
Hyponatremia and Oxidative Stress
by
 ,
,
Benedetta Fibbi
1,2, 
Giada Marroncini
1,2,
Cecilia Anceschi
2,
Laura Naldi
2 and
Alessandro Peri
1,2,* 1
Pituitary Diseases and Sodium Alterations Unit, AOU Careggi, 50139 Florence, Italy
2
Endocrinology, Department of Experimental and Clinical Biomedical Sciences “Mario Serio”, University of Florence, AOU Careggi, 50139 Florence, Italy
*
Author to whom correspondence should be addressed.
Antioxidants 2021, 10(11), 1768; https://doi.org/10.3390/antiox10111768
Submission received: 30 September 2021
/
Revised: 26 October 2021
/
Accepted: 3 November 2021
/
Published: 4 November 2021
(This article belongs to the Special Issue Oxidative Stress and Inflammation as Targets for Novel Preventive and Therapeutic Approaches in Non Communicable Diseases II)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Hyponatremia, i.e., the presence of a serum sodium concentration ([Na+]) < 136 mEq/L, is the most frequent electrolyte imbalance in the elderly and in hospitalized patients. Symptoms of acute hyponatremia, whose main target is the central nervous system, are explained by the “osmotic theory” and the neuronal swelling secondary to decreased extracellular osmolality, which determines cerebral oedema. Following the description of neurological and systemic manifestations even in mild and chronic hyponatremia, in the last decade reduced extracellular [Na+] was associated with detrimental effects on cellular homeostasis independently of hypoosmolality. Most of these alterations appeared to be elicited by oxidative stress. In this review, we focus on the role of oxidative stress on both osmolality-dependent and -independent impairment of cell and tissue functions observed in hyponatremic conditions. Furthermore, basic and clinical research suggested that oxidative stress appears to be a common denominator of the degenerative processes related to aging, cancer progression, and hyponatremia. Of note, low [Na+] is able to exacerbate multiple manifestations of senescence and to decrease progression-free and overall survival in oncologic patients.
1. Introduction
Hyponatremia, defined as a serum sodium concentration ([Na+]) < 136 mEq/L, is the most frequent electrolyte disorder encountered in clinical practice, especially in hospitalized patients and in the elderly [1]. Since hyponatremia is associated with increased morbidity and mortality even in mildly affected patients, it represents an economic burden in terms of hospitalization and health care costs [2,3,4,5,6,7,8]. From this view, a prompt and appropriate correction of this electrolytic imbalance is critical to prevent short- and long-term complications. However, treating hyponatremia is not always perceived as crucial by clinicians, especially when [Na+] is only slightly reduced, but it is potentially associated with negative impacts on body functions [9]. For this reason, the comprehension of molecular mechanisms involved in the pathogenesis of symptoms related to hyponatremia might help to raise the awareness about the importance of correcting even chronic and mild reductions of [Na+].
2. Hyponatremia and Health
Although it has long been thought that persistently but slightly reduced [Na+] was completely inconsequential on health, and therefore did not require any correction [1], nowadays chronic hyponatremia is known to have adverse outcomes on several organs and systems [10].
If prolonged over time, the perturbation of internal homeostasis can lead to permanent injuries of biological functions and potentially life-threatening events, as demonstrated by the association of [Na+] even mildly below the normal range with increased mortality [4,11,12,13,14]. This correlation was confirmed in cross-sectional studies performed on both inpatient and outpatient cohorts [11,12], and in a meta-analysis including 81 studies and 147,948 participants, which estimated an overall mortality risk ratio of 2.60 (95% confidence interval [CI], 2.31–2.93) in hyponatremic compared to normonatremic patients [3]. A worse prognosis was observed in patients affected by mild hyponatremia and heart, liver, brain, kidney and lung diseases [8,15,16,17], but this electrolyte imbalance was initially considered as a mere marker of disease severity rather than accelerating patient deterioration [18]. In some clinical settings, such as heart failure [19] and cirrhosis [20], inadequate circulation determines a non-osmotic trigger to vasopressin secretion, aimed to preserve blood pressure and circulating volume; moreover, the release of antidiuretic hormone can be stimulated in response to stress and hypothalamic-pituitary-adrenal axis activation [21]. The simultaneous measurement of plasma sodium and copeptin (a molecule co-released with vasopressin) in 6962 patients, revealed a significant association of all cause 30-day mortality with hyponatremia even independently of copeptin (and consequently of vasopressin) levels [22].
Besides the presence of a correlation between hyponatremia and mortality, clinical manifestations of chronic hyponatremia also include neurocognitive deficits and bone fractures/osteoporosis. In a cohort of 122 patients admitted to an emergency department and affected by apparently asymptomatic hyponatremia, the frequency of falls was significantly higher than age-matched normonatremic controls [9]; low [Na+] was demonstrated to induce gait disturbances similar to ethanol, which were more severe in patients older than 65 years than in younger subjects [23]. These alterations normalized after hyponatremia correction [9]. More recently, the analysis of data from 5435 patients included in the Osteoporotic Fractures in Men Study revealed a significant association between mild hyponatremia and cognitive impairment and decline at the baseline, evaluated by Mini-Mental Status Examination score or Trail Making Test Part B time. The correlation was much stronger for the second test, which measures executive functions (attention and inhibition control, cognitive flexibility, working memory) [24]. Even if dementia may alter vasopressin secretion [25], the maintenance of the association between cognitive impairment and hyponatremia in patients undergoing hemodialysis and peritoneal dialysis supports a mechanism independent of antidiuretic hormone release [26,27]. Bone fractures are more frequent in mild hyponatremic patients than in normonatremic ones [28,29,30], with a higher risk of hospitalization [12,20,31], increased lengths of stay in the hospital and mortality [32]. The increased rate of fractures associated with chronic hyponatremia is not only due to more frequent falls, but also to a higher prevalence of osteoporosis. Epidemiological data highlighted that the increased risk of bone demineralization depends on both severity and duration of the electrolyte imbalance: the lower the serum [Na+] is and the more it is maintained over time, the higher the patients risk of developing osteoporosis is [33]. Several studies have now confirmed a strong correlation between a decrease in serum [Na+] by as little as 2 to 4 mEq/L below the normal range and osteoporosis and fragility fractures, exceeding the risk related to the use of corticosteroid drugs or smoking [30,34,35,36]. The direct effect of low [Na+] on human health and body homeostasis is confirmed by the reversibility of clinical abnormalities secondary to mild and moderate chronic hyponatremia after appropriate correction. A statistically significant association between an increase in serum [Na+] in hyponatremic patients and a reduction in mortality, with a calculated odds ratio of 0.57 (95% CI, 0.40–0.81) was demonstrated in a meta-analysis of 15 studies [37]. Treatment of marked hyponatremia with the vasopressin antagonist tolvaptan improved mental health (SALT-1 and SALT-2) [38], psychomotor speed domain (INSIGHT) [39], Timed Up and Go test, nerve conduction velocities and F-wave latencies [40], and gait disturbances [9]. In the INSIGHT trial, the improvement of bone frailty after 22 days of tolvaptan treatment was also demonstrated [39]. Finally, the improvement of bone density was reported in a young man after removal of a vasopressin-secreting esthesioneuroblastoma of the maxillary sinus and normalization of [Na+] [41].
Pulmonary diseases are a frequent cause of hyponatremia, occurring in about 30% of patients affected by pneumonia [42]. Retrospective analysis of hyponatremia occurrence in COVID-19 patients during the first pandemic period demonstrated a prevalence of 22.9% at hospital admission, a worse respiratory performance (evaluated as P/F, i.e., the ratio of the partial pressure of oxygen in arterial blood PaO2 to the inspired oxygen fraction FiO2), and higher IL-6 levels in hyponatremic rather than in normonatremic hospitalized patients [43]. Since IL-6 is able to induce vasopressin secretion by a direct hypothalamic stimulation and by inducing alveolar basement membrane injury and pulmonary hypoxia and vasoconstriction [44,45,46,47], the pro-inflammatory cytokine may represent the common denominator of both acute respiratory insufficiency and syndrome of inappropriate antidiuresis (SIAD)-related hyponatremia. A very recent metanalysis of 8 studies and 11,493 patients showed a correlation of hyponatremia with COVID-19 poor outcomes (a composite of mortality, prolonged hospitalization and severe COVID-19, defined as severe pneumonia and/or needing intensive care unit support/invasive mechanical ventilation; OR 2.65 [1.89, 3.72], p < 0.001; I2: 67.2%, p = 0.003), with a 37% sensitivity and 82% specificity; while normal serum [Na+] is associated with a 16% post-test probability of a worse prognosis, the presence of hyponatremia increases this probability up to 33% [48].
An intriguing and unexpected association was also observed between chronic hyponatremia and overall and progression-free survival in cancer patients. An underlying tumor is responsible for about 14% of hyponatremias [49], whose prevalence in the oncologic setting varies with tumor type and treatment protocols, as well as serum [Na+] threshold employed. The most frequent cause of chronic hyponatremia in cancer patients is SIAD, mainly due to ectopic vasopressin secretion by cancer cells [50,51,52,53]. Several clinical evidences reported hyponatremia as an independent, negative prognostic factor in different types of blood and solid tumors (e.g., lymphoma [54], gastrointestinal cancers [55,56], hepatocellular carcinoma [57,58], mesothelioma [59], renal cell carcinoma [60,61], and small cell lung cancer [62,63]), and a concordant improvement of overall and progression-free survival after the appropriate correction of reduced serum [Na+] [24], even in patients with extensive and terminal disease [64]. Accordingly, hyponatremia has been proposed as a biomarker able to identify high-risk subjects affected by lung cancer [65].
