
Antimicrobial Random Peptide Mixtures Eradicate Acinetobacter baumannii Biofilms and Inhibit Mouse Models of Infection

Abstract
:1. Introduction
2. Results
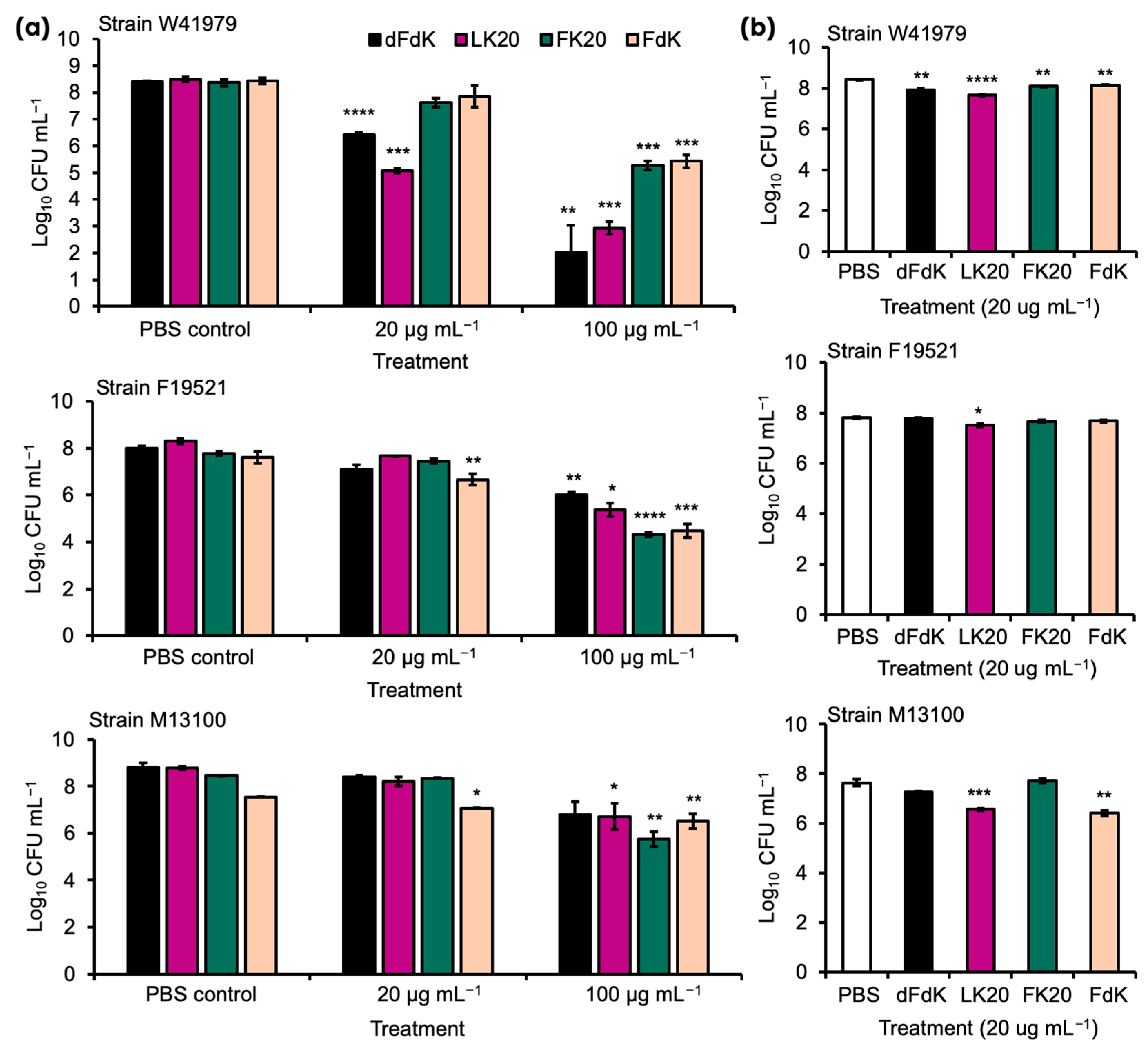
2.1. Random Peptide Mixtures Are Effective Antibacterial Agents against A. baumannii In Vitro
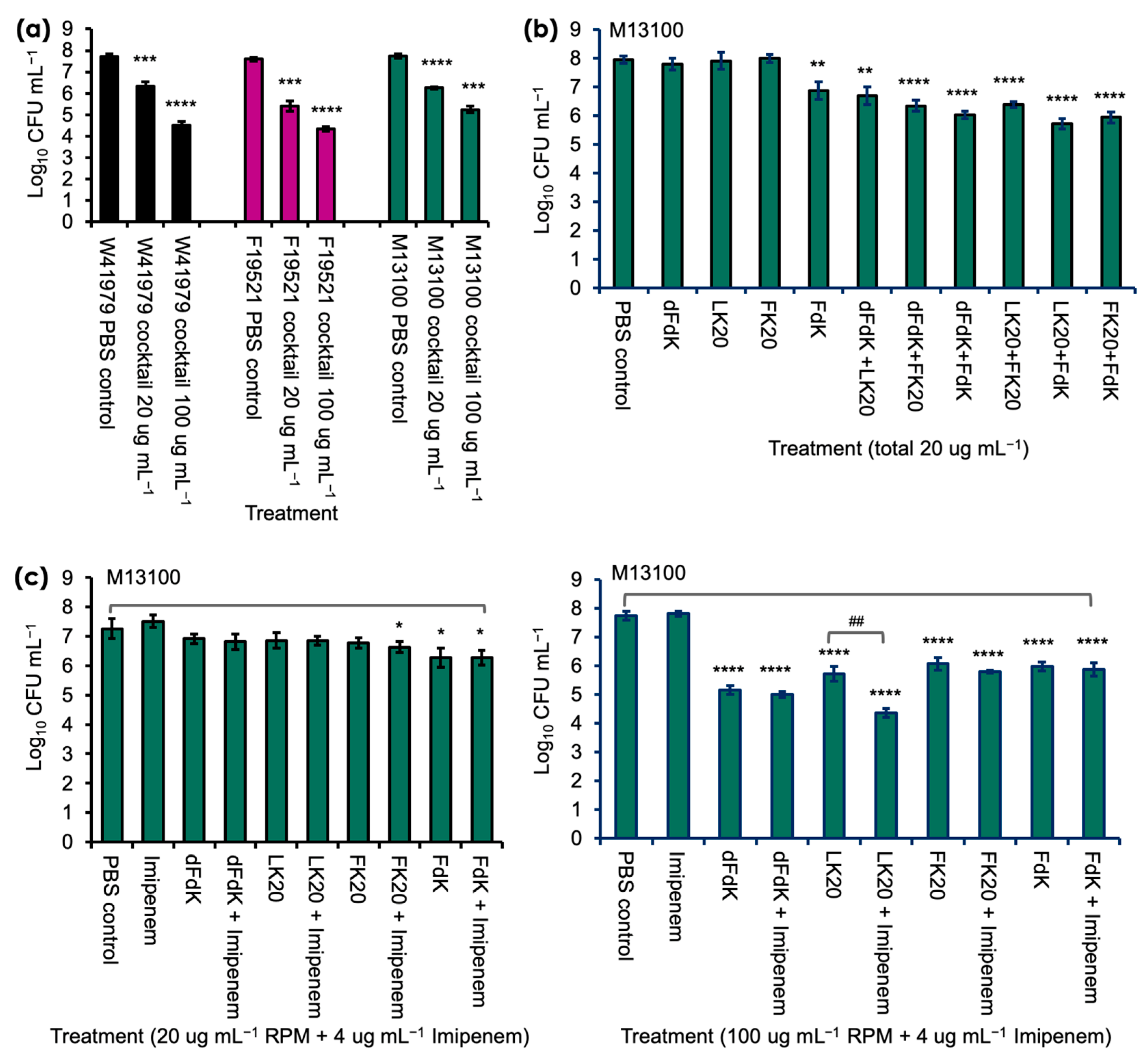
2.2. Random Peptide Mixtures in a Cocktail Are More Effective Than Single RPM and Show Additive Efficacy with Antibiotic in Killing A. baumannii In Vitro
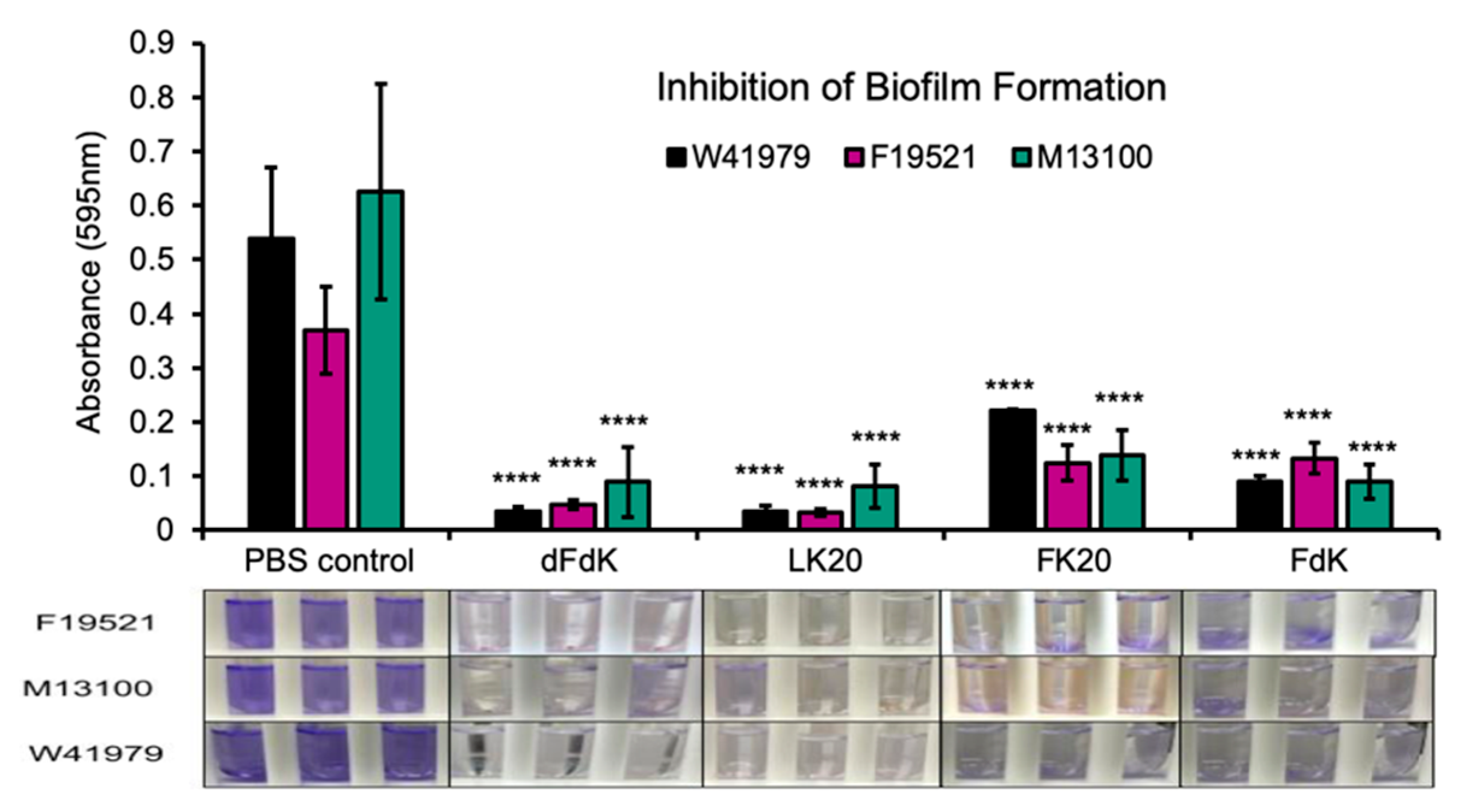
2.3. Random Peptide Mixtures Inhibit Biofilm Formation
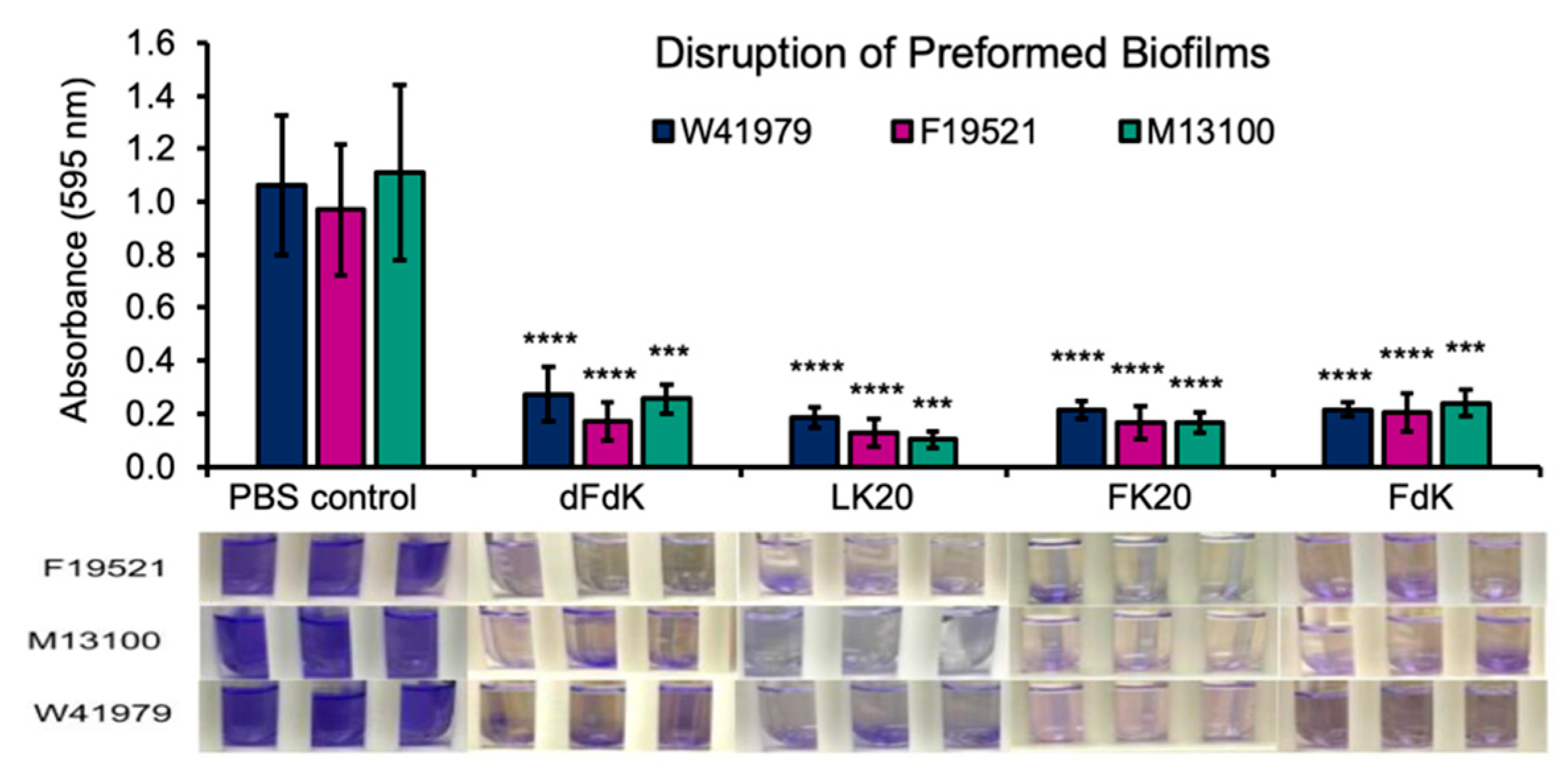
2.4. Random Peptide Mixtures Disrupt Preformed Biofilms
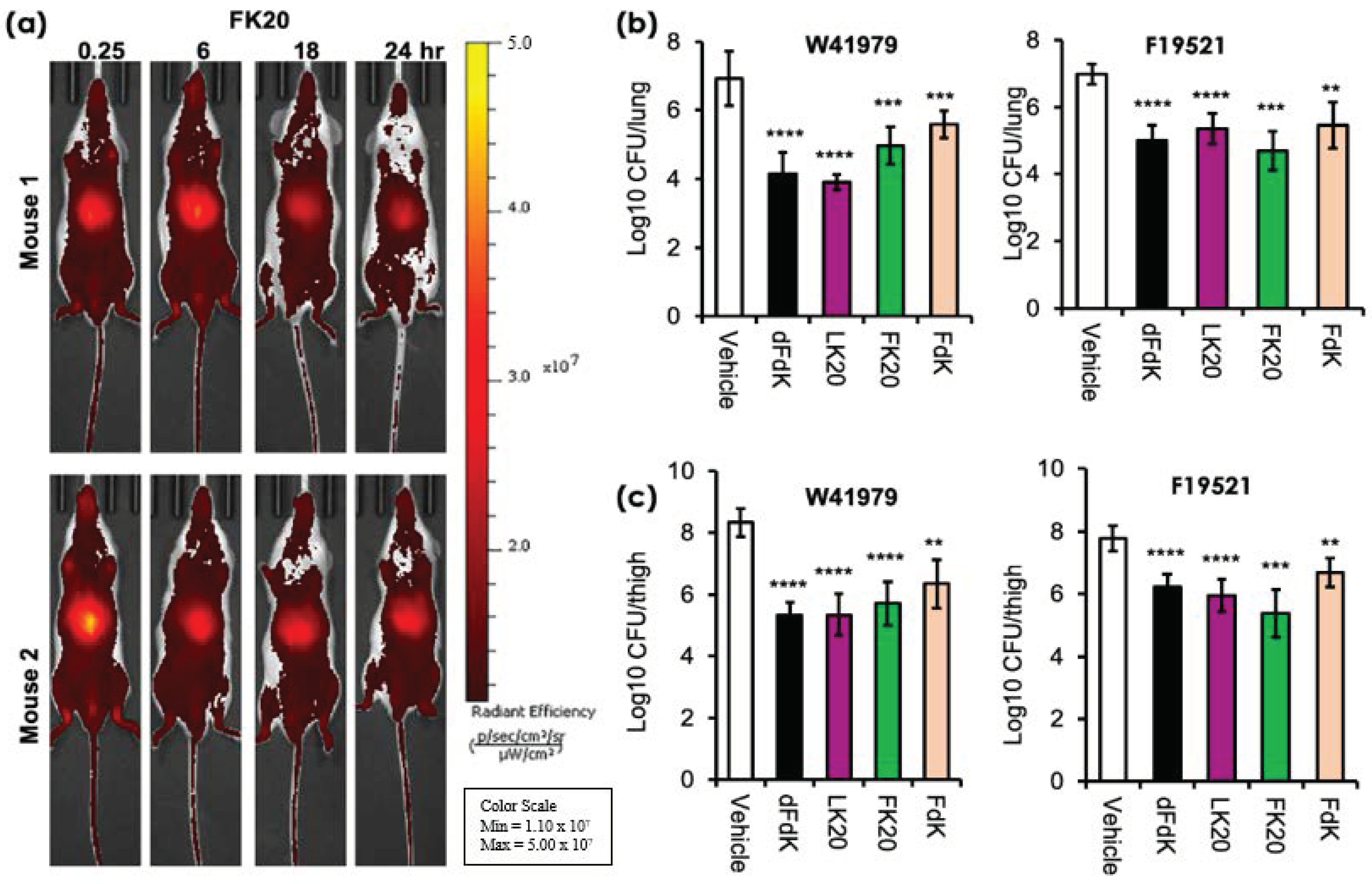
2.5. Random Peptide Mixtures Are Effective against Acute Pneumonia and Soft Tissue Infections in Mice
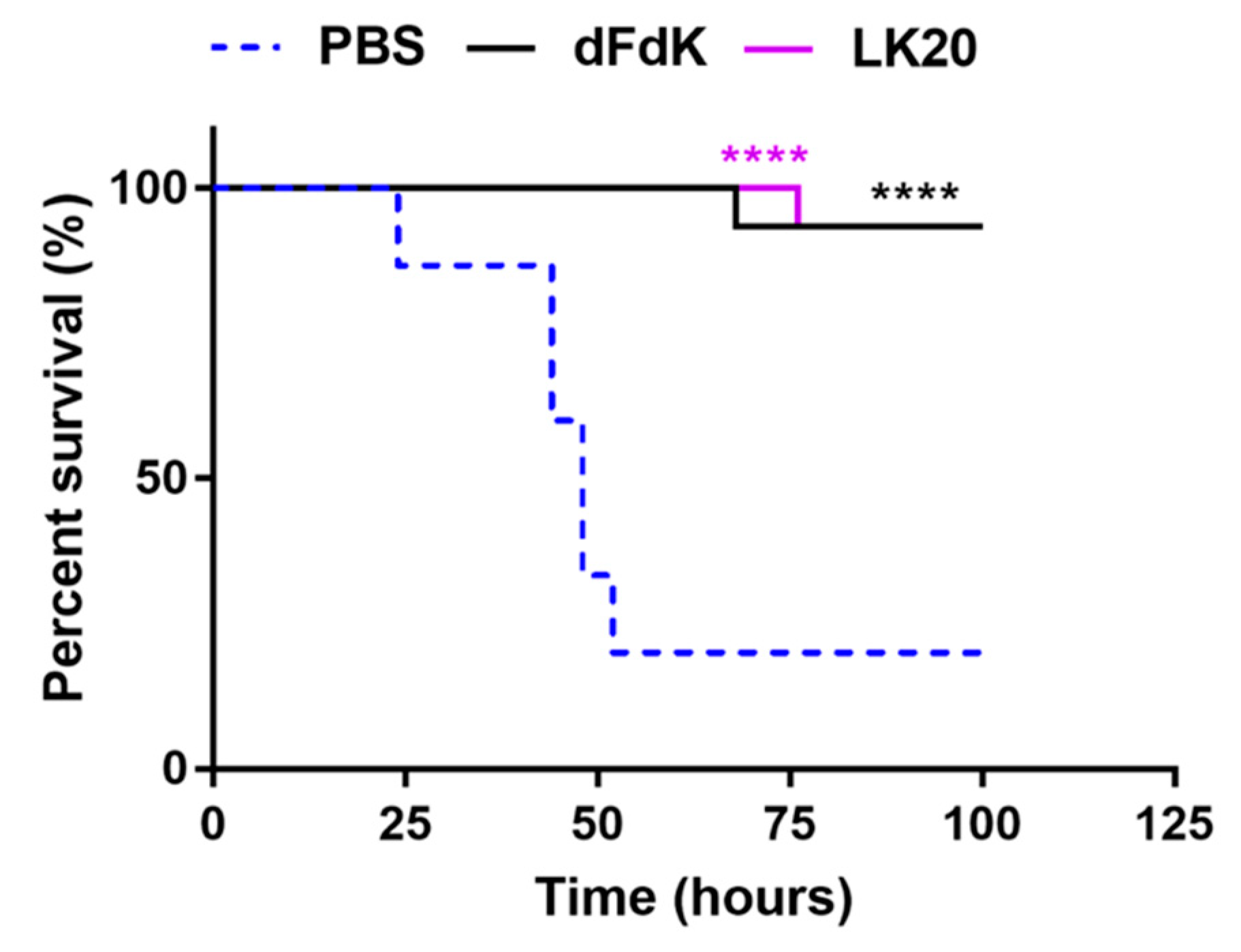
2.6. Random Peptide Mixtures Attenuate Mortality in a Mouse Model of Sepsis
3. Discussion
4. Materials and Methods
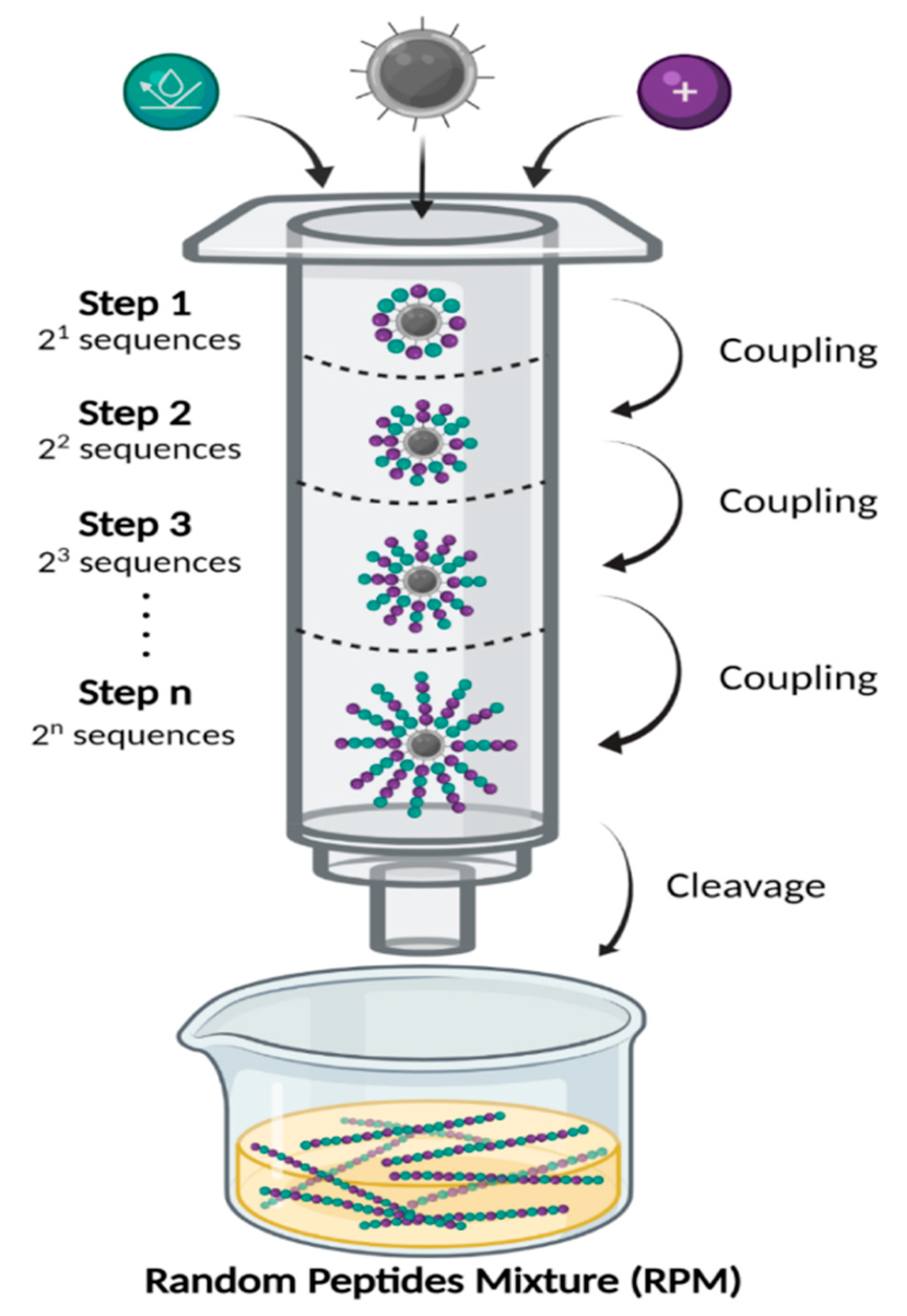
4.1. Synthesis and Storage of the Random Peptide Mixtures
4.2. Bacterial Cultures and MIC Determination
4.2.1. Bacterial Culture Conditions
4.2.2. Measurement of the Anti-Microbial Susceptibility Profile of Bacterial Isolates
4.3. In Vitro A. baumannii Killing Assays
4.4. Biofilm Inhibition and Disruption Assays
4.5. Mouse In Vivo Imaging and Infection Studies
4.5.1. In Vivo Imaging of FK20-Cy7.5 Pharmacodynamics
4.5.2. Mouse Model of Acute Pneumonia
4.5.3. Mouse Model of Neutropenic Soft Tissue Thigh Infection
4.5.4. Mouse Model of Bacterial Sepsis Survival
4.6. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hay, S.I.; Rao, P.C.; Dolecek, C.; Day, N.P.J.; Stergachis, A.; Lopez, A.D.; Murray, C.J.L. Measuring and mapping the global burden of antimicrobial resistance. BMC Med. 2018, 16, 78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Limmathurotsakul, D.; Dunachie, S.; Fukuda, K.; Feasey, N.A.; Okeke, I.N.; Holmes, A.H.; Moore, C.E.; Dolecek, C.; van Doorn, H.R.; Shetty, N.; et al. Improving the estimation of the global burden of antimicrobial resistant infections. Lancet Infect. Dis. 2019, 19, e392–e398. [Google Scholar] [CrossRef]
- Davies, J.; Davies, D. Origins and evolution of antibiotic resistance. Microbiol. Mol. Biol. Rev. 2010, 74, 417–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aslam, B.; Wang, W.; Arshad, M.I.; Khurshid, M.; Muzammil, S.; Rasool, M.H.; Nisar, M.A.; Alvi, R.F.; Aslam, M.A.; Qamar, M.U.; et al. Antibiotic resistance: A rundown of a global crisis. Infect. Drug Resist. 2018, 11, 1645–1658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.B.; Arnipalli, S.R.; Ziouzenkova, O. Antibiotics in Food Chain: The Consequences for Antibiotic Resistance. Antibiotics 2020, 9, 688. [Google Scholar] [CrossRef] [PubMed]
