
Synergistic Protection by Isoquercitrin and Quercetin against Glutamate-Induced Oxidative Cell Death in HT22 Cells via Activating Nrf2 and HO-1 Signaling Pathway: Neuroprotective Principles and Mechanisms of Dendropanax morbifera Leaves


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Plant Material
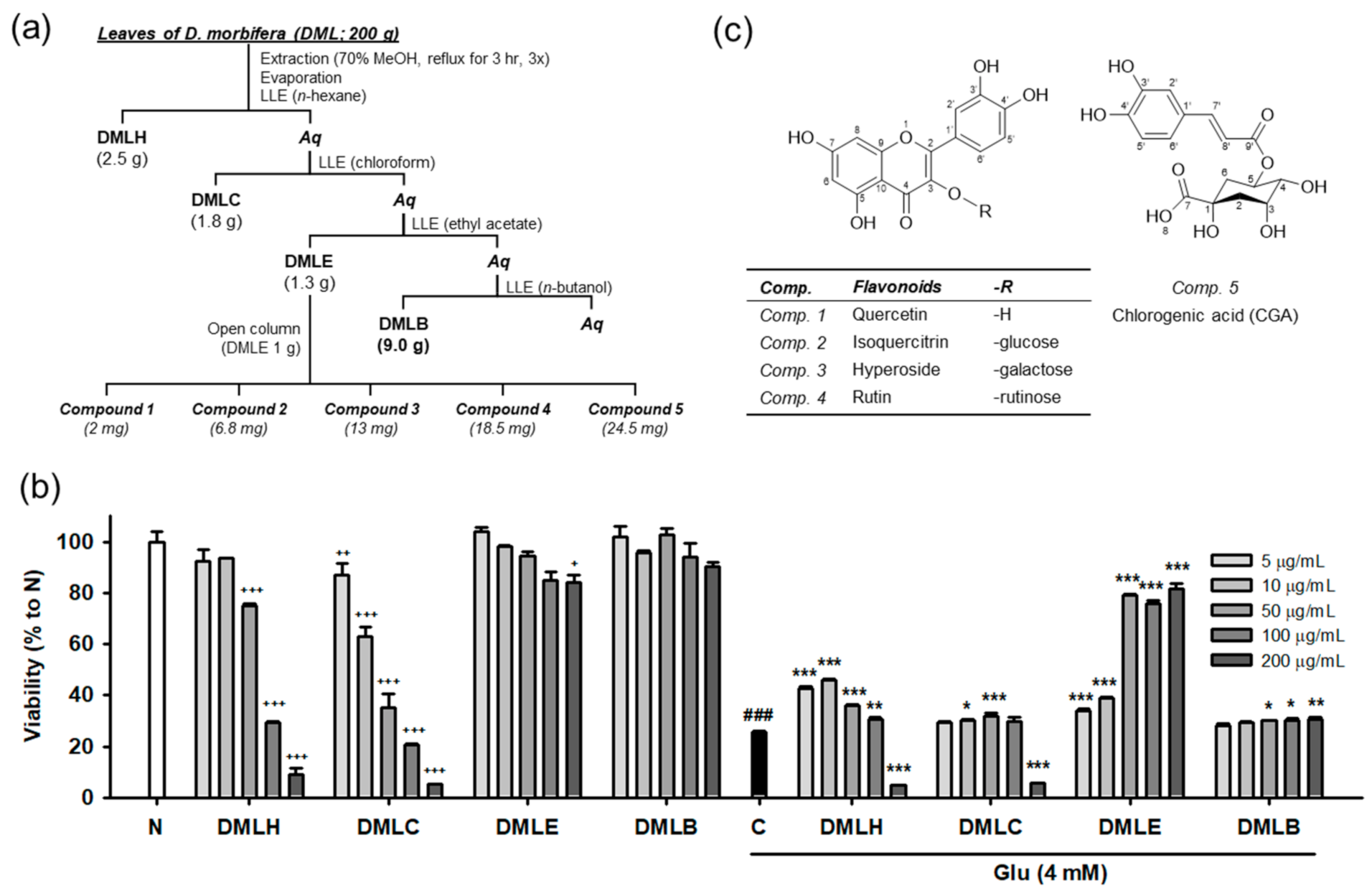
2.3. Extraction, Isolation, and Characterization of Compounds
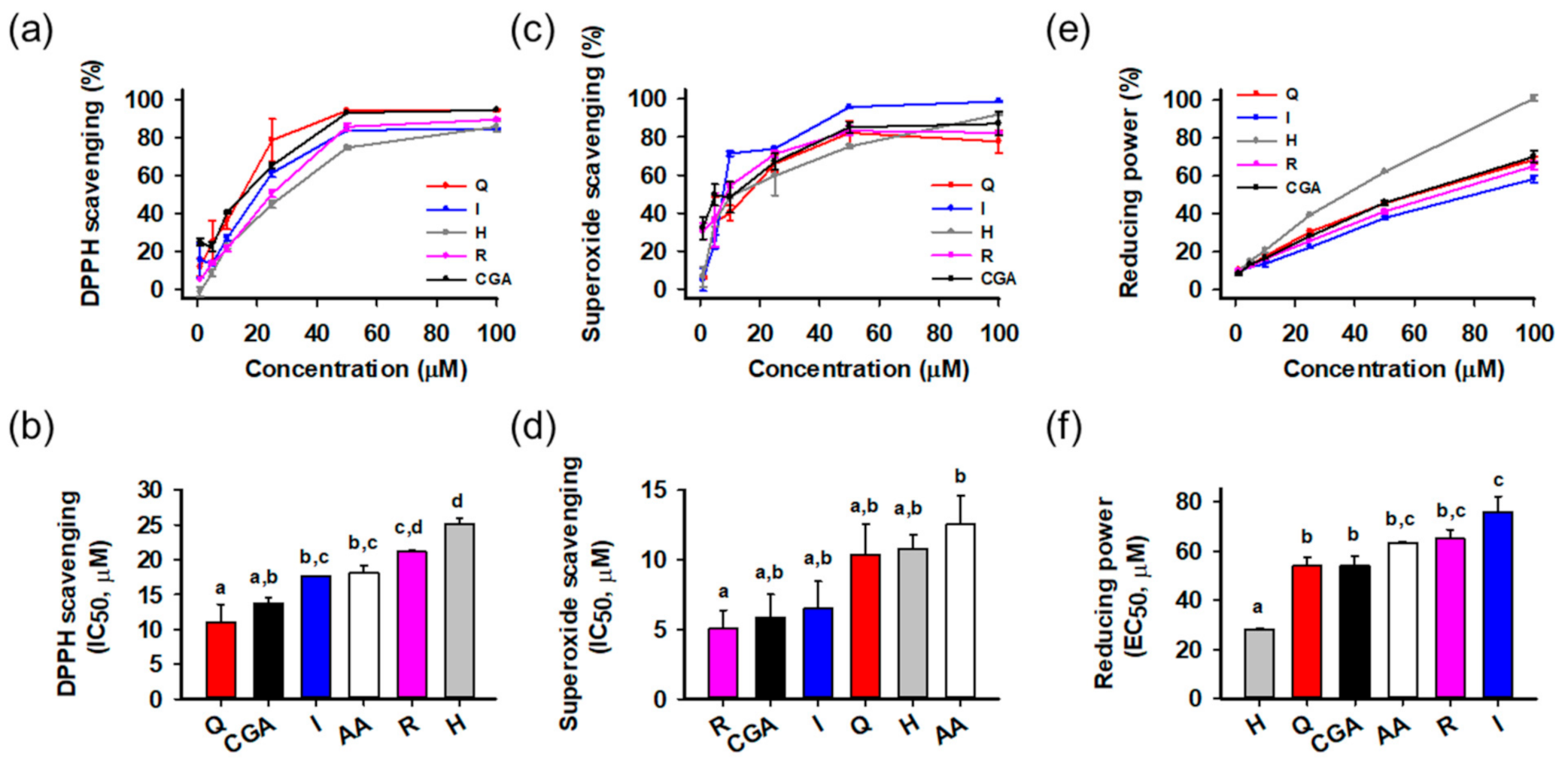
2.4. Antioxidant Activity Assays
2.4.1. DPPH Radical Scavenging Assay
2.4.2. Superoxide Radical Scavenging Assay
2.4.3. Reducing Power Assay
2.5. Cell Culture and Treatments
2.6. Cell Viability Assay
2.7. PI/Annexin V Apoptosis Analysis
2.8. Measurement of Intracellular and Mitochondrial ROS Generation
2.9. Measurement of Intracellular Calcium Levels
2.10. Measurement of Mitochondrial Membrane Potential
2.11. Measurement of Mitochondrial Membrane Permeabilization
2.12. Subcellular Fractionation
2.13. Western Blotting
2.14. Molecular Docking Simulation
2.15. Quantitative HPLC-UV Assay and Method Validation
2.16. Analysis of Synergistic Effect
2.17. Statistical Analysis
3. Results
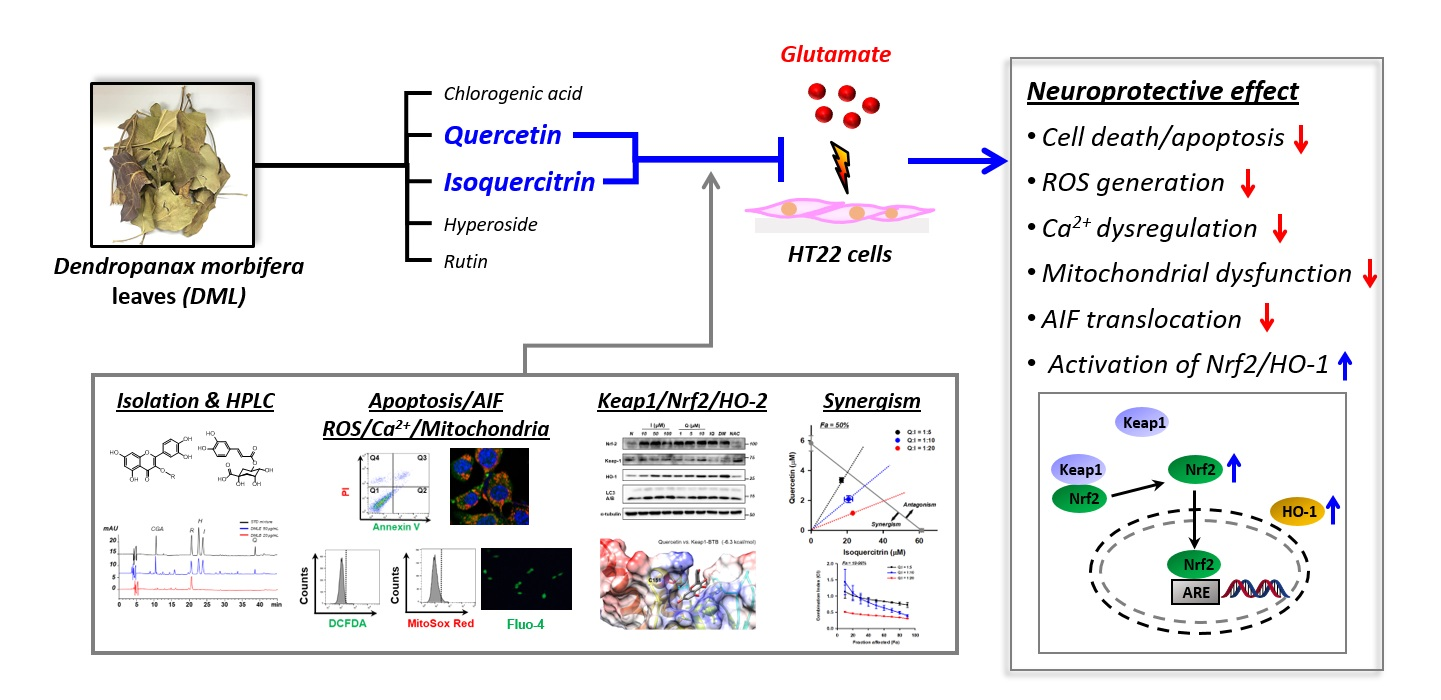
3.1. Extraction, Isolation, and Structual Characterization of Antioxidant Compounds from DML
3.2. Antioxidant Activities of Isolated Compounds
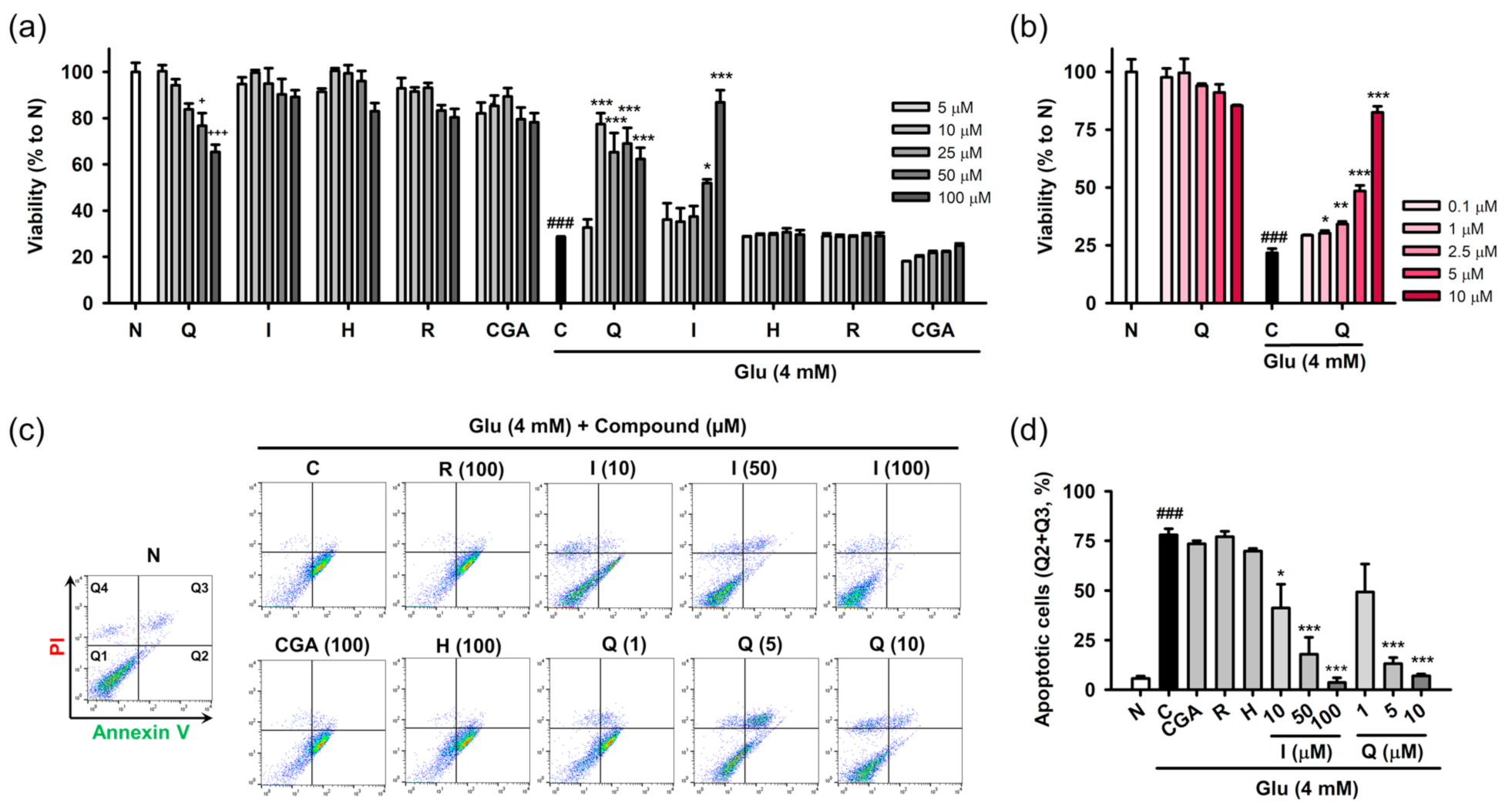
3.3. Isoquercitrin and Quercetin Inhibit Glu-Induced Apoptotic Cell Death in HT22 Cells
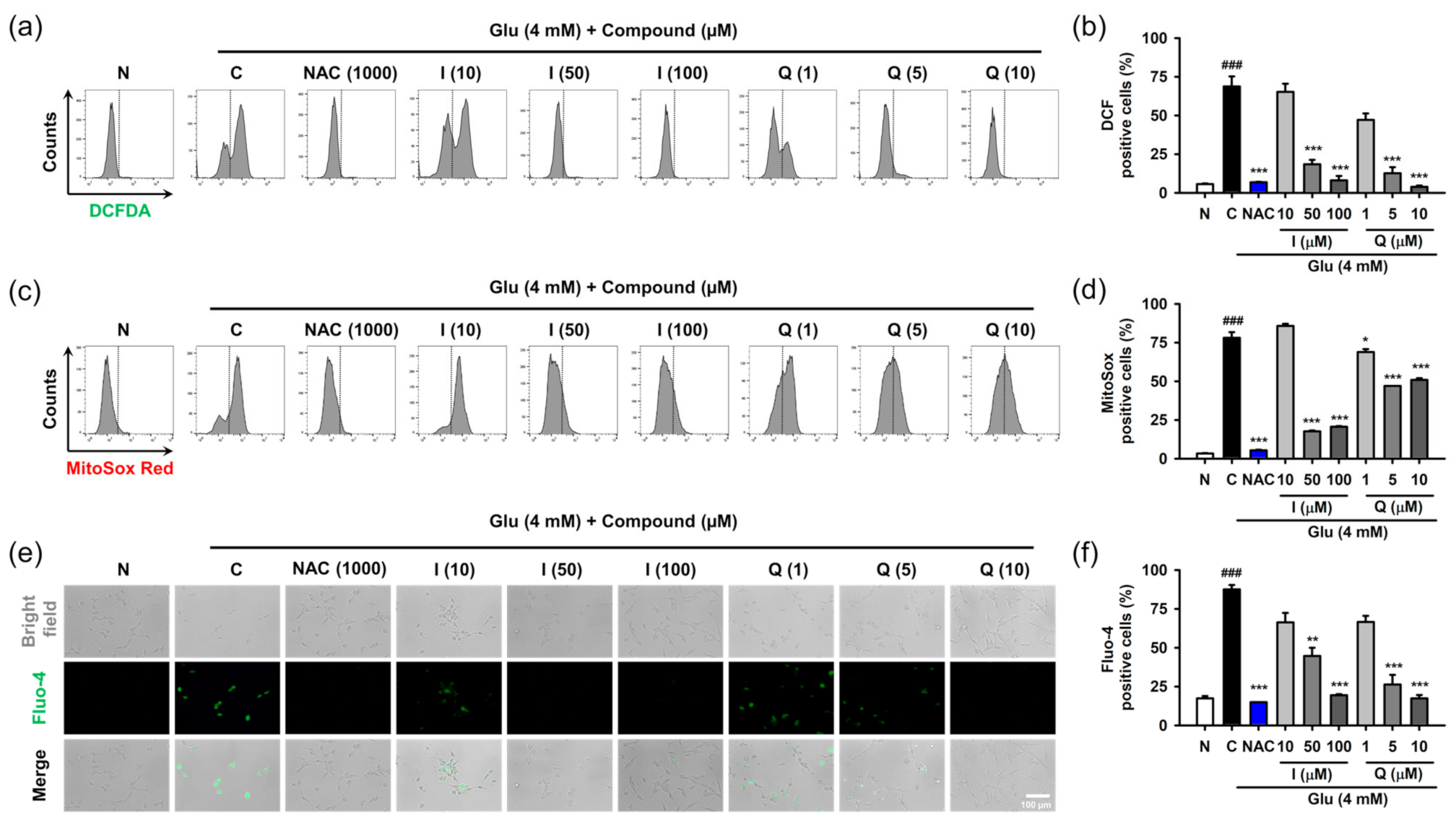
3.4. Isoquercitrin and Quercetin Attenuate Glu-Induced Intracellular/Mitochondrial ROS Generation and Ca2+ Dysregulation
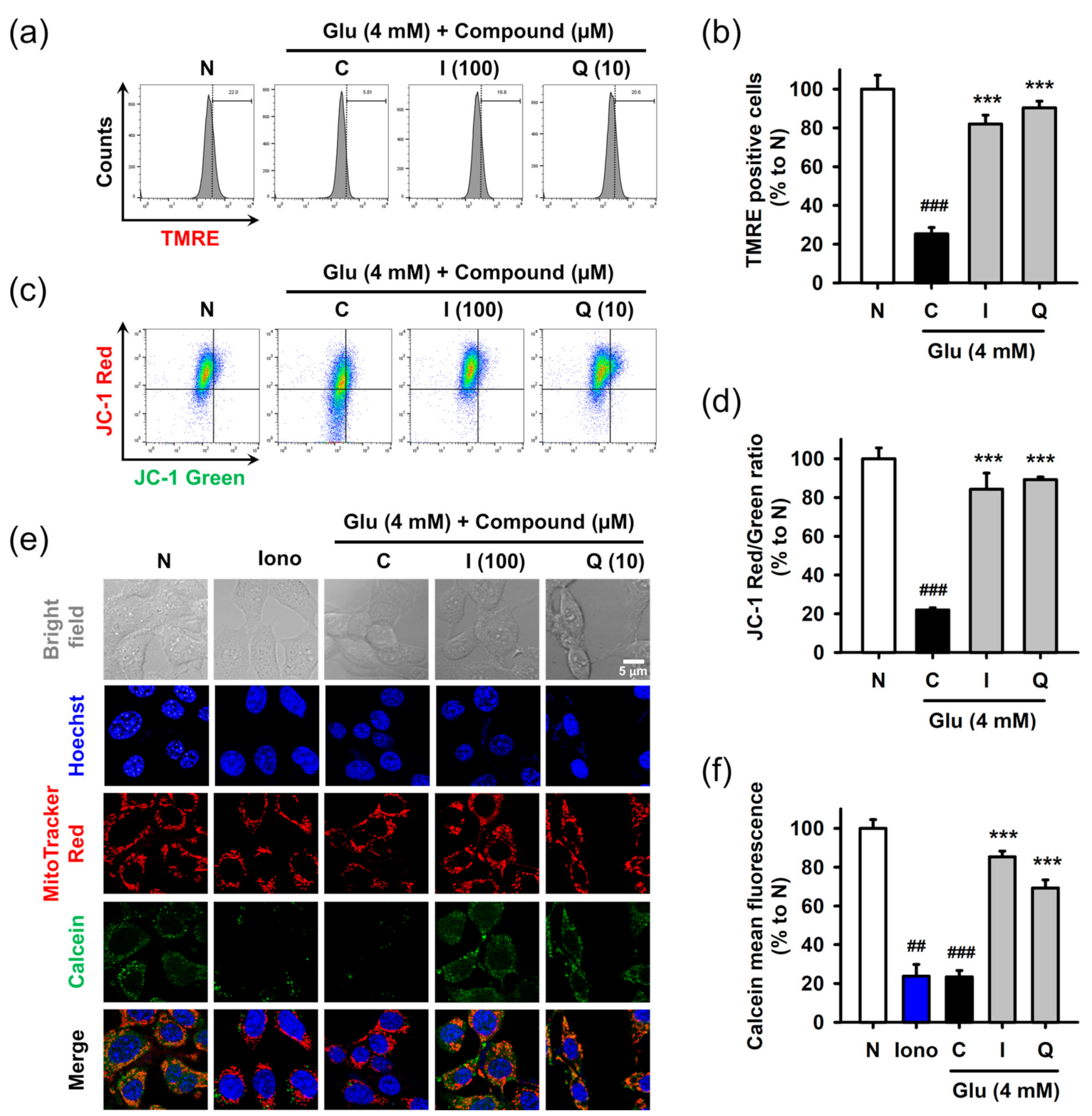
3.5. Isoquercitrin and Quercetin Restore Glu-Induced Mitochondrial Dysfunction
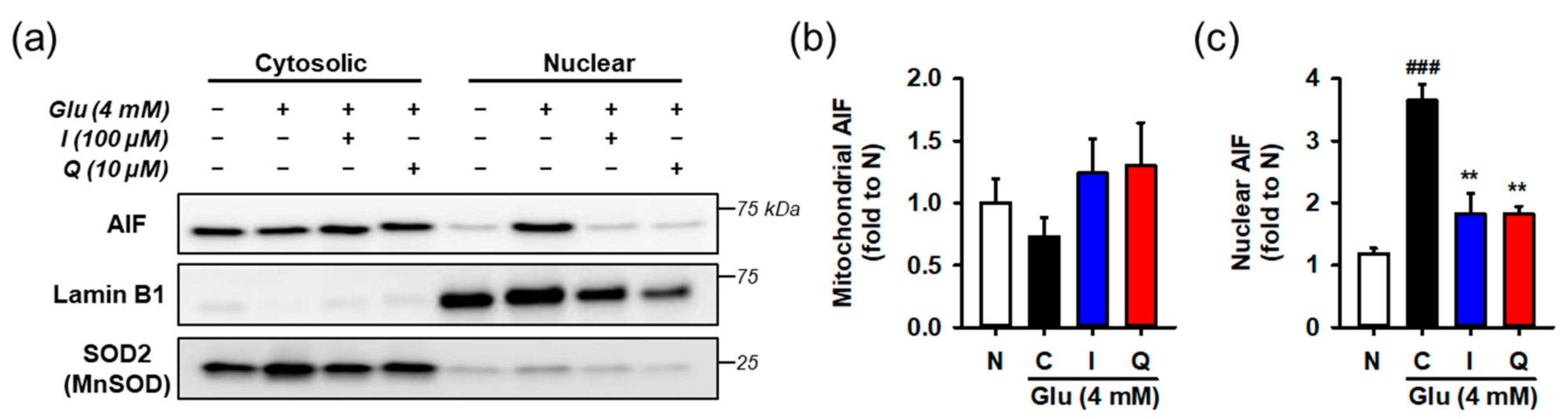
3.6. Isoquercitrin and Quercetin Inhibit Glu-Induced Nuclear Translocation of AIF
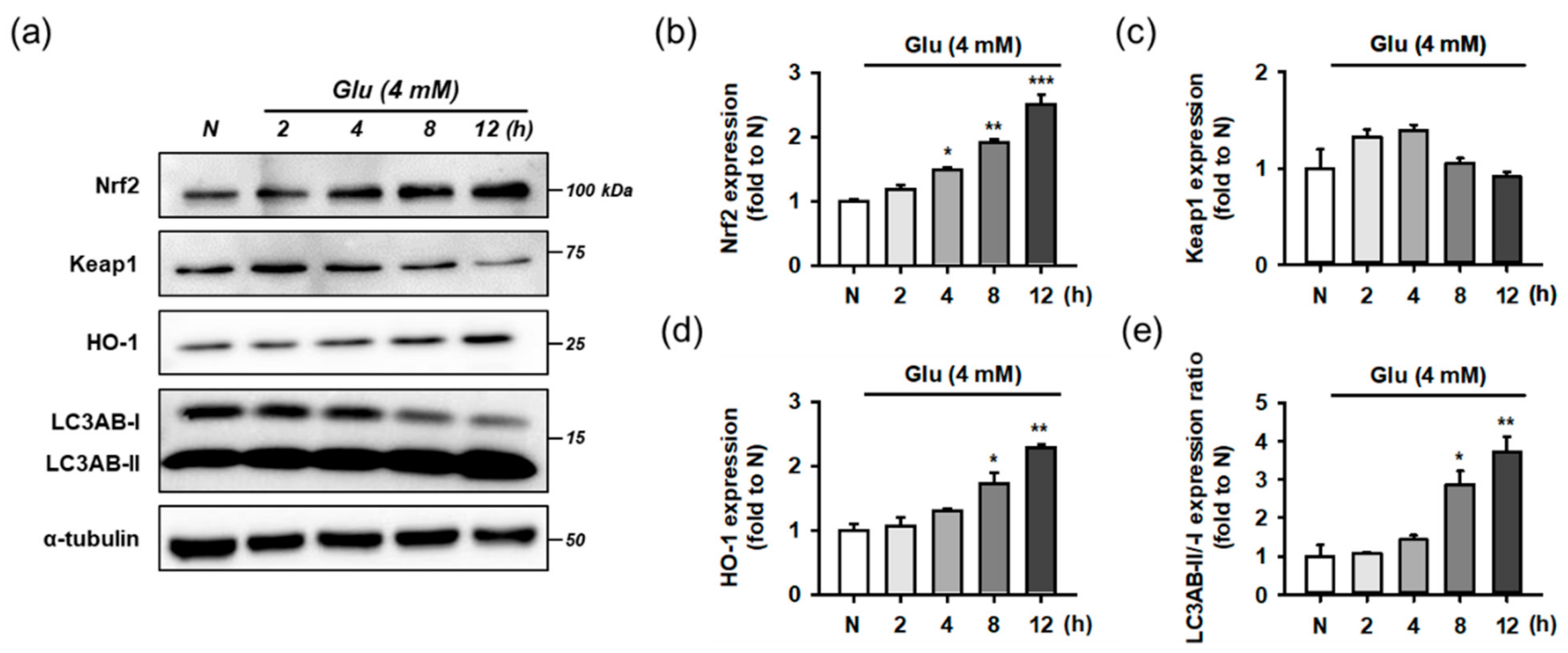
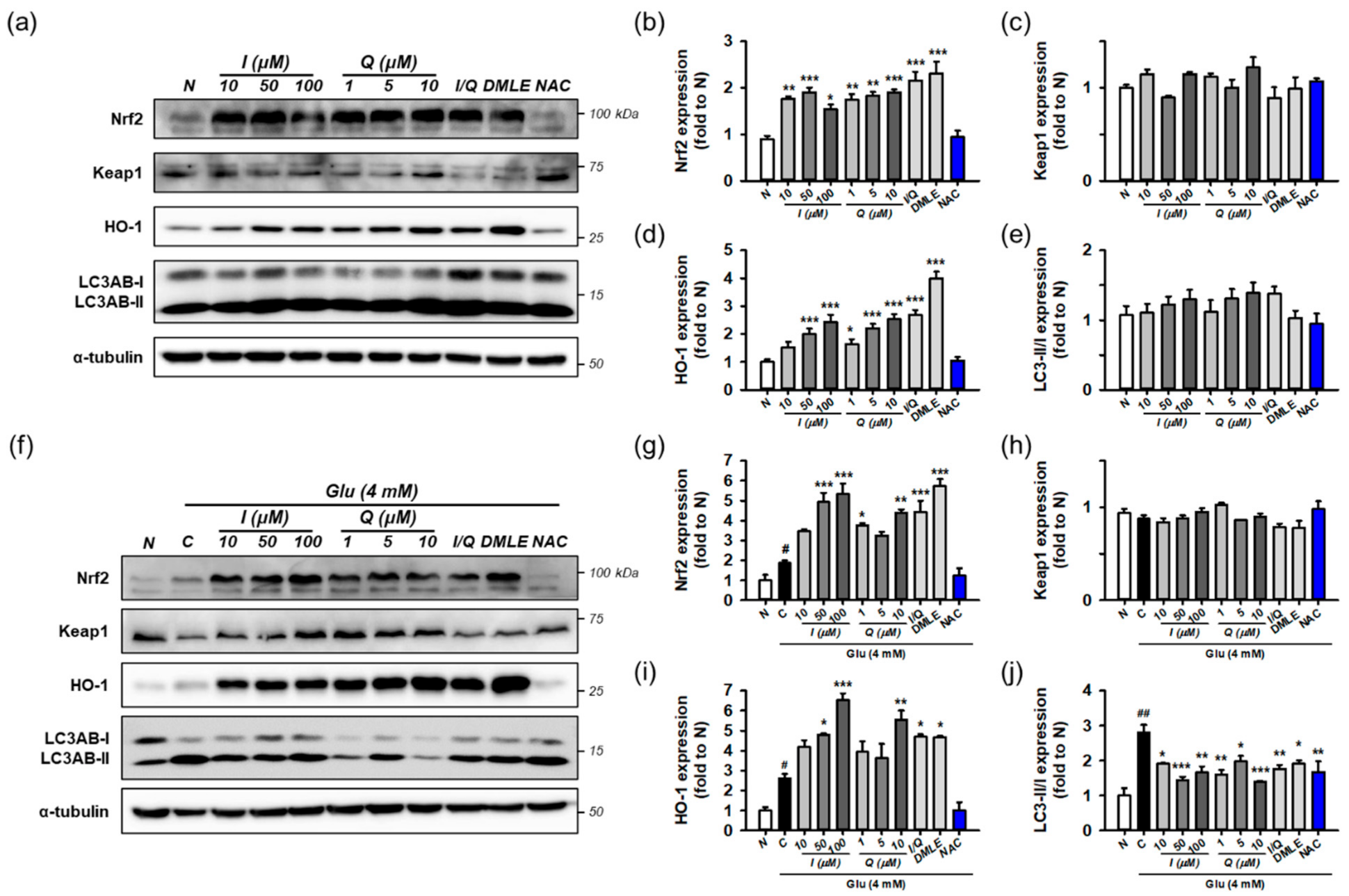
3.7. Isoquercitrin and Quercetin Activate Nrf2/HO-1 Signaling Pathway
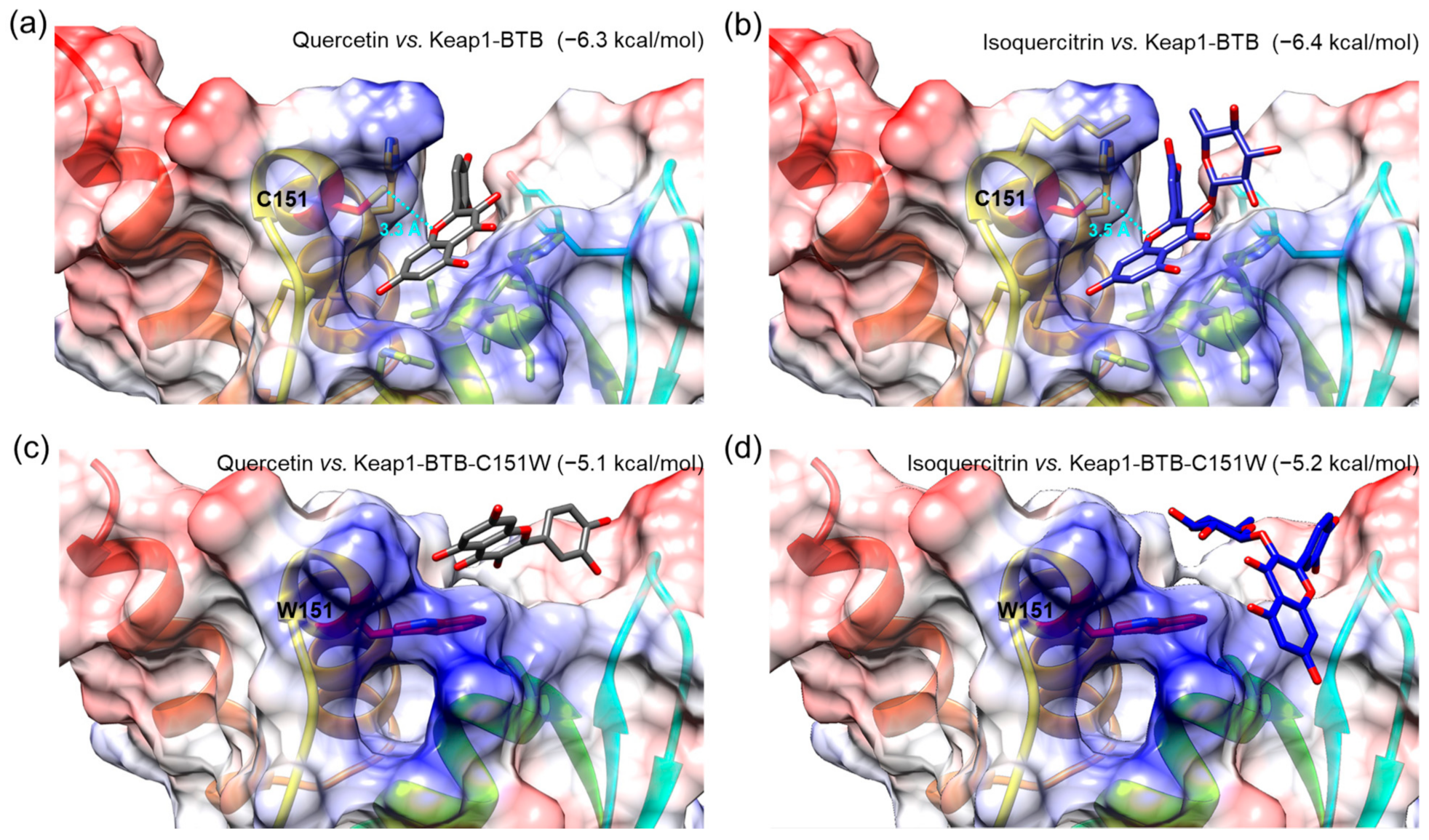
3.8. Molecular Docking Simulation of Isoquercitrin and Quercetin to Keap1-BTB Domain
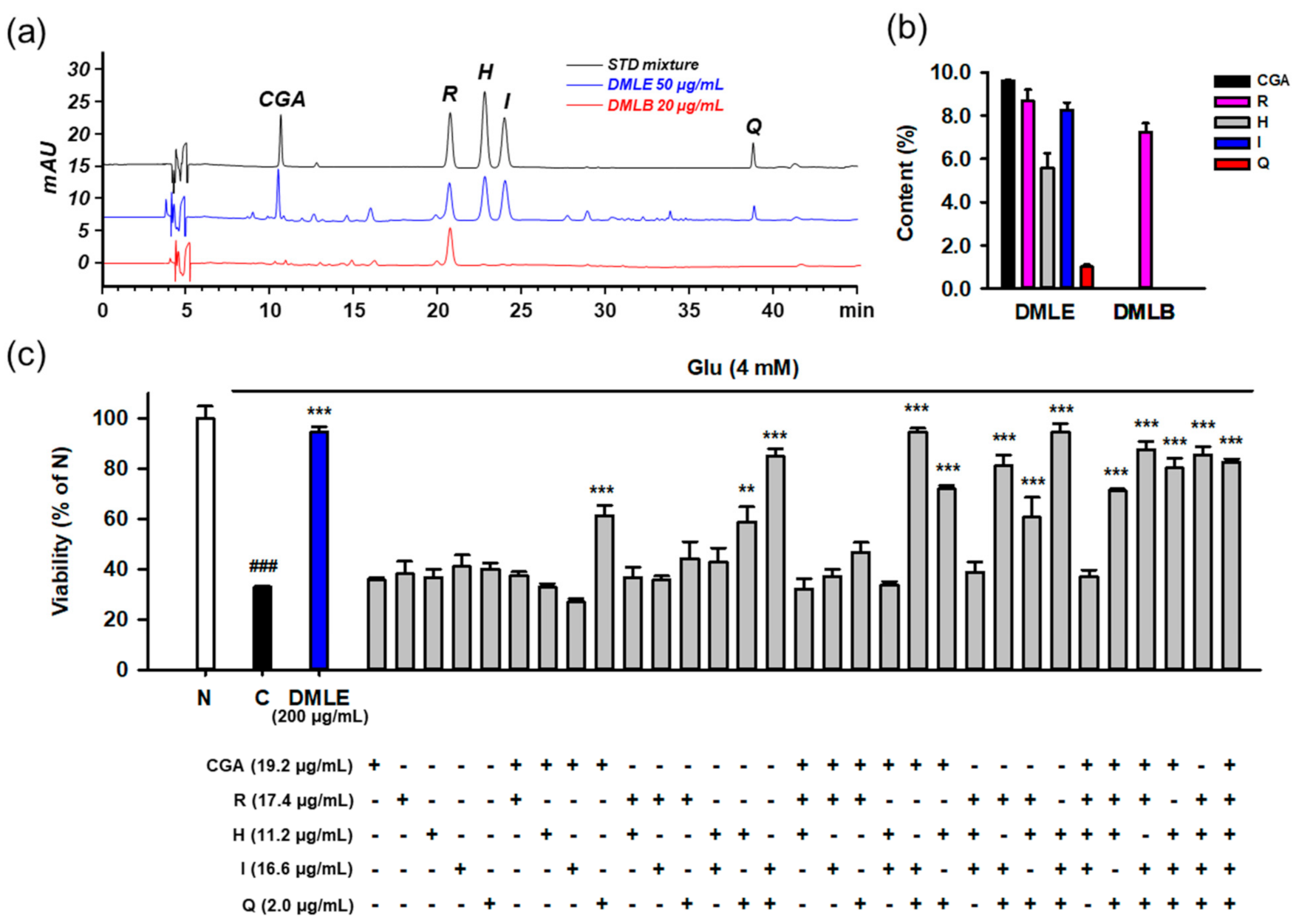