3. Osmotically-Induced Oxidative Stress
The “osmotic theory” was the first one formulated to explain neurologic symptoms associated with low extracellular [Na+]. When hyponatremia occurs, the resulting decrease in plasma osmolality (except for the rare cases of non-hypoosmotic hyponatremia) causes water movement into the brain by osmotic gradient, thus causing cerebral oedema [1,66]. The cellular elements most involved in swelling are astrocytes, namely glial cells which are a constituent of the blood-brain barrier and have a fundamental role in maintaining the fluid and electrolyte concentration of the extracellular space in the central nervous system [67]. In the brain, the intracellular/extracellular ionic homeostasis is particularly important, since excitatory and inhibitory synaptic events are driven by ionic gradients, which regulate the resting potential and the discharge pattern of neurons [68]. Sparing neurons from hypoosmolar stress is functional to preserve brain excitability, which is increased both directly (swelling-induced release of excitatory neurotransmitters) and indirectly (restrained diffusion of neurotransmitters and depolarizing agents due to the reduction of extracellular space volume) by swelling [68]. Therefore, an acute decrease in external osmolality determines an initial astrocyte swelling as a result of water movement from the extracellular to intracellular compartment, thus preventing the same phenomenon from occurring in neuronal cells [69] and limiting brain swelling. This first-line defense mechanism is quickly followed by a process known as volume regulatory decrease. This ancient adaptive mechanism, which is able to counteract cell volume alterations and consequently perturbations of cell functions (cell-cycle progression, proliferation, apoptosis, excitability and metabolism) [70], consists in extruding intracellular solutes (electrolytes and organic osmolytes) together with osmotically obligated water [71]. This phenomenon is crucial in the brain, in which the physical restriction of the skull limits the expansion and may determine a life-threatening increase in intracranial pressure. In the first hours, cells mainly lose inorganic ions (first Na+ and Cl−, then K+), as a result of an energy-dependent mechanism based on the activation of the Na+-K+ ATPase pump (the first signaling pathway of osmotransduction activated by cell swelling), Ca2+-dependent and -independent K+ channels, K+/Cl− co-transporters and volume-sensitive Cl− channels [66,71,72,73]. In cells exposed to sustained hyponatremia, a delayed loss of small organic osmolytes also starts: myoinisotol, betaine, creatine and amino acids (taurine, glycine, aspartate, glutamine and glutamate) are progressively extruded [74], and their efflux is maintained as long as low [Na+] persists as an essential adaptive mechanism in chronic hyponatremia. The completion of inorganic solute extrusion within 48 h defines the empirical threshold for acute (<48 h) and chronic (>48 h) hyponatremia [75].
While chronic hyponatremia has been traditionally defined as asymptomatic because of cell volume adaptation, in the last decade several studies demonstrated that even a mild chronic reduction of [Na+] may be associated with neurological signs and symptoms, i.e. gait impairment, attention and memory deficit, and increased risk of falls [3,9,28,76,77]. Accordingly, the correction of low [Na+] may effectively counteract the reduced cognitive performances observed in hyponatremic patients compared to normonatremic subjects [37,38,76]. The mechanisms that potentially cause these alterations are not completely understood, but the impairment of excitatory neurotransmitters may be involved. As previously mentioned, glutamate is one of the most important organic osmolytes involved in cellular adaptation to hyponatremia [71]. In physiologic conditions, the extracellular glutamate concentration is kept low to avoid an excessive activation of its receptors and glutamate neurotoxicity (GNT), a condition characterized by time-dependent damage of many cell components leading to cell death and prevented through the astrocytic re-uptake mediated by the Na+-dependent glial glutamate transporters GLT-1 and GLAST [78]. While the cerebral extracellular concentration of glutamate is increased under acute hypoosmotic conditions [79], its brain content decreases by about 40% after 14 days of sustained hyponatremia in rats [75], thus suggesting an impairment of synaptic excitatory neurotransmission due to chronic hyponatremia. Moreover, it was demonstrated that the sustained reduction of serum [Na+] induces gait disturbances and memory impairment in murine models by decreasing astrocytic glutamate re-uptake (through inhibition of GLT-1 and GLAST activities), and consequently long-term potentiation (LTP) at hippocampal synapses [80]. Nowadays, it is well established that GNT is a result of neuronal Ca2+ overloading, which is triggered by acute neuronal swelling (the cellular uptake of extracellular Na+ and Cl− causes plasma membrane depolarization, and subsequently Ca2+ channel opening) and initiates a cascade-like effect leading to cell death [81]. Beyond mitochondria accumulation of Ca2+, the generation of Ca2+-dependent reactive oxygen species (ROS) (e.g., hydrogen peroxides and superoxides, hydroxyl radicals and oxygen radicals) undoubtedly takes place in GNT [82,83,84,85,86], which is usually associated with marked oxidative stress [87,88]. ROS trigger peroxidative degradation of lipid membranes and modify the redox state of proteins involved in osmotransduction, specifically osmotically-activated tyrosine kinases (ERK1/2, p38, FAK, members of the Src family), which further increase their activity and alter cellular homeostasis [89,90]. Increased ROS formation after exposure to glutamate is divided in two phases: an early ROS production coupled to xanthyne-oxidase activation [91,92], and a later one mostly due to mitochondria as a by-product of glucose metabolism and ATP generation [85,93,94]. Therefore, some authors concluded that in the early GNT, non-mitochondrial ROS generation triggers a cell defense mechanism, while the delayed superoxide production, as well as in apoptosis, occurs secondary to a defect in mitochondrial electron transport and is a result of mitochondrial damage, which acts as a self-propagating process leading to cell dysfunction and death. In particular, the initial oxidative stress could impair mitochondrial energy production and promote depletion of energy stores, thus affecting intracellular homeostatic and protective mechanisms [84].
Beyond neuroactive solutes depletion by neurons, additional mechanisms are hypothesized to explain neurological alterations observed in chronically hyponatremic patients. It is noteworthy that the increased ROS production is also expected to deplete cellular antioxidant defenses, which in turn amplify oxidative stress and radical-mediated injury [95]. As Schultz et al. demonstrated for the first time in vivo, a disturbance of the antioxidant glutathione homeostasis is linked to both excitotoxic neuronal injury and neurodegeneration [96]. Among organic osmolytes, the antioxidants taurine and glutathione are also extruded by neuronal cells in response to extracellular hypoosmolality [97], and the adaptive decrease in their cell content was supposed to make neurons more susceptible to oxidative injury. In fact, glutathione depletion induced by treatment with buthionine sulfoximine or diethylmaleate exacerbates brain injury due respectively to middle cerebral artery ligation in rats [98], and hyperbaric hyperoxia in humans [99]. Using in vivo and in vitro murine and human models, Clark and colleagues demonstrated that brain tissues and cell cultures reduced their content of taurine and glutathione in response to hypoosmolality, and that the depletion was reverted by a slow normalization of serum [Na+] [100]. Regarding intracellular functions involved in the reduced availability of antioxidants, the authors observed an osmotically-induced decrease in the synthetic rate of glutathione (whose direct transport across the blood–brain barrier is supposed to be preserved), and an increased release of taurine from cells into the extracellular medium [100]. It has also been suggested that glutathione produced by astrocytes might be a disposal pathway for glutamate, and that decreased synthesis of the antioxidant due to hypoosmolality could exacerbate the injury induced by neurotransmitter accumulation [100,101]. In agreement with a role of the osmotic depletion of antioxidants in the pathogenesis of hyponatremia-related brain injury, the incidence of cerebral infarction in patients with subarachnoid hemorrhage who developed this electrolyte imbalance was significantly higher than in eunatremic subjects [102]. The same mechanism may also play a role in the pathogenesis of the osmotic demyelination syndrome [100].
In addition to inorganic and organic solutes extrusion, activation of phospholipases (particularly the isoforms A2 and D) is an intracellular pathway involved in osmotransduction signaling, as demonstrated by mobilization of arachidonic acid and lysophosphatidylcholine (LPC) in association with hypoosmotic swelling [37,38]. Arachidonic acid contributes to the regulation of K+ and Cl− channel activity and organic osmolyte efflux, and similarly to LPC, promotes the generation of ROS [91]. Interestingly, arachidonic acid and ROS were found to inhibit glutamate uptake in astrocytes [103].
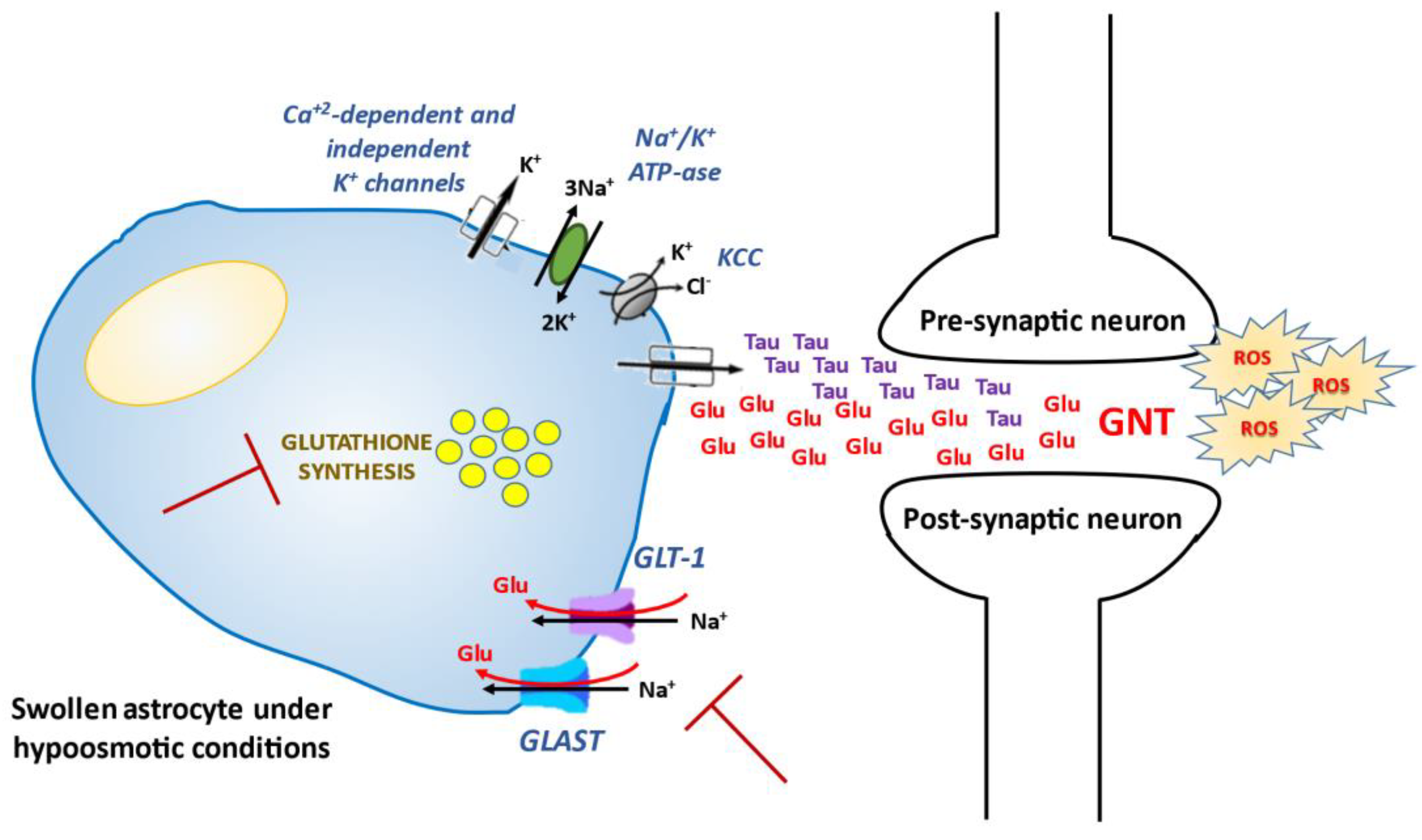
The main mechanisms triggered by hyponatremia and involved in osmotically-induced production of ROS are summarized in Figure 1.
