
5-(4-Nitrophenyl)furan-2-carboxylic Acid
by
 , , , and
, , , and
Matteo Mori
1,
Andrea Tresoldi
1,
Stefania Villa
1,
Giulia Cazzaniga
1,
Marco Bellinzoni
2 and
Fiorella Meneghetti
1,* 1
Department of Pharmaceutical Sciences, University of Milan, Via L. Mangiagalli 25, 20133 Milano, Italy
2
Unité de Microbiologie Structurale, Institut Pasteur, CNRS, Université de Paris, F-75015 Paris, France
*
Author to whom correspondence should be addressed.
Molbank 2022, 2022(4), M1515; https://doi.org/10.3390/M1515
Submission received: 11 November 2022
/
Revised: 22 November 2022
/
Accepted: 28 November 2022
/
Published: 2 December 2022
(This article belongs to the Section Structure Determination)
Abstract
:The ever-evolving research in the field of antitubercular agents has led to the identification of several new potential drug classes. Among them, 5-phenyl-furan-2-carboxylic acids have emerged as innovative potential therapeutics, targeting iron acquisition in mycobacterial species. In our efforts to characterize the molecular interactions between these compounds and their protein target (MbtI from M. tuberculosis) by means of co-crystallization experiments, we unexpectedly obtained the structure of 5-(4-nitrophenyl)furan-2-carboxylic acid (1). Herein, we describe the preparation of the compound and its analysis by 1H NMR, 13C NMR, HRMS, and SC-XRD.
Keywords:
furan; SC-XRD; synchrotron; Mycobacterium tuberculosis; antitubercular agent; iron acquisition; MbtI1. Introduction
Tuberculosis (TB) is an infectious disease caused by Mycobacterium tuberculosis (Mtb), an airborne pathogen that primarily affects the lungs. In 2020, TB was the second leading cause of death from a single infectious agent, after COVID-19 [1]. The socio-economic repercussions of the pandemic dramatically impacted the diagnosis and treatment of TB, resulting in an increase of the number of deaths after over a decade [1]. This uncontrolled spread of the disease also facilitated the emergence and diffusion of new resistant mycobacterial strains [1]. Therefore, advances in the development of novel therapeutic options are still critical to face the impending threat of TB. In this context, an innovative approach would be to design anti-virulence strategies targeting specific molecular pathways required for the establishment of the infection, although otherwise dispensable for the survival of the pathogen. Over the last decades, a considerable number of compounds, either derived from natural sources or obtained by chemical synthesis, have been investigated as new, potential anti-TB candidates [2,3,4].
Our research group has focused its attention on targeting the siderophore-mediated acquisition of iron in Mtb [5]. In detail, we identified a new class of furan-based inhibitors of the salicylate synthase MbtI [6], a Mg2+-dependent enzyme that converts chorismate to salicylate, which then acts as the building block for the biosynthesis of all siderophores. [7,8].
The furan scaffold is particularly common in antitubercular drugs. In detail, several 2,5-substituted furans have been identified as inhibitors of a variety of mycobacterial enzymes, including N-acetyl glutamate synthase (ArgA) [9], GlcNAc-Ins deacetylase (MshB) [10], lysine-ɛ-aminotransferase (LAT) [11], protein tyrosine phosphatase A (MptpA) [12,13], and thioredoxin reductases (TrxR) [14]. In this context, 5-substituted-2-nitrofurans deserve a particular mention because they have been widely investigated, despite, in some cases, their molecular target remaining unknown [15,16,17]. Interestingly, some of them also showed promising activities on other bacterial species, including Neisseria gonorrhoeae, Streptococcus pyogenes, and Staphylococcus aureus [18], and protozoan parasites, like Plasmodium falciparum [19].
Over the years, we optimized the scaffold of our furan-based compounds, identifying promising candidates active on mycobacterial models [20,21,22,23]. A key part of our investigations was the structural study of protein-ligand complexes, which led to the definition of the main interactions established by our compounds in the active site of the enzyme. During one of our data acquisition sessions at the European Synchrotron Radiation Facility (ESRF) in Grenoble (France), we ran across an unexpected diffraction pattern, which was incompatible with that of a protein or a simple inorganic salt. The analysis of the structure later revealed the crystal to be the organic ligand, namely 5-(4-nitrophenyl)furan-2-carboxylic acid (1). Despite being accidental and unsought, the obtainment of this structure was deemed a valuable result, especially considering that 5-phenyl-furan-2-carboxylic acids are intrinsically impervious to crystallization. This feature, which is confirmed by the absence of reported crystal structures for these compounds, is probably due to their limited solubility in organic and aqueous solvents, which results in their rapid precipitation and prevents the organization of the molecules in an ordered fashion to form a crystal lattice. Our previous results suggested that this issue could be partially overcome by derivatizing the compounds to form methyl esters. However, this strategy prevented the study of the molecules in their original form, essentially depriving them of the characteristic acidic function, which imparts unique features in terms of available hydrogen-bonding sites. Therefore, this apparently trivial crystal structure proved to be very important for the study of the intermolecular forces and interactions of this class of compounds.
2. Results and Discussion
2.1. Chemistry
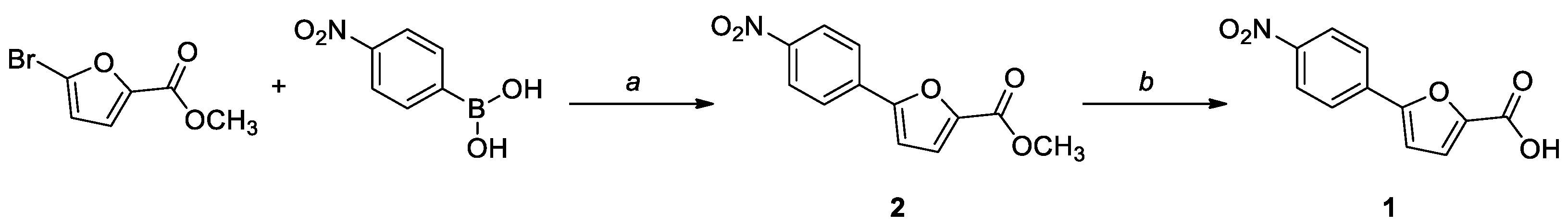
Compound 1 was prepared by an established procedure, involving a Suzuki coupling between methyl 5-bromofuran-2-carboxylate and (4-nitrophenyl)boronic acid, followed by a hydrolysis of the ester moiety in basic conditions (Scheme 1). The intermediate was characterized by 1H NMR and 13C NMR, while for the final compound, the HRMS spectrum was also acquired. The analytical data were consistent with the literature.
