunction
by
Yeonjae Kim
1,2,†, 
A Yeon Cho
1,†,
Hong Cheol Kim
1,†,
Dajung Ryu
1,2,
Sangmee Ahn Jo
3,4 and
Yi-Sook Jung
1,2,* 1
College of Pharmacy, Ajou University, Suwon 16499, Korea
2
Research Institute of Pharmaceutical Sciences and Technology, Ajou University, Suwon 16499, Korea
3
Department of Nanobiomedical Science & BK21 NBM Global Research Center for Regenerative Medicine, Dankook University, Cheonan 31116, Korea
4
Department of Pharmacology, College of Pharmacy, Dankook University, Cheonan 31116, Korea
*
Author to whom correspondence should be addressed.
†
These authors contributed equally to this work.
Antioxidants 2022, 11(2), 197; https://doi.org/10.3390/antiox11020197
Submission received: 18 November 2021
/
Revised: 14 January 2022
/
Accepted: 17 January 2022
/
Published: 20 January 2022
(This article belongs to the Special Issue Natural Antioxidant in Cardiovascular and Cerebrovascular Diseases)
Abstract
:The blood-brain barrier (BBB), which consists mainly of brain microvascular endothelial cells and astrocytes connected by tight junctions (TJs) and adhesion molecules (AMs), maintains the homeostatic balance between brain parenchyma and extracellular fluid. Accumulating evidence shows that BBB dysfunction is a common feature of neurodegenerative diseases, including stroke, traumatic brain injury, and Alzheimer’s disease. Among the various pathological pathways of BBB dysfunction, reactive oxygen species (ROS) are known to play a key role in inducing BBB disruption mediated via TJ modification, AM induction, cytoskeletal reorganization, and matrix metalloproteinase activation. Thus, antioxidants have been suggested to exert beneficial effects on BBB dysfunction-associated brain diseases. In this review, we summarized the sources of ROS production in multiple cells that constitute or surround the BBB, such as BBB endothelial cells, astrocytes, microglia, and neutrophils. We also reviewed various pathological mechanisms by which BBB disruption is caused by ROS in these cells. Finally, we summarized the effects of various natural polyphenols on BBB dysfunction to suggest a therapeutic strategy for BBB disruption-related brain diseases.
1. Introduction
The blood-brain barrier (BBB) is a structural membranous barrier that restricts the passage of molecules circulating in the blood into the brain and functions to maintain the homeostatic balance of the extracellular fluid in the brain under physiological conditions [1]. The BBB primarily comprises brain microvascular endothelial cells (ECs), astrocytes, and pericytes, which are characterized by a few pinocytic vesicles, abundant mitochondria, interendothelial tight junctions (TJs), and adherens junctions (AJs) [1]. While the low pinocytic activity of the BBB limits transcellular transport of molecules across it, paracellular permeability of the BBB can be regulated by TJ proteins, such as claudin and occludin, and AJ proteins, such as cadherin and catenin [2,3]. Occludin and claudins are known to be anchored to BBB endothelial cells by scaffolding proteins which consist of a family of zonula occludens (ZO) proteins, such as ZO-1, ZO-2, and ZO-3 [4]. The expression of TJ proteins and AJ proteins, such as claudin and cadherin, respectively, is closely associated with the permeability of the BBB. Indeed, increased expression of occludin has been shown to decrease permeability across the BBB by inducing a decrease in paracellular transport [5], and increased claudin-5 expression results in the reduction of the transit of large molecules [6].
Under pathological conditions, disruption of the BBB is common and plays a critical role in the pathological process of various cerebrovascular diseases, including stroke and Alzheimer’s disease (AD) [7,8]. Reactive oxygen species (ROS) play a major causative role in the disruption of the BBB by triggering signaling pathways that mediate the breakdown of TJ proteins and modification of AJ proteins. Accumulating evidence further demonstrates the role of ROS in the activation of matrix metalloproteinases (MMPs), a group of proteolytic enzymes that can degrade extracellular matrix components, leading to the disruption of the BBB [1,9]. In this context, it is suggested that antioxidants can act as potential neuroprotectants under disease conditions that induce damage to the BBB.
In the last decade, natural antioxidants have garnered significant interest in the development of promising therapeutics because of their safety, convenience, and bioactivities [10]. The most abundant types of these antioxidants are carotenoids (xanthophylls and carotenes), vitamins (vitamin E and C), and polyphenols (phenolic acids, flavonoids, anthocyanins, lignans, and stilbenes) [11,12]. Compared to carotenoids, the main scavengers of the ROS, such as singlet oxygen and peroxy radicals [13,14], and vitamin C, a chain-disrupting scavenger of peroxy radicals [15]. Polyphenols are the most numerous and widely distributed bioactive molecules, and predominantly contribute to the total antioxidant properties exhibited by various food types [16]. Polyphenols are advantageous, as they exhibit various antioxidant activities, such as inhibition of ROS generation, inactivation of ROS precursors, metal chelation, and ROS scavenging. Furthermore, polyphenols have been shown to exert beneficial effects against pathological conditions, such as cancer [17], type-2 diabetes mellitus, cardiovascular diseases, and cerebrovascular diseases [18,19,20].
In this review, we first summarize the ROS-generating pathways in the multiple cells that make up or surround the BBB, such as BBB ECs, astrocytes, microglia, and neutrophils. We also review the pathological mechanisms underlying BBB disruption is induced by ROS in those cells. We then provide an overview of the effects of various natural polyphenols on BBB dysfunction to suggest a therapeutic strategy for BBB-associated neurodegenerative diseases.
2. Oxidative Stress in Multiple Cells That Constitute or Surround the BBB
Under normal physiologic conditions, low levels of ROS formation within vascular cells are well controlled by the endogenous antioxidant system, which comprises important signaling molecules for normal vascular function. Increasing evidence demonstrates that the physiological impact of ROS depends on their intracellular levels as well as their chemical nature and subcellular localization [21,22]. Therefore, inappropriate scavenging of ROS may induce paradoxical reductive stress and, thereby, cause pathological disease [23,24,25]. Moreover, oxidative stress can be observed upon an imbalance between ROS formation and ROS removal, and plays a critical role in the pathogenesis of various diseases, including atherosclerosis, inflammatory diseases, and neurodegenerative disorders [26]. Under pathological conditions in BBB, excess ROS can activate a number of signaling molecules, such as hypoxia-inducible factor-1, nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), and MMPs, which result in loss of BBB integrity and BBB dysfunction.
The cells surrounding the BBB, including BBB ECs and astrocytes, are rich in mitochondria, one of the main sources of ROS, especially under pathological conditions. Although mitochondrial ROS have recently been suggested to be involved in cerebrovascular diseases [27], little is known about their roles in individual cells during the process of BBB disruption. In contrast, many studies have examined other ROS sources, including nitric oxide synthase (NOS) and nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (NOX) [28,29]. Therefore, this chapter summarizes the sources of ROS generation in each BBB surrounding cell by focusing on NOX, NOS, xanthine oxidase (XO), and cyclooxygenase (COX) rather than mitochondrial ROS.
2.1. Oxidative Stress in BBB Endothelial Cells during BBB Injury
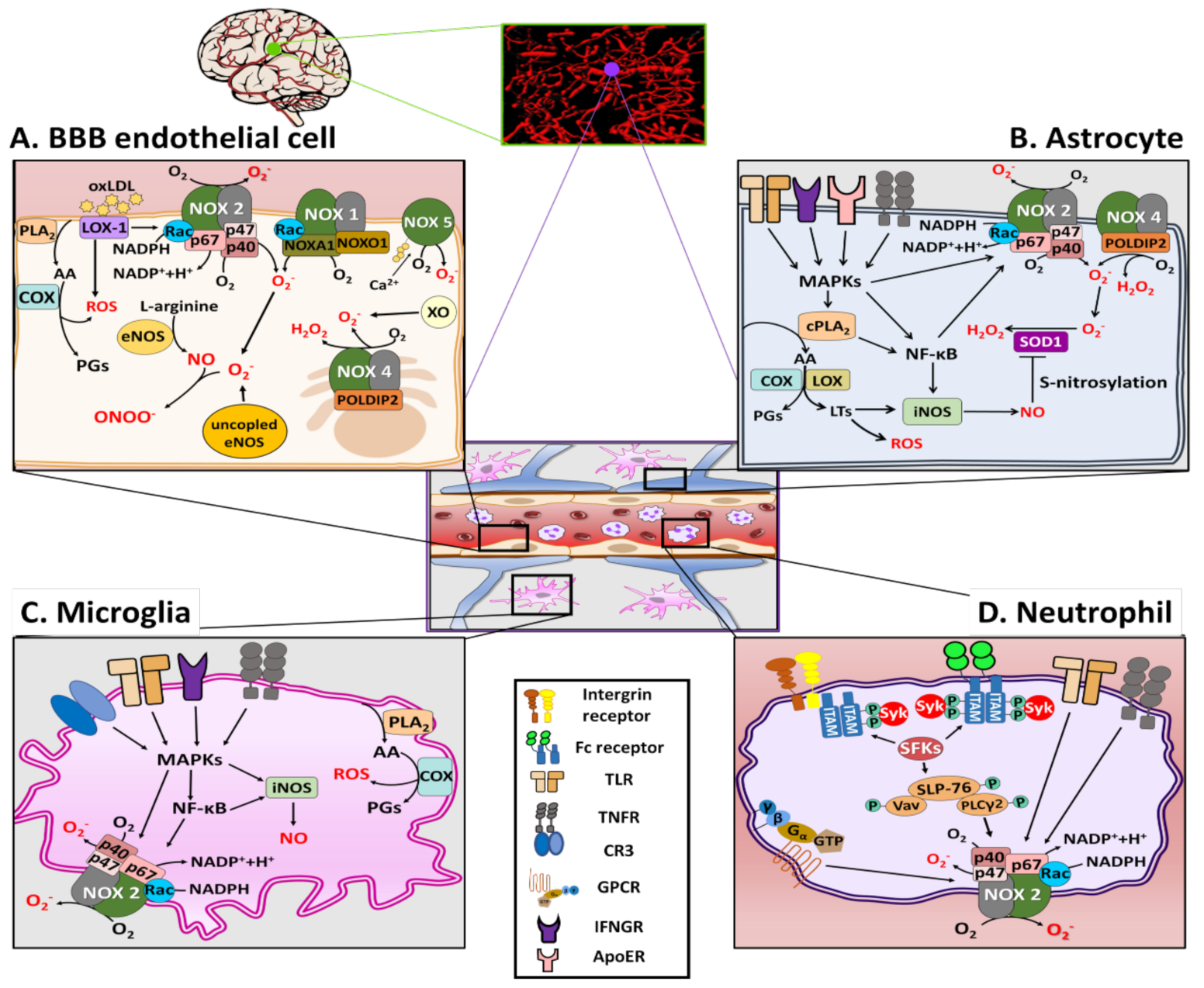
As shown in Figure 1A, there are several sources of ROS generation in BBB ECs, including NOS, XO, COX, and NOX family [28,29]. Among these, NOXs are known to be the primary source of ROS in the BBB ECs [30]. To date, seven isoforms of NOX have been identified: NOX1, NOX2, NOX3, NOX4, NOX5, DUOX1, and DUOX2. In general, all activated NOX isoforms can generate superoxide (O2−) by transferring electrons from the substrate NADPH via the catalytic subunit to molecular oxygen (O2) to form NADP+ and H+. Recent research provides a new perspective on the importance of the NOX family, particularly NOX1 and NOX2, in the generation of oxidative stress in BBB ECs [31,32]. Along with NOX1 and NOX2, NOX4 has also been reported as a major source of O2−, which induces apoptosis of BBB ECs exposed to the pro-inflammatory cytokines [33]. However, NOX4 preferentially generates H2O2 rather than O2− [34]. Accumulating evidence also shows that under condition of ischemia/reperfusion (I/R), increased levels of intracellular Ca2+ result in activation of NOX5 and subsequently increases the production of O2−, OONO−, and H2O2, leading to the disruption of the BBB [35,36]. Consistently, levels of ROS were increased following I/R injury in hippocampal brain slices from NOX-5 knock-in mice [36]. Some studies have shown that NOX inhibitors can improve the barrier function of the BBB in several disease models [37].
NOS is another possible source of ROS production. The NOS family consist of three members: neuronal NOS (nNOS), endothelial NOS (eNOS), both of which are constitutively expressed Ca2+-dependent enzymes, and inducible NOS (iNOS), whose expression can be induced in a Ca2+-independent manner [38]. Under pathophysiological conditions, aberrant eNOS expression can contribute to the development of vascular disease via increased oxidative stress (ONOO−) and conversion of eNOS to an O2−-producing enzyme (uncoupled eNOS), which further generates O2− rather than NO because of the decreased bioavailability of the tetrahydrobiopterin (BH4, cofactor) [38,39,40]. Hence, aberrant eNOS expression in BBB ECs is likely to account for increased protein tyrosine nitration by ONOO−, thereby inducing BBB disruption [40].
Furthermore, lectin-like low-density lipoprotein oxLDL receptor (LOX-1) is another possible source of ROS production in BBB ECs. LOX-1 regulates ROS production by influencing NOX activity in ECs. The binding of LOX-1 to ox-LDL significantly increases NOX activity via induction of NOX-2 and NOX-4 mRNA expression [41,42]. LOX-1 activation induced by oxLDL not only causes membrane translocation of p47phox and Rac, but also protein expression of gp91 and p22phox of NOX-2, which is mediated through protein kinase C (PKC) activation [43]. LOX-1 activation can also lead to the generation of ONOO−, causing BBB disruption [44].
COXs and XOs are also possible sources of ROS generation in BBB ECs. COXs are heme-containing enzymes that catalyze the conversion of arachidonic acid (AA) to prostaglandins (PGs), which are also important source of ROS in the brain and BBB ECs [45]. There are two main isoforms of COX: the COX-1 isoform is a housekeeping enzyme constitutively expressed in all tissues, and COX-2 can be expressed to cause inflammation, particularly expressed in the renal medulla and renal pelvis, the gastrointestinal tract, lung, thymus, and brain [46]. A recent study showed that angiotensin II (AII) increased ROS generation in bEnd3 cells via upregulation of COX-2 expression [47]. XO also generates ROS after oxygen deprivation in BBB ECs [48]. In a rat stroke model, XO inhibitor allopurinol decreased the infarct size, possibly through an antioxidant effect [49,50].
2.2. Oxidative Stress in Astrocytes during BBB Injury
Astrocytes play a vital role in maintaining the physiological functions of the central nervous system (CNS), such as nourishing neurons, maintaining BBB integrity, regulating synaptic activity, and processing cellular metabolites [51]. Physiologically, astrocytes can protect the CNS from oxidative stress-induced damage by exerting antioxidant effect, whereas they act as one of the main sources of detrimental ROS and reactive nitrogen species (RNS) under pathological conditions [52,53]. During brain injury, the state of astrocytes is altered from resting to reactive, and reactive astrocytes exert both protective and detrimental functions [54].
Among the NOXs, NOX2 and NOX4 are major isoforms responsible for the production of NOX-derived ROS in astrocytes [55]. In an AD model, amyloid-β (Aβ) could increase NOX2 activity and O2− levels in astrocytes, thereby inducing astrogliosis [56]. In rat brain, astrocyte-1 (RBA-1) cells activate NOX2 and increase ROS generation [57]. Activated cytosolic phospholipase A2 (cPLA2) in astrocytes is reported to interact with the mitochondrial antiviral-signaling protein and activate NF-κB, which may be involved in inducing the expression NOXs, thereby leading to ROS generation [58,59]. An in vitro study on astrocytes demonstrated that human immunodeficiency virus-1 glycoprotein 120-induced apoptotic cell death by mediating oxidative stress via NOX2 and NOX4 [60]. In an in vitro hypoosmotic swelling model, NOX2-mediated ROS production was increased in astrocytes [61], suggesting a causative role of ROS in astrocyte swelling [62].
Astrocytes also express iNOS, and the RNS generated by them are also key a component of astrocyte-induced oxidative stress [63,64]. In an in vitro astrocyte culture study, lipopolysaccharide (LPS) was shown to induce iNOS expression, leading to NO production [65]. Brain injury induced by severe systemic inflammation is associated with the NF-κB-mediated activation of iNOS in astrocytes [66]. Moreover, in an in vitro I/R model of astrocytes, NO produced by iNOS resulted in aggregation of superoxide dismutase 1 (SOD1) via S-nitrosylation of protein disulfide isomerase, which may be related to the pathogenesis of the injury [67]. Recently, ApoE4 has been reported to induce cPLA2 activation via the p38MAPK pathway followed by leukotriene B4 production via lipoxygenase activation, leading to iNOS activation and ROS generation, which results in oxidative stress and neuroinflammation [68]. Taken together, oxidative stress produced by NOXs and iNOS in astrocytes appear to play a crucial role in BBB injury, as shown in Figure 1B.
2.3. Oxidative Stress in Microglia during BBB Injury
Microglia, the brain-resident macrophages, are known to be dynamic mediators of cerebrovascular diseases. These phagocytic glial cells form a complex network that can respond toward damage and pathogen-associated stimuli, mediating either protective or deleterious responses to brain injury [69,70]. Microglia respond to damage-associated molecular patterns (DAMPs), molecules released from damaged neurons. This results in the activation of disease-associated microglia (DAM) [70]. There is a strong correlation between microglial activation and oxidative stress, ultimately leading to neurovascular injury [71,72].