3.9. Identification of Active Principles in DMLE Using HPLC Assay and Combinatory Treatment
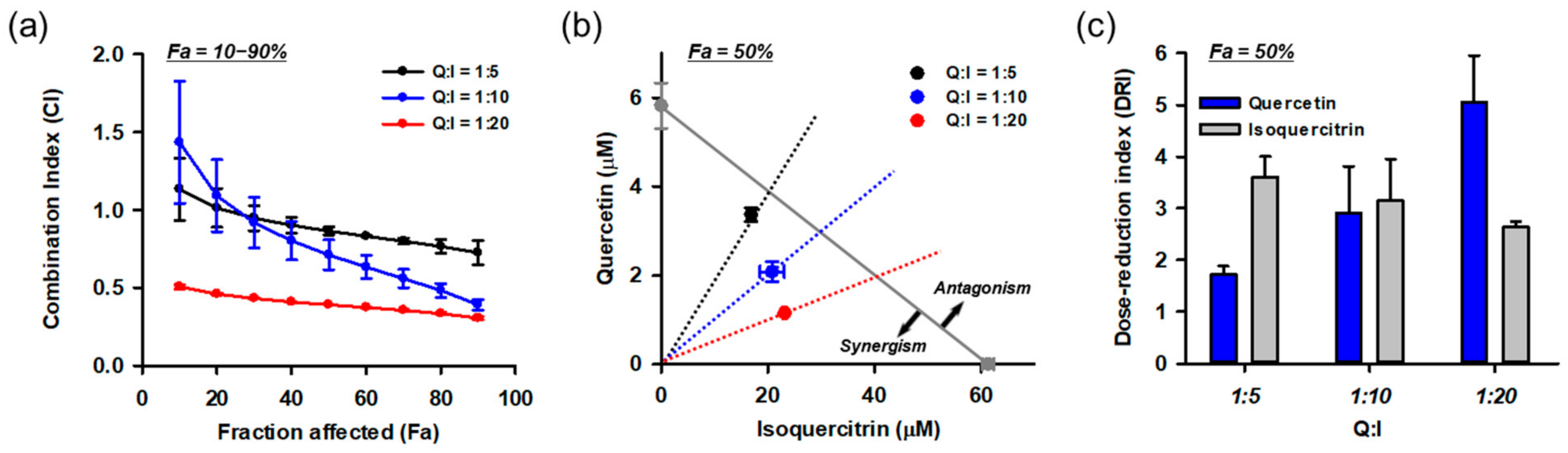
3.10. Combination of Isoquercitrin and Quercetin Synergistically Inhibited Glu-Induced Cell Death
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lai, T.W.; Zhang, S.; Wang, Y.T. Excitotoxicity and stroke: Identifying novel targets for neuroprotection. Prog. Neurobiol. 2014, 115, 157–188. [Google Scholar] [CrossRef] [Green Version]
- Dong, X.X.; Wang, Y.; Qin, Z.H. Molecular mechanisms of excitotoxicity and their relevance to pathogenesis of neurodegenerative diseases. Acta Pharmacol. Sin. 2009, 30, 379–387. [Google Scholar] [CrossRef] [Green Version]
- Avoli, M.; D’Antuono, M.; Louvel, J.; Köhling, R.; Biagini, G.; Pumain, R.; D’Arcangelo, G.; Tancredi, V. Network and pharmacological mechanisms leading to epileptiform synchronization in the limbic system in vitro. Prog. Neurobiol. 2002, 68, 167–207. [Google Scholar] [CrossRef]
- Curia, G.; Lucchi, C.; Vinet, J.; Gualtieri, F.; Marinelli, C.; Torsello, A.; Costantino, L.; Biagini, G. Pathophysiogenesis of mesial temporal lobe epilepsy: Is prevention of damage antiepileptogenic? Curr. Med. Chem. 2014, 21, 663–688. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Qin, Z.H. Molecular and cellular mechanisms of excitotoxic neuronal death. Apoptosis 2010, 15, 1382–1402. [Google Scholar] [CrossRef]
- Kritis, A.A.; Stamoula, E.G.; Paniskaki, K.A.; Vavilis, T.D. Researching glutamate-induced cytotoxicity in different cell lines: A comparative/collective analysis/study. Front. Cell. Neurosci. 2015, 9, 91. [Google Scholar] [CrossRef] [Green Version]
- Fricker, M.; Tolkovsky, A.M.; Borutaite, V.; Coleman, M.; Brown, G.C. Neuronal Cell Death. Physiol. Rev. 2018, 98, 813–880. [Google Scholar] [CrossRef]
- Tan, S.; Schubert, D.; Maher, P. Oxytosis: A novel form of programmed cell death. Curr. Top. Med. Chem. 2001, 1, 497–506. [Google Scholar] [CrossRef]
- Liu, B.; Teschemacher, A.G.; Kasparov, S. Astroglia as a cellular target for neuroprotection and treatment of neuro-psychiatric disorders. Glia 2017, 65, 1205–1226. [Google Scholar] [CrossRef] [Green Version]