2.2. Crystallization and Structure Determination
Compound 1 crystallized in the orthorhombic space group P212121; the asymmetric unit contained two crystallographically independent molecules of 1, along with two waters, and an ammonium ion. The ORTEP [24] diagram indicating the arbitrary atom-numbering scheme used in the discussion is reported in Figure 1.
The two molecules are almost completely planar; the angle between the best mean planes calculated for the furan and the phenyl ring is 4.1(1)° for the main molecule, and 6.1(1)° for molecule A. The torsion angles C4-C5-C6-C7 and C8-C9-N1-O4 are 3.7(4)° and 13.5(3)°, respectively, while the corresponding angles in molecule A are 5.6(4)° and 9.6(3)°. The overlay of the two molecules produced an RMSD of 0.1018 and a maximum distance between two equivalent atoms of 0.22(3) Å. All bond lengths, valence angles, and torsional angles are within the expected limits, as shown by CSD Mogul [25]. In detail, bond length values indicate an electron delocalization extending over the entire molecule. Interestingly, while molecule A has a protonated carboxylic function, the main molecule features a carboxylate anion. The significance of this discrepancy and its effects on the molecular arrangement in space are discussed in the next paragraph.
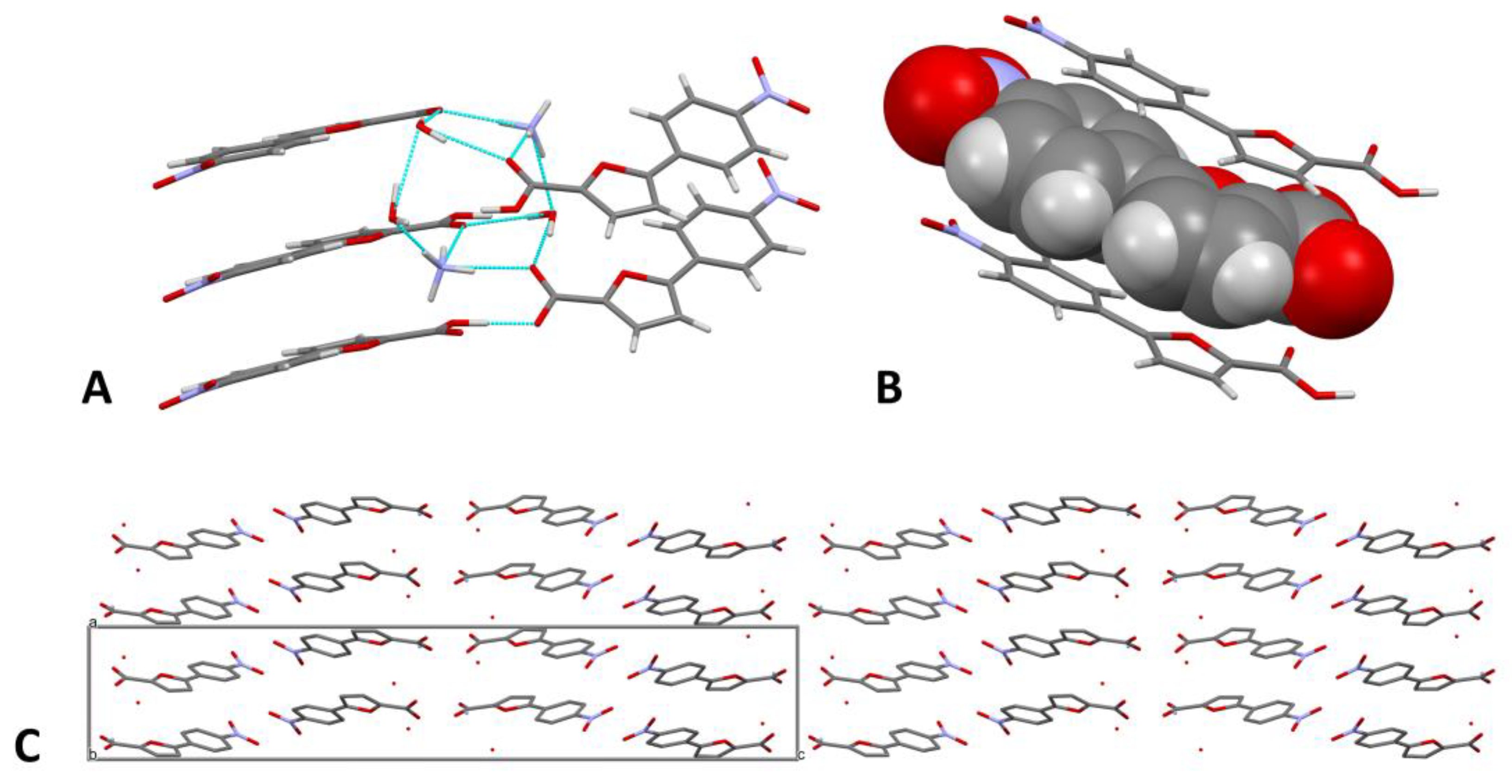
The crystal packing is governed by a dense network of H-bonds that connect the two molecules, the waters, and the NH4+ cation (Figure 2A). In detail, the acidic hydrogen of the carboxylic function of an A molecule bridges the O1 atom of a carboxylate; the two interacting molecules are inclined at 44.7(1)° with respect to each other, along the direction of the 21 helix. O1 also forms a H-bond with the O2W water, which, in turn, interacts with the other water molecule (O1W) and the NH4+ ion. The latter establishes contacts with both the waters and the carbonylic oxygens O2 and O2A, which are also bridged by the O1W water. Minor C-H···O interactions contribute to the stabilization of the network. A complete account of the H-bonds is provided in Table 1.
π-π Interactions between the aromatic portions of the compounds further contribute to the crystal packing (Figure 2B). The main molecule and molecule A are alternatingly stacked on planes inclined at 5.2(4)°; the centroid-centroid distances, calculated for the phenyl rings, are 3.88(1) Å and 3.60(1) Å. As shown in Figure 2C, the cell contains four pairs of molecules, which are repeated throughout the crystal; the empty spaces between them are occupied by the water molecules and by the NH4+ ion. The numerous intermolecular interactions consolidate the packing and stabilize the molecules in the solid state (Figure 2C). We may speculate that the presence of an ionic environment rich in solvation waters facilitated the solubilization of the compound and promoted the formation of a tight interaction network, which ultimately allowed the ordered organization of the molecules to form a crystal.
