Fast and Accurate Pneumocystis Pneumonia Diagnosis in Human Samples Using a Label-Free Plasmonic Biosensor

 , , ,
, , ,  and
and
Abstract
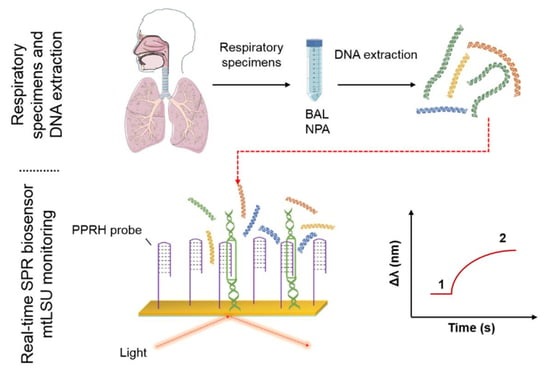
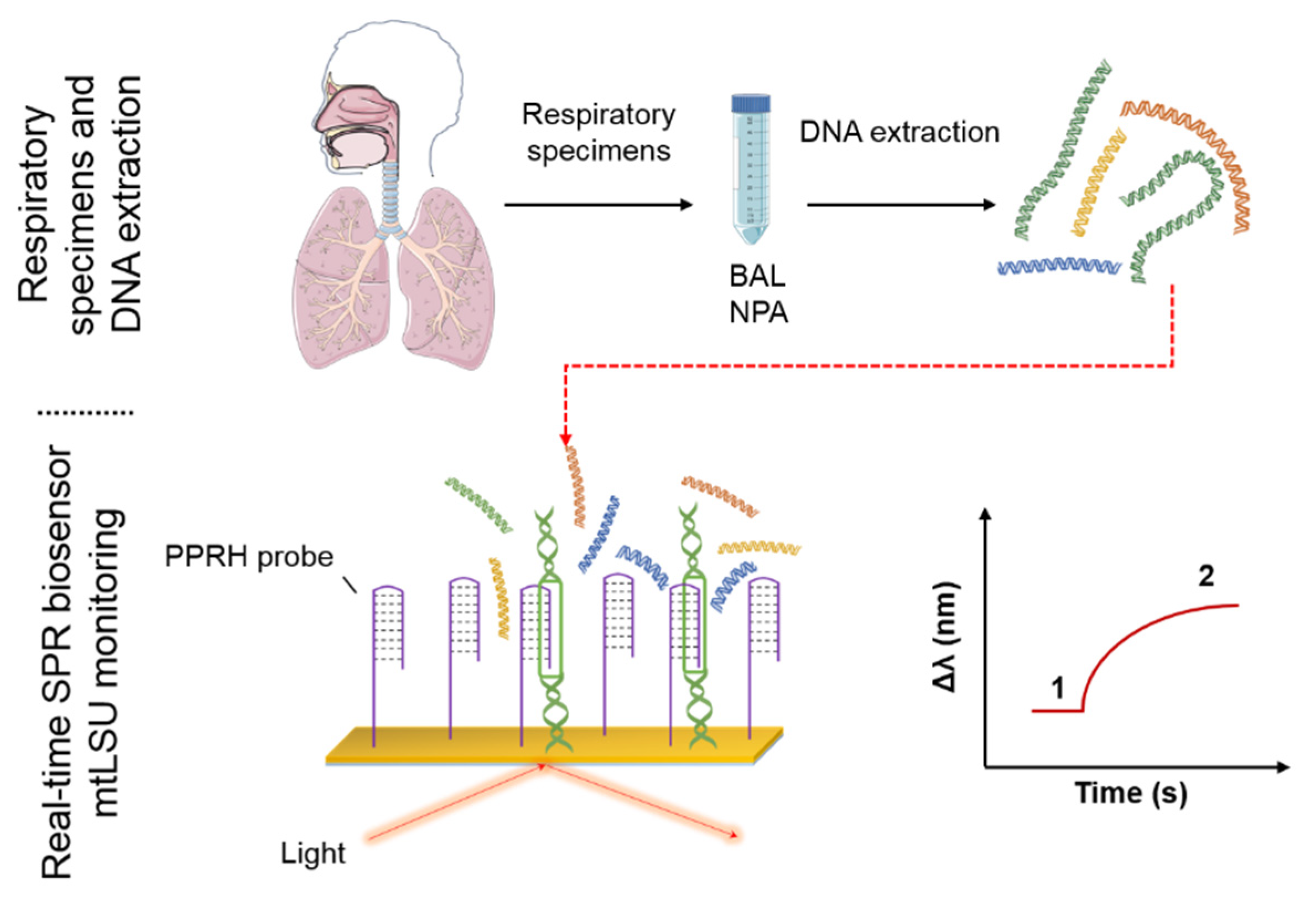
:
1. Introduction
2. Materials and Methods
2.1. Synthesis of the Oligonucleotides
2.2. Preparation of the Samples
2.3. Experimental Procedure for Detection Using the SPR Biosensor
2.3.1. Chemicals
2.3.2. SPR Biosensor Device
2.3.3. SPR Sensor Chips Fabrication and Cleaning
2.3.4. Bioreceptor Immobilization
2.3.5. Data Analysis
3. Results and Discussion
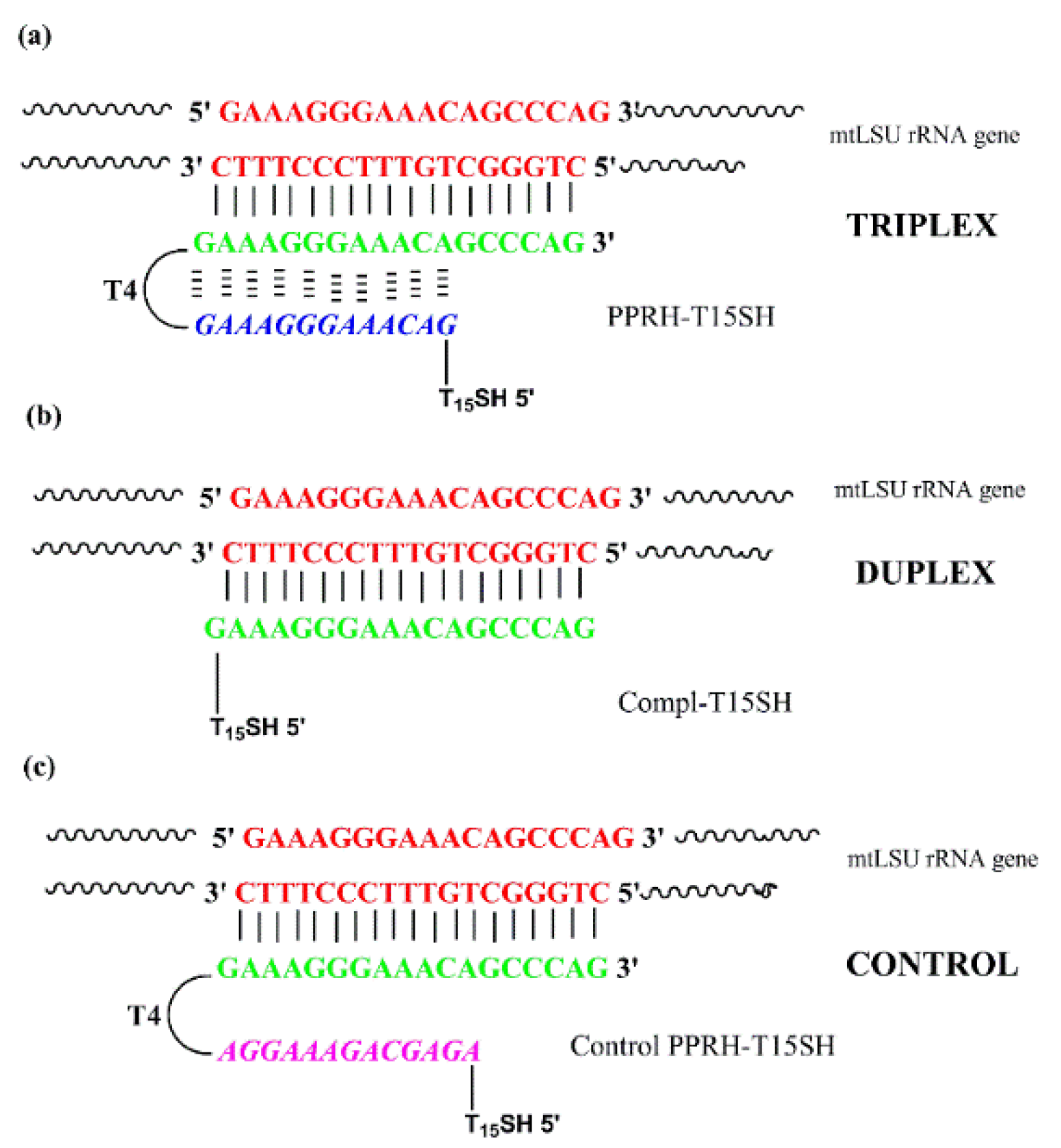
3.1. PPRH Probe Design
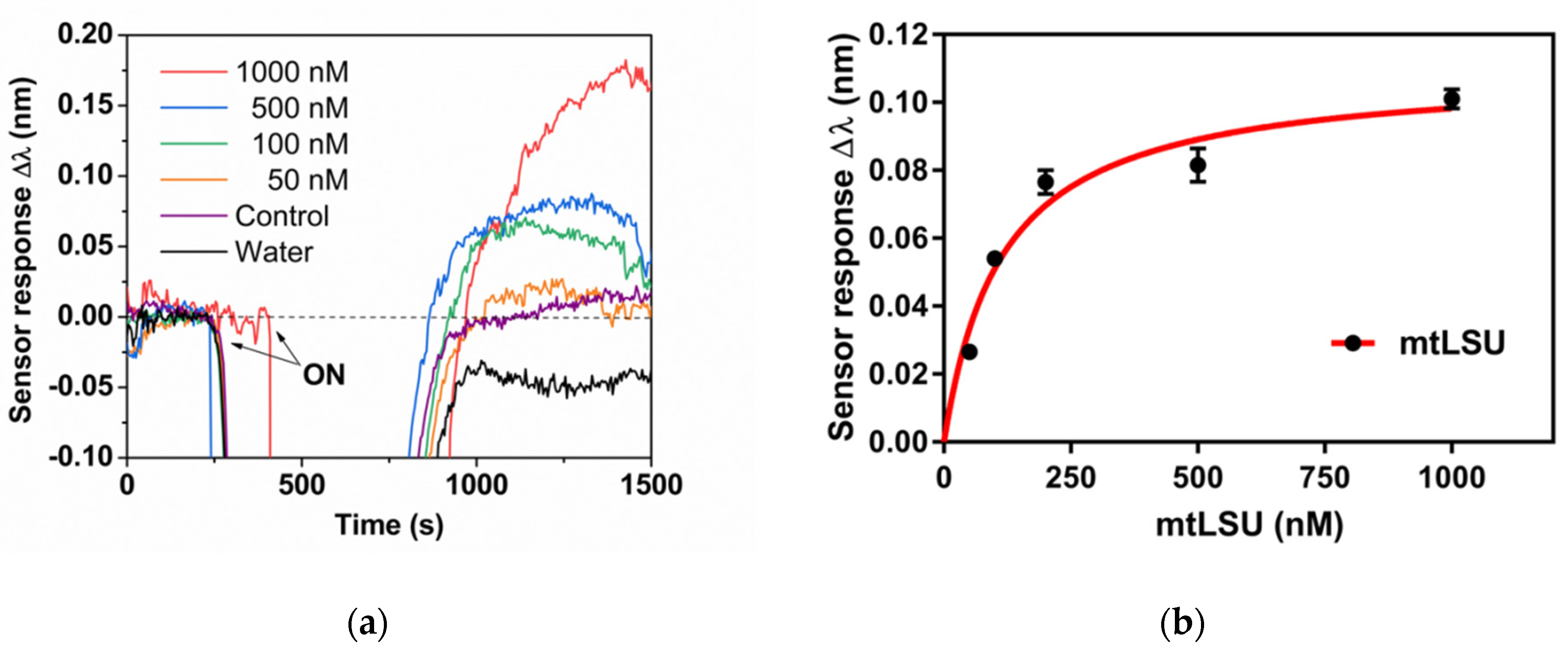
3.2. Biosensor for the Diagnosis of PcP
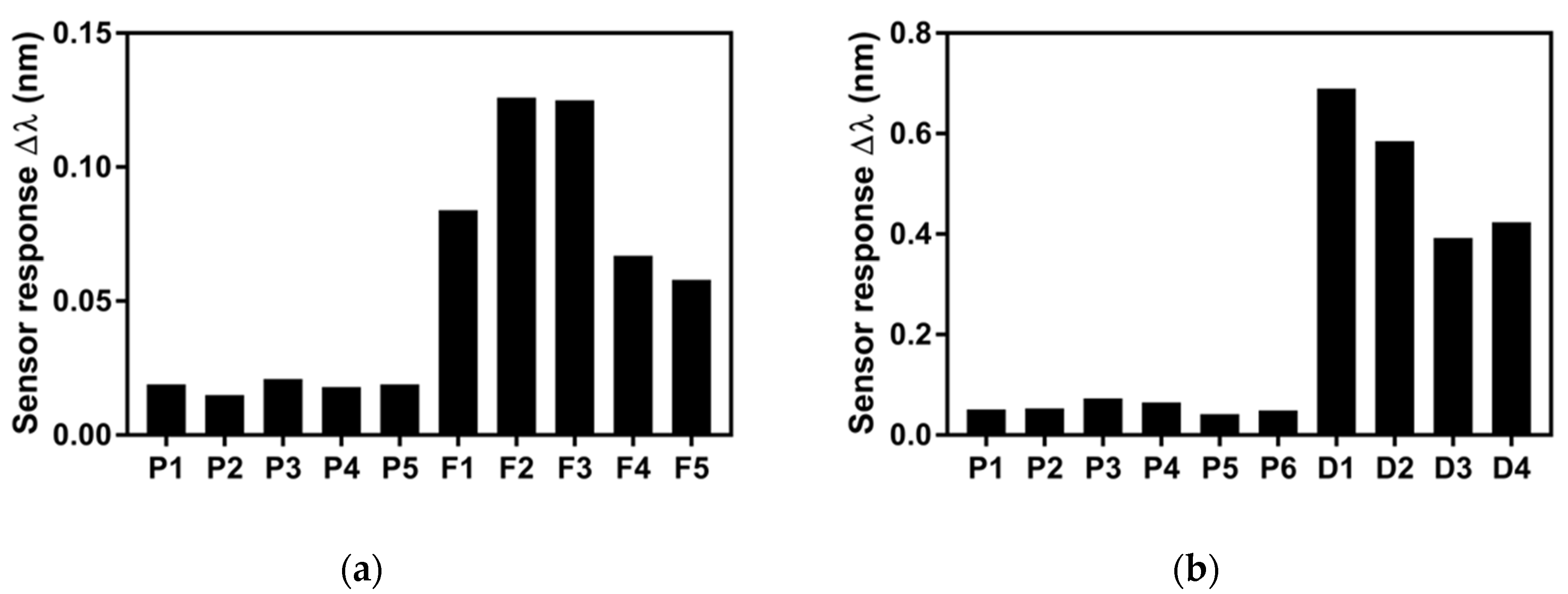
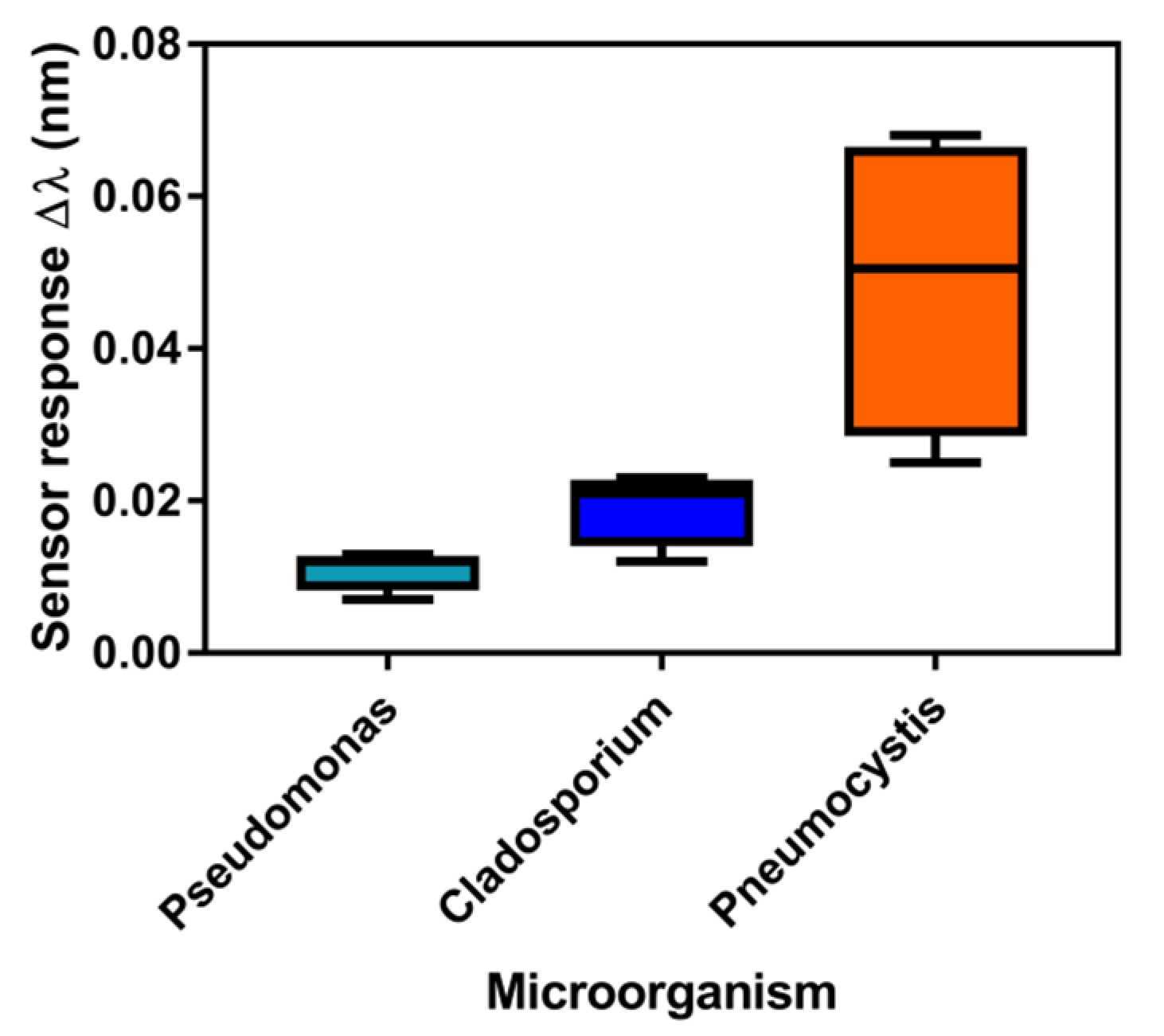
3.3. Analysis of mtLSU rRNA in Patients’ Samples
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A
Chemicals
Appendix B
Portable Custom-Made SPR Biosensor

Appendix C
SPR Sensor Chips Fabrication and Cleaning
Appendix D
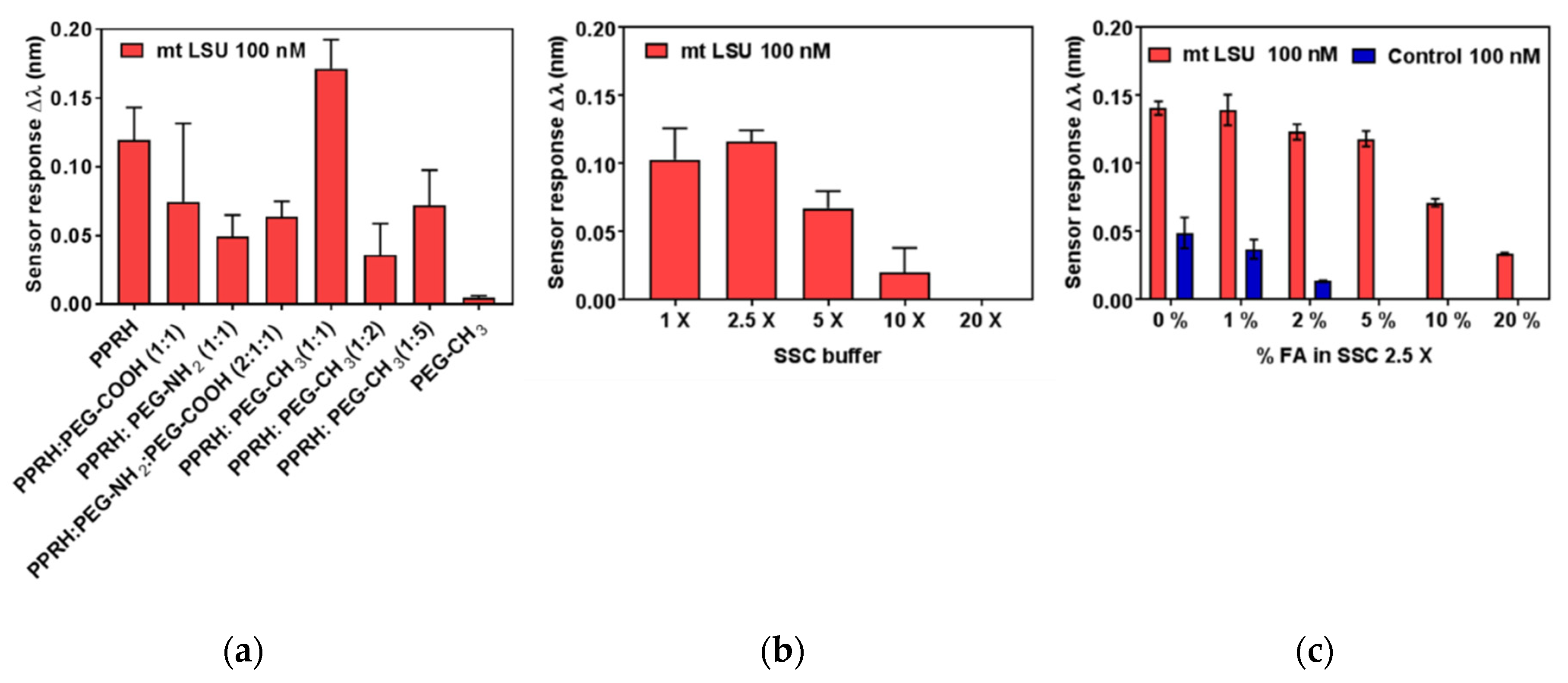
Bioassay Optimization

Appendix E
Data Analysis
Appendix F
Circular Dichroism (CD) Spectrometry

Appendix G
Reproducibility and Accuracy

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Concentration (nM) | Intra-Chip | |||||
|---|---|---|---|---|---|---|
| PPRH Probe | Control PPRH Probe | Complementary Probe | ||||
| Mean ± SD (n = 3) | % CV | Mean ± SD (n = 3) | % CV | Mean ± SD (n = 3) | % CV | |
