
Assessing Population Structure and Signatures of Selection in Wanbei Pigs Using Whole Genome Resequencing Data
 ,
, {kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Animals and Whole-Genome Resequencing
2.3. Read Mapping and Single Nucleotide Polymorphism (SNP) Calling
2.4. Population Structure and Linkage Disequilibrium Analysis
2.5. Identification of Selection Signatures
3. Results
3.1. Sequencing and Identification of SNPs
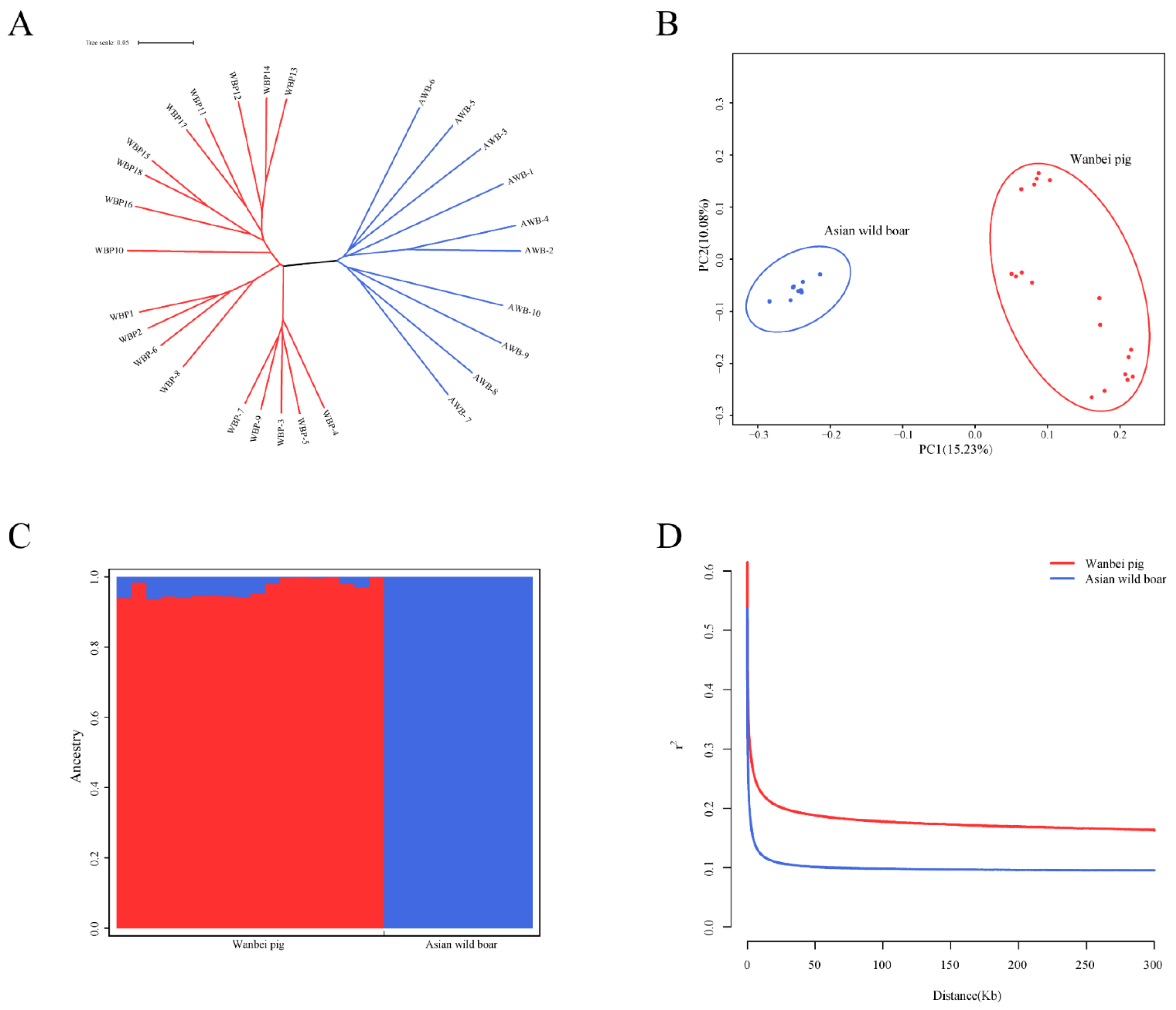
3.2. Population Structure and Linkage Disequilibrium Analysis
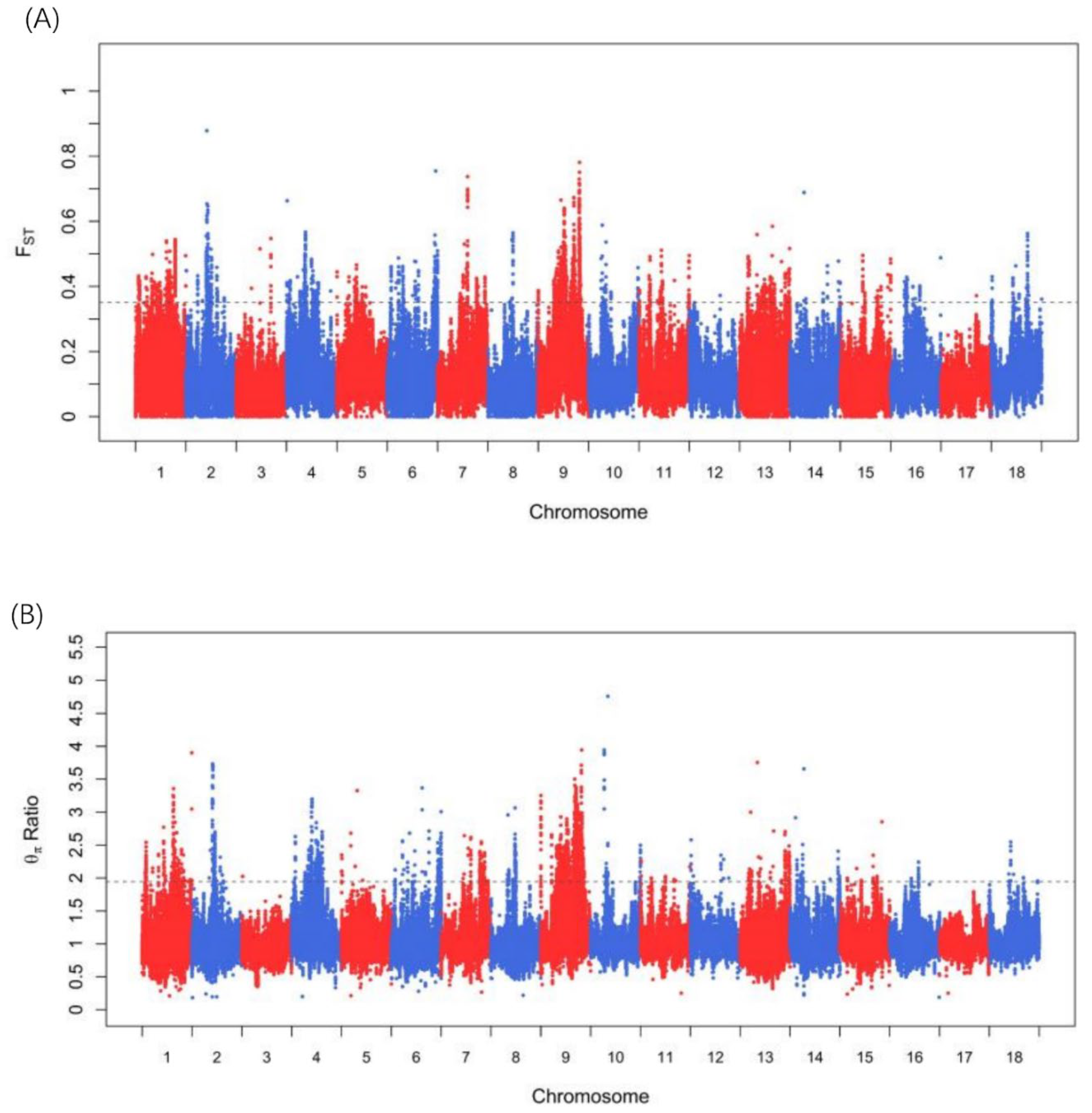
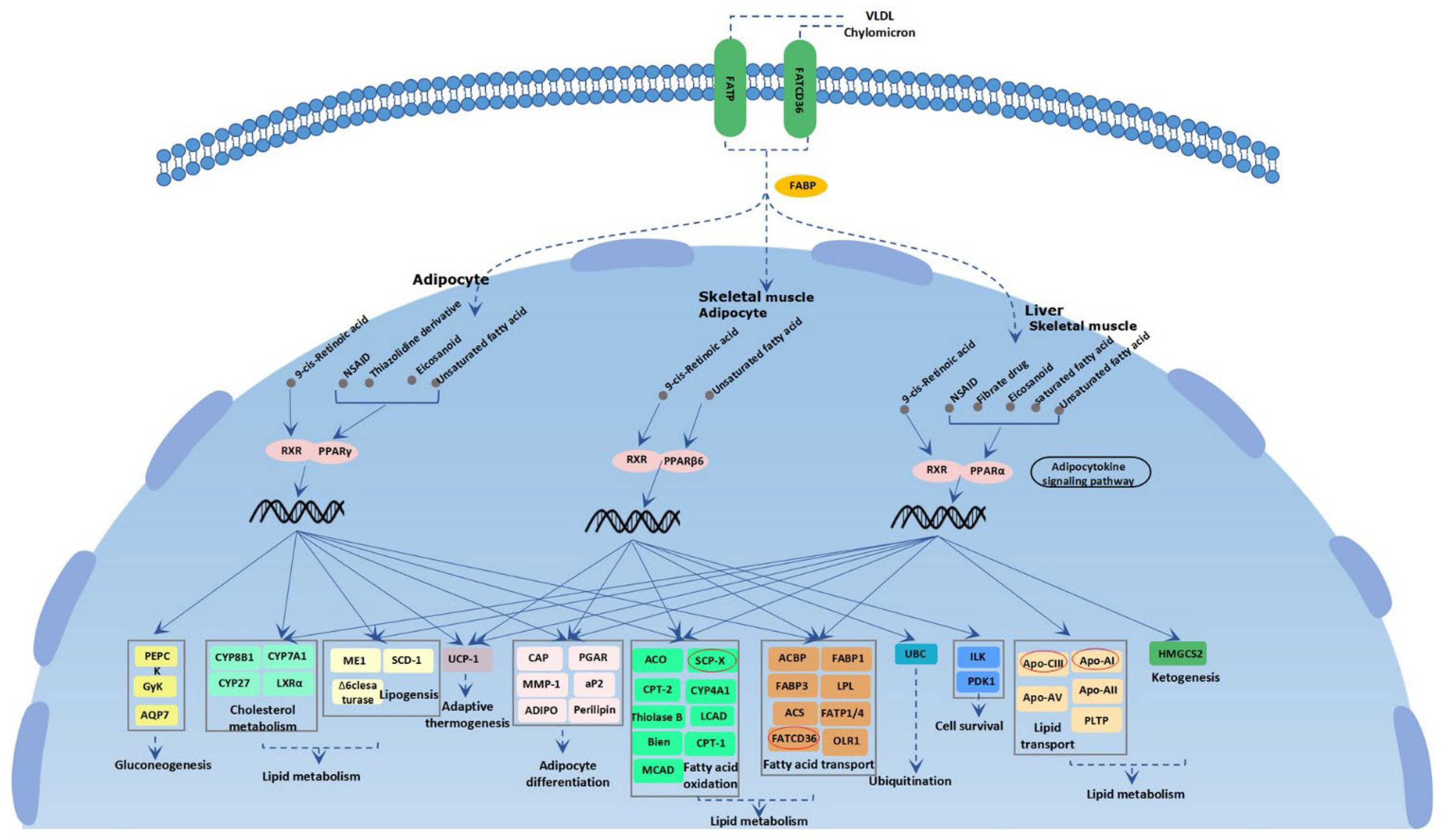
3.3. Candidate Genes under Selection Signatures
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Larson, G.; Dobney, K.; Albarella, U.; Fang, M.; Matisoo-Smith, E.; Robins, J.; Lowden, S.; Finlayson, H.; Brand, T.; Willerslev, E.; et al. Worldwide phylogeography of wild boar reveals multiple centers of pig domestication. Science 2005, 307, 1618–1621. [Google Scholar] [CrossRef] [Green Version]
- Veirano Fréchou, R. The state of the world’s animal genetic resources for food and agriculture. Acta Paediatr. 2007, 81, 21–24. [Google Scholar]
- Grossman, S.R.; Shylakhter, I.; Karlsson, E.K.; Byrne, E.H.; Morales, S.; Frieden, G.; Hostetter, E.; Angelino, E.; Garber, M.; Zuk, O.; et al. A composite of multiple signals distinguishes causal variants in regions of positive selection. Science 2010, 327, 883–886. [Google Scholar] [CrossRef] [Green Version]
- Lewontin, R.C.; Krakauer, J. Distribution of gene frequency as a test of the theory of the selective neutrality of polymorphisms. Genetics 1973, 74, 175–195. [Google Scholar] [CrossRef]
- Tajima, F. Evolutionary relationship of DNA sequences in finite populations. Genetics 1983, 105, 437–460. [Google Scholar] [CrossRef]
- Rubin, C.J.; Zody, M.C.; Eriksson, J.; Meadows, J.R.; Sherwood, E.; Webster, M.T.; Jiang, L.; Ingman, M.; Sharpe, T.; Ka, S.; et al. Whole-genome resequencing reveals loci under selection during chicken domestication. Nature 2010, 464, 587–591. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Sun, H.; Chen, Q.; Zhang, X.; Wang, Q.; Pan, Y. A genome scan for selection signatures in Taihu pig breeds using next-generation sequencing. Anim. Int. J. Anim. Biosci. 2019, 13, 683–693. [Google Scholar] [CrossRef]
- Zhao, P.; Yu, Y.; Feng, W.; Du, H.; Yu, J.; Kang, H.; Zheng, X.; Wang, Z.; Liu, G.E.; Ernst, C.W.; et al. Evidence of evolutionary history and selective sweeps in the genome of Meishan pig reveals its genetic and phenotypic characterization. GigaScience 2018, 7, giy058. [Google Scholar] [CrossRef] [Green Version]
