Integrating Single-Step GWAS and Bipartite Networks Reconstruction Provides Novel Insights into Yearling Weight and Carcass Traits in Hanwoo Beef Cattle


Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Phenotypic and Pedigree Data
2.3. Genotypic Data
2.4. Analysis of the Single-Step Genomic Association
2.5. Selection of Relevant SNP Windows and Putative Candidate Genes Identification
2.6. Functional Gene Set Annotation and Enrichment
2.7. Target Gene Prediction and Validation of Candidate miRNAs
2.8. Gene-Traits and miRNA–Gene Network Reconstruction
3. Results
3.1. Summary Statistics
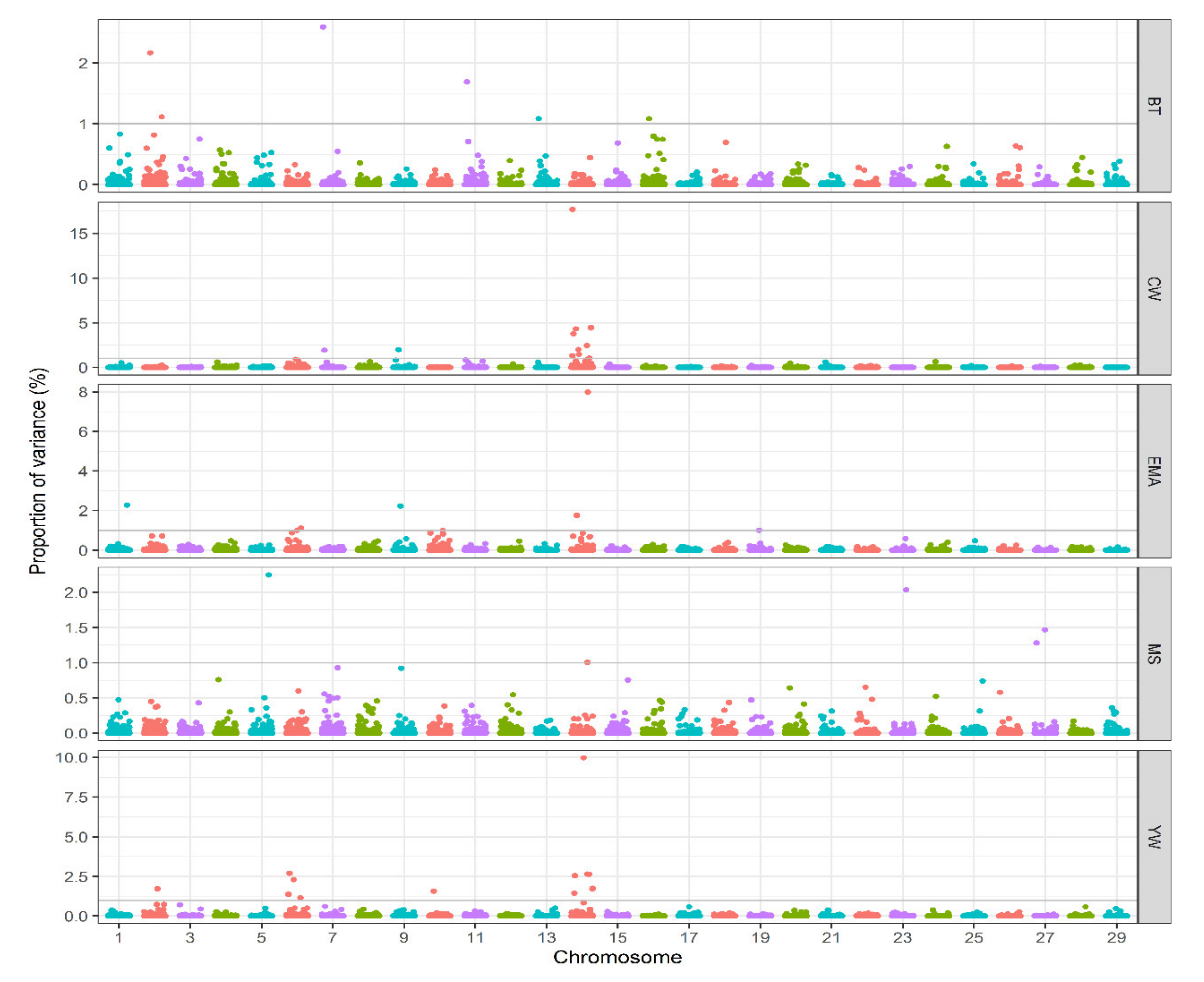
3.2. Association Analysis
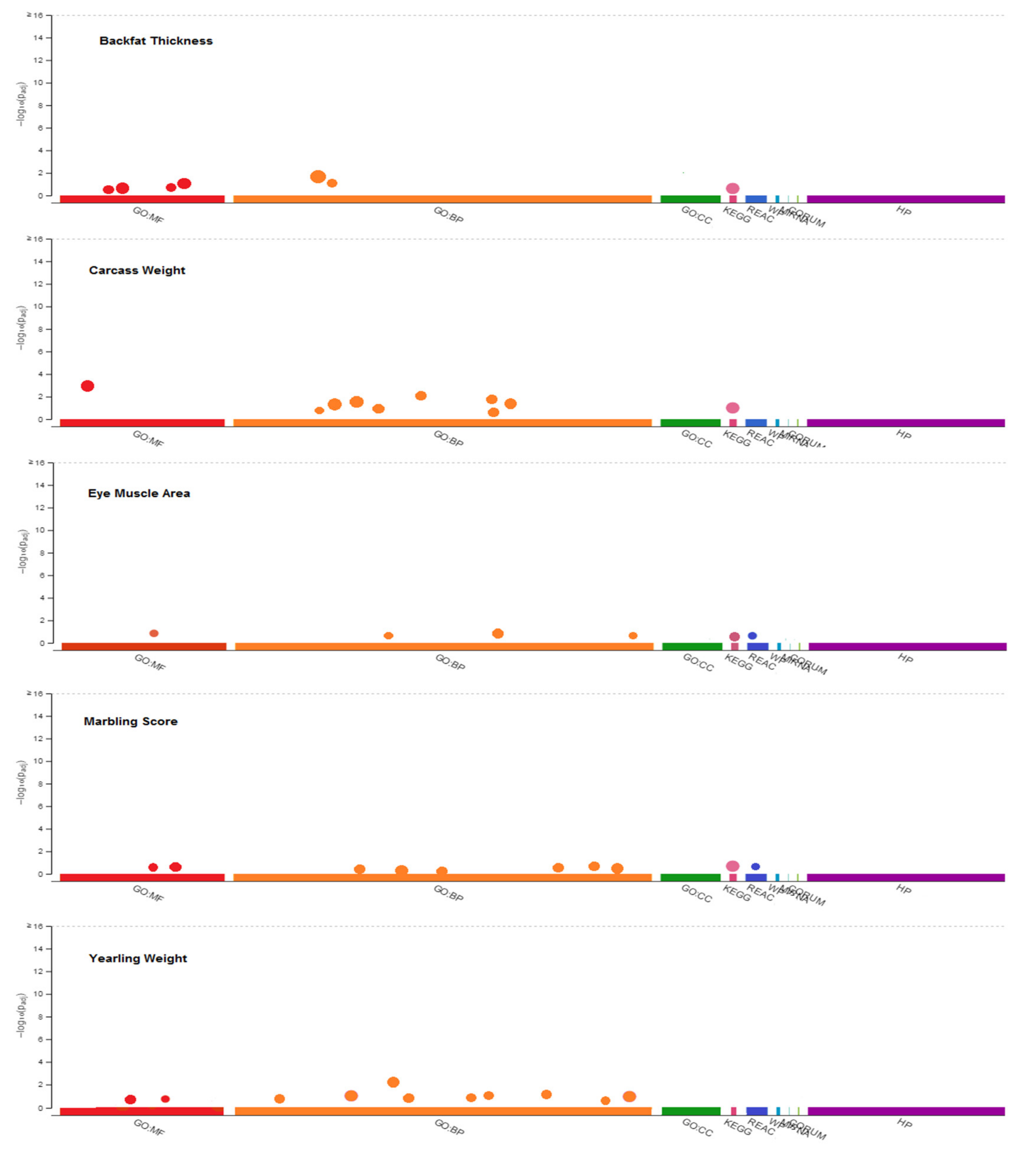
3.3. Functional Gene Set Annotation and Enrichment
3.4. Prediction of miRNA-Target Genes
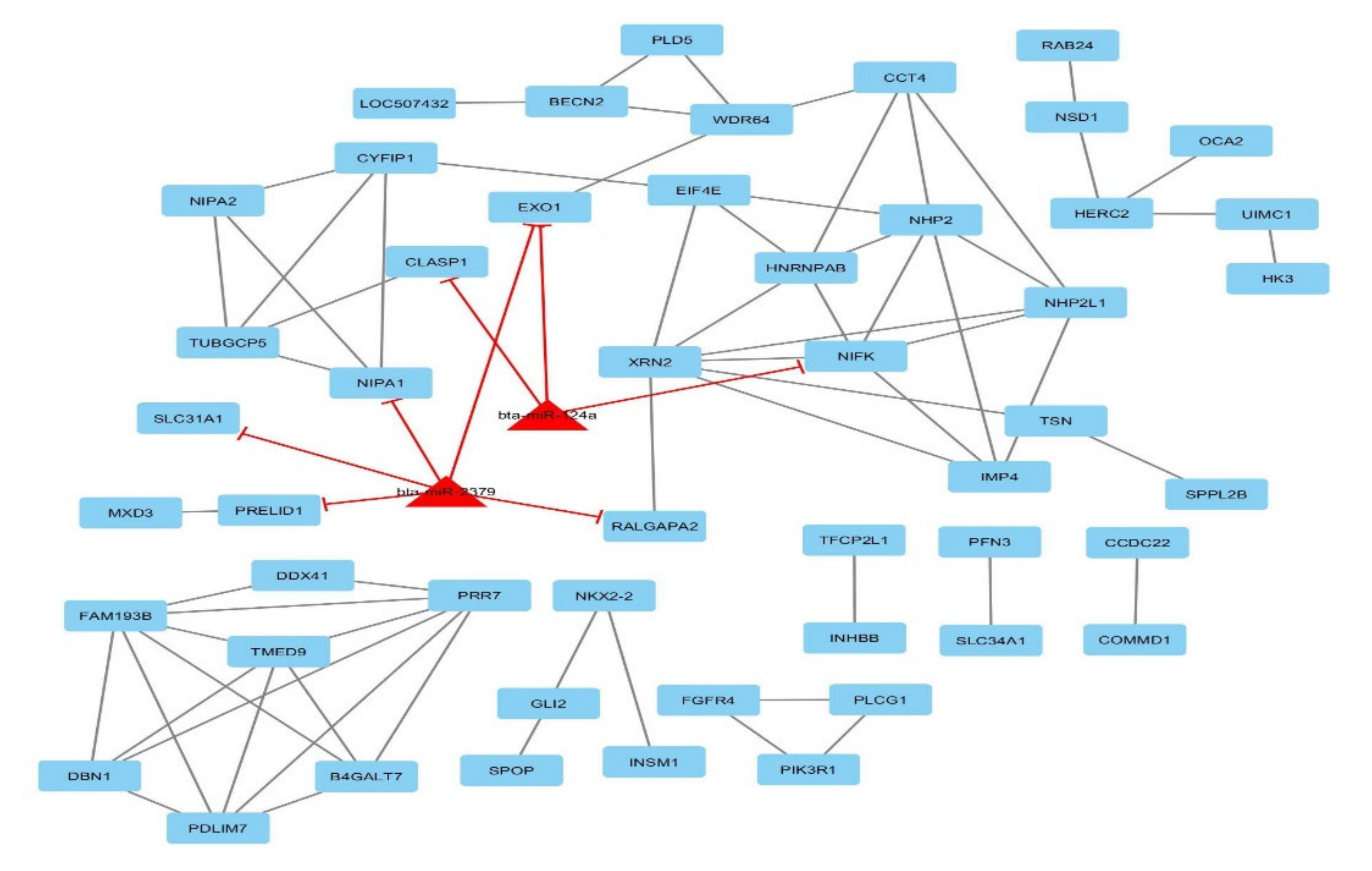
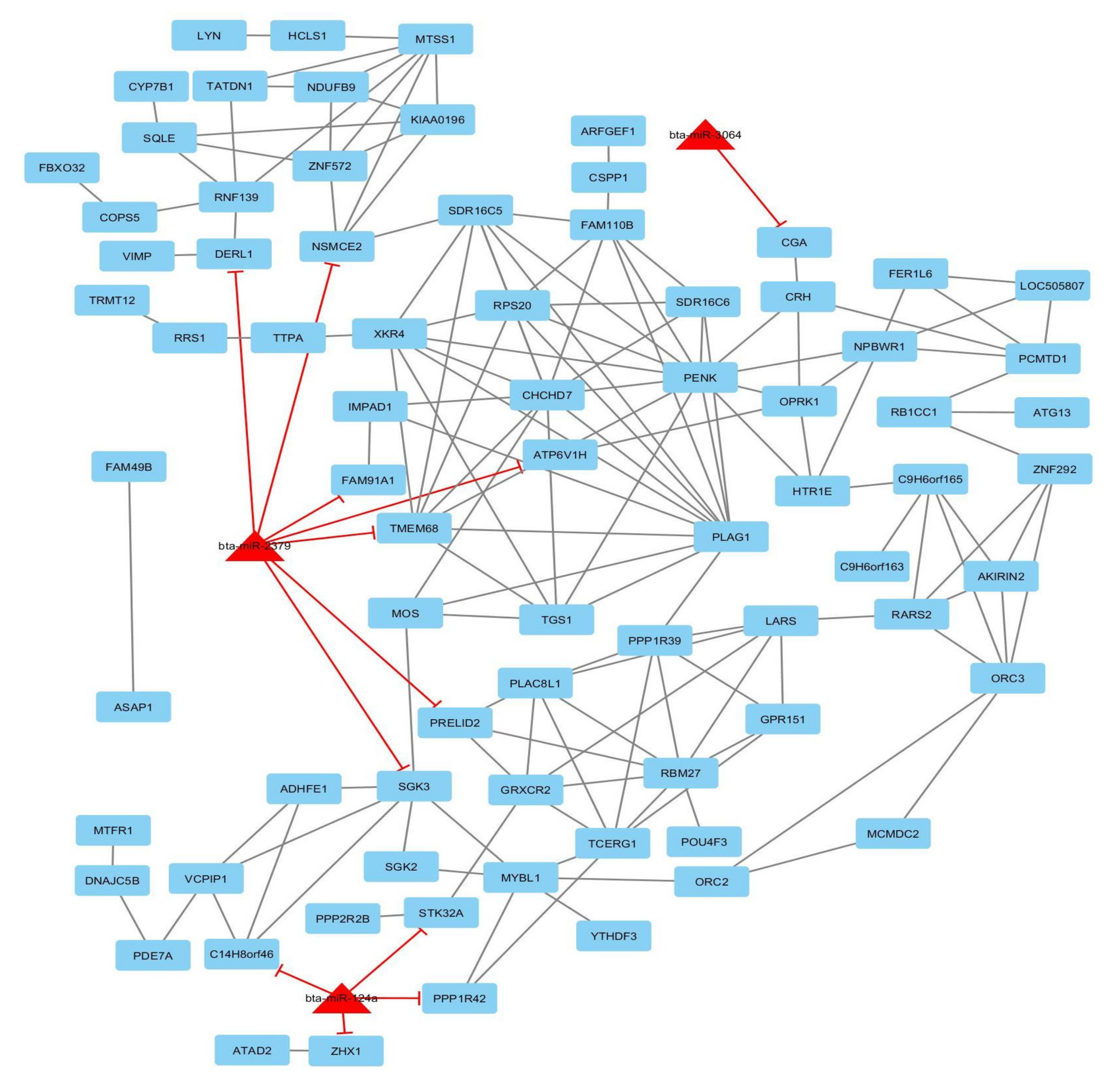
3.5. Gene-Traits and miRNA–Gene Network Reconstruction
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Jo, C.; Cho, S.; Chang, J.; Nam, K. Keys to production and processing of Hanwoo beef: A perspective of tradition and science. Anim. Front. 2012, 2, 32–38. [Google Scholar] [CrossRef]
- Kim, S.; Alam, M.; Park, M. Breeding initiatives for Hanwoo cattle to thrive as a beef industry—A review study. J. Anim. Breed. Genet. 2017, 1, 102–124. [Google Scholar]
- Matukumalli, L.K.; Lawley, C.T.; Schnabel, R.D.; Taylor, J.F.; Allan, M.F.; Heaton, M.P.; O’Connell, J.; Moore, S.S.; Smith, T.P.; Sonstegard, T.S.; et al. Development and characterization of a high density SNP genotyping assay for cattle. PLoS ONE 2009, 4, e5350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishimura, S.; Watanabe, T.; Mizoshita, K.; Tatsuda, K.; Fujita, T.; Watanabe, N.; Sugimoto, Y.; Takasuga, A. Genome-wide association study identified three major QTL for carcass weight including the PLAG1-CHCHD7 QTN for stature in Japanese Black cattle. BMC Genet. 2012, 13, 40. [Google Scholar] [CrossRef] [Green Version]
- Setoguchi, K.; Furuta, M.; Hirano, T.; Nagao, T.; Watanabe, T.; Sugimoto, Y.; Takasuga, A. Cross-breed comparisons identified a critical 591-kb region for bovine carcass weight QTL (CW-2) on chromosome 6 and the Ile-442-Met substitution in NCAPG as a positional candidate. BMC Genet. 2009, 10, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Weng, Z.; Su, H.; Saatchi, M.; Lee, J.; Thomas, M.G.; Dunkelberger, J.R.; Garrick, D.J. Genome-wide association study of growth and body composition traits in Brangus beef cattle. Livest. Sci. 2016, 183, 4–11. [Google Scholar] [CrossRef]
- Tizioto, P.C.; Decker, J.E.; Taylor, J.F.; Schnabel, R.D.; Mudadu, M.A.; Silva, F.L.; Mourão, G.B.; Coutinho, L.L.; Tholon, P.; Sonstegard, T.S.; et al. Genome scan for meat quality traits in Nelore beef cattle. Physiol. Genom. 2013, 45, 1012–1020. [Google Scholar] [CrossRef] [Green Version]
- Meyer, K.; Tier, B. “SNP Snappy”: A strategy for fast genome-wide association studies fitting a full mixed model. Genetics 2012, 190, 275–277. [Google Scholar] [CrossRef] [Green Version]
- Peters, S.; Kizilkaya, K.; Garrick, D.; Fernando, R.; Reecy, J.; Weaber, R.; Silver, G.; Thomas, M. Bayesian genome-wide association analysis of growth and yearling ultrasound measures of carcass traits in Brangus heifers. J. Anim. Sci. 2012, 90, 3398–3409. [Google Scholar] [CrossRef]
- Wang, H.; Misztal, I.; Aguilar, I.; Legarra, A.; Muir, W. Genome-wide association mapping including phenotypes from relatives without genotypes. Genet. Res. 2012, 94, 73–83. [Google Scholar] [CrossRef] [Green Version]
- Habier, D.; Fernando, R.L.; Kizilkaya, K.; Garrick, D.J. Extension of the Bayesian alphabet for genomic selection. BMC Bioinform. 2011, 12, 186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Lourenco, D.; Aguilar, I.; Legarra, A.; Misztal, I. Weighting strategies for single-step genomic BLUP: An iterative approach for accurate calculation of GEBV and GWAS. Front. Genet. 2016, 7, 151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magalhaes, A.F.; de Camargo, G.M.; Fernandes, G.A.; Gordo, D.G.; Tonussi, R.L.; Costa, R.B.; Espigolan, R.; Silva, R.M.d.O.; Bresolin, T.; de Andrade, W.B.; et al. Genome-wide association study of meat quality traits in Nellore cattle. PLoS ONE 2016, 11, e0157845. [Google Scholar] [CrossRef] [PubMed]
- Tiezzi, F.; Parker-Gaddis, K.L.; Cole, J.B.; Clay, J.S.; Maltecca, C. A genome-wide association study for clinical mastitis in first parity US Holstein cows using single-step approach and genomic matrix re-weighting procedure. PLoS ONE 2015, 10, e0114919. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Misztal, I.; Aguilar, I.; Legarra, A.; Fernando, R.L.; Vitezica, Z.; Okimoto, R.; Wing, T.; Hawken, R.; Muir, W.M. Genome-wide association mapping including phenotypes from relatives without genotypes in a single-step (ssGWAS) for 6-week body weight in broiler chickens. Front. Genet. 2014, 5, 134. [Google Scholar] [CrossRef] [Green Version]