| 100 | 0.126 ± 0.021 | 16 | 0.084 ± 0.010 | 12 | 0.095 ± 0.001 | 1 |
| 50 | 0.077 ± 0.014 | 18 | 0.050 ± 0.006 | 12 | 0.062 ± 0.010 | 11 |
| 25 | 0.038 ± 0.007 | 18 | 0.039 ± 0.002 | 5 | 0.048 ± 0.001 | 2 |
| 10 | 0.026 ± 0.002 | 8 | 0.025 ± 0.003 | 12 | 0.031 ± 0.01 | 36 |
| LOD | 2.852 ± 0.460 | 16 | 4.217 ± 0.583 | 14 | 3.139 ± 0.239 | 8 |
| Inter-chip | ||||||
| 100 | 0.120 ± 0.007 | 6 | 0.095 ± 0.023 | 24 | 0.097 ± 0.004 | 4 |
| 50 | 0.081 ± 0.005 | 6 | 0.070 ± 0.0003 | 0.5 | 0.061 ± 0.002 | 3 |
| 25 | 0.045 ± 0.013 | 28 | 0.024 ± 0.001 | 4 | 0.036 ± 0.016 | 44 |
| 10 | 0.027 ± 0.006 | 22 | 0.015 ± 0.005 | 33 | 0.025 ± 0.007 | 28 |
| LOD | 1.945 ± 0.270 | 14 | 3.581 ± 1.320 | 37 | 3.360 ± 0.313 | 9 |
| Concentration (nM) | Intra-Chip | Inter-Chip | ||||
|---|---|---|---|---|---|---|
| Mean ± SD (n = 3) | % CV | Mean ± SD | % CV | Mean ± SD (n = 3) | % CV | |
| 1000 | 0.101 ± 0.0028 | 3 | 0.120 ± 0.0021 | 2 | 0.111 ± 0.0134 | 12 |
| 500 | 0.081 ± 0.0049 | 6 | 0.085 ± 0.0049 | 6 | 0.083 ± 0.0028 | 3 |
| 200 | 0.076 ± 0.0035 | 5 | 0.070 ± 0.0092 | 13 | 0.073 ± 0.0042 | 6 |
| 100 | 0.054 ± 0.0014 | 3 | 0.046 ± 0.0042 | 9 | 0.050 ± 0.006 | 11 |
| LOD | 10.17 ± 0.6901 | 7 | 13.90 ± 0.6951 | 5 | 12.036 ± 2.642 | 22 |
| Sample | Sensor Response Δλ (nm) | Estimated Concentration (ng/µL) | PCR (ng/µL) |
|---|---|---|---|
| F1 | 0.084 | 2.773 | 5 ng/µL |
| F2 | 0.127 | 4.193 | 10 ng/µL |