- Bengtsson-Palme, J.; Kristiansson, E.; Larsson, D.G.J. Environmental factors influencing the development and spread of antibiotic resistance. FEMS Microbiol. Rev. 2018, 42, fux053. [Google Scholar] [CrossRef] [PubMed]
- Michael, C.A.; Dominey-Howes, D.; Labbate, M. The antimicrobial resistance crisis: Causes, consequences, and management. Front. Public Health 2014, 2, 145. [Google Scholar] [CrossRef]
- Rice, L.B. Federal funding for the study of antimicrobial resistance in nosocomial pathogens: No ESKAPE. J. Infect. Dis. 2008, 197, 1079–1081. [Google Scholar] [CrossRef]
- De Oliveira, D.M.P.; Forde, B.M.; Kidd, T.J.; Harris, P.N.A.; Schembri, M.A.; Beatson, S.A.; Paterson, D.L.; Walker, M.J. Antimicrobial Resistance in ESKAPE Pathogens. Clin. Microbiol. Rev. 2020, 33, e00181-19. [Google Scholar] [CrossRef]
- Sarshar, M.; Behzadi, P.; Scribano, D.; Palamara, A.T.; Ambrosi, C. Acinetobacter baumannii: An Ancient Commensal with Weapons of a Pathogen. Pathogens 2021, 10, 387. [Google Scholar] [CrossRef]
- Hamidian, M.; Nigro, S.J. Emergence, molecular mechanisms and global spread of carbapenem-resistant Acinetobacter baumannii. Microb. Genom. 2019, 5, e000306. [Google Scholar] [CrossRef]
- Kyriakidis, I.; Vasileiou, E.; Pana, Z.D.; Tragiannidis, A. Acinetobacter baumannii Antibiotic Resistance Mechanisms. Pathogens 2021, 10, 373. [Google Scholar] [CrossRef] [PubMed]
- Coyne, S.; Courvalin, P.; Perichon, B. Efflux-mediated antibiotic resistance in Acinetobacter spp. Antimicrob. Agents Chemother. 2011, 55, 947–953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, E.J.; Chabane, Y.N.; Goussard, S.; Snesrud, E.; Courvalin, P.; De, E.; Grillot-Courvalin, C. Contribution of resistance-nodulation-cell division efflux systems to antibiotic resistance and biofilm formation in Acinetobacter baumannii. mBio 2015, 6, e00309-15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uppalapati, S.R.; Sett, A.; Pathania, R. The Outer Membrane Proteins OmpA, CarO, and OprD of Acinetobacter baumannii Confer a Two-Pronged Defense in Facilitating Its Success as a Potent Human Pathogen. Front. Microbiol. 2020, 11, 589234. [Google Scholar] [CrossRef]
- Bertini, A.; Poirel, L.; Bernabeu, S.; Fortini, D.; Villa, L.; Nordmann, P.; Carattoli, A. Multicopy blaOXA-58 gene as a source of high-level resistance to carbapenems in Acinetobacter baumannii. Antimicrob. Agents Chemother. 2007, 51, 2324–2328. [Google Scholar] [CrossRef] [Green Version]
- Turton, J.F.; Ward, M.E.; Woodford, N.; Kaufmann, M.E.; Pike, R.; Livermore, D.M.; Pitt, T.L. The role of ISAba1 in expression of OXA carbapenemase genes in Acinetobacter baumannii. FEMS Microbiol. Lett. 2006, 258, 72–77. [Google Scholar] [CrossRef] [Green Version]
