1. Introduction
Interleukin-6 (IL-6) acts as a pleiotropic cytokine, due to its multiple roles in several tissues and organs. Typically, IL-6 has been investigated in immune cells, coordinating fundamental processes related to adaptive immune responses and being involved in acute phase reactions against pathogens and chronic inflammation. However, since it is secreted by many other cell types, IL-6 has been reported to play a role in other physiological processes [
1]. Recently, the multifunctional role of IL-6 has been investigated in regenerative processes, and it was observed that mesenchymal stem cells (MSCs) release extremely high levels of IL-6 compared to other cytokines. However, there is still little known about the precise IL-6 function in the homeostasis of MSCs and the role of these cells in immunoregulation under reparative processes [
2]. Xie and colleagues [
3] reported that MSCs from the bone marrow both secrete and respond to IL-6 and these high expression levels of IL-6 and its receptor promote MSCs osteogenic differentiation. In a clinical situation of fracture repair, the osteogenic commitment of MSCs can be complicated by the elevated levels of interleukins which specifically amplify the inflammatory environment [
4]. It has been shown that a rise in pro-inflammatory mediators such as interleukin (IL)-1β inhibits bone morphogenic protein 2 (BMP2)-induced osteogenic differentiation of MSCs [
5]. However, IL-6 seems to function differently because a synergetic effect with BMP-2 to enhance bone regeneration was shown in an animal model [
6].
A subpopulation of multipotent undifferentiated MSCs of neural crest origins also resides within the dental pulp of human teeth capable of undergoing odontogenic, adipogenic and neurogenic differentiation [
7]. Moreover, cells from the dental pulp express surface markers typically related to a mesenchymal origin [
8] and they own a peculiar genetic profile characterized by markers related to osteogenesis, such as BMP-2 and alkaline phosphatase (ALP) during standard procedures for odontogenic differentiation in vitro [
9]. The dental pulp is thus the residence of odontogenic MSCs, which are essential for the process of tooth development and injury repair through the secretion of reparative dentin. Nonetheless, the regenerative abilities of these cells are easily affected by the environmental conditions of their niche. As a matter of fact, dental pulp is vulnerable to trauma and bacterial infections, and the secretion of cytokines triggered by bacterial products such as lipopolysaccharide (LPS) may influence the mineralization potential of odontogenic pulp cells [
10,
11].
Therefore, investigating the close link between inflammation and the induction of regenerative processes in dental pulp is of fundamental importance to disclose whether this interplay may influence dentin and pulp regeneration. In a clinical situation, after injuries or bacterial infiltrations, cytokines are released from odontoblasts and probably mesenchymal pulp cells to finally initiate dental pulp inflammation as a mechanism of defense. These adaptive cell responses are related to oxidative and nitrosative stress, caused by reactive oxygen or nitrogen species (ROS or RNS, respectively). Dysregulated high levels of ROS or RNS may lead to apoptosis and cell cycle arrest in MSCs [
12,
13]. In parallel, several recent studies reported on the importance of a redox control of MSC differentiation [
14]. Our group has previously observed molecular pathways underlying the intracellular balance between redox homeostasis and extracellular matrix mineralization of human pulp cells (HPCs), emphasizing IL-6 as one of the major players involved [
15,
16]. Furthermore, a redox control through the ROS-activated MAPKs (mitogen-activated protein kinases) signaling has been highlighted under the osteogenic commitment of HPCs in vitro [
17].
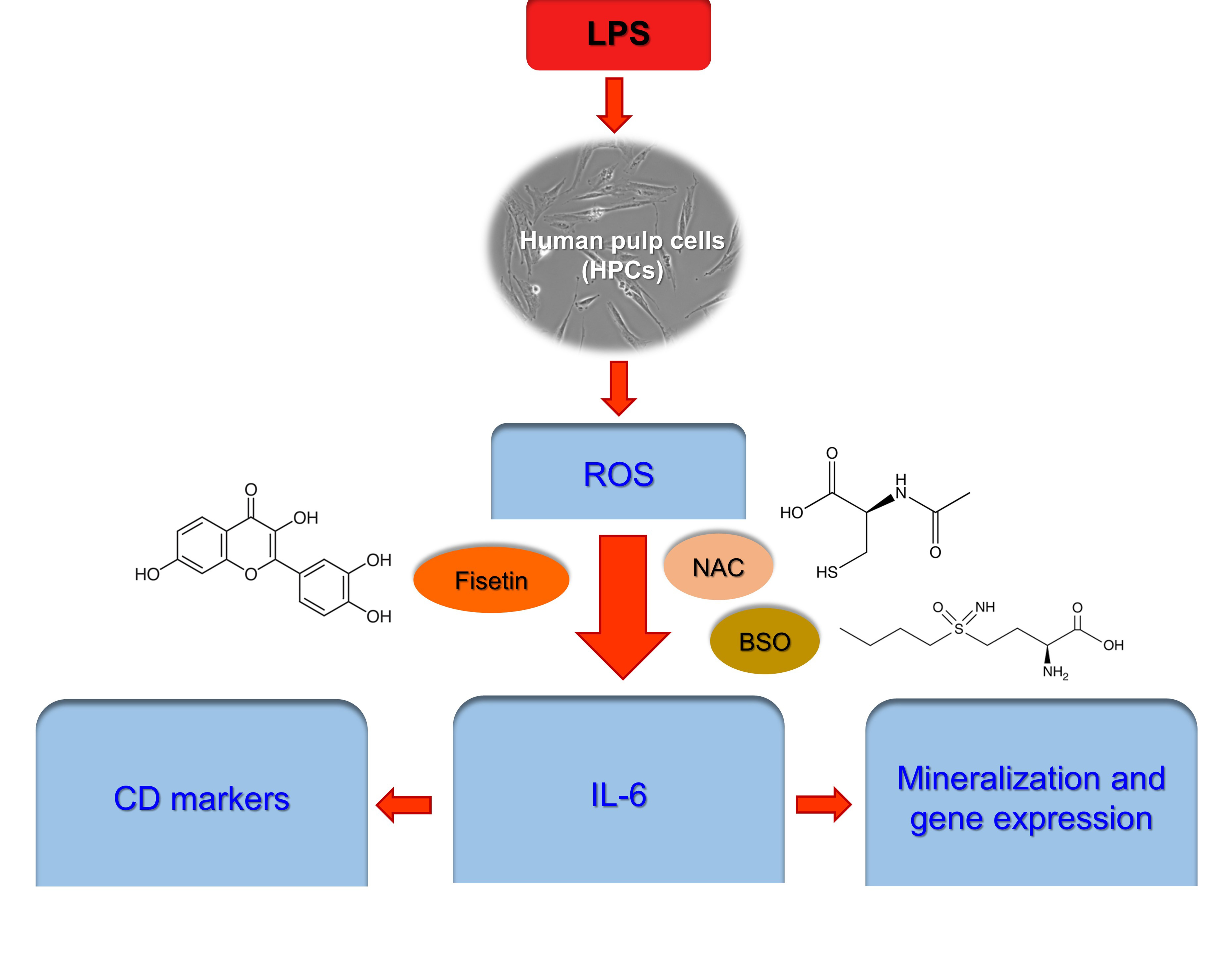
On this basis, the present study aims to demonstrate the relevance of redox homeostasis for vital HPC functions such as immune response and differentiation. It is assumed that redox control is crucial under the odontogenic commitment of mesenchymal pulp cells and that IL-6 secretion could be a link in the interplay between oxidative stress and mineralization pathways. To this end, primary HPCs were grown in normal and odontogenic media, stimulated with a low concentration of LPS capable of creating a redox homeostasis perturbance, and exposed to anti- or pro-oxidant substances up to 21 days. As an inhibitor of glutathione (GSH) synthesis, a major non-enzymatic antioxidant, buthionine sulfoximine (BSO) was used to further enhance oxidative stress. On the contrary, N-acetylcysteine (NAC) as an antioxidant by itself and as a substrate in GSH synthesis keeps GSH at high levels [