| F3 | 0.125 | 4.127 | 10 ng/µL |
| F4 | 0.067 | 2.212 | 5 ng/µL |
| F5 | 0.058 | 1.915 | 5 ng/µL |
| P1 | 0.019 | 0.627 | 3.69 ng/µL |
| P2 | 0.015 | 0.485 | 3.69 ng/µL |
| P3 | 0.021 | 0.693 | 3.69 ng/µL |
| P4 | 0.018 | 0.594 | 3.69 ng/µL |
| P5 | 0.019 | 0.627 |
References
- Pereira-Díaz, E.; Moreno-Verdejo, F.; de la Horra, C.; Guerrero, J.A.; Calderón, E.J.; Medrano, F.J. Changing Trends in the Epidemiology and Risk Factors of Pneumocystis Pneumonia in Spain. Front. Public Health 2019, 7, 275. [Google Scholar] [CrossRef] [PubMed]
- Morris, A.; Norris, K.A. Colonization by pneumocystis jirovecii and its role in disease. Clin. Microbiol. Rev. 2012, 25, 297–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.W.; Lin, C.C.; Kuo, C.F.; Liu, C.P.; Lee, C.M. Mortality predictors of Pneumocystis jirovecii pneumonia in human immunodeficiency virus-infected patients at presentation: Experience in a tertiary care hospital of northern Taiwan. J. Microbiol. Immunol. Infect. 2011, 44, 274–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calderón, E.J. Pneumocystis Infection: Seeing beyond the Tip of the Iceberg. Clin. Infect. Dis. 2010, 50, 354–356. [Google Scholar] [CrossRef]
- Tasaka, S. Recent Advances in the Diagnosis and Management of Pneumocystis Pneumonia. Tuberc. Respir. Dis. 2020, 83, 132–140. [Google Scholar] [CrossRef] [Green Version]
