
Sustained Energy Deficit Following Perinatal Asphyxia: A Shift towards the Fructose-2,6-bisphosphatase (TIGAR)-Dependent Pentose Phosphate Pathway and Postnatal Development

 , , and
, , and 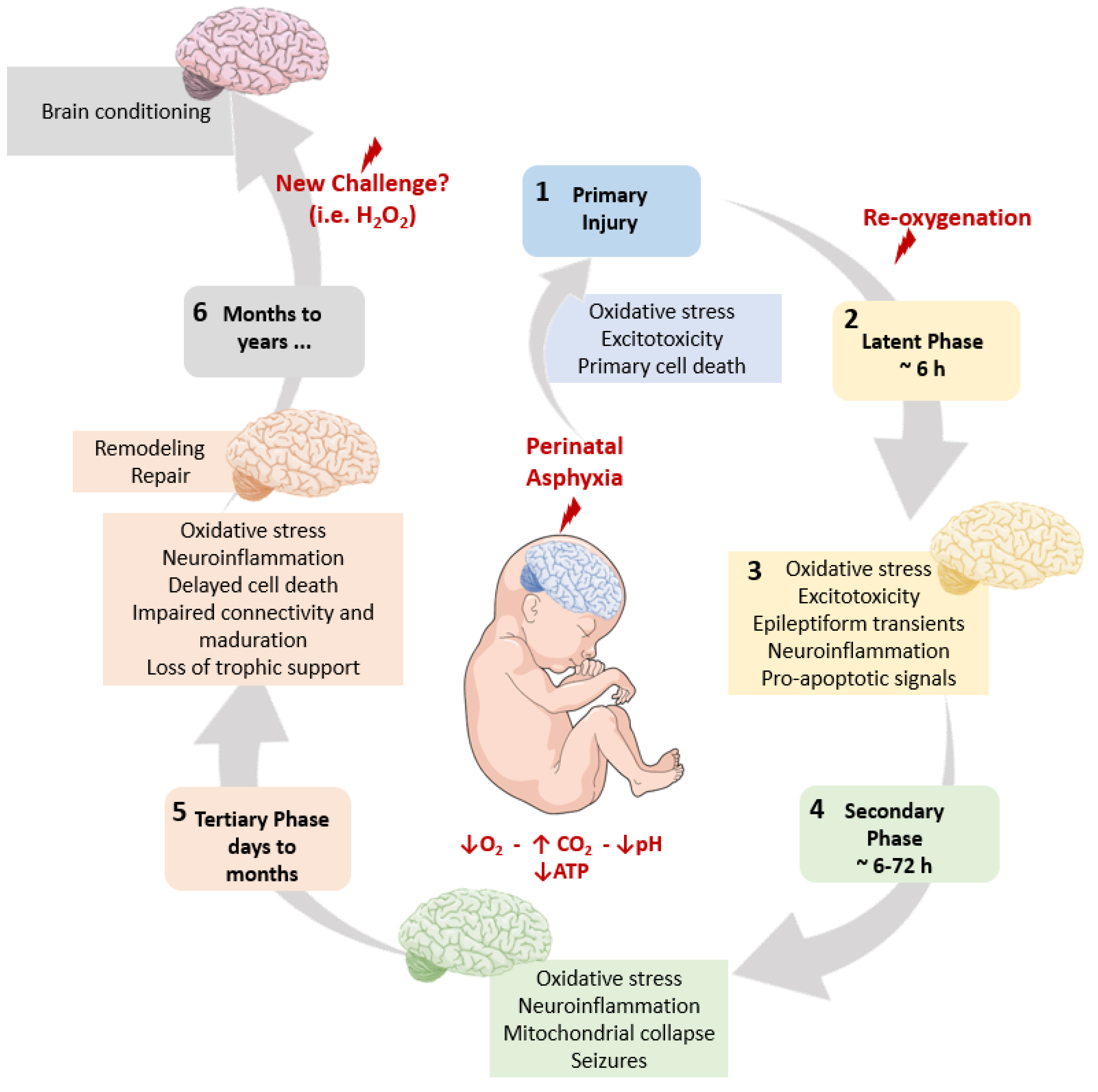{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
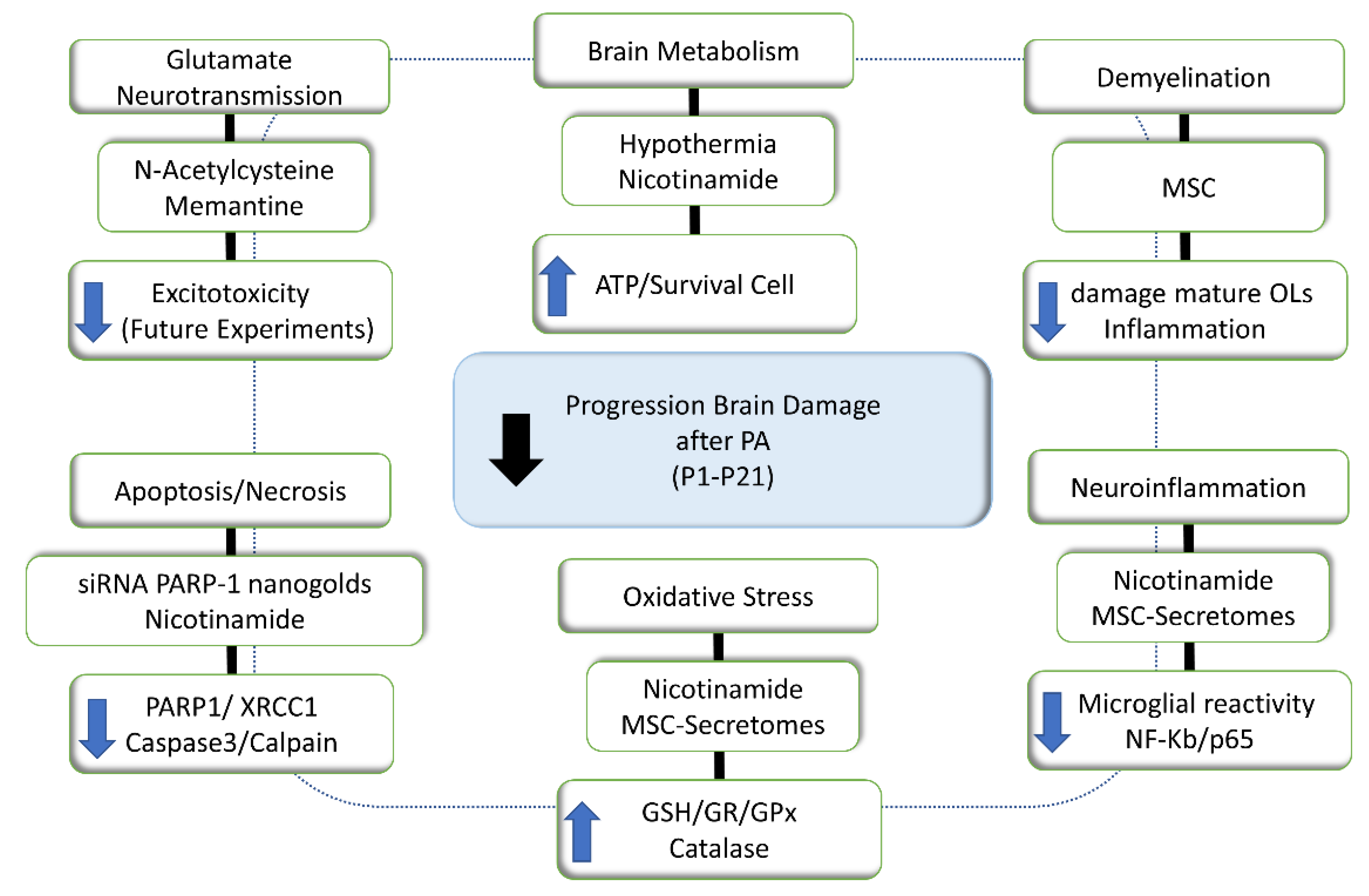
Abstract
:1. Pregnancy and Delivery
2. The Pathophysiological Cascade Elicited by PA
2.1. Free Radical Reactive Species
2.2. A Switch to Anaerobic Glycolysis
2.3. Glutamate and Extrasynaptic Glutamate Receptors
2.4. Mitochondrion: A Main Actor and a Vulnerable Target
2.5. Sentinel Proteins
2.5.1. Poly(ADP-ribose) Polymerases
2.5.2. X-ray Repair Cross Complementing 1 (XRCC1) Protein
2.6. A Schematic Summary of the Metabolic Cascade Elicited by PA
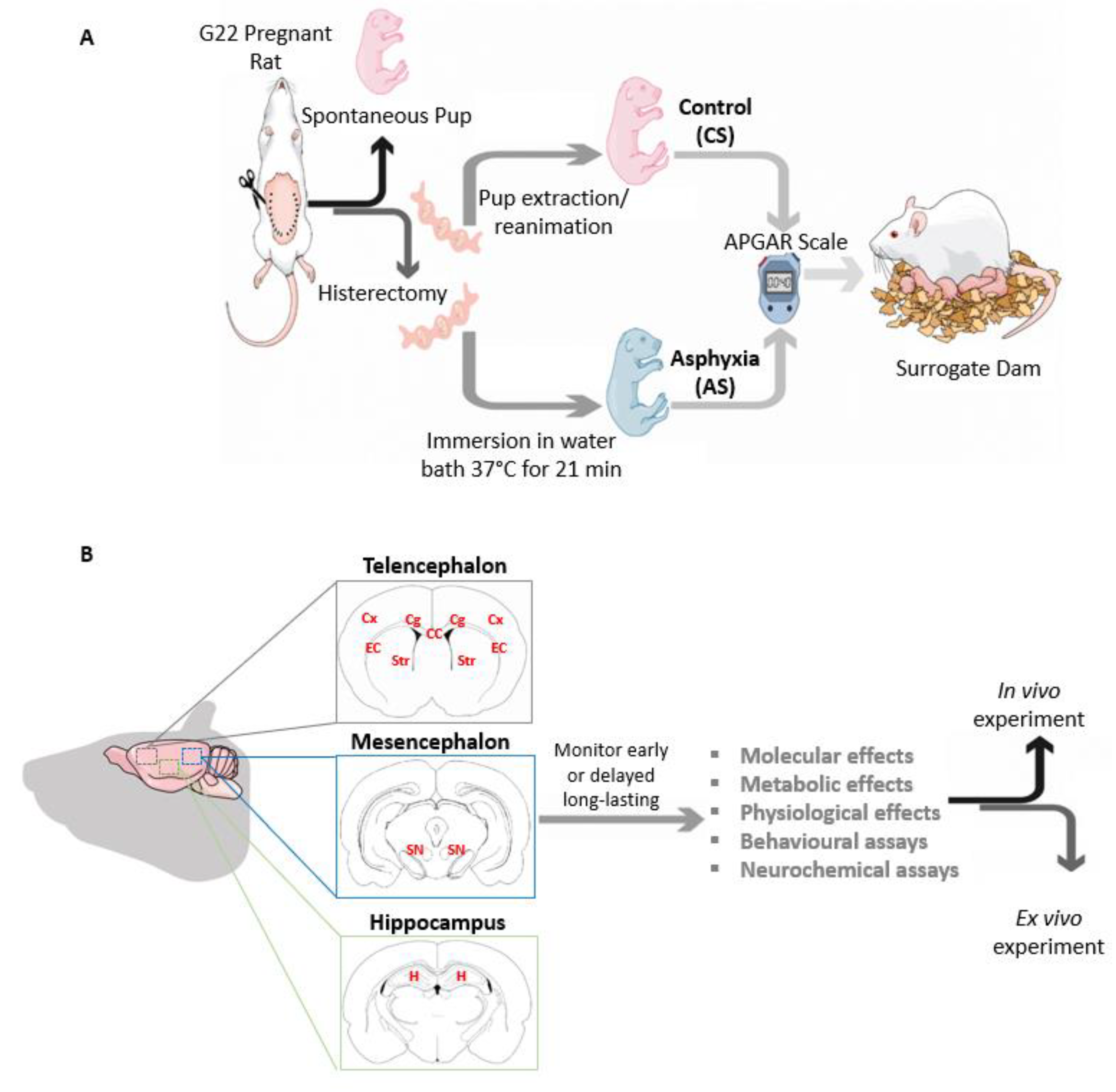
3. An Experimental Model of Global PA in Rats
4. The Energy Crisis Induced by Global PA
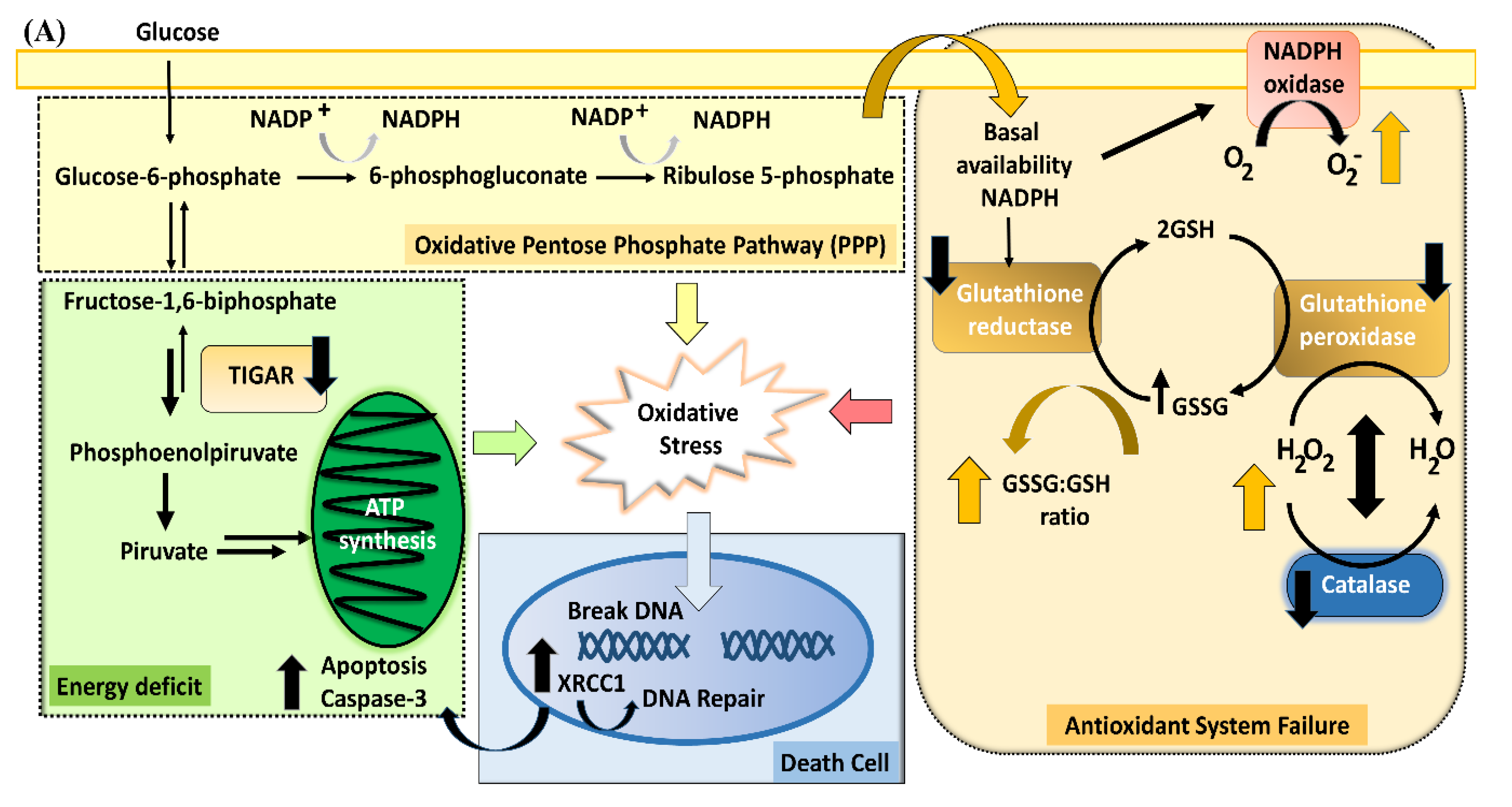
4.1. Redox Homeostasis
4.2. Glutathione (GSSG:GSH Ratio) and Glutathione Reductase
4.3. Impaired Control of Peroxidation
4.4. TIGAR Modulation of the Pentose Phosphate Pathway
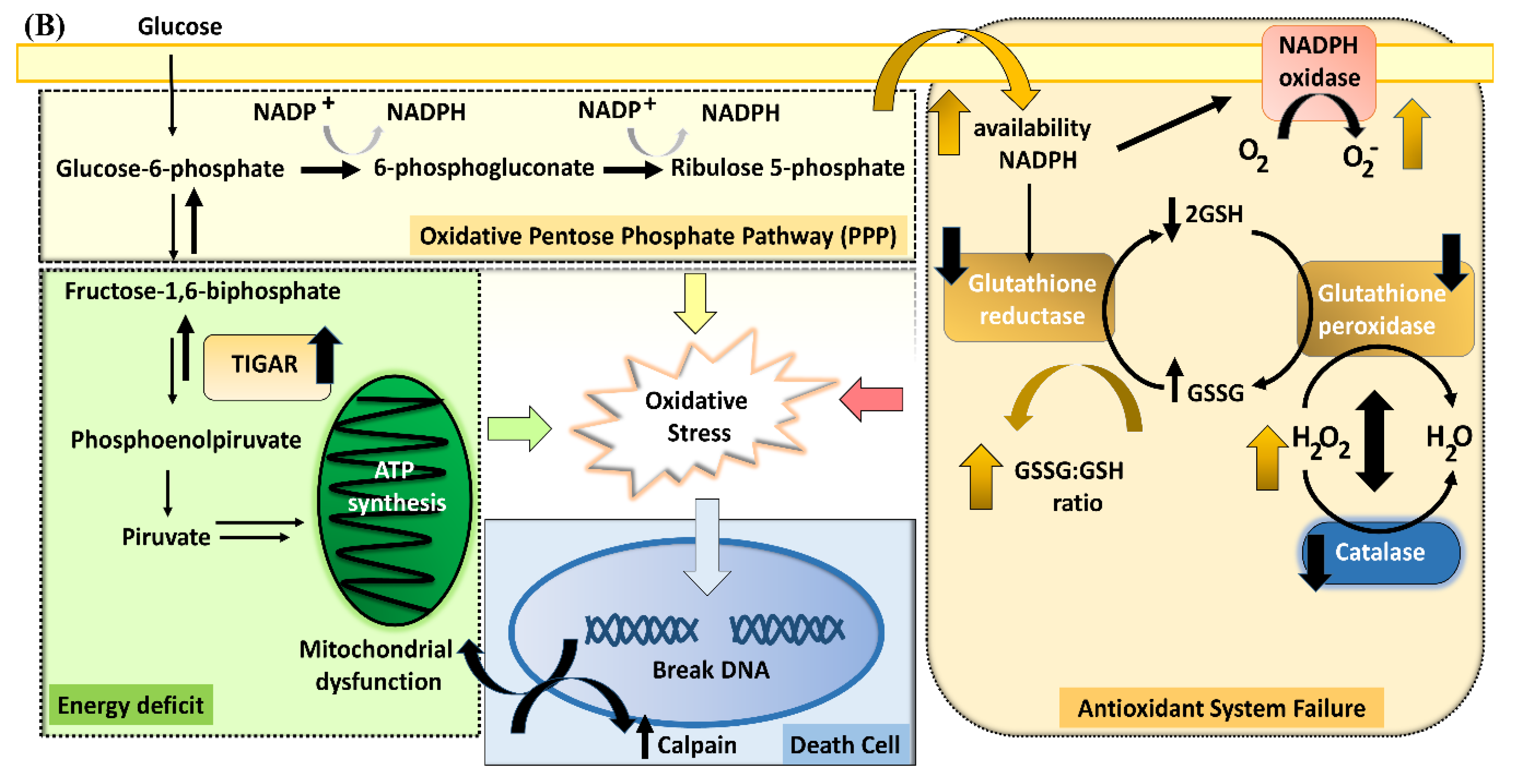
4.5. Delayed Cell Death as a Consequence of Long-Term Impaired Redox Homeostasis
4.6. A Schematic Summary of the Metabolic Cascade Elicited by PA
5. Vulnerability to Recurrent Metabolic Insults
5.1. Organotypic Cultures
5.2. Vulnerability to a Recurrent Metabolic Insult
6. Effect of PA on Oligodendrocyte Maturation
6.1. Effect of PA on Myelination
6.2. Effect of PA on Glial Cells in Telencephalic White Matter
7. Therapeutic Strategies to Prevent the Long-Term Effect of PA
7.1. Hypothermia
7.2. N-Acetylcysteine
7.3. Memantine as a Lead for a Neonatal Protecting Strategy
7.4. PARP-1 as a Target for Neuroprotection
7.5. The Vitamin B3 Family
7.6. Mesenchymal Stem Cell Secretomes (MSC-S)
7.7. A Schematic Summary of Proposed Therapeutic Strategies
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ADP | Adenosine diphosphate |
| AIF | Apoptosis-Inducing Factor |
| AMPA | α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid |
| AS | Asphyxia-exposed rat |
| ATP | Adenosine triphosphate |
| BRCT | breast cancer tumors |
| BER | base excision repair |
| Bcl-2 | B-cell lymphoma 2 |
| Bnip3 | Bcl-2/adenovirus E1B 19kDa interacting protein 3 |
| CLPFFD | peptide Cys-Leu-Pro-Phe-Phe-Asp |
| CNS | central nervous system |
| CS | cesarean-delivery control rat |
| D145 | 1-amino-3,5-dimethyladamantane |
| DBD | DNA-binding domain |
| DFX | deferoxamine |
| DNA | deoxyribonucleic acid |
| ERCC2 | Excision Repair Cross-Complementing Rodent Repair Group 2GluN2B, |
| FRAP | Potassium Ferricyanide Reducing Assay GSH, reduced glutathione |
| G6PGH | glucose-6-phosphate dehydrogenase |
| GFAP | Glial fibrillary acidic protein |
| GLAST (EAAT1) | glutamate transporters glutamate-aspartate transporter (excitatory amino acid transporter 1) |
| GLT-1 (EAAT2) | glutamate transporter-1 (excitatory amino acid transporter 2) |
| GluR2 | glutamate receptor 2 |
| GPx | glutathione peroxidase |
| GR | Glutathione Reductase |
| GSSG | oxidized glutathione |
| H2O2 | Hydrogen peroxide |
| HIF-1α | Hypoxia Inducible Factor-1alpha |
| HK2 | hexokinase 2 |
| HREs | hypoxia-responsive elements |
| Iba-1 | Ionized calcium-binding adaptor molecule 1 |
| i.c.v. | intra-cerebro-ventricular |
| IFN-γ | Interferon-γ |
| IL | interleukin |
| MAP-2 | microtubule-associated protein 2 |
| MBP | myelin basic protein |
| MOMP | outer membrane of the mitochondria |
| MSC | Mesenchymal Stem Cells |
| MSC-S | Mesenchymal Stem Cell Secretomes |
| NAD+ | nicotinamide adenine dinucleotide |
| NADP+ | nicotinamide adenine dinucleotide phosphate |
| NADPH | reduced form of nicotinamide adenine dinucleotide phosphate |
| Nix | NIP3-like protein X |
| NLD | nuclear localization signaling domain |
| NMDARs | N-methyl-D-aspartate receptors |
| nNOS | neuronal nitric oxide synthase |
| NO | nitric oxide |
| Noxa | Phorbol 12-myristate-13-acetate-induced protein 1 |
| NQO1 | NADPH quinone oxidoreductase 1 |
| Nrf2 | nuclear erythroid-related factor 2 |
| OL | oligodendrocyte |
| Olig-1 | oligodendroglial lineage-associated transcription factors-1 |
| P | postnatal day |
| p53 | cellular tumor antigen p53 |
| PA | perinatal asphyxia |
| pADPr | poly(ADP-ribosyl) |
| PARPs | poly(ADP-ribose) polymerases |
| PDGF | platelet-derived growth factor |
| PFK1 | phosphofructokinase 1 |
| PPP | pentose phosphate pathway |
| PTP | permeability transition pore |
| pVHL | von Hippel-Lindau tumor-suppressing factor RNS.1. reactive nitrogen species |
| ROS | reactive oxygen species |
| RT-qPCR | Quantitative reverse transcription PCR |
| SCAP | SREBP cleavage-activating protein |
| Smac/Diablo | Second mitochondria-derived activator of caspase protein/Direct Inhibitor of Apoptosis-Binding protein with Low isoelectric point |
| SREBP | sterol regulatory element binding protein |
| SSSs | small sharp spikes |
| TGF-β1 | Transforming growth factor beta 1 |
| TH | tyrosine hydroxylase |
| TIGAR | Tp53-inducible glycolysis and apoptosis regulator |
| TNFα | Tumor necrosis factor alpha |
| TUNEL | Terminal deoxynucleotidyl transferase dUTP nick end labeling |
| XRCC1 | X-ray Cross complementing Factor 1 |
| ΔΔCT method | Comparative CT (threshold cycles) method |
References
- Romero, R.; Tarca, A.L.; Tromp, G. Insights into the physiology of childbirth using transcriptomics. PLoS Med. 2006, 3, 739–742. [Google Scholar] [CrossRef]
- Soloff, M.S.; Jeng, Y.J.; Izban, M.G.; Sinha, M.; Luxon, B.A.; Stamnes, S.J.; England, S.K. Effects of progesterone treatment on expression of genes involved in uterine quiescence. Reprod. Sci. 2011, 18, 781–797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mondal, S.; Chaudhuri, A.; Kamilya, G.; Santra, D. Fetomaternal outcome in obstructed labor in a peripheral tertiary care hospital. Med. J. Dr. DY Patil Univ. 2013, 6, 146–150. [Google Scholar] [CrossRef]
- Kurinczuk, J.J.; White-koning, M.; Badawi, N. Early Human Development Epidemiology of neonatal encephalopathy and hypoxic—Ischaemic encephalopathy. Early Hum. Dev. 2010, 86, 329–338. [Google Scholar] [CrossRef]
- Basovich, S.N. The role of hypoxia in mental development and in the treatment of mental disorders: A review. Biosci. Trends 2010, 4, 288–296. [Google Scholar] [PubMed]
- Martinello, K.; Hart, A.R.; Mitra, S.; Robertson, N.J. Management and investigation of neonatal encephalopathy: 2017 update. Arch. Dis. Child. Fetal Neonatal Ed. 2017, 102, 346–358. [Google Scholar] [CrossRef]
- Ekwochi, U.; Asinobi, N.I.; Osuorah, C.D.; Ndu, I.K.; Ifediora, C.; Amadi, O.F.; Iheji, C.C.; Orjioke, C.J.; Okenwa, W.O.; Okeke, B.I. Incidence and Predictors of Mortality Among Newborns With Perinatal Asphyxia: A 4-Year Prospective Study of Newborns Delivered in Health Care Facilities in Enugu, South-East Nigeria. Clin. Med. Insights Pediatr. 2017, 11, 117955651774664. [Google Scholar] [CrossRef]
- Odd, D.; Lewis, G.; Whitelaw, A.; Gunnell, D. Resuscitation at birth and cognition at 8 years of age: A cohort study. Lancet 2009, 373, 1615–1622. [Google Scholar] [CrossRef]
- Douglas-Escobar, M.; Weiss, M.D. Hypoxic-Ischemic Encephalopathy. JAMA Pediatr. 2015, 169, 397. [Google Scholar] [CrossRef]
- Graham, H.K.; Rosenbaum, P.; Paneth, N.; Dan, B.; Lin, J. Cerebral palsy. Nat. Rev. Dis. Prim. 2016, 7, 15082. [Google Scholar] [CrossRef] [PubMed]
- Sahin, M.; Sur, M. Genes, circuits, and precision therapies for autism and related neurodevelopmental disorders. Science 2015, 350. [Google Scholar] [CrossRef] [Green Version]
- Jain, V.; Dhawan, A. Prognostic modeling in pediatric acute liver failure. Liver Transplant. 2016, 22, 1418–1430. [Google Scholar] [CrossRef] [PubMed]
- Pasternak, D.R. Scalp hypothermia resources sought. Oncol. Nurs. Forum 1991, 18, 476. [Google Scholar] [PubMed]
- Cowan, F.; Rutherford, M.; Groenendaal, F.; Eken, P.; Mercuri, E.; Bydder, G.M.; Meiners, L.C.; Dubowitz, L.M.S.; de Vries, L.S. Origin and timing of brain lesions in term infants with neonatal encephalopathy. Lancet 2003, 361, 736–742. [Google Scholar] [CrossRef]
- Miller, S.L.; Yan, E.B.; Castillo-Meléndez, M.; Jenkin, G.; Walker, D.W. Melatonin provides neuroprotection in the late-gestation fetal sheep brain in response to umbilical cord occlusion. Dev. Neurosci. 2005, 27, 200–210. [Google Scholar] [CrossRef] [PubMed]