- Loboda, A.; Damulewicz, M.; Pyza, E.; Jozkowicz, A.; Dulak, J. Role of Nrf2/HO-1 system in development, oxidative stress response and diseases: An evolutionarily conserved mechanism. Cell. Mol. Life Sci. 2016, 73, 3221–3247. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Locascio, L.M.; Doré, S. Critical Role of Nrf2 in Experimental Ischemic Stroke. Front. Pharmacol. 2019, 10, 153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, X.; Long, D. Nrf2 and Ferroptosis: A New Research Direction for Neurodegenerative Diseases. Front. Neurosci. 2020, 14, 267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robledinos-Antón, N.; Fernández-Ginés, R.; Manda, G.; Cuadrado, A. Activators and Inhibitors of NRF2: A Review of Their Potential for Clinical Development. Oxid. Med. Cell. Longev. 2019, 2019, 9372182. [Google Scholar] [CrossRef] [PubMed]
- Cuadrado, A.; Rojo, A.I.; Wells, G.; Hayes, J.D.; Cousin, S.P.; Rumsey, W.L.; Attucks, O.C.; Franklin, S.; Levonen, A.L.; Kensler, T.W.; et al. Therapeutic targeting of the NRF2 and KEAP1 partnership in chronic diseases. Nat. Rev. Drug Discov. 2019, 18, 295–317. [Google Scholar] [CrossRef] [Green Version]
- Scapagnini, G.; Vasto, S.; Abraham, N.G.; Caruso, C.; Zella, D.; Fabio, G. Modulation of Nrf2/ARE pathway by food polyphenols: A nutritional neuroprotective strategy for cognitive and neurodegenerative disorders. Mol. Neurobiol. 2011, 44, 192–201. [Google Scholar] [CrossRef] [Green Version]
- Kumar, H.; Kim, I.S.; More, S.V.; Kim, B.W.; Choi, D.K. Natural product-derived pharmacological modulators of Nrf2/ARE pathway for chronic diseases. Nat. Prod. Rep. 2014, 31, 109–139. [Google Scholar] [CrossRef]
- Park, H.J.; Kwak, M.; Baek, S.H. Neuroprotective effects of Dendropanax morbifera leaves on glutamate-induced oxidative cell death in HT22 mouse hippocampal neuronal cells. J. Ethnopharmacol. 2020, 251, 112518. [Google Scholar] [CrossRef]
- Balakrishnan, R.; Cho, D.Y.; Su-Kim, I.; Choi, D.K. Dendropanax Morbiferus and Other Species from the Genus Dendropanax: Therapeutic Potential of Its Traditional Uses, Phytochemistry, and Pharmacology. Antioxidants 2020, 9, 962. [Google Scholar] [CrossRef]