- Wu, P.; Yang, Q.; Wang, K.; Zhou, J.; Ma, J.; Tang, Q.; Jin, L.; Xiao, W.; Jiang, A.; Jiang, Y.; et al. Single step genome-wide association studies based on genotyping by sequence data reveals novel loci for the litter traits of domestic pigs. Genomics 2018, 110, 171–179. [Google Scholar] [CrossRef]
- Dadousis, C.; Pegolo, S.; Rosa, G.; Gianola, D.; Bittante, G.; Cecchinato, A. Pathway-based genome-wide association analysis of milk coagulation properties, curd firmness, cheese yield, and curd nutrient recovery in dairy cattle. J. Dairy Sci. 2017, 100, 1223–1231. [Google Scholar] [CrossRef]
- Fang, L.; Sahana, G.; Ma, P.; Su, G.; Yu, Y.; Zhang, S.; Lund, M.S.; Sørensen, P. Use of biological priors enhances understanding of genetic architecture and genomic prediction of complex traits within and between dairy cattle breeds. BMC Genom. 2017, 18, 604. [Google Scholar] [CrossRef]
- MacLeod, I.; Bowman, P.; Vander Jagt, C.; Haile-Mariam, M.; Kemper, K.; Chamberlain, A.; Schrooten, C.; Hayes, B.; Goddard, M. Exploiting biological priors and sequence variants enhances QTL discovery and genomic prediction of complex traits. BMC Genom. 2016, 17, 144. [Google Scholar] [CrossRef] [Green Version]
- Fang, L.; Jiang, J.; Li, B.; Zhou, Y.; Freebern, E.; Vanraden, P.M.; Cole, J.B.; Liu, G.E.; Ma, L. Genetic and epigenetic architecture of paternal origin contribute to gestation length in cattle. Commun. Biol. 2019, 2, 1–11. [Google Scholar] [CrossRef]
- Trynka, G.; Sandor, C.; Han, B.; Xu, H.; Stranger, B.E.; Liu, X.S.; Raychaudhuri, S. Chromatin marks identify critical cell types for fine mapping complex trait variants. Nat. Genet. 2013, 45, 124–130. [Google Scholar] [CrossRef] [PubMed]
- Xiang, R.; van den Berg, I.; MacLeod, I.M.; Hayes, B.J.; Prowse-Wilkins, C.P.; Wang, M.; Bolormaa, S.; Liu, Z.; Rochfort, S.J.; Reich, C.M.; et al. Quantifying the contribution of sequence variants with regulatory and evolutionary significance to 34 bovine complex traits. Proc. Natl. Acad. Sci. USA 2019, 116, 19398–19408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bahrami, A.; Miraie-Ashtiani, S.R.; Sadeghi, M.; Najafi, A. miRNA-mRNA network involved in folliculogenesis interactome: Systems biology approach. Reproduction 2017, 154, 51–65. [Google Scholar] [CrossRef] [PubMed]
- Bedhane, M.; van der Werf, J.; Gondro, C.; Duijvesteijn, N.; Lim, D.; Park, B.; Park, M.N.; Hee, R.S.; Clark, S. Genome-wide association study of meat quality traits in Hanwoo beef cattle using imputed whole-genome sequence data. Front. Genet. 2019, 10, 1235. [Google Scholar] [CrossRef] [PubMed]
- Bhuiyan, M.S.; Lim, D.; Park, M.; Lee, S.; Kim, Y.; Gondro, C.; Park, B.; Lee, S. Functional partitioning of genomic variance and genome-wide association study for carcass traits in Korean Hanwoo cattle using imputed sequence level SNP data. Front. Genet. 2018, 9, 217. [Google Scholar] [CrossRef]
- Dang, C.; Cho, S.; Sharma, A.; Kim, H.; Jeon, G.; Yeon, S.; Hong, S.; Park, B.; Kang, H.; Lee, S. Genome-wide association study for Warner-Bratzler shear force and sensory traits in Hanwoo (Korean cattle). Asian-Australas. J. Anim. Sci. 2014, 27, 1328. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.; Ryu, J.; Woo, J.; Kim, J.; Kim, C.; Lee, C. Genome-wide association study reveals five nucleotide sequence variants for carcass traits in beef cattle. Anim. Genet. 2011, 42, 361–365. [Google Scholar] [CrossRef]
- Lee, S.H.; Choi, B.H.; Lim, D.; Gondro, C.; Cho, Y.M.; Dang, C.G.; Sharma, A.; Jang, G.W.; Lee, K.T.; Yoon, D.; et al. Genome-wide association study identifies major loci for carcass weight on BTA14 in Hanwoo (Korean cattle). PLoS ONE 2013, 8, e74677. [Google Scholar] [CrossRef] [Green Version]
- Park, B.; Choi, T.; Kim, S.; Oh, S.H. National genetic evaluation (system) of Hanwoo (Korean native cattle). Asian-Australas. J. Anim. Sci. 2013, 26, 151. [Google Scholar] [CrossRef]
- Wiggans, G.; Sonstegard, T.; VanRaden, P.; Matukumalli, L.; Schnabel, R.; Taylor, J.; Schenkel, F.; van Tassell, C. Selection of single-nucleotide polymorphisms and quality of genotypes used in genomic evaluation of dairy cattle in the United States and Canada. J. Dairy Sci. 2009, 92, 3431–3436. [Google Scholar] [CrossRef] [Green Version]
- Mehrban, H.; Lee, D.H.; Naserkheil, M.; Moradi, M.H.; Ibáñez-Escriche, N. Comparison of conventional BLUP and single-step genomic BLUP evaluations for yearling weight and carcass traits in Hanwoo beef cattle using single trait and multi-trait models. PLoS ONE 2019, 14, e0223352. [Google Scholar] [CrossRef] [PubMed]
