
Potential Therapeutic Targets for Combination Antibody Therapy against Pseudomonas aeruginosa Infections

 ,
,
Abstract
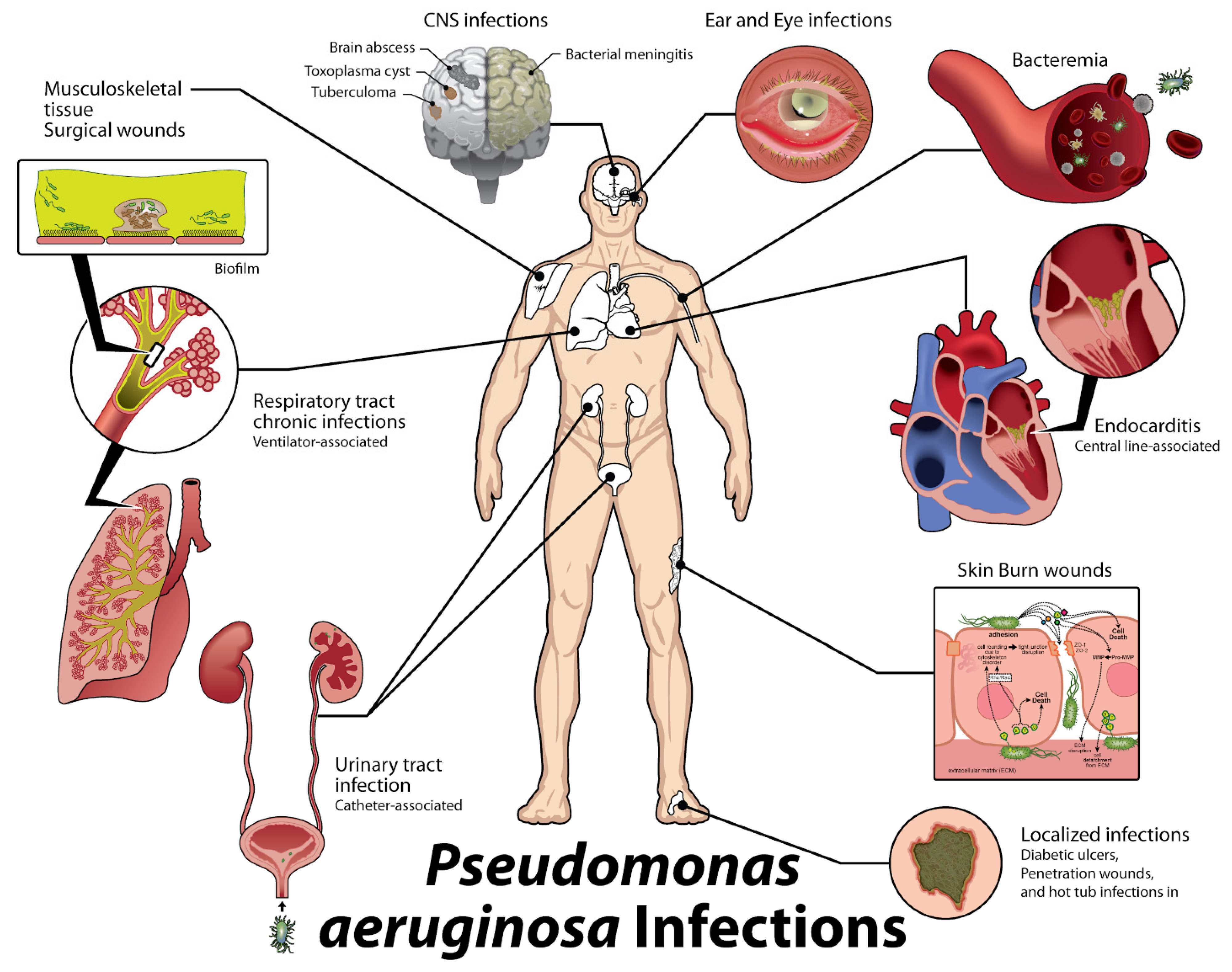
:1. Introduction
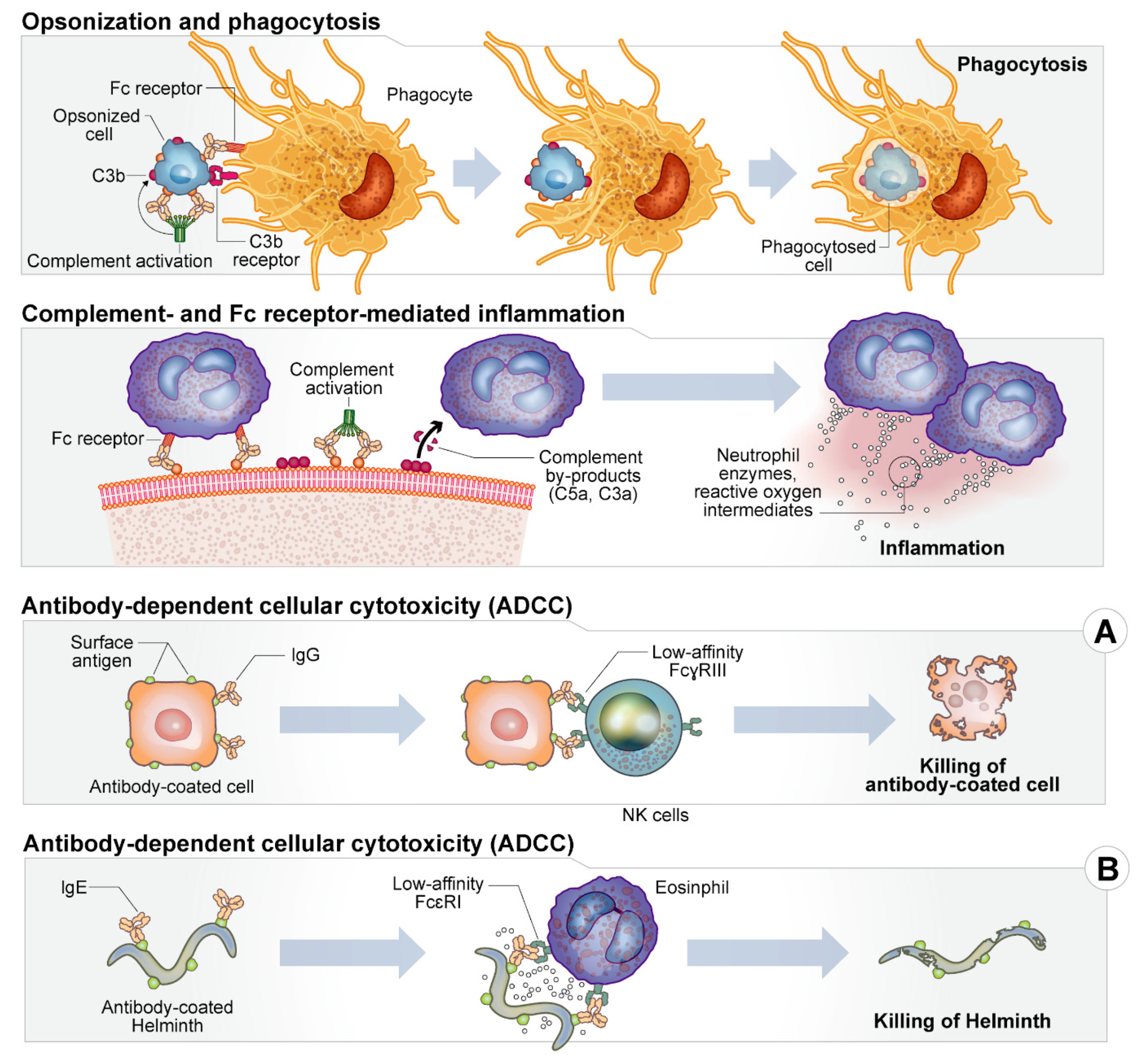
2. Host Immune Response
3. Description of Targets
3.1. Secreted Toxins and Invasins
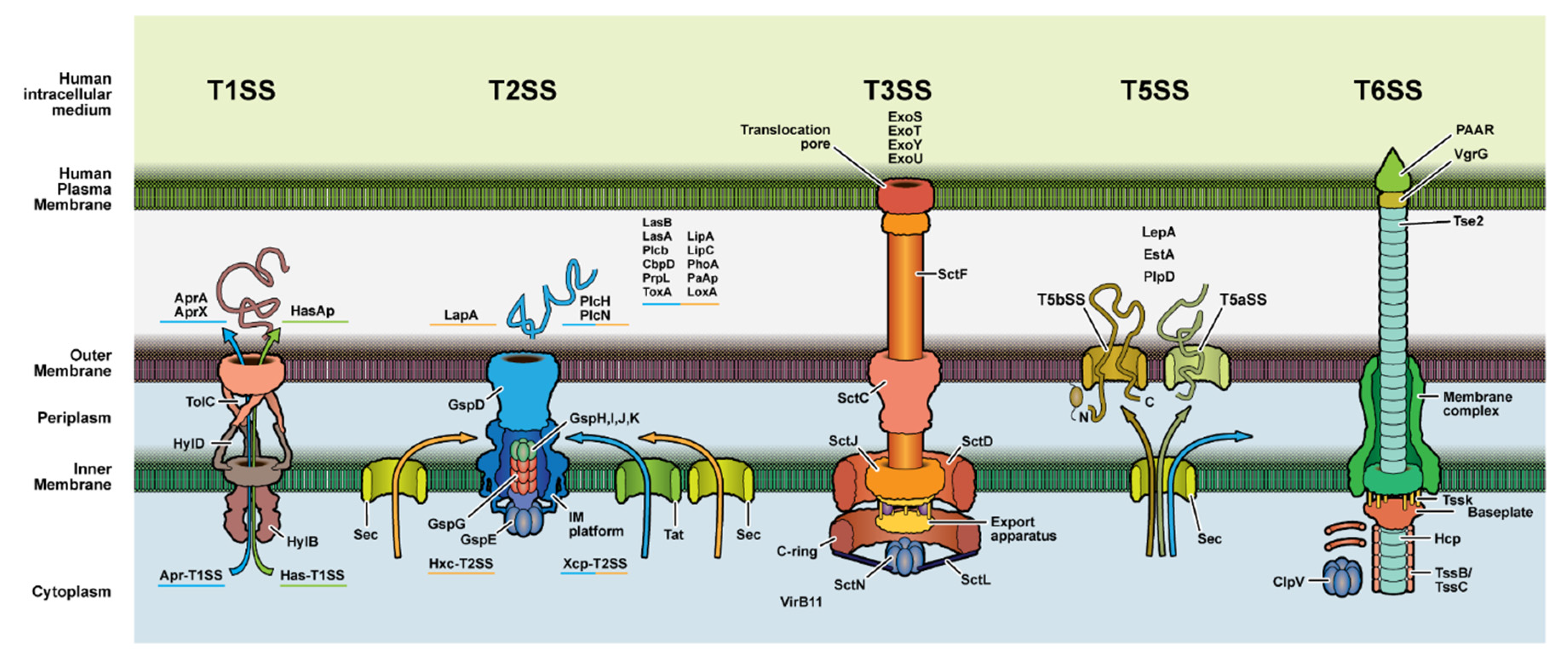
3.2. Secretion System Proteins
3.3. Quorum Sensing/Metabolites
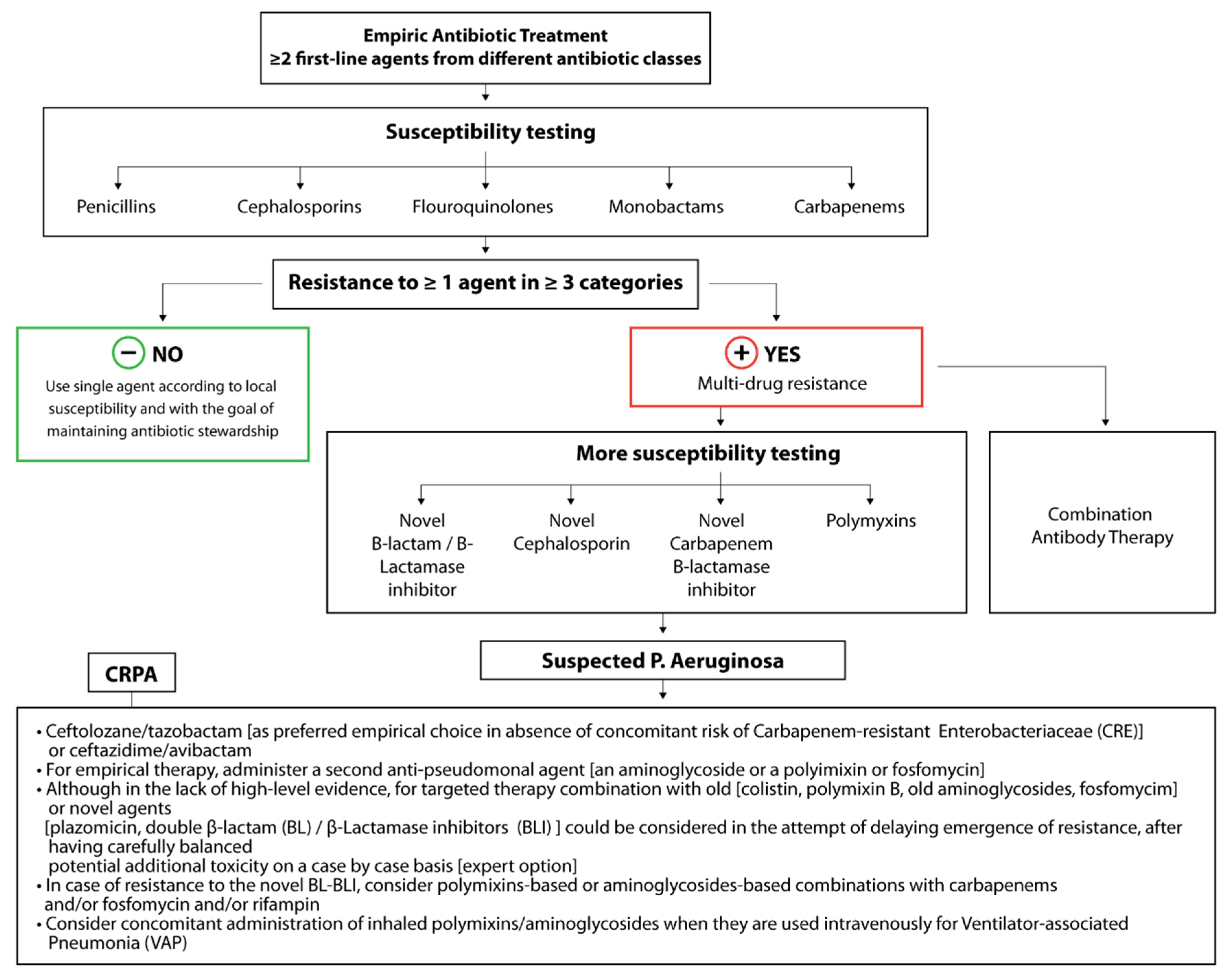
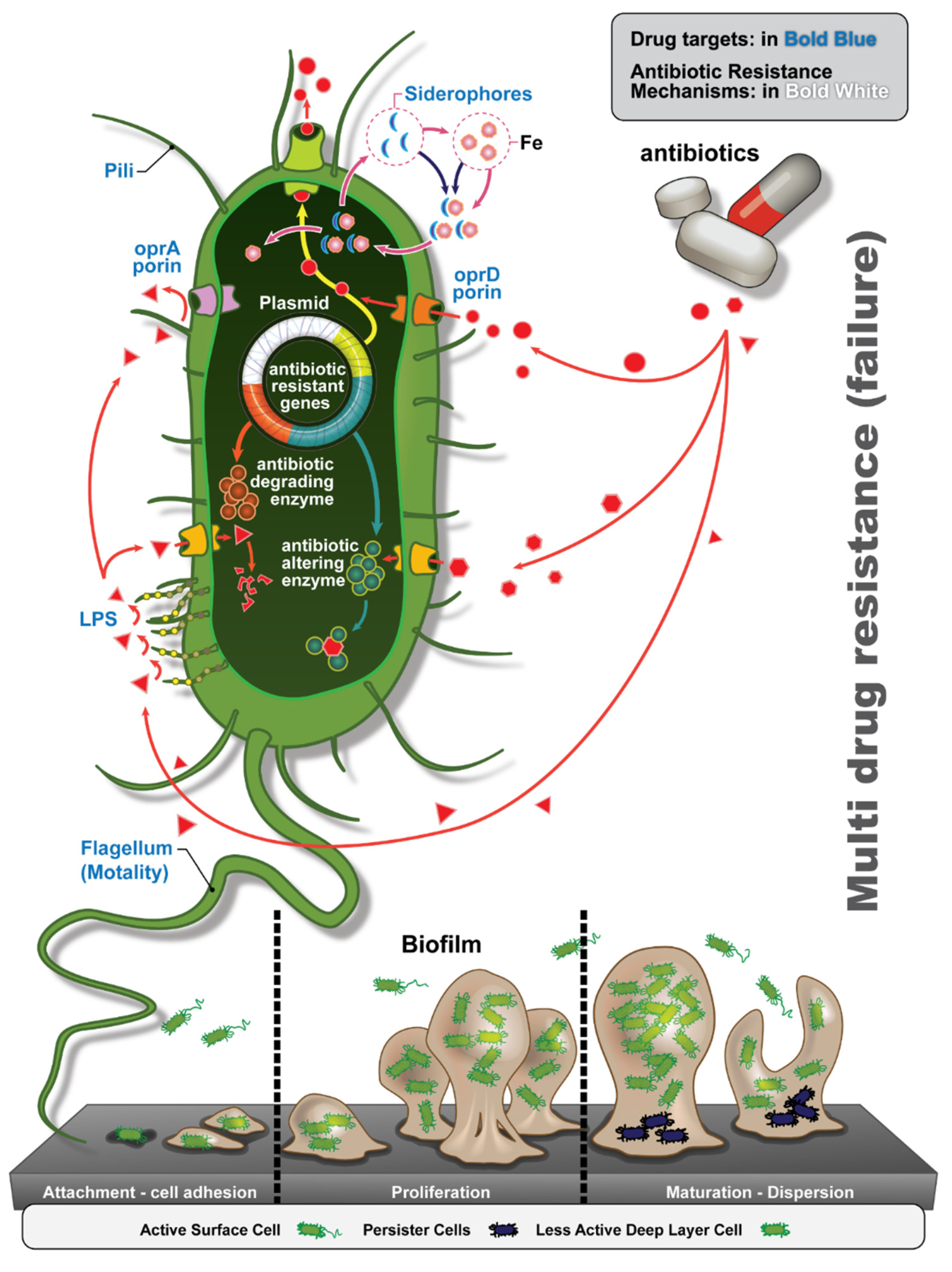
3.4. Antibiotic Resistance Determinants
3.5. Other Membrane Biomolecules
3.6. Motility Factors
3.7. Resource Scavenging Molecules
3.8. Immunomodulators
4. Antibodies as Therapeutics
Combinatorial Therapy
5. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AHL | Acyl Homoserine Lactone |
| AprA | Alkaline Protease A |
| CF | Cystic Fibrosis |
| CIF | CFTR Inhibiting Factor |
| CNS | Central Nervous System |
| CRPA | Carbapenem Resistant Pseudomonas aeruginosa |
| ExoU | Exotoxin U |
| ExoS | Exotoxin S |
| ExoT | Exotoxin T |
| ExoY | Exotoxin Y |
| FAb | antigen-binding region of an antibody |
| Fc | cell receptor region of an antibody |
| GlpF | Glycerol Uptake Facilitator Protein |
| GlpT | Glycerol-2-Phosphate Transporter |
| HCN | Hydrogen Cyanide |
| IDSA | Infectious Disease Society of America |