The Hirshfeld surface (HS) of 1 was mapped over the normalized contact distance (dnorm), according to the following equation:
- di: distance between the HS and the nearest nucleus inside the surface
- de: distance between the HS and the nearest nucleus outside the surface
- rvdW: van der Waals radius of the atom.
HSs were generated for both the molecules in the asymmetric unit; despite their being non-identical, the same general concepts can be applied to both. The structural details of the HSs are shown in Table 2.
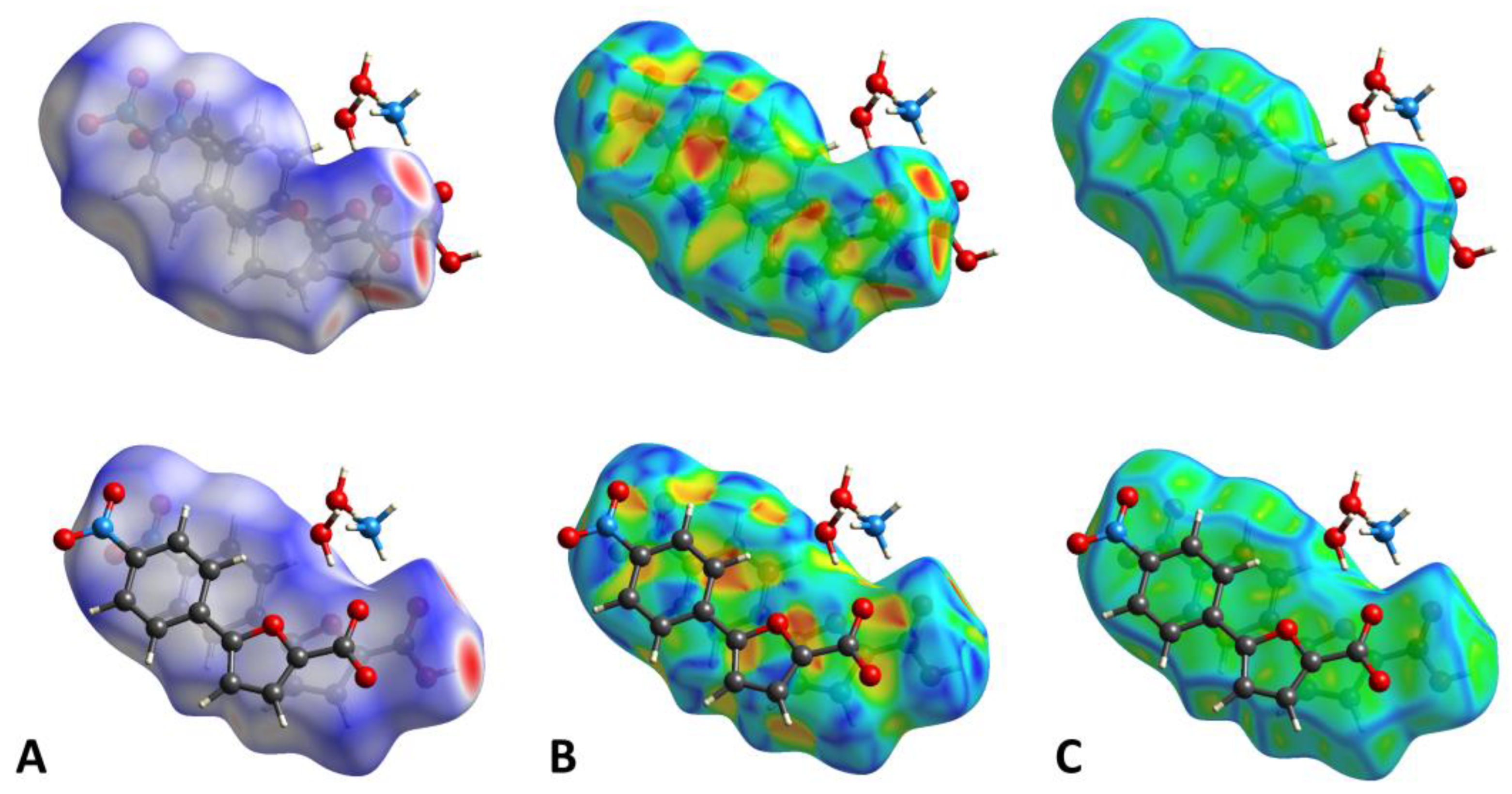
The dnorm property was visualized with a red-blue-white color scheme, based on the length of the intermolecular contacts with respect to the sum of the van der Waals radii (Figure 3A) [27]. The analysis of the surface revealed several intense red spots, corresponding to the strong, short-range H-bonds forming the network described above. The remaining, fainter red areas indicated the presence of minor C-H···O contacts. The shape index (SI, Figure 3B) property mapped over the HS evidenced a generally unperturbed structure, consistent with the planarity of the molecules. Moreover, the relevant stacking interactions were confirmed by the typical shape and color pattern on the aromatic portions of the SI. This observation was further supported by the curvedness plot (Figure 3C), which evidenced a large flat area extending over both the molecules.