- Misztal, I.; Tsuruta, S.; Lourenco, D.; Masuda, Y.; Aguilar, I.; Legarra, A.; Vitezica, Z. Manual for BLUPF90 Family of Programs; University of Georgia: Athens, GA, USA, 2018; Volume 2018. [Google Scholar]
- Aguilar, I.; Misztal, I.; Johnson, D.; Legarra, A.; Tsuruta, S.; Lawlor, T. Hot topic: A unified approach to utilize phenotypic, full pedigree, and genomic information for genetic evaluation of Holstein final score. J. Dairy Sci. 2010, 93, 743–752. [Google Scholar] [CrossRef] [PubMed]
- Aguilar, I.; Misztal, I.; Tsuruta, S.; Legarra, A.; Wang, H. PREGSF90–POSTGSF90: Computational tools for the implementation of single-step genomic selection and genome-wide association with ungenotyped individuals in BLUPF90 programs. In Proceedings of the 10th World Congress on Genetics Applied to Livestock Production (WCGALP), Vancouver, BC, Canada, 17–22 August 2014. [Google Scholar]
- VanRaden, P.M. Efficient methods to compute genomic predictions. J. Dairy Sci. 2008, 91, 4414–4423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lourenco, D.; Tsuruta, S.; Fragomeni, B.; Masuda, Y.; Aguilar, I.; Legarra, A.; Bertrand, J.; Amen, T.; Wang, L.; Moser, D.; et al. Genetic evaluation using single-step genomic best linear unbiased predictor in American Angus. J. Anim. Sci. 2015, 93, 2653–2662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valente, T.S.; Baldi, F.; Sant’Anna, A.C.; Albuquerque, L.G.; Paranhos da Costa, M.J.R. Genome-Wide Association Study between single nucleotide polymorphisms and flight speed in Nellore cattle. PLoS ONE 2016, 11, e0156956. [Google Scholar] [CrossRef]
- Lemos, M.V.; Chiaia, H.L.J.; Berton, M.P.; Feitosa, F.L.; Aboujaoud, C.; Camargo, G.M.; Pereira, A.S.; Albuquerque, L.G.; Ferrinho, A.M.; Mueller, L.F.; et al. Genome-wide association between single nucleotide polymorphisms with beef fatty acid profile in Nellore cattle using the single step procedure. BMC Genom. 2016, 17, 213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lourenco, D.; Legarra, A.; Tsuruta, S.; Masuda, Y.; Aguilar, I.; Misztal, I. Single-Step Genomic Evaluations from Theory to Practice: Using SNP Chips and Sequence Data in BLUPF90. Genes 2020, 11, 790. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment of Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2020. [Google Scholar]
- Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44. [Google Scholar]
- Mi, H.; Muruganujan, A.; Ebert, D.; Huang, X.; Thomas, P.D. PANTHER version 14: More genomes, a new PANTHER GO-slim and improvements in enrichment analysis tools. Nucleic Acids Res. 2019, 47, D419–D426. [Google Scholar] [CrossRef]
- Raudvere, U.; Kolberg, L.; Kuzmin, I.; Arak, T.; Adler, P.; Peterson, H.; Vilo, J. g: Profiler: A web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res. 2019, 47, W191–W198. [Google Scholar] [CrossRef] [Green Version]
- Sticht, C.; de La Torre, C.; Parveen, A.; Gretz, N. miRWalk: An online resource for prediction of microRNA binding sites. PLoS ONE 2018, 13, e0206239. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016, 44, D457–D462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bader, G.D.; Betel, D.; Hogue, C.W. BIND: The biomolecular interaction network database. Nucleic Acids Res. 2003, 31, 248–250. [Google Scholar] [CrossRef] [PubMed]
- Xenarios, I.; Salwinski, L.; Duan, X.J.; Higney, P.; Kim, S.M.; Eisenberg, D. DIP, the Database of Interacting Proteins: A research tool for studying cellular networks of protein interactions. Nucleic Acids Res. 2002, 30, 303–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chatr-Aryamontri, A.; Breitkreutz, B.-J.; Heinicke, S.; Boucher, L.; Winter, A.; Stark, C.; Nixon, J.; Ramage, L.; Kolas, N.; O’Donnell, L.; et al. The BioGRID interaction database: 2013 update. Nucleic Acids Res. 2012, 41, D816–D823. [Google Scholar] [CrossRef] [PubMed]
- Pagel, P.; Kovac, S.; Oesterheld, M.; Brauner, B.; Dunger-Kaltenbach, I.; Frishman, G.; Montrone, C.; Mark, P.; Stümpflen, V.; Mewes, H.W.; et al. The MIPS mammalian protein–protein interaction database. Bioinformatics 2005, 21, 832–834. [Google Scholar] [CrossRef] [Green Version]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.P.; et al. STRING v10: Protein–protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015, 43, D447–D452. [Google Scholar] [CrossRef] [PubMed]
- Najafi, A.; Bidkhori, G.; Bozorgmehr, J.H.; Koch, I.; Masoudi-Nejad, A. Genome scale modeling in systems biology: Algorithms and resources. Curr. Genom. 2014, 15, 130–159. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Jiang, Y.; An, Y.; Zhao, N.; Zhao, Y.; Yu, C. Soluble FGFR4 extracellular domain inhibits FGF19-induced activation of FGFR4 signaling and prevents nonalcoholic fatty liver disease. Biochem. Biophys. Res. Commun. 2011, 409, 651–656. [Google Scholar] [CrossRef]