- Kim, J.M.; Park, S.K.; Guo, T.J.; Kang, J.Y.; Ha, J.S.; Lee du, S.; Lee, U.; Heo, H.J. Anti-amnesic effect of Dendropanax morbifera via JNK signaling pathway on cognitive dysfunction in high-fat diet-induced diabetic mice. Behav. Brain Res. 2016, 312, 39–54. [Google Scholar] [CrossRef]
- Kim, W.; Yim, H.S.; Yoo, D.Y.; Jung, H.Y.; Kim, J.W.; Choi, J.H.; Yoon, Y.S.; Kim, D.W.; Hwang, I.K. Dendropanax morbifera Léveille extract ameliorates cadmium-induced impairment in memory and hippocampal neurogenesis in rats. BMC Complement. Altern. Med. 2016, 16, 452. [Google Scholar] [CrossRef] [Green Version]
- Kim, W.; Kim, D.W.; Yoo, D.Y.; Jung, H.Y.; Kim, J.W.; Kim, D.W.; Choi, J.H.; Moon, S.M.; Yoon, Y.S.; Hwang, I.K. Antioxidant effects of Dendropanax morbifera Léveille extract in the hippocampus of mercury-exposed rats. BMC Complement. Altern. Med. 2015, 15, 247. [Google Scholar] [CrossRef] [Green Version]
- Kim, W.; Kim, D.W.; Yoo, D.Y.; Jung, H.Y.; Nam, S.M.; Kim, J.W.; Hong, S.M.; Kim, D.W.; Choi, J.H.; Moon, S.M.; et al. Dendropanax morbifera Léveille extract facilitates cadmium excretion and prevents oxidative damage in the hippocampus by increasing antioxidant levels in cadmium-exposed rats. BMC Complement. Altern. Med. 2014, 14, 428. [Google Scholar] [CrossRef] [Green Version]
- Hou, Y.; Dan, X.; Babbar, M.; Wei, Y.; Hasselbalch, S.G.; Croteau, D.L.; Bohr, V.A. Ageing as a risk factor for neurodegenerative disease. Nat. Rev. Neurol. 2019, 15, 565–581. [Google Scholar] [CrossRef]
- Uttara, B.; Singh, A.V.; Zamboni, P.; Mahajan, R.T. Oxidative stress and neurodegenerative diseases: A review of upstream and downstream antioxidant therapeutic options. Curr. Neuropharmacol. 2009, 7, 65–74. [Google Scholar] [CrossRef] [Green Version]
- Barnham, K.J.; Masters, C.L.; Bush, A.I. Neurodegenerative diseases and oxidative stress. Nat. Rev. Drug Discov. 2004, 3, 205–214. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- International Conference on Harmonization, ICH Harmonised Guideline Q2 (R1). Validation of Analytical Procedures: Text and Methodology, ICH, Geneva, 2005. Available online: https://www.ich.org/ (accessed on 13 January 2020).
- Chou, T.C. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol. Rev. 2006, 58, 621–681. [Google Scholar] [CrossRef]