- Li, W.T.; Zhang, M.M.; Li, Q.G.; Tang, H.; Zhang, L.F.; Wang, K.J.; Zhu, M.Z.; Lu, Y.F.; Bao, H.G.; Zhang, Y.M.; et al. Whole-genome resequencing reveals candidate mutations for pig prolificacy. Proc. Biol. Sci. 2017, 284, 20172437. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Tian, S.; Jin, L.; Zhou, G.; Li, Y.; Zhang, Y.; Wang, T.; Yeung, C.K.; Chen, L.; Ma, J.; et al. Genomic analyses identify distinct patterns of selection in domesticated pigs and Tibetan wild boars. Nat. Genet. 2013, 45, 1431–1438. [Google Scholar] [CrossRef]
- Zhang, D.J.; He, X.M.; Wang, L.; Liu, D. Genomic sequence feature analysis of de novo assembly in Min pig. J. Jilin Agric. Univ. 2022. [Google Scholar] [CrossRef]
- Qin, M.; Li, C.; Li, Z.; Chen, W.; Zeng, Y. Genetic Diversities and Differentially Selected Regions Between Shandong Indigenous Pig Breeds and Western Pig Breeds. Front. Genet. 2020, 10, 1351. [Google Scholar] [CrossRef]
- Chen, M.; Wang, J.; Wang, Y.; Wu, Y.; Fu, J.; Liu, J.F. Genome-wide detection of selection signatures in Chinese indigenous Laiwu pigs revealed candidate genes regulating fat deposition in muscle. BMC Genet. 2018, 19, 31. [Google Scholar] [CrossRef] [Green Version]
- Yin, Z.J. Characteristics and protection and utilization of Local pig germplasm resources in Anhui Province. In Proceedings of the 11th Annual Conference of China Regional Pig Conservation and Utilization Collaborative Group, Beijing, China, 24 November 2015; Cambridge University Press: Cambridge, UK, 2015; pp. 13–17. [Google Scholar]
- Sambrook, J.; Russell, D.W. Molecular Cloning: A Laboratory Manual, 3rd ed.; Cold Spring Harbor Laboratory Press: New York, NY, USA, 2001. [Google Scholar]
- Wu, X.; Zhou, R.; Wang, Y.; Zhang, W.; Zheng, X.; Zhao, G.; Zhang, X.; Yin, Z.; Ding, Y. Genome-wide scan for runs of homozygosity in Asian wild boars and Anqing six-end-white pigs. Anim. Genet. 2022, 53, 867–871. [Google Scholar] [CrossRef]
- Groenen, M.A.; Archibald, A.L.; Uenishi, H.; Tuggle, C.K.; Takeuchi, Y.; Rothschild, M.F.; Rogel-Gaillard, C.; Park, C.; Milan, D.; Megens, H.J.; et al. Analyses of pig genomes provide insight into porcine demography and evolution. Nature 2012, 491, 393–398. [Google Scholar] [CrossRef] [Green Version]
- Patel, R.K.; Jain, M. NGS QC Toolkit: A Toolkit for Quality Control of Next Generation Sequencing Data. PLoS ONE 2012, 7, e30619. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. 1000 Genome Project Data Processing Subgroup. The sequence alignment/map format and SAM tools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2011, 38, e164. [Google Scholar] [CrossRef]
- Yang, J.; Lee, S.H.; Goddard, M.E.; Visscher, P.M. GCTA: A tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 2011, 88, 76–82. [Google Scholar] [CrossRef] [Green Version]