| IgA | Immunoglobulin A |
| IgE | Immunoglobulin E |
| IgG | Immunoglobulin G |
| IgM | Immunoglobulin M |
| IVIG | Intravenous Immunoglobulin |
| LOX | Lipoxygenase |
| mABs | Monoclonal Antibodies (is mABs in text) |
| MAM7 | Multivalent Adhesion Molecule 7 |
| MEP | Mucoid Exopolysaccharide |
| MIC | Minimum Inhibitory Concentration |
| MMP | Matrix Metallopeptide |
| LPS | Lipopolysaccharide |
| OMV | Outer Membrane Vesicle |
| NET | Neutrophil Extracellular Trap |
| PAB | Polyclonal Antibody |
| PC | Phosphatidylcholine |
| PCH | Pyochelin |
| PLC | Phospholipase C |
| PQS | Pseudomonas Quinolone Signal |
| QS | Quorum Sensing |
| ROS | Reactive Oxygen Species |
| TFP | Type IV Pili |
| TNF | Tumor Necrosis Factor |
| TLR | Toll-Like Receptor |
| T1SS | Type 1 Secretion System |
| T2SS | Type 2 Secretion System |
| T3SS | Type 3 Secretion System |
| UTI | Urinary Tract Infection |
References
- Kanj, S.; Sexton, M.D.; Daniel, M.D. Epidemiology, Microbiology, and Pathogenesis of Pseudomonas aeruginosa Infection. Available online: https://www.uptodate.com/contents/epidemiology-microbiology-and-pathogenesis-of-pseudomonas-aeruginosa-infection?search=pseudomonas&source=search_result&selectedTitle=2~150&usage_type=default&display_rank=2 (accessed on 21 September 2020).