18]. In addition, the dietary flavonoid fisetin was included in this study. It has shown strong anti-inflammatory and antioxidant effects in cell cultures and in animal models relevant to human diseases [
19]. Here, the expression of distinctive MSC-associated surface markers was analyzed and was shown to be dependent on redox homeostasis. In addition to the analysis of cell viability and the formation of oxidative stress, relevant parameters indicating mineralization have been analyzed. In parallel, a time-course of IL-6 secretion has been established to provide evidence for the simultaneous activation of immune responses.
2. Materials and Methods
2.1. Chemicals
Minimum essential medium (MEM) α (alpha modification) and fetal bovine serum (FBS) were purchased from Gibco (Thermo Fisher Scientific, Waltham, MA, USA). Lipopolysaccharide (LPS; E. coli, serotype 055:B5), L-buthionine sulfoximine (BSO; CAS-No. 83730-53-4), N-acetylcysteine (NAC; CAS-No. 616-91-1), 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), Alizarin red S (A5533), dexamethasone (D4902), ß-glycerophosphate (G9422), penicillin/streptomycin (P0781) came from Sigma Aldrich GmbH (Taufkirchen, Germany). Fisetin (S2298; CAS-No. 345909-34-4) was obtained from Selleck Chemicals (Biozol, Munich, Germany), 2′,7′-dichlorodihydrofluorescin diacetate (DCFH2-DA; CAS-No. 4091-99-0) came from MoBiTec (Göttingen, Germany), ascorbic acid was purchased from Merck (Darmstadt, Germany), and an interleukin 6 (IL-6) ELISA kit was obtained from BD Biosciences (San Diego, CA, USA). Anti-human CD90-FITC (clone 5E10) came from BD Biosciences (San Diego, CA, USA), a monoclonal human 5’-nucleotidase/CD73-PE-conjugated antibody (clone 606112, FAB5795P) was provided by R & D Systems (Minneapolis, MN, USA), monoclonal anti-human CD105 (Endoglin)/R-PE (326-050) was purchased from Ancell (Biomol GmbH, Hamburg, Germany), CD140a-FITC mouse monoclonal (130-115-336) was supplied by Miltenyi Biotec GmbH (Bergisch Gladbach, Germany), and CD45-FITC monoclonal antibody (HI30) (11-0459-42), mouse IgG1 kappa isotype control (FITC) (11-4714-42), mouse IgG1 isotype control (PE) (MA1-10415) and Stem Pro™ Accutase™ were purchased from Thermo Fisher Scientific (Waltham, MA, USA). All other chemicals used in the present study were of at least analytical grade.
2.2. Cell Culture
Cell cultures were established from a human third molar after informed consent and approval by the Ethics Committee of the Faculty of Medicine at the University of Regensburg (reference 16-101-0022, date of approval 16 March 2016). Human primary dental pulp cells (HPCs) from the root section of the pulp explant (HPC-10 m) were routinely cultivated in MEM α supplemented with 2.2 g/L of sodium bicarbonate (NaHCO
3), 10% fetal bovine serum (FBS), 500 U/mL of penicillin, and 0.5 mg/mL of streptomycin in a humidified atmosphere containing 5% CO
2 at 37 °C as described earlier [
20]. Cells were used up to the sixth passage after thawing.
2.3. MTT Assay
Cells from routine culture were seeded (7 × 10
3/well; 96-well plates) in 200 µL of routine culture medium (complete MEM α, M1) and incubated for 48 h at 37 °C and 5% CO
2. Next, cell cultures were exposed to LPS (0–50 µg/mL), NAC (0–20 mM), BSO (0–200 µM), or to fisetin (0–200 µM) for 24 h or 72 h in the routine culture medium M1, M1 supplemented with 10 nM dexamethasone and 10 mM β-glycerophosphate (M2), or M1 supplemented with 10 nM dexamethasone, 10 mM β-glycerophosphate and 50 µg/mL ascorbic acid (M3). Before administering NAC to cell cultures, solutions were conditioned at 37 °C and 5% CO
2 to allow for pH neutralization. After that, the exposure media were replaced by 200 µL/well of a solution of MTT (0.5 mg/mL) in M1, and cell cultures were incubated at 37 °C for 3 h in a humidified 5% CO
2-atmosphere. Then, the MTT solution was removed and replaced with 200 µL/well of DMSO (dimethyl sulfoxide) and gently swirled for 10 min. The optical density in each well was immediately measured using a spectrophotometer (Infinite F200, TECAN, Maennedorf, Zurich, Switzerland) at a wavelength of 540 nm. Each experiment was performed two times in duplicates per experimental condition (n = 4). The optical density readings obtained from treated cell cultures were summarized and normalized to untreated cell cultures (100%). The influence of the various supplements added to the routine culture medium (M2, M3) on cell viability was analyzed accordingly and is shown in the
Supplementary Materials (M1 = 100%).