- Felsenstein, J. PHYLIP-Phylogeny inference package (version 3.2). Cladistics 1989, 5, 164–166. [Google Scholar]
- Holsinger, K.E.; Weir, B.S. Genetics in geographically structured populations: Defining, estimating and interpreting FST. Nat. Rev. Genet. 2009, 10, 639–650. [Google Scholar] [CrossRef]
- Weir, B.C.; Cockerham, C.C. Estimating F-Statistics for the Analysis of Population Structure. Evolution 1984, 38, 1358–1370. [Google Scholar]
- Pfeifer, B.; Wittelsbürger, U.; Ramos-Onsins, S.E.; Lercher, M.J. PopGenome: An efficient Swiss army knife for population genomic analyses in R. Mol. Biol. Evol. 2014, 31, 1929–1936. [Google Scholar] [CrossRef] [Green Version]
- Kerstens, H.H.; Kollers, S.; Kommadath, A.; Del Rosario, M.; Dibbits, B.; Kinders, S.M.; Crooijmans, R.P.; Groenen, M.A. Mining for single nucleotide polymorphisms in pig genome sequence data. BMC Genom. 2009, 10, 4. [Google Scholar] [CrossRef] [Green Version]
- Starodub, O.; Jolly, C.A.; Atshaves, B.P.; Roths, J.B.; Murphy, E.J.; Kier, A.B.; Schroeder, F. Sterol carrier protein-2 localization in endoplasmic reticulum and role in phospholipid formation. Cell Physiol. 2000, 279, 1259–1269. [Google Scholar] [CrossRef] [Green Version]
- Stolowich, N.J.; Petrescu, A.D.; Huang, H.; Martin, G.G.; Scott, A.I.; Schroeder, F. Sterol carrier protein-2: Structure reveals function. Cell. Mol. Life Sci. 2002, 59, 193–212. [Google Scholar] [CrossRef]
- McLean, M.P.; Billheimer, J.T.; Warden, K.J.; Irby, R.B. Differential expression of hepatic sterol carrier proteins in the streptozotocin-treated diabetic rat. Endocrinology 1995, 136, 3360–3368. [Google Scholar] [CrossRef]
- Qin, W.; Liang, C.N.; Guo, X.; Chu, M.; Pei, J.; Bao, P.J.; Wu, X.Y.; Li, T.K.; Yan, P. PPARα signal pathway gene expression is associated with fatty acid content in yak and cattle longissimus dorsi muscle. Genet. Mol. Res. 2015, 14, 14469–14478. [Google Scholar] [CrossRef]
- Li, Y.; Lee, Y.M.; Kim, Y.S.; Park, S.P.; Kim, J.J. Identifying Loci Under Positive Selection in Yellow Korean Cattle (Hanwoo). Evol. Bioinform. 2019, 15, 1176934319859001. [Google Scholar] [CrossRef] [Green Version]
- Delgado-Lista, J.; Perez-Jimenez, F.; Ruano, J.; Perez-Martinez, P.; Fuentes, F.; Criado-Garcia, J.; Parnell, L.D.; Garcia-Rios, A.; Ordovas, J.M.; Lopez-Miranda, J. Effects of variations in the APOA1/C3/A4/A5 gene cluster on different parameters of postprandial lipid metabolism in healthy young men. J. Lipid Res. 2010, 51, 63–73. [Google Scholar] [CrossRef] [Green Version]