- Procop, G.W.; Haddad, S.; Quinn, J.; Wilson, M.L.; Henshaw, N.G.; Reller, L.B.; Artymyshyn, R.L.; Katanik, M.T.; Weinstein, M.P. Detection of pneumocystis jiroveci in respiratory specimens by four staining methods. J. Clin. Microbiol. 2004, 42, 3333–3335. [Google Scholar] [CrossRef] [Green Version]
- Durand-Joly, I.; Chabé, M.; Soula, F.; Delhaes, L.; Camus, D.; Dei-Cas, E. Molecular diagnosis of Pneumocystis pneumonia. FEMS Immunol. Med. Microbiol. 2005, 45, 405–410. [Google Scholar] [CrossRef] [Green Version]
- Respaldiza, N.; Montes-Cano, M.A.; Friaza, V.; Muñoz-Lobato, F.; Medrano, F.J.; Varela, J.M.; Calderón, E.J.; de la hora, C.N. Usefulness of oropharyngeal washings for identifying Pneumocystis jirovecii carriers. J. Eukaryot. Microbiol. 2006, 53, S100–S101. [Google Scholar] [CrossRef]
- Robberts, F.J.L.; Liebowitz, L.D.; Chalkley, L.J. Polymerase chain reaction detection of Pneumocystis jiroveci: Evaluation of 9 assays. Diagn. Microbiol. Infect. Dis. 2007, 58, 385–392. [Google Scholar] [CrossRef]
- Wakefield, A.E.; Pixley, F.J.; Banerji, S.; Sinclair, K.; Miller, R.F.; Moxon, E.R.; Hopkin, J.M. Amplification of mitochondrial ribosomal RNA sequences from Pneumocystis carinii DNA of rat and human origin. Mol. Biochem. Parasitol. 1990, 43, 69–76. [Google Scholar] [CrossRef]
- Brancart, F.; Rodriguez-Villalobosa, H.; Fonteynec, P.A.; Peres-Botab, D.; Liesnard, C. Quantitative TaqMan PCR for detection of Pneumocystis jiroveci. J. Microbiol. Meth. 2005, 61, 381–387. [Google Scholar] [CrossRef]