- Owlia, P.; Nosrati, R.; Alaghehbandan, R.; Lari, A.R. Antimicrobial susceptibility differences among mucoid and non-mucoid Pseudomonas aeruginosa isolates. GMS Hyg. Infect. Control. 2014, 9, 13. [Google Scholar] [CrossRef]
- Souha Kanj, M.D. Principles of Antimicrobial Therapy of Pseudomonas aeruginosa Infections. Available online: https://www-uptodate-com.proxy.rvu.edu/contents/principles-of-antimicrobial-therapy-of-pseudomonas-aeruginosa-infections#H6675731 (accessed on 21 September 2020).
- Centers for Disease Control and Prevention. Pseudomonas aeruginosa in Healthcare Settings. Available online: https://www.cdc.gov/hai/organisms/pseudomonas.html (accessed on 21 September 2020).
- Todar, K. Pseudomonas. Online Textbook of Bacteriology. Available online: http://textbookofbacteriology.net/pseudomonas_4.html (accessed on 21 September 2020).
- Nordmann, P.; Poirel, L. Epidemiology and Diagnostics of Carbapenem Resistance in Gram-negative Bacteria. Clin Infect Dis. 2019, 69 (Suppl. S7), S521–S528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamma, P.D.; Aitken, S.L.; Bonomo, R.A.; Mathers, A.J.; van Duin, D.; Clancy, C.J. Infectious Diseases Society of America Guidance on the Treatment of Extended-Spectrum β-lactamase Producing Enterobacterales (ESBL-E), Carbapenem-Resistant Enterobacterales (CRE), and Pseudomonas aeruginosa with Difficult-to-Treat Resistance (DTR-P. aeruginosa). Clin. Infect Dis. 2020, 72, e169–e183. [Google Scholar] [CrossRef]
- Pang, Z.; Raudonis, R.; Glick, B.R.; Lin, T.-J.; Cheng, Z. Antibiotic resistance in Pseudomonas aeruginosa: Mechanisms and alternative therapeutic strategies. Biotechnol. Adv. 2019, 37, 177–192. [Google Scholar] [CrossRef]
- Centers for Disease Control and Prevention. Antibiotic-Resistant Germs: New Threats. Available online: https://www.cdc.gov/drugresistance/biggest-threats.html (accessed on 6 May 2020).
- Kmeid, J.G.; Youssef, M.M.; Kanafani, Z.A.; Kanj, S.S. Combination therapy for Gram-negative bacteria: What is the evidence? Expert. Rev. Anti. Infect. Ther. 2013, 11, 1355–1362. [Google Scholar] [CrossRef]
- Grekov, I.; Thöming, J.G.; Kordes, A.; Häussler, S. Evolution of Pseudomonas aeruginosa toward higher fitness under standard laboratory conditions. ISME J. 2021, 15, 1165–1177. [Google Scholar] [CrossRef] [PubMed]
- Fothergill, J.L.; Panagea, S.; Hart, C.A.; Walshaw, M.J.; Pitt, T.L.; Winstanley, C. Widespread pyocyanin over-production among isolates of a cystic fibrosis epidemic strain. BMC Microbiol. 2007, 7, 45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathee, K.; Ciofu, O.; Sternberg, C.; Lindum, P.W.; Campbell, J.I.A.; Jensen, P.; Johnsen, A.H.; Givskov, M.; Ohman, D.E.; Søren, M.; et al. Mucoid conversion of Pseudomonas aeruginos by hydrogen peroxide: A mechanism for virulence activation in the cystic fibrosis lung. Microbiology 1999, 145, 1349–1357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Von Götz, F.; Häussler, S.; Jordan, D.; Saravanamuthu, S.S.; Wehmhöner, D.; Strüssmann, A.; Lauber, J.; Attree, I.; Buer, J.; Tümmler, B.; et al. Expression Analysis of a Highly Adherent and Cytotoxic Small Colony Variant of Pseudomonas aeruginosa Isolated from a Lung of a Patient with Cystic Fibrosis. J. Bacteriol. 2004, 186, 3837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Häußler, S.; Tümmler, B.; Weißbrodt, H.; Rohde, M.; Steinmetz, I. Small-Colony Variants of Pseudomonas aeruginosa in Cystic Fibrosis. Clin. Infect. Dis. 1999, 29, 621–625. [Google Scholar] [CrossRef]
- D’Argenio, D.A.; Calfee, M.W.; Rainey, P.B.; Pesci, E.C. Autolysis and Autoaggregation in Pseudomonas aeruginosa Colony Morphology Mutants. J. Bacteriol. 2002, 184, 6481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, E.E.; Buckley, D.G.; Wu, Z.; Saenphimmachak, C.; Hoffman, L.R.; D’Argenio, D.A.; Miller, S.I.; Ramsey, B.W.; Speert, D.P.; Moskowitz, S.M.; et al. Genetic adaptation by Pseudomonas aeruginosa to the airways of cystic fibrosis patients. Proc. Natl. Acad. Sci. USA 2006, 103, 8487–8492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subedi, D.; Vijay, A.K.; Kohli, G.S.; Rice, S.A.; Willcox, M. Comparative genomics of clinical strains of Pseudomonas aeruginosa strains isolated from different geographic sites. Sci. Rep. 2018, 8, 15668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiser, R.; Green, A.E.; Bull, M.J.; Cunningham-Oakes, E.; Jolley, K.A.; Maiden, M.C.J.; Hall, A.J.; Winstanley, C.; Weightman, A.J.; Donoghue, D.; et al. Not all Pseudomonas aeruginosa are equal: Strains from industrial sources possess uniquely large multireplicon genomes. Microb. Genom. 2019, 5, e000276. [Google Scholar] [CrossRef]
- Boucher, H.W.; Talbot, G.H.; Bradley, J.S.; Edwards, J.E.; Gilbert, D.; Rice, L.B.; Scheld, M.; Spellberg, B.; Bartlett, J. Bad bugs, no drugs: No ESKAPE! An update from the Infectious Diseases Society of America. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 2009, 48, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Bassetti, M.; Peghin, M.; Vena, A.; Giacobbe, D.R. Treatment of Infections Due to MDR Gram-Negative Bacteria. Front. Med. 2019, 6, 74. [Google Scholar] [CrossRef]
- Henrichfreise, B.; Wiegand, I.; Pfister, W.; Wiedemann, B. Resistance Mechanisms of Multiresistant Pseudomonas aeruginosa Strains from Germany and Correlation with Hypermutation. Antimicrob. Agents Chemother. 2007, 51, 4062–4070. [Google Scholar] [CrossRef] [Green Version]
- Song, Z.; Wu, H.; Ciofu, O.; Kong, K.-F.; Høiby, N.; Rygaard, J.; Kharazmi, A.; Mathee, K. Pseudomonas aeruginosa alginate is refractory to Th1 immune response and impedes host immune clearance in a mouse model of acute lung infection. J. Med. Microbiol. 2003, 52, 731–740. [Google Scholar] [CrossRef] [Green Version]
- Worlitzsch, D.; Tarran, R.; Ulrich, M.; Schwab, U.; Cekici, A.; Meyer, K.C.; Birrer, P.; Bellon, G.; Berger, J.; Weiss, T.; et al. Effects of reduced mucus oxygen concentration in airway Pseudomonas infections of cystic fibrosis patients. J. Clin. Investig. 2002, 109, 317–325. [Google Scholar] [CrossRef]
- Mauch, R.M.; Jensen, P.Ø.; Moser, C.; Levy, C.E.; Høiby, N. Mechanisms of humoral immune response against Pseudomonas aeruginosa biofilm infection in cystic fibrosis. J. Cyst. Fibros. 2018, 17, 143–152. [Google Scholar] [CrossRef] [Green Version]
- Fauvart, M.; De Groote, V.N.; Michiels, J. Role of persister cells in chronic infections: Clinical relevance and perspectives on anti-persister therapies. J. Med. Microbiol. 2011, 60, 699–709. [Google Scholar] [CrossRef]
- Moyed, H.S.; Bertrand, K.P. hipA, a newly recognized gene of Escherichia coli K-12 that affects frequency of persistence after inhibition of murein synthesis. J. Bacteriol. 1983, 155, 768–775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spoering, A.L.; Vulic, M.; Lewis, K. GlpD and PlsB participate in persister cell formation in Escherichia coli. J. Bacteriol. 2006, 188, 5136–5144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balaban, N.Q.; Merrin, J.; Chait, R.; Kowalik, L.; Leibler, S. Bacterial persistence as a phenotypic switch. Science 2004, 305, 1622–1625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wainwright, J.; Hobbs, G.; Nakouti, I. Persister cells: Formation, resuscitation and combative therapies. Arch Microbiol. 2021, 203, 5899–5906. [Google Scholar] [CrossRef] [PubMed]
- Louwagie, E.; Verstraete, L.; Michiels, J.; Verstraeten, N. Studying Bacterial Persistence: Established Methods and Current Advances. Methods Mol. Biol. 2021, 2357, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Kaldalu, N.; Hauryliuk, V.; Turnbull, K.J.; La Mensa, A.; Putrinš, M.; Tenson, T. In Vitro Studies of Persister Cells. Microbiol. Mol. Biol. Rev. 2020, 84, e00070-20. [Google Scholar] [CrossRef]
- Mulcahy, L.R.; Burns, J.L.; Lory, S.; Lewis, K. Emergence of Pseudomonas aeruginosa strains producing high levels of persister cells in patients with cystic fibrosis. J. Bacteriol. 2010, 192, 6191–6199. [Google Scholar] [CrossRef] [Green Version]
- Lewis, K. Multidrug tolerance of biofilms and persister cells. Curr. Top Microbiol. Immunol. 2008, 322, 107–131. [Google Scholar] [CrossRef] [PubMed]
- Möker, N.; Dean, C.R.; Tao, J. Pseudomonas aeruginosa increases formation of multidrug-tolerant persister cells in response to quorum-sensing signaling molecules. J. Bacteriol. 2010, 192, 1946–1955. [Google Scholar] [CrossRef] [Green Version]
- Robak, O.H.; Heimesaat, M.M.; Kruglov, A.A.; Prepens, S.; Ninnemann, J.; Gutbier, B.; Reppe, K.; Hochrein, H.; Suter, M.; Kirschning, C.J.; et al. Antibiotic treatment-induced secondary IgA deficiency enhances susceptibility to Pseudomonas aeruginosa pneumonia. J. Clin. Investig. 2018, 128, 3535–3545. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, M.; Brooks, B.D.; Brooks, A.E. The Complex Relationship between Virulence and Antibiotic Resistance. Genes 2017, 8, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meluleni, G.J.; Grout, M.; Evans, D.J.; Pier, G.B. Mucoid Pseudomonas aeruginosa growing in a biofilm in vitro are killed by opsonic antibodies to the mucoid exopolysaccharide capsule but not by antibodies produced during chronic lung infection in cystic fibrosis patients. J. Immunol. 1995, 155, 2029. [Google Scholar] [PubMed]