- Marriott, A.L.; Rojas-Mancilla, E.; Morales, P.; Herrera-Marschitz, M.; Tasker, R.A. Models of progressive neurological dysfunction originating early in life. Prog. Neurobiol. 2017, 155, 2–20. [Google Scholar] [CrossRef] [PubMed]
- Perez-Lobos, R.; Lespay-Rebolledo, C.; Tapia-Bustos, A.; Palacios, E.; Vío, V.; Bustamante, D.; Morales, P.; Herrera-Marschitz, M. Vulnerability to a Metabolic Challenge Following Perinatal Asphyxia Evaluated by Organotypic Cultures: Neonatal Nicotinamide Treatment. Neurotox. Res. 2017, 32, 426–443. [Google Scholar] [CrossRef]
- Low, J.A. Determining the contribution of asphyxia to brain damage in the neonate. J. Obs. Gynaecol. Res. 2004, 30, 276–286. [Google Scholar] [CrossRef]
- Wang, G.L.; Jiang, B.H.; Rue, E.A.; Semenza, G.L. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc. Natl. Acad. Sci. USA 1995, 92, 5510–5514. [Google Scholar] [CrossRef] [Green Version]
- Ke, Q.; Costa, M. Hypoxia-inducible factor-1 (HIF-1). Mol. Pharmacol. 2006, 70, 1469–1480. [Google Scholar] [CrossRef] [PubMed]
- Vangeison, G.; Carr, D.; Federoff, H.J.; Rempe, D.A. The good, the bad, and the cell type-specific roles of hypoxia inducible factor-1 alpha in neurons and astrocytes. J. Neurosci. 2008, 28, 1988–1993. [Google Scholar] [CrossRef]
- Bruick, R.K. Expression of the gene encoding the proapoptotic Nip3 protein is induced by hypoxia. Proc. Natl. Acad. Sci. USA 2000, 97, 9082–9087. [Google Scholar] [CrossRef] [Green Version]
- Sowter, H.M.; Ratcliffe, P.J.; Watson, P.; Greenberg, A.H.; Harris, A.L. HIF-1-dependent regulation of hypoxic induction of the cell death factors BNIP3 and NIX in human tumors. Cancer Res. 2001, 61, 6669–6673. [Google Scholar]
- Martí, J.M.; Garcia-Diaz, A.; Delgado-Bellido, D.; O’Valle, F.; González-Flores, A.; Carlevaris, O.; Rodríguez-Vargas, J.M.; Amé, J.C.; Dantzer, F.; King, G.L.; et al. Selective modulation by PARP-1 of HIF-1α-recruitment to chromatin during hypoxia is required for tumor adaptation to hypoxic conditions. Redox Biol. 2021, 41, 101885. [Google Scholar] [CrossRef] [PubMed]
- Robertson, S.J.; Kania, K.D.; Hladky, S.B.; Barrand, M.A. P-glycoprotein expression in immortalised rat brain endothelial cells: Comparisons following exogenously applied hydrogen peroxide and after hypoxia-reoxygenation. J. Neurochem. 2009, 111, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.A.; Zachary, J.F. Mechanisms and Morphology of Cellular Injury, Adaptation, and Death. Pathol. Basis Vet. Dis. 2017, 2–43.e19. [Google Scholar] [CrossRef]
- Herrera-Marschitz, M.; You, Z.B.; Goiny, M.; Meana, J.J.; Silveira, R.; Godukhin, O.V.; Chen, Y.; Espinoza, S.; Pettersson, E.; Loidl, C.F.; et al. On the origin of extracellular glutamate levels monitored in the basal ganglia of the rat by in vivo microdialysis. J. Neurochem. 1996, 66, 1726–1735. [Google Scholar] [CrossRef]
- Danbolt, N.C. Glutamate uptake. Prog. Neurobiol. 2001, 65, 1–105. [Google Scholar] [CrossRef]
- Rose, E.M.; Koo, J.C.P.; Antflick, J.E.; Ahmed, S.M.; Angers, S.; Hampson, D.R. Glutamate transporter coupling to Na,K-ATPase. J. Neurosci. 2009, 29, 8143–8155. [Google Scholar] [CrossRef]
- Trotti, D.; Rizzini, B.L.; Rossi, D.; Haugeto, O.; Racagni, G.; Danbolt, N.C.; Volterra, A. Neuronal and glial glutamate transporters possess an SH-based redox regulatory mechanism. Eur. J. Neurosci. 1997, 9, 1236–1243. [Google Scholar] [CrossRef]
- Petralia, R.S. Distribution of extrasynaptic NMDA receptors on neurons. Sci. World J. 2012, 2012, 267120. [Google Scholar] [CrossRef] [PubMed]
- Vizi, E.S.; Kisfali, M.; Lőrincz, T. Role of nonsynaptic GluN2B-containing NMDA receptors in excitotoxicity: Evidence that fluoxetine selectively inhibits these receptors and may have neuroprotective effects. Brain Res. Bull. 2013, 93, 32–38. [Google Scholar] [CrossRef]
- Stanika, R.I.; Pivovarova, N.B.; Brantner, C.A.; Watts, C.A.; Winters, C.A.; Andrews, S.B. Coupling diverse routes of calcium entry to mitochondrial dysfunction and glutamate excitotoxicity. Proc. Natl. Acad. Sci. USA 2009, 106, 9854–9859. [Google Scholar] [CrossRef] [Green Version]
- Moncada, S.; Bolaños, J.P. Nitric oxide, cell bioenergetics and neurodegeneration. J. Neurochem. 2006, 97, 1676–1689. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.-J.; Choi, S.-Y.; Koh, J.-Y. The role of NADPH oxidase, neuronal nitric oxide synthase and poly(ADP ribose) polymerase in oxidative neuronal death induced in cortical cultures by brain-derived neurotrophic factor and neurotrophin-4/5. J. Neurochem. 2002, 82, 894–902. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Pinzón, M.A.; Sick, T.J.; Rosenthal, M. Mechanism(s) of mitochondrial hyperoxidation after global cerebral ischemia. Adv. Exp. Med. Biol. 1999, 471, 175–180. [Google Scholar] [CrossRef]
- Parker, W.D.J.; Parks, J.K.; Swerdlow, R.H. Complex I deficiency in Parkinson’s disease frontal cortex. Brain Res. 2008, 1189, 215–218. [Google Scholar] [CrossRef] [Green Version]
- Circu, M.L.; Aw, T.Y. Reactive oxygen species, cellular redox systems, and apoptosis. Free Radic. Biol. Med. 2010, 48, 749–762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ying, W.; Alano, C.C.; Garnier, P.; Swanson, R.A. NAD+ as a metabolic link between DNA damage and cell death. J. Neurosci. Res. 2005, 79, 216–223. [Google Scholar] [CrossRef]
- Herrera-Marschitz, M.; Neira-Pena, T.; Rojas-Mancilla, E.; Espina-Marchant, P.; Esmar, D.; Perez, R.; Muñoz, V.; Gutierrez-Hernandez, M.; Rivera, B.; Simola, N.; et al. Perinatal asphyxia: CNS development and deficits with delayed onset. Front. Cell. Neurosci. 2014, 8, 47. [Google Scholar] [CrossRef] [Green Version]
- Alvarez-Gonzalez, R.; Jacobson, M.K. Characterization of polymers of adenosine diphosphate ribose generated in vitro and in vivo. Biochemistry 1987, 26, 3218–3224. [Google Scholar] [CrossRef] [PubMed]
- Amé, J.-C.; Spenlehauer, C.; de Murcia, G. The PARP superfamily. BioEssays 2004, 26, 882–893. [Google Scholar] [CrossRef] [PubMed]
- Althaus, F.R.; Richter, C. Poly-ADP-Ribosylation in the Recovery of Mammalian Cells from DNA Damage. In ADP-Ribosylation of Proteins: Enzymology and Biological Significance; Springer: Berlin/Heidelberg, Germany, 1987; pp. 66–92. ISBN 978-3-642-83077-8. [Google Scholar]
- Mazen, A.; Murcia, J.M.; Molinete, M.; Simonin, F.; Gradwohl, G.; Poirier, G.; de Murcia, G. Poly(ADP-ribose)polymerase: A novel finger protein. Nucleic Acids Res. 1989, 17, 4689–4698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, S.S.; Perez-Polo, J.R.; Noppens, K.M.; Grafe, M.R. Biphasic changes in the levels of poly(ADP-ribose) polymerase-1 and caspase 3 in the immature brain following hypoxia-ischemia. Int. J. Dev. Neurosci. Off. J. Int. Soc. Dev. Neurosci. 2005, 23, 673–686. [Google Scholar] [CrossRef]
- Zhang, H.; Somasundaram, K.; Peng, Y.; Tian, H.; Zhang, H.; Bi, D.; Weber, B.L.; El-Deiry, W.S. BRCA1 physically associates with p53 and stimulates its transcriptional activity. Oncogene 1998, 16, 1713–1721. [Google Scholar] [CrossRef] [Green Version]
- Langelier, M.-F.; Planck, J.L.; Roy, S.; Pascal, J.M. Structural Basis for DNA Damage—Dependent Poly(ADP-ribosyl) ation by Human PARP-1. Science 2012, 336, 728–732. [Google Scholar] [CrossRef] [Green Version]
- London, R.E. The structural basis of XRCC1-mediated DNA repair. DNA Repair 2015, 30, 90–103. [Google Scholar] [CrossRef] [Green Version]
- D’Amours, D.; Desnoyers, S.; D’Silva, I.; Poirier, G.G. Poly(ADP-ribosyl) ation reactions in the regulation of nuclear functions. Biochem. J. 1999, 342, 249–268. [Google Scholar] [CrossRef]
- Miwa, M.; Ida, C.; Yamashita, S.; Tanaka, M.; Fujisawa, J. Poly(ADP-ribose): Structure, Physicochemical Properties and Quantification In Vivo, with Special Reference to Poly(ADP-ribose) Binding Protein Modules. Curr. Protein Pept. Sci. 2016, 17, 683–692. [Google Scholar] [CrossRef]
- Berger, N.A. Poly(ADP-Ribose) in the Cellular Response to DNA Damage. Radiat. Res. 1985, 101, 4–15. [Google Scholar] [CrossRef]
- Hong, S.J.; Dawson, T.M.; Dawson, V.L. Nuclear and mitochondrial conversations in cell death: PARP-1 and AIF signaling. Trends Pharmacol. Sci. 2004, 25, 259–264. [Google Scholar] [CrossRef] [PubMed]