2.4. Detection of Reactive Oxygen Species (ROS)
Cells from routine culture were seeded (5 × 104/well) in 2 mL of M1 in 6-well plates and allowed to adhere for 48 h. Cell cultures to be treated with BSO were pre-incubated 24 h after seeding with 50 µM BSO for 18 h. Then, cell cultures were exposed to BSO (50 µM), NAC (2 mM or 5 mM) or fisetin (5 µM, 25 µM or 50 µM) both in the presence or absence of LPS (0.1 µg/mL).
The exposure of cultures was stopped after 3 h or 24 h by discarding the exposure media, and the intracellular production of ROS was measured by flow cytometry after staining the cells with the fluorescent dyes 2′,7′-dichlorodihydrofluorescin diacetate (DCFH2-DA). Favorable properties of this dye for the investigation of oxidative stress have been outlined previously [
21]. Cell cultures were incubated with 10 µM DCFH2-DA in culture medium 30 min prior to harvesting in phosphate-buffered saline (PBS)/5 mM ethylenediaminetetraacetic acid (EDTA). Then, the cells were collected by centrifugation and the cell pellet was resuspended in 200 μL calcium and magnesium-free PBS. 2′,7′-dichlorofluorescein (DCF) fluorescence was measured in the FL-1 channel (488/515-545) using a BD FACSCanto (Becton Dickinson) flow cytometer. Mean fluorescence intensities (MFIs) were established using histogram statistics and FACSDiva
TM 5.0.2 (Becton Dickinson) software. Individual fluorescence intensities measured in treated cell cultures were related to fluorescence quantified in unstained control cultures.
2.5. Immunophenotyping In Vitro by Flow Cytometry
The expression of surface markers in primary pulp cell cultures was analyzed by flow cytometry. Cells from routine cultures were seeded (5 × 104/well) in 3 mL of the M1 medium in 6-well plates and then they were let to adhere for 48 h. Cell cultures to be treated with BSO were pre-incubated 24 h after seeding with 50 µM BSO for 18 h in M1 culture medium. Cell cultures were then treated with BSO, NAC or fisetin in the presence or absence of LPS for 24 h or 3 days in the M1 or the M2 medium. After that, cells were harvested with 3 mL of Stem Pro™ Accutase™, collected by centrifugation in the cold, and washed once with FACS buffer made by 10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) buffer at pH 7.4, 140 mM sodium chloride (NaCl) and 2.5 mM calcium chloride (CaCl2. Cells were incubated with fluorochrome-conjugated antibodies (1:50 dilutions) in 50 μL of FACS buffer for 15 min in the dark. Cells were stained separately in each single screening tube with cluster of differentiation (CD)90-FITC, CD73-PE, CD105-PE, CD140a-FITC or isotype controls (FITC or PE). Then, the excess of antibodies was removed by adding fresh FACS buffer and centrifugation. After that, 20,000 events were run in a BD FACSCanto flow cytometer (BD Biosciences). Relative fluorescence emissions of gated cells by forward and side scatter properties (FSC/SSC) were analyzed with FACSDivaTM 5.0.2 (BD Bioscience) and expressed as mean fluorescence intensities (MFIs). The MFI ratio was calculated by dividing the fluorescence of the sample by the fluorescence of the related isotype control. Individual values obtained from independent experiments were summarized as means and standard deviations.
2.6. Odontogenic Differentiation In Vitro
Cells from routine culture were seeded (2 × 10
4/well) in 0.5 mL of M1 in 48-well plates and then let to adhere for 48 h. Cell cultures to be treated with BSO were pre-incubated after 24 h from the seeding with 50 µM BSO for 18 h in M1 medium. Then, the cultures were exposed to BSO, NAC or fisetin in the presence or absence of LPS in M1 or M2 medium. Fresh exposure media were provided every 72 h. The exposure of cultures was stopped after 1, 3, 7, 15 (data not shown), or 21 days by discarding the exposure media. Then, cell cultures were washed with PBS and fixed with 10% paraformaldehyde for 15 min at room temperature. Calcium precipitation was detected after Alizarin staining of cell cultures following an established procedure [
20].
2.7. Semi-Quantitative Real-Time PCR
Semiquantitative real-time PCR (TaqMan, Thermo Fisher Scientific, Waltham, MA, USA) was performed to analyze the expression of BMP-2 (Hs00154192_m1) and ALP (Hs01029144_m1) with RPS18 (Hs01375212_g1), 18S (Hs99999901_s1) and ACTB (Hs01060665_g1) as housekeeping genes. Cells from routine culture were seeded (2 × 104/well) in 0.5 mL of M1 in 48-well plates in triplicate and then they were let to adhere for 48 h. Cell cultures to be treated with BSO were pre-incubated after 24 h from the seeding with 50 µM BSO for 18 h in M1 medium. Then, the cultures were exposed to BSO, NAC or fisetin in the presence or absence of LPS in M1 or M2 medium. Fresh exposure media were provided every 72 h. The exposure of cultures was stopped after 1 (data not shown), 3, 7, 15 (data not shown), or 21 days by discarding the exposure media, and cell pellets were obtained by harvesting cells after incubation with Accutase™ and by centrifugation. RNA was isolated from cell pellets by RNeasy Mini Kit (Qiagen, Hilden, Germany), quantified spectrophotometrically, and 200 ng was transcribed into first strand cDNA in a volume of 20 µL (Omniscript RT Kit, Qiagen, Hilden, Germany). Real-time PCR was performed in a total volume of 20 µL containing 1 µL of TaqMan Assay, 10 µL TaqMan Fast Advanced Master Mix, 7 µL H2O and 2 µL cDNA (1:8 diluted). After denaturation at 95 °C (20 s), 40 sequential cycles were performed with denaturing at 95 °C (1 s) and annealing/elongation at 60 °C (20 s) in a QuantStudio 3 Real-Time PCR System (Thermo Fisher Scientific, Waltham, MA, USA). Delta Ct-values of BMP-2 and ALP were calculated by normalization on RPS18, 18 s and ACTB in average, and fold changes were presented relative to M1 or M2 at day 3 (Delta-Delta Ct-method). Results were summarized from two independent experiments performed in duplicates (n = 4).