4. Non Osmotically-Induced Oxidative Stress
Nowadays, it is well accepted that the central nervous system is not the only target of low [Na+]. Indeed, mild chronic hyponatremia has also been associated with detrimental effects on bone, specifically increased risk of osteoporosis and fractures independently of bone demineralization [12,30,31,36,104]. Bone matrix is a large reservoir of the body’s Na+, storing approximately one-third of this electrolyte [105]; in dogs, it is an osmotically inactive compartment from which Na+ is released during prolonged dietary deprivation [106]. As demonstrated in a rat model of SIAD, hyponatremia-related osteoporosis is due to increased osteoclastic activity, in the absence of other metabolic or hormonal alterations able to explain the accelerated bone resorption (i.e., sex steroid deficiency, metastasis-induced osteolysis, calcium-mediated signals, etc.) [36].
The described detrimental systemic effects secondary to chronic hyponatremia, traditionally defined as an asymptomatic or mildly symptomatic disorder, open a new scenario in understanding the pathophysiology of this condition and its clinical sequelae. In fact, neurological and extra-neurological alterations observed in chronic hyponatremia are explained in principle neither by the “osmotic theory” nor by the homeostatic mechanisms that counteract cell swelling in the presence of extracellular hypotonicity. Therefore, the intriguing hypothesis that hyponatremia could directly impair cellular homeostasis—and hence health status—was postulated. With regard to this point, Barsony et al. first showed that sustained low extracellular [Na+] activates osteoclastogenesis and osteoclastic bone matrix resorption in rats both in vivo and in vitro, independently of reduced osmolality [104]. In their view, this response is likely necessary to mobilize bone-stored Na+ in the attempt to restore normal extracellular [Na+]. Moreover, low [Na+] is able to stimulate the differentiation of early-stage osteoclast progenitors compared to normonatremic conditions, by increasing their sensitivity to growth factors (in particular M-CSF) through oxidative stress. These findings suggest the existence of a Na+-sensing mechanism or receptor on osteoclasts, as hypothesized also in the central nervous system and in the kidney. Interestingly, the authors found that the activity of the Na+-dependent vitamin C transporter is inhibited by low extracellular [Na+] in a dose-dependent manner, thus resulting in a reduced uptake of ascorbic acid. As well as playing a central role in setting the equilibrium between osteoclastogenenesis and osteoblastic functions, ascorbic acid is also a key scavenger of oxidative stress [107,108]. As expected, reduced ascorbic acid uptake observed in the above-mentioned model of chronic hyponatremia is associated with increased accumulation of ROS in osteoclastic cells and oxidative DNA damage product 8-OHdg in the sera of hyponatremic rats compared to controls, in agreement with the excessive production of free radicals and osteoclastic bone reabsorption observed in other forms of osteoporosis (e.g., estrogen/androgen deficiency and chronic inflammation) [104]. By developing an in vitro model able to mimic chronic hyponatremia, we further assessed the correlation between hyponatremia and bone health and analyzed the second process involved in bone remodeling alongside resorption, namely neoformation of bone matrix. We showed that reduced extracellular [Na+] disrupts gene expression, proliferation, migration, and cytokine production in human mesenchymal stromal cells (hMSC) [109], which are precursors of mesodermal cell types (including adipocytes and osteoblasts of bone matrix) exhibiting different degrees of stemness [110]. In post-menopausal osteoporosis and other conditions characterized by bone loss, the bone marrow shows an imbalance between adipogenesis and osteogenesis, with an accumulation of adipose tissue at the expense of the osteoblastic compartment [111]. In our in vitro model, low [Na+] impairs osteoblast activity and differentiation of hMSC, which are shifted toward the adipogenic phenotype at the expense of the osteogenic one [109].
Oxidative stress is also a well-recognized mediator of degenerative processes related to senescence, other than osteoporosis [112], especially in the brain [113]. It is then not inconceivable to speculate that chronic hyponatremia might play a direct role in the pathogenesis of degenerative diseases, in particular aging-related multi-organ pathologies, and that its combination with comorbidities in old people might critically weaken the defense against oxidative stress. As a consequence, sustained low [Na+] might accelerate the aging process and represent an independent risk factor for the development and progression of age-related infirmities. In fact, the prevalence of hyponatremia increases progressively with aging, and its major impact (in terms of morbidity and mortality) is exerted in the elderly [114]. The link between chronic hyponatremia and senescence is supported by evidence that chronic hyponatremia (also in this case regardless of hypoosmolality) accelerates and exacerbates multiple manifestations of senescence, including osteoporosis, hypogonadism with testicular fibrosis and arrest of spermatogenesis, reduced adiposity, cardiomyopathy with left ventricular hypertrophy and fibrosis, and sarcopenia, in male rats [115]. Consistently with these data, primary cultures of neonatal rat cardiomyocytes exposed to low extracellular [Na+] (but compensated hypoosmolality) and hearts isolated from hyponatremic animals showed increased ROS production and intracellular Ca2+ concentrations compared to control cells and tissues [116]. This results in a greater vulnerability of cells against oxidative stress and an exacerbation of myocardial injury due to ischemia/reperfusion, as evidenced by significantly larger infarct size and lower left ventricular developed pressure after exposure to global hypoxia in rats with hyponatremia compared to normonatremic ones [116]. Reoxygenation of cells triggers a burst of ROS, and their increment in low Na+ conditions may amplify mitochondrial permeability transition pore opening and induce cell death [117]. Swelling and enlargement of mitochondria and destruction of cristae in cardiomyocytes exposed to low [Na+] might be the result of increased ROS content, which in turn could be secondary to intracellular Ca2+ overload and activation of Ca2+-dependent ROS-generating enzymes [118].
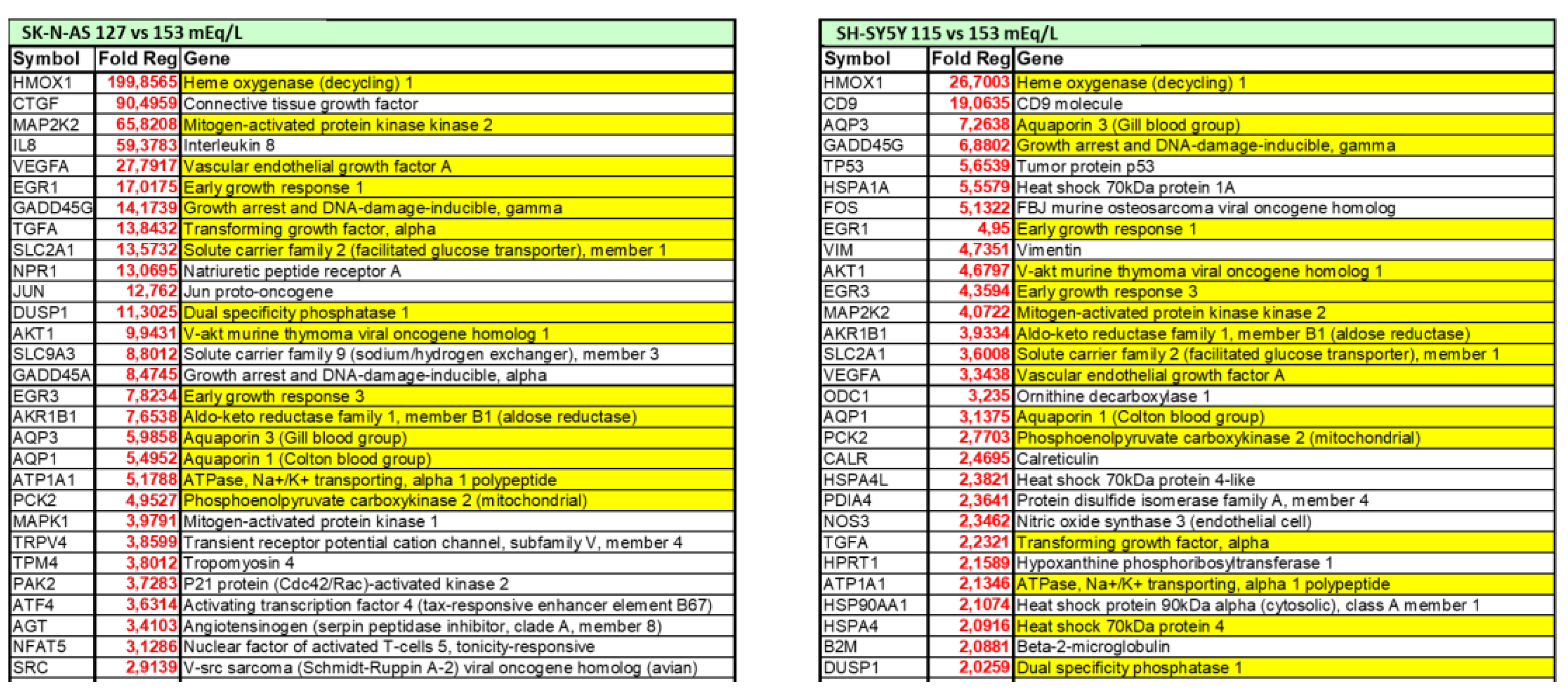
Understanding the potential direct effects of low extracellular [Na+] is of particular interest also in the brain, which is one of the main targets of both chronic hyponatremia and senescence. In the last decade, our laboratory demonstrated that low extracellular [Na+] directly impairs cellular homeostasis in an in vitro neuronal model of chronic hyponatremia [119]. Sustained low extracellular [Na+] was demonstrated to induce cell distress by affecting cell viability and adhesion, expression of anti-apoptotic genes (Bcl-2, DHCR24) and ability to differentiate into a mature neuronal phenotype, even in the presence of compensated osmolality. As a result of a comprehensive microarray analysis, we showed that cell functions involved in “cell death and survival” are the most altered in the presence of reduced [Na+] compared to controls, and that the expression of the heme oxygenase-1 (HMOX-1) gene is the most increased [119] (Figure 2).