- Martin-Oliva, D.; Aguilar-Quesada, R.; O’Valle, F.; Muñoz-Gámez, J.A.; Martínez-Romero, R.; García del Moral, R.; Ruiz de Almodóvar, J.M.; Villuendas, R.; Piris, M.A.; Oliver, F.J. Inhibition of Poly(ADP-Ribose) Polymerase Modulates Tumor-Related Gene Expression, Including Hypoxia-Inducible Factor-1 Activation, during Skin Carcinogenesis. Cancer Res. 2006, 66, 5744–5756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez-Flores, A.; Aguilar-Quesada, R.; Siles, E.; Pozo, S.; Rodríguez-Lara, M.I.; López –Jiménez, L.; López-Rodríguez, M.; Peralta-Leal, A.; Villar, D.; Martín-Oliva, D.; et al. Interaction between PARP-1 and HIF-2α in the hypoxic response. Oncogene 2014, 33, 891–898. [Google Scholar] [CrossRef] [Green Version]
- El-Khamisy, S.F.; Masutani, M.; Suzuki, H.; Caldecott, K.W. A requirement for PARP-1 for the assembly or stability of XRCC1 nuclear foci at sites of oxidative DNA damage. Nucleic Acids Res. 2003, 31, 5526–5533. [Google Scholar] [CrossRef] [Green Version]
- Wei, L.; Nakajima, S.; Hsieh, C.-L.; Kanno, S.; Masutani, M.; Levine, A.S.; Yasui, A.; Lan, L. Damage response of XRCC1 at sites of DNA single strand breaks is regulated by phosphorylation and ubiquitylation after degradation of poly(ADP-ribose). J. Cell Sci. 2013, 126, 4414–4423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiappe-Gutierrez, M.; Kitzmueller, E.; Labudova, O.; Fuerst, G.; Hoeger, H.; Hardmeier, R.; Nohl, H.; Gille, L.; Lubec, B. mRNA levels of the hypoxia inducible factor (HIF-1) and DNA repair genes in perinatal asphyxia of the rat. Life Sci. 1998, 63, 1157–1167. [Google Scholar] [CrossRef]
- Fujimura, M.; Morita-Fujimura, Y.; Sugawara, T.; Chan, P.H. Early Decrease of XRCC1, a DNA Base Excision Repair Protein, May Contribute to DNA Fragmentation After Transient Focal Cerebral Ischemia in Mice. Stroke 1999, 30, 2456–2463. [Google Scholar] [CrossRef] [Green Version]
- Barkhuizen, M.; van den Hove, D.L.A.; Vles, J.S.H.; Steinbusch, H.W.M.; Kramer, B.W.; Gavilanes, A.W.D. 25 Years of ReseArch. on Global Asphyxia in the Immature Rat Brain. Neurosci. Biobehav. Rev. 2017, 75, 166–182. [Google Scholar] [CrossRef]
- Hamdy, N.; Eide, S.; Sun, H.-S.; Feng, Z.-P. Animal models for neonatal brain injury induced by hypoxic ischemic conditions in rodents. Exp. Neurol. 2020, 334, 113457. [Google Scholar] [CrossRef]
- Llanos, A.J.; Ebensperger, G.; Herrera, E.A.; Reyes, R.V.; Cabello, G.; Díaz, M.; Giussani, D.A.; Parer, J.T. The heme oxygenase–carbon monoxide system in the regulation of cardiorespiratory function at high altitude. Respir. Physiol. Neurobiol. 2012, 184, 186–191. [Google Scholar] [CrossRef] [PubMed]
- Bjelke, B.; Andersson, K.; Ögren, S.O.; Bolme, P. Asphyctic lesion: Proliferation of tyrosine hydroxylase-immunoreactive nerve cell bodies in the rat substantia nigra and functional changes in dopamine neurotransmission. Brain Res. 1991, 543, 1–9. [Google Scholar] [CrossRef]
- Andersson, K.; Bjelke, B.; Bolme, P.; Ögren, S.O. Asphyxia-induced lesion of the rat hippocampus (CA1, CA3) and the nigro-striatal dopamine system. In Hypoxia and Ischemia. CNS; Humboldt University of Berlin: Berlin, Germany, 1992; Volume 41, pp. 71–76. [Google Scholar]
- Herrera-Marschitz, M.; Loidl, C.F.; Andersson, K.; Ungerstedt, U. Prevention of mortality induced by perinatal asphyxia: Hypothermia or glutamate antagonism? Amino Acids 1993, 5, 413–419. [Google Scholar] [CrossRef] [PubMed]
- Herrera-Marschitz, M.; Morales, P.; Leyton, L.; Bustamante, D.; Klawitter, V.; Espina-Marchant, P.; Allende, C.; Lisboa, F.; Cunich, G.; Jara-Cavieres, A.; et al. Perinatal asphyxia: Current status and approaches towards neuroprotective strategies, with focus on sentinel proteins. Neurotox. Res. 2011, 19, 603–627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dell’Anna, E.; Chen, Y.; Engidawork, E.; Andersson, K.; Lubec, G.; Luthman, J.; Herrera-Marschitz, M. Delayed neuronal death following perinatal asphyxia in rat. Exp. Brain Res. 1997, 115, 105–115. [Google Scholar] [CrossRef]
- Morales, P.; Simola, N.; Bustamante, D.; Lisboa, F.; Fiedler, J.; Gebicke-Haerter, P.J.; Morelli, M.; Tasker, R.A.; Herrera-Marschitz, M. Nicotinamide prevents the long-term effects of perinatal asphyxia on apoptosis, non-spatial working memory and anxiety in rats. Exp. Brain Res. 2010, 202, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Tapia-bustos, A.; Lespay-rebolledo, C.; Vío, V.; Pérez-lobos, R.; Casanova-ortiz, E.; Ezquer, F.; Herrera-marschitz, M.; Morales, P. Neonatal mesenchymal stem cell treatment improves myelination impaired by global perinatal asphyxia in rats. Int. J. Mol. Sci. 2021, 22, 3275. [Google Scholar] [CrossRef]
- Lespay-Rebolledo, C.; Perez-Lobos, R.; Tapia-Bustos, A.; Vio, V.; Morales, P.; Herrera-Marschitz, M. Regionally Impaired Redox Homeostasis in the Brain of Rats Subjected to Global Perinatal Asphyxia: Sustained Effect up to 14 Postnatal Days. Neurotox. Res. 2018, 34, 660–676. [Google Scholar] [CrossRef]
- Morales, P.; Klawitter, V.; Johansson, S.; Huaiquín, P.; Barros, V.G.; Avalos, A.M.; Fiedler, J.; Bustamante, D.; Gomez-Urquijo, S.; Goiny, M.; et al. Perinatal asphyxia impairs connectivity and dopamine neurite branching in organotypic triple culture from rat substantia nigra, neostriatum and neocortex. Neurosci. Lett. 2003, 348, 175–179. [Google Scholar] [CrossRef]
- Klawitter, V.; Morales, P.; Bustamante, D.; Gomez-Urquijo, S.; Hökfelt, T.; Herrera-Marschitz, M. Plasticity of basal ganglia neurocircuitries following perinatal asphyxia: Effect of nicotinamide. Exp. Brain Res. 2007, 180, 139–152. [Google Scholar] [CrossRef] [Green Version]
- Romijn, H.J.; Hofman, M.A.; Gramsbergen, A. At what age is the developing cerebral cortex of the rat comparable to that of the full-term newborn human baby? Early Hum. Dev. 1991, 26, 61–67. [Google Scholar] [CrossRef]
- Craig, A.; Ling Luo, N.; Beardsley, D.J.; Wingate-Pearse, N.; Walker, D.W.; Hohimer, A.R.; Back, S.A. Quantitative analysis of perinatal rodent oligodendrocyte lineage progression and its correlation with human. Exp. Neurol. 2003, 181, 231–240. [Google Scholar] [CrossRef]
- Engidawork, E.; Chen, Y.; Dell’Anna, E.; Goiny, M.; Lubec, G.; Ungerstedt, U.; Andersson, K.; Herrera-Marschitz, M. Effect of Perinatal Asphyxia on Systemic and Intracerebral pH and Glycolysis Metabolism in the Rat. Exp. Neurol. 1997, 145, 390–396. [Google Scholar] [CrossRef]
- Engidawork, E.; Loidl, F.; Chen, Y.; Kohlhauser, C.; Stoeckler, S.; Dell’Anna, E.; Lubec, B.; Lubec, G.; Goiny, M.; Gross, J.; et al. Comparison between hypothermia and glutamate antagonism treatments on the immediate outcome of perinatal asphyxia. Exp. Brain Res. 2001, 138, 375–383. [Google Scholar] [CrossRef] [PubMed]
- Lijun Lin, M.; Perryman, B.; Friedman, D.; Roberts, R.; Ma, T.S. Determination of the catalytic site of creatine kinase by site-directed mutagenesis. Biochim. Biophys. Acta-Protein Struct. Mol. Enzymol. 1994, 1206, 97–104. [Google Scholar] [CrossRef]
- Lubec, B.; Chiappe-Gutierrez, M.; Hoeger, H.; Kitzmueller, E.; Lubec, G. Glucose Transporters, Hexokinase, and Phosphofructokinase in Brain of Rats with Perinatal Asphyxia. Pediatr. Res. 2000, 47, 84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tachikawa, M.; Hosoya, K.-I.; Ohtsuki, S.; Terasaki, T. A Novel Relationship Between Creatine Transport at the Blood-Brain and Blood-Retinal Barriers, Creatine Biosynthesis, And its Use for Brain and Retinal Energy Homeostasis. In Creatine and Creatine Kinase in Health and Disease; Salomons, G.S., Wyss, M., Eds.; Springer: Dordrecht, The Netherlands, 2007; pp. 83–98. ISBN 978-1-4020-6486-9. [Google Scholar]
- Ireland, Z.; Dickinson, H.; Snow, R.; Walker, D.W. Maternal creatine: Does it reach the fetus and improve survival after an acute hypoxic episode in the spiny mouse (Acomys cahirinus)? Am. J. Obstet. Gynecol. 2008, 198, 431.e1–431.e6. [Google Scholar] [CrossRef]
- Ireland, Z.; Castillo-Melendez, M.; Dickinson, H.; Snow, R.; Walker, D.W. A maternal diet supplemented with creatine from mid-pregnancy protects the newborn spiny mouse brain from birth hypoxia. Neuroscience 2011, 194, 372–379. [Google Scholar] [CrossRef]
- Bågenholm, R.; Nilsson, U.A.; Kjellmer, I. Formation of free radicals in hypoxic ischemic brain damage in the neonatal rat, assessed by an endogenous spin trap and lipid peroxidation. Brain Res. 1997, 773, 132–138. [Google Scholar] [CrossRef]