2.8. Determination of IL-6
Exposure media collected every 72 h from 48-well plates for semi-quantitative real-time PCR were used for the analysis of interleukin-6 (IL-6). IL-6 secretion was quantified using a standard ELISA kit containing biotinylated anti-human monoclonal antibodies following the manufacturer’s instructions (BD Biosciences). The absorbance was read at 450 nm by means of a spectrophotometer (Infinite F200, TECAN, Mainz, Germany), optical densities were collected (Magellan software; version 6.2) and IL-6 concentrations were calculated fitting the obtained optical densities in a standard curve ranging from 4.7 to 300 pg/mL. Detectable amounts of IL-6 were summarized from duplicate measurements in independent experiments.
2.9. Statistical Analysis
Statistics were performed using one-way analysis of variance (ANOVA) followed by Tukey’s multiple comparison test by means of the Prism 5.0 software (GraphPad, San Diego, CA, USA). Results are presented as mean values ± standard deviations. Values of p ≤ 0.05 were considered statistically significant.
4. Discussion
Being functionally responsible for the maintenance and repair of tissues of the dentin-pulp complex and its associated immune system, pulp cells own a high regenerative capacity and respond to various types of damage. Consequently, dental MSCs proliferate and migrate into the damaged tissue, differentiate into odontoblast-like cells, and form reparative dentin as the main mechanism leading to reparative dentinogenesis [
23]. LPS is a glycolipid and a pathogenic molecule of Gram-negative cariogenic bacteria capable of inducing dental pulp inflammation. The endotoxin binds to its Toll-like receptor (TLR)-4 and triggers the activation of the redox-sensitive transcription factor nuclear factor kappa-light-chain-enhancer of activated B cells (NFκB) or MAPKs, thus resulting in the release of pro- or anti-inflammatory cytokines, such as IL-6 [
24].
In this study, we investigated the relevance of the cellular redox balance of vital dental pulp cells functions. To this end, pulp cells were stimulated with a low concentration of LPS (0.1 µg/mL) to induce a perturbation of the intracellular redox equilibrium and mimic inflammation. In addition, cells were exposed to BSO as an irreversible inhibitor of γ-glutamylcysteine synthetase (γ-GCS) to exacerbate the intracellular oxidative stress [