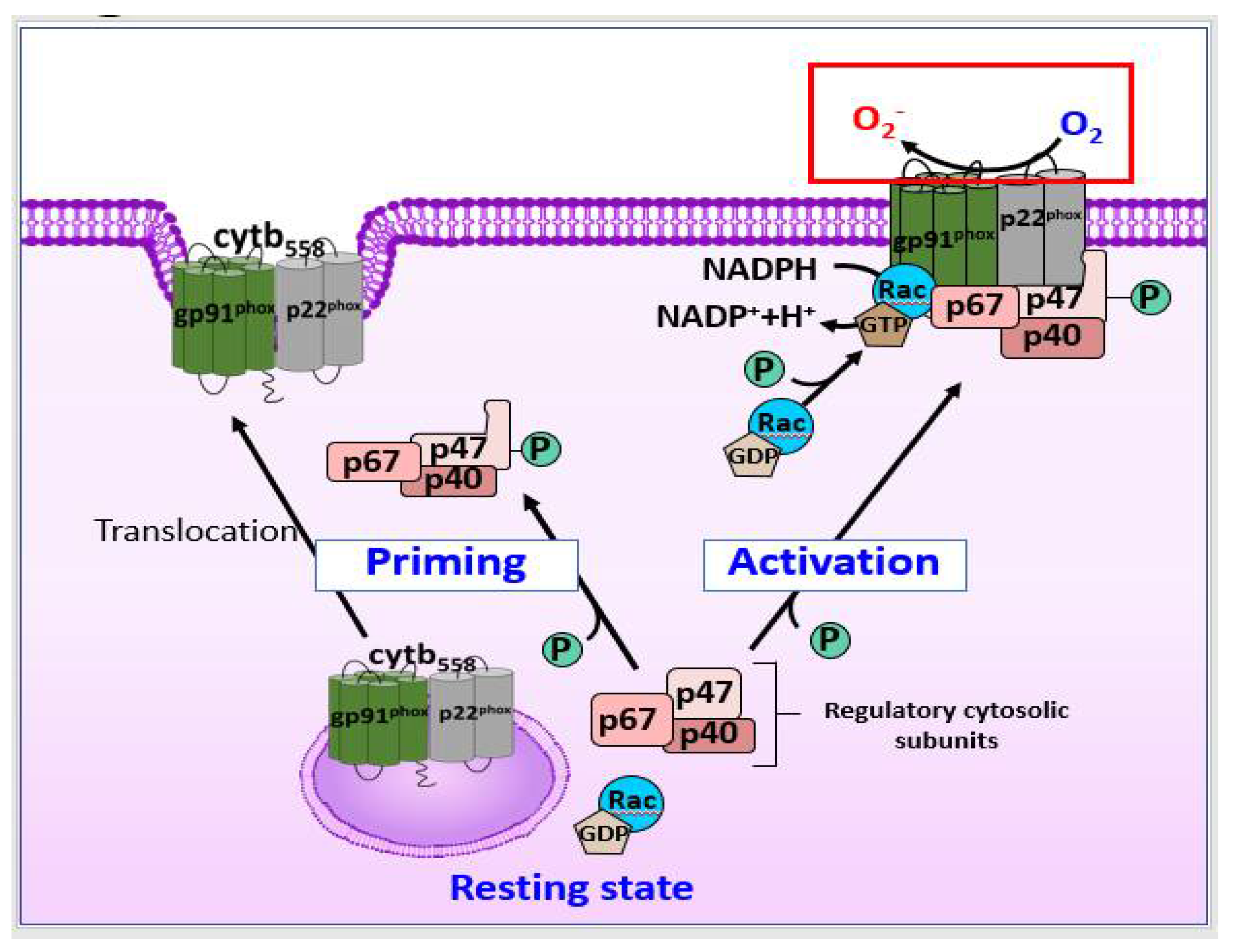
As shown in Figure 1C, in microglia, ROS that induce BBB dysfunction are primarily generated by NOX2, and the activation of NOX2 in DAM is associated with DAMPs, such as Aβ, high mobility group box 1 (HMGB1), and fibrinogen, during both acute and chronic neuroinflammation [73,74]. DAMP stimulation of pattern recognition receptors on the microglia, such as complement receptor 3 and Toll-like receptor 4 (TLR4), mediates activation of the NLRP3 (nucleotide-binding oligomerization domain leucine rich repeat and pyrin domain-containing protein 3) inflammasome, along with that of NF-κB and mitogen-activated protein kinases (MAPKs), such as the extracellular signal-regulated kinases (ERK) [75,76,77]. As shown in Figure 2, upon microglial activation, signaling via pro-inflammatory stimuli mediated by interferon (IFN)-γ results in the activation of cytosolic regulatory subunits of NOX2, resulting in their translocation to the membrane to assemble the active enzyme complex with flavocytochrome b558 (cytb558) [78]. Serine-threonine kinases, including the MAPK family and protein kinase C, are also capable of catalyzing NOX2 assembly. The rate-limiting step of NOX2 activation involves serine-threonine phosphorylation of p47phox by p21 (cdc42/Rac1)-activated kinase-1 [79]. The activated small GTPase Rac1/2 can also translocate to the membrane subunit, cytb558.
Microglia can also generate RNS via NOS. Activation of microglia includes its polarization, which can lead to persistent RNS production via expression of iNOS [80,81], subsequently inducing damage to healthy host cells [82]. In addition, crosstalk between the pathways of NOS and NOX has been demonstrated. ONOO− is an RNS family originated principally by the enzyme NOX through the reaction of NO and O2−. These NOs are generated via stimuli produced by LPS, IFN-γ, tumor necrosis factor (TNF)-α, interleukin (IL)-1β, arachidonate, and ATP in microglia [83]. LPS and IFN-γ lead to the activation of cPLA2 and MAPKs, and thereafter, activation of iNOS and NOX, resulting in the production of ROS and NO [84].
Recent studies have shown that the expression of prostaglandin-endoperoxide synthase 2 (PTGS2), also known as COX2, is highly upregulated in response to LPS and is associated with ROS in microglia [85]. PTGS2 plays a very important role in arachidonic acid metabolism, codifying COX2 enzymes, and inducing PG production. COX2 is rapidly expressed in response to various pro-inflammatory cytokines, such as IL-1β, IL-6, and TNF-α, and thereafter produces PGE2 and free radicals. PGE2, in turn, adversely affects the brain by inducing inflammation, oxidative stress, and excitotoxicity [86].
2.4. Oxidative Stress in Neutrophil during BBB Injury
Neutrophils, the hallmark of acute inflammation, are phagocytic immune cells found in the bloodstream. During the acute phase of inflammation, neutrophils transmigrate to the site of injury within minutes in response to chemical signals, such as IL-8, complement component 5a, N-formyl methionyl-leucyl-phenylalanine, and H2O2, in a process called chemotaxis [87,88]. In the ischemic brain, neutrophils perform several functions, including ROS production, phagocytosis, degranulation, and release of neutrophil extracellular traps (NETs), resulting in increased BBB permeability [89,90].
There are various sources of ROS in neutrophils, including NOSs and NOXs, and NOX2 may be the most important source of ROS [91]. The binding of corresponding ligands to some members of G protein-coupled receptors (GPCRs) can transform neutrophils into a “primed” state for robust activation of the NOX2 complex, as shown in Figure 2 [87]. Signals from cytokine receptors, such as TNF receptors (TNFRs) and TLRs, can also prime neutrophils, inducing phosphorylation of p47phox [92,93]. When neutrophils are activated, NOX2 is activated through the similar pathways of those described previously in astrocytes, and the activated NOX produces ROS [87].
Contrary to some GPCRs, Fc receptors and some integrin receptors can directly activate NOX2 and generate ROS [87]. As shown in Figure 1D, the ligand binding of ligands to Fc receptors leads to the phosphorylation of the immunoreceptor tyrosine-based activation motifs (ITAMs) by Src family kinases (SFKs), leading to the recruitment and the phosphorylation of the Src homology domain of spleen tyrosine kinase (Syk), which further results in activation of the SH2-domain-containing leukocyte protein of 76 kDa (SLP76) signaling complex, and consequently, NOX activation [87,94].
3. Role of Oxidative Stress in BBB Dysfunction
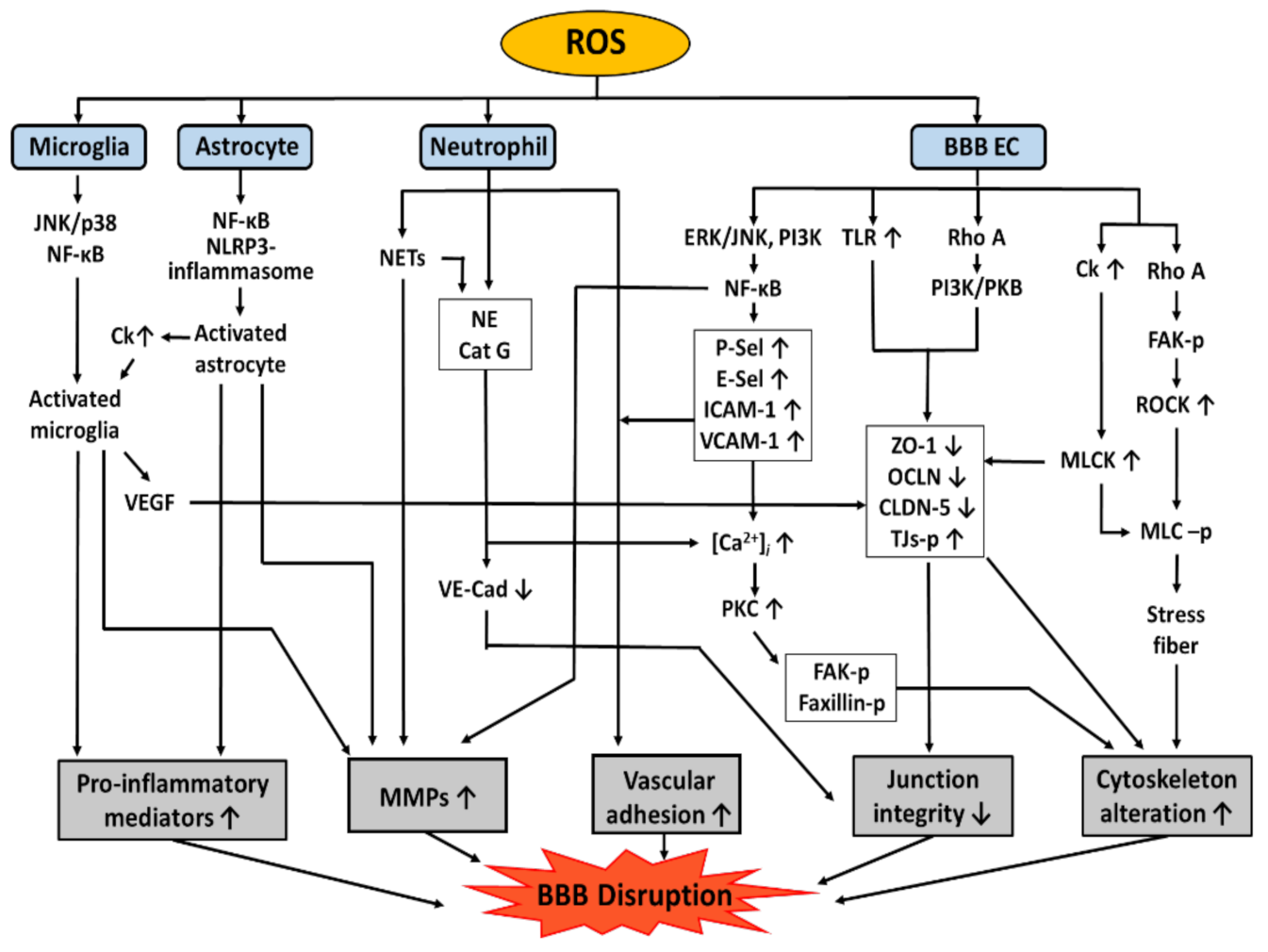
As mentioned above, oxidative stress is one of the most critical causes of BBB dysfunction [1]. A variety of pathways are involved in mediating BBB dysfunction, including modification of TJ proteins, induction of the expression of adhesion molecules (AMs), cytoskeletal reorganization, MMP activation, and NET formation and release of pro-inflammatory mediators, as shown in Figure 3. AMs are a subset of cell surface proteins that are involved in the binding of cells with other cells or with the extracellular matrix (ECM) via a process called cell adhesion [95]. There are several types of AMs on BBB ECs, such as ICAM-1, VCAM-1, platelet endothelial cell adhesion molecule-1 (PECAM), and selectins (P-Sel, E-Sel), which will be discussed in more detail in this chapter.
3.1. TJ Proteins
The barrier function of the BBB is mainly determined by TJ proteins, which act as gatekeepers of the paracellular space between BBB ECs, thereby governing the passage of water-soluble molecules and ions across the BBB [96]. TJs are mainly composed of three proteins: claudins, occludins, and ZO proteins [97]. Among more than 24 members of claudins (20–24-kDa), the main isoforms in brain ECs include claudin-1, -3, -5, and -12, and claudin-5 is especially known to prevent paracellular diffusion of large particles through the BBB [6]. Occludin is the main structural protein of the TJs, and its expression level can represent the structural integrity of the BBB; for instance, lower levels of occludin can reflect an increased BBB permeability. ZOs (ZO-1, ZO-2, and ZO-3) are scaffolding proteins that interact with intracellular components such as F-actin to influence cytoskeleton mobility and other functions [98]. Claudins and occludin bind to the cytoplasmic C-terminal domain of the actin cytoskeleton in BBB ECs via ZOs as accessory proteins [99].
Numerous studies have indicated a correlation between oxidative stress and alterations in TJ complexes [100]. ROS can affect TJ proteins through the following mechanisms: (1) decreased expression of TJ proteins, (2) induction of TJ proteins redistribution, and (3) phosphorylation of TJ proteins [101]. ROS increase the expression of TLRs in BBB ECs, resulting in downregulation of the expression of occludin and claudin-5 and subsequently increasing BBB permeability [102]. An exposure of BBB ECs to hypoxia reduced the expression of claudin-5, occludin, and ZO-1 [1,103]. According to Haorah J et al. [104], ONOO− can induce a reduction in claudin-5 expression. Other studies have shown that LPS attenuated the expression of claudin-5 and ZO-1 in bEnd3 cells [105]. In addition to the expression level of TJ proteins, ROS can regulate the distribution of TJ proteins via the RhoA/PI3 kinase (PI3K)/protein kinase B (PKB/Akt) signaling pathway [106]. A study performed using bEnd3 has shown that the exposure to LPS changed the distribution of claudin-5 and F-actin [105]. Exposure to H2O2 leads to the redistribution of ZO-1 from the TJ sites to the cytosol, resulting in decreased transepithelial electrical resistance and increased BBB permeability [100,107]. Furthermore, ROS have also been implicated in the phosphorylation of TJ proteins that can alter their interactions with transmembrane proteins and the actin cytoskeleton, resulting in changes to the structure and barrier function of the BBB [99]. Since there are many serine/threonine and tyrosine residues that act as phosphorylation sites of TJ proteins, the states of TJ protein phosphorylation could have different effects on BBB permeability, depending on the phosphorylation type or the signaling pathway [108]. Previous research has shown that ROS induce the activation of RhoA/RhoK in human immunodeficiency virus-infected mice and, subsequently, mediate the phosphorylation of claudin-5 and occludin, leading to a decrease in BBB tightness [109]. Tyrosine phosphorylation of BBB TJ complexes induced by ROS increases intracellular gap formation and vascular permeability [99]. Changes in the phosphorylation of occludin and ZO-1 also affect the distribution of these TJ proteins [104,110].
3.2. AMs
While the expression of AMs is repressed under conditions not involving any type of stimulation, it is temporarily coordinated to ensure that the processes of leukocyte rolling, and firm adhesion/emigration can occur for several hours after the initiation of an inflammatory response [111]. Oxidative stress has been shown to induce the expression of AMs that affect BBB permeability by mediating leukocyte-vascular adhesion and infiltration to the brain. Therefore, inhibition of AMs expression may prevent BBB dysfunction. Oxidative stress also activates NF-κB, resulting in the expression of ICAM-1 and VCAM-1 [112], and the crosslinking of ICAM-1 activates the Ca2+ signaling pathways, leading to cytoskeletal alterations, thereby disrupting the BBB [113]. Thus, blocking ICAM-1 with an antibody reduces ischemic brain injury in Wistar rats [114]. Similarly, in a mouse stroke model, elimination of ICAM-1 or neutrophils reduced infarct volume, decreased mortality, and improved BBB dysfunction [115]. Other AMs, such as PECAM-1 [116], E-Sel [117], and P-Sel [118], have also been reported to be redox-regulated. ROS can increase the expression of P-Sel and E-Sel. These selectins promote neutrophil tethering and rolling adhesion by binding to their ligand P-Sel glycoprotein-1 (PSGL-1) on neutrophils [119], similar to that observed with ICAM and VCAM. In another study, stroke-induced infarct size was decreased in P-Sel knockout mice, as evidenced by BBB disruption and granulocyte infiltration [120]. This means that AMs can play a role in BBB opening, and the inhibition of AMs can prevent BBB dysfunction [121].
The receptors for AMs, such as PSGL-1, β2 integrin CD18, and α integrin CD11b, on neutrophils and microglia were also redox-dependent [122,123] and have been shown to participate in the initial movement of leukocytes into the ischemic region [124]. A recent report further showed that the NOX2/ROS signaling pathway can affect very late antigen-4 (VLA-4) and lymphocyte function-associated antigen 1 (LFA-1) expression in monocytes [125]. This concludes that inhibiting NOX2/ROS signaling pathways might be an effective strategy to reduce monocyte-endothelial adherence. Recruited leukocytes, such as neutrophils and macrophages, may further exacerbate injury to the BBB by producing more ROS.
3.3. Cytoskeletal Reorganization
Under physiological conditions, the TJ proteins (occludin and claudin) and AJ protein (cadherin) in BBB ECs are anchored to the actin cytoskeleton by multiple accessory proteins (ZO-1, ZO-2, and ZO-3), indicating that dynamic interactions between the junctional proteins (JPs) and cytoskeleton are essential for the maintenance of BBB integrity [2]. While actin is normally organized at the periphery of ECs, it reorganizes into cytoplasmic filaments called stress fibers upon encountering pathological stimuli, leading to an increase in BBB permeability [126].