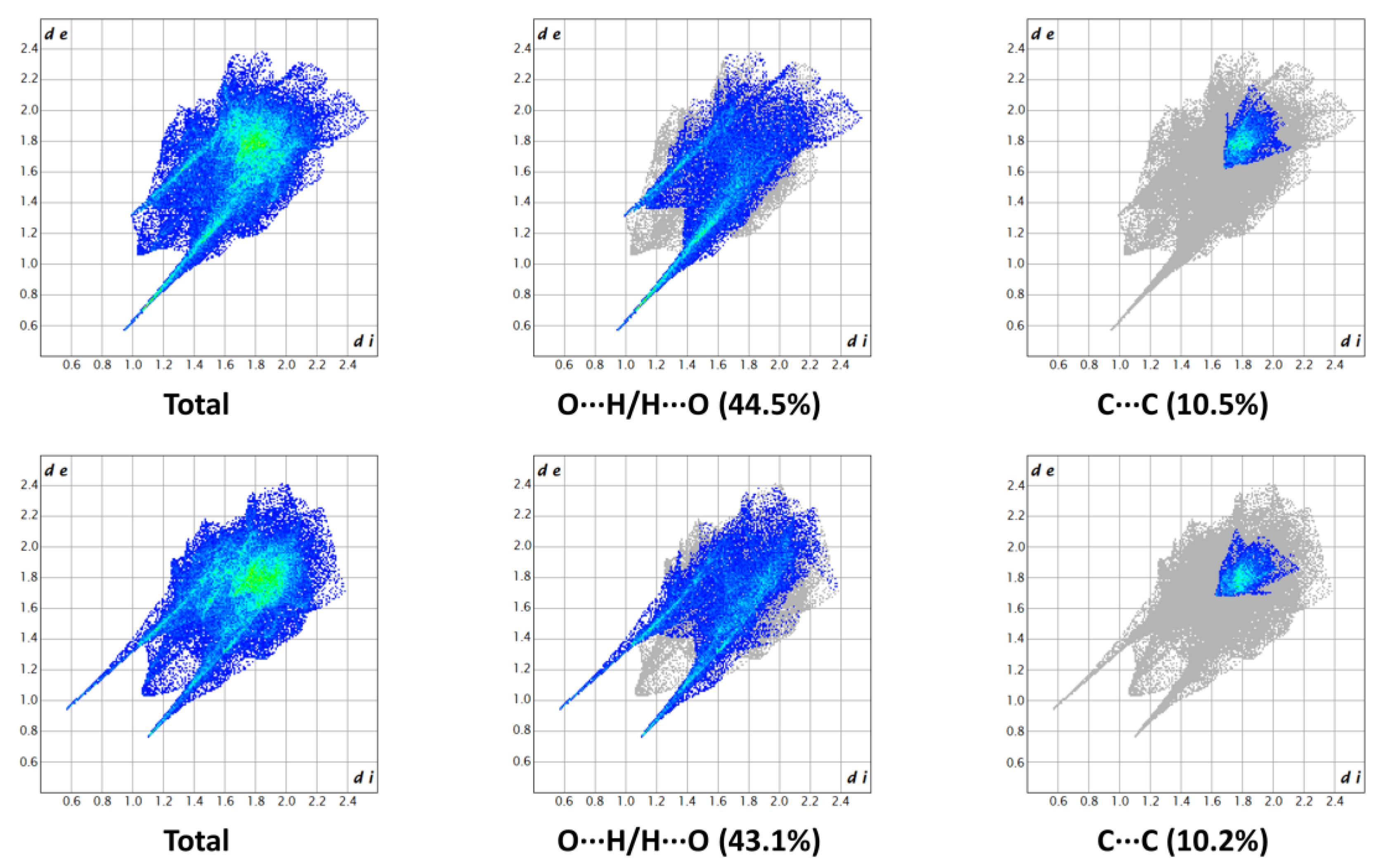
The two-dimensional (2D) fingerprints of the two HSs, providing a visual summary of the contribution of each contact type and the relative area of the surface corresponding to it, revealed the importance of the many short-range O···H/H···O contacts, represented by the long spikes protruding towards the lower left part of the graph. The presence of C···C contacts, indicating the π-π stacking of the aromatic rings, was confirmed by the characteristic arrow-shaped region at the center of the plot. The high density of green spots at 1.8–1.9 Å de/di confirmed the high contribution of these points on the HS. The remaining significant portions of the surface were attributed to unspecific van der Waals contacts (H···H and C···H/H···C). The main fingerprint plots related to the two HSs are reported in Figure 4. The enrichment ratios, calculated according to Jelsch et al. [28], showed that O···H/H···O and C···C contacts were enriched (Exy > 1), further substantiating the previous observations.
3. Materials and Methods
3.1. Chemistry
All reagents and solvents were purchased from Sigma-Aldrich (St. Louis, MO, USA). Air-sensitive reactions were performed in an inert N2 atmosphere. The course of the reactions was followed by thin-layer chromatography (TLC) using aluminum-backed silica gel 60 plates (0.2 mm; Merck, Darmstadt, Germany). Flash column chromatography was performed for the purification of the crude products, when necessary, using silica gel 60 (40–63 μm; Merck, Darmstadt, Germany) in the indicated solvent system. The melting points were determined in open capillary tubes, with a Stuart SMP30 Melting Point Apparatus (Cole-Parmer Stuart, Stone, UK). 1H and 13C NMR experiments were performed on a Varian Oxford 300 MHz instrument (Varian, Palo Alto, CA, USA), operating at 300 MHz for 1H and 75 MHz for 13C (T = 20 °C). Chemical shifts are expressed in ppm (δ), and J-couplings are given in Hertz. 1H data are reported in the following order: ppm, multiplicity (s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet), number of protons, and assignment. For the sake of clarity, the arbitrary atom numbering scheme indicated in Figure 1 for the main molecule was also used to define the signal attributions. The high-resolution mass spectrometry (HRMS) analysis was carried out on a Q-ToF Synapt G2-Si HDMS system (Waters, Milford, MA, USA). All relevant spectra are reported in the Supplementary Materials.
Methyl 5-(4-nitrophenyl)furan-2-carboxylate (2). Methyl 5-bromofuran-2-carboxylate (250 mg, 1.2 mmol), (4-nitrophenyl)boronic acid (267 mg, 1.6 mmol), and bis(triphenylphosphine)palladium (II) dichloride (42 mg, 5% mol) were dissolved in dry 1,4-dioxane (10 mL), under a N2 atmosphere. A 2 M Na2CO3 solution (254 mg, 2.4 mmol, 1.2 mL) was then added, and the resulting mixture was stirred overnight at 90 °C. After completion, the solution was cooled to room temperature and filtered on a celite pad. The filtrate was diluted with water and extracted with ethyl acetate (3 × 4 mL). The organic layer was dried over anhydrous Na2SO4, filtered, and concentrated in vacuo. The crude product was purified by flash column chromatography (cyclohexane–EtOAc–DCM 8:1:1) to afford a yellow solid. Yield: 20%. TLC (cyclohexane–EtOAc 8:2): Rf = 0.30. Mp: 194 °C (dec.). 1H NMR (300 MHz, CDCl3) δ (ppm): 8.28 (d, J = 8.5 Hz, 2H, H8,10), 7.93 (d, J = 8.5 Hz, 2H, H7,11), 7.28 (d, J = 3.7 Hz, 1H, H3), 6.95 (d, J = 3.7 Hz, 1H, H4), 3.94 (s, 3H, OCH3). 13C NMR (75 MHz, CDCl3) δ (ppm): 159.06 (C1), 154.94 (C5), 147.70 (C9), 145.44 (C2), 135.23 (C6), 125.46 (C7,11), 124.57 (C8,10), 120.12 (C3), 110.36 (C4), 52.42 (OCH3).