18], and to the well-known reducing agent NAC, capable of successfully counteracting oxidation in several cell models [
18,
25]. Moreover, the natural flavonol fisetin was administered to HPCs. Fisetin (3,3′,4′,7-tetrahydroxyflavone) is a dietary flavonoid found in various fruits and vegetables and has shown strong anti-inflammatory, antioxidant, anti-tumorigenic, anti-invasive, anti-angiogenic, anti-diabetic, neuroprotective, and cardioprotective effects in cell cultures and in animal models relevant to human diseases [
19]. However, its mechanism of action is still under investigation and its potential role in the mineralization process has been only recently disclosed [
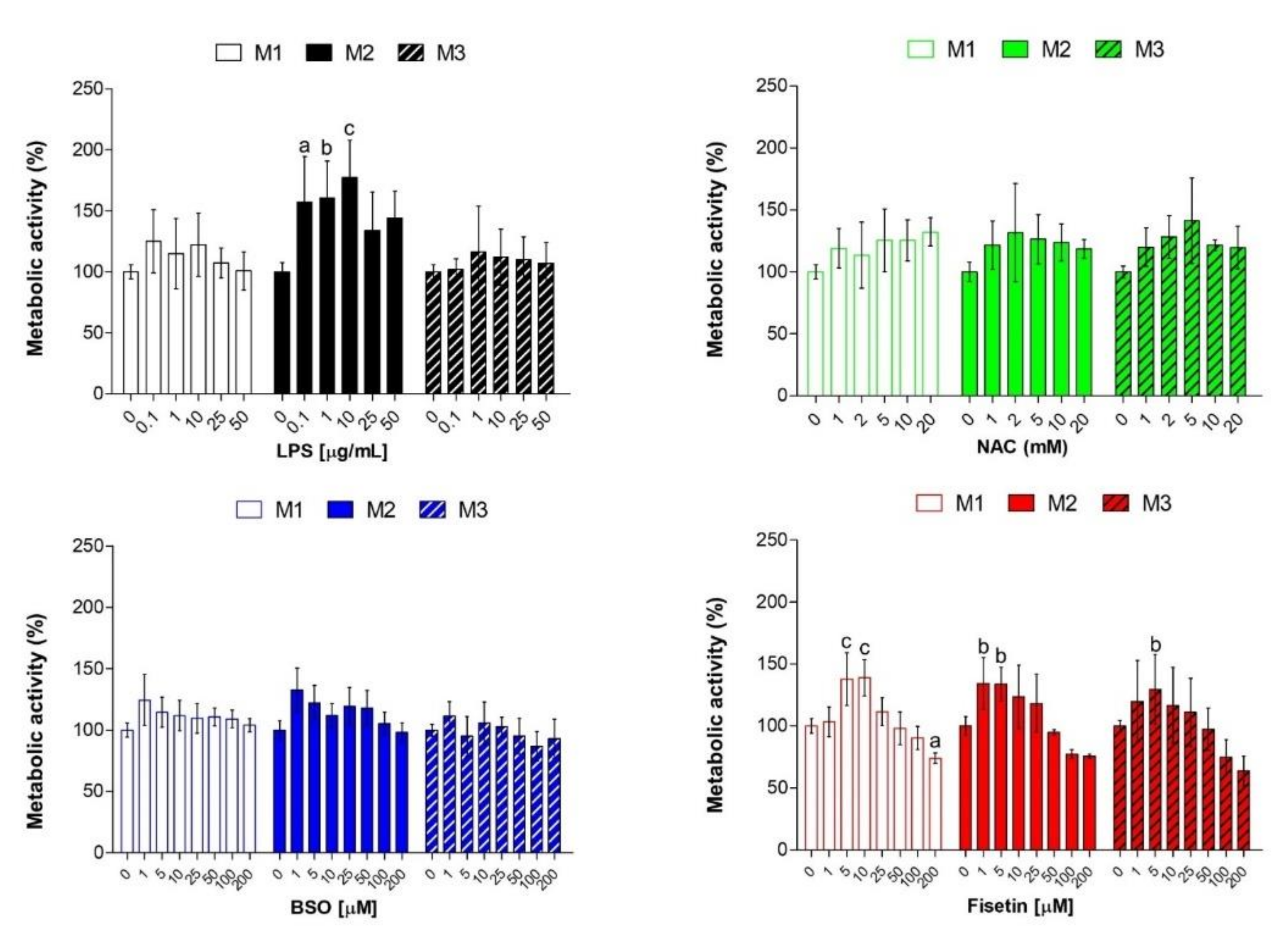
26]. Preliminary analyses on metabolic activity and the generation of ROS were performed to establish the accuracy of our experimental model (
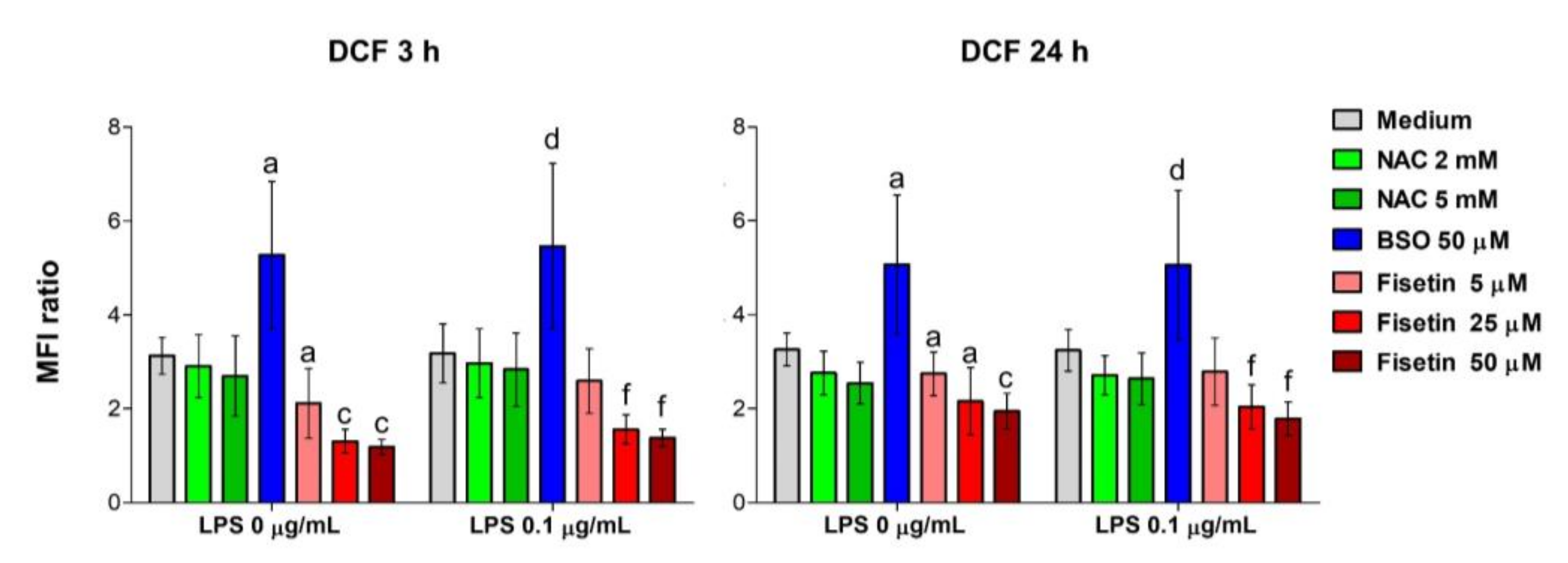
Figure 1,
Figure 2 and
Figure S2). Only the highest concentrations of NAC decrease cell metabolic activity mainly after 3 days; whereas, in the presence of fisetin, a bell-shaped trend is registered, and this flavonoid starts to be cytotoxic at concentrations higher than 50 µM. This could be ascribed to the so-called antioxidant-paradox, broadly discussed elsewhere [
27]. In light of this, concentrations of fisetin tested non-cytotoxic under the current experimental conditions were used for further analyses in the present investigation. Moreover, considering that cell metabolic activities towards increasing concentrations of the various substances were comparable in the M2 and M3 media, and that ascorbic acid contained in the M3 medium could influence the redox homeostasis of cells reacting with radical compounds [
28], only the M1 and the M2 media were used for further analyses. ROS measurements were instead performed only in complete medium M1 to detect the influence of the single substances also in combination with LPS, avoiding any other interference from additives used in the odontogenic media M2 or M3. As expected, BSO increases the intracellular concentration of reactive oxygen species measured by DCF already at the earliest exposure time (3 h), while the two antioxidants exert their proper activity reducing them in a dose-dependent manner over the time of the experiment. Thus, these suitable experimental conditions were used to analyze the relevance of parameters related to mineralization and odontoblastic differentiation.