- Baroukh, N.; Bauge, E.; Akiyama, J.; Chang, J.; Afzal, V.; Fruchart, J.C.; Rubin, E.M.; Fruchart-Najib, J.; Pennacchio, L.A. Analysis of apolipoprotein A5, c3, and plasma triglyceride concentrations in genetically engineered mice. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1297–1302. [Google Scholar] [CrossRef]
- Li, J.; Xing, S.; Zhao, G.; Zheng, M.; Yang, X.; Sun, J.; Wen, J.; Liu, R. Identification of diverse cell populations in skeletal muscles and biomarkers for intramuscular fat of chicken by single-cell RNA sequencing. BMC Genom. 2020, 21, 752. [Google Scholar] [CrossRef]
- Liu, R.; Wang, H.; Liu, J.; Wang, J.; Zheng, M.; Tan, X.; Xing, S.; Cui, H.; Li, Q.; Zhao, G.; et al. Uncovering the embryonic development-related proteome and metabolome signatures in breast muscle and intramuscular fat of fast-and slow-growing chickens. BMC Genom. 2017, 18, 816. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Xing, K.; Jiang, Y.; Liu, Y.; Wang, C.; Ding, X. Using Machine Learning to Identify Biomarkers Affecting Fat Deposition in Pigs by Integrating Multisource Transcriptome Information. J. Agric. Food Chem. 2022, 70, 10359–10370. [Google Scholar] [CrossRef]
- Wang, Z.; Shang, P.; Li, Q.; Wang, L.; Chamba, Y.; Zhang, B.; Zhang, H.; Wu, C. iTRAQ-based proteomic analysis reveals key proteins affecting muscle growth and lipid deposition in pigs. Sci. Rep. 2017, 7, 46717. [Google Scholar] [CrossRef]
- Hodis, H.N.; Mack, W.J. Triglyceride-rich lipoproteins and the progression of coronary artery disease. Curr. Opin. Lipidol. 1995, 6, 209–214. [Google Scholar] [CrossRef]
- Ito, Y.; Azrolan, N.; O’Connell, A.; Walsh, A.; Breslow, J.L. Hypertriglyceridemia as a result of human apo CIII gene expression in transgenic mice. Science 1990, 249, 790–793. [Google Scholar] [CrossRef]
- Maeda, N.; Li, H.; Lee, D.; Oliver, P.; Quarfordt, S.H.; Osada, J. Targeted disruption of the apolipoprotein C-III gene in mice results in hypotriglyceridemia and protection from postprandial hypertriglyceridemia. J. Biol. Chem. 1994, 269, 23610–23616. [Google Scholar] [CrossRef]
- Glatz, J.F.; Luiken, J.J.; Bonen, A. Membrane fatty acid transporters as regulators of lipid metabolism: Implications for metabolic disease. Physiol. Behav. 2010, 90, 367–417. [Google Scholar] [CrossRef] [Green Version]
- Jeong, J.; Kwon, E.; Im, S.; Seo, K.; Baik, M. Expression of fat deposition and fat removal genes is associated with intramuscular fat content in longissimus dorsi muscle of Korean cattle steers. J. Anim. Sci. 2012, 90, 2044–2053. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Hanif, Q.; Cao, Y.; Yu, Y.; Lei, C.; Zhang, G.; Zhao, Y. Whole Genome Scan and Selection Signatures for Climate Adaption in Yanbian Cattle. Front. Genet. 2020, 11, 94. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Wang, J.; Ma, C.; Wang, Z.; Chen, H.; Su, H.; Wan, Y.; Jiang, R. Genome-wide re-sequencing and transcriptome analysis reveal candidate genes associated with the pendulous comb phenotype in domestic chickens. Anim. Sci. J. Nihon Chikusan Gakkaiho 2020, 91, e13308. [Google Scholar] [CrossRef] [PubMed]