- Ikeda, T.; Koo, H.; Xia, Y.X.; Ikenoue, T.; Choi, B.H. Bimodal upregulation of glial cell line-derived neurotrophic factor (GDNF) in the neonatal rat brain following ischemic/hypoxic injury. Int. J. Dev. Neurosci. 2002, 20, 555–562. [Google Scholar] [CrossRef]
- Vannucci, S.J.; Hagberg, H. Hypoxia–ischemia in the immature brain. J. Exp. Biol. 2004, 207, 3149–3154. [Google Scholar] [CrossRef] [Green Version]
- Capani, F.; Loidl, C.F.; Aguirre, F.; Piehl, L.; Facorro, G.; Hager, A.; De Paoli, T.; Farach, H.; Pecci-Saavedra, J. Changes in reactive oxygen species (ROS) production in rat brain during global perinatal asphyxia: An ESR study. Brain Res. 2001, 914, 204–207. [Google Scholar] [CrossRef]
- Capani, F.; Loidl, C.F.; Piehl, L.L.; Facorro, G.; De Paoli, T.; Hager, A. Long term production of reactive oxygen species during perinatal asphyxia in the rat central nervous system: Effects of hypothermia. Int. J. Neurosci. 2003, 113, 641–654. [Google Scholar] [CrossRef]
- Kumar, A.; Pant, P.; Basu, S.; Rao, G.R.K.; Khanna, H.D. Oxidative Stress in Neonatal Hyperbilirubinemia. J. Trop. Pediatr. 2007, 53, 69–71. [Google Scholar] [CrossRef]
- Seema, S.; Goel, A.; Mamta, P.; Sumitra, B.; Shah, S. Correlation of oxidative stress biomarker and serum marker of brain injury in hypoxic ischemic encephalopathy. Int. J. Med. Appl. Sci. 2014, 3, 106–115. [Google Scholar]
- Perrone, S.; Negro, S.; Tataranno, M.L.; Buonocore, G. Oxidative stress and antioxidant strategies in newborns. J. Matern. Neonatal Med. 2010, 23, 63–65. [Google Scholar] [CrossRef] [PubMed]
- Gane, B.; Nandakumar, S.; Bhat, B.; Rao, R.; Adhisivam, B.; Joy, R.; Prasad, P.; Shruti, S. Biochemical marker as predictor of outcome in perinatal asphyxia. Curr. Pediatr. Res. 2013, 17, 63–66. [Google Scholar]
- Qin, X.; Cheng, J.; Zhong, Y.; Mahgoub, O.K.; Akter, F.; Fan, Y.; Aldughaim, M.; Xie, Q.; Qin, L.; Gu, L.; et al. Mechanism and treatment related to oxidative stress in neonatal hypoxic-ischemic encephalopathy. Front. Mol. Neurosci. 2019, 12, 88. [Google Scholar] [CrossRef] [PubMed]
- Morales, P.; Fiedler, J.L.; Andrés, S.; Berrios, C.; Huaiquín, P.; Bustamante, D.; Cardenas, S.; Parra, E.; Herrera-Marschitz, M. Plasticity of hippocampus following perinatal asphyxia: Effects on postnatal apoptosis and neurogenesis. J. Neurosci. Res. 2008, 86, 2650–2662. [Google Scholar] [CrossRef]
- Homi, H.M.; Freitas, J.J.S.; Curi, R.; Velasco, I.T.; Junior, B.A.S. Changes in superoxide dismutase and catalase activities of rat brain regions during early global transient ischemia/reperfusion. Neurosci. Lett. 2002, 333, 37–40. [Google Scholar] [CrossRef]
- Aquilano, K.; Baldelli, S.; Ciriolo, M.R. Glutathione: New roles in redox signalling for an old antioxidant. Front. Pharmacol. 2014, 5, e1-12. [Google Scholar] [CrossRef] [Green Version]
- Lespay-Rebolledo, C.; Tapia-Bustos, A.; Bustamante, D.; Morales, P.; Herrera-Marschitz, M. The Long-Term Impairment in Redox Homeostasis Observed in the Hippocampus of Rats Subjected to Global Perinatal Asphyxia (PA) Implies Changes in Glutathione-Dependent Antioxidant Enzymes and TIGAR-Dependent Shift Towards the Pentose Phosphate Pathways: Ef. Neurotox. Res. 2019, 36, 472–490. [Google Scholar] [CrossRef]
- Farfán, N.; Carril, J.; Redel, M.; Zamorano, M.; Araya, M.; Monzón, E.; Alvarado, R.; Contreras, N.; Tapia-Bustos, A.; Quintanilla, M.E.; et al. Intranasal administration of mesenchymal stem cell secretome reduces hippocampal oxidative stress, neuroinflammation and cell death, improving the behavioral outcome following perinatal asphyxia. Int. J. Mol. Sci. 2020, 21, 7800. [Google Scholar] [CrossRef] [PubMed]
- Day, B.J. Antioxidant therapeutics: Pandora’s box. Free Radic. Biol. Med. 2014, 66, 58–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, M.; An, C.; Gao, Y.; Leak, R.K.; Chen, J.; Zhang, F. Emerging roles of Nrf2 and phase II antioxidant enzymes in neuroprotection. Prog. Neurobiol. 2013, 100, 30–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bryan, H.K.; Olayanju, A.; Goldring, C.E.; Park, B.K. The Nrf2 cell defence pathway: Keap1-dependent and -independent mechanisms of regulation. Biochem. Pharmacol. 2013, 85, 705–717. [Google Scholar] [CrossRef] [Green Version]
- Dringen, R.; Gutterer, J.M.; Hirrlinger, J. Glutathione metabolism in brain. Eur. J. Biochem. 2000, 267, 4912–4916. [Google Scholar] [CrossRef]
- Franco, J.L.; Posser, T.; Dunkley, P.R.; Dickson, P.W.; Mattos, J.J.; Martins, R.; Bainy, A.C.D.; Marques, M.R.; Dafre, A.L.; Farina, M. Methylmercury neurotoxicity is associated with inhibition of the antioxidant enzyme glutathione peroxidase. Free Radic. Biol. Med. 2009, 47, 449–457. [Google Scholar] [CrossRef]
- Bolaños, J.P.; Delgado-Esteban, M.; Herrero-Mendez, A.; Fernandez-Fernandez, S.; Almeida, A. Regulation of glycolysis and pentose–phosphate pathway by nitric oxide: Impact on neuronal survival. Biochim. Biophys. Acta-Bioenerg. 2008, 1777, 789–793. [Google Scholar] [CrossRef] [Green Version]
- Brekke, E.M.F.; Morken, T.S.; Widerøe, M.; Håberg, A.K.; Brubakk, A.-M.; Sonnewald, U. The Pentose Phosphate Pathway and Pyruvate Carboxylation after Neonatal Hypoxic-Ischemic Brain Injury. J. Cereb. Blood Flow Metab. 2014, 34, 724–734. [Google Scholar] [CrossRef] [Green Version]
- Fullerton, H.J.; Ditelberg, J.S.; Chen, S.F.; Sarco, D.P.; Chan, P.H.; Epstein, C.J.; Ferriero, D.M. Copper/zinc superoxide dismutase transgenic brain accumulates hydrogen peroxide after perinatal hypoxia ischemia. Ann. Neurol. 1998, 44, 357–364. [Google Scholar] [CrossRef]
- Puka-Sundvall, M.; Wallin, C.; Gilland, E.; Hallin, U.; Wang, X.; Sandberg, M.; Karlsson, J.-O.; Blomgren, K.; Hagberg, H. Impairment of mitochondrial respiration after cerebral hypoxia-ischemia in immature rats: Relationship to activation of caspase-3 and neuronal injury. Dev. Brain Res. 2000, 125, 43–50. [Google Scholar] [CrossRef]
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; Igarashi, K.; Engel, J.D.; Yamamoto, M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999, 13, 76–86. [Google Scholar] [CrossRef] [Green Version]
- Hertz, L.; Zielke, H.R. Astrocytic control of glutamatergic activity: Astrocytes as stars of the show. Trends Neurosci. 2004, 27, 735–743. [Google Scholar] [CrossRef]
- Benzie, I.F.F.; Strain, J.J. The Ferric Reducing Ability of Plasma (FRAP) as a Measure of “Antioxidant Power”: The FRAP Assay. Anal. Biochem. 1996, 239, 70–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voorn, P.; Kalsbeek, A.; Jorritsma-Byham, B.; Groenewegen, H.J. The pre- and postnatal development of the dopaminergic cell groups in the ventral mesencephalon and the dopaminergic innervation of the striatum of the rat. Neuroscience 1988, 25, 857–887. [Google Scholar] [CrossRef]
- Veal, E.A.; Day, A.M.; Morgan, B.A. Hydrogen Peroxide Sensing and Signaling. Mol. Cell 2007, 26, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, P. Classical catalase: Ancient and modern. Arch. Biochem. Biophys. 2012, 525, 95–101. [Google Scholar] [CrossRef]
- Armogida, M.; Spalloni, A.; Amantea, D.; Nutini, M.; Petrelli, F.; Longone, P.; Bagetta, G.; Nisticò, R.; Mercuri, N.B. The Protective Role of Catalase against Cerebral Ischemia in vitro and in vivo. Int. J. Immunopathol. Pharmacol. 2011, 24, 735–747. [Google Scholar] [CrossRef]
- Spolarics, Z.; Wu, J.-X. Role of glutathione and catalase in H2O2detoxification in LPS-activated hepatic endothelial and Kupffer cells. Am. J. Physiol. Liver Physiol. 1997, 273, G1304–G1311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ibi, M.; Sawada, H.; Kume, T.; Katsuki, H.; Kaneko, S.; Shimohama, S.; Akaike, A. Depletion of Intracellular Glutathione Increases Susceptibility to Nitric Oxide in Mesencephalic Dopaminergic Neurons. J. Neurochem. 1999, 73, 1696–1703. [Google Scholar] [CrossRef] [Green Version]
- Hohnholt, M.C.; Dringen, R. Short time exposure to hydrogen peroxide induces sustained glutathione export from cultured neurons. Free Radic. Biol. Med. 2014, 70, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Baud, O.; Greene, A.E.; Li, J.; Wang, H.; Volpe, J.J.; Rosenberg, P.A. Glutathione Peroxidase-Catalase Cooperativity Is Required for Resistance to Hydrogen Peroxide by Mature Rat Oligodendrocytes. J. Neurosci. 2004, 24, 1531–1540. [Google Scholar] [CrossRef] [Green Version]