- Chou, T.C.; Talalay, P. Quantitative analysis of dose-effect relationships: The combined effects of multiple drugs or enzyme inhibitors. Adv. Enzym. Regul. 1984, 22, 27–55. [Google Scholar] [CrossRef]
- El-Sayed, A.M.; Ezzat, S.M.; Salama, M.M.; Sleem, A.A. Hepatoprotective and cytotoxic activities of Delonix regia flower extracts. Pharm. J. 2011, 3, 49–56. [Google Scholar] [CrossRef] [Green Version]
- Sukito, A.; Tachibana, S. Isolation of hyperoside and isoquercitrin from Camellia sasanqua as antioxidant agents. Pak. J. Biol. Sci. 2014, 17, 999–1006. [Google Scholar] [CrossRef]
- Olszewska, M. Flavonoids from Prunus serotina Ehrh. Acta Pol. Pharm. 2005, 62, 127–133. [Google Scholar]
- Yang, J.; Kwon, Y.S.; Kim, M.J. Isolation and characterization of bioactive compounds from Lepisorus thunbergianus (Kaulf.). Arab. J. Chem. 2015, 8, 407–413. [Google Scholar] [CrossRef]
- Görlach, A.; Bertram, K.; Hudecova, S.; Krizanova, O. Calcium and ROS: A mutual interplay. Redox Biol. 2015, 6, 260–271. [Google Scholar] [CrossRef] [Green Version]
- Castro, J.P.; Wardelmann, K.; Grune, T.; Kleinridders, A. Mitochondrial Chaperones in the Brain: Safeguarding Brain Health and Metabolism? Front. Endocrinol. 2018, 9, 196. [Google Scholar] [CrossRef] [Green Version]
- Picard, M.; McEwen, B.S. Mitochondria impact brain function and cognition. Proc. Natl. Acad. Sci. USA 2014, 111, 7–8. [Google Scholar] [CrossRef] [Green Version]
- Tait, S.W.; Green, D.R. Mitochondrial regulation of cell death. Cold Spring Harb. Perspect. Biol. 2013, 5. [Google Scholar] [CrossRef] [Green Version]
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787–795. [Google Scholar] [CrossRef]
- Tait, S.W.; Green, D.R. Mitochondria and cell death: Outer membrane permeabilization and beyond. Nat. Rev. Mol. Cell Biol. 2010, 11, 621–632. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Blomgren, K.; Kroemer, G. Mitochondrial membrane permeabilization in neuronal injury. Nat. Rev. Neurosci. 2009, 10, 481–494. [Google Scholar] [CrossRef] [PubMed]