- Prodanović, R.; Korićanac, G.; Vujanac, I.; Djordjević, A.; Pantelić, M.; Romić, S.; Stanimirović, Z.; Kirovski, D. Obesity-driven prepartal hepatic lipid accumulation in dairy cows is associated with increased CD36 and SREBP-1 expression. Res. Vet. Sci. 2016, 107, 16–19. [Google Scholar] [CrossRef]
- Nickerson, J.G.; Alkhateeb, H.; Benton, C.R.; Lally, J.; Nickerson, J.; Han, X.X.; Wilson, M.H.; Jain, S.S.; Snook, L.A.; Glatz, J.; et al. Greater transport efficiencies of the membrane fatty acid transporters FAT/CD36 and FATP4 compared with FABPpm and FATP1 and differential effects on fatty acid esterification and oxidation in rat skeletal muscle. J. Biol. Chem. 2009, 284, 16522–16530. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Kohan, A.B.; Lo, C.M.; Liu, M.; Howles, P.; Tso, P. Apolipoprotein A-IV: A protein intimately involved in metabolism. J. Lipid Res. 2015, 56, 1403–1418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreira, G.; Boschiero, C.; Cesar, A.; Reecy, J.M.; Godoy, T.F.; Trevisoli, P.A.; Cantão, M.E.; Ledur, M.C.; Ibelli, A.; Peixoto, J.O.; et al. A genome-wide association study reveals novel genomic regions and positional candidate genes for fat deposition in broiler chickens. BMC Genom. 2018, 19, 374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlegel, G.; Ringseis, R.; Windisch, W.; Schwarz, F.J.; Eder, K. Effects of a rumen-protected mixture of conjugated linoleic acids on hepatic expression of genes involved in lipid metabolism in dairy cows. J. Dairy Sci. 2012, 95, 3905–3918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, L.; Shi, L.; Liu, L.; Liang, R.; Li, Q.; Li, J.; Han, B.; Sun, D. Analysis of Liver Proteome and Identification of Critical Proteins Affecting Milk Fat, Protein, and Lactose Metabolism in Dairy Cattle with iTRAQ. Proteomics 2019, 19, e1800387. [Google Scholar] [CrossRef]
- Zhou, J.; Zhang, Y.; Wu, J.; Qiao, M.; Xu, Z.; Peng, X.; Mei, S. Proteomic and lipidomic analyses reveal saturated fatty acids, phosphatidylinositol, phosphatidylserine, and associated proteins contributing to intramuscular fat deposition. J. Proteom. 2021, 241, 104235. [Google Scholar] [CrossRef]