HMOX-1 is an inducible stress protein with a metabolic function in heme turnover [120] and potent anti-apoptotic and antioxidant activities in different cells, including neurons [121]. In the brain, induction of HMOX-1 by intracellular factors that directly or indirectly generate ROS, preserves neurons from oxidative injury secondary to cerebral ischemia [122] or ethanol intoxication [123]. Elicitation of oxidative stress in the presence of low [Na+] was confirmed by cytofluorimetric analysis of total intracellular ROS and ROS-induced lipid peroxidation [124]. These findings reinforce the hypothesis that chronic hyponatremia, through increased oxidative stress and ROS generation, may have a role in brain distress and aging by reducing neuronal differentiation ability, a well-known co-factor in the etiopathogenesis of neurodegenerative diseases such as Alzheimer’s disease [125]. Finally, we also demonstrated that the correction of sustained low extracellular [Na+] may not be able to revert all the cell alterations associated with reduced [Na+], specifically the expression level of the anti-apoptotic genes Bcl-2 and DHCR24 or of the HMOX-1 gene, even when [Na+] was gradually increased [124]. Admittedly, these data appear to reinforce the recommendation to carefully diagnose and treat patients with hyponatremia because a prompt intervention aimed to correct serum [Na+] might prevent possible residual abnormalities.
It is now widely accepted that hyponatremia represents a negative independent prognostic factor in oncologic patients, and is associated with poor progression-free and overall survival in several cancers [54,55,56,57,58,59,60,61,62,63]. The direct contribution of this electrolyte imbalance (which cannot be considered a mere surrogate marker of the severity of clinical conditions) is supported by the observation that the correction of serum [Na+] may reduce the overall mortality rate in hyponatremic patients [37]. We recently demonstrated, for the first time, that the reduction of extracellular [Na+] is able to alter the homeostasis of different human cancer cell lines, thus affecting cell functions (i.e., proliferation, adhesion and invasion) distinctive of a more malignant behavior able to increase cell tumorigenicity [126]. The three steps of carcinogenesis (initiation, promotion, and progression) and the resistance to treatment are strongly impaired by an imbalance between ROS and antioxidant production [127,128]. In fact, oxidative stress regulates cell growth, cytoskeleton remodeling and migration, excitability, exocytosis and endocytosis, autophagy, hormone signaling, necrosis, and apoptosis, namely cell properties deregulated in cancer [127,129]. Furthermore, ROS involvement in carcinogenesis, local invasiveness and metastatization is displayed by their ability to induce genomic instability and/or transcriptional errors [130], and to activate pro-survival and pro-metastatic pathways [129]. Our demonstration of an increased expression of HMOX-1 in cancer cell lines cultured in low extracellular [Na+], compared to normal Na+ conditions, validates the role of oxidative stress as the molecular basis of hyponatremia-associated poorer outcomes in oncologic patients [126]. Cancer cells have great abilities to adapt to perturbation of cellular homeostasis, including the imbalanced redox status secondary to their high metabolism and local hypoxia. Through a fine regulation of both ROS production and ROS scavenging pathways (the theory of ROS rheostat), they show a high antioxidant capacity, allowing oxidative stress levels compatible with cellular functions even if higher than in normal cells [131]. Recent studies reported an increased expression of ROS scavengers and low ROS levels in liver and breast cancer stem cells [132,133], whose maintenance is crucial for the survival of pre-neoplastic foci. In this view, chemotherapy and radiotherapy, which strongly induce ROS synthesis, are often able to eliminate the bulk of cancer cells but not to definitely cure cancer, because of the up-regulated levels of antioxidants in stem cells, which are thus spared and selected for in the presence of high ROS. An additional mechanism responsible for therapeutic failure is ROS-dependent accumulation of DNA mutations, leading to drug resistance [131]. In this very complex scenario, antioxidant inhibitors are considered a promising therapeutic tool in cancer treatment, especially regarding glutathione metabolism. Since glutathione is a key regulator of the redox balance and protects cancer cells from stress due to hypoxia and nutrient deficiency in solid tumors, the combination of glutathione inhibitors with radiotherapy or chemotherapy could improve the effects of radiation or drugs. However, other enzymes with a scavenging effect on oxidative stress (HSP90, thioredoxin, enzyme poly-ADP-ribose polymerase or PARP) may be targeted for anticancer treatments, and are currently under study [131]. Since cancer-related hyponatremia adversely affects the response to chemotherapy and everolimus in metastatic renal cell carcinoma [61,134], correction of low serum [Na+] may exert its role in improving cancer survival [135,136] by regulating cancer cell ROS rheostat.
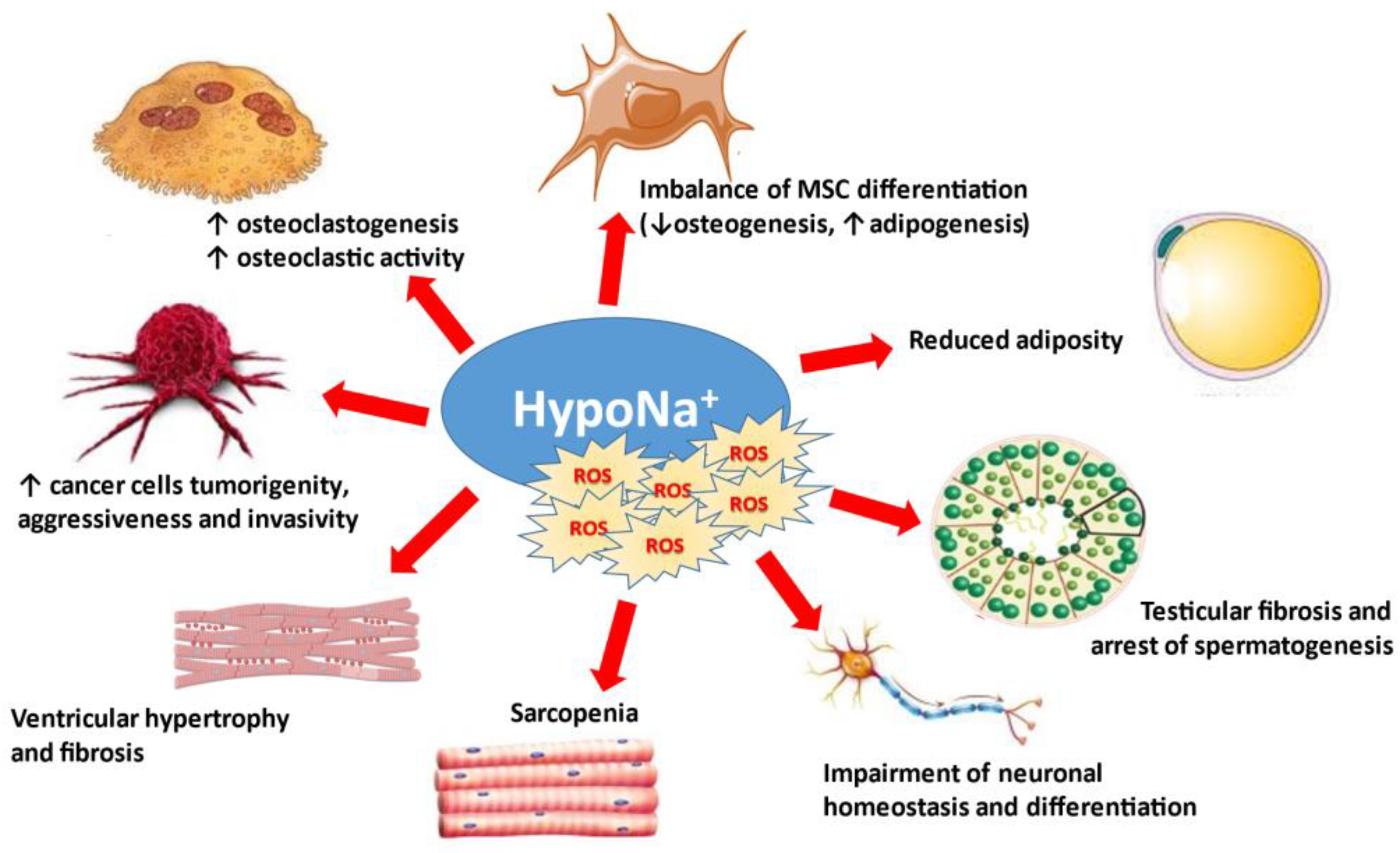
The direct effects of reduced extracellular [Na+] on cells and tissues are summarized in Figure 3.
5. Conclusions
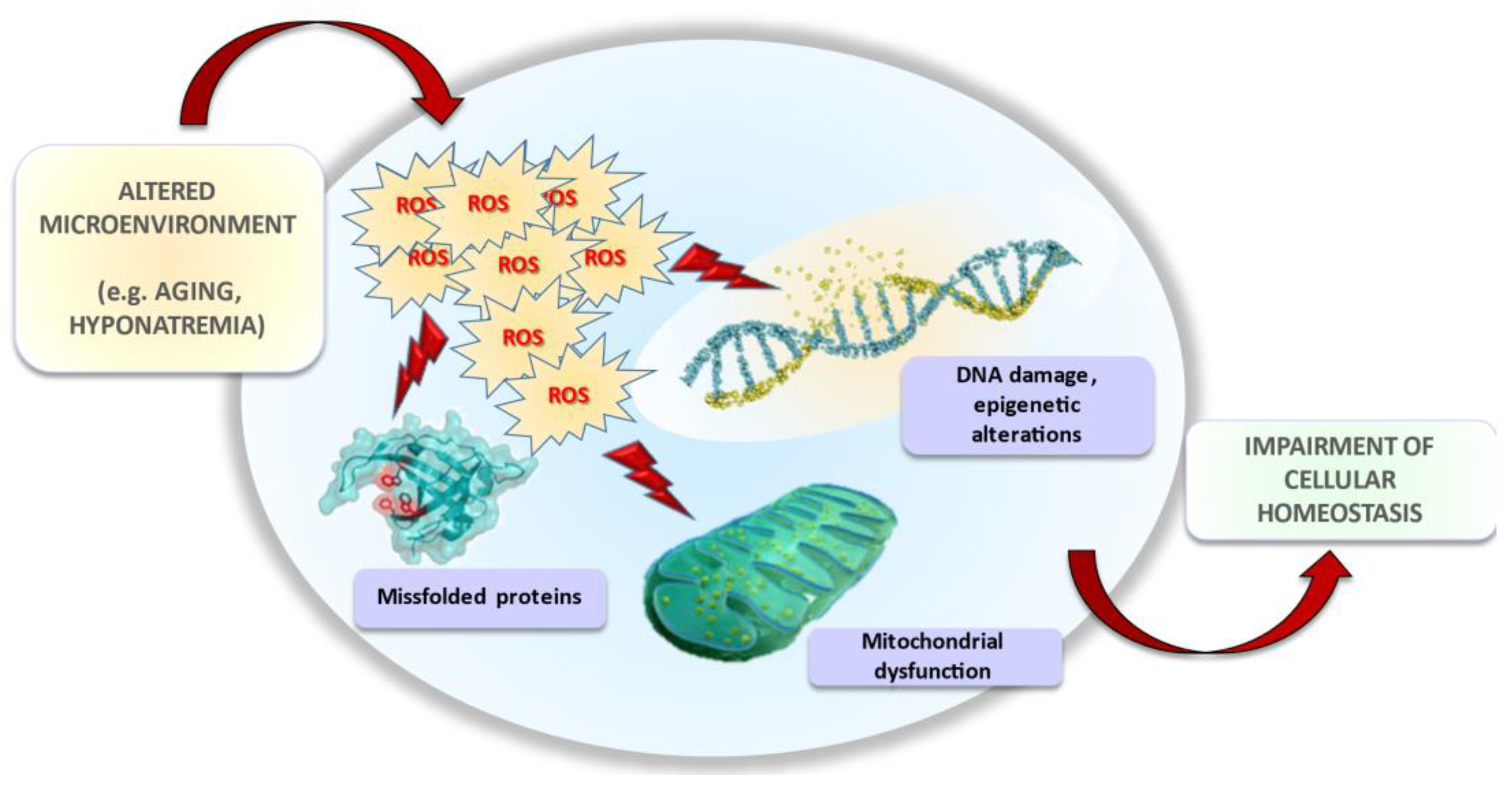
In the last decade, several in vitro and in vivo studies suggested that neurological and extra-neurological symptoms observed in hyponatremic patients might be dependent on a perturbation of cellular homeostasis. Specifically, low extracellular [Na+] impairs cellular functions (e.g., gene protein expression, proliferation, migration, and invasion) involved in senescence and carcinogenesis, thus amplifying tissue injuries related to aging and promoting cancer progression. In this scenario, oxidative stress seems to be the common denominator of intracellular events common to both processes (Figure 4). The studies published in recent years opened a new, unpredicted scenario. Further data will be necessary to fully elucidate the specific molecular pathways triggered by reduced extracellular [Na+] and responsible for oxidative damage, and to comprehensively understand all potential implications of long-term exposure to hyponatremic conditions.