- White, P.L.; Backx, M.; Barnes, R.A. Diagnosis and management of Pneumocystis jirovecii infection. Expert Rev. Anti Infect. Ther. 2017, 15, 435–447. [Google Scholar] [CrossRef] [PubMed]
- Masson, J.F. Surface plasmon resonance clinical biosensors for medical diagnostics. ACS Sens. 2017, 2, 16–30. [Google Scholar] [CrossRef]
- Soler, M.; Huertas, C.S.; Lechuga, L.M. Label-free plasmonic biosensors for point-of-care diagnostics: A review. Expert Rev. Mol. Diagn. 2019, 19, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Šípová, H.; Homola, J. Surface plasmon resonance sensing of nucleic acids: A review. Anal. Chim. Acta. 2013, 773, 9–23. [Google Scholar] [CrossRef] [PubMed]
- Huertas, C.S.; Calvo-Lozano, O.; Mitchell, A.; Lechuga, L.M. Advanced evanescent-wave optical biosensors for the detection of nucleic acids: An analytic perspective. Front. Chem. 2019, 7, 724. [Google Scholar] [CrossRef] [Green Version]
- Aviñó, A.; Jorge, A.F.; Huertas, C.S.; Cova, T.F.; Pais, A.; Lechuga, L.M.; Eritja, R.; Fabrega, C. Aptamer-peptide conjugates as a new strategy to modulate human α-thrombin binding affinity. Biochim. Biophys. Acta. Gen. Subj. 2019, 1863, 1619–1630. [Google Scholar]
- Wang, S.; Dong, Y.; Liang, X. Development of a SPR aptasensor containing oriented aptamer for direct capture and detection of tetracycline in multiple honey samples. Biosens. Bioelectron. 2018, 109, 1–7. [Google Scholar] [CrossRef]