Cytoskeletons of BBB ECs are known to be altered by oxidative stress via various pathways. One is the Rho-dependent pathway [106]; once elevated level of superoxide activates the Rho-dependent pathway, Rho can phosphorylate several proteins, including focal adhesion kinase (FAK) [106,127,128], mammalian diaphanous-related formin, and Rho-associated protein kinase (ROCK) [106,129]. Activated ROCK then increases phosphorylation of the myosin light chain (MLC) by either directly or indirectly inhibiting MLC phosphatases [129]. Oxidative stress can also increase the expression of chemokine receptor type 5, which subsequently activates MLC phosphorylation by MLC kinase (MLCK), leading to rearrangement of the actin structure [130]. In addition, MLCK can also phosphorylate TJ proteins, increasing the cytoskeleton reorganization. The second is the RhoA/PI3K pathway; oxidative stress selectively triggers signaling cascades, including RhoA, PI3K, and PKB/Akt. As a result, the actin cytoskeleton is rearranged, while occludin and claudin 5 are spatially distributed and disappear [106]. The third is a PKC-dependent pathway. Oxidative stress induces an increase in the expression of adhesion molecules such as ICAM-1 and VCAM-1, which can elevate intracellular Ca2+, and subsequently leads to PKC activation and phosphorylation of FAK and paxillin [113,131]. As a result, these pathways induce stress fiber formation, which reduces BBB integrity.
3.4. MMPs
MMPs are zinc-dependent endopeptidases that degrade TJ proteins and the ECM. They are usually activated by cleavage induced by other MMPs or proteases [132,133]. Physiologically, MMP expression is very low in the brain; however, it is rapidly upregulated during injury [134]. The proteolytic activity of MMPs has been demonstrated to induce BBB disruption, and therefore, the inhibition of MMP activity by tissue inhibitors of MMPs (TIMPs) may prevent BBB dysfunction [132,135]. MMP levels are increased by ROS in BBB ECs as well as in other cells of the neurovascular unit such as neurons, astrocytes, and microglia under inflammatory conditions [134]. ROS regulate the activity of MMPs through oxidation or S-nitrosylation of MMPs, leading to the subsequent activation of MMPs [136,137]. Additionally, MMP expression can be induced by ROS via ERK/c-Jun N-terminal kinase activation and NF-κB pathways [138]. Expression of MMP-1, MMP-2, and MMP-9 can also be increased by protein tyrosine kinase (PTK), which is activated by ROS. Furthermore, the levels of TIMP-1 and -2 in human BBB ECs were reduced by ROS in a PTK-dependent manner [99]. Increased activity of MMPs degrades the basement membrane and disrupts TJ assembly, resulting in increased BBB permeability [139,140]. Recent research has shown that the activation of MMP leads to cleavage of TJ proteins, which leads to detachment of ECs from the ECM and, thus, BBB disruption [141]. In addition, activation of MMP-2 and MMP-9 is also associated with BBB disruption characterized by leukocyte infiltration and microglial activation [142]
Oxidative stress-induced production of cytokines, such as IL-1β and TNF-α, can activate signaling pathways, including ERK, P38, and PI3K, increasing the binding activity of NF-κB to the promoter region of MMP-9 and, thereby, inducing the transcription of MMP-9 [143]. In mice exposed to prolonged hypoxia, the activation of MMP-9 and subsequent decrease in the expression of occludin and ZO-1, resulting in BBB permeability, has been observed [144]. Overexpression of MMP-9 is reported to cause significant degradation of occludin and claudin-5 [145]. MMP-2, which is constitutively expressed by astrocytes, provides the basis for the first local event of proteolysis, leading to basal lamina degradation and damage to BBB [134]. MMP-2 also degrades occludin and claudin-5 in a rat model of ischemic stroke [132]. Recent research has shown that MMP-3 can increase BBB permeability, and this effect may be at least partially mediated by the ERK pathway and ERK-induced disruption of inter-EC junctions (ZO-1, claudin-5, and occludin) [146].
3.5. NETs and Pro-Inflammatory Mediators
As explained above, neutrophils can be recruited by oxidative stress, and they adhere to activated vasculature, via the interaction of ICAM-2 and LFA-1, which can trigger the generation of ROS [131,147]. ROS generated from extravasated granulocytes can induce lipid peroxidation, which preferentially oxidizes polyunsaturated fatty acids. Indeed, since lipids are a major component of cell membranes and play a critical role in maintaining cell integrity, excessive lipid peroxidation can cause irreversible changes in the physical structure of cell membranes, which may result in BBB disruption and cell death [148,149,150]. During the acute phase of neuroinflammation, neutrophils transmigrate into the injury site by releasing granular contents, such as MMP and elastase [132,151]. For instance, TNF-α stimulates neutrophils to release elastase and cathepsin G, which can cleave VE-cadherin and damage junctional integrity [152]. Other permeability-increasing cytokines include IL-1β and chemokines (CXCL1, 2, 3, and 8) [153,154]. In addition, NETs have recently been highlighted as BBB-altering factors. NETs are web-like structures of extracellular fibers, composed of DNA produced by neutrophils, which primarily bind to pathogens [155]. NETs can effectively trap invading pathogens and disrupt their virulence by presenting a high concentration of local antimicrobial peptides, such as cathelicidins [156,157] and proteases (myeloperoxidase and neutrophil elastase) [158]. NETs are also reported to finally damage the BBB and cause harm to adjacent neurons and other brain cells [155]. In a mechanistic study, phorbol 12-myristate 13-acetate (PMA)-induced NOX have been reported to activate ERK and p38 MAPK, which result in the release of NETs from neutrophils, suggesting the involvement of ROS in NET formation [159]. The role of ROS in NET formation is supported by studies in patients with AD, which demonstrated that Aβ-induced NOX is associated with NET development and BBB disruption. Intravascular NETs can also trigger the coagulation cascade and promote thrombosis, thereby exacerbating cerebrovascular diseases [160]. Furthermore, intravascular NETs release proteolytic proteins, such as cathepsin G and MMPs, which are associated with the disruption of junctional complexes and retraction of endothelial cells.
In addition to intravascular neutrophils, extravasated granulocytes can also release NETs in the perivascular spaces as well as the brain parenchyma, which in turn can lead to microglial activation and neurotoxicity [160]. Furthermore, it is suggested that pro-inflammatory mediators released by microglial cells, such as TNF-α, IL-1, and IL-8, can induce activation of neutrophils to enhance NET release, which further activates microglial cells [151]. LPS-activated microglia induce mRNA expression as well as secretion of pro-inflammatory mediators, such as TNF-α, IL-1α, IL-1β, and IL-6 [161]. These pro-inflammatory cytokines, together with NO, further lead to increased permeability of BBB ECs and upregulation of AMs, facilitating leukocyte recruitment into the brain, and consequently, leading to brain injury [162]. Activated microglia can also induce upregulation of vascular endothelial growth factor (VEGF) [163], which renders the BBB partially open and causes leakage of serum proteins, such as fibrinogen and albumin from the blood stream into the brain parenchyma [164]. Microglia are attracted to these proteins, and perivascular microglia migrate to the damaged blood vessels, causing further damage by phagocytosis of ECs [165]. VEGF has been found to change the distribution, expression, and phosphorylation of TJ proteins such as ZO-1 and occludin [166,167], and thereby, to enhance the permeability of BBB ECs [168].
Similar to microglia, astrocytes can also be activated by ROS to induce BBB disruption. In RBA-1 cells, LPS induces phosphorylation of NF-κB p65 via an increase in the production of ROS mediated via NOX activation, which leads to increased MMP-9 mRNA and protein levels [57]. Another study in astrocytes reported that mitochondrial ROS mediates the classical NLRP3 inflammasome activation, which cleaves pro-inflammatory cytokines induced by LPS [169]. DAMPs and HMGB1 bind to receptors such as TLR4 or RAGE on astrocytes, subsequently activating astrocytes. Activated astrocytes can secrete TNF-α, IL-1β, IL-6, MMPs, and chemokines, which may indirectly and directly impact BBB integrity by disrupting claudin-5 and occludin, thereby inducing the breakdown of the BBB and immune cell infiltration [170,171]
4. Effects of Natural Polyphenols on BBB Dysfunction
Natural polyphenols are secondary metabolites that are produced by plants to protect themselves from other organisms. Polyphenols demonstrate a “typical polyphenol structure” (i.e., several hydroxyl groups on aromatic rings) and are classified as flavonoids (flavonols, flavones, flavanols, flavanones, isoflavones, and anthocyanins) and non-flavonoids (stilbenes, phenolic acids lignans, tannins, and hydroxycinnamic acids) [12,172]. Polyphenols have been reported to be strong antioxidants that can neutralize ROS by donating an electron or hydrogen atom, or suppress the generation of ROS by inhibiting the formation of ROS. They also act as direct radical scavengers of the lipid peroxidation chain reactions. As natural antioxidants, various polyphenols have recently been found to exert beneficial effects on neurodegenerative diseases by ameliorating BBB destruction [173]. In this chapter, we summarize the effects of two subgroups of polyphenols, namely flavonoids and non-flavonoids, on BBB dysfunction.
4.1. Flavonoids
Flavonoids are phenolic compounds found in various vegetables, fruits, seeds, nuts, grains, spices, wine, and tea [174,175]. They contribute to colors of the flowers and reduce the stress response in plants by scavenging ROS and UV absorption [176]. Moreover, they can exhibit pharmacological activities, such as antioxidant, anti-inflammatory, and anticancer effects [177]. Flavonoids are subdivided into subgroups such as flavonols, flavones, flavanols, flavanones, isoflavones, and chalcones [178]. This review summarizes the effects of various flavonoids on BBB dysfunction and the underlying mechanisms (Table 1).
4.1.1. Flavonols-Quercetin/Kaempferol/Rutin
Numerous studies performed using various experimental models have reported that flavonols such as quercetin, kaempferol, and rutin ameliorate BBB disruption [180,184,185].
Quercetin can prevent BBB damage by regulating several ROS production pathways. In fibrillar Aβ-induced BBB damage in human BBB EC, quercetin not only prevented BBB disruption but also attenuated increased BBB permeability by preventing the overproduction of ROS and maintaining SOD activity [179]. In a rat model of cerebral I/R injury, quercetin attenuated the increased BBB permeability, upregulated the expression of tight junctions, such as claudin-5 and ZO-1, and inhibited MMP-9 expression, which is associated with the Wnt/β-catenin signaling pathway, thereby reducing brain edema and ameliorating BBB dysfunction [180]. In female rats with oxidative stress induced by polychlorinated biphenyl, the administration of quercetin exerted protective effects on the BBB by upregulating the mRNA expression of transmembrane TJ proteins (occludin, claudin-5, and JAM) and cytoplasmic accessory TJ proteins (ZO-1, ZO-2) [181]. In murine bEnd3 cells under high glucose conditions, quercetin showed antioxidant effects by reducing ROS generation and increasing relative gene expression of Nrf2 and antioxidant enzymes, including MnSOD and HO-1 [182]. Under the same conditions, quercetin ameliorates BBB disruption by reducing BBB permeability, increasing the expression of claudin-5 and inhibiting the expression of NF-κB, iNOS, TNF-α, and IL-6. Quercetin also inhibits monocyte adhesion and transmigration on cerebral ECs, and the underlying mechanisms are associated with the inhibition of E-selectin [183].
Kaempferol attenuates LPS-induced BBB dysfunction by inhibiting the increase of BBB permeability and the degradation of TJ proteins such as occludin-1, claudin-1, and CX-43 in mice [184].
Rutin has been reported to exhibit protective effects in relation to antioxidant enzymes, such as SOD in ischemic tissue injury [198]. In an in vivo rat stroke model, rutin was reported to exert a protective effect on BBB dysfunction against cerebral ischemic injury by ameliorating BBB permeability and MMP-9 activity [185].
Therefore, it can be suggested that quercetin, kaempferol, and rutin can act as protective agents against BBB dysfunction-associated diseases.
4.1.2. Flavones-Baicalein/Vitexin/Luteolin
Baicalein, luteolin, and vitexin are flavones that were reported to ameliorate BBB dysfunction under oxidative stress [187,189,199].
In the human BBB EC model, baicalein, which is a known LOX inhibitor, attenuated H2O2-induced cell injury and 12/15-LOX activity. In the mouse ischemia model, baicalein reduced brain edema and BBB permeability by inhibiting the degradation of claudin-5. Therefore, it can be suggested that baicalein protects the brain from oxidative stress via the inhibition of 12/15-LOX [186]. In another rat intracerebral hemorrhage (ICH) model, baicalein contributed to the reduction of brain edema and BBB permeability and increasing ZO-1 protein levels, and also inhibited MAPK and NF-κB signaling pathways, leading to the downregulation of iNOS and ONOO− production and preventing BBB disruption. Furthermore, baicalein inhibited cell apoptosis by reducing the level of caspase-3 protein [187].
In an in vitro Aβ-induced BBB damage model, luteolin protected human BBB ECs and human astrocytes by improving cell viability and ameliorated BBB dysfunction by reducing COX-2 expression. These effects may be attributed to the inhibition of p38 MAPK activation and the NF-κB signaling pathway. Furthermore, luteolin exhibited a slight decrease in ROS generation [188]. Moreover, in another in vivo model of Aβ damage, luteolin was shown to maintain BBB integrity by ROS scavenging and redox balancing [200].
A study of hypoxic injury showed that vitexin ameliorated BBB dysfunction by increasing cell viability and reducing BBB permeability. The underlying mechanism for improving BBB permeability includes upregulation of the expression of TJ proteins (ZO-1 and claudin-5) and downregulation of the expression of MMP-2 and MMP-9. Furthermore, vitexin could reduce ONOO− generation and NO level by inhibiting iNOS activity. Vitexin also increased eNOS activity through the PI3K/Akt pathway [189].
4.1.3. Flavanols-Catechin/EGCG/Theaflavin
Flavonols, including catechin, epigallocatechin gallate (EGCG), and theaflavin, have been demonstrated to attenuate BBB dysfunction under oxidative stress [190,191,192].
In an in vivo rat model of traumatic brain injury, catechin attenuated the increase in BBB leakage by inhibiting the loss of TJ proteins by upregulating the mRNA levels of ZO-1 and occludin. It also reduced the brain water content and infarct volume [190].
In the LPS-induced human BBB EC damage model, EGCG attenuated the increase in BBB leakage and the loss of TJ proteins, such as claudin-5 and occludin, by upregulating their mRNA levels. In addition, EGCG inhibited the increase in the expression of adhesion molecule (ICAM-1, VCAM-1) expression by downregulating their mRNA levels and attenuating monocyte adhesion on human BBB EC [191].
In a rat ICH model, theaflavin ameliorated the increase in BBB leakage, brain edema volume, and oxidative stress by scavenging ROS [192].
4.1.4. Flavanones-Pinocembrin/Hesperidin
Flavanones are a class of flavonoids that include pinocembrin and hesperidin. They can attenuate BBB dysfunction under oxidative stress [194,195].
In an in vivo study involving the global cerebral I/R(CI/R) rat model, pinocembrin attenuated BBB dysfunction by inhibiting the increase in BBB permeability and reducing the development of brain edema [193]. In a rat model of cerebral ischemia, pinocembrin exerted protective effects on cerebrovascular units by maintaining the integrity of the BBB and increasing the mRNA levels of TJ proteins, such as occludin and ZO-1. In addition, pinocembrin also inhibited the activation of microglia and astrocytes and the initial leukocyte migration by downregulating the expression of adhesion molecules, such as ICAM-1 and VCAM-1. Furthermore, pinocembrin reduced the expression of MMP-9. The inhibition of MMP-9 expression may be due to a reduction in ROS levels [194].
Hesperidin ameliorated brain edema in a mouse stroke model. Hesperidin also inhibited BBB disruption by inhibiting the degradation of claudin-5 and the redistribution of ZO-1 in mouse brain and bEnd3 cells. In bEnd3 cells, hesperidin exerted antioxidative effects by scavenging ROS and inhibiting FoxO3a translocation, which was reported to regulate the expression of genes related to apoptosis and oxidative stress. Therefore, ROS can contribute to the regulation of BBB integrity as an upstream signaling pathway for FoxO3a/MMP3 and MMP9-mediated degradation of claudin-5 and redistribution of ZO-1 [195].
4.1.5. Isoflavones and Chalcones-Puerarin/Isoliquiritigenin
Other flavonoid antioxidants include puerarin and isoliquiritigenin, which can also be characterized as isoflavones and chalcones [197,201].
In the subarachnoid hemorrhage mouse model, puerarin protected the brain by attenuating the increase in BBB permeability and brain edema volume. In addition, puerarin exerts antioxidant effects by reducing ROS and activating antioxidative proteins, such as SOD2 and SIRT3 [196].
In a rat model of ICH, isoliquiritigenin protected the brain by reducing brain edema and attenuating BBB dysfunction by inhibiting the increase in BBB permeability and exerting antioxidant effects by reducing ROS levels and increasing Nrf2 levels and antioxidant enzyme activities, such as SOD and HO-1. In addition, isoliquiritigenin reduced the infiltration of neutrophils and the recruitment of microglia to the injury site of the brain, consequently inhibiting brain edema [197].
4.2. Non-Flavonoid Polyphenols
As non-flavonoid polyphenols, stilbenes and phenolic acids have been found in various plants [178,202]. They have been reported to exhibit pharmacological activities, such as antioxidant and anti-inflammatory effects [203,204]. In this section, we focus on the effects of non-flavonoid polyphenols including resveratrol, caffeic acid, and gallic acid on BBB dysfunction and the underlying mechanisms (Table 2).
4.2.1. Stilbenes-Resveratrol
Stilbenes are polyphenols that have defensive effects, such as antifungal phytoalexins against infection or injury in many plant species [202]. A well-known stilbene, resveratrol is found in grapes, red wine, berries, knotweed, peanuts, and other plants, and is reported to have anticancer, anti-inflammatory, and antioxidant properties [205]. Furthermore, numerous studies have reported that resveratrol exerts protective effects on the BBB [206,207].