Author Contributions
Writing—review and editing, B.F., G.M., C.A., L.N., A.P. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding. The APC was funded by PRIN 2017R5ZE2C.
Acknowledgments
The authors wish to thank other collaborators, who have been involved in past years in the studies presented in this review, and in particular Susanna Benvenuti, Paola Luciani, Cristiana Deledda, Alice Errico, Federica Baldanzi.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Adrogué, H.J.; Madias, N.E. Hyponatremia. N. Engl. J. Med. 2000, 342, 1581–1589. [Google Scholar] [CrossRef]
- Wald, R.; Jaber, B.L.; Price, L.L.; Upadhyay, A.; Madias, N.E. Impact of hospital-associated hyponatremia on selected outcomes. Arch. Intern. Med. 2010, 170, 294–302. [Google Scholar] [CrossRef]
- Corona, G.; Giuliani, C.; Parenti, G.; Norello, D.; Verbalis, J.G.; Forti, G.; Maggi, M.; Peri, A. Moderate hyponatremia is associated with increased risk of mortality: Evidence from a meta-analysis. PLoS ONE 2013, 8, e80451. [Google Scholar] [CrossRef] [PubMed]
- Mohan, S.; Gu, S.; Parikh, A.; Radhakrishnan, J. Prevalence of hyponatremia and association with mortality: Results from NHANES. Am. J. Med. 2013, 126, 1127–1137. [Google Scholar] [CrossRef] [Green Version]
- Luca, A.; Angermayr, B.; Bertolini, G.; Koenig, F.; Vizzini, G.; Ploner, M.; Peck-Radosavljevic, M.; Gridelli, B.; Bosch, J. An integrated MELD model including serum sodium and age improves the prediction of early mortality in patients with cirrhosis. Liver Transpl. 2007, 13, 1174–1180. [Google Scholar] [CrossRef] [PubMed]
- Grodin, J.L. Pharmacologic approaches to electrolyte abnormalities in heart failure. Curr. Heart Fail. Rep. 2016, 13, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Rossi, J.; Bayram, M.; Udelson, J.E.; Lloyd-Jones, D.; Adams, K.F.; Oconnor, C.M.; Gattis Stough, W.; Ouyang, J.; Shin, D.D.; Orlandi, C.; et al. Improvement in hyponatremia during hospitalization for worsening heart failure is associated with improved outcomes: Insights from the Acute and Chronic Therapeutic Impact of a Vasopressin Antagonist in Chronic Heart Failure (ACTIV in CHF) trial. Acute Card. Care 2007, 9, 82–86. [Google Scholar] [CrossRef] [PubMed]
- Nair, V.; Niederman, M.S.; Masani, N.; Fishbane, S. Hyponatremia in community-acquired pneumonia. Am. J. Nephrol. 2007, 27, 184–190. [Google Scholar] [CrossRef]
- Renneboog, B.; Musch, W.; Vandemergel, X.; Manto, M.U.; Decaux, G. Mild Chronic Hyponatremia is Associated with Falls, Unsteadiness, and Attention Deficits. Am. J. Med. 2006, 119, 71.e1–71.e8. [Google Scholar] [CrossRef] [PubMed]
- Decaux, G. Is asymptomatic hyponatremia really asymptomatic? Am. J. Med. 2006, 119, S79–S82. [Google Scholar] [CrossRef]
- Rondon-Berrios, H.; Berl, T. Mild chronic hyponatremia in the ambulatory setting: Significance and management. Clin. J. Am. Soc. Nephrol. 2015, 10, 2268–2278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoorn, E.J.; Rivadeneira, F.; van Meurs, J.B.; Ziere, G.; Stricker, B.H.; Hofman, A.; Pols, H.A.; Zietse, R.; Uitterlinden, A.G.; Zillikens, M.C. Mild hyponatremia as a risk factor for fractures: The Rotterdam Study. J. Bone Miner. Res. 2011, 26, 1822–1828. [Google Scholar] [CrossRef]
- Sajadieh, A.; Binici, Z.; Mouridsen, M.R.; Nielsen, O.W.; Hansen, J.F.; Haugaard, S.B. Mild hyponatremia carries a poor prognosis in community subjects. Am. J. Med. 2009, 122, 679–686. [Google Scholar] [CrossRef]
- Holland-Bill, L.; Christiansen, C.F.; Heide-Jørgensen, U.; Ulrichsen, S.P.; Ring, T.; Jørgensen, J.O.L.; Sørensen, H.T. Hyponatremia and mortality risk: A Danish cohort study of 279 508 acutely hospitalized patients. Eur. J. Endocrinol. 2015, 173, 71–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Q.Q.; Fan, X.D.; Li, T.; Hao, Y.Y.; Ma, F. Short- and long-term prognostic value of hyponatremia in patients with acute coronary syndrome: A systematic review and meta-analysis. PLoS ONE 2018, 13, e0193857. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, B.; Staff, I.; Fortunato, G.; McCullough, L.D. Hyponatremia in the prognosis of acute ischemic stroke. J. Stroke Cerebrovasc. Dis. 2014, 23, 850–854. [Google Scholar] [CrossRef]
- Curhan, G.C.; Brunelli, S.M. Mortality associated with low serum sodium concentration in maintenance hemodialysis. Am. J. Med. 2011, 124, 77–84. [Google Scholar]
- Chawla, A.; Sterns, R.H.; Nigwekar, S.U.; Cappuccio, J.D. Mortality and serum sodium: Do patients die from or with hyponatremia? Clin. J. Am. Soc. Nephrol. 2011, 6, 960–965. [Google Scholar] [CrossRef] [Green Version]
- Velavan, P.; Khan, N.K.; Goode, K.; Rigby, A.S.; Loh, P.H.; Komajda, M.; Follath, F.; Swedberg, K.; Madeira, H.; Cleland, J.G.F. Predictors of short term mortality in hert failure—insights from the Euro Heart Failure Survey. Int. J. Cardiol. 2010, 138, 63–69. [Google Scholar] [CrossRef]
- Arroyo, V.; Rodes, J.; Gutierrez-Lizarraga, M.A.; Revert, L. Prognostic value of spontaneous hyponatremia in cirrhosis with ascites. Am. J. Dig. Dis. 1976, 21, 249–256. [Google Scholar] [CrossRef]
- Schrier, R.W. Water and sodium retention in edematous disorders: Role of vasopressin and aldosterone. Am. J. Med. 2006, 119, S47–S53. [Google Scholar] [CrossRef]
- Eckart, A.; Hausfater, P.; Amin, D.; Amin, A.; Haubitz, S.; Bernard, M.; Baumgartner, A.; Struja, T.; Kutz, A.; Christ-Crain, M.; et al. Hyponatremia and activation of vasopressin secretion are both independently associated with 30-day mortality: Results of a multicenter, observational study. J. Intern. Med. 2018, 284, 270–281. [Google Scholar] [CrossRef]
- Renneboog, B.; Sattar, L.; Decaux, G. Attention and postural balance are much more affected in older than in younger adults with mild or moderate chronic hyponatremia. Eur. J. Intern. Med. 2017, 41, e25–e26. [Google Scholar] [CrossRef]
- Nowak, K.L.; Yaffe, K.; Orwoll, E.S.; Ix, J.H.; You, Z.; Barrett-Connor, E.; Hoffman, A.R.; Chonchol, M. Serum sodium and cognition in older community-dwelling men. Clin. J. Am. Soc. Nephrol. 2018, 13, 366–374. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, E.D.; Melander, O.; Elmståhl, S.; Lethagen, E.; Minthon, L.; Pihlsgård, M.; Nägga, K. Copeptin, a marker of vasopressin, predicts vascular dementia but not Alzheimer’s disease. J. Alzheimers Dis. 2016, 52, 1047–1053. [Google Scholar] [CrossRef] [PubMed]
- Shavit, L.; Mikeladze, I.; Torem, C.; Slotki, I. Mild hyponatremia is associated with functional and cognitive decline in chronic hemodialysis patients. Clin. Nephrol. 2014, 82, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Duan, L.P.; Dong, J. Hyponatremia and cognitive impairment in patients treated with peritoneal dialysis. Clin. J. Am. Soc. Nephrol. 2015, 10, 1806–1813. [Google Scholar]
- Corona, G.; Norello, D.; Parenti, G.; Sforza, A.; Maggi, M.; Peri, A. Hyponatremia, falls and bone fractures: A systematic review and meta-analysis. Clin. Endocrinol. 2018, 89, 505–513. [Google Scholar] [CrossRef]
- Negri, A.L.; Ayus, J.C. Hyponatremia and bone disease. Rev. Endocr. Metab. Disord. 2017, 18, 67–78. [Google Scholar] [CrossRef]
- Gankam, K.F.; Andres, C.; Sattar, L.; Melot, C.; Decaux, G. Mild hyponatremia and risk of fracture in the ambulatory elderly. QJM Int. J. Med. 2008, 101, 583–588. [Google Scholar] [CrossRef]
- Kinsella, S.; Moran, S.; Sullivan, M.O.; Molloy, M.G.; Eustace, J.A. Hyponatremia independent of osteoporosis is associated with fracture occurrence. Clin. J. Am. Soc. Nephrol. 2010, 5, 275–280. [Google Scholar] [CrossRef] [Green Version]
- Kuo, S.C.; Kuo, P.; Rau, C.; Wu, S.; Hsu, S.; Hsieh, C. Hyponatremia is associated with worse outcomes from fall injuries in the elderly. Int. J. Environ. Res. Public Health 2017, 14, 460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Usala, R.L.; Fernandez, S.J.; Mete, M.; Cowen, L.; Shara, N.M.; Barsony, J.; Verbalis, J.G. Hyponatremia is associated with increased osteoporosis and bone fractures in a large US health system population. J. Clin. Endocrinol. Metab. 2015, 100, 3021–3031. [Google Scholar] [CrossRef]
- Van Staa, T.; Leufkens, H.; Cooper, C. The epidemiology of corticosteroid-induced osteoporosis: A meta-analysis. Osteoporos. Int. 2002, 13, 777–787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kruse, C.; Eiken, P.; Verbalis, J.; Vestergaard, P. The effect of chronic mild hyponatremia on bone mineral loss evaluated by retrospective national Danish patient data. Bone 2016, 84, 9–14. [Google Scholar] [CrossRef]