- Hu, Y.; Cecconello, A.; Idili, A.; Ricci, F.; Willner, I. Triplex DNA nanostructures: From basic properties to applications. Angew. Chem. Int. Ed. 2017, 56, 15210–15233. [Google Scholar] [CrossRef]
- Aviñó, A.; Eritja, R.; Ciudad, C.J.; Noé, V. Parallel Clamps and Polypurine Hairpins (PPRH) for Gene Silencing and Triplex-Affinity Capture: Design, Synthesis, and Use. Curr. Protoc. Nucleic Acid Chem. 2019, 77, e78. [Google Scholar] [CrossRef]
- Rodríguez, L.; Villalobos, X.; Solé, A.; Lliberos, C.; Ciudad, C.J.; Noe, V. Improved design of PPRHs for gene silencing. Mol. Pharm. 2015, 12, 867–877. [Google Scholar] [CrossRef] [PubMed]
- Aviñó, A.; Huertas, C.S.; Lechuga, L.M.; Eritja, R. Sensitive and label-free detection of miRNA-145 by triplex formation. Anal. Bioanal. Chem. 2016, 408, 885–893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nadal, A.; Coll, A.; Aviñó, A.; Esteve, T.; Eritja, R.; Pla, M. Efficient Sequence-Specific Purification of Listeria innocua mRNA Species by Triplex Affinity Capture with Parallel Tail-Clamps. ChemBioChem 2006, 7, 1039–1047. [Google Scholar] [CrossRef] [PubMed]
- Carrascosa, L.G.; Gómez-Montes, S.; Aviñó, A.; Nadal, A.; Pla, M.; Eritja, R.; Lechuga, L.M. Sensitive and label-free biosensing of RNA with predicted secondary structures by a triplex affinity capture method. Nucleic Acids Res. 2012, 40, e56. [Google Scholar] [CrossRef]