- Ohno, H. Pathogenetic and clinical implications of non-immunoglobulin: BCL6 translocations in B-cell non-Hodgkin’s lymphoma. J. Clin. Exp. Hematop. 2006, 46, 43–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LaPensee, C.R.; Lin, G.; Dent, A.L.; Schwartz, J. Deficiency of the transcriptional repressor B cell lymphoma 6 (Bcl6) is accompanied by dysregulated lipid metabolism. PLoS ONE 2014, 9, e97090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, X.; Zhou, Y.; Yang, Y.; Peng, J.; Song, T.; Xu, T.; Wei, H.; Jiang, S.; Peng, J. Identification of zinc finger protein Bcl6 as a novel regulator of early adipose commitment. Open Biol. 2016, 6, 160065. [Google Scholar] [CrossRef] [PubMed]
- Takeshita, S.; Moritani, M.; Kunika, K.; Inoue, H.; Itakura, M. Diabetic modifer QTLs identifed in F2 intercrosses between Akita and A/J mice. Mamm. Genome 2006, 17, 927–940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kose, H.; Bando, Y.; Izumi, K.; Yamada, T.; Matsumoto, K. Epistasis between hyperglycemic QTLs revealed in a double congenic of the OLETF rat. Mamm. Genome 2007, 18, 609–615. [Google Scholar] [CrossRef] [PubMed]
- Su, Z.; Tsaih, S.W.; Szatkiewicz, J.; Shen, Y.; Paigen, B. Candidate genes for plasma triglyceride, FFA, and glucose revealed from an intercross between inbred mouse strains NZB/B1NJ and NZW/LacJ. J. Lipid Res. 2008, 49, 1500–1510. [Google Scholar] [CrossRef] [Green Version]
- Moradian, H.; Esmailizadeh, K.A.; Mohammadabadi, M.; Asadi, F.M. Whole genome detection of recent selection signatures in Sarabi cattle: A unique Iranian taurine breed. Genes Genom. 2020, 42, 203–215. [Google Scholar] [CrossRef]
- Guo, D.; Zhou, Y.; Wei, X.; Zhang, S.; Jin, T.; Zhang, Y.; Lin, M.; Zhou, X.; Xie, Y.; He, C.; et al. Preliminary study of genome-wide association identifies novel susceptibility genes for serum mineral elements in the Chinese Han population. Biol. Trace Elem. Res. 2022, 200, 2549–2555. [Google Scholar] [CrossRef]
- Silver, M.; Chen, P.; Li, R.; Cheng, C.Y.; Wong, T.Y.; Tai, E.S.; Teo, Y.Y.; Montana, G. Pathways-driven sparse regression identifies pathways and genes associated with highdensity lipoprotein cholesterol in two Asian cohorts. PLoS Genet. 2013, 9, e1003939. [Google Scholar] [CrossRef]
- Bovo, S.; Mazzoni, G.; Bertolini, F.; Schiavo, G.; Galimberti, G.; Gallo, M.; Dall’Olio, S.; Fontanesi, L. Genome-wide association studies for 30 haematological and blood clinical-biochemical traits in Large White pigs reveal genomic regions affecting intermediate phenotypes. Sci. Rep. 2019, 9, 7003. [Google Scholar] [CrossRef] [Green Version]
- Fortes, M.R.; Reverter, A.; Kelly, M.; McCulloch, R.; Lehnert, S.A. Genome-wide association study for inhibin, luteinizing hormone, insulin-like growth factor 1, testicular size and semen traits in bovine species. Andrology 2013, 1, 644–650. [Google Scholar] [CrossRef] [PubMed]