- Verbalis, J.G.; Barsony, J.; Sugimura, Y.; Tian, Y.; Adams, D.J.; Carter, E.A.; Resnick, H.E. Hyponatremia-induced osteoporosis. J. Bone Miner. Res. 2010, 25, 554–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corona, G.; Giuliani, C.; Verbalis, J.G.; Forti, G.; Maggi, M.; Peri, A. Hyponatremia improvement is associated with a reduced risk of mortality: Evidence from a meta-analysis. PLoS ONE 2015, 10, e0124105. [Google Scholar] [CrossRef]
- Schrier, R.W.; Gross, P.; Gheorghiade, M.; Berl, T.; Verbalis, J.G.; Czerwiec, F.S.; Orlandi, C.; SALT Investigators. Tolvaptan, a selective oral vasopressin V2-receptor antagonist, for hyponatremia. N. Engl. J. Med. 2006, 355, 2099–2112. [Google Scholar] [CrossRef] [Green Version]
- Verbalis, J.G.; Ellison, H.; Hobart, M.; Krasa, H.; Ouyang, J.; Czerwiec, F.S.; Investigation of the Neurocognitive Impact of Sodium Improvement in Geriatric Hyponatremia: Efficacy and Safety of Tolvaptan (INSIGHT) Investigators. Tolvaptan and neurocognitive function in mild to moderate chronic hyponatremia: A randomized trial (INSIGHT). Am. J. Kidney Dis. 2016, 67, 893–901. [Google Scholar] [CrossRef]
- Gosch, M.; Joosten-Gstrein, B.; Heppner, H.J.; Lechleitner, M. Hyponatremia in geriatric inhospital patients: Effects on results of a comprehensive geriatric assessment. Gerontology 2012, 58, 430–440. [Google Scholar] [CrossRef] [Green Version]
- Sejling, A.; Thorsteinsson, A.; Pedersen-Bjergaard, U.; Eiken, P. Recovery from SIADH-associated osteoporosis: A case report. J. Clin. Endocrinol. Metab. 2014, 99, 3527–3530. [Google Scholar] [CrossRef] [Green Version]
- Cuesta, M.; Slattery, D.; Goulden, E.L.; Gupta, S.; Tatro, E.; Sherlock, M.; Tormey, W.; O’Neill, S.; Thompson, C.J. Hyponatraemia in patients with community-acquired pneumonia; prevalence and aetiology, and natural history of SIAD. Clin. Endocrinol. 2019, 90, 744–752. [Google Scholar] [CrossRef]
- Berni, A.; Malandrino, D.; Corona, G.; Maggi, M.; Parenti, G.; Fibbi, B.; Poggesi, L.; Bartoloni, A.; Lavorini, F.; Fanelli, A.; et al. Serum sodium alterations in SARS CoV-2 (COVID-19) infection: Impact on patient outcome. Eur. J. Endocrinol. 2021, 185, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Mehta, P.; McAuley, D.F.; Brown, M.; Sanchez, E.; Tattersall, R.S.; Manson, J.J.; HLH Across Speciality Collaboration, UK. COVID19: Consider cytokine storm syndromes and immunosuppression. Lancet 2020, 395, 1033–1034. [Google Scholar] [CrossRef]
- Park, S.J.; Shin, J.I. Inflammation and hyponatremia: An underrecognized condition? Korean, J. Pediatr. 2013, 56, 519–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, C.; Zhou, L.; Hu, Z.; Zhang, S.; Yang, S.; Tao, Y.; Xie, C.; Ma, K.; Shang, K.; Wang, W.; et al. Dysregulation of immune response in patients with COVID-19 in Wuhan, China. Clin. Infect. Dis. 2020, 71, 762–768. [Google Scholar] [CrossRef]
- Mastorakos, G.; Weber, J.S.; Magiakou, M.A.; Gunn, H.; Chrousos, G.P. Hypothalamic-pituitary-adrenal axis activation and stimulation of systemic vasopressin secretion by recombinant interleukin-6 in humans: Potential implications for the syndrome of inappropriate vasopressin secretion. J. Clin. Endocrinol. Metab. 1994, 79, 934–939. [Google Scholar]
- Akbar, M.R.; Pranata, R.; Wibowo, A.; Irvan; Sihite, T.A.; Martha, J.W. The Prognostic Value of Hyponatremia for Predicting Poor Outcome in Patients With COVID-19: A Systematic Review and Meta-Analysis. Front. Med. 2021, 14, 666949. [Google Scholar] [CrossRef]
- Castillo, J.J.; Vincent, M.; Justice, E. Diagnosis and management of hyponatremia in cancer patients. Oncologist 2012, 17, 756–765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zogheri, A.; Di Mambro, A.; Mannelli, M.; Serio, M.; Forti, G.; Peri, A. Hyponatremia and pituitary adenoma: Think twice about the etiopathogenesis. J. Endocrinol. Investig. 2006, 29, 750–753. [Google Scholar] [CrossRef]
- Padfield, P.L.; Morton, J.J.; Brown, J.J.; Lever, A.F.; Robertson, J.I.; Wood, M.; Fox, R. Plasma arginine vasopressin in the syndrome of antidiuretic hormone excess associated with bronchogenic carcinoma. Am. J. Med. 1976, 61, 825–831. [Google Scholar] [CrossRef]
- Shapiro, J.; Richardson, G.E. Hyponatremia of malignancy. Crit. Rev. Oncol. Hematol. 1995, 18, 129–135. [Google Scholar] [CrossRef]
- Sorensen, J.B.; Andersen, M.K.; Hansen, H.H. Syndrome of inappropriate secretion of antidiuretic hormone (SIADH) in malignant disease. J. Intern. Med. 1995, 238, 97–110. [Google Scholar] [CrossRef]
- Dhaliwal, H.S.; Rohatiner, A.Z.; Gregory, W.; Richards, M.A.; Johnson, P.W.; Whelan, J.S.; Gallagher, C.J.; Matthews, J.; Ganesan, T.S.; Barnett, M.J. Combination chemotherapy for intermediate and high grade non-Hodgkin’s lymphoma. Br. J. Cancer 1993, 68, 767–774. [Google Scholar] [CrossRef] [Green Version]
- Zhou, M.H.; Wang, Z.H.; Zhou, H.W.; Liu, M.; Gu, Y.J.; Sun, J.Z. Clinical outcome of 30 patients with bone marrow metastases. J. Cancer Res. Ther. 2018, 14, S512–S515. [Google Scholar]
- Choi, J.S.; Bae, E.H.; Ma, S.K.; Kweon, S.S.; Kim, S.W. Prognostic impact of hyponatraemia in patients with colorectal cancer. Colorectal Dis. 2015, 17, 409–416. [Google Scholar] [CrossRef] [PubMed]
- Gines, P.; Guevara, M. Hyponatremia in cirrhosis: Pathogenesis, clinical significance, and management. Hepatology 2008, 48, 1002–1010. [Google Scholar] [CrossRef]
- Cescon, M.; Cucchetti, A.; Grazi, G.L.; Ferrero, A.; Viganò, L.; Ercolani, G.; Zanello, M.; Ravaioli, M.; Capussotti, L.; Pinna, A.D. Indication of the extent of hepatectomy for hepatocellular carcinoma on cirrhosis by a simple algorithm based on preoperative variables. Arch. Surg. 2009, 144, 57–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berardi, R.; Caramanti, M.; Fiordoliva, I.; Morgese, F.; Savini, A.; Rinaldi, S.; Torniai, M.; Tiberi, M.; Ferrini, C.; Castagnani, M.; et al. Hyponatraemia is a predictor of clinical outcome for malignant pleural mesothelioma. Support Care Cancer 2015, 23, 621–626. [Google Scholar] [CrossRef]
- Vasudev, N.S.; Brown, J.E.; Brown, S.R.; Rafiq, R.; Morgan, R.; Patel, P.M.; O’Donnell, D.; Harnden, P.; Rogers, M.; Cocks, K.; et al. Prognostic factors in renal cell carcinoma: Association of preoperative sodium concentration with survival. Clin. Cancer Res. 2008, 14, 1775–1781. [Google Scholar] [CrossRef] [Green Version]
- Schutz, F.A.; Xie, W.; Donskov, F.; Sircar, M.; McDermott, D.F.; Rini, B.I.; Agarwal, N.; Pal, S.K.; Srinivas, S.; Kollmannsberger, C.; et al. The impact of low serum sodium on treatment outcome of targeted therapy in metastatic renal cell carcinoma: Results from the International Metastatic Renal Cell Cancer Database Consortium. Eur. Urol. 2014, 65, 723–730. [Google Scholar] [CrossRef] [Green Version]
- Gandhi, L.; Johnson, B.E. Paraneoplastic syndromes associated with small cell lung cancer. J. Natl. Compr. Canc. Netw. 2006, 4, 631–638. [Google Scholar] [CrossRef]
- Rawson, N.S.; Peto, J. An overview of prognostic factors in small cell lung cancer. A report from the Subcommittee for the Management of Lung Cancer of the United Kingdom Coordinating Committee on Cancer Research. Br. J. Cancer 1990, 61, 597–604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balachandran, K.; Okines, A.; Gunapala, R.; Morganstein, D.; Popat, S. Resolution of severe hyponatraemia is associated with improved survival in patients with cancer. BMC Cancer 2015, 15, 163. [Google Scholar] [CrossRef] [Green Version]
- Kasi, P.M. Proposing the use of hyponatremia as a marker to help identify high risk individuals for lung cancer. Med. Hypotheses 2012, 79, 327–328. [Google Scholar] [CrossRef]
- Ayus, C.; Achinger, S.G.; Arieff, A. Brain cell volume regulation in hyponatremia: Role of sex, age, vasopressin, and hypoxia. Am. J. Physiol. Renal Physiol. 2008, 295, F619–F624. [Google Scholar] [CrossRef] [PubMed]
- Kimelberg, H.K. Water homeostasis in the brain: Basic concepts. Neuroscience 2004, 129, 851–860. [Google Scholar] [CrossRef] [PubMed]
- Pasantes-Morales, H.; Tuz, K. Volume changes in neurons: Hyperexcitability and neuronal death. Contrib. Nephrol. 2006, 152, 221–240. [Google Scholar]
- Simard, M.; Nedergaard, M. The neurobiology of glia in the context of water and ion homeostasis. Neuroscience 2004, 129, 877–896. [Google Scholar] [CrossRef] [PubMed]