- Rello, J.; Krenn, C.-G.; Locker, G.; Pilger, E.; Madl, C.; Balica, L.; Dugernier, T.; Laterre, P.-F.; Spapen, H.; Depuydt, P.; et al. A randomized placebo-controlled phase II study of a Pseudomonas vaccine in ventilated ICU patients. Crit. Care Lond. Engl. 2017, 21, 22. [Google Scholar] [CrossRef] [Green Version]
- Adlbrecht, C.; Wurm, R.; Depuydt, P.; Spapen, H.; Lorente, J.A.; Staudinger, T.; Creteur, J.; Zauner, C.; Meier-Hellmann, A.; Eller, P.; et al. Efficacy, immunogenicity, and safety of IC43 recombinant Pseudomonas aeruginosa vaccine in mechanically ventilated intensive care patients—A randomized clinical trial. Crit. Care 2020, 24, 74. [Google Scholar] [CrossRef] [Green Version]
- Georgescu, M.; Gheorghe, I.; Curutiu, C.; Lazar, V.; Bleotu, C.; Chifiriuc, M.-C. Virulence and resistance features of Pseudomonas aeruginosa strains isolated from chronic leg ulcers. BMC Infect. Dis. 2016, 16, 3–9. [Google Scholar] [CrossRef] [Green Version]
- Michalska, M.; Wolf, P. Pseudomonas Exotoxin A: Optimized by evolution for effective killing. Front. Microbiol. 2015, 6, 963. [Google Scholar] [CrossRef] [Green Version]
- Morlon-Guyot, J.; Méré, J.; Bonhoure, A.; Beaumelle, B. Processing of Pseudomonas aeruginosa Exotoxin A Is Dispensable for Cell Intoxication. Infect. Immun. 2009, 77, 3090–3099. [Google Scholar] [CrossRef] [Green Version]
- Hertle, R.; Mrsny, R.; Fitzgerald, D.J. Dual-Function Vaccine for Pseudomonas aeruginosa: Characterization of Chimeric Exotoxin A-Pilin Protein. Infect. Immun. 2001, 69, 6962–6969. [Google Scholar] [CrossRef] [Green Version]
- Saint-Criq, V.; Villeret, B.; Bastaert, F.; Kheir, S.; Hatton, A.; Cazes, A.; Xing, Z.; Sermet-Gaudelus, I.; Garcia-Verdugo, I.; Edelman, A.; et al. Pseudomonas aeruginosa LasB protease impairs innate immunity in mice and humans by targeting a lung epithelial cystic fibrosis transmembrane regulator-IL-6-antimicrobial-repair pathway. Thorax 2018, 73, 49–61. [Google Scholar] [CrossRef] [Green Version]
- Engel, L.S.; Hill, J.M.; Caballero, A.R.; Green, L.C.; O’Callaghan, R.J. Protease IV, a Unique Extracellular Protease and Virulence Factor from Pseudomonas aeruginosa. J. Biol. Chem. 1998, 273, 16792–16797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, J.; Li, X.-H.; Kim, S.-K.; Lee, J.-H. Post-secretional activation of Protease IV by quorum sensing in Pseudomonas aeruginosa. Sci. Rep. 2017, 7, 4416. [Google Scholar] [CrossRef] [Green Version]
- Sharma, A.; Krause, A.; Worgall, S. Recent developments for Pseudomonas vaccines. Hum. Vaccin. 2011, 7, 999–1011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thibodeaux, B.A.; Caballero, A.R.; Dajcs, J.J.; Marquart, M.E.; Engel, L.S.; O’Callaghan, R.J. Pseudomonas aeruginosa Protease IV: A Corneal Virulence Factor of Low Immunogenicity. Ocul. Immunol. Inflamm. 2005, 13, 169–182. [Google Scholar] [CrossRef] [PubMed]
- Tielen, P.; Kuhn, H.; Rosenau, F.; Jaeger, K.-E.; Flemming, H.-C.; Wingender, J. Interaction between extracellular lipase LipA and the polysaccharide alginate of Pseudomonas aeruginosa. BMC Microbiol. 2013, 13, 159. [Google Scholar] [CrossRef] [Green Version]
- Funken, H.; Knapp, A.; Vasil, M.L.; Wilhelm, S.; Jaeger, K.-E.; Rosenau, F. The lipase LipA (PA2862) but not LipC (PA4813) from Pseudomonas aeruginosa influences regulation of pyoverdine production and expression of the sigma factor PvdS. J. Bacteriol. 2011, 193, 5858–5860. [Google Scholar] [CrossRef] [Green Version]
- Jaeger, K.-E.; Kharazmi, A.; Høiby, N. Extracellular lipase of Pseudomonas aeruginosa: Biochemical characterization and effect on human neutrophil and monocyte function in vitro. Microb. Pathog. 1991, 10, 173–182. [Google Scholar] [CrossRef]
- Terada, L.S.; Johansen, K.A.; Nowbar, S.; Vasil, A.I.; Vasil, M.L. Pseudomonas aeruginosa Hemolytic Phospholipase C Suppresses Neutrophil Respiratory Burst Activity. Orndorff, P.E., Ed. Infect. Immun. 1999, 67, 2371–2376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Z.; Kang, Y.; Norris, M.; Troyer, R.M.; Son, M.S.; Schweizer, H.P.; Dow, S.W.; Hoang, T.T. Blocking phosphatidylcholine utilization in Pseudomonas aeruginosa, via mutagenesis of fatty acid, glycerol and choline degradation pathways, confirms the importance of this nutrient source in vivo. PLoS ONE 2014, 9, e103778. [Google Scholar] [CrossRef] [Green Version]
- Maroui, I.; Aboulkacem, A.; Mohammed, T.; Belhaj, A. Virulence profiles of clinical and environmental Pseudomonas aeruginosa isolates from Central Morocco. Afr. J. Microbiol. Res. 2016, 10, 473–480. [Google Scholar] [CrossRef] [Green Version]
- Granström, M.; Ericsson, A.; Strandvik, B.; Wretlind, B.; Pavlovskis, O.R.; Berka, R.; Vasil, M.L. Relation between Antibody Response to Pseudomonas aeruginosa Exoproteins and Colonization/Infection in Patients with Cystic Fibrosis. Acta. Paediatr. 1984, 73, 772–777. [Google Scholar] [CrossRef]
- Yu, Y.-J.; Wang, X.-H.; Fan, G.-C. Versatile effects of bacterium-released membrane vesicles on mammalian cells and infectious/inflammatory diseases. Acta Pharmacol. Sin. 2018, 39, 514–533. [Google Scholar] [CrossRef] [PubMed]
- Döring, G.; Dalhoff, A.; Vogel, O.; Brunner, H.; Dröge, U.; Botzenhart, K. In Vivo Activity of Proteases of Pseudomonas aeruginosa in a Rat Model. J. Infect. Dis. 1984, 149, 532–537. [Google Scholar] [CrossRef] [PubMed]
- Fick, R.B., Jr.; Baltimore, R.S.; Squier, S.U.; Reynolds, H.Y. IgG Proteolytic Activity of Pseudomonas aeruginosa in Cystic Fibrosis. J. Infect. Dis. 1985, 151, 589–598. [Google Scholar] [CrossRef] [PubMed]
- Parmely, M.; Gale, A.; Clabaugh, M.; Horvat, R.; Zhou, W.W. Proteolytic inactivation of cytokines by Pseudomonas aeruginosa. Infect. Immun. 1990, 58, 3009. [Google Scholar] [CrossRef] [Green Version]
- Horvat, R.T.; Parmely, M.J. Pseudomonas aeruginosa alkaline protease degrades human gamma interferon and inhibits its bioactivity. Infect. Immun. 1988, 56, 2925. [Google Scholar] [CrossRef] [Green Version]
- Kharazmi, A.; Eriksen, H.O.; DÖRing, G.; Goldstein, W.; HØIby, N. Effect of pseudomonas aeruginosa proteases on human leukocyte phagocytosis and bactericidal activity. Acta. Pathol. Microbiol. Scand. Ser. C Immunol. 1986, 94, 175–179. [Google Scholar] [CrossRef]
- Theander, T.G.; Kharazmi, A.; Pedersen, B.K.; Christensen, L.D.; Tvede, N.; Poulsen, L.K.; Odum, N.; Svenson, M.; Bendtzen, K. Inhibition of human lymphocyte proliferation and cleavage of interleukin-2 by Pseudomonas aeruginosa proteases. Infect. Immun. 1988, 56, 1673. [Google Scholar] [CrossRef] [Green Version]
- Pedersen, B.K.; Kharazmi, A. Inhibition of human natural killer cell activity by Pseudomonas aeruginosa alkaline protease and elastase. Infect. Immun. 1987, 55, 986. [Google Scholar] [CrossRef] [Green Version]
- Nomura, K.; Obata, K.; Keira, T.; Miyata, R.; Hirakawa, S.; Takano, K.-I.; Kohno, T.; Sawada, N.; Himi, T.; Kojima, T. Pseudomonas aeruginosa elastase causes transient disruption of tight junctions and downregulation of PAR-2 in human nasal epithelial cells. Respir. Res. 2014, 15, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kessler, E.; Safrin, M.; Gustin, J.K.; Ohman, D.E. Elastase and the LasA Protease of Pseudomonas aeruginosa Are Secreted with Their Propeptides. J. Biol. Chem. 1998, 273, 30225–30231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Homma, J.Y.; Abe, C.; Tanamoto, K.; Hirao, Y.; Morihara, K.; Tsuzuki, H.; Yanagawa, R.; Honda, E.; Aoi, Y.; Fujimoto, Y.; et al. Effectiveness of immunization with single and multi-component vaccines prepared from a common antigen (OEP), protease and elastase toxoids of Pseudomonas aeruginosa on protection against hemorrhagic pneumonia in mink due to P. aeruginosa. Jpn. J. Exp. Med. 1978, 48, 111–133. [Google Scholar] [PubMed]
- Hirao, Y.; Homma, J. Therapeutic effect of immunization with OEP, protease toxoid and elastase toxoid on corneal ulcers in mice due to Pseudomonas aeruginosa infection. Jpn. J. Exp. Med. 1978, 48, 41–51. [Google Scholar]
- Sokol, P.A.; Kooi, C.; Hodges, R.S.; Cachia, P.; Woods, D.E. Immunization with a Pseudomonas aeruginosa Elastase Peptide Reduces Severity of Experimental Lung Infections Due to P. aeruginosa or Burkholderia cepacia. J. Infect. Dis. 2000, 181, 1682–1692. [Google Scholar] [CrossRef] [Green Version]
- Kawaharajo, K.; Homma, J. Effects of elastase, protease and common antigen (OEP) from Pseudomonas aeruginosa on protection against burns in mice. Jpn. J. Exp. Med. 1977, 47, 495–500. [Google Scholar] [PubMed]