- Miliara, X.; Tatsuta, T.; Berry, J.L.; Rouse, S.L.; Solak, K.; Chorev, D.S.; Wu, D.; Robinson, C.V.; Matthews, S.; Langer, T. Structural determinants of lipid specificity within Ups/PRELI lipid transfer proteins. Nat. Commun. 2019, 10, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Cruz, R.; Plant, P.J.; Pablo, L.A.; Lin, S.; Chackowicz, J.; Correa, J.; Bain, J.; Batt, J. PDLIM7 is a novel target of the ubiquitin ligase Nedd4-1 in skeletal muscle. Biochem. J. 2016, 473, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Sjöholm, K.; Palming, J.; Lystig, T.C.; Jennische, E.; Woodruff, T.K.; Carlsson, B.; Carlsson, L.M. The expression of inhibin beta B is high in human adipocytes, reduced by weight loss, and correlates to factors implicated in metabolic disease. Biochem. Biophys. Res. Commun. 2006, 344, 1308–1314. [Google Scholar] [CrossRef] [PubMed]
- Mokry, F.B.; Higa, R.H.; de Alvarenga Mudadu, M.; de Lima, A.O.; Meirelles, S.L.C.; da Silva, M.V.G.B.; Cardoso, F.F.; de Oliveira, M.M.; Urbinati, I.; Niciura, S.C.M.; et al. Genome-wide association study for backfat thickness in Canchim beef cattle using Random Forest approach. BMC Genet. 2013, 14, 47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.H.; van der Werf, J.; Lee, S.H.; Park, E.W.; Gondro, C.; Yoon, D.; Oh, S.J.; Kim, O.H.; Gibson, J.; Thompson, J.; et al. Genome wide QTL mapping to identify candidate genes for carcass traits in Hanwoo (Korean Cattle). Genes Genom. 2012, 34, 43–49. [Google Scholar] [CrossRef]
- Roberts, A. Genome-wide association study for carcass traits in a composite beef cattle breed. Livest. Sci. 2018, 213, 35–43. [Google Scholar]
- Karim, L.; Takeda, H.; Lin, L.; Druet, T.; Arias, J.A.; Baurain, D.; Cambisano, N.; Davis, S.R.; Farnir, F.; Grisart, B.; et al. Variants modulating the expression of a chromosome domain encompassing PLAG1 influence bovine stature. Nat. Genet. 2011, 43, 405–413. [Google Scholar] [CrossRef]
- Utsunomiya, Y.T.; Carmo, A.S.D.; Carvalheiro, R.; Neves, H.H.; Matos, M.C.; Zavarez, L.B.; O’Brien, A.M.P.; Sölkner, J.; McEwan, J.C.; Cole, J.B.; et al. Genome-wide association study for birth weight in Nellore cattle points to previously described orthologous genes affecting human and bovine height. BMC Genet. 2013, 14, 52. [Google Scholar] [CrossRef] [Green Version]
- Littlejohn, M.; Grala, T.; Sanders, K.; Walker, C.; Waghorn, G.; Macdonald, K.; Coppieters, W.; Georges, M.; Spelman, R.; Hillerton, E.; et al. Genetic variation in PLAG1 associates with early life body weight and peripubertal weight and growth in Bos taurus. Anim. Genet. 2012, 43, 591–594. [Google Scholar] [CrossRef]
- Lindholm-Perry, A.; Kuehn, L.; Smith, T.; Ferrell, C.; Jenkins, T.; Freetly, H.; Snelling, W. A region on BTA14 that includes the positional candidate genes LYPLA1, XKR4 and TMEM68 is associated with feed intake and growth phenotypes in cattle 1. Anim. Genet. 2012, 43, 216–219. [Google Scholar] [CrossRef]
- Porto-Neto, L.; Bunch, R.; Harrison, B.; Barendse, W. Variation in the XKR4 gene was significantly associated with subcutaneous rump fat thickness in indicine and composite cattle. Anim. Genet. 2012, 43, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Ramayo-Caldas, Y.; Fortes, M.; Hudson, N.; Porto-Neto, L.; Bolormaa, S.; Barendse, W.; Kelly, M.; Moore, S.; Goddard, M.; Lehnert, S.; et al. A marker-derived gene network reveals the regulatory role of PPARGC1A, HNF4G, and FOXP3 in intramuscular fat deposition of beef cattle. J. Anim. Sci. 2014, 92, 2832–2845. [Google Scholar] [CrossRef] [PubMed]
- Ramayo-Caldas, Y.; Renand, G.; Ballester, M.; Saintilan, R.; Rocha, D. Multi-breed and multi-trait co-association analysis of meat tenderness and other meat quality traits in three French beef cattle breeds. Genet. Sel. Evol. 2016, 48, 37. [Google Scholar] [CrossRef] [Green Version]
- Seong, J.; Kong, H. Association between polymorphisms of the CRH and POMC genes with economic traits in Korean cattle (Hanwoo). Genet. Mol. Res. 2015, 14, 10415–10421. [Google Scholar] [CrossRef] [PubMed]
- Saatchi, M.; Schnabel, R.D.; Taylor, J.F.; Garrick, D.J. Large-effect pleiotropic or closely linked QTL segregate within and across ten US cattle breeds. BMC Genom. 2014, 15, 442. [Google Scholar] [CrossRef] [Green Version]
- Bolormaa, S.; Pryce, J.E.; Kemper, K.E.; Hayes, B.J.; Zhang, Y.; Tier, B.; Barendse, W.; Reverter, A.; Goddard, M.E. Detection of quantitative trait loci in Bos indicus and Bos taurus cattle using genome-wide association studies. Genet. Sel. Evol. 2013, 45, 43. [Google Scholar] [CrossRef] [Green Version]