- Thal, S.E.; Zhu, C.; Thal, S.C.; Blomgren, K.; Plesnila, N. Role of apoptosis inducing factor (AIF) for hippocampal neuronal cell death following global cerebral ischemia in mice. Neurosci. Lett. 2011, 499, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Cao, G.; Clark, R.S.; Pei, W.; Yin, W.; Zhang, F.; Sun, F.Y.; Graham, S.H.; Chen, J. Translocation of apoptosis-inducing factor in vulnerable neurons after transient cerebral ischemia and in neuronal cultures after oxygen-glucose deprivation. J. Cereb. Blood Flow Metab. 2003, 23, 1137–1150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukui, M.; Song, J.H.; Choi, J.; Choi, H.J.; Zhu, B.T. Mechanism of glutamate-induced neurotoxicity in HT22 mouse hippocampal cells. Eur. J. Pharmacol. 2009, 617, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q. Role of nrf2 in oxidative stress and toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef] [Green Version]
- Vomund, S.; Schäfer, A.; Parnham, M.J.; Brüne, B.; von Knethen, A. Nrf2, the Master Regulator of Anti-Oxidative Responses. Int. J. Mol. Sci. 2017, 18, 2772. [Google Scholar] [CrossRef] [Green Version]
- Rusten, T.E.; Stenmark, H. p62, an autophagy hero or culprit? Nat. Cell Biol. 2010, 12, 207–209. [Google Scholar] [CrossRef]
- Taguchi, K.; Fujikawa, N.; Komatsu, M.; Ishii, T.; Unno, M.; Akaike, T.; Motohashi, H.; Yamamoto, M. Keap1 degradation by autophagy for the maintenance of redox homeostasis. Proc. Natl. Acad. Sci. USA 2012, 109, 13561–13566. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; Choi, J.; Ryu, J.; Park, S.G.; Cho, S.; Park, B.C.; Lee, D.H. Activation of autophagy during glutamate-induced HT22 cell death. Biochem. Biophys. Res. Commun. 2009, 388, 339–344. [Google Scholar] [CrossRef]
- Seo, J.Y.; Kim, S.K.; Nguyen, P.H.; Lee, J.Y.; Tung, P.H.T.; Sung, S.H.; Oh, W.K. Chemical constituents from a Gynostemma laxum and their antioxidant and neuroprotective activities. Chin. Med. 2017, 12, 15. [Google Scholar] [CrossRef] [Green Version]
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs from 1981 to 2014. J. Nat. Prod. 2016, 79, 629–661. [Google Scholar] [CrossRef] [Green Version]
- Valentová, K.; Vrba, J.; Bancířová, M.; Ulrichová, J.; Křen, V. Isoquercitrin: Pharmacology, toxicology, and metabolism. Food Chem. Toxicol. 2014, 68, 267–282. [Google Scholar] [CrossRef]