5-(4-Nitrophenyl)furan-2-carboxylic acid (1). To a solution of methyl 5-(4-nitrophenyl)furan-2-carboxylate (2, 50 mg, 0.2 mmol) in a mixture of H2O (2.6 mL) and MeOH (1.3 mL) was added NaOH (24 mg, 0.6 mmol). The reaction mixture was stirred at reflux for 3 h and partially concentered in vacuo to remove the solvent; then, 1 M HCl was added to adjust the pH to 3–4. The aqueous phase was extracted with EtOAc (3 × 5 mL), the combined organic phases were dried over anhydrous Na2SO4, filtered, and evaporated under reduced pressure to obtain the desired product as a yellow solid. No further purification was necessary. Yield: 80%. TLC (DCM–MeOH 8:2): Rf = 0.17. Mp: 256 °C (dec.). 1H NMR (300 MHz, DMSO-d6) δ (ppm): 13.35 (br s exch. D2O, 1H, COOH), 8.31 (d, J = 9.0 Hz, 2H, H8,10), 8.04 (d, J = 9.0 Hz, 2H, H7,11), 7.44 (d, J = 3.7 Hz, 1H, H3), 7.37 (d, J = 3.7 Hz, 1H, H4). 13C NMR (75 MHz, CDCl3) δ (ppm): 159.52 (C1), 154.21 (C5), 147.34 (C9), 146.21 (C2), 135.26 (C6), 125.67 (C7,11), 124.93 (C8,10), 120.29 (C3), 112.08 (C4). HRMS (ESI/Q-ToF): calcd. for C11H6NO5 232.0246, found 232.0248 [M + H]+.
3.2. X-ray Diffraction
Compound 1 crystallized in a 96-well protein crystallization plate, set up using the sitting-drop vapor diffusion method, according to established protocols [29]. A nice rod formed after about 2 weeks at 4 °C in the following conditions: 1.26 M (NH4)2SO4, 0.1 M NaOAc buffer pH 4.5, 0.2 M NaCl. The solution also contained the target protein MbtI, at a concentration of 23 mg/mL in 25 mM Hepes·NaOH pH 8.0, 150 mM NaCl, 1% glycerol. The crystal was collected at 4 °C and cryoprotected using parathion oil, before being frozen in liquid nitrogen. Diffraction data were acquired at the macromolecular crystallography beamline ID23-2 of the European Synchrotron Radiation Facility (ESRF) in Grenoble (France) [30]. ID23-2 features a focused microbeam (10 × 4 μm) at fixed energy (14.2 keV), an MD3-UP microdiffractometer (Arinax, Moirans, France), and a Dectris Pilatus 3 X 2M detector (Dectris AG, Baden, Switzerland). Data were collected at 100 K, using scan angles of 1° (delta_phi) up to a resolution of 0.75 Å. Data processing was performed with XDS [31], and the structure was solved based on the output file XDS_ASCII.HKL by direct methods using SIR-2019 [32]. Iterative cycles of full-matrix least-squares refinement on F02 and ΔF synthesis were carried out by SHELXL-18/3 [33] on the WinGX v.2021.3 suite [24]. The structure was analyzed by PARST [26], Mogul [25], and Mercury 2022.2.0 [34]; the graphical representations were rendered with ORTEP-3 v.2020.1 and Mercury [34]. HS analysis was performed with CrystalExplorer21.5 [35]. CCDC entry 2218901 contains the supplementary crystallographic data for this paper. A summary of the data collection and refinement statistics is reported in Table 3; further details are reported in the Supplementary Materials.
4. Conclusions
During a data collection session at the European Synchrotron Radiation Facility (ESRF) in the framework of our studies on MbtI-ligand complexes, we unexpectedly obtained the structure of 5-(4-nitrophenyl)furan-2-carboxylic acid (1). This is one of the lead compounds in our series of furan-based inhibitors of MbtI. Considering the difficulty of growing high-quality crystals of these compounds, we welcomed the opportunity to study the structure of 1 by means of synchrotron diffraction data. Our analysis revealed an abundance of strong intermolecular contacts, a complex network of H-bonds and tight π-π stacking forces. The environment, rich in ions and water molecules, probably facilitated the interaction of the molecules and promoted their organization in the solid state. To complete the description of compound 1, its synthesis and analytical characterization by means of NMR and HRMS were also described.
Supplementary Materials
Table S1. Atomic coordinates and equivalent isotropic displacement parameters. Table S2. Bond lengths [Å] for 1. Table S3. Bond angles [°] for 1. Table S4. Anisotropic displacement parameters. Figure S1. 1H NMR spectrum of 2. Figure S2. 13C NMR spectrum of 2. Figure S3. 1H NMR spectrum of 1. Figure S4. 13C NMR spectrum of 1. Figure S5. HRMS spectra of 1.
Author Contributions
Conceptualization, M.M., F.M. and M.B.; investigation, M.M., G.C., A.T. and M.B.; writing—original draft preparation, M.M. and F.M.; writing—review and editing, M.M., S.V., F.M. and M.B.; supervision, S.V., F.M. and M.B. All authors have read and agreed to the published version of the manuscript.
Funding
This work was funded by the University of Milan, “Linea B”.
Data Availability Statement
Crystallographic data (CCDC entry: 2218901) can be obtained free of charge from the Cambridge Crystallographic Data Centre, 12, Union Road, Cambridge CB21EZ, UK; fax: +44 1223 336 033 ([email protected]).
Conflicts of Interest
The authors declare no conflict of interest.
References
- World Health Organization. Global Tuberculosis Report 2021; World Health Organization: Geneva, Switzerland, 2021; ISBN 9789240037021.