- Corvec, S.; Poirel, L.; Naas, T.; Drugeon, H.; Nordmann, P. Genetics and expression of the carbapenem-hydrolyzing oxacillinase gene blaOXA-23 in Acinetobacter baumannii. Antimicrob. Agents Chemother. 2007, 51, 1530–1533. [Google Scholar] [CrossRef] [Green Version]
- Mugnier, P.D.; Poirel, L.; Nordmann, P. Functional analysis of insertion sequence ISAba1, responsible for genomic plasticity of Acinetobacter baumannii. J. Bacteriol. 2009, 191, 2414–2418. [Google Scholar] [CrossRef] [Green Version]
- Tsioutis, C.; Kritsotakis, E.I.; Karageorgos, S.A.; Stratakou, S.; Psarologakis, C.; Kokkini, S.; Gikas, A. Clinical epidemiology, treatment and prognostic factors of extensively drug-resistant Acinetobacter baumannii ventilator-associated pneumonia in critically ill patients. Int. J. Antimicrob. Agents 2016, 48, 492–497. [Google Scholar] [CrossRef]
- Brotfain, E.; Borer, A.; Koyfman, L.; Saidel-Odes, L.; Frenkel, A.; Gruenbaum, S.E.; Rosenzweig, V.; Zlotnik, A.; Klein, M. Multidrug Resistance Acinetobacter Bacteremia Secondary to Ventilator-Associated Pneumonia: Risk Factors and Outcome. J. Intensive Care Med. 2017, 32, 528–534. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.J.; Gallo, R.L. Antimicrobial peptides. Curr. Biol. 2016, 26, R14–R19. [Google Scholar] [CrossRef] [PubMed]
- Lei, J.; Sun, L.; Huang, S.; Zhu, C.; Li, P.; He, J.; Mackey, V.; Coy, D.H.; He, Q. The antimicrobial peptides and their potential clinical applications. Am. J. Transl. Res. 2019, 11, 3919–3931. [Google Scholar] [PubMed]
- Huan, Y.; Kong, Q.; Mou, H.; Yi, H. Antimicrobial Peptides: Classification, Design, Application and Research Progress in Multiple Fields. Front. Microbiol. 2020, 11, 582779. [Google Scholar] [CrossRef] [PubMed]
- Oren, Z.; Ramesh, J.; Avrahami, D.; Suryaprakash, N.; Shai, Y.; Jelinek, R. Structures and mode of membrane interaction of a short alpha helical lytic peptide and its diastereomer determined by NMR, FTIR, and fluorescence spectroscopy. Eur. J. Biochem. 2002, 269, 3869–3880. [Google Scholar] [CrossRef] [Green Version]
- Rathinakumar, R.; Walkenhorst, W.F.; Wimley, W.C. Broad-spectrum antimicrobial peptides by rational combinatorial design and high-throughput screening: The importance of interfacial activity. J. Am. Chem. Soc. 2009, 131, 7609–7617. [Google Scholar] [CrossRef] [Green Version]
- Zasloff, M. Antimicrobial peptides, innate immunity, and the normally sterile urinary tract. J. Am. Soc. Nephrol. 2007, 18, 2810–2816. [Google Scholar] [CrossRef] [Green Version]