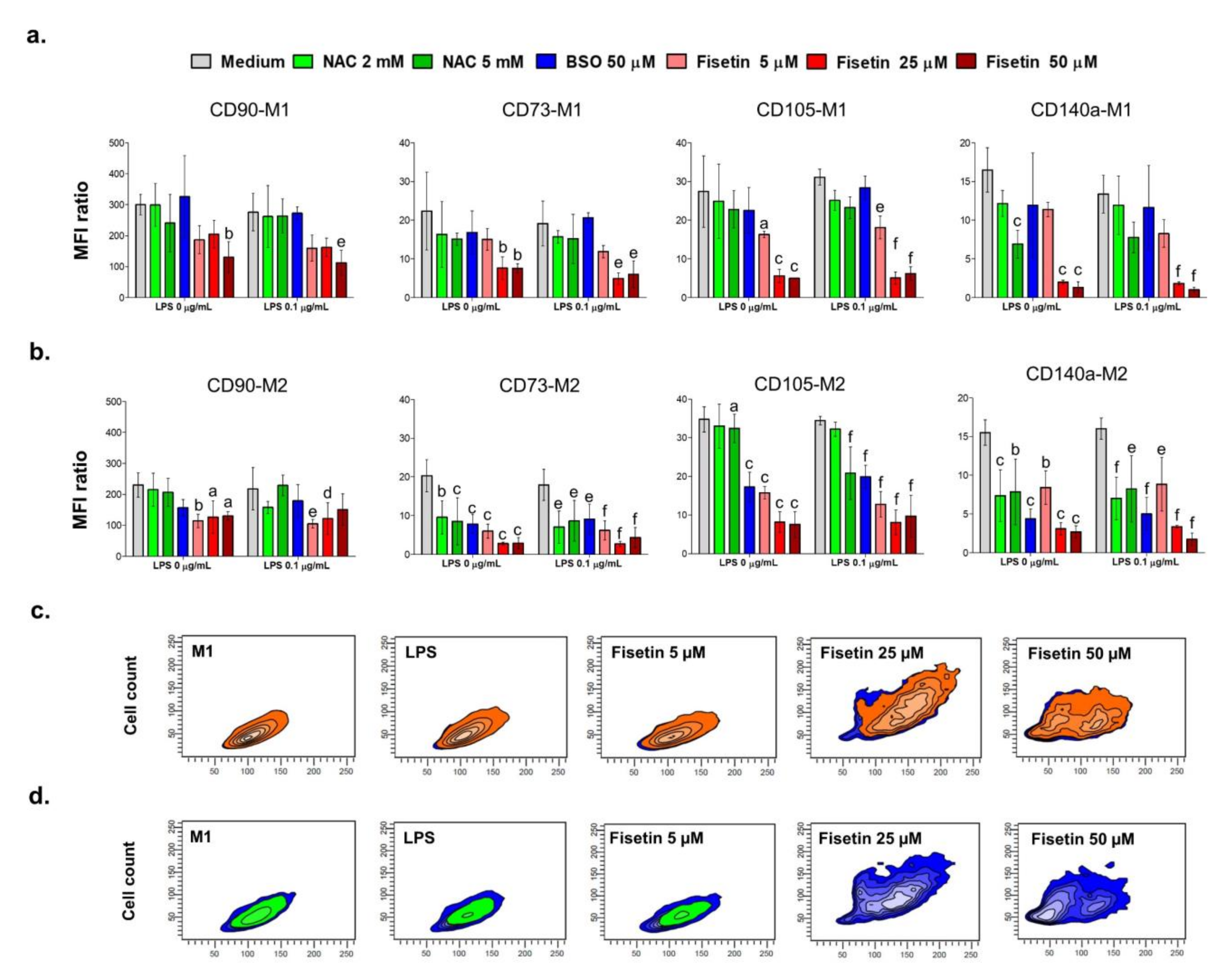
Since the present paper aims to investigate the relationship underlying oxidative stress and mineralization occurrence, evaluating the modulation of CD markers involved in the HPC odontogenic commitment under pro- and antioxidant conditions is crucial. A minimal phenotypic pattern for the identification of MSCs requires them to be positive for CD90, CD73 and CD105 expression, while being negative for CD34 or CD45 [
8]. As for CD140a (platelet-derived growth factor receptor A, PDGFRα), it is reported to identify a population of progenitor cells of neural crest origin and it has been included in this investigation due to its weak expression found in cells previously isolated from the dental pulp-interface and immunophenotypically characterized [
20]. The positivity of MSCs from the dental pulp to the various cell markers is differentially modulated under the odontogenic commitment [
17]. Firstly, CD90 (Thy-1) is found decreased in a time-dependent manner, meaning that cell stemness is reduced towards the osteogenic differentiation [
17]. Secondly, the ecto-5′-nucleotidase CD73 is upregulated under osteogenic conditions and highly expressed in odontoblasts, as it is involved in adenosine production and in the enhancement of bone metabolism through the Wntβ-catenin signaling [
29]. Thirdly, expression levels of the co-receptor for ligands of the transforming growth factor (TGF)-β superfamily (endoglin or CD105), have been found to increase rapidly in freshly harvested MSCs after 4–7 days of culture, and afterwards they gradually decrease during differentiation [
30]. Regarding our experimental model, we show that the stimulation with a low LPS concentration does not affect the immunophenotypic profile of HPCs compared to controls. This is in accordance with the heavily calcium deposition shown with the Alizarin red staining at 21 days in M2. Similar findings were previously reported [
31]. This intriguing observation can be plausibly related to a positive role of low concentration of nitric oxide (NO) in the stimulation of odontogenic differentiation [
32], because LPS is an enhancer of NO release through the activation of NFκB and the subsequent induction of nitric oxide synthase (iNOS) [
33]. On the other hand, a massive NFκB activation consequent to an exaggerated NO production has been reported to impair differentiation of MSCs by promoting β-catenin degradation [
34]. Thus, pathways underlying the promotion of HPC odontogenic differentiation under sub-toxic LPS-stimulation should be further investigated, along with the potential control of mineralization not only through oxygen radicals but also through the modulation of nitrosative species.
In parallel, in the presence of BSO, a decrease in CD73 and CD140a is registered, as well as a lack of mineralization. It is plausible to assume that the administration of BSO, causing a burst of oxidative stress, leads to cytotoxicity and does not allow for the odontogenic commitment of HPCs. Moreover, BSO, depleting the GSH pool and thus presumably impairing the GSH/GSSG ratio, can affect osteogenic activity as reported elsewhere [
35]. Next, the presence of NAC affects the enhanced expression of CD73 found in controls mainly at the highest concentration. In parallel, mineralization is decreased with 5 mM NAC in the M2 medium, especially in the absence of LPS. NAC has been widely described as a direct ROS scavenger [
36] and it has been demonstrated that the MSC osteogenic commitment is under redox control with low amounts of oxygen radicals acting as molecular signals [
14,
17]. The negative effect on the expression of CD markers and even more on the mineralized matrix deposition can be observed with fisetin in a dose-dependent manner, in accordance with the recent work of Lorthongpanich and colleagues [
26]. This is extremely evident when one considers the decrease in positivity of cell populations to CD73 and CD140a (
Figure 3c,d, respectively) and the lack of Alizarin red staining when cells are exposed to 25 and 50 µM fisetin independently from the presence of LPS. Moreover, the microscopic observation of treated cell cultures reveals a fisetin-dependent decrease in cell density and attachment, which are crucial in triggering mineralization. Notably, this observation lays the grounds for further investigations regarding the role of flavonoids in the mineralization process. Indeed, a low dose-fisetin administration could be beneficial, if ROS is properly decreased and the calcium deposition allowed. It is currently believed that a regulated and moderated ROS production promotes essential signaling pathways such as Erk1/2 phosphorylation, which modulates cell functions such as differentiation. Moreover, numerous reports describe the importance of a redox control of MSC differentiation [
37]. Furthermore, fisetin and other polyphenolic structures that own a catechol ring are particularly prone to specific aromatic electron delocalization which may happen as a result of contact with hydrogen acceptors, quinone, and vicinal diketones [
38]. In addition, multihydroxyflavonols can undergo several metabolic reactions faster than other substances in a large plethora of culture media producing metabolites in different amounts whose effects on cell viability are not clarified [
39]. Again, the administration of exogenous antioxidants can be a double-edged sword in the cellular redox state, due to a subtle equilibrium existing between beneficial effects at physiologic doses versus deleterious effects at high doses [
40].