- Casilag, F.; Lorenz, A.; Krueger, J.; Klawonn, F.; Weiss, S.; Häussler, S. The LasB Elastase of Pseudomonas aeruginosa Acts in Concert with Alkaline Protease AprA To Prevent Flagellin-Mediated Immune Recognition. Infect. Immun. 2016, 84, 162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poole, K.; Hancock, R.E.W. Secretion of alkaline phosphatase and phsopholipase C in Pseudomonas aeruginosa is specific and does not involve an increase in outer membrane permeability. FEMS Microbiol. Lett. 1983, 16, 25–29. [Google Scholar] [CrossRef]
- Liu, X.; Long, D.; You, H.; Yang, D.; Zhou, S.; Zhang, S.; Li, M.; He, M.; Xiong, M.; Wang, X. Phosphatidylcholine affects the secretion of the alkaline phosphatase PhoA in Pseudomonas strains. Microbiol. Res. 2016, 192, 21–29. [Google Scholar] [CrossRef]
- Miyoshi, S.-I.; Shinoda, S. Bacterial Metalloprotease as the Toxic Factor in Infection. J. Toxicol. Toxin. Rev. 1997, 16, 177–194. [Google Scholar] [CrossRef]
- Zhang, C.; Bijl, E.; Svensson, B.; Hettinga, K. The Extracellular Protease AprX from Pseudomonas and its Spoilage Potential for UHT Milk: A Review. Compr. Rev. Food Sci. Food Saf. 2019, 18, 834–852. [Google Scholar] [CrossRef] [Green Version]
- Filloux, A. Protein Secretion Systems in Pseudomonas aeruginosa: An Essay on Diversity, Evolution, and Function. Front. Microbiol. 2011, 2, 155. [Google Scholar] [CrossRef] [Green Version]
- Higgins, C.F. ABC transporters: Physiology, structure and mechanism—An overview. Res. Microbiol. 2001, 152, 205–210. [Google Scholar] [CrossRef]
- Duong, F.; Lazdunski, A.; Carni, B.; Murgier, M. Sequence of a cluster of genes controlling synthesis and secretion of alkaline protease in Pseudomonas aeruginosa: Relationships to other secretory pathways. Gene 1992, 121, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Caza, M.; Kronstad, J.W. Shared and distinct mechanisms of iron acquisition by bacterial and fungal pathogens of humans. Front. Cell Infect. Microbiol. 2013, 3, 80. [Google Scholar] [CrossRef] [PubMed]
- Ball, G.; Durand, É.; Lazdunski, A.; Filloux, A. A novel type II secretion system in Pseudomonas aeruginosa. Mol. Microbiol. 2002, 43, 475–485. [Google Scholar] [CrossRef]
- Cornelis, G.R. The type III secretion injectisome, a complex nanomachine for intracellular ‘toxin’ delivery. Biol. Chem. 2010, 391, 745–751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fleiszig, S.M.; Zaidi, T.S.; Preston, M.J.; Grout, M.; Evans, D.J.; Pier, G.B. Relationship between cytotoxicity and corneal epithelial cell invasion by clinical isolates of Pseudomonas aeruginosa. Infect. Immun. 1996, 64, 2288–2294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finck-Barbançon, V.; Goranson, J.; Zhu, L.; Sawa, T.; Wiener-Kronish, J.P.; Fleiszig, S.M.J.; Wu, C.; Mende-Mueller, L.; Frank, D.W. ExoU expression by Pseudomonas aeruginosa correlates with acute cytotoxicity and epithelial injury. Mol. Microbiol. 1997, 25, 547–557. [Google Scholar] [CrossRef]
- Roy-Burman, A.; Savel, R.H.; Racine, S.; Swanson, B.L.; Revadigar, N.S.; Fujimoto, J.; Sawa, T.; Frank, D.W.; Wiener-Kronish, J.P. Type III Protein Secretion Is Associated with Death in Lower Respiratory and Systemic Pseudomonas aeruginosa Infections. J. Infect. Dis. 2001, 183, 1767–1774. [Google Scholar] [CrossRef] [Green Version]
- Hauser, A.R. The type III secretion system of Pseudomonas aeruginosa: Infection by injection. Nat. Rev. Microbiol. 2009, 7, 654–665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anantharajah, A.; Mingeot-Leclercq, M.-P.; Van Bambeke, F. Targeting the Type Three Secretion System in Pseudomonas aeruginosa. Trends Pharmacol. Sci. 2016, 37, 734–749. [Google Scholar] [CrossRef]
- Sato, H.; Frank, D.W. Multi-Functional Characteristics of the Pseudomonas aeruginosa Type III Needle-Tip Protein, PcrV Comparison to Orthologs in other Gram-negative Bacteria. Front. Microbiol. 2011, 2, 142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sawa, T.; Ito, E.; Nguyen, V.H.; Haight, M. Anti-PcrV antibody strategies against virulent Pseudomonas aeruginosa. Hum. Vaccines Immunother. 2014, 10, 2843–2852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lynch, S.V.; Flanagan, J.; Sawa, T.; Fang, A.; Baek, M.S.; Rubio-Mills, A.; Ajayi, T.; Yanagihara, K.; Hirakata, Y.; Kohno, S.; et al. Polymorphisms in the Pseudomonas aeruginosa type III secretion protein, PcrV-implications for anti-PcrV immunotherapy. Microb. Pathog. 2010, 48, 197–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moriyama, K.; Wiener-Kronish, J.P.; Sawa, T. Protective effects of affinity-purified antibody and truncated vaccines against Pseudomonas aeruginosa V-antigen in neutropenic mice. Microbiol. Immunol. 2009, 53, 587–594. [Google Scholar] [CrossRef] [PubMed]
- Sawa, T.; Yahr, T.; Ohara, M.; Kurahashi, K.; Gropper, M.A.; Wiener-Kronish, J.P.; Frank, D.W. Active and passive immunization with the Pseudomonas V antigen protects against type III intoxication and lung injury. Nat. Med. 1999, 5, 392–398. [Google Scholar] [CrossRef]
- Baer, M.; Sawa, T.; Flynn, P.; Luehrsen, K.; Martinez, D.; Wiener-Kronish, J.P.; Yarranton, G.; Bebbington, C. An engineered human antibody fab fragment specific for Pseudomonas aeruginosa PcrV antigen has potent antibacterial activity. Infect. Immun. 2009, 77, 1083–1090. [Google Scholar] [CrossRef] [Green Version]
- Faure, K.; Fujimoto, J.; Shimabukuro, D.W.; Ajayi, T.; Shime, N.; Moriyama, K.; Spack, E.G.; Wiener-Kronish, J.P.; Sawa, T. Effects of monoclonal anti-PcrV antibody on Pseudomonas aeruginosa-induced acute lung injury in a rat model. J. Immun. Based Ther. Vaccines. 2003, 1, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frank, D.W.; Vallis, A.; Wiener-Kronish, J.P.; Roy-Burman, A.; Spack, E.G.; Mullaney, B.P.; Megdoud, M.; Marks, J.D.; Fritz, R.; Sawa, T. Generation and Characterization of a Protective Monoclonal Antibody to Pseudomonas aeruginosa PcrV. J. Infect. Dis. 2002, 186, 64–73. [Google Scholar] [CrossRef] [Green Version]
- Imamura, Y.; Yanagihara, K.; Fukuda, Y.; Kaneko, Y.; Seki, M.; Izumikawa, K.; Miyazaki, Y.; Hirakata, Y.; Sawa, T.; Wiener-Kronish, J.P.; et al. Effect of anti-PcrV antibody in a murine chronic airway Pseudomonas aeruginosa infection model. Eur. Respir. J. 2007, 29, 965. [Google Scholar] [CrossRef] [Green Version]
- Shime, N.; Sawa, T.; Fujimoto, J.; Faure, K.; Allmond, L.R.; Karaca, T.; Swanson, B.L.; Spack, E.G.; Wiener-Kronish, J.P. Therapeutic Administration of Anti-PcrV F(ab’)2 in Sepsis Associated with Pseudomonas aeruginosa. J. Immunol. 2001, 167, 5880. [Google Scholar] [CrossRef] [Green Version]
- Song, Y.; Baer, M.; Srinivasan, R.; Lima, J.; Yarranton, G.; Bebbington, C.; Lynch, S.V. PcrV antibody–antibiotic combination improves survival in Pseudomonas aeruginosa-infected mice. Eur. J. Clin. Microbiol. Infect. Dis. 2012, 31, 1837–1845. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Li, H.; Zhou, J.; Zhong, M.; Zhu, D.; Feng, N.; Liu, F.; Bai, C.; Song, Y. PcrV antibody protects multi-drug resistant Pseudomonas aeruginosa induced acute lung injury. Respir. Physiol. Neurobiol. 2014, 193, 21–28. [Google Scholar] [CrossRef]
- Hosseinidoust, Z.; van de Ven, T.G.M.; Tufenkji, N. Evolution of Pseudomonas aeruginosa virulence as a result of phage predation. Appl. Environ. Microbiol. 2013, 79, 6110–6116. [Google Scholar] [CrossRef] [Green Version]
- Vance, R.E.; Rietsch, A.; Mekalanos, J.J. Role of the Type III Secreted Exoenzymes S, T, and Y in Systemic Spread of Pseudomonas aeruginosa PAO1 In Vivo. Infect. Immun. 2005, 73, 1706–1713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hauser, A.R.; Cobb, E.; Bodí, M.; Mariscal, D.; Vallés, J.; Engel, J.N.; Rello, J. Type III protein secretion is associated with poor clinical outcomes in patients with ventilator-associated pneumonia caused by Pseudomonas aeruginosa. Crit. Care Med. 2002, 30, 521–528. [Google Scholar] [CrossRef]
- Leo, J.C.; Grin, I.; Linke, D. Type V secretion: Mechanism(s) of autotransport through the bacterial outer membrane. Philos. Trans. R. Soc. B Biol. Sci. 2012, 367, 1088–1101. [Google Scholar] [CrossRef] [Green Version]
- Salacha, R.; Kovacic, F.; Brochier-Armanet, C.; Wilhelm, S.; Tommassen, J.; Filloux, A.; Voulhoux, R.; Bleves, S. The Pseudomonas aeruginosa patatin-like protein PlpD is the archetype of a novel Type V secretion system. Environ. Microbiol. 2010, 12, 1498–1512. [Google Scholar] [CrossRef]
- Chen, L.; Zou, Y.; She, P.; Wu, Y. Composition, function, and regulation of T6SS in Pseudomonas aeruginosa. Microbiol. Res. 2015, 172, 19–25. [Google Scholar] [CrossRef] [PubMed]
- The Type VI Secretion System: A Dynamic System for Bacterial Communication? Available online: https://www-ncbi-nlm-nih-gov.proxy.rvu.edu/pmc/articles/PMC5532429/ (accessed on 20 August 2021).