- Bouwman, A.C.; Bovenhuis, H.; Visker, M.H.; van Arendonk, J.A. Genome-wide association of milk fatty acids in Dutch dairy cattle. BMC Genet. 2011, 12, 43. [Google Scholar] [CrossRef] [Green Version]
- Conte, G.; Mele, M.; Chessa, S.; Castiglioni, B.; Serra, A.; Pagnacco, G.; Secchiari, P. Diacylglycerol acyltransferase 1, stearoyl-CoA desaturase 1, and sterol regulatory element binding protein 1 gene polymorphisms and milk fatty acid composition in Italian Brown cattle. J. Dairy Sci. 2010, 93, 753–763. [Google Scholar] [CrossRef] [Green Version]
- Boitard, S.; Rocha, D. Detection of signatures of selective sweeps in the Blonde d’Aquitaine cattle breed. Anim. Genet. 2013, 44, 579–583. [Google Scholar] [CrossRef]
- Russell, P.J.; Hertz, P.E.; McMillan, B. Biology: The Dynamic Science; Cengage Learning: Boston, MA, USA, 2012. [Google Scholar]
- Von der Heyde, S.; Fromm-Dornieden, C.; Salinas-Riester, G.; Beissbarth, T.; Baumgartner, B.G. Dynamics of mRNA and polysomal abundance in early 3T3-L1 adipogenesis. BMC Genom. 2014, 15, 381. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Huang, Z.; Jia, G.; Wu, X.; Wu, C. Molecular cloning, tissue distribution, and functional analysis of porcine Akirin2. Anim. Biotechnol. 2012, 23, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Lee, S.; Hong, M.; Park, S.; Lee, Y.; Kim, J.; Lee, H.; Jeong, D.; Song, Y.; Lee, S. Association of a single nucleotide polymorphism in the akirin 2 gene with economically important traits in Korean native cattle. Anim. Genet. 2013, 44, 750–753. [Google Scholar] [CrossRef] [PubMed]
- Sukegawa, S.; Miyake, T.; Ibi, T.; Takahagi, Y.; Murakami, H.; Morimatsu, F.; Yamada, T. Multiple marker effects of single nucleotide polymorphisms in three genes, AKIRIN2, EDG1 and RPL27A, for marbling development in Japanese Black cattle. Anim. Sci. J. 2014, 85, 193–197. [Google Scholar] [CrossRef] [PubMed]
- Debiec-Rychter, M.; van Valckenborgh, I.; van den Broeck, C.; Hagemeijer, A.; van de Ven, W.J.; Kas, K.; van Damme, B.; Voz, M.L. Histologic localization of PLAG1 (pleomorphic adenoma gene 1) in pleomorphic adenoma of the salivary gland: Cytogenetic evidence of common origin of phenotypically diverse cells. Lab. Investig. 2001, 81, 1289–1297. [Google Scholar] [CrossRef] [Green Version]
- Ranson, M. Epidermal growth factor receptor tyrosine kinase inhibitors. Br. J. Cancer 2004, 90, 2250–2255. [Google Scholar] [CrossRef] [Green Version]
- Aggarwal, B.B.; Surh, Y.J.; Shishodia, S. The Molecular Targets and Therapeutic Uses of Curcumin in Health and Disease; Springer: Berlin/Heidelberg, Germany, 2007; Volume 595. [Google Scholar]
- Bahrami, A.; Miraie-Ashtiani, S.R.; Sadeghi, M.; Najafi, A.; Ranjbar, R. Dynamic modeling of folliculogenesis signaling pathways in the presence of miRNAs expression. J. Ovarian Res. 2017, 10, 76. [Google Scholar] [CrossRef] [Green Version]
- Greenawalt, D.M.; Sieberts, S.K.; Cornelis, M.C.; Girman, C.J.; Zhong, H.; Yang, X.; Guinney, J.; Qi, L.; Hu, F.B. Integrating genetic association, genetics of gene expression, and single nucleotide polymorphism set analysis to identify susceptibility Loci for type 2 diabetes mellitus. Am. J. Epidemiol. 2012, 176, 423–430. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Zan, L.; Zhao, S.; Xin, Y.; Li, L.; Cui, W.; Tang, Z.; Li, K. Molecular characterization, polymorphism of bovine ZBTB38 gene and association with body measurement traits in native Chinese cattle breeds. Mol. Biol. Rep. 2010, 37, 4041–4049. [Google Scholar] [CrossRef]
- McClure, M.; Morsci, N.; Schnabel, R.; Kim, J.; Yao, P.; Rolf, M.; McKay, S.; Gregg, S.; Chapple, R.; Northcutt, S.; et al. A genome scan for quantitative trait loci influencing carcass, post-natal growth and reproductive traits in commercial Angus cattle. Anim. Genet. 2010, 41, 597–607. [Google Scholar] [CrossRef]
- Lu, G.; Ren, S.; Korge, P.; Choi, J.; Dong, Y.; Weiss, J.; Koehler, C.; Chen, J.N.; Wang, Y. A novel mitochondrial matrix serine/threonine protein phosphatase regulates the mitochondria permeability transition pore and is essential for cellular survival and development. Genes Dev. 2007, 21, 784–796. [Google Scholar] [CrossRef] [Green Version]