- Okada, Y.; Sato, K.; Numata, T. Pathophysiology and puzzles of the volume-sensitive outwardly rectifying anion channel. J. Physiol. 2009, 15, 2141–2149. [Google Scholar]
- Verbalis, J.G. Brain volume regulation in response to changes in osmolality. Neuroscience 2010, 168, 862–870. [Google Scholar] [CrossRef] [PubMed]
- Pasantes-Morales, H.; Franco, R.; Ordaz, B.; Ochoa, L.D. Mechanisms Counteracting Swelling in Brain Cells During Hyponatremia. Arch. Med. Res. 2002, 33, 237–244. [Google Scholar] [CrossRef] [Green Version]
- Okada, Y. Volume expansion-sensing outward-rectifier Cl-channel: Fresh start to the molecular identity and volume sensor. Am. J. Physiol. 1997, 273, C755–C789. [Google Scholar] [CrossRef]
- Fisher, S.K.; Heacock, A.M.; Keep, R.F.; Foster, D.J. Receptor regulation of osmolyte homeostasis in neural cells. J. Physiol. 2010, 18, 3355–3364. [Google Scholar] [CrossRef]
- Verbalis, J.G.; Gullans, S.R. Hyponatremia causes large sustained reductions in brain content of multiple organic osmolytes in rats. Brain Res. 1991, 567, 274–282. [Google Scholar] [CrossRef]
- Miyazaki, T.; Ohmoto, K.; Hirose, T.; Fujiki, H. Chronic hyponatremia impairs memory in rats: Effects of vasopressin antagonist tolvaptan. J. Endocrinol. 2010, 206, 105–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suárez, V.; Norello, D.; Sen, E.; Todorova, P.; Hackl, M.J.; Hüser, C.; Grundmann, F.; Kubacki, T.; Becker, I.; Peri, A.; et al. Impairment of Neurocognitive Functioning, Motor Performance, and Mood Stability in Hospitalized Patients with Euvolemic Moderate and Profound Hyponatremia. Am. J. Med. 2020, 133, 986–993.e5. [Google Scholar] [CrossRef]
- Danbolt, N.C. Glutamate uptake. Prog. Neurobiol. 2001, 65, 1–105. [Google Scholar] [CrossRef]
- Haskew-Layton, R.E.; Rudkouskaya, A.; Jin, Y.; Feustel, P.J.; Kimelberg, H.K.; Mongin, A.A. Two distinct modes of hypoosmotic medium-induced release of excitatory amino acids and taurine in the rat brain in vivo. PLoS ONE 2008, 3, e3543. [Google Scholar] [CrossRef] [Green Version]
- Fujisawa, H.; Sugimura, Y.; Takagi, H.; Mizoguchi, H.; Takeuchi, H.; Izumida, H.; Nakashima, K.; Ochiai, H.; Takeuchi, S.; Kiyota, A.; et al. Chronic Hyponatremia Causes Neurologic and Psychologic Impairments. J. Am. Soc. Nephrol. 2016, 27, 766–780. [Google Scholar] [CrossRef] [Green Version]
- Nicholls, D.G.; Budd, S.L. Mitochondria and neuronal survival. Physiol. Rev. 2000, 80, 315–360. [Google Scholar] [CrossRef] [Green Version]
- Lafon-Cazal, M.; Pietri, S.; Culcasi, M.; Boeckaert, J. NMDA-dependent superoxide production and neurotoxicity. Nature 1993, 364, 535–537. [Google Scholar] [CrossRef]
- Budd, S.L.; Nicholls, D.G. Mitochondria, calcium regulation, and acute glutamate excitotoxicity in cultured cerebellar granule cells. J. Neurochem. 1996, 67, 2282–2291. [Google Scholar] [CrossRef] [PubMed]
- Atlante, A.; Gagliardi, S.; Minervini, G.M.; Ciotti, M.T.; Marra, E.; Calissano, P. Glutamate neurotoxicity in rat cerebellar granule cells: A major role for xanthine oxidase in oxygen radical formation. J. Neurochem. 1997, 68, 2038–2045. [Google Scholar] [CrossRef] [Green Version]
- Castilho, R.F.; Nicholls, D.G. Oxidative stress, mitochondrial function, and acute glutamate excitotoxicity in cultured cerebellar granule cells. J. Neurochem. 1999, 72, 1394–1401. [Google Scholar] [CrossRef] [Green Version]
- Pereira, C.F.; Oliveira, C.R. Oxidative glutamate toxicity involves mitochondrial dysfunction and perturbation of intracellular Ca2+ homeostasis. Neurosci. Res. 2000, 37, 227–236. [Google Scholar] [CrossRef] [Green Version]
- Cadenas, E.; Davies, K.J. Mitochondrial free radical generation, oxidative stress, and aging. Free Radic. Biol. Med. 2000, 29, 222–230. [Google Scholar] [CrossRef]
- Chan, P.H. Reactive oxygen radicals in signaling and damage in the ischemic brain. J. Cereb. Blood Flow Metab. 2001, 21, 2–14. [Google Scholar] [CrossRef]
- Pasantes-Morales, H.; Lezama, R.; Ramos-Mandujano, G.; Tuz, K.L. Mechanisms of cell volume regulation in hypo-osmolality. Am. J. Med. 2006, 119, S4–S11. [Google Scholar] [CrossRef] [PubMed]
- Lambert, I.H. Regulation of the cellular content of the organic osmolyte taurine in mammalian cells. Neurochem. Res. 2004, 29, 27–63. [Google Scholar] [CrossRef]
- Dykens, J.A.; Stern, A.; Trenkner, E. Mechanism of kainate toxicity to cerebellar neurons in vitro is analogous to reperfusion tissue injury. J. Neurochem. 1987, 49, 1222–1228. [Google Scholar] [CrossRef] [PubMed]
- Dykens, J.A. Isolated cerebral and cerebellar mitochondria produce free radicals when exposed to elevated CA2+ and Na+: Implications for neurodegeneration. J. Neurochem. 1994, 63, 584–591. [Google Scholar] [CrossRef]
- Ankarcrona, M.; Dypbukt, J.M.; Bonfoco, E.; Zhivotovsky, B.; Orrenius, S.; Lipton, S.A.; Nicotera, P. Glutamate-induced neuronal death: A succession of necrosis or apoptosis depending on mitochondrial function. Neuron 1995, 15, 961–973. [Google Scholar] [CrossRef] [Green Version]
- Carriedo, S.G.; Yin, H.Z.; Sensi, S.L.; Weiss, J.H. Rapid Ca2+ entry through Ca2+-permeable AMPA/Kainate channels triggers marked intracellular Ca2+ rises and consequent oxygen radical production. J. Neurosci. 1998, 18, 7727–7738. [Google Scholar] [CrossRef] [Green Version]
- Ciani, E.; Groneng, L.; Voltattorni, M.; Rolseth, V.; Contestabile, A.; Paulsen, R.E. Inhibition of free radical production or free radical scavenging protects from the excitotoxic cell death mediated by glutamate in cultures of cerebellar granule neurons. Brain Res. 1996, 728, 1–6. [Google Scholar] [CrossRef]
- Schulz, J.B.; Lindenau, J.; Seyfried, J.; Dichgans, J. Glutathione, oxidative stress and neurodegeneration. Eur. J. Biochem. 2000, 267, 4904–4911. [Google Scholar] [CrossRef] [PubMed]
- Lien, Y.H.; Shapiro, J.I.; Chan, L. Study of brain electrolytes and organic osmolytes during correction of chronic hyponatremia. J. Clin. Invest. 1991, 88, 303–309. [Google Scholar] [CrossRef] [Green Version]
- Mizui, T.; Kinouchi, H.; Chan, P.H. Depletion of brain glutathione by buthionine sulfoximine enhances cerebral ischemic injury in rats. Am. J. Physiol. 1992, 262, H313–H317. [Google Scholar] [CrossRef]
- Weber, C.A.; Duncan, C.A.; Lyons, M.J.; Jenkinson, S.G. Depletion of tissue glutathione with diethyl maleate enhances hyperbaric oxygen toxicity. Am. J. Physiol. 1990, 258, L308–L312. [Google Scholar] [CrossRef]
- Clark, E.C.; Thomas, D.; Baer, J.; Sterns, R.H. Depletion of glutathione from brain cells in hyponatremia. Kidney Int. 1996, 49, 470–476. [Google Scholar] [CrossRef] [Green Version]
- Yudkoff, M.; Pleasure, D.; Cregar, L.; Lin, Z.P.; Nissim, I.; Stern, J.; Nissim, I. Glutathione turnover in cultured astrocytes: Studies with [15N]glutamate. J. Neurochem. 1990, 55, 137–145. [Google Scholar] [CrossRef]
- Hasan, D.; Wudicks, E.F.M.; Vermeulen, M. Hyponatremia is associated with cerebral ischemia in patients with aneurysmal subarachnoid hemorrhage. Ann. Neurol. 1990, 27, 106–108. [Google Scholar] [CrossRef]
- Volterra, A.; Trotti, D.; Racagni, G. Glutamate uptake is inhibited by arachidonic acid and oxygen radicals via two distinct and additive mechanisms. Mol. Pharmacol. 1994, 46, 986–992. [Google Scholar]
- Barsony, J.; Sugimura, Y.; Verbalis, J.G. Osteoclast response to low extracellular sodium and the mechanism of hyponatremia-induced bone loss. J. Biol. Chem. 2011, 286, 10864–10875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergstrom, W.H.; Wallace, W.M. Bone as a sodium and potassium reservoir. J. Clin. Investig. 1954, 33, 867–873. [Google Scholar] [CrossRef] [PubMed]
- Seeliger, E.; Ladwig, M.; Reinhardt, H.W. Are large amounts of sodium stored in an osmotically inactive form during sodium retention? Balance studies in freely moving dogs. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2006, 290, R1429–R1435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, X.; Itoh, N.; Taniguchi, T.; Nakanishi, T.; Tatsu, Y.; Yumoto, N.; Tanaka, K. Zinc-induced sodium-dependent vitamin C transporter 2 expression: Potent roles in osteoblast differentiation. Arch. Biochem. Biophys. 2003, 420, 114–120. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.H.; Liao, E.Y.; Zhou, H.D.; Dai, R.C.; Yuan, L.Q.; Wu, X.P. Ascorbic acid inhibits osteoclastogenesis of RAW264.7 cells induced by receptor activated nuclear factor kappaB ligand (RANKL) in vitro. J. Endocrinol. Investig. 2005, 28, 253–260. [Google Scholar] [CrossRef]