- Lardinois, O.M.; Mestdagh, M.M.; Rouxhet, P.G. Reversible inhibition and irreversible inactivation of catalase in presence of hydrogen peroxide. Biochim. Biophys. Acta-Protein Struct. Mol. Enzymol. 1996, 1295, 222–238. [Google Scholar] [CrossRef]
- Deponte, M. Glutathione catalysis and the reaction mechanisms of glutathione-dependent enzymes. Biochim. Biophys. Acta-Gen. Subj. 2013, 1830, 3217–3266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, J.Y.; Black, S.M. Developmental Changes in Murine Brain Antioxidant Enzymes. Pediatr. Res. 2003, 54, 77–82. [Google Scholar] [CrossRef] [Green Version]
- Panfoli, I.; Candiano, G.; Malova, M.; De Angelis, L.; Cardiello, V.; Buonocore, G.; Ramenghi, L.A. Oxidative Stress as a Primary Risk Factor for Brain Damage in Preterm Newborns. Front. Pediatr. 2018, 6, 369. [Google Scholar] [CrossRef] [Green Version]
- Lafemina, M.J.; Sheldon, R.A.; Ferriero, D.M. Acute Hypoxia-Ischemia Results in Hydrogen Peroxide Accumulation in Neonatal But Not Adult Mouse Brain. Pediatr. Res. 2006, 59, 680–683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weis, S.N.; Schunck, R.V.A.; Pettenuzzo, L.F.; Krolow, R.; Matté, C.; Manfredini, V.; Maria do Carmo, R.P.; Vargas, C.R.; Dalmaz, C.; Wyse, A.T.S.; et al. Early biochemical effects after unilateral hypoxia–ischemia in the immature rat brain. Int. J. Dev. Neurosci. 2011, 29, 115–120. [Google Scholar] [CrossRef] [Green Version]
- Chakravarti, R.; Gupta, K.; Majors, A.; Ruple, L.; Aronica, M.; Stuehr, D.J. Novel insights in mammalian catalase heme maturation: Effect of NO and thioredoxin-1. Free Radic. Biol. Med. 2015, 82, 105–113. [Google Scholar] [CrossRef] [Green Version]
- Krych-Madej, J.; Gebicka, L. Do pH and flavonoids influence hypochlorous acid-induced catalase inhibition and heme modification? Int. J. Biol. Macromol. 2015, 80, 162–169. [Google Scholar] [CrossRef]
- Ghosh, S.; Janocha, A.J.; Aronica, M.A.; Swaidani, S.; Comhair, S.A.A.; Xu, W.; Zheng, L.; Kaveti, S.; Kinter, M.; Hazen, S.L.; et al. Nitrotyrosine proteome survey in asthma identifies oxidative mechanism of catalase inactivation. J. Immunol. 2006, 176, 5587–5597. [Google Scholar] [CrossRef] [Green Version]
- Bensaad, K.; Tsuruta, A.; Selak, M.A.; Vidal, M.N.C.; Nakano, K.; Bartrons, R.; Gottlieb, E.; Vousden, K.H. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell 2006, 126, 107–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.; Sun, M.; Cao, L.; Gu, J.; Ge, J.; Chen, J.; Han, R.; Qin, Y.-Y.; Zhou, Z.-P.; Ding, Y.; et al. A TIGAR-regulated metabolic pathway is critical for protection of brain ischemia. J. Neurosci. 2014, 34, 7458–7471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stincone, A.; Prigione, A.; Cramer, T.; Wamelink, M.M.C.; Campbell, K.; Cheung, E.; Olin-Sandoval, V.; Grüning, N.-M.; Krüger, A.; Tauqeer Alam, M.; et al. The return of metabolism: Biochemistry and physiology of the pentose phosphate pathway. Biol. Rev. Camb. Philos. Soc. 2015, 90, 927–963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ros, S.; Schulze, A. Balancing glycolytic flux: The role of 6-phosphofructo-2-kinase/fructose 2,6-bisphosphatases in cancer metabolism. Cancer Metab. 2013, 1, 8. [Google Scholar] [CrossRef] [Green Version]
- Okar, D.A.; Manzano, A.; Navarro-Sabatè, A.; Riera, L.; Bartrons, R.; Lange, A.J. PFK-2/FBPase-2: Maker and breaker of the essential biofactor fructose-2,6-bisphosphate. Trends Biochem. Sci. 2001, 26, 30–35. [Google Scholar] [CrossRef]
- Fico, A.; Paglialunga, F.; Cigliano, L.; Abrescia, P.; Verde, P.; Martini, G.; Iaccarino, I.; Filosa, S. Glucose-6-phosphate dehydrogenase plays a crucial role in protection from redox-stress-induced apoptosis. Cell Death Differ. 2004, 11, 823–831. [Google Scholar] [CrossRef] [PubMed]
- Cheung, E.C.; Ludwig, R.L.; Vousden, K.H. Mitochondrial localization of TIGAR under hypoxia stimulates HK2 and lowers ROS and cell death. Proc. Natl. Acad. Sci. USA 2012, 109, 20491–20496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- da-Silva, W.S.; Gómez-Puyou, A.; de Gómez-Puyou, M.T.; Moreno-Sanchez, R.; De Felice, F.G.; de Meis, L.; Oliveira, M.F.; Galina, A. Mitochondrial bound hexokinase activity as a preventive antioxidant defense: Steady-state ADP formation as a regulatory mechanism of membrane potential and reactive oxygen species generation in mitochondria. J. Biol. Chem. 2004, 279, 39846–39855. [Google Scholar] [CrossRef] [Green Version]
- Cao, L.; Chen, J.; Li, M.; Qin, Y.-Y.; Sun, M.; Sheng, R.; Han, F.; Wang, G.; Qin, Z.-H. Endogenous level of TIGAR in brain is associated with vulnerability of neurons to ischemic injury. Neurosci. Bull. 2015, 31, 527–540. [Google Scholar] [CrossRef] [Green Version]
- Lu, Q.; Wainwright, M.S.; Harris, V.A.; Aggarwal, S.; Hou, Y.; Rau, T.; Poulsen, D.J.; Black, S.M. Increased NADPH oxidase-derived superoxide is involved in the neuronal cell death induced by hypoxia–ischemia in neonatal hippocampal slice cultures. Free Radic. Biol. Med. 2012, 53, 1139–1151. [Google Scholar] [CrossRef] [Green Version]
- Kleikers, P.W.M.; Wingler, K.; Hermans, J.J.R.; Diebold, I.; Altenhöfer, S.; Radermacher, K.A.; Janssen, B.; Görlach, A.; Schmidt, H.H.H.W. NADPH oxidases as a source of oxidative stress and molecular target in ischemia/reperfusion injury. J. Mol. Med. 2012, 90, 1391–1406. [Google Scholar] [CrossRef] [PubMed]
- Gupte, S.A.; Levine, R.J.; Gupte, R.S.; Young, M.E.; Lionetti, V.; Labinskyy, V.; Floyd, B.C.; Ojaimi, C.; Bellomo, M.; Wolin, M.S.; et al. Glucose-6-phosphate dehydrogenase-derived NADPH fuels superoxide production in the failing heart. J. Mol. Cell. Cardiol. 2006, 41, 340–349. [Google Scholar] [CrossRef] [Green Version]
- Balteau, M.; Tajeddine, N.; de Meester, C.; Ginion, A.; Des Rosiers, C.; Brady, N.R.; Sommereyns, C.; Horman, S.; Vanoverschelde, J.-L.; Gailly, P.; et al. NADPH oxidase activation by hyperglycaemia in cardiomyocytes is independent of glucose metabolism but requires SGLT1. Cardiovasc. Res. 2011, 92, 237–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suh, S.W.; Shin, B.S.; Ma, H.; Van Hoecke, M.; Brennan, A.M.; Yenari, M.A.; Swanson, R.A. Glucose and NADPH oxidase drive neuronal superoxide formation in stroke. Ann. Neurol. 2008, 64, 654–663. [Google Scholar] [CrossRef]
- Kuehne, A.; Emmert, H.; Soehle, J.; Winnefeld, M.; Fischer, F.; Wenck, H.; Gallinat, S.; Terstegen, L.; Lucius, R.; Hildebrand, J.; et al. Acute Activation of Oxidative Pentose Phosphate Pathway as First-Line Response to Oxidative Stress in Human Skin Cells. Mol. Cell 2015, 59, 359–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lorek, A.; Takei, Y.; Cady, E.B.; Wyatt, J.S.; Penrice, J.; Edwards, A.D.; Peebles, D.; Wylezinska, M.; Owen-Reece, H.; Kirkbride, V. Delayed (“secondary”) cerebral energy failure after acute hypoxia-ischemia in the newborn piglet: Continuous 48-hour studies by phosphorus magnetic resonance spectroscopy. Pediatr. Res. 1994, 36, 699–706. [Google Scholar] [CrossRef] [Green Version]
- Thoresen, M.; Penrice, J.; Lorek, A.; Cady, E.B.; Wylezinska, M.; Kirkbride, V.; Cooper, C.E.; Brown, G.C.; Edwards, A.D.; Wyatt, J.S.; et al. Mild Hypothermia after Severe Transient Hypoxia-Ischemia Ameliorates Delayed Cerebral Energy Failure in the Newborn Piglet. Pediatr. Res. 1995, 37, 667–670. [Google Scholar] [CrossRef] [Green Version]
- Benjelloun, N.; Renolleau, S.; Represa, A.; Ben-Ari, Y.; Charriaut-Marlangue, C. Inflammatory responses in the cerebral cortex after ischemia in the P7 neonatal Rat. Stroke 1999, 30, 1914–1916. [Google Scholar] [CrossRef] [Green Version]
- Hudome, S.; Palmer, C.; Roberts, R.L.; Mauger, D.; Housman, C.; Towfighi, J. The role of neutrophils in the production of hypoxic-ischemic brain injury in the neonatal rat. Pediatr. Res. 1997, 41, 607–616. [Google Scholar] [CrossRef]
- Winerdal, M.; Winerdal, M.E.; Kinn, J.; Urmaliya, V.; Winqvist, O.; Adén, U. Long lasting local and systemic inflammation after cerebral hypoxic ischemia in newborn mice. PLoS ONE 2012, 7, e36422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Northington, F.J.; Ferriero, D.M.; Graham, E.M.; Traystman, R.J.; Martin, L.J. Early Neurodegeneration after Hypoxia-Ischemia in Neonatal Rat Is Necrosis while Delayed Neuronal Death Is Apoptosis. Neurobiol. Dis. 2001, 8, 207–219. [Google Scholar] [CrossRef] [Green Version]