- Thilakarathna, S.H.; Rupasinghe, H.P. Flavonoid bioavailability and attempts for bioavailability enhancement. Nutrients 2013, 5, 3367–3387. [Google Scholar] [CrossRef]
- Gonzales, G.B.; Van Camp, J.; Vissenaekens, H.; Raes, K.; Smagghe, G.; Grootaert, C. Review on the Use of Cell Cultures to Study Metabolism, Transport, and Accumulation of Flavonoids: From Mono-Cultures to Co-Culture Systems. Compr. Rev. Food Sci. Food Saf. 2015, 14, 741–754. [Google Scholar] [CrossRef]
- Arredondo, F.; Echeverry, C.; Abin-Carriquiry, J.A.; Blasina, F.; Antúnez, K.; Jones, D.P.; Go, Y.M.; Liang, Y.L.; Dajas, F. After cellular internalization, quercetin causes Nrf2 nuclear translocation, increases glutathione levels, and prevents neuronal death against an oxidative insult. Free Radic. Biol. Med. 2010, 49, 738–747. [Google Scholar] [CrossRef]
- Zuo, Z.; Zhang, L.; Zhou, L.; Chang, Q.; Chow, M. Intestinal absorption of hawthorn flavonoids--in vitro, in situ and in vivo correlations. Life Sci. 2006, 79, 2455–2462. [Google Scholar] [CrossRef]
- Boyer, J.; Brown, D.; Liu, R.H. Uptake of quercetin and quercetin 3-glucoside from whole onion and apple peel extracts by Caco-2 cell monolayers. J. Agric. Food Chem. 2004, 52, 7172–7179. [Google Scholar] [CrossRef]
- Tian, X.J.; Yang, X.W.; Yang, X.; Wang, K. Studies of intestinal permeability of 36 flavonoids using Caco-2 cell monolayer model. Int. J. Pharm. 2009, 367, 58–64. [Google Scholar] [CrossRef]
- Ferenczyova, K.; Kalocayova, B.; Bartekova, M. Potential Implications of Quercetin and its Derivatives in Cardioprotection. Int. J. Mol. Sci. 2020, 21, 1585. [Google Scholar] [CrossRef] [Green Version]
- Paulke, A.; Eckert, G.P.; Schubert-Zsilavecz, M.; Wurglics, M. Isoquercitrin provides better bioavailability than quercetin: Comparison of quercetin metabolites in body tissue and brain sections after six days administration of isoquercitrin and quercetin. Pharmazie 2012, 67, 991–996. [Google Scholar]