- Olsen, H.G.; Nilsen, H.; Hayes, B.; Berg, P.R.; Svendsen, M.; Lien, S.; Meuwissen, T. Genetic support for a quantitative trait nucleotide in the ABCG2 gene affecting milk composition of dairy cattle. BMC Genet. 2007, 8, 32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Mamun, H.A.; Kwan, P.; Clark, S.A.; Ferdosi, M.H.; Tellam, R.; Gondro, C. Genome-wide association study of body weight in Australian Merino sheep reveals an orthologous region on OAR6 to human and bovine genomic regions affecting height and weight. Genet. Sel. Evol. 2015, 47, 66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, S.; Casas, E.; Allan, M.; Keele, J.; Snelling, W.; Wheeler, T.; Shackelford, S.; Koohmaraie, M.; Smith, T. Evaluation in beef cattle of six deoxyribonucleic acid markers developed for dairy traits reveals an osteopontin polymorphism associated with postweaning growth. J. Anim. Sci. 2007, 85, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cánovas, A.; Reverter, A.; DeAtley, K.L.; Ashley, R.L.; Colgrave, M.L.; Fortes, M.R.; Islas-Trejo, A.; Lehnert, S.; Porto-Neto, L.; Rincón, G.; et al. Multi-tissue omics analyses reveal molecular regulatory networks for puberty in composite beef cattle. PLoS ONE 2014, 9, e102551. [Google Scholar] [CrossRef] [Green Version]
- Shin, S.; Chung, E. Novel SNPs in the bovine ADIPOQ and PPARGC1A genes are associated with carcass traits in Hanwoo (Korean cattle). Mol. Biol. Rep. 2013, 40, 4651–4660. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Liu, M.; Liu, D.; Lan, X.; Lei, C.; Chen, H. The novel coding region SNPs of PPARGC1A gene and their associations with growth traits in Chinese native cattle. Mol. Biol. Rep. 2014, 41, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Weikard, R.; Kuhn, C.; Goldammer, T.; Freyer, G.; Schwerin, M. The bovine PPARGC1A gene: Molecular characterization and association of an SNP with variation of milk fat synthesis. Physiol. Genom. 2005, 21, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Soria, L.; Corva, P.; Sica, A.B.; Villarreal, E.; Melucci, L.; Mezzadra, C.; Mazzucco, J.P.; Macedo, G.F.; Silvestro, C.; Schor, A.; et al. Association of a novel polymorphism in the bovine PPARGC1A gene with growth, slaughter and meat quality traits in Brangus steers. Mol. Cell. Probes 2009, 23, 304–308. [Google Scholar] [CrossRef]
- Snelling, W.; Allan, M.; Keele, J.; Kuehn, L.; Mcdaneld, T.; Smith, T.; Sonstegard, T.; Thallman, R.; Bennett, G. Genome-wide association study of growth in crossbred beef cattle. J. Anim. Sci. 2010, 88, 837–848. [Google Scholar] [CrossRef] [Green Version]
- Santana, M.; Utsunomiya, Y.; Neves, H.; Gomes, R.; Garcia, J.; Fukumasu, H.; Silva, S.; Leme, P.; Coutinho, L.; Eler, J.; et al. Genome-wide association study for feedlot average daily gain in Nellore cattle (Bos indicus). J. Anim. Breed. Genet. 2014, 131, 210–216. [Google Scholar] [CrossRef]
- Olsen, H.G.; Lien, S.; Gautier, M.; Nilsen, H.; Roseth, A.; Berg, P.R.; Sundsaasen, K.K.; Svendsen, M.; Meuwissen, T.H. Mapping of a milk production quantitative trait locus to a 420-kb region on bovine chromosome 6. Genetics 2005, 169, 275–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daetwyler, H.D.; Schenkel, F.S.; Sargolzaei, M.; Robinson, J.A.B. A genome scan to detect quantitative trait loci for economically important traits in Holstein cattle using two methods and a dense single nucleotide polymorphism map. J. Dairy Sci. 2008, 91, 3225–3236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, L.; Liu, J.; Sun, D.; Ma, P.; Ding, X.; Yu, Y.; Zhang, Q. Genome wide association studies for milk production traits in Chinese Holstein population. PLoS ONE 2010, 5, e13661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beenken, A.; Mohammadi, M. The FGF family: Biology, pathophysiology and therapy. Nat. Rev. Drug Discov. 2009, 8, 235–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Trait (Units) | Number of Animals with Record (and Genotype) | Mean (SE) | Min. | Max. | SD | CV% |
|---|---|---|---|---|---|---|
| BT (mm) | 5824 (1151) | 8.71 (0.05) | 1.00 | 30.00 | 3.71 | 42.61 |
| CW (kg) | 5824 (1151) | 343.96 (0.60) | 158.00 | 519.00 | 45.61 | 13.26 |
| EMA (cm2) | 5821 (1151) | 78.90 (0.12) | 40.00 | 123.00 | 9.12 | 11.56 |
| MS (score) | 3991 (1151) | 3.33 (0.03) | 1.00 | 9.00 | 1.61 | 48.46 |
| YW (kg) | 15,279 (1540) | 342.06 (0.38) | 133.86 | 535.90 | 47.48 | 13.88 |
| Trait a | Chr b | QTL Region (Mb) c | GV% d | Candidate Genes e |
|---|---|---|---|---|
| BT | 2 | 0.35–1.28 | 2.16 | LOC104971094, LOC107132231, LOC107132232, LOC784948, LGSN, OCA2, LOC783772, LOC100301143, HERC2, LOC104971093, TRNAE-CUC, NIPA1, NIPA2, LOC107132230, CYFIP1, TUBGCP5, TRNAE-UUC, CCDC115, IMP4, PTPN18 |
| BT | 2 | 72.46–73.83 | 1.11 | INHBB, LOC781979, LOC104971258, GLI2, LOC104971259, LOC100335292, TRNAL-CAA, TFCP2L1, CLASP1, NIFK, TSN |
| BT | 7 | 39.69–40.83 | 2.59 | HK3, UIMC1, LOC510252, LOC100141185, LOC101905975, LOC784341, LOC533921, LOC782447, TRNAR-CCU, ZNF346, FGFR4, LOC100336707, LOC540197, RAB24, PRELID1, MXD3, LMAN2, RGS14, SLC34A1, PFN3, F12, GRK6, PRR7, DBN1, PDLIM7, DOK3, LOC104969157, DDX41, FAM193B, LOC509184, LOC100139419, TMED9, B4GALT7, LOC107132625, N4BP3, RMND5B, LOC101905866, NHP2, HNRNPAB, PHYKPL, COL23A1 |