Resveratrol protected ECs by reducing oxidative stress and inhibiting the release of VEGF. VEGF is a mediator that can impact the integrity of the BBB. Moreover, resveratrol attenuated monocyte adhesion on human BBB EC and inhibited the expression of adhesion molecules, such as PECAM and VCAM-1 [208]. Under condition of high glucose levels, resveratrol inhibited cell apoptosis and attenuated the increase in ROS levels by inhibiting the activation of NOX1 and the increase of NOX1 mRNA via the inhibition of NF-κB activation in bEnd3 cells [209]. In a mouse model of aging, resveratrol protected the brain by reducing ROS levels and the mRNA expression of NOX1, NOX2, and NOX4. Resveratrol exerts antioxidant activity by reducing ROS levels in both astrocytes and BBB ECs, whereas its inhibitory effects on mRNA of NOX2 and NOX4 were shown in BBB ECs [210].
An in vitro study of BBB ECs further showed that resveratrol increased cell viability and ameliorated BBB dysfunction by reducing BBB permeability and ROS levels and inhibiting oxLDL-induced disruption of TJs, such as occludin and ZO-1. Resveratrol also improved the disruption of F-actin and cytoskeleton and attenuated cerebral endothelial cell apoptosis by reducing cytochrome C release and caspase-3 and -9 activation and by regulating the Bcl-2/Bax ratio [207]. In a mouse model of autoimmune encephalomyelitis, resveratrol protected the BBB by inhibiting BBB leakage and the loss of TJs, such as ZO-1, claudin-5, and occludin, as well as the expression of AMs, such as ICAM-1 and VCAM-1. In addition, resveratrol exhibited antioxidant properties by reducing the mRNA levels of NOX2 and NOX4 [206].

{kind=link}
{kind=link}
{kind=link}
Table 2.
Effects of non-flavonoid polyphenols on oxidative stress-induced BBB dysfunction.
| Antioxidants | Model | Insult | Findings | Reference | |
|---|---|---|---|---|---|
| stilbenes | resveratrol | HCMEC | CSE | VEGF↓, PECAM↓, VCAM-1↓, monocyte adhesion↓ | [208] |
| bEnd3 | high glucose | cell apoptosis↓, NF-kB↓, NOX1↓, ROS↓ | [209] | ||
| mouse aged | aging | cortical tissue: ROS↓, NOX1↓, NOX2↓, NOX4↓ | [210] | ||
| rat aged | aging | CMVEC: ROS↓, NOX2↓, NOX4↓, | |||
| astrocyte: ROS↓ | |||||
| mouse cEC | oxLDL | BBB permeability↓, occludin↑, ZO-1↑, cell viability↑ ROS↓, cytC↓, F-actin↑, Bcl-2/Bax ratio↑, caspase-3↓, caspase-9↓, apoptosis↓ | [207] | ||
| mouse + EAE | MOG | BBB permeability↓, occludin↑, ZO-1↑, claudin-5↑, ICAM-1↓, VCAM-1↓, iNOS↓, NOX2↓, NOX4↓ | [206] | ||
| phenolic acid | caffeic acid | bEnd3 | high glucose | NF-kB↓, ROS↓, NOX4↓ Nrf2↑ | [211] |
| mouse CI/R | MCAO | VE-cadherin↑, MPO↓, infarct volume↓ | |||
| bEnd3 | high glucose | NF-kB↓, ROS↓, NOX4↓, Cu/ZnSOD↑, Nrf2↑ | [182] | ||
| bEnd3 | high glucose | claudin-5↑, occludin↑, ZO-1↑, ZO-2↑, NF-kB↓, COX-2↓, | [183] | ||
| gallic acid | rat CI/R | 4VO-I/R | BBB permeability↓, lipid peroxydation↓, SOD↑ | [212] | |
| rat CI/R | 4VO-I/R + DPM | lipid peroxydation↓, SOD↑ | |||
| bEnd3 | high glucose | IL-6↓, ROS↓, NOX4↓, Cu/ZnSOD↑, Nrf2↑ | [182] | ||
| bEnd3 | high glucose | claudin-5↑, occludin↑, ZO-2↑, NF-kB↓, iNOS↓ | [183] | ||
4VO, 4-vessel transient occlusion; BBB, blood-brain-barrier; Bcl-2/Bax, B-cell lymphoma-2/-associated; bEnd3, mouse brain endothelial cell; cEC, Cerebrovascular endothelial cell; CI/R, cerebral ischemia/reperfusion; CMVEC, cerebromicrovascular endothelial cell; COX-2, cyclooxygenase-2; CSE, cigarette smoke extract; Cu/ZnSOD, copper-zinc superoxide dismutase; cytC, cytochrome C; DPM, diesel particular matter; EAE, enchephalomyelitis; HCMEC, human cerebral microvascular endothelial cell; I/R, ischemia/reperfusion; ICAM-1, intercellular adhesion molecule-1; MCAO, middle cerebral artery occlusion; MOG, myelin oligodendroglial glycoprotein; MPO, myeloperoxidase; NF-kB, nuclear factor kappa-light-chain-enhancer of activated B cells; NOS, nitric oxide synthase; NOX, NADPH oxidase; Nrf2, nuclear respiratory factor 2; oxLDL, oxidized LDL; PECAM, platelet endothelial cell adhesion molecule; ROS, reactive oxygen species; SOD, superoxide dismutase; VCAM-1, vascular cell adhesion molecule-1; VE, vascular endothelial; VEGF, vascular endothelial growth factor; ZO, zonula occludin.
4.2.2. Phenolic Acid-Caffeic Acid/Gallic Acid
Caffeic acid and gallic acid are phenolic acids present in many fruits and vegetables. These compounds exert beneficial effects, such as antioxidant effects [204]. Furthermore, they can attenuate BBB dysfunction under conditions involving oxidative stress [182,183].
In an in vitro study of bEnd3 cells, high glucose induced a decrease in Nrf2 gene expression and following elevation in ROS level [211]. In contrast, Nrf2 expression levels were increased in an in vivo hyperglycemic model. This discrepancy in Nrf2 expression in vivo and in vitro may be explained by various cell populations in brain tissue, and different durations of hyperglycemic condition. Caffeic acid abolished these effects of high glucose on Nrf2 expression and ROS level [211].
In a rat model of cerebral IR, gallic acid attenuated oxidative stress by upregulating the activity of SOD, downregulating lipid peroxidation and reducing BBB permeability, thereby preventing BBB disruption. However, there was no effect on BBB permeability when rats were subjected to I/R following exposure to air pollution, which may be due to the severity of the damage [212].
Caffeic acid, in particular, also inhibits NF-κB [182]. They also improved gene expression of TJ proteins, such as claudin-5, occludin, ZO-2; and inhibited NF-κB activation [183]. Caffeic acid particularly increased ZO-1 protein expression. In addition, the phenolic acids ameliorated BBB disruption by attenuating an increase in BBB permeability. Moreover, these compounds inhibited monocyte chemoattractant protein-1 (MCP-1) expression, suggesting that these compounds can inhibit monocyte adhesion and transmigration in BBB ECs [183].
5. Conclusions
The BBB plays an important role in maintaining the homeostatic balance between brain parenchyma and systemic circulation under physiological conditions, whereas it can be disrupted under pathological conditions. Accumulating evidence shows that BBB disruption is a crucial process in various neuroinflammatory diseases such as stroke and AD. Since BBB dysfunction has been reported to be closely related to oxidative stress, targeting oxidative stress and BBB disruption may be a promising therapeutic strategy for neuroinflammatory diseases.
In this review, we focused on the understanding of the pathways involved in the generation of oxidative stress in cells that constitute or surround the BBB under pathological conditions. We also showed that ROS can trigger a variety of signaling pathways in cells associated with the BBB, leading to TJ activation, AJ modification, cytoskeletal reorganization, and MMP activation. All of these pathological processes result in BBB dysfunction and eventually neuroinflammatory diseases. Therefore, antioxidants are likely to act as potential protective agents against BBB-related diseases. Various natural polyphenols have been reported to exert beneficial anti-oxidant effects on BBB disruption. In addition, we summarized the potential of natural polyphenols against oxidative stress and BBB disruptive pathology. In conclusion, this review suggests that a number of natural polyphenols can act as promising neuroprotective agents, especially for BBB dysfunction-related brain diseases.
Author Contributions
Conceptualization, Y.-S.J.; writing—original draft preparation Y.K., A.Y.C., H.C.K. and D.R.; writing—review and editing, Y.K., A.Y.C., H.C.K., D.R., S.A.J. and Y.-S.J.; supervision, Y.-S.J.; project administration, Y.-S.J.; funding acquisition, Y.-S.J. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Commercialization Promotion Agency for R&D Outcomes (COMPA) funded by the Ministry of Science and ICT (MSIT) (2020-JDH-2-CG-1), Republic of Korea.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Pun, P.B.; Lu, J.; Moochhala, S. Involvement of ROS in BBB dysfunction. Free Radic. Res. 2009, 43, 348–364. [Google Scholar] [CrossRef] [PubMed]
- Daneman, R.; Prat, A. The blood–brain barrier. Cold Spring Harb. Perspect. Biol. 2015, 7, a020412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deeken, J.F.; Löscher, W. The blood-brain barrier and cancer: Transporters, treatment, and Trojan horses. Clin. Cancer Res. 2007, 13, 1663–1674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abbott, N.J.; Patabendige, A.A.; Dolman, D.E.; Yusof, S.R.; Begley, D.J. Structure and function of the blood-brain barrier. Neurobiol. Dis. 2010, 37, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Balda, M.S.; Flores-Maldonado, C.; Cereijido, M.; Matter, K. Multiple domains of occludin are involved in the regulation of paracellular permeability. J. Cell. Biochem. 2000, 78, 85–96. [Google Scholar] [CrossRef]
- Nitta, T.; Hata, M.; Gotoh, S.; Seo, Y.; Sasaki, H.; Hashimoto, N.; Furuse, M.; Tsukita, S. Size-selective loosening of the blood-brain barrier in claudin-5-deficient mice. J. Cell Biol. 2003, 161, 653–660. [Google Scholar] [CrossRef] [PubMed]
- Perrelli, A.; Retta, S.F. Polymorphisms in genes related to oxidative stress and inflammation: Emerging links with the pathogenesis and severity of Cerebral Cavernous Malformation disease. Free Radic. Biol. Med. 2021, 172, 403–417. [Google Scholar] [CrossRef]
- Song, K.; Li, Y.; Zhang, H.; An, N.; Wei, Y.; Wang, L.; Tian, C.; Yuan, M.; Sun, Y.; Xing, Y. Oxidative stress-mediated blood-brain barrier (BBB) disruption in neurological diseases. Oxid. Med. Cell. Longev. 2020, 2020, 4356386. [Google Scholar] [CrossRef]
- Chiurchiù, V.; Orlacchio, A.; Maccarrone, M. Is modulation of oxidative stress an answer? The state of the art of redox therapeutic actions in neurodegenerative diseases. Oxid. Med. Cell. Longev. 2016, 2016, 7909380. [Google Scholar] [CrossRef] [Green Version]
- Shalaby, E.; Azzam, G.M. Antioxidants in Foods and Its Applications; IntechOpen Limited: London, UK, 2018. [Google Scholar]
- Baiano, A.; Del Nobile, M.A. Antioxidant Compounds from Vegetable Matrices: Biosynthesis, Occurrence, and Extraction Systems. Crit. Rev. Food Sci. Nutr. 2016, 56, 2053–2068. [Google Scholar] [CrossRef]
- Manach, C.; Scalbert, A.; Morand, C.; Rémésy, C.; Jiménez, L. Polyphenols: Food sources and bioavailability. Am. J. Clin. Nutr. 2004, 79, 727–747. [Google Scholar] [CrossRef] [Green Version]
- Stahl, W.; Sies, H. Bioactivity and protective effects of natural carotenoids. Biochim. Biophys. Acta 2005, 1740, 101–107. [Google Scholar] [CrossRef] [Green Version]
- Rao, A.V.; Rao, L.G. Carotenoids and human health. Pharmacol. Res. 2007, 55, 207–216. [Google Scholar] [CrossRef]
- Rice-Evans, C.A.; Miller, N.J. Antioxidant activities of flavonoids as bioactive components of food. Biochem. Soc. Trans. 1996, 24, 790–795. [Google Scholar] [CrossRef]
- Wang, H.; Cao, G.; Prior, R.L. Total antioxidant capacity of fruits. J. Agric. Food Chem. 1996, 44, 701–705. [Google Scholar] [CrossRef]
- Finetti, F.; Moglia, A.; Schiavo, I.; Donnini, S.; Berta, G.N.; Di Scipio, F.; Perrelli, A.; Fornelli, C.; Trabalzini, L.; Retta, S.F. Yeast-Derived Recombinant Avenanthramides Inhibit Proliferation, Migration and Epithelial Mesenchymal Transition of Colon Cancer Cells. Nutrients 2018, 10, 1159. [Google Scholar] [CrossRef] [Green Version]
- Bucciantini, M.; Leri, M.; Nardiello, P.; Casamenti, F.; Stefani, M. Olive Polyphenols: Antioxidant and Anti-Inflammatory Properties. Antioxidants 2021, 10, 1044. [Google Scholar] [CrossRef]
- Goitre, L.; DiStefano, P.V.; Moglia, A.; Nobiletti, N.; Baldini, E.; Trabalzini, L.; Keubel, J.; Trapani, E.; Shuvaev, V.V.; Muzykantov, V.R.; et al. Up-regulation of NADPH oxidase-mediated redox signaling contributes to the loss of barrier function in KRIT1 deficient endothelium. Sci. Rep. 2017, 7, 8296. [Google Scholar] [CrossRef] [Green Version]
- Perrelli, A.; Goitre, L.; Salzano, A.M.; Moglia, A.; Scaloni, A.; Retta, S.F. Biological Activities, Health Benefits, and Therapeutic Properties of Avenanthramides: From Skin Protection to Prevention and Treatment of Cerebrovascular Diseases. Oxid. Med. Cell. Longev. 2018, 2018, 6015351. [Google Scholar] [CrossRef]
- Retta, S.F.; Chiarugi, P.; Trabalzini, L.; Pinton, P.; Belkin, A.M. Reactive oxygen species: Friends and foes of signal transduction. J. Signal Transduct. 2012, 2012, 534029. [Google Scholar] [CrossRef]
- Goitre, L.; Pergolizzi, B.; Ferro, E.; Trabalzini, L.; Retta, S.F. Molecular Crosstalk between Integrins and Cadherins: Do Reactive Oxygen Species Set the Talk? J. Signal Transduct. 2012, 2012, 807682. [Google Scholar] [CrossRef]
- Holmstrom, K.M.; Finkel, T. Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat. Rev. Mol. Cell Biol. 2014, 15, 411–421. [Google Scholar] [CrossRef]
- Espinosa-Diez, C.; Miguel, V.; Mennerich, D.; Kietzmann, T.; Sanchez-Perez, P.; Cadenas, S.; Lamas, S. Antioxidant responses and cellular adjustments to oxidative stress. Redox Biol. 2015, 6, 183–197. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, H.H.; Stocker, R.; Vollbracht, C.; Paulsen, G.; Riley, D.; Daiber, A.; Cuadrado, A. Antioxidants in Translational Medicine. Antioxid. Redox Signal. 2015, 23, 1130–1143. [Google Scholar] [CrossRef] [Green Version]
- Miwa, S.; Beckman, K.B.; Muller, F. Oxidative Stress in Aging: From Model Systems to Human Diseases; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2008. [Google Scholar]
- Antognelli, C.; Trapani, E.; Delle Monache, S.; Perrelli, A.; Daga, M.; Pizzimenti, S.; Barrera, G.; Cassoni, P.; Angelucci, A.; Trabalzini, L.; et al. KRIT1 loss-of-function induces a chronic Nrf2-mediated adaptive homeostasis that sensitizes cells to oxidative stress: Implication for Cerebral Cavernous Malformation disease. Free Radic. Biol. Med. 2018, 115, 202–218. [Google Scholar] [CrossRef]
- Bedard, K.; Krause, K.-H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef]
- Brandes, R.P.; Weissmann, N.; Schröder, K. NADPH oxidases in cardiovascular disease. Free Radic. Biol. Med. 2010, 49, 687–706. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, C.; Moreira, P.I. Oxidative Stress: A Major Player in Cerebrovascular Alterations Associated to Neurodegenerative Events. Front. Physiol. 2018, 9, 806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Girouard, H.; Park, L.; Anrather, J.; Zhou, P.; Iadecola, C. Angiotensin II attenuates endothelium-dependent responses in the cerebral microcirculation through nox-2-derived radicals. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 826–832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuriakose, M.; Younger, D.; Ravula, A.R.; Alay, E.; Rama Rao, K.V.; Chandra, N. Synergistic Role of Oxidative Stress and Blood-Brain Barrier Permeability as Injury Mechanisms in the Acute Pathophysiology of Blast-induced Neurotrauma. Sci. Rep. 2019, 9, 7717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basuroy, S.; Bhattacharya, S.; Leffler, C.W.; Parfenova, H. Nox4 NADPH oxidase mediates oxidative stress and apoptosis caused by TNF-α in cerebral vascular endothelial cells. Am. J. Physiol.-Cell Physiol. 2009, 296, C422–C432. [Google Scholar] [CrossRef] [Green Version]
- Schröder, K.; Zhang, M.; Benkhoff, S.; Mieth, A.; Pliquett, R.; Kosowski, J.; Kruse, C.; Luedike, P.; Michaelis, U.R.; Weissmann, N. Nox4 is a protective reactive oxygen species generating vascular NADPH oxidase. Circ. Res. 2012, 110, 1217–1225. [Google Scholar] [CrossRef] [Green Version]