- Gudbjartsson, D.F.; Walters, G.B.; Thorleifsson, G.; Stefansson, H.; Halldorsson, B.V.; Zusmanovich, P.; Sulem, P.; Thorlacius, S.; Gylfason, A.; Steinberg, S.; et al. Many sequence variants affecting diversity of adult human height. Nat. Genet. 2008, 40, 609–615. [Google Scholar] [CrossRef] [PubMed]
- Fortes, M.R.; Kemper, K.; Sasazaki, S.; Reverter, A.; Pryce, J.E.; Barendse, W.; Bunch, R.; McCulloch, R.; Harrison, B.; Bolormaa, S.; et al. Evidence for pleiotropism and recent selection in the PLAG1 region in Australian Beef cattle. Anim. Genet. 2013, 44, 636–647. [Google Scholar] [CrossRef] [PubMed]
- Karim, L.; Takeda, H.; Lin, L.; Druet, T.; Arias, J.A.; Baurain, D.; Cambisano, N.; Davis, S.R.; Farnir, F.; Grisart, B.; et al. Variants modulating the expression of a chromosome domain encompassing PLAG1 influence bovine stature. Nat. Genet. 2011, 43, 405–415. [Google Scholar] [CrossRef] [PubMed]
- Ghoreishifar, S.M.; Eriksson, S.; Johansson, A.M.; Khansefid, M.; Moghaddaszadeh-Ahrabi, S.; Parna, N.; Davoudi, P.; Javanmard, A. Signatures of selection reveal candidate genes involved in economic traits and cold acclimation in five Swedish cattle breeds. Genet. Sel. Evol. GSE 2020, 52, 52. [Google Scholar] [CrossRef]
- Fortes, M.R.S.; Reverter, A.; Hawken, R.J.; Bolormaa, S.; Lehnert, S.A. Candidate genes associated with testicular development, sperm quality, and hormone levels of inhibin, luteinizing hormone, and insulin-like growth factor 1 in Brahman bulls. Biol. Reprod. 2012, 87, 58. [Google Scholar] [CrossRef]
- Rubin, C.J.; Megens, H.J.; Martinez Barrio, A.; Maqbool, K.; Sayyab, S.; Schwochow, D.; Wang, C.; Carlborg, Ö.; Jern, P.; Jørgensen, C.B.; et al. Strong signatures of selection in the domestic pig genome. Proc. Natl. Acad. Sci. USA 2012, 109, 19529–19536. [Google Scholar] [CrossRef] [Green Version]
- Xu, P.; Li, D.; Wu, Z.; Ni, L.; Liu, J.; Tang, Y.; Yu, T.; Ren, J.; Zhao, X.; Huang, M. An imputation-based genome-wide association study for growth and fatness traits in Sujiang pigs. Anim. Int. J. Anim. Biosci. 2022, 16, 100591. [Google Scholar] [CrossRef]
- Yuan, Z.R.; Li, J.; Liu, L.; Zhang, L.P.; Zhang, L.M.; Chen, C.; Chen, X.J.; Gao, X.; Li, J.Y.; Chen, J.B.; et al. Single nucleotide polymorphism of CACNA2D1 gene and its association with milk somatic cell score in cattle. Mol. Boil. Rep. 2011, 38, 5179–5183. [Google Scholar] [CrossRef]
- Yuan, Z.R.; Xu, S.Z. Novel SNPs of the bovine CACNA2D1 gene and their association with carcass and meat quality traits. Mol. Biol. Rep. 2011, 38, 365–370. [Google Scholar] [CrossRef]
- Ferri, P.; Barbieri, E.; Burattini, S.; Guescini, M.; D’Emilio, A.; Biagiotti, L.; Del Grande, P.; De Luca, A.; Stocchi, V.; Falcieri, E. Expression and subcellular localization of myogenic regulatory factors during the differentiation of skeletal muscle C2C12 myoblasts. J. Cell. Biochem. 2009, 108, 1302–1317. [Google Scholar] [CrossRef]