- Huertas, C.S.; Aviñó, A.; Kurachi, C.; Piqué, A.; Sandoval, J.; Eritja, R.; Esteller, M.; Lechuga, L.M. Label-free DNA-methylation detection by direct ds-DNA fragment screening using poly-purine hairpins. Biosens. Bioelectron. 2018, 120, 47–54. [Google Scholar] [CrossRef]
- Frank-Kamenetskii, M.D.; Mirkin, S.M. Triplex DNA structures. Annu. Rev. Biochem. 1995, 64, 65–95. [Google Scholar] [CrossRef]
- Merino-Casallo, I.; Friaza, V.; Menao, S.; Domingo, J.M.; Olivera, S.; Calderón, E.J.; Torralba, M.Á. Pneumocystis jirovecii in Spanish Patients With Heart Failure. Front. Public Health 2019, 7, 289. [Google Scholar] [CrossRef]
- Schoch, C.L.; Seifert, K.A.; Huhndorf, S.; Robert, V.; Spouge, J.L.; Levesque, C.A.; Chen, W. Fungal Barcoding Consortium. Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for Fungi. Proc. Natl. Acad. Sci. USA 2012, 109, 6241–6246. [Google Scholar] [CrossRef] [Green Version]
- Deschaght, P.; De Baere, T.; Van Simaey, L.; De Baets, F.; De Vos, D.; Pirnay, J.P.; Vaneechoutte, M. Comparison of the sensitivity of culture, PCR and quantitative real-time PCR for the detection of Pseudomonas aeruginosain sputum of cystic fibrosis patients. BMC Microbiol. 2009, 9, 244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The Triplex-Forming Oligonucleotide Target Sequence Search Home Page. Available online: http://utw10685.utweb.utexas.edu/tfo/about.php (accessed on 25 June 2020).