- do Carmo, L.S.; Berk, B.C.; Harrison, D.G. NOX5 as a therapeutic target in cerebral ischemic injury. J. Clin. Investig. 2019, 129, 1530–1532. [Google Scholar] [CrossRef]
- Casas, A.I.; Kleikers, P.W.; Geuss, E.; Langhauser, F.; Adler, T.; Busch, D.H.; Gailus-Durner, V.; de Angelis, M.H.; Egea, J.; Lopez, M.G. Calcium-dependent blood-brain barrier breakdown by NOX5 limits postreperfusion benefit in stroke. J. Clin. Investig. 2019, 129, 1772–1778. [Google Scholar] [CrossRef]
- Kahles, T.; Luedike, P.; Endres, M.; Galla, H.-J.; Steinmetz, H.; Busse, R.; Neumann-Haefelin, T.; Brandes, R.P. NADPH oxidase plays a central role in blood-brain barrier damage in experimental stroke. Stroke 2007, 38, 3000–3006. [Google Scholar] [CrossRef] [Green Version]
- Han, F.; Fukunaga, K. β-Amyloid Accumulation in Neurovascular Units Following Brain Embolism. J. Pharmacol. Sci. 2009, 111, 101–109. [Google Scholar] [CrossRef] [Green Version]
- Van Den Tweel, E.R.; Nijboer, C.; Kavelaars, A.; Heijnen, C.J.; Groenendaal, F.; Van Bel, F. Expression of nitric oxide synthase isoforms and nitrotyrosine formation after hypoxia–ischemia in the neonatal rat brain. J. Neuroimmunol. 2005, 167, 64–71. [Google Scholar] [CrossRef]
- Han, F.; Shirasaki, Y.; Fukunaga, K. Microsphere embolism-induced endothelial nitric oxide synthase expression mediates disruption of the blood–brain barrier in rat brain. J. Neurochem. 2006, 99, 97–106. [Google Scholar] [CrossRef]
- Hong, D.; Bai, Y.P.; Gao, H.C.; Wang, X.; Li, L.F.; Zhang, G.G.; Hu, C.P. Ox-LDL induces endothelial cell apoptosis via the LOX-1-dependent endoplasmic reticulum stress pathway. Atherosclerosis 2014, 235, 310–317. [Google Scholar] [CrossRef]
- Perez, L.; Vallejos, A.; Echeverria, C.; Varela, D.; Cabello-Verrugio, C.; Simon, F. OxHDL controls LOX-1 expression and plasma membrane localization through a mechanism dependent on NOX/ROS/NF-kappaB pathway on endothelial cells. Lab. Investig. 2019, 99, 421–437. [Google Scholar] [CrossRef]
- Tsai, K.L.; Chen, L.H.; Chiou, S.H.; Chiou, G.Y.; Chen, Y.C.; Chou, H.Y.; Chen, L.K.; Chen, H.Y.; Chiu, T.H.; Tsai, C.S.; et al. Coenzyme Q10 suppresses oxLDL-induced endothelial oxidative injuries by the modulation of LOX-1-mediated ROS generation via the AMPK/PKC/NADPH oxidase signaling pathway. Mol. Nutr. Food Res. 2011, 55 (Suppl. S2), S227–S240. [Google Scholar] [CrossRef]
- Schreurs, M.P.; Hubel, C.A.; Bernstein, I.M.; Jeyabalan, A.; Cipolla, M.J. Increased oxidized low-density lipoprotein causes blood-brain barrier disruption in early-onset preeclampsia through LOX-1. FASEB J. 2013, 27, 1254–1263. [Google Scholar] [CrossRef] [Green Version]
- Sobey, C.G.; Heistad, D.D.; Faraci, F.M. Potassium channels mediate dilatation of cerebral arterioles in response to arachidonate. Am. J. Physiol. 1998, 275, H1606–H1612. [Google Scholar] [CrossRef]
- Kirkby, N.S.; Chan, M.V.; Zaiss, A.K.; Garcia-Vaz, E.; Jiao, J.; Berglund, L.M.; Verdu, E.F.; Ahmetaj-Shala, B.; Wallace, J.L.; Herschman, H.R.; et al. Systematic study of constitutive cyclooxygenase-2 expression: Role of NF-kappaB and NFAT transcriptional pathways. Proc. Natl. Acad. Sci. USA 2016, 113, 434–439. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.W.; Hao, R.J.; Wei, Y.Y.; Yu, G.R. The protective effect of harpagoside on angiotensin II (Ang II)-induced blood–brain barrier leakage in vitro. Phytother. Res. 2021, 35, 6241–6254. [Google Scholar] [CrossRef]
- Beetsch, J.W.; Park, T.S.; Dugan, L.L.; Shah, A.R.; Gidday, J.M. Xanthine oxidase-derived superoxide causes reoxygenation injury of ischemic cerebral endothelial cells. Brain Res. 1998, 786, 89–95. [Google Scholar] [CrossRef]
- Martz, D.; Rayos, G.; Schielke, G.P.; Betz, A.L. Allopurinol and dimethylthiourea reduce brain infarction following middle cerebral artery occlusion in rats. Stroke 1989, 20, 488–494. [Google Scholar] [CrossRef] [Green Version]
- Ono, T.; Tsuruta, R.; Fujita, M.; Aki, H.S.; Kutsuna, S.; Kawamura, Y.; Wakatsuki, J.; Aoki, T.; Kobayashi, C.; Kasaoka, S. Xanthine oxidase is one of the major sources of superoxide anion radicals in blood after reperfusion in rats with forebrain ischemia/reperfusion. Brain Res. 2009, 1305, 158–167. [Google Scholar] [CrossRef] [Green Version]
- Verkhratsky, A.; Parpura, V.; Vardjan, N.; Zorec, R. Physiology of astroglia. Neurogl. Neurodegener. Dis. 2019, 1175, 45–91. [Google Scholar]
- Fernandez-Fernandez, S.; Almeida, A.; Bolaños, J.P. Antioxidant and bioenergetic coupling between neurons and astrocytes. Biochem. J. 2012, 443, 3–11. [Google Scholar] [CrossRef] [Green Version]
- Sofroniew, M.V. Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci. 2009, 32, 638–647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michinaga, S.; Koyama, Y. Pathophysiological Responses and Roles of Astrocytes in Traumatic Brain Injury. Int. J. Mol. Sci. 2021, 22, 6418. [Google Scholar] [CrossRef] [PubMed]
- Sorce, S.; Stocker, R.; Seredenina, T.; Holmdahl, R.; Aguzzi, A.; Chio, A.; Depaulis, A.; Heitz, F.; Olofsson, P.; Olsson, T.; et al. NADPH oxidases as drug targets and biomarkers in neurodegenerative diseases: What is the evidence? Free Radic. Biol. Med. 2017, 112, 387–396. [Google Scholar] [CrossRef] [PubMed]
- Chay, K.O.; Nam Koong, K.Y.; Hwang, S.; Kim, J.K.; Bae, C.S. NADPH Oxidase Mediates β-Amyloid Peptide-Induced Neuronal Death in Mouse Cortical Cultures. Chonnam Med. J. 2017, 53, 196–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, C.C.; Hsiao, L.D.; Tseng, H.C.; Kuo, C.M.; Yang, C.M. Pristimerin Inhibits MMP-9 Expression and Cell Migration Through Attenuating NOX/ROS-Dependent NF-κB Activation in Rat Brain Astrocytes Challenged with LPS. J. Inflamm. Res. 2020, 13, 325–341. [Google Scholar] [CrossRef] [PubMed]
- Chao, C.C.; Gutierrez-Vazquez, C.; Rothhammer, V.; Mayo, L.; Wheeler, M.A.; Tjon, E.C.; Zandee, S.E.J.; Blain, M.; de Lima, K.A.; Takenaka, M.C.; et al. Metabolic Control of Astrocyte Pathogenic Activity via cPLA2-MAVS. Cell 2019, 179, 1483–1498.e1422. [Google Scholar] [CrossRef] [PubMed]
- Morgan, M.J.; Liu, Z.G. Crosstalk of reactive oxygen species and NF-kappaB signaling. Cell Res. 2011, 21, 103–115. [Google Scholar] [CrossRef] [Green Version]
- Shah, A.; Kumar, S.; Simon, S.D.; Singh, D.P.; Kumar, A. HIV gp120- and methamphetamine-mediated oxidative stress induces astrocyte apoptosis via cytochrome P450 2E1. Cell Death Dis. 2013, 4, e850. [Google Scholar] [CrossRef] [Green Version]
- Reinehr, R.; Görg, B.; Becker, S.; Qvartskhava, N.; Bidmon, H.J.; Selbach, O.; Haas, H.L.; Schliess, F.; Häussinger, D. Hypoosmotic swelling and ammonia increase oxidative stress by NADPH oxidase in cultured astrocytes and vital brain slices. Glia 2007, 55, 758–771. [Google Scholar] [CrossRef]
- Jayakumar, A.R.; Rama Rao, K.V.; Tong, X.Y.; Norenberg, M.D. Calcium in the mechanism of ammonia-induced astrocyte swelling. J. Neurochem. 2009, 109 (Suppl. S1), 252–257. [Google Scholar] [CrossRef] [Green Version]
- Gabbott, P.L.; Bacon, S.J. Localisation of NADPH diaphorase activity and NOS immunoreactivity in astroglia in normal adult rat brain. Brain Res. 1996, 714, 135–144. [Google Scholar] [CrossRef]
- Galea, E.; Feinstein, D.L.; Reis, D.J. Induction of calcium-independent nitric oxide synthase activity in primary rat glial cultures. Proc. Natl. Acad. Sci. USA 1992, 89, 10945–10949. [Google Scholar] [CrossRef] [Green Version]
- Moriyama, M.; Fujitsuka, S.; Kawabe, K.; Takano, K.; Nakamura, Y. Zinc Potentiates Lipopolysaccharide-induced Nitric Oxide Production in Cultured Primary Rat Astrocytes. Neurochem. Res. 2018, 43, 363–374. [Google Scholar] [CrossRef]
- Bellaver, B.; Dos Santos, J.P.; Leffa, D.T.; Bobermin, L.D.; Roppa, P.H.A.; da Silva Torres, I.L.; Gonçalves, C.A.; Souza, D.O.; Quincozes-Santos, A. Systemic Inflammation as a Driver of Brain Injury: The Astrocyte as an Emerging Player. Mol. Neurobiol. 2018, 55, 2685–2695. [Google Scholar] [CrossRef]
- Chen, X.; Guan, T.; Li, C.; Shang, H.; Cui, L.; Li, X.M.; Kong, J. SOD1 aggregation in astrocytes following ischemia/reperfusion injury: A role of NO-mediated S-nitrosylation of protein disulfide isomerase (PDI). J. Neuroinflam. 2012, 9, 237. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Li, B.; Solomon, V.; Fonteh, A.; Rapoport, S.I.; Bennett, D.A.; Arvanitakis, Z.; Chui, H.C.; Miller, C.; Sullivan, P.M.; et al. Calcium-dependent cytosolic phospholipase A2 activation is implicated in neuroinflammation and oxidative stress associated with ApoE4. Mol. Neurodegener. 2021, 16, 26. [Google Scholar] [CrossRef]
- Sankowski, R.; Böttcher, C.; Masuda, T.; Geirsdottir, L.; Sagar; Sindram, E.; Seredenina, T.; Muhs, A.; Scheiwe, C.; Shah, M.J.; et al. Mapping microglia states in the human brain through the integration of high-dimensional techniques. Nat. Neurosci. 2019, 22, 2098–2110. [Google Scholar] [CrossRef]
- Deczkowska, A.; Keren-Shaul, H.; Weiner, A.; Colonna, M.; Schwartz, M.; Amit, I. Disease-Associated Microglia: A Universal Immune Sensor of Neurodegeneration. Cell 2018, 173, 1073–1081. [Google Scholar] [CrossRef] [Green Version]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef] [Green Version]
- Reiss, A.B.; Arain, H.A.; Stecker, M.M.; Siegart, N.M.; Kasselman, L.J. Amyloid toxicity in Alzheimer’s disease. Rev. Neurosci. 2018, 29, 613–627. [Google Scholar] [CrossRef]
- Gülke, E.; Gelderblom, M.; Magnus, T. Danger signals in stroke and their role on microglia activation after ischemia. Ther. Adv. Neurol. Disord. 2018, 11, 1756286418774254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubartelli, A. DAMP-Mediated Activation of NLRP3-Inflammasome in Brain Sterile Inflammation: The Fine Line between Healing and Neurodegeneration. Front. Immunol. 2014, 5, 99. [Google Scholar] [CrossRef] [PubMed]
- Hou, L.; Wang, K.; Zhang, C.; Sun, F.; Che, Y.; Zhao, X.; Zhang, D.; Li, H.; Wang, Q. Complement receptor 3 mediates NADPH oxidase activation and dopaminergic neurodegeneration through a Src-Erk-dependent pathway. Redox Biol. 2018, 14, 250–260. [Google Scholar] [CrossRef] [PubMed]
- Savage, C.D.; Lopez-Castejon, G.; Denes, A.; Brough, D. NLRP3-Inflammasome Activating DAMPs Stimulate an Inflammatory Response in Glia in the Absence of Priming Which Contributes to Brain Inflammation after Injury. Front. Immunol. 2012, 3, 288. [Google Scholar] [CrossRef] [Green Version]
- Kawai, T.; Akira, S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity 2011, 34, 637–650. [Google Scholar] [CrossRef] [Green Version]
- Diebold, B.A.; Bokoch, G.M. Molecular basis for Rac2 regulation of phagocyte NADPH oxidase. Nat. Immunol. 2001, 2, 211–215. [Google Scholar] [CrossRef]
- Roepstorff, K.; Rasmussen, I.; Sawada, M.; Cudre-Maroux, C.; Salmon, P.; Bokoch, G.; van Deurs, B.; Vilhardt, F. Stimulus-dependent regulation of the phagocyte NADPH oxidase by a VAV1, Rac1, and PAK1 signaling axis. J. Biol. Chem. 2008, 283, 7983–7993. [Google Scholar] [CrossRef] [Green Version]
- Jorens, P.G.; Matthys, K.E.; Bult, H. Modulation of nitric oxide synthase activity in macrophages. Mediat. Inflamm. 1995, 4, 75–89. [Google Scholar] [CrossRef] [Green Version]
- Marques, C.P.; Cheeran, M.C.; Palmquist, J.M.; Hu, S.; Lokensgard, J.R. Microglia are the major cellular source of inducible nitric oxide synthase during experimental herpes encephalitis. J. Neurovirol. 2008, 14, 229–238. [Google Scholar] [CrossRef] [Green Version]
- Weidinger, A.; Kozlov, A.V. Biological Activities of Reactive Oxygen and Nitrogen Species: Oxidative Stress versus Signal Transduction. Biomolecules 2015, 5, 472–484. [Google Scholar] [CrossRef] [Green Version]
- Min, K.J.; Pyo, H.K.; Yang, M.S.; Ji, K.A.; Jou, I.; Joe, E.H. Gangliosides activate microglia via protein kinase C and NADPH oxidase. Glia 2004, 48, 197–206. [Google Scholar] [CrossRef]
- Chuang, D.Y.; Simonyi, A.; Kotzbauer, P.T.; Gu, Z.; Sun, G.Y. Cytosolic phospholipase A2 plays a crucial role in ROS/NO signaling during microglial activation through the lipoxygenase pathway. J. Neuroinflam. 2015, 12, 199. [Google Scholar] [CrossRef] [Green Version]
- Krasemann, S.; Madore, C.; Cialic, R.; Baufeld, C.; Calcagno, N.; El Fatimy, R.; Beckers, L.; O’Loughlin, E.; Xu, Y.; Fanek, Z.; et al. The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity 2017, 47, 566–581.e9. [Google Scholar] [CrossRef] [Green Version]
- Minghetti, L. Cyclooxygenase-2 (COX-2) in inflammatory and degenerative brain diseases. J. Neuropathol. Exp. Neurol. 2004, 63, 901–910. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, G.T.; Green, E.R.; Mecsas, J. Neutrophils to the ROScue: Mechanisms of NADPH Oxidase Activation and Bacterial Resistance. Front. Cell. Infect. Microbiol. 2017, 7, 373. [Google Scholar] [CrossRef]
- Yoo, S.K.; Starnes, T.W.; Deng, Q.; Huttenlocher, A. Lyn is a redox sensor that mediates leukocyte wound attraction in vivo. Nature 2011, 480, 109–112. [Google Scholar] [CrossRef] [Green Version]
- Mortaz, E.; Alipoor, S.D.; Adcock, I.M.; Mumby, S.; Koenderman, L. Update on Neutrophil Function in Severe Inflammation. Front. Immunol. 2018, 9, 2171. [Google Scholar] [CrossRef] [Green Version]
- Joice, S.L.; Mydeen, F.; Couraud, P.O.; Weksler, B.B.; Romero, I.A.; Fraser, P.A.; Easton, A.S. Modulation of blood-brain barrier permeability by neutrophils: In vitro and in vivo studies. Brain Res. 2009, 1298, 13–23. [Google Scholar] [CrossRef] [Green Version]
- Dupré-Crochet, S.; Erard, M.; Nüβe, O. ROS production in phagocytes: Why, when, and where? J. Leukoc. Biol. 2013, 94, 657–670. [Google Scholar] [CrossRef]
- El-Benna, J.; Hurtado-Nedelec, M.; Marzaioli, V.; Marie, J.C.; Gougerot-Pocidalo, M.A.; Dang, P.M. Priming of the neutrophil respiratory burst: Role in host defense and inflammation. Immunol. Rev. 2016, 273, 180–193. [Google Scholar] [CrossRef]
- El-Benna, J.; Dang, P.M.; Gougerot-Pocidalo, M.A. Priming of the neutrophil NADPH oxidase activation: Role of p47phox phosphorylation and NOX2 mobilization to the plasma membrane. Semin. Immunopathol. 2008, 30, 279–289. [Google Scholar] [CrossRef]
- Bruhns, P. Properties of mouse and human IgG receptors and their contribution to disease models. Blood 2012, 119, 5640–5649. [Google Scholar] [CrossRef]
- Chothia, C.; Jones, E.Y. The molecular structure of cell adhesion molecules. Annu. Rev. Biochem. 1997, 66, 823–862. [Google Scholar] [CrossRef] [Green Version]
- Reese, T.S.; Karnovsky, M.J. Fine structural localization of a blood-brain barrier to exogenous peroxidase. J. Cell Biol. 1967, 34, 207–217. [Google Scholar] [CrossRef] [Green Version]
- Nian, K.; Harding, I.C.; Herman, I.M.; Ebong, E.E. Blood-Brain Barrier Damage in Ischemic Stroke and Its Regulation by Endothelial Mechanotransduction. Front. Physiol. 2020, 11, 605398. [Google Scholar] [CrossRef] [PubMed]
- Furuse, M. Molecular basis of the core structure of tight junctions. Cold Spring Harb. Perspect. Biol. 2010, 2, a002907. [Google Scholar] [CrossRef] [PubMed]
- Haorah, J.; Ramirez, S.H.; Schall, K.; Smith, D.; Pandya, R.; Persidsky, Y. Oxidative stress activates protein tyrosine kinase and matrix metalloproteinases leading to blood-brain barrier dysfunction. J. Neurochem. 2007, 101, 566–576. [Google Scholar] [CrossRef] [PubMed]
- Lochhead, J.J.; Ronaldson, P.T.; Davis, T.P. Hypoxic Stress and Inflammatory Pain Disrupt Blood-Brain Barrier Tight Junctions: Implications for Drug Delivery to the Central Nervous System. AAPS J. 2017, 19, 910–920. [Google Scholar] [CrossRef]