- Chen, M.; Dai, L.H.; Fei, A.; Pan, S.M.; Wang, H.R. Isoquercetin activates the ERK1/2-Nrf2 pathway and protects against cerebral ischemia-reperfusion injury in vivo and in vitro. Exp. Ther. Med. 2017, 13, 1353–1359. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, A.; Khan, M.M.; Hoda, M.N.; Raza, S.S.; Khan, M.B.; Javed, H.; Ishrat, T.; Ashafaq, M.; Ahmad, M.E.; Safhi, M.M.; et al. Quercetin protects against oxidative stress associated damages in a rat model of transient focal cerebral ischemia and reperfusion. Neurochem. Res. 2011, 36, 1360–1371. [Google Scholar] [CrossRef] [PubMed]
- Suganthy, N.; Devi, K.P.; Nabavi, S.F.; Braidy, N.; Nabavi, S.M. Bioactive effects of quercetin in the central nervous system: Focusing on the mechanisms of actions. Biomed. Pharmacother. 2016, 84, 892–908. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.; Abdel-Aziz, A.; Sánchez-Alcázar, J.; Wang, Z.; Rios Rossi Lima, T.; Morales-Alamo, D.; Salimi Dafsari, H.; Garg, A.; Önal, G.; Ghosh, M.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition). Autophagy 2021, 17, 1–382. [Google Scholar] [CrossRef] [PubMed]
- Codogno, P.; Meijer, A.J. Autophagy and signaling: Their role in cell survival and cell death. Cell Death Differ. 2005, 12 (Suppl. 2), 1509–1518. [Google Scholar] [CrossRef]











Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, H.-J.; Kim, H.-N.; Kim, C.Y.; Seo, M.-D.; Baek, S.-H. Synergistic Protection by Isoquercitrin and Quercetin against Glutamate-Induced Oxidative Cell Death in HT22 Cells via Activating Nrf2 and HO-1 Signaling Pathway: Neuroprotective Principles and Mechanisms of Dendropanax morbifera Leaves. Antioxidants 2021, 10, 554. https://doi.org/10.3390/antiox10040554
Park H-J, Kim H-N, Kim CY, Seo M-D, Baek S-H. Synergistic Protection by Isoquercitrin and Quercetin against Glutamate-Induced Oxidative Cell Death in HT22 Cells via Activating Nrf2 and HO-1 Signaling Pathway: Neuroprotective Principles and Mechanisms of Dendropanax morbifera Leaves. Antioxidants. 2021; 10(4):554. https://doi.org/10.3390/antiox10040554
Chicago/Turabian StylePark, Hye-Jin, Ha-Neul Kim, Chul Young Kim, Min-Duk Seo, and Seung-Hoon Baek. 2021. "Synergistic Protection by Isoquercitrin and Quercetin against Glutamate-Induced Oxidative Cell Death in HT22 Cells via Activating Nrf2 and HO-1 Signaling Pathway: Neuroprotective Principles and Mechanisms of Dendropanax morbifera Leaves" Antioxidants 10, no. 4: 554. https://doi.org/10.3390/antiox10040554