- Northington, F.J.; Chavez-Valdez, R.; Graham, E.M.; Razdan, S.; Gauda, E.B.; Martin, L.J. Necrostatin decreases oxidative damage, inflammation, and injury after neonatal HI. J. Cereb. Blood Flow Metab. 2011, 31, 178–189. [Google Scholar] [CrossRef] [PubMed]
- Neira-Peña, T.; Rojas-Mancilla, E.; Munoz-Vio, V.; Perez, R.; Gutierrez-Hernandez, M.; Bustamante, D.; Morales, P.; Hermoso, M.A.; Gebicke-Haerter, P.; Herrera-Marschitz, M. Perinatal Asphyxia Leads to PARP-1 Overactivity, p65 Translocation, IL-1β and TNF-α Overexpression, and Apoptotic-Like Cell Death in Mesencephalon of Neonatal Rats: Prevention by Systemic Neonatal Nicotinamide Administration. Neurotox. Res. 2015, 27, 453–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tapia-Bustos, A.; Perez-Lobos, R.; Vío, V.; Lespay-Rebolledo, C.; Palacios, E.; Chiti-Morales, A.; Bustamante, D.; Herrera-Marschitz, M.; Morales, P. Modulation of Postnatal Neurogenesis by Perinatal Asphyxia: Effect of D1 and D2 Dopamine Receptor Agonists. Neurotox. Res. 2017, 31, 109–121. [Google Scholar] [CrossRef]
- Chen, B.; Tang, L. Protective effects of catalase on retinal ischemia/reperfusion injury in rats. Exp. Eye Res. 2011, 93, 599–606. [Google Scholar] [CrossRef]
- Ikonomidou, C.; Kaindl, A.M. Neuronal death and oxidative stress in the developing brain. Antioxid. Redox Signal. 2011, 14, 1535–1550. [Google Scholar] [CrossRef]
- Ghosh, S.; Canugovi, C.; Yoon, J.S.; Wilson, D.M., 3rd; Croteau, D.L.; Mattson, M.P.; Bohr, V.A. Partial loss of the DNA repair scaffolding protein, Xrcc1, results in increased brain damage and reduced recovery from ischemic stroke in mice. Neurobiol. Aging 2015, 36, 2319–2330. [Google Scholar] [CrossRef] [Green Version]
- Neira-Peña, T.; Espina-Marchant, P.; Rojas-Mancilla, E.; Esmar, D.; Kraus, C.; Munoz, V.; Perez, R.; Rivera, B.; Bustamante, D.; Valdes, J.L.; et al. Molecular, Cellular, and Behavioural Effects Produced by Perinatal Asphyxia: Protection by Poly (ADP-Ribose) Polymerase 1 (PARP-1) Inhibition. In Handbook of Neurotoxicity; Springer: New York, NY, USA, 2014; pp. 2075–2098. ISBN 0301-0082. [Google Scholar]
- Neumar, R.W.; Meng, F.H.; Mills, A.M.; Xu, Y.A.; Zhang, C.; Welsh, F.A.; Siman, R. Calpain activity in the rat brain after transient forebrain ischemia. Exp. Neurol. 2001, 170, 27–35. [Google Scholar] [CrossRef] [Green Version]
- Ostwald, K.; Hagberg, H.; Andiné, P.; Karlsson, J.O. Upregulation of calpain activity in neonatal rat brain after hypoxic-ischemia. Brain Res. 1993, 630, 289–294. [Google Scholar] [CrossRef]
- Blomgren, K.; Hallin, U.; Andersson, A.L.; Puka-Sundvall, M.; Bahr, B.A.; McRae, A.; Saido, T.C.; Kawashima, S.; Hagberg, H. Calpastatin is up-regulated in response to hypoxia and is a suicide substrate to calpain after neonatal cerebral hypoxia-ischemia. J. Biol. Chem. 1999, 274, 14046–14052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chua, B.T.; Guo, K.; Li, P. Direct cleavage by the calcium-activated protease calpain can lead to inactivation of caspases. J. Biol. Chem. 2000, 275, 5131–5135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neumar, R.W.; Xu, Y.A.; Gada, H.; Guttmann, R.P.; Siman, R. Cross-talk between calpain and caspase proteolytic systems during neuronal apoptosis. J. Biol. Chem. 2003, 278, 14162–14167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bordone, L.; Campbell, C. DNA ligase III is degraded by calpain during cell death induced by DNA-damaging agents. J. Biol. Chem. 2002, 277, 26673–26680. [Google Scholar] [CrossRef] [Green Version]
- Cheng, S.-Y.; Wang, S.-C.; Lei, M.; Wang, Z.; Xiong, K. Regulatory role of calpain in neuronal death. Neural Regen. Res. 2018, 13, 556–562. [Google Scholar] [CrossRef] [PubMed]
- Brown, H.K.; Speechley, K.N.; Macnab, J.; Natale, R.; Campbell, M.K. Maternal, fetal, and placental conditions associated with medically indicated late preterm and early term delivery: A retrospective study. BJOG Int. J. Obstet. Gynaecol. 2016, 123, 763–770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flores-Balter, G.; Cordova-Jadue, H.; Chiti-Morales, A.; Lespay, C.; Espina-Marchant, P.; Falcon, R.; Grinspun, N.; Sanchez, J.; Bustamante, D.; Morales, P.; et al. Effect of perinatal asphyxia on tuberomammillary nucleus neuronal density and object recognition memory: A possible role for histamine? Behav. Brain Res. 2016, 313, 226–232. [Google Scholar] [CrossRef]
- Mattson, M.P. Mitochondrial regulation of neuronal plasticity. Neurochem. Res. 2007, 32, 707–715. [Google Scholar] [CrossRef]
- Deng, W. Neurobiology of injury to the developing brain. Nat. Rev. Neurol. 2010, 6, 328–336. [Google Scholar] [CrossRef] [PubMed]
- Orrenius, S.; Nicotera, P.; Zhivotovsky, B. Cell death mechanisms and their implications in toxicology. Toxicol. Sci. 2011, 119, 3–19. [Google Scholar] [CrossRef] [Green Version]
- Sies, H. Hydrogen peroxide as a central redox signaling molecule in physiological oxidative stress: Oxidative eustress. Redox Biol. 2017, 11, 613–619. [Google Scholar] [CrossRef] [PubMed]
- Aschbacher, K.; O’Donovan, A.; Wolkowitz, O.M.; Dhabhar, F.S.; Su, Y.; Epel, E. Good stress, bad stress and oxidative stress: Insights from anticipatory cortisol reactivity. Psychoneuroendocrinology 2013, 38, 1698–1708. [Google Scholar] [CrossRef] [Green Version]
- Plenz, D.; Kitai, S.T. Organotypic cortex-striatum-mesencephalon cultures: The nigrostriatal pathway. Neurosci. Lett. 1996, 209, 177–180. [Google Scholar] [CrossRef]
- Plenz, D.; Herrera-Marschitz, M.; Kitai, S.T. Morphological organization of the globus pallidus-subthalamic nucleus system studied in organotypic cultures. J. Comp. Neurol. 1998, 397, 437–457. [Google Scholar] [CrossRef]
- Gomez-Urquijo, S.M.; Hökfelt, T.; Ubink, R.; Lubec, G.; Herrera-Marschitz, M. Neurocircuitries of the basal ganglia studied in organotypic cultures: Focus on tyrosine hydroxylase, nitric oxide synthase and neuropeptide immunocytochemistry. Neuroscience 1999, 94, 1133–1151. [Google Scholar] [CrossRef]
- Skulachev, V.P. Role of uncoupled and non-coupled oxidations in maintenance of safely low levels of oxygen and its one-electron reductants. Q. Rev. Biophys. 1996, 29, 169–202. [Google Scholar] [CrossRef] [PubMed]
- Hoeger, H.; Engelmann, M.; Bernert, G.; Seidl, R.; Bubna-Littitz, H.; Mosgoeller, W.; Lubec, B.; Lubec, G. Long term neurological and behavioral effects of graded perinatal asphyxia in the rat. Life Sci. 2000, 66, 947–962. [Google Scholar] [CrossRef]
- Reemst, K.; Noctor, S.C.; Lucassen, P.J.; Hol, E.M. The Indispensable Roles of Microglia and Astrocytes during Brain Development. Front. Hum. Neurosci. 2016, 10, 566. [Google Scholar] [CrossRef] [Green Version]
- Back, S.A.; Han, B.H.; Luo, N.L.; Chricton, C.A.; Xanthoudakis, S.; Tam, J.; Arvin, K.L.; Holtzman, D.M. Selective vulnerability of late oligodendrocyte progenitors to hypoxia-ischemia. J. Neurosci. 2002, 22, 455–463. [Google Scholar] [CrossRef] [Green Version]
- McTigue, D.M.; Tripathi, R.B. The life, death, and replacement of oligodendrocytes in the adult CNS. J. Neurochem. 2008, 107, 1–19. [Google Scholar] [CrossRef]
- Benarroch, E.E. Oligodendrocytes: Susceptibility to injury and involvement in neurologic disease. Neurology 2009, 72, 1779–1785. [Google Scholar] [CrossRef]
- Bradl, M.; Lassmann, H. Oligodendrocytes: Biology and pathology. Acta Neuropathol. 2010, 119, 37–53. [Google Scholar] [CrossRef] [Green Version]
- Miron, V.E.; Kuhlmann, T.; Antel, J.P. Cells of the oligodendroglial lineage, myelination, and remyelination. Biochim. Biophys. Acta 2011, 1812, 184–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilkins, A.; Majed, H.; Layfield, R.; Compston, A.; Chandran, S. Oligodendrocytes promote neuronal survival and axonal length by distinct intracellular mechanisms: A novel role for oligodendrocyte-derived glial cell line-derived neurotrophic factor. J. Neurosci. 2003, 23, 4967–4974. [Google Scholar] [CrossRef] [Green Version]
- Simons, M.; Nave, K.-A. Oligodendrocytes: Myelination and Axonal Support. Cold Spring Harb. Perspect. Biol. 2015, 8, a020479. [Google Scholar] [CrossRef]
- Guardia Clausi, M.; Pasquini, L.A.; Soto, E.F.; Pasquini, J.M. Apotransferrin-induced recovery after hypoxic/ischaemic injury on myelination. ASN Neuro 2010, 2, e00048. [Google Scholar] [CrossRef]