- Liu, H.; Qiu, K.; He, Q.; Lei, Q.; Lu, W. Mechanisms of Blood-Brain Barrier Disruption in Herpes Simplex Encephalitis. J. Neuroimmune Pharmacol. 2019, 14, 157–172. [Google Scholar] [CrossRef]
- Nagyoszi, P.; Wilhelm, I.; Farkas, A.E.; Fazakas, C.; Dung, N.T.; Haskó, J.; Krizbai, I.A. Expression and regulation of toll-like receptors in cerebral endothelial cells. Neurochem. Int. 2010, 57, 556–564. [Google Scholar] [CrossRef]
- Won, S.; Sayeed, I.; Peterson, B.L.; Wali, B.; Kahn, J.S.; Stein, D.G. Vitamin D prevents hypoxia/reoxygenation-induced blood-brain barrier disruption via vitamin D receptor-mediated NF-kB signaling pathways. PLoS ONE 2015, 10, e0122821. [Google Scholar] [CrossRef] [Green Version]
- Haorah, J.; Knipe, B.; Leibhart, J.; Ghorpade, A.; Persidsky, Y. Alcohol-induced oxidative stress in brain endothelial cells causes blood-brain barrier dysfunction. J. Leukoc. Biol. 2005, 78, 1223–1232. [Google Scholar] [CrossRef] [Green Version]
- He, F.; Peng, J.; Deng, X.L.; Yang, L.F.; Wu, L.W.; Zhang, C.L.; Yin, F. RhoA and NF-kappaB are involved in lipopolysaccharide-induced brain microvascular cell line hyperpermeability. Neuroscience 2011, 188, 35–47. [Google Scholar] [CrossRef]
- Schreibelt, G.; Kooij, G.; Reijerkerk, A.; van Doorn, R.; Gringhuis, S.I.; van der Pol, S.; Weksler, B.B.; Romero, I.A.; Couraud, P.O.; Piontek, J.; et al. Reactive oxygen species alter brain endothelial tight junction dynamics via RhoA, PI3 kinase, and PKB signaling. FASEB J. 2007, 21, 3666–3676. [Google Scholar] [CrossRef] [Green Version]
- Seok, S.M.; Kim, J.M.; Park, T.Y.; Baik, E.J.; Lee, S.H. Fructose-1,6-bisphosphate ameliorates lipopolysaccharide-induced dysfunction of blood-brain barrier. Arch. Pharm. Res. 2013, 36, 1149–1159. [Google Scholar] [CrossRef]
- Yuan, S.; Liu, K.J.; Qi, Z. Occludin regulation of blood-brain barrier and potential therapeutic target in ischemic stroke. Brain Circ. 2020, 6, 152–162. [Google Scholar] [CrossRef]
- Yamamoto, M.; Ramirez, S.H.; Sato, S.; Kiyota, T.; Cerny, R.L.; Kaibuchi, K.; Persidsky, Y.; Ikezu, T. Phosphorylation of claudin-5 and occludin by rho kinase in brain endothelial cells. Am. J. Pathol. 2008, 172, 521–533. [Google Scholar] [CrossRef] [Green Version]
- Wong, V. Phosphorylation of occludin correlates with occludin localization and function at the tight junction. Am. J. Physiol. 1997, 273, C1859–C1867. [Google Scholar] [CrossRef]
- Horie, Y.; Wolf, R.; Anderson, D.C.; Granger, D.N. Hepatic leukostasis and hypoxic stress in adhesion molecule-deficient mice after gut ischemia/reperfusion. J. Clin. Investig. 1997, 99, 781–788. [Google Scholar] [CrossRef] [Green Version]
- Yilmaz, G.; Granger, D.N. Leukocyte recruitment and ischemic brain injury. Neuromol. Med. 2010, 12, 193–204. [Google Scholar] [CrossRef] [Green Version]
- Etienne-Manneville, S.; Manneville, J.B.; Adamson, P.; Wilbourn, B.; Greenwood, J.; Couraud, P.O. ICAM-1-coupled cytoskeletal rearrangements and transendothelial lymphocyte migration involve intracellular calcium signaling in brain endothelial cell lines. J. Immunol. 2000, 165, 3375–3383. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.L.; Chopp, M.; Jiang, N.; Tang, W.X.; Prostak, J.; Manning, A.M.; Anderson, D.C. Anti-intercellular adhesion molecule-1 antibody reduces ischemic cell damage after transient but not permanent middle cerebral artery occlusion in the Wistar rat. Stroke 1995, 26, 1438–1443. [Google Scholar] [CrossRef]
- Kitagawa, K.; Matsumoto, M.; Mabuchi, T.; Yagita, Y.; Ohtsuki, T.; Hori, M.; Yanagihara, T. Deficiency of intercellular adhesion molecule 1 attenuates microcirculatory disturbance and infarction size in focal cerebral ischemia. J. Cereb. Blood Flow Metab. 1998, 18, 1336–1345. [Google Scholar] [CrossRef] [Green Version]
- Rattan, V.; Sultana, C.; Shen, Y.; Kalra, V.K. Oxidant stress-induced transendothelial migration of monocytes is linked to phosphorylation of PECAM-1. Am. J. Physiol. 1997, 273, E453–E461. [Google Scholar] [CrossRef]
- Shen, J.S.; Meng, X.L.; Moore, D.F.; Quirk, J.M.; Shayman, J.A.; Schiffmann, R.; Kaneski, C.R. Globotriaosylceramide induces oxidative stress and up-regulates cell adhesion molecule expression in Fabry disease endothelial cells. Mol. Genet. Metab. 2008, 95, 163–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takano, M.; Meneshian, A.; Sheikh, E.; Yamakawa, Y.; Wilkins, K.B.; Hopkins, E.A.; Bulkley, G.B. Rapid upregulation of endothelial P-selectin expression via reactive oxygen species generation. Am. J. Physiol. Heart Circ. Physiol. 2002, 283, H2054–H2061. [Google Scholar] [CrossRef] [PubMed]
- Dimasi, D.; Sun, W.Y.; Bonder, C.S. Neutrophil interactions with the vascular endothelium. Int. Immunopharmacol. 2013, 17, 1167–1175. [Google Scholar] [CrossRef]
- Jin, A.Y.; Tuor, U.I.; Rushforth, D.; Kaur, J.; Muller, R.N.; Petterson, J.L.; Boutry, S.; Barber, P.A. Reduced blood brain barrier breakdown in P-selectin deficient mice following transient ischemic stroke: A future therapeutic target for treatment of stroke. BMC Neurosci. 2010, 11, 12. [Google Scholar] [CrossRef] [Green Version]
- Fabene, P.F.; Navarro Mora, G.; Martinello, M.; Rossi, B.; Merigo, F.; Ottoboni, L.; Bach, S.; Angiari, S.; Benati, D.; Chakir, A.; et al. A role for leukocyte-endothelial adhesion mechanisms in epilepsy. Nat. Med. 2008, 14, 1377–1383. [Google Scholar] [CrossRef]
- Roy, A.; Jana, A.; Yatish, K.; Freidt, M.B.; Fung, Y.K.; Martinson, J.A.; Pahan, K. Reactive oxygen species up-regulate CD11b in microglia via nitric oxide: Implications for neurodegenerative diseases. Free Radic. Biol. Med. 2008, 45, 686–699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsukimori, K.; Tsushima, A.; Fukushima, K.; Nakano, H.; Wake, N. Neutrophil-derived reactive oxygen species can modulate neutrophil adhesion to endothelial cells in preeclampsia. Am. J. Hypertens 2008, 21, 587–591. [Google Scholar] [CrossRef] [Green Version]
- del Zoppo, G.J.; Mabuchi, T. Cerebral microvessel responses to focal ischemia. J. Cereb. Blood Flow Metab. 2003, 23, 879–894. [Google Scholar] [CrossRef]
- Chai, Y.; Cao, Z.; Yu, R.; Liu, Y.; Yuan, D.; Lei, L. Dexmedetomidine Attenuates LPS-Induced Monocyte-Endothelial Adherence via Inhibiting Cx43/PKC-α/NOX2/ROS Signaling Pathway in Monocytes. Oxid. Med. Cell. Longev. 2020, 2020, 2930463. [Google Scholar] [CrossRef]
- Petrache, I.; Birukov, K.; Zaiman, A.L.; Crow, M.T.; Deng, H.; Wadgaonkar, R.; Romer, L.H.; Garcia, J.G. Caspase-dependent cleavage of myosin light chain kinase (MLCK) is involved in TNF-alpha-mediated bovine pulmonary endothelial cell apoptosis. FASEB J. 2003, 17, 407–416. [Google Scholar] [CrossRef]
- Ridley, A.J.; Hall, A. Signal transduction pathways regulating Rho-mediated stress fibre formation: Requirement for a tyrosine kinase. EMBO J. 1994, 13, 2600–2610. [Google Scholar] [CrossRef]
- Schreibelt, G.; Musters, R.J.; Reijerkerk, A.; de Groot, L.R.; van der Pol, S.M.; Hendrikx, E.M.; Dopp, E.D.; Dijkstra, C.D.; Drukarch, B.; de Vries, H.E. Lipoic acid affects cellular migration into the central nervous system and stabilizes blood-brain barrier integrity. J. Immunol. 2006, 177, 2630–2637. [Google Scholar] [CrossRef] [Green Version]
- Pellegrin, S.; Mellor, H. Actin stress fibres. J. Cell Sci. 2007, 120, 3491–3499. [Google Scholar] [CrossRef] [Green Version]
- Shiu, C.; Barbier, E.; Di Cello, F.; Choi, H.J.; Stins, M. HIV-1 gp120 as well as alcohol affect blood-brain barrier permeability and stress fiber formation: Involvement of reactive oxygen species. Alcohol. Clin. Exp. Res. 2007, 31, 130–137. [Google Scholar] [CrossRef]
- Kim, S.R.; Bae, Y.H.; Bae, S.K.; Choi, K.S.; Yoon, K.H.; Koo, T.H.; Jang, H.O.; Yun, I.; Kim, K.W.; Kwon, Y.G.; et al. Visfatin enhances ICAM-1 and VCAM-1 expression through ROS-dependent NF-kappaB activation in endothelial cells. Biochim. Biophys. Acta 2008, 1783, 886–895. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Estrada, E.Y.; Thompson, J.F.; Liu, W.; Rosenberg, G.A. Matrix metalloproteinase-mediated disruption of tight junction proteins in cerebral vessels is reversed by synthetic matrix metalloproteinase inhibitor in focal ischemia in rat. J. Cereb. Blood Flow Metab. 2007, 27, 697–709. [Google Scholar] [CrossRef]
- Sternlicht, M.D.; Werb, Z. How matrix metalloproteinases regulate cell behavior. Annu. Rev. Cell Dev. Biol 2001, 17, 463–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Candelario-Jalil, E.; Yang, Y.; Rosenberg, G.A. Diverse roles of matrix metalloproteinases and tissue inhibitors of metalloproteinases in neuroinflammation and cerebral ischemia. Neuroscience 2009, 158, 983–994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cunningham, L.A.; Wetzel, M.; Rosenberg, G.A. Multiple roles for MMPs and TIMPs in cerebral ischemia. Glia 2005, 50, 329–339. [Google Scholar] [CrossRef] [PubMed]
- Gu, Z.; Kaul, M.; Yan, B.; Kridel, S.J.; Cui, J.; Strongin, A.; Smith, J.W.; Liddington, R.C.; Lipton, S.A. S-nitrosylation of matrix metalloproteinases: Signaling pathway to neuronal cell death. Science 2002, 297, 1186–1190. [Google Scholar] [CrossRef]
- Meli, D.N.; Christen, S.; Leib, S.L. Matrix metalloproteinase-9 in pneumococcal meningitis: Activation via an oxidative pathway. J. Infect. Dis. 2003, 187, 1411–1415. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, H.L.; Wang, H.H.; Wu, W.B.; Chu, P.J.; Yang, C.M. Transforming growth factor-β1 induces matrix metalloproteinase-9 and cell migration in astrocytes: Roles of ROS-dependent ERK- and JNK-NF-κB pathways. J. Neuroinflam. 2010, 7, 88. [Google Scholar] [CrossRef] [Green Version]
- Gürsoy-Ozdemir, Y.; Can, A.; Dalkara, T. Reperfusion-induced oxidative/nitrative injury to neurovascular unit after focal cerebral ischemia. Stroke 2004, 35, 1449–1453. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Thompson, J.F.; Taheri, S.; Salayandia, V.M.; McAvoy, T.A.; Hill, J.W.; Yang, Y.; Estrada, E.Y.; Rosenberg, G.A. Early inhibition of MMP activity in ischemic rat brain promotes expression of tight junction proteins and angiogenesis during recovery. J. Cereb. Blood Flow Metab. 2013, 33, 1104–1114. [Google Scholar] [CrossRef] [Green Version]
- Cummins, P.M. Occludin: One protein, many forms. Mol. Cell. Biol. 2012, 32, 242–250. [Google Scholar] [CrossRef] [Green Version]
- Rosell, A.; Ortega-Aznar, A.; Alvarez-Sabín, J.; Fernández-Cadenas, I.; Ribó, M.; Molina, C.A.; Lo, E.H.; Montaner, J. Increased brain expression of matrix metalloproteinase-9 after ischemic and hemorrhagic human stroke. Stroke 2006, 37, 1399–1406. [Google Scholar] [CrossRef] [Green Version]
- Lehner, C.; Gehwolf, R.; Tempfer, H.; Krizbai, I.; Hennig, B.; Bauer, H.C.; Bauer, H. Oxidative stress and blood-brain barrier dysfunction under particular consideration of matrix metalloproteinases. Antioxid. Redox Signal. 2011, 15, 1305–1323. [Google Scholar] [CrossRef]
- Bauer, A.T.; Burgers, H.F.; Rabie, T.; Marti, H.H. Matrix metalloproteinase-9 mediates hypoxia-induced vascular leakage in the brain via tight junction rearrangement. J. Cereb. Blood Flow Metab. 2010, 30, 837–848. [Google Scholar] [CrossRef] [Green Version]
- Chen, F.; Ohashi, N.; Li, W.; Eckman, C.; Nguyen, J.H. Disruptions of occludin and claudin-5 in brain endothelial cells in vitro and in brains of mice with acute liver failure. Hepatology 2009, 50, 1914–1923. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Zheng, M.; Betancourt, C.E.; Liu, L.; Sitikov, A.; Sladojevic, N.; Zhao, Q.; Zhang, J.H.; Liao, J.K.; Wu, R. Increase in Blood-Brain Barrier (BBB) Permeability Is Regulated by MMP3 via the ERK Signaling Pathway. Oxid. Med. Cell. Longev. 2021, 2021, 6655122. [Google Scholar] [CrossRef]
- Gorina, R.; Lyck, R.; Vestweber, D.; Engelhardt, B. beta2 integrin-mediated crawling on endothelial ICAM-1 and ICAM-2 is a prerequisite for transcellular neutrophil diapedesis across the inflamed blood-brain barrier. J. Immunol. 2014, 192, 324–337. [Google Scholar] [CrossRef] [Green Version]
- Gaschler, M.M.; Stockwell, B.R. Lipid peroxidation in cell death. Biochem. Biophys. Res. Commun. 2017, 482, 419–425. [Google Scholar] [CrossRef]
- Su, L.J.; Zhang, J.H.; Gomez, H.; Murugan, R.; Hong, X.; Xu, D.; Jiang, F.; Peng, Z.Y. Reactive Oxygen Species-Induced Lipid Peroxidation in Apoptosis, Autophagy, and Ferroptosis. Oxid. Med. Cell. Longev. 2019, 2019, 5080843. [Google Scholar] [CrossRef] [Green Version]
- Bahcekapili, N.; Uzum, G.; Gokkusu, C.; Kuru, A.; Ziylan, Y.Z. The relationship between erythropoietin pretreatment with blood-brain barrier and lipid peroxidation after ischemia/reperfusion in rats. Life Sci 2007, 80, 1245–1251. [Google Scholar] [CrossRef]
- Pietronigro, E.C.; Della Bianca, V.; Zenaro, E.; Constantin, G. NETosis in Alzheimer’s Disease. Front. Immunol. 2017, 8, 211. [Google Scholar] [CrossRef] [Green Version]
- Hermant, B.; Bibert, S.; Concord, E.; Dublet, B.; Weidenhaupt, M.; Vernet, T.; Gulino-Debrac, D. Identification of proteases involved in the proteolysis of vascular endothelium cadherin during neutrophil transmigration. J. Biol. Chem. 2003, 278, 14002–14012. [Google Scholar] [CrossRef]
- DiStasi, M.R.; Ley, K. Opening the flood-gates: How neutrophil-endothelial interactions regulate permeability. Trends Immunol. 2009, 30, 547–556. [Google Scholar] [CrossRef] [Green Version]
- Rigor, R.R.; Beard, R.S., Jr.; Litovka, O.P.; Yuan, S.Y. Interleukin-1β-induced barrier dysfunction is signaled through PKC-θ in human brain microvascular endothelium. Am. J. Physiol. Cell Physiol. 2012, 302, C1513–C1522. [Google Scholar] [CrossRef] [Green Version]
- Allen, C.; Thornton, P.; Denes, A.; McColl, B.W.; Pierozynski, A.; Monestier, M.; Pinteaux, E.; Rothwell, N.J.; Allan, S.M. Neutrophil cerebrovascular transmigration triggers rapid neurotoxicity through release of proteases associated with decondensed DNA. J. Immunol. 2012, 189, 381–392. [Google Scholar] [CrossRef]
- Barnado, A.; Crofford, L.J.; Oates, J.C. At the Bedside: Neutrophil extracellular traps (NETs) as targets for biomarkers and therapies in autoimmune diseases. J. Leukoc. Biol. 2016, 99, 265–278. [Google Scholar] [CrossRef]
- Brinkmann, V.; Zychlinsky, A. Beneficial suicide: Why neutrophils die to make NETs. Nat. Rev. Microbiol. 2007, 5, 577–582. [Google Scholar] [CrossRef]
- Papayannopoulos, V.; Metzler, K.D.; Hakkim, A.; Zychlinsky, A. Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J. Cell Biol. 2010, 191, 677–691. [Google Scholar] [CrossRef] [Green Version]
- Zeng, M.Y.; Miralda, I.; Armstrong, C.L.; Uriarte, S.M.; Bagaitkar, J. The roles of NADPH oxidase in modulating neutrophil effector responses. Mol. Oral Microbiol. 2019, 34, 27–38. [Google Scholar] [CrossRef]
- Manda-Handzlik, A.; Demkow, U. The Brain Entangled: The Contribution of Neutrophil Extracellular Traps to the Diseases of the Central Nervous System. Cells 2019, 8, 1477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, H.; Wang, H.L.; Pu, H.J.; Shi, Y.J.; Zhang, J.; Zhang, W.T.; Wang, G.H.; Hu, X.M.; Leak, R.K.; Chen, J.; et al. Ethyl pyruvate protects against blood-brain barrier damage and improves long-term neurological outcomes in a rat model of traumatic brain injury. CNS Neurosci. Ther. 2015, 21, 374–384. [Google Scholar] [CrossRef] [PubMed]