- Guo, L.; Sun, H.; Zhao, Q.; Xu, Z.; Zhang, Z.; Liu, D.; Qadri, Q.R.; Ma, P.; Wang, Q.; Pan, Y. Positive selection signatures in Anqing six-end-white pig population based on reduced-representation genome sequencing data. Anim. Genet. 2021, 52, 143–154. [Google Scholar] [CrossRef] [PubMed]
- Fabbri, M.C.; Dadousis, C.; Tiezzi, F.; Maltecca, C.; Lozada-Soto, E.; Biffani, S.; Bozzi, R. Genetic diversity and population history of eight Italian beef cattle breeds using measures of autozygosity. PLoS ONE 2021, 16, e0248087. [Google Scholar] [CrossRef]
- Xue, H.L.; Zhou, Z.X. Effects of the MyoG Gene on the Partial Growth Traits in Pigs. Acta Genet. Sin. 2006, 33, 992–997. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Zhang, G.X.; Zhang, T.; Wang, J.Y.; Fan, Q.C.; Tang, Y.; Ding, F.X.; Zhang, L. Myf5 and MyoG gene SNPs associated with Bian chicken growth trait. Genet. Mol. Res. 2016, 15, 15037043. [Google Scholar] [CrossRef]
- Fraga, C.G.; Oteiza, P.I.; Keen, C.L. Trace elements and human health. Mol. Asp. Med. 2005, 26, 233–234. [Google Scholar] [CrossRef]
- Fraga, C.G. Relevance, essentiality and toxicity of trace elements in human health. Mol. Asp. Med. 2005, 26, 235–244. [Google Scholar] [CrossRef]
- Kawashima, A.; Osman, B.A.; Takashima, M.; Kikuchi, A.; Kohchi, S.; Satoh, E.; Tamba, M.; Matsuda, M.; Okamura, N. CABS1 is a novel calcium-binding protein specifically expressed in elongate spermatids of mice. Biol. Reprod. 2009, 80, 1293–1304. [Google Scholar] [CrossRef] [Green Version]
- Shawki, H.H.; Kigoshi, T.; Katoh, Y.; Matsuda, M.; Ugboma, C.M.; Takahashi, S.; Oishi, H.; Kawashima, A. Identification, localization, and functional analysis of the homologues of mouse CABS1 protein in porcine testis. Exp. Anim. 2016, 65, 253–265. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, W.; Liu, L.; Zhou, M.; Su, S.; Dong, L.; Meng, X.; Li, X.; Wang, C. Assessing Population Structure and Signatures of Selection in Wanbei Pigs Using Whole Genome Resequencing Data. Animals 2023, 13, 13. https://doi.org/10.3390/ani13010013
Zhang W, Liu L, Zhou M, Su S, Dong L, Meng X, Li X, Wang C. Assessing Population Structure and Signatures of Selection in Wanbei Pigs Using Whole Genome Resequencing Data. Animals. 2023; 13(1):13. https://doi.org/10.3390/ani13010013
Chicago/Turabian StyleZhang, Wei, Linqing Liu, Mei Zhou, Shiguang Su, Lin Dong, Xinxin Meng, Xueting Li, and Chonglong Wang. 2023. "Assessing Population Structure and Signatures of Selection in Wanbei Pigs Using Whole Genome Resequencing Data" Animals 13, no. 1: 13. https://doi.org/10.3390/ani13010013