| BT | 11 | 60.34–61.55 | 1.69 | FAM161A, CCT4, COMMD1, B3GNT2, TMEM17, EHBP1, OTX1 |
| BT | 13 | 40.02–41.40 | 1.08 | CFAP61, INSM1, RALGAPA2, LOC104973781, LOC104973782, KIZ, LOC100140493, XRN2, NKX2-4, NKX2-2, LOC614124, PAX1 |
| BT | 16 | 34.80–35.91 | 1.08 | LOC100297170, PLD5, LOC104974409, LOC104974410, BECN2, MAP1LC3C, EXO1, WDR64, LOC101907876 |
| CW | 7 | 58.66–60.49 | 1.91 | PRELID2, LOC107132647, LOC100138092, LOC788619, GRXCR2, SH3RF2, PLAC8L1, LARS, RBM27, POU4F3, TCERG1, GPR151, PPP2R2B, TRNAC-GCA, STK32A |
| CW | 9 | 62.93–64.58 | 1.98 | TRNAE-UUC, LOC100336843, AKIRIN2, ORC3, RARS2, SLC35A1, CFAP206, C9H6orf163, SMIM8, LOC104969583, GJB7, LOC107132774, ZNF292, LOC509829, CGA, HTR1E |
| CW | 14 | 11.23–12.26 | 1.04 | ASAP1, FAM49B, GSDMC |
| CW | 14 | 16.55–17.80 | 4.46 | NSMCE2, KIAA0196, SQLE, ZNF572, MTSS1, NDUFB9, TATDN1, LOC104968469, RNF139, TRMT12, LOC531462, TMEM65, FER1L6, LOC101907615, FAM91A1, ANXA13 |
| CW | 14 | 17.85–19.46 | 2.00 | LOC100848930, FBXO32, WDYHV1, ATAD2, ZHX1, C14H8orf76, FAM83A, TRNAM-CAU, LOC104974006, TBC1D31, DERL1, ZHX2, LOC104974007, LOC100139328 |
| CW | 14 | 22.09–23.61 | 4.31 | SNTG1, LOC614437, PCMTD1, LOC101906226, LOC104974020, ST18, LOC100141260, LOC101906592, FAM150A, RB1CC1, LOC104974017, NPBWR1, OPRK1, ATP6V1H, RGS20 |
| CW | 14 | 24.58–25.33 | 17.66 | XKR4, TMEM68, TGS1, LYN, RPS20, MOS, PLAG1, CHCHD7, SDR16C5, SDR16C6, PENK, LOC101907667 |
| CW | 14 | 25.36–26.15 | 2.43 | LOC101907667, IMPAD1, FAM110B |
| CW | 14 | 29.43–30.44 | 3.75 | NKAIN3, LOC107133118, GGH, TTPA, YTHDF3, LOC101907975 |
| CW | 14 | 30.54–32.16 | 1.30 | MIR124A-2, BHLHE22, CYP7B1, LOC104974032, ARMC1, MTFR1, LOC104974034, PDE7A, LOC101902754, LOC100299601, LOC104974036, DNAJC5B, TRNAY-GUA, TRNAA-AGC, TRIM55 |
| CW | 14 | 32.25–33.90 | 1.43 | CRH, LOC790324, ZSCAN5B, RRS1, ADHFE1, C14H8orf46, MYBL1, VCPIP1, SGK3, LOC104974037, MCMDC2, LOC784087, LOC100847363, TCF24, PPP1R42, COPS5, CSPP1, ARFGEF1, TRNAC-GCA, CPA6, LOC101902584 |
| EMA | 1 | 127.77–128.74 | 2.27 | GK5, TFDP2, LOC101903974, LOC511302, ATP1B3, GRK7, RNF7, LOC104968752, RASA2, LOC100294923, ZBTB38, LOC107131348, LOC104971030, LOC104971031, PXYLP1, LOC104971032 |
| EMA | 6 | 0.1–1.03 | 1.00 | APELA, LOC101905490, LOC513842, LOC101907917 |
| EMA | 6 | 54.52–55.64 | 1.11 | NOT_FOUND |
| EMA | 9 | 57.18–58.10 | 2.22 | TRNAC-ACA, LOC782527, EPHA7 |
| EMA | 14 | 22.09–23.61 | 1.75 | SNTG1, LOC614437, PCMTD1, LOC101906226, LOC104974020, ST18, LOC100141260, LOC101906592, FAM150A, RB1CC1, LOC104974017, NPBWR1, OPRK1, ATP6V1H, RGS20 |
| EMA | 14 | 24.58–25.33 | 7.98 | XKR4, TMEM68, TGS1, LYN, RPS20, MOS, PLAG1, CHCHD7, SDR16C5, SDR16C6, PENK, LOC101907667 |
| EMA | 19 | 48.90–50.02 | 1.00 | LOC100140873, TEX2, TRNAG-UCC, LOC104975109, LOC101902037, PECAM1, MILR1, POLG2, DDX5, MIR3064, CEP95, SMURF2, TRNAE-CUC, KPNA2, TRNAR-CCG, C19H17orf58, BPTF, TRNAE-UUC, NOL11, TRNAS-AGA, PITPNC1, LOC101905668 |
| MS | 5 | 95.87–97.74 | 2.24 | ATF7IP, LOC100139060, GRIN2B, EMP1, GSG1, FAM234B, HEBP1, GPRC5D, GPRC5A, DDX47, APOLD1, CDKN1B, LOC101901926, GPR19, CREBL2, LOC101902028, LOC107132517, DUSP16 |
| MS | 14 | 5.01–5.69 | 1.00 | LOC100850800, COL22A1, FAM135B |
| MS | 23 | 30.27–31.28 | 2.03 | ZNF389, ZSCAN16, ZNF165, OR2B6, HIST1H2BB, HIST1H2AG, bta-mir-2379, ZNF184, ZNF391, POM121L2, PRSS16, HIST1H2BN, ZNF322, ABT1 |
| MS | 27 | 16.11–17.15 | 1.28 | LOC101905556, LOC101905700, ZFP42, TRNAG-UCC, TRIML2, TRNAF-AAA, TRIML1, LOC507011 |
| MS | 27 | 19.17–20.48 | 1.46 | MICU3, FGF20, LOC104976064, TRNAC-ACA, MSR1, LOC104976066, TUSC3 |
| YW | 2 | 42.77–43.75 | 1.71 | LOC785568, LOC615401, ARL6IP6, TRNAY-GUA, PRPF40A, FMNL2, LOC101902790 |
| YW | 6 | 37.26–38.45 | 1.36 | FAM13A, LOC104972724, LOC100847719, HERC3, NAP1L5, PYURF, PIGY, HERC5, HERC6, PPM1K, ABCG2, LOC781421, PKD2, SPP1, MEPE, IBSP, LOC104972726 |
| YW | 6 | 39.50–40.67 | 2.68 | LOC782905 |
| YW | 6 | 44.67–45.42 | 1.14 | PPARGC1A |
| YW | 6 | 48.80–49.97 | 2.30 | LOC107132565 |
| YW | 10 | 50.75–51.96 | 1.56 | LOC107132854, FAM81A, MYO1E, CCNB2, RNF111, SLTM, FAM63B, LOC533308, ADAM10, LIPC, LOC101904602, LOC101903685, TRNAE-UUC |
| YW | 14 | 16.55–17.80 | 1.70 | NSMCE2, KIAA0196, SQLE, ZNF572, MTSS1, NDUFB9, TATDN1, LOC104968469, RNF139, TRMT12, LOC531462, TMEM65, FER1L6, LOC101907615, FAM91A1, ANXA13 |
| YW | 14 | 22.09–23.61 | 2.54 | SNTG1, LOC614437, PCMTD1, LOC101906226, LOC104974020, ST18, LOC100141260, LOC101906592, FAM150A, RB1CC1, LOC104974017, NPBWR1, OPRK1, ATP6V1H, RGS20 |
| YW | 14 | 24.58–25.33 | 9.96 | XKR4, TMEM68, TGS1, LYN, RPS20, MOS, PLAG1, CHCHD7, SDR16C5, SDR16C6, PENK, LOC101907667 |
| YW | 14 | 25.36–26.15 | 1.74 | LOC101907667, IMPAD1, FAM110B |
| YW | 14 | 29.43–30.44 | 2.64 | NKAIN3, LOC107133118, GGH, TTPA, YTHDF3, LOC101907975 |