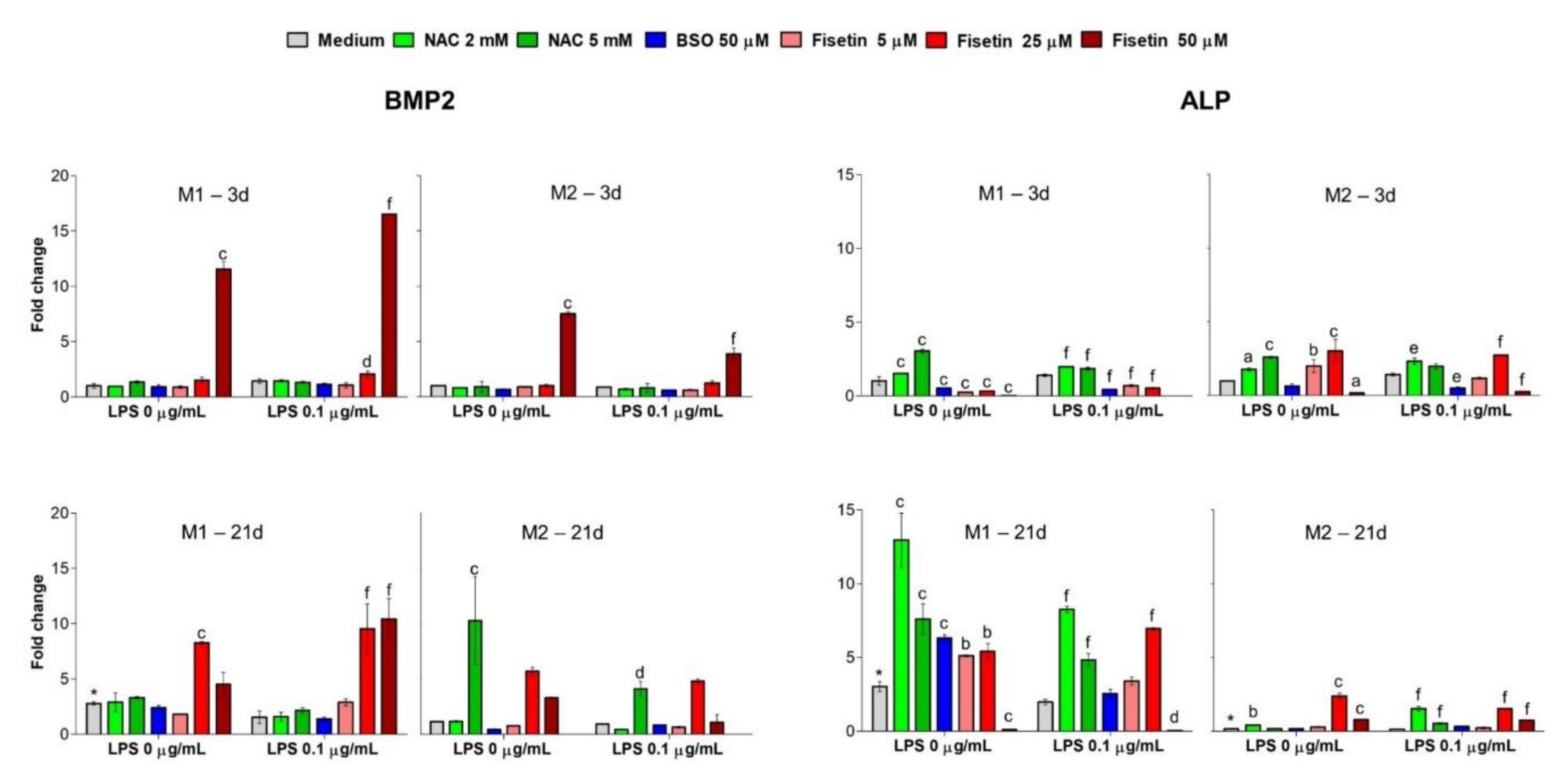
Bone morphogenic protein 2 (BMP2) is widely known as a potent growth factor early expressed in odontogenic precursors. To differentiate MSCs, BMP-2 binds to type I and type II serine/threonine kinase receptors on target cells, activating Smad (canonical) and non-Smad (non-canonical) signaling pathways, which ultimately activates odontogenic genes such as Runt-related transcription factor 2 (RUNX2) and osterix (Osx) [
41]. In parallel, alkaline phosphatase (ALP) is highly expressed in cells of mineralized tissue and is critical in the formation of hard tissue. ALP functions as an inorganic phosphate rate enhancer and facilitates mineralization as well as reducing the concentration of extracellular pyrophosphate, an inhibitor of mineral formation [
42]. Surprisingly, gene expression levels of BMP2 in our experimental model are clearly enhanced in samples treated with substances which do not allow for the achievement of mineralization after 21 days, for instance, 5 mM NAC (21 days in M2), 25 µM fisetin (after 21 days) and 50 µM fisetin (in all the experimental conditions, except for 21 days M2). It is plausible to assume that the observed gene upregulation is a positive feedback mechanism in a continuous effort of HPCs to achieve hard tissue formation. Simultaneously, upregulation of the ALP gene is registered for NAC at 21 days only in M1, which plausibly does not lead to a functional enhanced activity of the enzyme due to the decreased mineralization observed at the same experimental time. In parallel, ALP expression is not enhanced in the M2 conditions. The optimal pH for the ALP activity ranged between 8.5 and 9.5 [
43]. In light of this, it can be speculated that, as for BMP2, the ALP gene is continuously up regulated but the activity of the enzyme is not optimal, due to the acidity of the environment caused by the high concentration of NAC. This is not observed in the M2 medium, resulting indeed in a better deposition of calcium nodules. As for fisetin, a strong induction is registered after 21 days in the M1 medium, and a weak one can be observed also in the presence of M2. Once again, although the expression of the ALP gene is triggered, there is a failure in its functional activity due to the lack of mineralized matrix deposition.