- Buick, A.R.; Doig, M.V.; Jeal, S.C.; Land, G.S.; McDowall, R.D. Method validation in the bioanalytical laboratory. J. Pharm. Biomed. Anal. 1990, 8, 629–637. [Google Scholar] [CrossRef]







| Target | Sequence | Length | GC Content (%) |
|---|---|---|---|
| mtLSU | 5′-CTGGGCTGTTTCCCTTTC-3′ | 18 | 55.55 |
| Control | 5′-TTCCGTGGCTGTTCTCCT-3′ | 18 | 55.55 |
| PPRH Probe | Control PPRH Probe | Complementary Probe | |
|---|---|---|---|
| Kd (nm) | 44.06 | 78.76 | 43.36 |
| Bmax (nm/nM) | 0.1593 | 0.1473 | 0.1306 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Calvo-Lozano, O.; Aviñó, A.; Friaza, V.; Medina-Escuela, A.; S. Huertas, C.; Calderón, E.J.; Eritja, R.; Lechuga, L.M. Fast and Accurate Pneumocystis Pneumonia Diagnosis in Human Samples Using a Label-Free Plasmonic Biosensor. Nanomaterials 2020, 10, 1246. https://doi.org/10.3390/nano10061246
Calvo-Lozano O, Aviñó A, Friaza V, Medina-Escuela A, S. Huertas C, Calderón EJ, Eritja R, Lechuga LM. Fast and Accurate Pneumocystis Pneumonia Diagnosis in Human Samples Using a Label-Free Plasmonic Biosensor. Nanomaterials. 2020; 10(6):1246. https://doi.org/10.3390/nano10061246
Chicago/Turabian StyleCalvo-Lozano, Olalla, Anna Aviñó, Vicente Friaza, Alfonso Medina-Escuela, César S. Huertas, Enrique J. Calderón, Ramón Eritja, and Laura M. Lechuga. 2020. "Fast and Accurate Pneumocystis Pneumonia Diagnosis in Human Samples Using a Label-Free Plasmonic Biosensor" Nanomaterials 10, no. 6: 1246. https://doi.org/10.3390/nano10061246