| YW | 14 | 30.54–32.16 | 2.62 | MIR124A-2, BHLHE22, CYP7B1, LOC104974032, ARMC1, MTFR1, LOC104974034, PDE7A, LOC101902754, LOC100299601, LOC104974036, DNAJC5B, TRNAY-GUA, TRNAA-AGC, TRIM55 |
| YW | 14 | 32.25–33.90 | 1.43 | CRH, LOC790324, ZSCAN5B, RRS1, ADHFE1, C14H8orf46, MYBL1, VCPIP1, SGK3, LOC104974037, MCMDC2, LOC784087, LOC100847363, TCF24, PPP1R42, COPS5, CSPP1, ARFGEF1, TRNAC-GCA, CPA6, LOC101902584 |
| Traits a | Term ID | Term Name | Genes | p-Value |
|---|---|---|---|---|
| BT | GO:0006869 | lipid transport | PRELID1 | 0.010233961 |
| GO:0010876 | lipid localization | PRELID1 | 0.014172362 | |
| GO:0005319 | lipid transporter activity | PRELID1 | 0.019846208 | |
| GO:0006629 | lipid metabolic process | FGFR4 | 0.025006995 | |
| GO:0008289 | lipid binding | COMMD1, PFN3 | 0.047311441 | |
| GO:0008610 | lipid biosynthetic process | FGFR4 | 0.035303187 | |
| KEGG:04015 | Rap1 signaling pathway | RGS14 | 0.047961872 | |
| CW | GO:0001558 | regulation of cell growth | SGK3 | 0.044446017 |
| GO:0007173 | epidermal growth factor receptor signaling pathway | FAM83A | 0.039752933 | |
| GO:0016049 | cell growth | POU4F3 | 0.035059849 | |
| GO:0035264 | multicellular organism growth | PLAG1 | 0.030366765 | |
| GO:0035265 | organ growth | PLAG1, CGA | 0.025673681 | |
| GO:0040007 | growth | PLAG1, POU4F3, CGA, SGK3 | 0.020980597 | |
| GO:0048589 | developmental growth | PLAG1, POU4F3, CGA | 0.016287514 | |
| GO:0060560 | developmental growth involved in morphogenesis | POU4F3 | 0.01159443 | |
| GO:0005515 | protein binding | ARFGEF1, ATAD2, LYN, MYBL1, RB1CC1, AKIRIN2, CRH, CGA, NPBWR1, PENK, PPP1R42, RGS20, TCERG1, ZHX1, ZHX2 | 0.006901346 | |
| KEGG:04080 | Neuroactive ligand-receptor interaction | CGA, HTR1E, NPBWR1, OPRK1, PENK, CRH | 0.039862933 | |
| EMA | GO:0006629 | lipid metabolic process | LYN, SDR16C5 | 0.049805624 |
| GO:0019216 | regulation of lipid metabolic process | LYN | 0.042803519 | |
| GO:0033993 | response to lipid | DDX5 | 0.043747312 | |
| GO:0008289 | lipid binding | TEX2 | 0.049508111 | |
| KEGG:04514 | Cell adhesion molecules | LOC100140873, PECAM1 | 0.04067771 | |
| REAC:R-BTA-210990 | PECAM1 interactions | LYN, PECAM1 | 0.049805624 | |
| MS | GO:0006629 | lipid metabolic process | CREBL2 | 0.01889403 |
| GO:0006869 | lipid transport | APOLD1, MSR1 | 0.041826385 | |
| GO:0008610 | lipid biosynthetic process | CREBL2 | 0.021157549 | |
| GO:0010876 | lipid localization | APOLD1 | 0.016791777 | |
| GO:0019915 | lipid storage | MSR1 | 0.025742261 | |
| GO:0045444 | fat cell differentiation | CREBL2 | 0.019486141 | |
| GO:0008289 | lipid transport | APOLD1 | 0.017945142 | |
| GO:0071814 | protein-lipid complex binding | MSR1 | 0.016404143 | |
| KEGG:04010 | MAPK signaling pathway | DUSP16, FGF20 | 0.014863145 | |
| REAC:R-BTA-5673001 | RAF/MAP kinase cascade | DUSP16, FGF20, GRIN2B | 0.016791777 | |
| YW | GO:0001558 | regulation of cell growth | ADAM10, SGK3 | 0.046130564 |
| GO:0016049 | cell growth | ADAM10 | 0.043928692 | |
| GO:0035264 | multicellular organism growth | PLAG1 | 0.04172682 | |
| GO:0035265 | organ growth | PLAG1 | 0.039524949 | |
| GO:0040007 | growth | ADAM10, PLAG1, SGK3 | 0.037323077 | |
| GO:0040008 | regulation of growth | ADAM10, SGK3, MYO1E | 0.035121205 | |
| GO:0048589 | developmental growth | PLAG1 | 0.032919333 | |
| GO:0070848 | response to growth factor | IBSP, RNF111 | 0.030717462 | |
| GO:0071363 | cellular response to growth factor stimulus | RNF111 | 0.02851559 | |
| GO:0005515 | protein binding | ADAM10, ARFGEF1, LYN, MYBL1, PPARGC1A, RB1CC1, CRH, MEPE, NPBWR1, PKD2, PENK, PPP1R24, RGS20, SPP1 | 0.026313718 | |
| GO:0140096 | catalytic activity, acting on a protein | ADAM10, CCNB2, COPS5, CPA6, GGH, HERC3, HERC5, HERC6, LYN, MOS, PCMTD1, PPM1K, RNF111, RNF139, SGK3, VCPIP1, NSMCE2 | 0.048130564 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Naserkheil, M.; Bahrami, A.; Lee, D.; Mehrban, H. Integrating Single-Step GWAS and Bipartite Networks Reconstruction Provides Novel Insights into Yearling Weight and Carcass Traits in Hanwoo Beef Cattle. Animals 2020, 10, 1836. https://doi.org/10.3390/ani10101836
Naserkheil M, Bahrami A, Lee D, Mehrban H. Integrating Single-Step GWAS and Bipartite Networks Reconstruction Provides Novel Insights into Yearling Weight and Carcass Traits in Hanwoo Beef Cattle. Animals. 2020; 10(10):1836. https://doi.org/10.3390/ani10101836
Chicago/Turabian StyleNaserkheil, Masoumeh, Abolfazl Bahrami, Deukhwan Lee, and Hossein Mehrban. 2020. "Integrating Single-Step GWAS and Bipartite Networks Reconstruction Provides Novel Insights into Yearling Weight and Carcass Traits in Hanwoo Beef Cattle" Animals 10, no. 10: 1836. https://doi.org/10.3390/ani10101836