- Dean, J.M.; Moravec, M.D.; Grafe, M.; Abend, N.; Ren, J.; Gong, X.; Volpe, J.J.; Jensen, F.E.; Hohimer, A.R.; Back, S.A. Strain-specific differences in perinatal rodent oligodendrocyte lineage progression and its correlation with human. Dev. Neurosci. 2011, 33, 251–260. [Google Scholar] [CrossRef] [Green Version]
- Dimou, L.; Simon, C.; Kirchhoff, F.; Takebayashi, H.; Götz, M. Progeny of Olig2-expressing progenitors in the gray and white matter of the adult mouse cerebral cortex. J. Neurosci. 2008, 28, 10434–10442. [Google Scholar] [CrossRef] [PubMed]
- Young, K.M.; Psachoulia, K.; Tripathi, R.B.; Dunn, S.-J.; Cossell, L.; Attwell, D.; Tohyama, K.; Richardson, W.D. Oligodendrocyte dynamics in the healthy adult CNS: Evidence for myelin remodeling. Neuron 2013, 77, 873–885. [Google Scholar] [CrossRef] [Green Version]
- Labombarda, F.; González, S.L.; Lima, A.; Roig, P.; Guennoun, R.; Schumacher, M.; de Nicola, A.F. Effects of progesterone on oligodendrocyte progenitors, oligodendrocyte transcription factors, and myelin proteins following spinal cord injury. Glia 2009, 57, 884–897. [Google Scholar] [CrossRef] [PubMed]
- Nait-Oumesmar, B.; Picard-Riéra, N.; Kerninon, C.; Baron-Van Evercooren, A. The role of SVZ-derived neural precursors in demyelinating diseases: From animal models to multiple sclerosis. J. Neurol. Sci. 2008, 265, 26–31. [Google Scholar] [CrossRef]
- Nicholas, R.S.; Wing, M.G.; Compston, A. Nonactivated microglia promote oligodendrocyte precursor survival and maturation through the transcription factor NF-kappa B. Eur. J. Neurosci. 2001, 13, 959–967. [Google Scholar] [CrossRef] [PubMed]
- Camargo, N.; Goudriaan, A.; van Deijk, A.-L.F.; Otte, W.M.; Brouwers, J.F.; Lodder, H.; Gutmann, D.H.; Nave, K.-A.; Dijkhuizen, R.M.; Mansvelder, H.D.; et al. Oligodendroglial myelination requires astrocyte-derived lipids. PLoS Biol. 2017, 15, e1002605. [Google Scholar] [CrossRef] [PubMed]
- Thoresen, M.; Tooley, J.; Liu, X.; Jary, S.; Fleming, P.; Luyt, K.; Jain, A.; Cairns, P.; Harding, D.; Sabir, H. Time is brain: Starting therapeutic hypothermia within three hours after birth improves motor outcome in asphyxiated newborns. Neonatology 2013, 104, 228–233. [Google Scholar] [CrossRef] [PubMed]
- Newborn, C.O.N.F.A.N.D. Respiratory Support in Preterm Infants at Birth. Pediatrics 2014, 133, 171–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shankaran, S.; Laptook, A.R.; Pappas, A.; McDonald, S.A.; Das, A.; Tyson, J.E.; Poindexter, B.B.; Schibler, K.; Bell, E.F.; Heyne, R.J.; et al. Effect of depth and duration of cooling on deaths in the NICU among neonates with hypoxic ischemic encephalopathy: A randomized clinical trial. JAMA 2014, 312, 2629–2639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabir, H.; Cowan, F.M. Prediction of outcome methods assessing short- and long-term outcome after therapeutic hypothermia. Semin. Fetal Neonatal Med. 2015, 20, 115–121. [Google Scholar] [CrossRef]
- Ahearne, C.E.; Boylan, G.B.; Murray, D.M. Short and long term prognosis in perinatal asphyxia: An update. World J. Clin. Pediatr. 2016, 5, 67–74. [Google Scholar] [CrossRef] [PubMed]
- McDouall, A.; Wassink, G.; Bennet, L.; Gunn, A.; Davidson, J. Challenges in developing therapeutic strategies for mild neonatal encephalopathy. Neural Regen. Res. 2022, 17, 277–282. [Google Scholar] [CrossRef] [PubMed]
- Laptook, A.R.; Corbett, R.J.; Sterett, R.; Garcia, D.; Tollefsbol, G. Quantitative relationship between brain temperature and energy utilization rate measured in vivo using 31P and 1H magnetic resonance spectroscopy. Pediatr. Res. 1995, 38, 919–925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erecinska, M.; Thoresen, M.; Silver, I.A. Effects of Hypothermia on Energy Metabolism in Mammalian Central Nervous System. J. Cereb. Blood Flow Metab. 2003, 23, 513–530. [Google Scholar] [CrossRef]
- Thoresen, M.; Satas, S.; Puka-Sundvall, M.; Whitelaw, A.; Hallström, A.; Løberg, E.M.; Ungerstedt, U.; Steen, P.A.; Hagberg, H. Post-hypoxic hypothermia reduces cerebrocortical release of NO and excitotoxins. Neuroreport 1997, 8, 3359–3362. [Google Scholar] [CrossRef]
- Nakashima, K.; Todd, M.M. Effects of hypothermia on the rate of excitatory amino acid release after ischemic depolarization. Stroke 1996, 27, 913–918. [Google Scholar] [CrossRef]
- Rostami, E.; Rocksén, D.; Ekberg, N.R.; Goiny, M.; Ungerstedt, U. Brain metabolism and oxygenation in healthy pigs receiving hypoventilation and hyperoxia. Respir. Physiol. Neurobiol. 2013, 189, 537–542. [Google Scholar] [CrossRef]
- Nurse, S.; Corbett, D. Neuroprotection after several days of mild, drug-induced hypothermia. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 1996, 16, 474–480. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.B.; Jackson, J.G. Astroglial glutamate transporters coordinate excitatory signaling and brain energetics. Neurochem. Int. 2016, 98, 56–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berk, M.; Malhi, G.S.; Gray, L.J.; Dean, O.M. The promise of N-acetylcysteine in neuropsychiatry. Trends Pharmacol. Sci. 2013, 34, 167–177. [Google Scholar] [CrossRef]
- Wink, L.K.; Adams, R.; Wang, Z.; Klaunig, J.E.; Plawecki, M.H.; Posey, D.J.; McDougle, C.J.; Erickson, C.A. A randomized placebo-controlled pilot study of N-acetylcysteine in youth with autism spectrum disorder. Mol. Autism 2016, 7, 26. [Google Scholar] [CrossRef] [Green Version]
- Bavarsad Shahripour, R.; Harrigan, M.R.; Alexandrov, A. V N-acetylcysteine (NAC) in neurological disorders: Mechanisms of action and therapeutic opportunities. Brain Behav. 2014, 4, 108–122. [Google Scholar] [CrossRef] [PubMed]
- Quintanilla, M.E.; Rivera-Meza, M.; Berríos-Cárcamo, P.; Salinas-Luypaert, C.; Herrera-Marschitz, M.; Israel, Y. Beyond the “First Hit”: Marked Inhibition by N-Acetyl Cysteine of Chronic Ethanol Intake But Not of Early Ethanol Intake. Parallel Effects on Ethanol-Induced Saccharin Motivation. Alcohol. Clin. Exp. Res. 2016, 40, 1044–1051. [Google Scholar] [CrossRef] [PubMed]
- Herrera-Marschitz, M.; Arbuthnott, G.; Ungerstedt, U. The rotational model and microdialysis: Significance for dopamine signalling, clinical studies, and beyond. Prog. Neurobiol. 2010, 90, 176–189. [Google Scholar] [CrossRef]
- Krieglstein, J.; el Nasr, M.S.; Lippert, K. Neuroprotection by memantine as increased by hypothermia and nimodipine, Dedicated to Professor Dr. G. Seitz on the occasion of his 60th birthday. Eur. J. Pharm. Sci. 1997, 5, 71–77. [Google Scholar] [CrossRef]
- Eliasson, M.J.; Sampei, K.; Mandir, A.S.; Hurn, P.D.; Traystman, R.J.; Bao, J.; Pieper, A.; Wang, Z.Q.; Dawson, T.M.; Snyder, S.H.; et al. Poly(ADP-ribose) polymerase gene disruption renders mice resistant to cerebral ischemia. Nat. Med. 1997, 3, 1089–1095. [Google Scholar] [CrossRef]
- Seeman, P.; Caruso, C.; Lasaga, M. Memantine agonist action at dopamine D2High receptors. Synapse 2008, 62, 149–153. [Google Scholar] [CrossRef] [PubMed]
- Danysz, W.; Parsons, C.G.; Kornhuber, J.; Schmidt, W.J.; Quack, G. Aminoadamantanes as NMDA receptor antagonists and antiparkinsonian agents—Preclinical studies. Neurosci. Biobehav. Rev. 1997, 21, 455–468. [Google Scholar] [CrossRef]
- Volbracht, C.; Van Beek, J.; Zhu, C.; Blomgren, K.; Leist, M. Neuroprotective properties of memantine in different in vitro and in vivo models of excitotoxicity. Eur. J. Neurosci. 2006, 23, 2611–2622. [Google Scholar] [CrossRef] [Green Version]
- Rammes, G.; Zieglgänsberger, W.; Parsons, C.G. The fraction of activated N-methyl-D-aspartate receptors during synaptic transmission remains constant in the presence of the glutamate release inhibitor riluzole. J. Neural Transm. 2008, 115, 1119–1126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Muñoz, M.; Lopez-Huerta, V.G.; Carrillo-Reid, L.; Arbuthnott, G.W. Extrasynaptic glutamate NMDA receptors: Key players in striatal function. Neuropharmacology 2015, 89, 54–63. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.-N.; Johnson, S.W. Memantine selectively blocks extrasynaptic NMDA receptors in rat substantia nigra dopamine neurons. Brain Res. 2015, 1603, 1–7. [Google Scholar] [CrossRef]
- Manning, S.M.; Boll, G.; Fitzgerald, E.; Selip, D.B.; Volpe, J.J.; Jensen, F.E. The clinically available NMDA receptor antagonist, memantine, exhibits relative safety in the developing rat brain. Int. J. Dev. Neurosci. 2011, 29, 767–773. [Google Scholar] [CrossRef] [Green Version]