- Cazzaniga, G.; Mori, M.; Chiarelli, L.R.; Gelain, A.; Meneghetti, F.; Villa, S. Natural products against key Mycobacterium tuberculosis enzymatic targets: Emerging opportunities for drug discovery. Eur. J. Med. Chem. 2021, 224, 113732. [Google Scholar] [CrossRef] [PubMed]
- Ejalonibu, M.A.; Ogundare, S.A.; Elrashedy, A.A.; Ejalonibu, M.A.; Lawal, M.M.; Mhlongo, N.N.; Kumalo, H.M. Drug Discovery for Mycobacterium tuberculosis Using Structure-Based Computer-Aided Drug Design Approach. Int. J. Mol. Sci. 2021, 22, 13259. [Google Scholar] [CrossRef] [PubMed]
- Motamen, S.; Quinn, R.J. Analysis of Approaches to Anti-tuberculosis Compounds. ACS Omega 2020, 5, 28529–28540. [Google Scholar] [CrossRef] [PubMed]
- Buroni, S.; Chiarelli, L.R. Antivirulence compounds: A future direction to overcome antibiotic resistance? Future Microbiol. 2020, 15, 299–301. [Google Scholar] [CrossRef]
- Chiarelli, L.R.; Mori, M.; Barlocco, D.; Beretta, G.; Gelain, A.; Pini, E.; Porcino, M.; Mori, G.; Stelitano, G.; Costantino, L.; et al. Discovery and Development of Novel Salicylate Synthase (MbtI) Furanic Inhibitors as Antitubercular Agents. Eur. J. Med. Chem. 2018, 155, 754–763. [Google Scholar] [CrossRef]
- Chao, A.; Sieminski, P.J.; Owens, C.P.; Goulding, C.W. Iron Acquisition in Mycobacterium tuberculosis. Chem. Rev. 2019, 119, 1193–1220. [Google Scholar] [CrossRef]
- Shyam, M.; Shilkar, D.; Rakshit, G.; Jayaprakash, V. Approaches for targeting the mycobactin biosynthesis pathway for novel anti-tubercular drug discovery: Where we stand. Expert Opin. Drug Discov. 2022, 17, 699–715. [Google Scholar] [CrossRef]
- Khurana, H.; Srivastava, M.; Chaudhary, D.; Gosain, T.P.; Kumari, R.; Bean, A.C.; Chugh, S.; Maiti, T.K.; Stephens, C.E.; Asthana, S.; et al. Identification of diphenyl furan derivatives via high throughput and computational studies as ArgA inhibitors of Mycobacterium tuberculosis. Int. J. Biol. Macromol. 2021, 193, 1845–1858. [Google Scholar] [CrossRef]
- Metaferia, B.B.; Fetterolf, B.J.; Shazad-ul-Hussan, S.; Moravec, M.; Smith, J.A.; Ray, S.; Gutierrez-Lugo, M.T.; Bewley, C.A. Synthesis of natural product-inspired inhibitors of Mycobacterium tuberculosis mycothiol-associated enzymes: The first inhibitors of GlcNAc-Ins deacetylase. J. Med. Chem. 2007, 50, 6326–6336. [Google Scholar] [CrossRef]
- Reshma, R.S.; Jeankumar, V.U.; Kapoor, N.; Saxena, S.; Bobesh, K.A.; Vachaspathy, A.R.; Kolattukudy, P.E.; Sriram, D. Mycobacterium tuberculosis lysine-ɛ-aminotransferase a potential target in dormancy: Benzothiazole based inhibitors. Bioorg. Med. Chem. 2017, 25, 2761–2771. [Google Scholar] [CrossRef]
- Manger, M.; Scheck, M.; Prinz, H.; Von Kries, J.P.; Langer, T.; Saxena, K.; Schwalbe, H.; Fürstner, A.; Rademann, J.; Waldmann, H. Discovery of Mycobacterium Tuberculosis Protein Tyrosine Phosphatase A (MptpA) Inhibitors Based on Natural Products and a Fragment-Based Approach. ChemBioChem 2005, 6, 1749–1753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sens, L.; de Souza, A.C.A.; Pacheco, L.A.; Menegatti, A.C.O.; Mori, M.; Mascarello, A.; Nunes, R.J.; Terenzi, H. Synthetic thiosemicarbazones as a new class of Mycobacterium tuberculosis protein tyrosine phosphatase A inhibitors. Bioorg. Med. Chem. 2018, 26, 5742–5750. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, N.L.; Lipker, L.; Hanson, A.M.; Bohl, C.J.; Engel, K.E.; Kalous, K.S.; Stemper, M.E.; Sem, D.S.; Schwan, W.R. Docking into Mycobacterium tuberculosis Thioredoxin Reductase Protein Yields Pyrazolone Lead Molecules for Methicillin-Resistant Staphylococcus aureus. Antibiotics 2017, 6, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verbitskiy, E.V.; Baskakova, S.A.; Belyaev, D.V.; Vakhrusheva, D.V.; Eremeeva, N.I.; Rusinov, G.L.; Charushin, V.N. Renaissance of 4-(5-nitrofuran-2-yl)-5-arylamino substituted pyrimidines: Microwave-assisted synthesis and antitubercular activity. Mendeleev Commun. 2021, 31, 210–212. [Google Scholar] [CrossRef]
- Elsaman, T.; Mohamed, M.S.; Mohamed, M.A. Current development of 5-nitrofuran-2-yl derivatives as antitubercular agents. Bioorg. Chem. 2019, 88, 102969. [Google Scholar] [CrossRef]
- Faazil, S.; Malik, M.S.; Ahmed, S.A.; Alsantali, R.I.; Yedla, P.; Alsharif, M.A.; Shaikh, I.N.; Kamal, A. Novel linezolid-based oxazolidinones as potent anticandidiasis and antitubercular agents. Bioorg. Chem. 2022, 126, 105869. [Google Scholar] [CrossRef]
- Verbitskiy, E.V.; Baskakova, S.A.; Gerasimova, N.A.; Evstigneeva, N.P.; Zil’berberg, N.V.; Kungurov, N.V.; Kravchenko, M.A.; Rusinov, G.L.; Chupakhina, O.N.; Charushin, V.N. New 5-arylamino-4-(5-nitrofuran-2-yl)pyrimidines as promising antibacterial agents. Mendeleev Commun. 2018, 28, 393–395. [Google Scholar] [CrossRef]
- Kaur, S.; Nieto, N.S.; McDonald, P.; Beck, J.R.; Honzatko, R.B.; Roy, A.; Nelson, S.W. Discovery of small molecule inhibitors of Plasmodium falciparum apicoplast DNA polymerase. J. Enzyme Inhib. Med. Chem. 2022, 37, 1320–1326. [Google Scholar] [CrossRef]