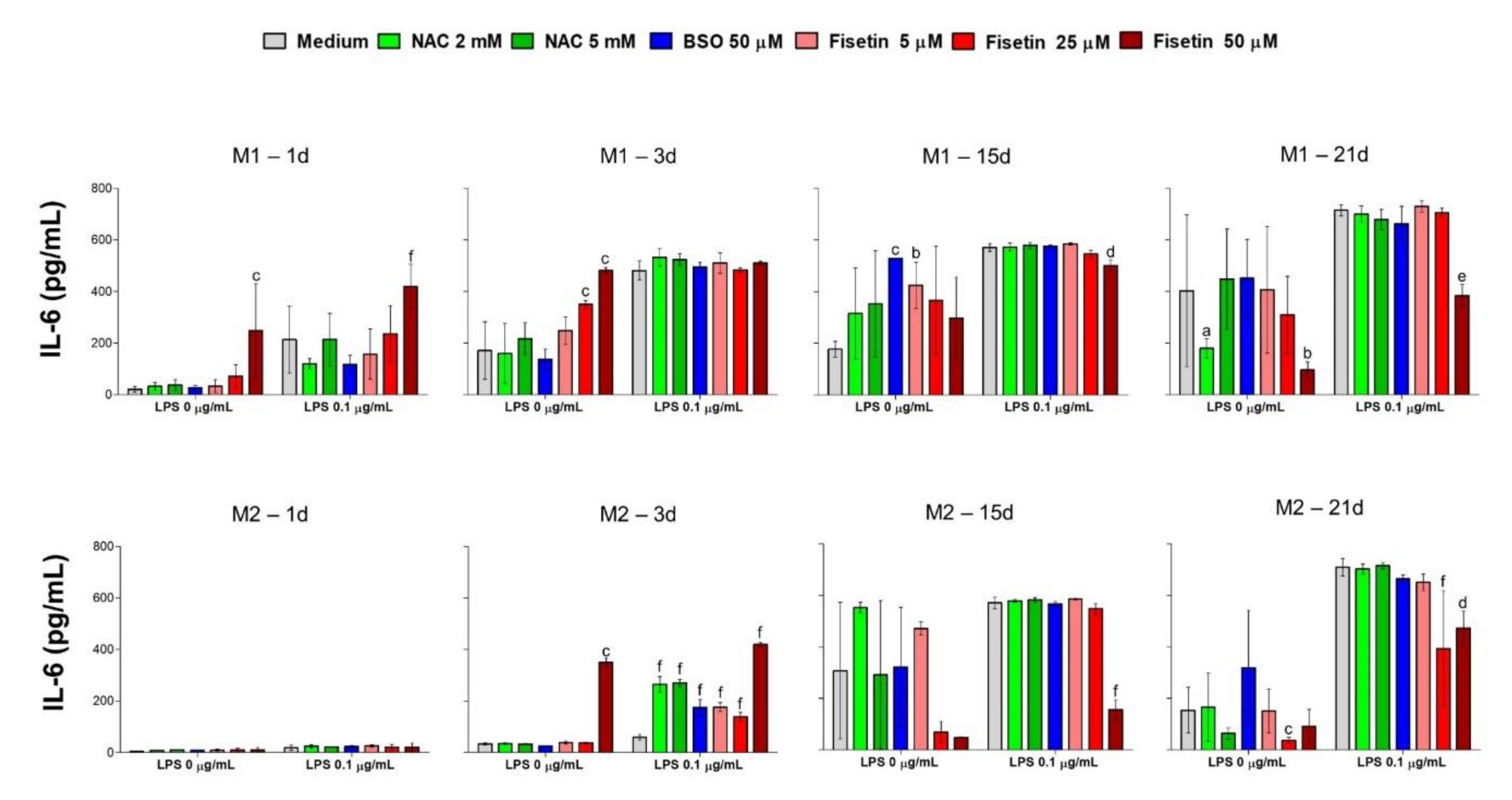
The term osteoimmunology refers to the existing link between the immune and the skeletal system. Pro-inflammatory cytokines, such as tumor necrosis factor (TNF)-α and IL-6 play important roles in immune responses and bone metabolism. LPS-induced secretion of TNF-α and IL-6 in macrophages is well documented, while IL-6 was recently found to be a crucial mediator of the actions of osteoblasts and osteoclasts through sophisticated mechanisms, which reflect a dual effect of this cytokine [
44]. It was shown that IL-6 contributes to bone remodeling in the early stages of fracture healing, and after fracture surgery, is involved in bone remodeling activation [
4]. In the present data set, a significant induction of LPS-mediated IL-6 secretion can be observed in HPCs already after one day in the M1 medium, and it is maintained up to day 21, independently from other substance administered in parallel. Contrariwise, cells grown in the M2 medium strongly secrete IL-6 in the presence of LPS only after 15 days of culture. This anti-inflammatory effect could be caused by the presence of the low concentration of dexamethasone in the odontogenic medium M2 [
45]. Moreover, it could be speculated that in the M1 conditions, an early IL-6 secretion has a canonical inflammatory function, being involved in the recruitment of activated immune cells in the oral mucosa, which in turn secrete IL-17 contributing to the eradication of oral bacteria [
46]. The latest secretion of IL-6 from HPCs grown in the osteogenic medium M2, could be related to a cytokine-mediated control of cell odontogenic commitment, as already reported in the presence of bone grafts for regenerative purposes. In this experimental model, IL-6 increased in a time-dependent manner started from day 15 in parallel with the activation of antioxidant proteins such as nuclear factor erythroid 2–related factor 2 (Nrf2), phosphorylated extracellular signal-regulated kinase (Erk 1/2) and catalase [
15,
47], thus indicating a redox-control of the odontogenic commitment through IL-6. In addition to Nrf2-related antioxidant enzymes, a spontaneous upregulation of silent information regulator type 1 (SIRT1) and glutathione peroxidase 1 has been reported during MSC osteogenic commitment, which accounted for the enhanced resistance to oxidative stress upon osteogenic differentiation without the administration of antioxidant supplementations [
48].


 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