- Zasloff, M.; Martin, B.; Chen, H.C. Antimicrobial activity of synthetic magainin peptides and several analogues. Proc. Natl. Acad. Sci. USA 1988, 85, 910–913. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Liu, Y.; Shen, T.; Chen, L.; Zhang, C.; Cai, K.; Liao, C.; Wang, C. Antimicrobial activity of the antibacterial peptide PMAP-36 and its analogues. Microb. Pathog. 2019, 136, 103712. [Google Scholar] [CrossRef]
- Gorr, S.U.; Flory, C.M.; Schumacher, R.J. In vivo activity and low toxicity of the second-generation antimicrobial peptide DGL13K. PLoS ONE 2019, 14, e0216669. [Google Scholar] [CrossRef] [Green Version]
- Mourtada, R.; Herce, H.D.; Yin, D.J.; Moroco, J.A.; Wales, T.E.; Engen, J.R.; Walensky, L.D. Design of stapled antimicrobial peptides that are stable, nontoxic and kill antibiotic-resistant bacteria in mice. Nat. Biotechnol. 2019, 37, 1186–1197. [Google Scholar] [CrossRef] [PubMed]
- Fjell, C.D.; Hiss, J.A.; Hancock, R.E.; Schneider, G. Designing antimicrobial peptides: Form follows function. Nat. Rev. Drug Discov. 2011, 11, 37–51. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Koh, J.J.; Liu, S.; Lakshminarayanan, R.; Verma, C.S.; Beuerman, R.W. Membrane Active Antimicrobial Peptides: Translating Mechanistic Insights to Design. Front. Neurosci. 2017, 11, 73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greber, K.E.; Dawgul, M. Antimicrobial Peptides Under Clinical Trials. Curr. Top. Med. Chem. 2017, 17, 620–628. [Google Scholar] [CrossRef] [PubMed]
- Neuhaus, F.C.; Baddiley, J. A continuum of anionic charge: Structures and functions of D-alanyl-teichoic acids in gram-positive bacteria. Microbiol. Mol. Biol. Rev. 2003, 67, 686–723. [Google Scholar] [CrossRef] [Green Version]
- Gunn, J.S.; Ryan, S.S.; Van Velkinburgh, J.C.; Ernst, R.K.; Miller, S.I. Genetic and functional analysis of a PmrA-PmrB-regulated locus necessary for lipopolysaccharide modification, antimicrobial peptide resistance, and oral virulence of Salmonella enterica serovar typhimurium. Infect. Immun. 2000, 68, 6139–6146. [Google Scholar] [CrossRef] [PubMed]
- Perron, G.G.; Zasloff, M.; Bell, G. Experimental evolution of resistance to an antimicrobial peptide. Proc. Biol. Sci. 2006, 273, 251–256. [Google Scholar] [CrossRef] [Green Version]
- Epand, R.M. Host Defense Peptides and Their Potential as Therapeutic Agents. Anticancer. Res. 2016, 36, 4375. [Google Scholar]
- Bayat, M.; Zargar, M.; Chudinova, E.; Astarkhanova, T.; Pakina, E. In Vitro Evaluation of Antibacterial and Antifungal Activity of Biogenic Silver and Copper Nanoparticles: The First Report of Applying Biogenic Nanoparticles against Pilidium concavum and Pestalotia sp. Fungi. Molecules 2021, 26, 5402. [Google Scholar] [CrossRef]