- Chiarelli, L.R.; Mori, M.; Beretta, G.; Gelain, A.; Pini, E.; Sammartino, J.C.; Stelitano, G.; Barlocco, D.; Costantino, L.; Lapillo, M.; et al. New Insight into Structure-Activity of Furan-based Salicylate Synthase (MbtI) Inhibitors as Potential Antitubercular Agents. J. Enzyme Inhib. Med. Chem. 2019, 34, 823–828. [Google Scholar] [CrossRef] [Green Version]
- Mori, M.; Stelitano, G.; Gelain, A.; Pini, E.; Chiarelli, L.R.; Sammartino, J.C.; Poli, G.; Tuccinardi, T.; Beretta, G.; Porta, A.; et al. Shedding X-ray Light on the Role of Magnesium in the Activity of M. tuberculosis Salicylate Synthase (MbtI) for Drug Design. J. Med. Chem. 2020, 63, 7066–7080. [Google Scholar] [CrossRef]
- Mori, M.; Stelitano, G.; Chiarelli, L.R.; Cazzaniga, G.; Gelain, A.; Barlocco, D.; Pini, E.; Meneghetti, F.; Villa, S. Synthesis, Characterization, and Biological Evaluation of New Derivatives Targeting MbtI as Antitubercular Agents. Pharmaceuticals 2021, 14, 155. [Google Scholar] [CrossRef] [PubMed]
- Mori, M.; Stelitano, G.; Griego, A.; Chiarelli, L.R.; Cazzaniga, G.; Gelain, A.; Pini, E.; Camera, M.; Canzano, P.; Fumagalli, A.; et al. Synthesis and Assessment of the In Vitro and Ex Vivo Activity of Salicylate Synthase (Mbti) Inhibitors as New Candidates for the Treatment of Mycobacterial Infections. Pharmaceuticals 2022, 15, 992. [Google Scholar] [CrossRef] [PubMed]
- Farrugia, L.J. WinGX and ORTEP for Windows: An update. J. Appl. Crystallogr. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Bruno, I.J.; Cole, J.C.; Kessler, M.; Luo, J.; Motherwell, W.D.S.; Purkis, L.H.; Smith, B.R.; Taylor, R.; Cooper, R.I.; Harris, S.E.; et al. Retrieval of Crystallographically-Derived Molecular Geometry Information. J. Chem. Inf. Comput. Sci. 2004, 44, 2133–2144. [Google Scholar] [CrossRef] [PubMed]
- Nardelli, M. PARST 95—An update to PARST: A system of Fortran routines for calculating molecular structure parameters from the results of crystal structure analyses. J. Appl. Crystallogr. 1995, 28, 659. [Google Scholar] [CrossRef]
- Cazzaniga, G.; Mori, M.; Meneghetti, F.; Chiarelli, L.R.; Stelitano, G.; Caligiuri, I.; Rizzolio, F.; Ciceri, S.; Poli, G.; Staver, D.; et al. Virtual screening and crystallographic studies reveal an unexpected γ-lactone derivative active against MptpB as a potential antitubercular agent. Eur. J. Med. Chem. 2022, 234, 114235. [Google Scholar] [CrossRef]
- Jelsch, C.; Ejsmont, K.; Huder, L. The enrichment ratio of atomic contacts in crystals, an indicator derived from the Hirshfeld surface analysis. IUCrJ 2014, 1, 119–128. [Google Scholar] [CrossRef]
- Weber, P.; Pissis, C.; Navaza, R.; Mechaly, A.E.; Saul, F.; Alzari, P.M.; Haouz, A. High-Throughput Crystallization Pipeline at the Crystallography Core Facility of the Institut Pasteur. Molecules 2019, 24, 4451. [Google Scholar] [CrossRef] [Green Version]
- Nanao, M.; Basu, S.; Zander, U.; Giraud, T.; Surr, J.; Guijarro, M.; Lentini, M.; Felisaz, F.; Sinoir, J.; Morawe, C.; et al. ID23-2: An automated and high-performance microfocus beamline for macromolecular crystallography at the ESRF. J. Synchrotron Radiat. 2022, 29, 581–590. [Google Scholar] [CrossRef]
- Kabsch, W. IUCr XDS. Acta Crystallogr. Sect. D Struct. Biol. 2010, 66, 125–132. [Google Scholar] [CrossRef]
- Burla, M.C.; Caliandro, R.; Carrozzini, B.; Cascarano, G.L.; Cuocci, C.; Giacovazzo, C.; Mallamo, M.; Mazzone, A.; Polidori, G. Crystal structure determination and refinement via SIR2014. J. Appl. Crystallogr. 2015, 48, 306–309. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacRae, C.F.; Sovago, I.; Cottrell, S.J.; Galek, P.T.A.; McCabe, P.; Pidcock, E.; Platings, M.; Shields, G.P.; Stevens, J.S.; Towler, M.; et al. Mercury 4.0: From visualization to analysis, design and prediction. J. Appl. Crystallogr. 2020, 53, 226–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spackman, P.R.; Turner, M.J.; McKinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer: A program for Hirshfeld surface analysis, visualization and quantitative analysis of molecular crystals. J. Appl. Crystallogr. 2021, 54, 1006–1011. [Google Scholar] [CrossRef]
Scheme 1.
Synthetic procedure for the preparation of 1. Reagents and conditions: (a) Pd(PPh3)2Cl2, 2 M Na2CO3, 1,4-dioxane, 90 °C, overnight, N2; (b) NaOH, H2O/MeOH 2:1, reflux, 3 h.
Scheme 1.
Synthetic procedure for the preparation of 1. Reagents and conditions: (a) Pd(PPh3)2Cl2, 2 M Na2CO3, 1,4-dioxane, 90 °C, overnight, N2; (b) NaOH, H2O/MeOH 2:1, reflux, 3 h.

Figure 1.
ORTEP diagram of 1, with the arbitrary atom numbering scheme. Displacement ellipsoids are drawn at the 40% probability level.
Figure 1.
ORTEP diagram of 1, with the arbitrary atom numbering scheme. Displacement ellipsoids are drawn at the 40% probability level.

Figure 2.