- Wang, M.; Schaefer, A.L.; Dandekar, A.A.; Greenberg, E.P. Quorum sensing and policing of Pseudomonas aeruginosa social cheaters. Proc. Natl. Acad. Sci. USA 2015, 112, 2187. [Google Scholar] [CrossRef] [Green Version]
- Häussler, S.; Becker, T. The Pseudomonas Quinolone Signal (PQS) Balances Life and Death in Pseudomonas aeruginosa Populations. Ausubel, F.M., Ed. PLoS Pathog. 2008, 4, e1000166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, J.; Cheng, J.; Wang, Y.; Shen, X. The Pseudomonas Quinolone Signal (PQS): Not Just for Quorum Sensing Anymore. Front. Cell Infect. Microbiol. 2018, 8, 230. [Google Scholar] [CrossRef] [PubMed]
- Castañeda-Tamez, P.; Ramírez-Peris, J.; Pérez-Velázquez, J.; Kuttler, C.; Jalalimanesh, A.; Saucedo-Mora, M.Á.; Jiménez-Cortés, J.G.; Maeda, T.; González, Y.; Tomás, M.; et al. Pyocyanin Restricts Social Cheating in Pseudomonas aeruginosa. Front. Microbiol. 2018, 9, 1348. [Google Scholar] [CrossRef] [PubMed]
- Hall, S.; McDermott, C.; Anoopkumar-Dukie, S.; McFarland, A.J.; Forbes, A.; Perkins, A.V.; Davey, A.K.; Chess-Williams, R.; Kiefel, M.J.; Arora, D.; et al. Cellular Effects of Pyocyanin, a Secreted Virulence Factor of Pseudomonas aeruginosa. Toxins 2016, 8, 236. [Google Scholar] [CrossRef]
- Branzk, N.; Lubojemska, A.; Hardison, S.E.; Wang, Q.; Gutierrez, M.G.; Brown, G.D.; Papayannopoulos, V. Neutrophils sense microbe size and selectively release neutrophil extracellular traps in response to large pathogens. Nat. Immunol. 2014, 15, 1017–1025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Susilowati, H.; Artanto, S.; Yulianto, H.; Sosroseno, W.; Hutomo, S. The protective effects of antigen-specific IgY on pyocyanin-treated human lymphoma Raji cells [version 2; peer review: 2 approved]. F1000Research 2019, 8, 1008. [Google Scholar] [CrossRef]
- Telford, G.; Wheeler, D.; Williams, P.; Tomkins, P.T.; Appleby, P.; Sewell, H.; Stewart, G.S.A.B.; Bycroft, B.W.; Pritchard, D.I. The Pseudomonas aeruginosa Quorum-Sensing Signal MoleculeN-(3-Oxododecanoyl)-l-Homoserine Lactone Has Immunomodulatory Activity. Infect. Immun. 1998, 66, 36–42. [Google Scholar] [CrossRef] [Green Version]
- Zhu, H.; Conibear, T.C.R.; Thuruthyil, S.J.; Willcox, M.D.P. Pseudomonas aeruginosa Quorum-Sensing Signal Molecules Induce IL-8 Production by Human Corneal Epithelial Cells. Eye Contact Lens Sci. Clin. Pract. 2008, 34, 179–181. [Google Scholar] [CrossRef] [PubMed]
- Gaisford, W.; Pritchard, D.I.; Cooke, A. OdDHL Inhibits T Cell Subset Differentiation and Delays Diabetes Onset in NOD Mice. Clin. Vaccine Immunol. 2011, 18, 1213–1220. [Google Scholar] [CrossRef]
- Borges, A.; Sousa, P.; Gaspar, A.; Vilar, S.; Borges, F.; Simões, M. Furvina inhibits the 3-oxo-C12-HSL-based quorum sensing system of Pseudomonas aeruginosa and QS-dependent phenotypes. Biofouling 2017, 33, 156–168. [Google Scholar] [CrossRef]
- Hooi, D.S.W.; Bycroft, B.W.; Chhabra, S.R.; Williams, P.; Pritchard, D.I. Differential Immune Modulatory Activity of Pseudomonas aeruginosa Quorum-Sensing Signal Molecules. Infect. Immun. 2004, 72, 6463–6470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooley, M.A.; Whittall, C.; Rolph, M.S. Pseudomonas signal molecule 3-oxo-C12-homoserine lactone interferes with binding of rosiglitazone to human PPARγ. Microbes. Infect. 2010, 12, 231–237. [Google Scholar] [CrossRef] [PubMed]
- Jarosz, L.M.; Ovchinnikova, E.S.; Meijler, M.M.; Krom, B.P. Microbial Spy Games and Host Response: Roles of a Pseudomonas aeruginosa Small Molecule in Communication with Other Species. PLoS Pathog. 2011, 7, e1002312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyairi, S.; Tateda, K.; Fuse, E.T.; Ueda, C.; Saito, H.; Takabatake, T.; Ishii, Y.; Horikawa, M.; Ishiguro, M.; Standiford, T.J.; et al. Immunization with 3-oxododecanoyl-l-homoserine lactone–protein conjugate protects mice from lethal Pseudomonas aeruginosa lung infection. J. Med. Microbiol. 2006, 55, 1381–1387. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Asfahl, K.L.; Li, N.; Sun, F.; Xiao, J.; Shen, D.; Dandekar, A.A.; Wang, M. Conditional quorum-sensing induction of a cyanide-insensitive terminal oxidase stabilizes cooperating populations of Pseudomonas aeruginosa. Nat. Commun. 2019, 10, 4999. [Google Scholar] [CrossRef] [PubMed]
- Ryall, B.; Davies, J.C.; Wilson, R.; Shoemark, A.; Williams, H.D. Pseudomonas aeruginosa, cyanide accumulation and lung function in CF and non-CF bronchiectasis patients. Eur. Respir. J. 2008, 32, 740. [Google Scholar] [CrossRef] [Green Version]
- Pessi, G.; Haas, D. Transcriptional control of the hydrogen cyanide biosynthetic genes hcnABC by the anaerobic regulator ANR and the quorum-sensing regulators LasR and RhlR in Pseudomonas aeruginosa. J. Bacteriol. 2000, 182, 6940–6949. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez-Rojas, A.; Mena, A.; Martín, S.; Borrell, N.; Oliver, A.; Blázquez, J. Inactivation of the hmgA gene of Pseudomonas aeruginosa leads to pyomelanin hyperproduction, stress resistance and increased persistence in chronic lung infection. Microbiology 2009, 155, 1050–1057. [Google Scholar] [CrossRef] [Green Version]
- Hunter, R.C.; Newman, D.K. A Putative ABC Transporter, HatABCDE, Is among Molecular Determinants of Pyomelanin Production in Pseudomonas aeruginosa. J. Bacteriol. 2010, 192, 5962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Defoirdt, T. Quorum-Sensing Systems as Targets for Antivirulence Therapy. Trends. Microbiol. 2018, 26, 313–328. [Google Scholar] [CrossRef]
- Gupta, R.K.; Chhibber, S.; Harjai, K. Acyl Homoserine Lactones from Culture Supernatants of Pseudomonas aeruginosa Accelerate Host Immunomodulation. PLoS ONE 2011, 6, e20860. [Google Scholar] [CrossRef]
- Morita, Y.; Tomida, J.; Kawamura, Y. MexXY multidrug efflux system of Pseudomonas aeruginosa. Front. Microbiol. 2012, 3, 408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.-Z.; Plésiat, P.; Nikaido, H. The Challenge of Efflux-Mediated Antibiotic Resistance in Gram-Negative Bacteria. Clin. Microbiol. Rev. 2015, 28, 337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brooks, B.D.; Brooks, A.E. Therapeutic strategies to combat antibiotic resistance. Adv. Drug. Deliv. Rev. 2014, 78, 14–27. [Google Scholar] [CrossRef]
- Yang, L.; Chen, L.; Shen, L.; Surette, M.; Duan, K. Inactivation of MuxABC-OpmB transporter system in Pseudomonas aeruginosa leads to increased ampicillin and carbenicillin resistance and decreased virulence. J. Microbiol. 2011, 49, 107–114. [Google Scholar] [CrossRef]
- Ma, D.; Cook, D.N.; Hearst, J.E.; Nikaido, H. Efflux pumps and drug resistance in Gram-negative bacteria. Trends. Microbiol. 1994, 2, 489–493. [Google Scholar] [CrossRef]
- Bianco, N.; Neshat, S.; Poole, K. Conservation of the multidrug resistance efflux gene oprM in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 1997, 41, 853–856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chemani, C.; Imberty, A.; de Bentzmann, S.; Pierre, M.; Wimmerová, M.; Guery, B.P.; Faure, K. Role of LecA and LecB Lectins in Pseudomonas aeruginosa-Induced Lung Injury and Effect of Carbohydrate Ligands. Infect. Immun. 2009, 77, 2065–2075. [Google Scholar] [CrossRef] [Green Version]
- Son, M.S.; Matthews, W.J., Jr.; Kang, Y.; Nguyen, D.T.; Hoang, T.T. In vivo evidence of Pseudomonas aeruginosa nutrient acquisition and pathogenesis in the lungs of cystic fibrosis patients. Infect. Immun. 2007, 75, 5313–5324. [Google Scholar] [CrossRef] [Green Version]
- Malek, A.A.; Chen, C.; Wargo, M.J.; Beattie, G.A.; Hogan, D.A. Roles of three transporters, CbcXWV, BetT1, and BetT3, in Pseudomonas aeruginosa choline uptake for catabolism. J. Bacteriol. 2011, 193, 3033–3041. [Google Scholar] [CrossRef] [Green Version]
- Huebinger, R.M.; Stones, D.H.; Santos, M.D.S.; Carlson, D.L.; Song, J.; Vaz, D.P.; Keen, E.; Wolf, S.; Orth, K.; Krachler, A.M. Targeting bacterial adherence inhibits multidrug-resistant Pseudomonas aeruginosa infection following burn injury. Sci. Rep. 2016, 6, 39341. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Estrada, O.; Zaborina, O.; Bains, M.; Shen, L.; Kohler, J.E.; Patel, N.; Musch, M.W.; Chang, E.B.; Fu, Y.-X.; et al. Recognition of Host Immune Activation by Pseudomonas aeruginosa. Science 2005, 309, 774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Metruccio, M.M.E.; Evans, D.J.; Gabriel, M.M.; Kadurugamuwa, J.L.; Fleiszig, S.M.J. Pseudomonas aeruginosa Outer Membrane Vesicles Triggered by Human Mucosal Fluid and Lysozyme Can Prime Host Tissue Surfaces for Bacterial Adhesion. Front. Microbiol. 2016, 7, 871. [Google Scholar] [CrossRef] [Green Version]