- Rozhin, A.; Batasheva, S.; Kruychkova, M.; Cherednichenko, Y.; Rozhina, E.; Fakhrullin, R. Biogenic Silver Nanoparticles: Synthesis and Application as Antibacterial and Antifungal Agents. Micromachines 2021, 12, 1480. [Google Scholar] [CrossRef]
- Moran Diaz, J.R.; Guevara-Salazar, J.A.; Cuevas Hernandez, R.I.; Trujillo Ferrara, J.G. A more specific concept of a pharmacophore to better rationalize drug design, tailor patient therapy, and tackle bacterial resistance to antibiotics. Expert Opin. Drug Discov. 2022, 17, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Seidel, T.; Schuetz, D.A.; Garon, A.; Langer, T. The Pharmacophore Concept and Its Applications in Computer-Aided Drug Design. Prog. Chem. Org. Nat. Prod. 2019, 110, 99–141. [Google Scholar] [PubMed]
- Seidel, T.; Wieder, O.; Garon, A.; Langer, T. Applications of the Pharmacophore Concept in Natural Product inspired Drug Design. Mol. Inform. 2020, 39, e2000059. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Cho, M.; Son, H.S. Simulation Model of Bacterial Resistance to Antibiotics Using Individual-Based Modeling. J. Comput. Biol. 2018, 25, 1059–1070. [Google Scholar] [CrossRef]
- Dobson, A.J.; Purves, J.; Kamysz, W.; Rolff, J. Comparing selection on S. aureus between antimicrobial peptides and common antibiotics. PLoS ONE 2013, 8, e76521. [Google Scholar]
- Hayouka, Z.; Bella, A.; Stern, T.; Ray, S.; Jiang, H.; Grovenor, C.R.M.; Ryadnov, M.G. Binary Encoding of Random Peptide Sequences for Selective and Differential Antimicrobial Mechanisms. Angew. Chem. Int. Ed. Engl. 2017, 56, 8099–8103. [Google Scholar] [CrossRef]
- Hayouka, Z.; Chakraborty, S.; Liu, R.; Boersma, M.D.; Weisblum, B.; Gellman, S.H. Interplay among subunit identity, subunit proportion, chain length, and stereochemistry in the activity profile of sequence-random peptide mixtures. J. Am. Chem. Soc. 2013, 135, 11748–11751. [Google Scholar] [CrossRef] [Green Version]
- Hayouka, Z.; Mortenson, D.E.; Kreitler, D.F.; Weisblum, B.; Forest, K.T.; Gellman, S.H. Evidence for Phenylalanine Zipper-Mediated Dimerization in the X-ray Crystal Structure of a Magainin 2 Analogue. J. Am. Chem. Soc. 2013, 135, 15738–15741. [Google Scholar] [CrossRef] [Green Version]
- Amso, Z.; Hayouka, Z. Antimicrobial random peptide cocktails: A new approach to fight pathogenic bacteria. Chem. Commun. 2019, 55, 2007–2014. [Google Scholar] [CrossRef]
- Stern, T.; Zelinger, E.; Hayouka, Z. Random peptide mixtures inhibit and eradicate methicillin-resistant Staphylococcus aureus biofilms. Chem. Commun. 2016, 52, 7102–7105. [Google Scholar] [CrossRef]
- Topman, S.; Tamir-Ariel, D.; Bochnic-Tamir, H.; Stern Bauer, T.; Shafir, S.; Burdman, S.; Hayouka, Z. Random peptide mixtures as new crop protection agents. Microb. Biotechnol. 2018, 11, 1027–1036. [Google Scholar] [CrossRef] [PubMed]