- Fibbi, B.; Benvenuti, S.; Giuliani, C.; Deledda, C.; Luciani, P.; Monici, M.; Mazzanti, B.; Ballerini, C.; Peri, A. Low extracellular sodium promotes adipogenic commitment of human mesenchymal stromal cells: A novel mechanism for chronic hyponatremia-induced bone loss. Endocrine 2016, 52, 73–85. [Google Scholar] [CrossRef]
- Baksh, D.; Song, L.; Tuan, R.S. Adult mesenchymal stem cells: Characterization, differentiation, and application in cell and gene therapy. J. Cell. Mol. Med. 2004, 8, 301–316. [Google Scholar] [CrossRef]
- Rosen, C.J.; Ackert-Bicknell, C.; Rodriguez, J.P.; Pino, A.M. Marrow fat and the bone microenvironment: Developmental, functional and pathological implications. Crit. Rev. Eukariot. Gene Expr. 2009, 19, 109–124. [Google Scholar] [CrossRef] [Green Version]
- Sohal, R.S.; Weindruch, R. Oxidative stress, caloric restriction, and aging. Science 1996, 273, 59–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yakner, B.A.; Lu, T.; Loerch, P. The Aging Brain. Annu. Rev. Pathol. 2008, 3, 41–66. [Google Scholar] [CrossRef] [PubMed]
- Upadhyay, A.; Jaber, B.L.; Madias, N.E. Incidence and prevalence of hyponatremia. Am. J. Med. 2006, 119, S30–S35. [Google Scholar] [CrossRef]
- Barsony, J.; Manigrasso, M.B.; Xu, Q.; Tam, H.; Verbalis, J.G. Chronic hyponatremia exacerbates multiple manifestations of senescence in male rats. Age 2013, 35, 271–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oniki, T.; Teshima, Y.; Nishio, S.; Ishii, Y.; Kira, S.; Abe, I.; Yufu, K.; Takahashi, N. Hyponatremia aggravates cardiac susceptibility to ischemia/reperfusion injury. Int. J. Exp. Path. 2019, 100, 350–358. [Google Scholar] [CrossRef]
- Crompton, M. The mitochondrial permeability transition pore and its role in cell death. Biochem. J. 1999, 341, 233–249. [Google Scholar] [CrossRef]
- Goordeva, A.V.; Zvyagilskaya, R.A.; Labas, Y.A. Cross-talk between reactive oxygen species and calcium in living cells. Biochemistry 2003, 68:1077-1080; Brookes PS. Mitochondrial nitric oxide synthase. Mitochondrion 2004, 3, 187–204. [Google Scholar]
- Benvenuti, S.; Deledda, C.; Luciani, P.; Modi, G.; Bossio, A.; Giuliani, C.; Fibbi, B.; Peri, A. Low extracellular sodium causes neuronal distress independently of reduced osmolality in an experimental model of chronic hyponatremia. Neuromol. Med. 2013, 15, 493–503. [Google Scholar] [CrossRef]
- Mancuso, C. Heme oxygenase and its products in the nervous system. Antioxid. Redox Signal 2004, 6, 878–887. [Google Scholar]
- Chen, K.; Gunter, K.; Maines, M.D. Neurons overexpressing heme oxygenase-1 resist oxidative stress-mediated cell death. J. Neurochem. 2000, 75, 304–313. [Google Scholar] [CrossRef]
- Takizawa, S.; Hirabayashi, H.; Matsushima, K.; Tokuoka, K.; Shinohara, Y. Induction of heme oxygenase protein protects neurons in cortex and striatum, but not in hippocampus, against transient forebrain ischemia. J. Cereb. Blood Flow Metab. 1998, 18, 559–569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ku, B.M.; Joo, Y.; Mun, J.; Roh, G.S.; Kang, S.S.; Cho, G.J. Heme oxygenase protects hippocampal neurons from ethanol-induced neurotoxicity. Neurosci. Lett. 2006, 405, 168–171. [Google Scholar] [CrossRef] [PubMed]
- Benvenuti, S.; Deledda, C.; Luciani, P.; Giuliani, C.; Fibbi, B.; Muratori, M.; Peri, A. Neuronal distress induced by low extracellular sodium in vitro is partially reverted by the return to normal sodium. J. Endocrinol. Investig. 2016, 39, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Shruster, A.; Melamed, E.; Offen, D. Neurogenesis in the aged and neurodegenerative brain. Apoptosis 2010, 15, 1415–1421. [Google Scholar] [CrossRef]
- Marroncini, G.; Fibbi, B.; Errico, A.; Grappone, C.; Maggi, M.; Peri, A. Effects of low extracellular sodium on proliferation and invasive activity of cancer cells in vitro. Endocrine 2020, 67, 473–484. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Panieri, E.; Santoro, M.M. ROS homeostasis and metabolism: A dangerous liason in cancer cells. Cell Death Dis. 2016, 7, e2253. [Google Scholar] [CrossRef]
- Zelenka, J.; Koncosova, M.; Ruml, T. Targeting of stress response pathways in the prevention and treatment of cancer. Biotechnol. Adv. 2018, 36, 583–602. [Google Scholar] [CrossRef]
- Marnett, L.J. Oxyradicals and DNA damage. Carcinogenesis 2000, 21, 361–370. [Google Scholar] [CrossRef] [Green Version]
- Gorrini, C.; Harris, I.S.; Mak, T.W. Modulation of oxidative stress as an anticancer strategy. Nat. Rev. Drug Discov. 2013, 12, 931–947. [Google Scholar] [CrossRef]
- Diehn, M.; Cho, R.W.; Lobo, N.A.; Kalisky, T.; Dorie, M.J.; Kulp, A.N.; Qian, D.; Lam, J.S.; Ailles, L.E.; Wong, M.; et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature 2009, 458, 780–783. [Google Scholar] [CrossRef]
- Kim, H.M.; Haraguchi, N.; Ishii, H.; Ohkuma, M.; Okano, M.; Mimori, K.; Eguchi, H.; Yamamoto, H.; Nagano, H.; Sekimoto, M.; et al. Increased CD13 expression reduces reactive oxygen species, promoting survival of liver cancer stem cells via an epithelial-mesenchymal transition-like phenomenon. Ann. Surg. Oncol. 2012, 19, 539–548. [Google Scholar] [CrossRef]
- Jeppesen, A.N.; Jensen, H.K.; Donskov, F.; Marcussen, N.; von der Maase, H. Hyponatremia as a prognostic and predictive factor in metastatic renal cell carcinoma. Br. J. Cancer 2010, 102, 867–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Penttila, P.; Bono, P.; Peltola, K.; Donskov, F. Hyponatremia associates with poor outcome in metastatic renal cell carcinoma patients treated with everolimus: Prognostic impact. Acta Oncol. 2018, 57, 1580–1585. [Google Scholar] [CrossRef] [PubMed]
- Berardi, R.; Santoni, M.; Newsom-Davis, T.; Caramanti, M.; Rinaldi, S.; Tiberi, M.; Morgese, F.; Torniai, M.; Pistelli, M.; Onofri, A.; et al. Hyponatremia normalization as an independent prognostic factor in patients with advanced non-small cell lung cancer treated with first-line therapy. Oncotarget 2017, 8, 23871–23879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
Non osmotically-induced effects of hyponatremia and oxidative stress. GLT-1 and GLAST: Na+-dependent glial glutamate transporters; ROS: reactive oxygen species; Glu: glutamate; Tau: taurine; GNT: glutamate neurotoxicity; KCC: K+/Cl− co-transporters.
Figure 1.
Non osmotically-induced effects of hyponatremia and oxidative stress. GLT-1 and GLAST: Na+-dependent glial glutamate transporters; ROS: reactive oxygen species; Glu: glutamate; Tau: taurine; GNT: glutamate neurotoxicity; KCC: K+/Cl− co-transporters.

Figure 2.
List of differentially expressed genes in two in vitro neuronal models (SH-SY5Y and SKN-AS cell lines), maintained at reduced (115 mmol/L and 127 mmol/L, respectively) or normal (153 mmol/L) [Na+], as assessed by microarray analysis. Positive fold-regulations are reported in red, negative fold-regulations are in blue. Yellow marked genes are commonly regulated in both cell lines.
Figure 2.
List of differentially expressed genes in two in vitro neuronal models (SH-SY5Y and SKN-AS cell lines), maintained at reduced (115 mmol/L and 127 mmol/L, respectively) or normal (153 mmol/L) [Na+], as assessed by microarray analysis. Positive fold-regulations are reported in red, negative fold-regulations are in blue. Yellow marked genes are commonly regulated in both cell lines.

Figure 3.
Osmotically-independent effects of hyponatremia and oxidative stress. MSC: mesenchymal stromal cells.
Figure 3.
Osmotically-independent effects of hyponatremia and oxidative stress. MSC: mesenchymal stromal cells.

Figure 4.
Effects of hyponatremia on cell and tissue homeostasis.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Fibbi, B.; Marroncini, G.; Anceschi, C.; Naldi, L.; Peri, A. Hyponatremia and Oxidative Stress. Antioxidants 2021, 10, 1768. https://doi.org/10.3390/antiox10111768
AMA Style
Fibbi B, Marroncini G, Anceschi C, Naldi L, Peri A. Hyponatremia and Oxidative Stress. Antioxidants. 2021; 10(11):1768. https://doi.org/10.3390/antiox10111768
Chicago/Turabian StyleFibbi, Benedetta, Giada Marroncini, Cecilia Anceschi, Laura Naldi, and Alessandro Peri. 2021. "Hyponatremia and Oxidative Stress" Antioxidants 10, no. 11: 1768. https://doi.org/10.3390/antiox10111768
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.