- Shabbir, U.; Tyagi, A.; Elahi, F.; Aloo, S.O.; Oh, D.-H. The Potential Role of Polyphenols in Oxidative Stress and Inflammation Induced by Gut Microbiota in Alzheimer’s Disease. Antioxidants 2021, 10, 1370. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.; Gu, R.; Zhang, M.; Ren, H.; Shu, Q.; Xu, G.; Wu, H. Microglia enhanced the angiogenesis, migration and proliferation of co-cultured RMECs. BMC Ophthalmol. 2018, 18, 249. [Google Scholar] [CrossRef]
- Jolivel, V.; Bicker, F.; Binamé, F.; Ploen, R.; Keller, S.; Gollan, R.; Jurek, B.; Birkenstock, J.; Poisa-Beiro, L.; Bruttger, J.; et al. Perivascular microglia promote blood vessel disintegration in the ischemic penumbra. Acta Neuropathol. 2015, 129, 279–295. [Google Scholar] [CrossRef]
- Dudvarski Stankovic, N.; Teodorczyk, M.; Ploen, R.; Zipp, F.; Schmidt, M.H.H. Microglia-blood vessel interactions: A double-edged sword in brain pathologies. Acta Neuropathol. 2016, 131, 347–363. [Google Scholar] [CrossRef]
- Wang, W.; Dentler, W.L.; Borchardt, R.T. VEGF increases BMEC monolayer permeability by affecting occludin expression and tight junction assembly. Am. J. Physiol. Heart Circ. Physiol. 2001, 280, H434–H440. [Google Scholar] [CrossRef]
- DeMaio, L.; Rouhanizadeh, M.; Reddy, S.; Sevanian, A.; Hwang, J.; Hsiai, T.K. Oxidized phospholipids mediate occludin expression and phosphorylation in vascular endothelial cells. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H674–H683. [Google Scholar] [CrossRef]
- Wang, W.; Merrill, M.J.; Borchardt, R.T. Vascular endothelial growth factor affects permeability of brain microvessel endothelial cells in vitro. Am. J. Physiol. 1996, 271, C1973–C1980. [Google Scholar] [CrossRef]
- Alfonso-Loecches, S.; Ureña-Peralta, J.R.; Morillo-Bargues, M.J.; Oliver-De La Cruz, J.; Guerri, C. Role of mitochondria ROS generation in ethanol-induced NLRP3 inflammasome activation and cell death in astroglial cells. Front. Cell. Neurosci. 2014, 8, 216. [Google Scholar] [CrossRef]
- Archie, S.R.; Al Shoyaib, A.; Cucullo, L. Blood-Brain Barrier Dysfunction in CNS Disorders and Putative Therapeutic Targets: An Overview. Pharmaceutics 2021, 13, 1779. [Google Scholar] [CrossRef]
- Liu, L.R.; Liu, J.C.; Bao, J.S.; Bai, Q.Q.; Wang, G.Q. Interaction of Microglia and Astrocytes in the Neurovascular Unit. Front. Immunol. 2020, 11, 1024. [Google Scholar] [CrossRef]
- Di Lorenzo, C.; Colombo, F.; Biella, S.; Stockley, C.; Restani, P. Polyphenols and Human Health: The Role of Bioavailability. Nutrients 2021, 13, 273. [Google Scholar] [CrossRef]
- Lam, V.; Hackett, M.; Takechi, R. Antioxidants and Dementia Risk: Consideration through a Cerebrovascular Perspective. Nutrients 2016, 8, 828. [Google Scholar] [CrossRef] [Green Version]
- Brouillard, R.; Cheminat, A. Flavonoids and plant color. Prog. Clin. Biol. Res. 1988, 280, 93–106. [Google Scholar]
- Kühnau, J. The flavonoids. A class of semi-essential food components: Their role in human nutrition. World Rev. Nutr. Diet. 1976, 24, 117–191. [Google Scholar]
- Heller, W.; Forkmann, G. The Flavonoids: Advances in Research since 1986. Secondary Plant Products. Encyclopedia of Plant Physiology; Harborne, J.B., Ed.; Chapman & Hall: London, UK, 1994. [Google Scholar]
- Kumar, S.; Pandey, A.K. Chemistry and biological activities of flavonoids: An overview. Sci. World J. 2013, 2013, 162750. [Google Scholar] [CrossRef] [Green Version]
- Pietta, P.G. Flavonoids as antioxidants. J. Nat. Prod. 2000, 63, 1035–1042. [Google Scholar] [CrossRef]
- Li, Y.; Zhou, S.; Li, J.; Sun, Y.; Hasimu, H.; Liu, R.; Zhang, T. Quercetin protects human brain microvascular endothelial cells from fibrillar β-amyloid1-40-induced toxicity. Acta Pharm. Sin. B 2015, 5, 47–54. [Google Scholar] [CrossRef] [Green Version]
- Jin, Z.; Ke, J.; Guo, P.; Wang, Y.; Wu, H. Quercetin improves blood-brain barrier dysfunction in rats with cerebral ischemia reperfusion via Wnt signaling pathway. Am. J. Transl. Res. 2019, 11, 4683–4695. [Google Scholar]
- Selvakumar, K.; Prabha, R.L.; Saranya, K.; Bavithra, S.; Krishnamoorthy, G.; Arunakaran, J. Polychlorinated biphenyls impair blood-brain barrier integrity via disruption of tight junction proteins in cerebrum, cerebellum and hippocampus of female Wistar rats: Neuropotential role of quercetin. Hum. Exp. Toxicol. 2013, 32, 706–720. [Google Scholar] [CrossRef]
- Taïlé, J.; Arcambal, A.; Clerc, P.; Gauvin-Bialecki, A.; Gonthier, M.P. Medicinal Plant Polyphenols Attenuate Oxidative Stress and Improve Inflammatory and Vasoactive Markers in Cerebral Endothelial Cells during Hyperglycemic Condition. Antioxidants 2020, 9, 573. [Google Scholar] [CrossRef]
- Taïlé, J.; Patché, J.; Veeren, B.; Gonthier, M.P. Hyperglycemic Condition Causes Pro-Inflammatory and Permeability Alterations Associated with Monocyte Recruitment and Deregulated NFκB/PPARγ Pathways on Cerebral Endothelial Cells: Evidence for Polyphenols Uptake and Protective Effect. Int. J. Mol. Sci. 2021, 22, 1385. [Google Scholar] [CrossRef]
- Cheng, X.; Yang, Y.L.; Yang, H.; Wang, Y.H.; Du, G.H. Kaempferol alleviates LPS-induced neuroinflammation and BBB dysfunction in mice via inhibiting HMGB1 release and down-regulating TLR4/MyD88 pathway. Int. Immunopharmacol. 2018, 56, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Jang, J.W.; Lee, J.K.; Hur, H.; Kim, T.W.; Joo, S.P.; Piao, M.S. Rutin improves functional outcome via reducing the elevated matrix metalloproteinase-9 level in a photothrombotic focal ischemic model of rats. J. Neurol. Sci. 2014, 339, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Jin, G.; Arai, K.; Murata, Y.; Wang, S.; Stins, M.F.; Lo, E.H.; van Leyen, K. Protecting against cerebrovascular injury: Contributions of 12/15-lipoxygenase to edema formation after transient focal ischemia. Stroke 2008, 39, 2538–2543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, M.; Lai, L.; Li, X.; Zhang, X.; He, X.; Liu, W.; Li, R.; Ke, X.; Fu, C.; Huang, Z.; et al. Baicalein Attenuates Neurological Deficits and Preserves Blood-Brain Barrier Integrity in a Rat Model of Intracerebral Hemorrhage. Neurochem. Res. 2016, 41, 3095–3102. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.X.; Xing, J.G.; Wang, L.L.; Jiang, H.L.; Guo, S.L.; Liu, R. Luteolin Inhibits Fibrillary β-Amyloid(1-40)-Induced Inflammation in a Human Blood-Brain Barrier Model by Suppressing the p38 MAPK-Mediated NF-κB Signaling Pathways. Molecules 2017, 22, 334. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.H.; Zhang, X.Q.; Wang, N.D.; Zheng, M.D.; Yan, J. Vitexin protects against ischemia/reperfusion-induced brain endothelial permeability. Eur. J. Pharmacol. 2019, 853, 210–219. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Zhang, J.; Cai, Y.; Huang, J.; You, L. Catechin attenuates traumatic brain injury-induced blood-brain barrier damage and improves longer-term neurological outcomes in rats. Exp. Physiol. 2017, 102, 1269–1277. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Ye, L.; Wang, X.; Liu, J.; Wang, Y.; Zhou, Y.; Ho, W. (-)-Epigallocatechin gallate inhibits endotoxin-induced expression of inflammatory cytokines in human cerebral microvascular endothelial cells. J. Neuroinflam. 2012, 9, 161. [Google Scholar] [CrossRef] [Green Version]
- Fu, G.; Wang, H.; Cai, Y.; Zhao, H.; Fu, W. Theaflavin alleviates inflammatory response and brain injury induced by cerebral hemorrhage via inhibiting the nuclear transcription factor kappa β-related pathway in rats. Drug Des. Dev. Ther. 2018, 12, 1609–1619. [Google Scholar] [CrossRef] [Green Version]
- Meng, F.; Liu, R.; Gao, M.; Wang, Y.; Yu, X.; Xuan, Z.; Sun, J.; Yang, F.; Wu, C.; Du, G. Pinocembrin attenuates blood-brain barrier injury induced by global cerebral ischemia-reperfusion in rats. Brain Res. 2011, 1391, 93–101. [Google Scholar] [CrossRef]
- Gao, M.; Zhu, S.Y.; Tan, C.B.; Xu, B.; Zhang, W.C.; Du, G.H. Pinocembrin protects the neurovascular unit by reducing inflammation and extracellular proteolysis in MCAO rats. J. Asian Nat. Prod. Res. 2010, 12, 407–418. [Google Scholar] [CrossRef]
- Lee, B.K.; Hyun, S.W.; Jung, Y.S. Yuzu and Hesperidin Ameliorate Blood-Brain Barrier Disruption during Hypoxia via Antioxidant Activity. Antioxidants 2020, 9, 843. [Google Scholar] [CrossRef]
- Zhang, Y.; Yang, X.; Ge, X.; Zhang, F. Puerarin attenuates neurological deficits via Bcl-2/Bax/cleaved caspase-3 and Sirt3/SOD2 apoptotic pathways in subarachnoid hemorrhage mice. Biomed. Pharmacother. 2019, 109, 726–733. [Google Scholar] [CrossRef]
- Zeng, J.; Chen, Y.; Ding, R.; Feng, L.; Fu, Z.; Yang, S.; Deng, X.; Xie, Z.; Zheng, S. Isoliquiritigenin alleviates early brain injury after experimental intracerebral hemorrhage via suppressing ROS- and/or NF-κB-mediated NLRP3 inflammasome activation by promoting Nrf2 antioxidant pathway. J. Neuroinflam. 2017, 14, 119. [Google Scholar] [CrossRef] [Green Version]
- Wei, S.M.; Yan, Z.Z.; Zhou, J. Protective effect of rutin on testicular ischemia-reperfusion injury. J. Pediatr. Surg. 2011, 46, 1419–1424. [Google Scholar] [CrossRef]
- Tahanian, E.; Sanchez, L.A.; Shiao, T.C.; Roy, R.; Annabi, B. Flavonoids targeting of IκB phosphorylation abrogates carcinogen-induced MMP-9 and COX-2 expression in human brain endothelial cells. Drug Des. Dev. Ther. 2011, 5, 299–309. [Google Scholar] [CrossRef] [Green Version]
- Liu, R.; Gao, M.; Qiang, G.F.; Zhang, T.T.; Lan, X.; Ying, J.; Du, G.H. The anti-amnesic effects of luteolin against amyloid beta(25-35) peptide-induced toxicity in mice involve the protection of neurovascular unit. Neuroscience 2009, 162, 1232–1243. [Google Scholar] [CrossRef]
- Song, Q.; Zhao, Y.; Li, Q.; Han, X.; Duan, J. Puerarin protects against iron overload-induced retinal injury through regulation of iron-handling proteins. Biomed. Pharmacother. 2020, 122, 109690. [Google Scholar] [CrossRef]
- Lall, R.K.; Syed, D.N.; Adhami, V.M.; Khan, M.I.; Mukhtar, H. Dietary polyphenols in prevention and treatment of prostate cancer. Int. J. Mol. Sci. 2015, 16, 3350–3376. [Google Scholar] [CrossRef]
- Cucciolla, V.; Borriello, A.; Oliva, A.; Galletti, P.; Zappia, V.; Della Ragione, F. Resveratrol: From basic science to the clinic. Cell Cycle 2007, 6, 2495–2510. [Google Scholar] [CrossRef]
- Kumar, N.; Goel, N. Phenolic acids: Natural versatile molecules with promising therapeutic applications. Biotechnol. Rep. 2019, 24, e00370. [Google Scholar] [CrossRef]
- Espinoza, J.L.; Trung, L.Q.; Inaoka, P.T.; Yamada, K.; An, D.T.; Mizuno, S.; Nakao, S.; Takami, A. The Repeated Administration of Resveratrol Has Measurable Effects on Circulating T-Cell Subsets in Humans. Oxid. Med. Cell. Longev. 2017, 2017, 6781872. [Google Scholar] [CrossRef]
- Wang, D.; Li, S.P.; Fu, J.S.; Zhang, S.; Bai, L.; Guo, L. Resveratrol defends blood-brain barrier integrity in experimental autoimmune encephalomyelitis mice. J. Neurophysiol. 2016, 116, 2173–2179. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.L.; Chang, H.C.; Chen, T.L.; Chang, J.H.; Chiu, W.T.; Lin, J.W.; Chen, R.M. Resveratrol protects against oxidized LDL-induced breakage of the blood-brain barrier by lessening disruption of tight junctions and apoptotic insults to mouse cerebrovascular endothelial cells. J. Nutr. 2010, 140, 2187–2192. [Google Scholar] [CrossRef] [Green Version]
- Kaisar, M.A.; Prasad, S.; Cucullo, L. Protecting the BBB endothelium against cigarette smoke-induced oxidative stress using popular antioxidants: Are they really beneficial? Brain Res. 2015, 1627, 90–100. [Google Scholar] [CrossRef] [Green Version]
- Chen, F.; Qian, L.H.; Deng, B.; Liu, Z.M.; Zhao, Y.; Le, Y.Y. Resveratrol protects vascular endothelial cells from high glucose-induced apoptosis through inhibition of NADPH oxidase activation-driven oxidative stress. CNS Neurosci. Ther. 2013, 19, 675–681. [Google Scholar] [CrossRef]
- Toth, P.; Tarantini, S.; Tucsek, Z.; Ashpole, N.M.; Sosnowska, D.; Gautam, T.; Ballabh, P.; Koller, A.; Sonntag, W.E.; Csiszar, A.; et al. Resveratrol treatment rescues neurovascular coupling in aged mice: Role of improved cerebromicrovascular endothelial function and downregulation of NADPH oxidase. Am. J. Physiol. Heart Circ. Physiol. 2014, 306, H299–H308. [Google Scholar] [CrossRef] [Green Version]
- Arcambal, A.; Taïlé, J.; Couret, D.; Planesse, C.; Veeren, B.; Diotel, N.; Gauvin-Bialecki, A.; Meilhac, O.; Gonthier, M.P. Protective Effects of Antioxidant Polyphenols against Hyperglycemia-Mediated Alterations in Cerebral Endothelial Cells and a Mouse Stroke Model. Mol. Nutr. Food Res. 2020, 64, e1900779. [Google Scholar] [CrossRef]
- Mirshekari Jahangiri, H.; Sarkaki, A.; Farbood, Y.; Dianat, M.; Goudarzi, G. Gallic acid affects blood-brain barrier permeability, behaviors, hippocampus local EEG, and brain oxidative stress in ischemic rats exposed to dusty particulate matter. Environ. Sci. Pollut. Res. Int. 2020, 27, 5281–5292. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Schematic illustration of ROS production in multiple cells that constitute or surround the BBB. (A) ROS production in BBB ECs. BBB ECs generate ROS via XO, COX, NOS, and NOX. The NOX family primarily reduces O2 to O2−. COX generates ROS by catalyzing the conversion of AA to PGs. LOX-1 activates NOX2 through the binding of oxLDL, resulting in the production of ROS. Under pathophysiological conditions, eNOS can produce ONOO− with L-arginine and also generate O2− via conversion to uncoupled eNOS. (B) ROS production in astrocytes. Under a pathophysiological condition, increased activity of NOX2 and NOX4 can result in the generation of O2− and H2O2. ApoE4, LPS, and IFN induce cPLA2 activation via p38MAPK, leading to iNOS activation and ROS generation. Increased expression of iNOS results in the generation of NO, with subsequent aggregation of SOD1 via S-nitrosylation of protein disulfide isomerase. (C) ROS production in microglia. Microglia generate ROS primarily by NOX2. DAMP mediated stimulation of PRRs and pro-inflammatory stimuli can activate MAPKs and NF-κB, resulting in NOX2 activation. Microglia also generate NO via iNOS. The expression of COX in microglia can mediate ROS generation by catalyzing the conversion of AA to PGs. (D) ROS production in neutrophils. Under pathophysiological condition, neutrophil generates ROS primarily by NOX2. Ligation of FcγRs phosphorylates ITAM by SFKs resulting in activation of Syk, which induces SLP76 activation, leading to the activation of NOX2. AA, arachidonic acid; ApoE, Apolipoprotein E; BBB EC, blood-brain barrier endothelial cell; COX, cyclooxygenase; CR3, complement receptor3; cPLA2, cytosolic PLA2 DAMPs: damage-associated molecular patterns; eNOS: endothelial NOS; FcγRs, Fcγ receptors; GPCRs, G protein-coupled receptors; IFN, interferon; iNOS, inducible NOS; ITAM, immunoreceptor tyrosine-based activation motif; LOX, lipoxygenase; LOX-1, lectin-like oxidized low-density lipoprotein receptor-1; LPS, lipopolysaccharide; MAPK, mitogen-activated protein kinase; NADPH, nicotinamide adenine dinucleotide phosphate; NF-kB, nuclear factor kappa-light-chain-enhancer of activated B cells; NOS, nitric oxide synthase; NOX, NADPH oxidase; oxLDL, oxidized low-density lipoprotein; p40, p40phox; p47, p47phox; p67, p67phox; PGs, bioactive prostaglandins; PLA2, phospholipase A2; PLCγ2, phospholipase C γ2; PRRs, pattern recognition receptors; SFK, Src family kinase; SLP76, SH2-domain- containing leukocyte protein of 76 kDa; SOD1, superoxide dismutase; Syk, spleen tyrosine kinase; TLR4, Toll-like receptor 4; TNFR, TNF receptor; XO, xanthine oxidase.