- Bennett, R.C.; Oh, M.W.; Kuo, S.H.; Belo, Y.; Maron, B.; Malach, E.; Lin, J.; Hayouka, Z.; Lau, G.W. Random Peptide Mixtures as Safe and Effective Antimicrobials against Pseudomonas aeruginosa and MRSA in Mouse Models of Bacteremia and Pneumonia. ACS Infect. Dis. 2021, 7, 672–680. [Google Scholar] [CrossRef] [PubMed]
- Del Pozo, J.L. Biofilm-related disease. Expert Rev. Anti Infect. Ther. 2018, 16, 51–65. [Google Scholar] [CrossRef] [PubMed]
- Capoor, M.N.; Ruzicka, F.; Schmitz, J.E.; James, G.A.; Machackova, T.; Jancalek, R.; Smrcka, M.; Lipina, R.; Ahmed, F.S.; Alamin, T.F.; et al. Propionibacterium acnes biofilm is present in intervertebral discs of patients undergoing microdiscectomy. PLoS ONE 2017, 12, e0174518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parsek, M.R.; Singh, P.K. Bacterial biofilms: An emerging link to disease pathogenesis. Annu. Rev. Microbiol. 2003, 57, 677–701. [Google Scholar] [CrossRef] [PubMed]
- Gominet, M.; Compain, F.; Beloin, C.; Lebeaux, D. Central venous catheters and biofilms: Where do we stand in 2017? APMIS 2017, 125, 365–375. [Google Scholar] [CrossRef] [Green Version]
- Yousif, A.; Jamal, M.A.; Raad, I. Biofilm-based central line-associated bloodstream infections. Adv. Exp. Med. Biol. 2015, 830, 157–179. [Google Scholar]
- Arciola, C.R.; Campoccia, D.; Montanaro, L. Implant infections: Adhesion, biofilm formation and immune evasion. Nat. Rev. Microbiol. 2018, 16, 397–409. [Google Scholar] [CrossRef]
- Haney, E.F.; Trimble, M.J.; Cheng, J.T.; Valle, Q.; Hancock, R.E.W. Critical Assessment of Methods to Quantify Biofilm Growth and Evaluate Antibiofilm Activity of Host Defence Peptides. Biomolecules 2018, 8, 29. [Google Scholar] [CrossRef] [Green Version]
- Lewis, K. Multidrug tolerance of biofilms and persister cells. Curr. Top. Microbiol. Immunol. 2008, 322, 107–131. [Google Scholar]
- Yan, J.; Bassler, B.L. Surviving as a Community: Antibiotic Tolerance and Persistence in Bacterial Biofilms. Cell Host Microbe 2019, 26, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Lewis, K. Persister cells. Annu. Rev. Microbiol. 2010, 64, 357–372. [Google Scholar] [CrossRef]
- Tan, R.M.; Kuang, Z.; Hao, Y.; Lee, F.; Lee, T.; Lee, R.J.; Lau, G.W. Type IV pilus glycosylation mediates resistance of Pseudomonas aeruginosa to opsonic activities of the pulmonary surfactant protein A. Infect. Immun. 2015, 83, 1339–1346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, L.; Lin, J.; Kuang, Z.; Vidal, J.E.; Lau, G.W. Deletion analysis of Streptococcus pneumoniae late competence genes distinguishes virulence determinants that are dependent or independent of competence induction. Mol. Microbiol. 2015, 97, 151–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, J.; Park, P.; Li, H.; Oh, M.W.; Dobrucki, I.T.; Dobrucki, W.; Lau, G.W. Streptococcus pneumoniae Elaborates Persistent and Prolonged Competent State during Pneumonia-Derived Sepsis. Infect. Immun. 2020, 88, e00919-19. [Google Scholar] [CrossRef] [PubMed]