- Moghaddam, E.K.; Owlia, P.; Jahangiri, A.; Rasooli, I.; Rahbar, M.R.; Aghajani, M. Conserved OprF as a Selective Immunogen against Pseudomonas aeruginosa. Iran J. Pathol. 2017, 12, 165–170. [Google Scholar] [CrossRef]
- Ezhang, W.; Esun, J.; Eding, W.; Lin, J.; Tian, R.; Elu, L.; Eliu, X.; Eshen, X.; Eqian, P.-Y. Extracellular matrix-associated proteins form an integral and dynamic system during Pseudomonas aeruginosa biofilm development. Front. Cell. Infect. Microbiol. 2015, 5, 40. [Google Scholar] [CrossRef] [Green Version]
- Pier, G. Pseudomonas aeruginosa lipopolysaccharide: A major virulence factor, initiator of inflammation and target for effective immunity. Int. J. Med. Microbiol. 2007, 297, 277–295. [Google Scholar] [CrossRef] [Green Version]
- Kronborg, G.; Fomsgaard, A.; Galanos, C.; Freudenberg, M.A.; Høiby, N. Antibody responses to lipid A, core, and O sugars of the Pseudomonas aeruginosa lipopolysaccharide in chronically infected cystic fibrosis patients. J. Clin. Microbiol. 1992, 30, 1848–1855. [Google Scholar] [CrossRef] [Green Version]
- Cryz, S.J.; Sadoff, J.C.; Fürer, E. Octavalent Pseudomonas aeruginosa O-polysaccharide-toxin A conjugate vaccine. Microb. Pathog. 1989, 6, 75–80. [Google Scholar] [CrossRef]
- Hastie, A.T.; Hingley, S.T.; Higgins, M.L.; Kueppers, F.; Shryock, T. Rhamnolipid from Pseudomonas aeruginosa inactivates mammalian tracheal ciliary axonemes. Cell. Motil. Cytoskeleton. 1986, 6, 502–509. [Google Scholar] [CrossRef]
- Caiazza, N.C.; Shanks, R.M.Q.; O’Toole, G.A. Rhamnolipids Modulate Swarming Motility Patterns of Pseudomonas aeruginosa. J. Bacteriol. 2005, 187, 7351–7361. [Google Scholar] [CrossRef] [Green Version]
- Zhu, H.; Bandara, R.; Conibear, T.C.R.; Thuruthyil, S.J.; Rice, S.A.; Kjelleberg, S.; Givskov, M.; Willcox, M.D.P. Pseudomonas aeruginosa with LasI Quorum-Sensing Deficiency during Corneal Infection. Investig. Ophthalmol. Vis. Sci. 2004, 45, 1897–1903. [Google Scholar] [CrossRef] [Green Version]
- Alarcon, I.; Kwan, L.; Yu, C.; Evans, D.J.; Fleiszig, S.M.J. Role of the corneal epithelial basement membrane in ocular defense against Pseudomonas aeruginosa. Infect. Immun. 2009, 77, 3264–3271. [Google Scholar] [CrossRef] [Green Version]
- Giagkas, D.C.; Choli-Papadopoulou, T.; Pantazaki, A.A. Development of an Antibody for Detection of Rhamnolipids Characterized as a Major Bacterial Virulence Factor. Antibodies 2013, 2, 501–516. [Google Scholar] [CrossRef] [Green Version]
- Ma, L.; Lu, H.; Sprinkle, A.; Parsek, M.R.; Wozniak, D.J. Pseudomonas aeruginosa Psl Is a Galactose- and Mannose-Rich Exopolysaccharide. J. Bacteriol. 2007, 189, 8353–8356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pier, G.B.; Matthews, W.J., Jr.; Eardley, D.D. Immunochemical Characterization of the Mucoid Exopolysaccharide of Pseudomonas aeruginosa. J. Infect. Dis. 1983, 147, 494–503. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Pop, L.M.; Vitetta, E.S. Engineering therapeutic monoclonal antibodies. Immunol. Rev. 2008, 222, 9–27. [Google Scholar] [CrossRef] [PubMed]
- May, T.B.; Shinabarger, D.; Maharaj, R.; Kato, J.; Chu, L.; DeVault, J.D.; Roychoudhury, S.; Zielinski, N.A.; Berry, A.; Rothmel, R.K. Alginate synthesis by Pseudomonas aeruginosa: A key pathogenic factor in chronic pulmonary infections of cystic fibrosis patients. Clin. Microbiol. Rev. 1991, 4, 191–206. [Google Scholar] [CrossRef]
- McCaslin, C.A.; Petrusca, D.N.; Poirier, C.; Serban, K.A.; Anderson, G.G.; Petrache, I. Impact of alginate-producing Pseudomonas aeruginosa on alveolar macrophage apoptotic cell clearance. J. Cyst. Fibros. Off. J. Eur. Cyst. Fibros. Soc. 2015, 14, 70–77. [Google Scholar] [CrossRef] [Green Version]
- Ellis, T.N.; Kuehn, M.J. Virulence and Immunomodulatory Roles of Bacterial Outer Membrane Vesicles. Microbiol. Mol. Biol. Rev. 2010, 74, 81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Augustin, D.K.; Heimer, S.R.; Tam, C.; Li, W.Y.; Le Due, J.M.; Evans, D.J.; Fleiszig, S.M.J. Role of defensins in corneal epithelial barrier function against Pseudomonas aeruginosa traversal. Infect. Immun. 2011, 79, 595–605. [Google Scholar] [CrossRef] [Green Version]
- Baumgarten, T.; Sperling, S.; Seifert, J.; Von Bergen, M.; Steiniger, F.; Wick, L.Y.; Heipieper, H.J. Membrane vesicle formation as a multiple-stress response mechanism enhances Pseudomonas putida DOT-T1E cell surface hydrophobicity and biofilm formation. Appl. Environ. Microbiol. 2012, 78, 6217–6224. [Google Scholar] [CrossRef] [Green Version]
- Bloemberg, G.V.; O’Toole, G.A.; Lugtenberg, B.J.; Kolter, R. Green fluorescent protein as a marker for Pseudomonas spp. Appl. Environ. Microbiol. 1997, 63, 4543–4551. [Google Scholar] [CrossRef] [Green Version]
- Bomberger, J.M.; Maceachran, D.P.; Coutermarsh, B.A.; Ye, S.; O’Toole, G.A.; Stanton, B.A. Long-distance delivery of bacterial virulence factors by Pseudomonas aeruginosa outer membrane vesicles. PLoS Pathog. 2009, 5, e1000382. [Google Scholar] [CrossRef] [Green Version]
- Koeppen, K.; Barnaby, R.; Jackson, A.A.; Gerber, S.A.; Hogan, D.A.; Stanton, B.A. Tobramycin reduces key virulence determinants in the proteome of Pseudomonas aeruginosa outer membrane vesicles. PLoS ONE 2019, 14, e0211290. [Google Scholar] [CrossRef] [Green Version]
- Potvin, E.; Lehoux, D.E.; Kukavica-Ibrulj, I.; Richard, K.L.; Lau, G.W.; Levesque, R.C. In vivo functional genomics of Pseudomonas aeruginosa for high-throughput screening of new virulence factors and antibacterial targets. Environ. Microbiol. 2003, 5, 1294–1308. [Google Scholar] [CrossRef]
- Comolli, J.C.; Hauser, A.R.; Waite, L.; Whitchurch, C.B.; Mattick, J.S.; Engel, J.N. Pseudomonas aeruginosa Gene Products PilT and PilU Are Required for Cytotoxicity In Vitro and Virulence in a Mouse Model of Acute Pneumonia. Infect. Immun. 1999, 67, 3625. [Google Scholar] [CrossRef] [Green Version]
- Burrows, L.L. Pseudomonas aeruginosa Twitching Motility: Type IV Pili in Action. Annu. Rev. Microbiol. 2012, 66, 493–520. [Google Scholar] [CrossRef] [Green Version]
- Leighton, T.L.; Mok, M.C.; Junop, M.S.; Howell, P.L.; Burrows, L.L. Conserved, unstructured regions in Pseudomonas aeruginosa PilO are important for type IVa pilus function. Sci. Rep. 2018, 8, 2600. [Google Scholar] [CrossRef] [Green Version]
- Curran, C.S.; Bolig, T.; Torabi-Parizi, P. Mechanisms and Targeted Therapies for Pseudomonas aeruginosa Lung Infection. Am. J. Respir. Crit. Care Med. 2018, 197, 708–727. [Google Scholar] [CrossRef]
- Campodónico, V.L.; Llosa, N.J.; Grout, M.; Döring, G.; Maira-Litrán, T.; Pier, G.B. Evaluation of Flagella and Flagellin of Pseudomonas aeruginosa as Vaccines. Infect. Immun. 2010, 78, 746–755. [Google Scholar] [CrossRef] [Green Version]
- Yeung, Y.A.; Foletti, D.; Deng, X.; Abdiche, Y.; Strop, P.; Glanville, J.; Pitts, S.; Lindquist, K.; Sundar, P.D.; Sirota, M.; et al. Germline-encoded neutralization of a Staphylococcus aureus virulence factor by the human antibody repertoire. Nat. Commun. 2016, 7, 13376. [Google Scholar] [CrossRef] [PubMed]
- Peek, M.E.; Bhatnagar, A.; McCarty, N.A.; Zughaier, S.M. Pyoverdine, the Major Siderophore in Pseudomonas aeruginosa, Evades NGAL Recognition. Interdiscip. Perspect. Infect. Dis. 2012, 2012, 843509. [Google Scholar] [CrossRef] [Green Version]
- Kang, D.; Kirienko, D.R.; Webster, P.; Fisher, A.L.; Kirienko, N.V. Pyoverdine, a siderophore from Pseudomonas aeruginosa, translocates into C. elegans, removes iron, and activates a distinct host response. Virulence 2018, 9, 804–817. [Google Scholar] [CrossRef] [Green Version]
- Cezard, C.; Farvacques, N.; Sonnet, P. Sonnet Chemistry and Biology of Pyoverdines, Pseudomonas Primary Siderophores. Curr. Med. Chem. 2015, 22, 165–186. [Google Scholar] [CrossRef]
- Orazi, G.; O’Toole, G.A. Pseudomonas aeruginosa Alters Staphylococcus aureus Sensitivity to Vancomycin in a Biofilm Model of Cystic Fibrosis Infection. mBio 2017, 8, e00873-17. [Google Scholar] [CrossRef] [Green Version]
- Braud, A.; Hannauer, M.; Mislin, G.L.A.; Schalk, I.J. The Pseudomonas aeruginosa Pyochelin-Iron Uptake Pathway and Its Metal Specificity. J. Bacteriol. 2009, 191, 3517. [Google Scholar] [CrossRef] [Green Version]
- Cox, M.J.; Allgaier, M.; Taylor, B.; Baek, M.S.; Huang, Y.J.; Daly, R.A.; Karaoz, U.; Andersen, G.L.; Brown, R.; Fujimura, K.E.; et al. Airway microbiota and pathogen abundance in age-stratified cystic fibrosis patients. PLoS ONE 2010, 5, e11044. [Google Scholar] [CrossRef]