Figure 1.
Schematic illustration of ROS production in multiple cells that constitute or surround the BBB. (A) ROS production in BBB ECs. BBB ECs generate ROS via XO, COX, NOS, and NOX. The NOX family primarily reduces O2 to O2−. COX generates ROS by catalyzing the conversion of AA to PGs. LOX-1 activates NOX2 through the binding of oxLDL, resulting in the production of ROS. Under pathophysiological conditions, eNOS can produce ONOO− with L-arginine and also generate O2− via conversion to uncoupled eNOS. (B) ROS production in astrocytes. Under a pathophysiological condition, increased activity of NOX2 and NOX4 can result in the generation of O2− and H2O2. ApoE4, LPS, and IFN induce cPLA2 activation via p38MAPK, leading to iNOS activation and ROS generation. Increased expression of iNOS results in the generation of NO, with subsequent aggregation of SOD1 via S-nitrosylation of protein disulfide isomerase. (C) ROS production in microglia. Microglia generate ROS primarily by NOX2. DAMP mediated stimulation of PRRs and pro-inflammatory stimuli can activate MAPKs and NF-κB, resulting in NOX2 activation. Microglia also generate NO via iNOS. The expression of COX in microglia can mediate ROS generation by catalyzing the conversion of AA to PGs. (D) ROS production in neutrophils. Under pathophysiological condition, neutrophil generates ROS primarily by NOX2. Ligation of FcγRs phosphorylates ITAM by SFKs resulting in activation of Syk, which induces SLP76 activation, leading to the activation of NOX2. AA, arachidonic acid; ApoE, Apolipoprotein E; BBB EC, blood-brain barrier endothelial cell; COX, cyclooxygenase; CR3, complement receptor3; cPLA2, cytosolic PLA2 DAMPs: damage-associated molecular patterns; eNOS: endothelial NOS; FcγRs, Fcγ receptors; GPCRs, G protein-coupled receptors; IFN, interferon; iNOS, inducible NOS; ITAM, immunoreceptor tyrosine-based activation motif; LOX, lipoxygenase; LOX-1, lectin-like oxidized low-density lipoprotein receptor-1; LPS, lipopolysaccharide; MAPK, mitogen-activated protein kinase; NADPH, nicotinamide adenine dinucleotide phosphate; NF-kB, nuclear factor kappa-light-chain-enhancer of activated B cells; NOS, nitric oxide synthase; NOX, NADPH oxidase; oxLDL, oxidized low-density lipoprotein; p40, p40phox; p47, p47phox; p67, p67phox; PGs, bioactive prostaglandins; PLA2, phospholipase A2; PLCγ2, phospholipase C γ2; PRRs, pattern recognition receptors; SFK, Src family kinase; SLP76, SH2-domain- containing leukocyte protein of 76 kDa; SOD1, superoxide dismutase; Syk, spleen tyrosine kinase; TLR4, Toll-like receptor 4; TNFR, TNF receptor; XO, xanthine oxidase.

Figure 2.
Schematic illustration of NOX2 in priming and activation state. NOX2 is a multi-protein electron transfer system that is made up five components, i.e., gp91phox, p22phox, p40phox, p47phox, and p67phox. In the resting state, the membrane-bound catalytic core of NOX2, composed of gp91phox and p22phox, forms a cytb558, and the regulatory trimeric complex which is composed of p40phox, p47phox, and p67phox resides in the cytosol of cells. Priming state induces translocation of cytb558 to the plasma membrane via granule exocytosis and partial phosphorylation of p47phox, leading to conformational changes. Upon activation, Rac exchanges GDP to GTP for direct binding to p67phox and gp91phox. The regulatory cytosolic subunits translocate to the membranes and binds to cytb588, leading to NOX activation. cytb558, flavocytochrome b558; GDP, guanosine diphosphate; GTP, Guanosine triphosphate; NADP/NADPH, nicotinamide adenine dinucleotide phosphate; NOX, NADPH oxidase; phox, phagocyte oxidase; p40, p40phox; p47, p47phox; p67, p67phox.
Figure 2.
Schematic illustration of NOX2 in priming and activation state. NOX2 is a multi-protein electron transfer system that is made up five components, i.e., gp91phox, p22phox, p40phox, p47phox, and p67phox. In the resting state, the membrane-bound catalytic core of NOX2, composed of gp91phox and p22phox, forms a cytb558, and the regulatory trimeric complex which is composed of p40phox, p47phox, and p67phox resides in the cytosol of cells. Priming state induces translocation of cytb558 to the plasma membrane via granule exocytosis and partial phosphorylation of p47phox, leading to conformational changes. Upon activation, Rac exchanges GDP to GTP for direct binding to p67phox and gp91phox. The regulatory cytosolic subunits translocate to the membranes and binds to cytb588, leading to NOX activation. cytb558, flavocytochrome b558; GDP, guanosine diphosphate; GTP, Guanosine triphosphate; NADP/NADPH, nicotinamide adenine dinucleotide phosphate; NOX, NADPH oxidase; phox, phagocyte oxidase; p40, p40phox; p47, p47phox; p67, p67phox.

Figure 3.
Pathways involved in ROS-induced BBB disruption. ROS can disrupt BBB by pro-inflammatory mediators, MMP activation, cytoskeleton rearrangement, and downregulation of junction integrity. Junction integrity can be downregulated by cleavage of VE-cadherin, phosphorylation, redistribution, and downregulated expression of TJ proteins. BBB disruption can also occur by cytoskeleton rearrangement caused by stress fiber formation, phosphorylation of TJ proteins, FAK, and Faxillin. Pro-inflammatory mediators can be released by adherent neutrophils, activated astrocytes and VEGF from activated microglia and activated astrocytes. MMPs can be released by NETs, activated astrocytes, VEGF, and ECs. BBB, Blood-brain barrier; Cat G, cathepsin G; Ck, chemokine; CLDN-5, cloudin-5; [Ca2+]i, intracellular calcium level; E-Sel, E-selectin; ERK, extracellular signal-regulated kinases; EC, endothelial cell; FAK, focal adhesion kinase; ICAM-1, intra-cellular adhesion molecule-1; JNK, c-Jun N-terminal kinase; MLC, myosin light chain; MLCK, MLC kinase; MMP, matrix metalloproteinase; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; NE, neutrophil elastase; NETs, neutrophil extracellular traps; NLRP3, nucleotide-binding oligomerization domain leucine rich repeat and pyrin do-main-containing protein 3; OCLN, occludin; p38, p38 mitogen-activated protein kinases; PI3K, phosphatidylinositol-3-kinase; P-Sel, P-selectin; PKB, protein kinase B; PKC, protein kinase C; -p, Phosphorylation; ROCK, Rho-associated protein kinase; ROS, reactive oxygen species; TJ, tight junction; TLR4, Toll like receptor 4; VCAM-1, vascular cellular adhesion molecule-1; VE-Cad, VE-cadherin; VEGF, vascular endothelial growth factor; ZO-1, zona occluden-1.
Figure 3.
Pathways involved in ROS-induced BBB disruption. ROS can disrupt BBB by pro-inflammatory mediators, MMP activation, cytoskeleton rearrangement, and downregulation of junction integrity. Junction integrity can be downregulated by cleavage of VE-cadherin, phosphorylation, redistribution, and downregulated expression of TJ proteins. BBB disruption can also occur by cytoskeleton rearrangement caused by stress fiber formation, phosphorylation of TJ proteins, FAK, and Faxillin. Pro-inflammatory mediators can be released by adherent neutrophils, activated astrocytes and VEGF from activated microglia and activated astrocytes. MMPs can be released by NETs, activated astrocytes, VEGF, and ECs. BBB, Blood-brain barrier; Cat G, cathepsin G; Ck, chemokine; CLDN-5, cloudin-5; [Ca2+]i, intracellular calcium level; E-Sel, E-selectin; ERK, extracellular signal-regulated kinases; EC, endothelial cell; FAK, focal adhesion kinase; ICAM-1, intra-cellular adhesion molecule-1; JNK, c-Jun N-terminal kinase; MLC, myosin light chain; MLCK, MLC kinase; MMP, matrix metalloproteinase; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; NE, neutrophil elastase; NETs, neutrophil extracellular traps; NLRP3, nucleotide-binding oligomerization domain leucine rich repeat and pyrin do-main-containing protein 3; OCLN, occludin; p38, p38 mitogen-activated protein kinases; PI3K, phosphatidylinositol-3-kinase; P-Sel, P-selectin; PKB, protein kinase B; PKC, protein kinase C; -p, Phosphorylation; ROCK, Rho-associated protein kinase; ROS, reactive oxygen species; TJ, tight junction; TLR4, Toll like receptor 4; VCAM-1, vascular cellular adhesion molecule-1; VE-Cad, VE-cadherin; VEGF, vascular endothelial growth factor; ZO-1, zona occluden-1.

Table 1.
Effects of flavonoids on oxidative stress-induced BBB dysfunction.
| Antioxidants | Model (Animal/Cell) | Insult | Findings | Reference | |
|---|---|---|---|---|---|
| flavonol | quercetin | HBMEC | Aβ1-40 | BBB permeability↓, ROS↓, SOD↑ | [179] |
| rat CI/R | BCCAO | BBB permeability↓, claudin-5↑, ZO-1↑, MMP-9↓, brain edema↓, β-catenin↑ | [180] | ||
| rat | PCB | occludin↑, claudin-5↑, JAM↑, ZO-1↑, ZO-2↑, | [181] | ||
| bEnd3 | high glucose | NF-kB↓, ROS↓, HO-1↑, MnSOD↑ | [182] | ||
| bEnd3 | high glucose | BBB permeability↓, claudin-5↑, MCP-1↓, E-selectin↓, monocyte adhesion↓, NF-kB↓, iNOS↓, | [183] | ||
| kaempferol | Mouse | LPS | BBB permeability↓, claudin-1↑, occludin↑, CX43↑, MCP-1↓, COX-2↓, iNOS↓, | [184] | |
| rutin | rat CI | photothrombosis | BBB permeability↓, MMP-9↓, SOD↑ | [185] | |
| flavone | baicalein | HMBEC | H2O2 | cell injury↓ | [186] |
| rat CI | MCAO | claudin-5↑, BBB permeability↓, brain edema↓ | |||
| rat ICH | collagenase (IV) | ZO-1↑, BBB permeability↓, ONOO−↓, iNOS↓, brain edema↓, NF-kB↓ | [187] | ||
| luteolin | HBMEC | Aβ1-40 | BBB permeability↓, cell viability↑, NF-kB↓, p38-MAPK↓, COX-2↓, ROS↓ | [188] | |
| vitexin | HBMEC | OGD/R | claudin-5↑, ZO-1↑, MMP-9↓, MMP-2↓, BBB permeability↓, caspase-3↓, ONOO-↓, iNOS↓, eNOS↑ | [189] | |
| flavanols | catechin | rat TBI | CCI | BBB permeability↓, occludin↑, ZO-1↑, iNOS↓, brain edema↓, infarct volume↓ | [190] |
| (-)-EGCG | HCMEC | LPS | BBB permeability↓, claudin-5↑, occludin↑, ICAM-1↓, VCAM-1↓, monocyte adhesion↓, NF-kB↓ | [191] | |
| theaflavin | rat ICH | collagenase (VII) | BBB permeability↓, ROS↓, brain edema↓, CXCL1↓, caspase-1↓, NF-kB↓ | [192] | |
| flavanones | pinocembrin | rat CI/R | OGD/R | BBB permeability↓, brain edema↓ | [193] |
| rat CI | MCAO | occludin↑, ZO-1↑, MMP-9↓, ICAM-1↓, VCAM-1↓, iNOS↓, astrocyte activation | [194] | ||
| hesperidin | mouse CI | MCAO | BBB permeability↓, claudin-5↑, ZO-1↑, brain edema↓ | [195] | |
| bEnd3 | N2 gas | BBB permeability↓, claudin-5↑, ZO-1↑, MMP-3↓, MMP-9↓, ROS↓, FoxO3a↓ | |||
| isoflavone | puerarin | mouse SAH | endovascular perforation | BBB permeability↓, brain edema↓, ROS↓, SIRT3↑, SOD2↑ | [196] |
| chalcone | isoliquiritigenin | rat ICH | collagenase (VI) | BBB leakage↓, ROS↓, Nrf2↑, SOD↑, HO-1↑, brain edema↓, NF-kB↓ | [197] |
Aβ, amyloid beta; BBB, blood-brain barrier; ROS, reactive oxygen species; BCCAO, bilateral common carotid artery occlusion; Bcl-2/Bax, B-cell lymphoma-2/-associated X; bEnd3, mouse brain endothelial cell; CCI, controlled cortical impact; CI/R, cerebral ischemia/reperfusion; CI, cerebral ischemia; COX-2, cyclooxygenase-2; CX43, connexin 43; CXCL, C-X-C motif chemokine ligand; EGCG, epigallocatechin-3-gallate; eNOS; endothelial nitric oxide synthase; FoxO3a, Forkhead box O 3a; GSH, glutathione; hAs, human astrocyte; HBMEC, human brain microvascular endothelial cells; HCMEC, human cerebral microvascular endothelial cell; HO-1, heme oxygenase-1; ICAM-1, intercellular adhesion molecule 1; ICH, intracerebral hemorrhage; iNOS, inducible nitric oxide synthase; JAM, junctional adhesion molecules; LPS, lipopolysaccharide; MAPK, mitogen-activated protein kinases; MCAO, middle cerebral artery occlusion; MCP-1, monocyte chemoattractant protein1; MMP, matrix metalloproteinase; MMP9, Matrix metalloproteinase 9; MnSOD, manganese superoxide dismutase; NF-kB, nuclear factor kappa-light-chain-enhancer of activated B cells; OGD/R, oxygen and glucose deprivation/reoxygenation; PCB, polychlorinated biphenyl; SAH, subarachnoid hemorrhage mice SIRT3, Sirtuin 3; SOD, superoxide dismutase; TBI, traumatic brain injury; VCAM-1, vascular cell adhesion molecules; ZO, zonula occludens.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Kim, Y.; Cho, A.Y.; Kim, H.C.; Ryu, D.; Jo, S.A.; Jung, Y.-S. Effects of Natural Polyphenols on Oxidative Stress-Mediated Blood-Brain Barrier Dysfunction. Antioxidants 2022, 11, 197. https://doi.org/10.3390/antiox11020197
AMA Style
Kim Y, Cho AY, Kim HC, Ryu D, Jo SA, Jung Y-S. Effects of Natural Polyphenols on Oxidative Stress-Mediated Blood-Brain Barrier Dysfunction. Antioxidants. 2022; 11(2):197. https://doi.org/10.3390/antiox11020197
Chicago/Turabian StyleKim, Yeonjae, A Yeon Cho, Hong Cheol Kim, Dajung Ryu, Sangmee Ahn Jo, and Yi-Sook Jung. 2022. "Effects of Natural Polyphenols on Oxidative Stress-Mediated Blood-Brain Barrier Dysfunction" Antioxidants 11, no. 2: 197. https://doi.org/10.3390/antiox11020197
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.