- Motika, S.E.; Ulrich, R.J.; Geddes, E.J.; Lee, H.Y.; Lau, G.W.; Hergenrother, P.J. Gram-Negative Antibiotic Active Through Inhibition of an Essential Riboswitch. J. Am. Chem. Soc. 2020, 142, 10856–10862. [Google Scholar] [CrossRef] [PubMed]
- Parker, E.N.; Drown, B.S.; Geddes, E.J.; Lee, H.Y.; Ismail, N.; Lau, G.W.; Hergenrother, P.J. Implementation of permeation rules leads to a FabI inhibitor with activity against Gram-negative pathogens. Nat. Microbiol. 2020, 5, 67–75. [Google Scholar] [CrossRef]
- Garcia Chavez, M.; Garcia, A.; Lee, H.Y.; Lau, G.W.; Parker, E.N.; Komnick, K.E.; Hergenrother, P.J. Synthesis of Fusidic Acid Derivatives Yields a Potent Antibiotic with an Improved Resistance Profile. ACS Infect. Dis. 2021, 7, 493–505. [Google Scholar] [CrossRef]
- Boylan, C.J.; Campanale, K.; Iversen, P.W.; Phillips, D.L.; Zeckel, M.L.; Parr, T.R. Pharmacodynamics of oritavancin (LY333328) in a neutropenic-mouse thigh model of Staphylococcus aureus infection. Antimicrob. Agents Chemother. 2003, 47, 1700–1706. [Google Scholar] [CrossRef] [Green Version]
- Cheah, S.E.; Wang, J.; Nguyen, V.T.; Turnidge, J.D.; Li, J.; Nation, R.L. New pharmacokinetic/pharmacodynamic studies of systemically administered colistin against Pseudomonas aeruginosa and Acinetobacter baumannii in mouse thigh and lung infection models: Smaller response in lung infection. J. Antimicrob. Chemother. 2015, 70, 3291–3297. [Google Scholar]
- Stern Bauer, T.; Hayouka, Z. Random mixtures of antimicrobial peptides inhibit bacteria associated with pasteurized bovine milk. J. Pept. Sci. 2018, 24, e3088. [Google Scholar] [CrossRef] [PubMed]
- Topman-Rakover, S.; Malach, E.; Burdman, S.; Hayouka, Z. Antibacterial lipo-random peptide mixtures exhibit high selectivity and synergistic interactions. Chem. Commun. 2020, 56, 12053–12056. [Google Scholar] [CrossRef] [PubMed]
- Spellberg, B.; Rex, J.H. The value of single-pathogen antibacterial agents. Nat. Rev. Drug. Discov. 2013, 12, 963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falagas, M.E.; Rafailidis, P.I. Attributable mortality of Acinetobacter baumannii: No longer a controversial issue. Crit. Care 2007, 11, 134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dijkshoorn, L.; Nemec, A.; Seifert, H. An increasing threat in hospitals: Multidrug-resistant Acinetobacter baumannii. Nat. Rev. Microbiol. 2007, 5, 939–951. [Google Scholar] [CrossRef] [PubMed]