- Arevalo-Ferro, C.; Hentzer, M.; Reil, G.; Görg, A.; Kjelleberg, S.; Givskov, M.; Riedel, K.; Eberl, L. Identification of quorum-sensing regulated proteins in the opportunistic pathogen Pseudomonas aeruginosa by proteomics. Environ. Microbiol. 2003, 5, 1350–1369. [Google Scholar] [CrossRef]
- Wandersman, C.; Delepelaire, P. Bacterial Iron Sources: From Siderophores to Hemophores. Annu. Rev. Microbiol. 2004, 58, 611–647. [Google Scholar] [CrossRef]
- Létoffé, S.; Redeker, V.; Wandersman, C. Isolation and characterization of an extracellular haem-binding protein from Pseudomonas aeruginosa that shares function and sequence similarities with the Serratia marcescens HasA haemophore. Mol. Microbiol. 1998, 28, 1223–1234. [Google Scholar] [CrossRef] [Green Version]
- Bhagirath, A.Y.; Li, Y.; Somayajula, D.; Dadashi, M.; Badr, S.; Duan, K. Cystic fibrosis lung environment and Pseudomonas aeruginosa infection. BMC Pulm. Med. 2016, 16, 174. [Google Scholar] [CrossRef] [Green Version]
- Tam, V.C. Lipidomic profiling of bioactive lipids by mass spectrometry during microbial infections. Semin. Immunol. 2013, 25, 240–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dennis, E.A.; Norris, P.C. Eicosanoid storm in infection and inflammation. Nat. Rev. Immunol. 2015, 15, 511–523. [Google Scholar] [CrossRef] [Green Version]
- Morello, E.; Pérez-Berezo, T.; Boisseau, C.; Baranek, T.; Guillon, A.; Bréa, D.; Lanotte, P.; Carpena, X.; Pietrancosta, N.; Hervé, V.; et al. Pseudomonas aeruginosa Lipoxygenase LoxA Contributes to Lung Infection by Altering the Host Immune Lipid Signaling. Front. Microbiol. 2019, 10, 1826. [Google Scholar] [CrossRef] [Green Version]
- Serhan, C.N.; Chiang, N.; Van Dyke, T.E. Resolving inflammation: Dual anti-inflammatory and pro-resolution lipid mediators. Nat. Rev. Immunol. 2008, 8, 349–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alonzo, F., 3rd; Torres, V.J. The bicomponent pore-forming leucocidins of Staphylococcus aureus. Microbiol. Mol. Biol. Rev. MMBR 2014, 78, 199–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scharmann, W. Cytotoxic effects of leukocidin from Pseudomonas aeruginosa on polymorphonuclear leukocytes from cattle. Infect. Immun. 1976, 13, 836–843. [Google Scholar] [CrossRef] [Green Version]
- Abbas, A.; Lichtman, A.; Pillai, S. Basic Immunology: Functions and Disorders of the Immune System, 5th ed.; Elsevier: Amsterdam, The Netherlands, 2016. [Google Scholar]
- Development of Antibody-Based Therapeutics—Translational Considerations. Available online: http://www.springer.com/biomed/pharmacology+%26+toxicology/book/978-1-4419-5953-9 (accessed on 24 September 2014).
- Hoffman, W.; Lakkis, F.G.; Chalasani, G. B Cells, Antibodies, and More. Clin. J. Am. Soc. Nephrol. CJASN 2016, 11, 137–154. [Google Scholar] [CrossRef]
- Ascoli, C.A.; Aggeler, B. Overlooked benefits of using polyclonal antibodies. BioTechniques 2018, 65, 127–136. [Google Scholar] [CrossRef] [Green Version]
- Arnold, J.N.; Wormald, M.R.; Sim, R.B.; Rudd, P.M.; Dwek, R.A. The Impact of Glycosylation on the Biological Function and Structure of Human Immunoglobulins. Annu. Rev. Immunol. 2007, 25, 21–50. [Google Scholar] [CrossRef]
- Justiz Vaillant, A.A.; Ramphul, K. Immunoglobulin. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2020. Available online: http://www.ncbi.nlm.nih.gov/books/NBK513460/ (accessed on 10 August 2020).
- Domenech, M.; Sempere, J.; de Miguel, S.; Yuste, J. Combination of Antibodies and Antibiotics as a Promising Strategy Against Multidrug-Resistant Pathogens of the Respiratory Tract. Front. Immunol. 2018, 9, 2700. [Google Scholar] [CrossRef] [Green Version]
- Mariathasan, S.; Tan, M.-W. Antibody–Antibiotic Conjugates: A Novel Therapeutic Platform against Bacterial Infections. Trends Mol. Med. 2017, 23, 135–149. [Google Scholar] [CrossRef] [PubMed]
- Yuste, J.; Botto, M.; Bottoms, S.E.; Brown, J.S. Serum Amyloid P Aids Complement-Mediated Immunity to Streptococcus pneumoniae. PLoS Pathog. 2007, 3, 1208–1219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subrt, N.; Mesak, L.R.; Davies, J. Modulation of virulence gene expression by cell wall active antibiotics in Staphylococcus aureus. J. Antimicrob. Chemother. 2011, 66, 979–984. [Google Scholar] [CrossRef]
- Lehar, S.M.; Pillow, T.; Xu, M.; Staben, L.; Kajihara, K.K.; Vandlen, R.; DePalatis, L.; Raab, H.; Hazenbos, W.L.; Morisaki, J.H.; et al. Novel antibody–antibiotic conjugate eliminates intracellular S. aureus. Nature 2015, 527, 323–328. [Google Scholar] [CrossRef]
- Hansel, T.T.; Kropshofer, H.; Singer, T.; Mitchell, J.A.; George, A.J.T. The safety and side effects of monoclonal antibodies. Nat. Rev. Drug. Discov. 2010, 9, 325–338. [Google Scholar] [CrossRef] [PubMed]
- Lu, R.-M.; Hwang, Y.-C.; Liu, I.-J.; Lee, C.-C.; Tsai, H.-Z.; Li, H.-J.; Wu, H.-C. Development of therapeutic antibodies for the treatment of diseases. J. Biomed. Sci. 2020, 27, 1–30. [Google Scholar] [CrossRef]
- Mulcahy, L.R.; Isabella, V.M.; Lewis, K. Pseudomonas aeruginosa biofilms in disease. Microb. Ecol. 2014, 68, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Vonarburg, C.; Loetscher, M.; Spycher, M.O.; Kropf, A.; Illi, M.; Salmon, S.; Roberts, S.; Steinfuehrer, K.; Campbell, I.; Koernig, S.; et al. Topical application of nebulized human IgG, IgA and IgAM in the lungs of rats and non-human primates. Respir. Res. 2019, 20, 99. [Google Scholar] [CrossRef] [Green Version]
- Jones, R.G.A.; Martino, A. Targeted localized use of therapeutic antibodies: A review of non-systemic, topical and oral applications. Crit. Rev. Biotechnol. 2015, 36, 506–520. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Location or Class | Examples | Activity/Effects on Host |
|---|---|---|
| Cell surface | Alginate | Antiphagocytic, resists opsonic killing |
| Lipopolysaccharide | Endotoxic, antiphagocytic, avoids preformed antibody to previously encountered O antigens | |
| Pili (produced by type IV secretion) | Twitching motility, biofilm formation, adherence to host tissues | |
| Flagella | Motility, biofilm formation, adherence to host tissues and mucin components | |
| Injection of type III secretion factors | PcrG, PcrV, PcrH, PopB, and PopD proteins form injection bridge for type III effectors | |
| Outer membrane | Siderophore receptors | Provides iron for microbial growth and survival |
| Efflux pumps | Remove antibiotics | |
| Secretion systems | ||
| Elastase, lipase, phospholipases, chitin-binding protein, exotoxin A, and others | Variety of proteolytic, lipolytic, and toxic factors; degrade host immune effectors |
| ExoS, ExoT, ExoU, ExoY | Intoxicates cells (ExoS, ExoT); cytotoxic (ExoU); disrupts actin cytoskeleton |
| Cytoplasmic and membrane-associated proteins, ATPases, lipoproteins, Hcp1 protein | Poorly characterized but found in animal studies to be needed for optimal virulence, particularly in chronic infection |
| Iron acquisition | Pyoverdin, pyochelin, HasAP | Scavenge iron from the host for bacterial use |
| Secreted toxins | Hemolysins, rhamnolipid phospholipases | Kill leukocytes, hemolysis of red cells, degrade host cell surface glycolipids |
| Secreted oxidative factors | Pyocyanin, ferric pyochelin, HCN | Produce reactive oxygen species: H2O2, O2− Inflammatory, disrupts epithelial cell function |
| Quorum sensing | LasR/LasI, RhlR/RhlI, PQS | Biofilm formation, regulation of virulence factor secretion |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Proctor, L.L.; Ward, W.L.; Roggy, C.S.; Koontz, A.G.; Clark, K.M.; Quinn, A.P.; Schroeder, M.; Brooks, A.E.; Small, J.M.; Towne, F.D.; et al. Potential Therapeutic Targets for Combination Antibody Therapy against Pseudomonas aeruginosa Infections. Antibiotics 2021, 10, 1530. https://doi.org/10.3390/antibiotics10121530
Proctor LL, Ward WL, Roggy CS, Koontz AG, Clark KM, Quinn AP, Schroeder M, Brooks AE, Small JM, Towne FD, et al. Potential Therapeutic Targets for Combination Antibody Therapy against Pseudomonas aeruginosa Infections. Antibiotics. 2021; 10(12):1530. https://doi.org/10.3390/antibiotics10121530
Chicago/Turabian StyleProctor, Luke L., Whitney L. Ward, Conner S. Roggy, Alexandra G. Koontz, Katie M. Clark, Alyssa P. Quinn, Meredith Schroeder, Amanda E. Brooks, James M. Small, Francina D. Towne, and et al. 2021. "Potential Therapeutic Targets for Combination Antibody Therapy against Pseudomonas aeruginosa Infections" Antibiotics 10, no. 12: 1530. https://doi.org/10.3390/antibiotics10121530