- Ducrocq, S.; Benjelloun, N.; Plotkine, M.; Ben-Ari, Y.; Charriaut-Marlangue, C. Poly (ADP-Ribose) Synthase Inhibition Reduces Ischemic Injury and Inflammation in Neonatal Rat Brain. J. Neurochem. 2000, 74, 2504–2511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakakibara, Y.; Mitha, A.P.; Ogilvy, C.S.; Maynard, K.I. Post-treatment with nicotinamide (vitamin B3) reduces the infarct volume following permanent focal cerebral ischemia in female Sprague-Dawley and Wistar rats. Neurosci. Lett. 2000, 281, 111–114. [Google Scholar] [CrossRef]
- Nagayama, T.; Simon, R.P.; Chen, D.; Henshall, D.C.; Pei, W.; Stetler, R.A.; Chen, J. Activation of poly(ADP-ribose) polymerase in the rat hippocampus may contribute to cellular recovery following sublethal transient global ischemia. J. Neurochem. 2000, 74, 1636–1645. [Google Scholar] [CrossRef]
- Saldeen, J.; Welsh, N. Nicotinamide-induced apoptosis in insulin producing cells is associated with cleavage of poly(ADP-ribose) polymerase. Mol. Cell. Endocrinol. 1998, 139, 99–107. [Google Scholar] [CrossRef]
- Bustamante, D.; Goiny, M.; Aström, G.; Gross, J.; Andersson, K.; Herrera-Marschitz, M. Nicotinamide prevents the long-term effects of perinatal asphyxia on basal ganglia monoamine systems in the rat. Exp. Brain Res. 2003, 148, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Bustamante, D.; Morales, P.; Pereyra, J.T.; Goiny, M.; Herrera-Marschitz, M. Nicotinamide prevents the effect of perinatal asphyxia on dopamine release evaluated with in vivo microdialysis 3 months after birth. Exp. Brain Res. 2007, 177, 358–369. [Google Scholar] [CrossRef]
- Virág, L.; Szabó, C. The therapeutic potential of poly(ADP-ribose) polymerase inhibitors. Pharmacol. Rev. 2002, 54, 375–429. [Google Scholar] [CrossRef] [PubMed]
- Klawitter, V.; Morales, P.; Bustamante, D.; Goiny, M.; Herrera-Marschitz, M. Plasticity of the central nervous system (CNS) following perinatal asphyxia: Does nicotinamide provide neuroprotection? Amino Acids 2006, 31, 377–384. [Google Scholar] [CrossRef]
- Hortobágyi, T.; Görlach, C.; Benyó, Z.; Lacza, Z.; Hortobágyi, S.; Wahl, M.; Harkany, T. Inhibition of neuronal nitric oxide synthase-mediated activation of poly(ADP-ribose) polymerase in traumatic brain injury: Neuroprotection by 3-aminobenzamide. Neuroscience 2003, 121, 983–990. [Google Scholar] [CrossRef]
- Allende-Castro, C.; Espina-Marchant, P.; Bustamante, D.; Rojas-Mancilla, E.; Neira, T.; Gutierrez-Hernandez, M.A.; Esmar, D.; Valdes, J.L.; Morales, P.; Gebicke-Haerter, P.J.; et al. Further studies on the hypothesis of PARP-1 inhibition as a strategy for lessening the long-term effects produced by perinatal asphyxia: Effects of nicotinamide and theophylline on PARP-1 activity in brain and peripheral tissue. Neurotox. Res. 2012, 22, 79–90. [Google Scholar] [CrossRef]
- Kamat, J.P.; Devasagayam, T.P. Nicotinamide (vitamin B3) as an effective antioxidant against oxidative damage in rat brain mitochondria. Redox Rep. 1999, 4, 179–184. [Google Scholar] [CrossRef] [Green Version]
- Chong, Z.Z.; Lin, S.-H.; Maiese, K. The NAD+ precursor nicotinamide governs neuronal survival during oxidative stress through protein kinase B coupled to FOXO3a and mitochondrial membrane potential. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 2004, 24, 728–743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawamura, T.; Mori, N.; Shibata, K. β-Nicotinamide Mononucleotide, an Anti-Aging Candidate Compound, Is Retained in the Body for Longer than Nicotinamide in Rats. J. Nutr. Sci. Vitaminol. 2016, 62, 272–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Hu, X.; Yang, Y.; Takata, T.; Sakurai, T. Nicotinamide mononucleotide protects against β-amyloid oligomer-induced cognitive impairment and neuronal death. Brain Res. 2016, 1643, 1–9. [Google Scholar] [CrossRef]
- Turunc Bayrakdar, E.; Uyanikgil, Y.; Kanit, L.; Koylu, E.; Yalcin, A. Nicotinamide treatment reduces the levels of oxidative stress, apoptosis, and PARP-1 activity in Aβ(1-42)-induced rat model of Alzheimer’s disease. Free Radic. Res. 2014, 48, 146–158. [Google Scholar] [CrossRef]
- Vio, V.; Riveros, A.L.; Tapia-Bustos, A.; Lespay-Rebolledo, C.; Perez-Lobos, R.; Muñoz, L.; Pismante, P.; Morales, P.; Araya, E.; Hassan, N.; et al. Gold nanorods/siRNA complex administration for knockdown of PARP-1: A potential treatment for perinatal asphyxia. Int. J. Nanomed. 2018, 13, 6839–6854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehmel, M.; Jovanović, N.; Spitz, U. Nicotinamide Riboside-The Current State of ReseArch. and Therapeutic Uses. Nutrients 2020, 12, 1616. [Google Scholar] [CrossRef] [PubMed]
- Van Velthoven, C.T.J.; Kavelaars, A.; Heijnen, C.J. Mesenchymal stem cells as a treatment for neonatal ischemic brain damage. Pediatr. Res. 2012, 71, 474–481. [Google Scholar] [CrossRef] [PubMed]
- Castillo-Melendez, M.; Yawno, T.; Jenkin, G.; Miller, S.L. Stem cell therapy to protect and repair the developing brain: A review of mechanisms of action of cord blood and amnion epithelial derived cells. Front. Neurosci. 2013, 7, 194. [Google Scholar] [CrossRef] [Green Version]
- De Gregorio, C.; Contador, D.; Díaz, D.; Cárcamo, C.; Santapau, D.; Lobos-Gonzalez, L.; Acosta, C.; Campero, M.; Carpio, D.; Gabriele, C.; et al. Human adipose-derived mesenchymal stem cell-conditioned medium ameliorates polyneuropathy and foot ulceration in diabetic BKS db/db mice. Stem Cell Res. Ther. 2020, 11, 168. [Google Scholar] [CrossRef]
- Vizoso, F.; Eiro, N.; Cid, S.; Schneider, J.; Perez-Fernandez, R. Mesenchymal Stem Cell Secretome: Toward Cell-Free Therapeutic Strategies in Regenerative Medicine. Int. J. Mol. Sci. 2017, 18, 1852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, P.; Kandoi, S.; Misra, R.; Vijayalakshmi, S.; Rajagopal, K.; Verma, R.S. The mesenchymal stem cell secretome: A new paradigm towards cell-free therapeutic mode in regenerative medicine. Cytokine Growth Factor Rev. 2019, 46, 1–9. [Google Scholar] [CrossRef]
- De Cássia Noronha, N.; Mizukami, A.; Caliári-Oliveira, C.; Cominal, J.G.; Rocha, J.L.M.; Covas, D.T.; Swiech, K.; Malmegrim, K.C.R. Priming approaches to improve the efficacy of mesenchymal stromal cell-based therapies. Stem Cell Res. Ther. 2019, 10, 131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quintanilla, M.E.; Ezquer, F.; Morales, P.; Santapau, D.; Berríos-Cárcamo, P.; Ezquer, M.; Herrera-Marschitz, M.; Israel, Y. Intranasal mesenchymal stem cell secretome administration markedly inhibits alcohol and nicotine self-administration and blocks relapse-intake: Mechanism and translational options. Stem Cell Res. Ther. 2019, 10, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Oses, C.; Olivares, B.; Ezquer, M.; Acosta, C.; Bosch, P.; Donoso, M.; Léniz, P.; Ezquer, F. Preconditioning of adipose tissue-derived mesenchymal stem cells with deferoxamine increases the production of pro-angiogenic, neuroprotective and anti-inflammatory factors: Potential application in the treatment of diabetic neuropathy. PLoS ONE 2017, 12, e017. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lespay-Rebolledo, C.; Tapia-Bustos, A.; Perez-Lobos, R.; Vio, V.; Casanova-Ortiz, E.; Farfan-Troncoso, N.; Zamorano-Cataldo, M.; Redel-Villarroel, M.; Ezquer, F.; Quintanilla, M.E.; et al. Sustained Energy Deficit Following Perinatal Asphyxia: A Shift towards the Fructose-2,6-bisphosphatase (TIGAR)-Dependent Pentose Phosphate Pathway and Postnatal Development. Antioxidants 2022, 11, 74. https://doi.org/10.3390/antiox11010074
Lespay-Rebolledo C, Tapia-Bustos A, Perez-Lobos R, Vio V, Casanova-Ortiz E, Farfan-Troncoso N, Zamorano-Cataldo M, Redel-Villarroel M, Ezquer F, Quintanilla ME, et al. Sustained Energy Deficit Following Perinatal Asphyxia: A Shift towards the Fructose-2,6-bisphosphatase (TIGAR)-Dependent Pentose Phosphate Pathway and Postnatal Development. Antioxidants. 2022; 11(1):74. https://doi.org/10.3390/antiox11010074
Chicago/Turabian StyleLespay-Rebolledo, Carolyne, Andrea Tapia-Bustos, Ronald Perez-Lobos, Valentina Vio, Emmanuel Casanova-Ortiz, Nancy Farfan-Troncoso, Marta Zamorano-Cataldo, Martina Redel-Villarroel, Fernando Ezquer, Maria Elena Quintanilla, and et al. 2022. "Sustained Energy Deficit Following Perinatal Asphyxia: A Shift towards the Fructose-2,6-bisphosphatase (TIGAR)-Dependent Pentose Phosphate Pathway and Postnatal Development" Antioxidants 11, no. 1: 74. https://doi.org/10.3390/antiox11010074