- Flemming, H.C.; Wuertz, S. Bacteria and archaea on Earth and their abundance in biofilms. Nat. Rev. Microbiol. 2019, 17, 247–260. [Google Scholar] [CrossRef] [PubMed]
- Galie, S.; Garcia-Gutierrez, C.; Miguelez, E.M.; Villar, C.J.; Lombo, F. Biofilms in the Food Industry: Health Aspects and Control Methods. Front. Microbiol. 2018, 9, 898. [Google Scholar] [CrossRef]
- Donlan, R.M.; Costerton, J.W. Biofilms: Survival mechanisms of clinically relevant microorganisms. Clin. Microbiol. Rev. 2002, 15, 167–193. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibiotic | MIC (μg mL−1) | Interpretation 1 | |
|---|---|---|---|
| Strain W41979 | Ceftazidime | >64 | R |
| Cefepime | 32 | R | |
| Gentamycin | >16 | R | |
| Levofloxacin | 4 | I | |
| Pipperacillin/Tazobactam | >128 | R | |
| Ciprofloxacin | >2 | R | |
| Tobramycin | >16 | R | |
| Imipenem | >16 | R | |
| Meropenem | >8 | R | |
| Colistin | 0.19 | S | |
| Strain F19521 | Ceftazidime | >64 | R |
| Cefepime | >64 | R | |
| Gentamycin | >16 | R | |
| Levofloxacin | 4 | I | |
| Pipperacillin/Tazobactam | >128 | R | |
| Ciprofloxacin | >2 | R | |
| Tobramycin | >16 | R | |
| Imipenem | >16 | R | |
| Meropenem | >8 | R | |
| Colistin | 0.125 | S | |
| Strain M13100 | Amikacin | 4 | S |
| Cefazolin | >32 | R | |
| Ceftazidime | >64 | R | |
| Cefepime | >64 | R | |
| Ceftriaxone | >64 | R | |
| Gentamycin | >16 | R | |
| Levofloxacin | 4 | I | |
| Pipperacillin/Tazobactam | >128 | R | |
| Trimethoprim/sulphamethox | >320 | R | |
| Ampicillin/sulbactam | >32/16 | R | |
| Tobramycin | >16 | R | |
| Imipenem | 4 | R | |
| Erapenem | 8 | R |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Caraway, H.E.; Lau, J.Z.; Maron, B.; Oh, M.W.; Belo, Y.; Brill, A.; Malach, E.; Ismail, N.; Hayouka, Z.; Lau, G.W. Antimicrobial Random Peptide Mixtures Eradicate Acinetobacter baumannii Biofilms and Inhibit Mouse Models of Infection. Antibiotics 2022, 11, 413. https://doi.org/10.3390/antibiotics11030413
Caraway HE, Lau JZ, Maron B, Oh MW, Belo Y, Brill A, Malach E, Ismail N, Hayouka Z, Lau GW. Antimicrobial Random Peptide Mixtures Eradicate Acinetobacter baumannii Biofilms and Inhibit Mouse Models of Infection. Antibiotics. 2022; 11(3):413. https://doi.org/10.3390/antibiotics11030413
Chicago/Turabian StyleCaraway, Hannah E., Jonathan Z. Lau, Bar Maron, Myung Whan Oh, Yael Belo, Aya Brill, Einav Malach, Nahed Ismail, Zvi Hayouka, and Gee W. Lau. 2022. "Antimicrobial Random Peptide Mixtures Eradicate Acinetobacter baumannii Biofilms and Inhibit Mouse Models of Infection" Antibiotics 11, no. 3: 413. https://doi.org/10.3390/antibiotics11030413