(A). Simplified graphical representation of the H-bond network. (B). Stick-spacefilling model showing the π-π stacking interactions between the molecules. (C). Crystal packing viewed along the b axis. Hydrogen atoms are omitted for the sake of clarity.
Figure 2.
(A). Simplified graphical representation of the H-bond network. (B). Stick-spacefilling model showing the π-π stacking interactions between the molecules. (C). Crystal packing viewed along the b axis. Hydrogen atoms are omitted for the sake of clarity.

Figure 3.
(A). HS mapped over dnorm with a fixed color scale in the range −0.8453 au (red)—1.2461 au (blue) for the main molecule (above) and −0.8481 au (red)—1.2111 au (blue) for molecule A (below), based on the length of the intermolecular contacts with respect to the sum of the van der Waals radii (red: shorter; blue: longer; white: same). (B). HS mapped over the shape-index (color scale: −0.9957 au—0.9994 au for the main molecule; −0.9882 au—0.9924 au for molecule A). Blue areas indicate bumps, while red regions represent hollows. (C). HS mapped over the curvedness (color scale: −4.0490 au—0.4420 au for the main molecule; −3.8574 au—0.3755 au for molecule A). Flat regions are colored green, while edges are blue.
Figure 3.
(A). HS mapped over dnorm with a fixed color scale in the range −0.8453 au (red)—1.2461 au (blue) for the main molecule (above) and −0.8481 au (red)—1.2111 au (blue) for molecule A (below), based on the length of the intermolecular contacts with respect to the sum of the van der Waals radii (red: shorter; blue: longer; white: same). (B). HS mapped over the shape-index (color scale: −0.9957 au—0.9994 au for the main molecule; −0.9882 au—0.9924 au for molecule A). Blue areas indicate bumps, while red regions represent hollows. (C). HS mapped over the curvedness (color scale: −4.0490 au—0.4420 au for the main molecule; −3.8574 au—0.3755 au for molecule A). Flat regions are colored green, while edges are blue.

Figure 4.
Main 2D fingerprint plots of the HSs of 1 (above: main molecule; below: molecule A). the graphical representation provides a summary of the frequency of each combination of de and di across the HS. Points with a lower contribution to the surface are colored blue, while green indicates a larger contribution.
Figure 4.
Main 2D fingerprint plots of the HSs of 1 (above: main molecule; below: molecule A). the graphical representation provides a summary of the frequency of each combination of de and di across the HS. Points with a lower contribution to the surface are colored blue, while green indicates a larger contribution.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
H-bond geometry (D: Donor, A: Acceptor), as calculated by PARST [26].
Table 1.
H-bond geometry (D: Donor, A: Acceptor), as calculated by PARST [26].
| H-Bond | D-H/Å | H∙∙∙A/Å | D∙∙∙A/Å | D-H∙∙∙A/° |
|---|---|---|---|---|
| O1W-H1W···O2 0 | 0.976(4) | 1.788(4) | 2.753(2) | 169(4) |
| C11-H11···O1W 0 | 0.930(2) | 2.731(2) | 3.657(3) | 174(1) |
| N2-H2A···O2W 0 | 0.88(3) | 1.89(3) | 2.773(3) | 176(3) |
| N2-H2B···O1W 0 | 0.99(3) | 1.89(3) | 2.862(3) | 166(3) |
| N2-H2C···O2A 0 | 0.94(4) | 1.95(4) | 2.859(3) | 162(3) |
| C11A-H11A···O2W 0 | 0.930(2) | 2.527(2) | 3.304(3) | 141(1) |
| C3-H3···O2W I | 0.930(2) | 2.459(2) | 3.270(3) | 146(1) |
| C4-H4···O5A I | 0.930(2) | 2.536(2) | 3.357(3) | 147(1) |
| O1A-H1A···O1 II | 0.96(4) | 1.55(4) | 2.498(2) | 171(3) |
| N2-H2D···O2 III | 0.94(3) | 1.83(3) | 2.765(3) | 168(3) |
| O1W-H2W···O2A IV | 0.85(4) | 2.03(4) | 2.856(2) | 165(4) |
| O2W-H3W···O1 V | 0.87(4) | 1.88(4) | 2.742(2) | 167(4) |
| O2W-H4W···O1W VI | 0.98(4) | 1.89(4) | 2.833(3) | 161(4) |
Equivalent positions: 0 x,y,z; I x−1,y + 1,z; II x + ½,3/2−y,−z; III x + ½,½−y,−z; IV x−½,½−y,−z; V x + 1,y−1,z; VI x + 1,y,z.
Table 2.
Details of the two HSs generated for 1 (HS-0: main molecule; HS-A: molecule A).
| 1 | V (Å3) | A (Å2) | G | Ω |
|---|---|---|---|---|
| HS-0 | 233.21 | 241.21 | 0.760 | 0.314 |
| HS-A | 244.00 | 247.39 | 0.763 | 0.341 |
Table 3.
Summary of the crystallographic data and refinement statistics for 1.
| Parameter | Data | |
|---|---|---|
| Identification code | 1 | |
| Empirical formula | C22H21N3O12 | |
| Formula weight | 519.42 | |
| Temperature | 100(2) K | |
| Crystal system | Orthorhombic | |
| Space group | P212121 | |
| Unit cell dimensions | a = 7.34 Å | α = 90° |
| b = 7.97 Å | β = 90° | |
| c = 39.21 Å | γ = 90° | |
| Volume | 2293.8 Å3 | |
| Z | 4 | |
| Density (calculated) | 1.504 Mg/m3 | |
| Absorption coefficient | 0.125 mm−1 | |
| F(000) | 1080 | |
| Crystal size | 0.1 × 0.02 × 0.01 mm3 | |
| θ range for data collection | 1.039 to 28.431° | |
| Index ranges | −8 ≤ h ≤ 8, −10 ≤ k ≤ 10, −47 ≤ l ≤ 47 | |
| Reflections collected | 29,316 | |
| Independent reflections | 5065 [R(int) = 0.0553] | |
| Refinement method | Full-matrix least-squares on F2 | |
| Data/restraints/parameters | 5065/0/370 | |
| Goodness-of-fit on F2 | 1.039 | |
| Final R indices [I > 2σ(I)] | R1 = 0.0348, wR2 = 0.0836 | |
| R indices (all data) | R1 = 0.0407, wR2 = 0.0878 | |
| Largest diff. peak and hole | 0.319 and −0.214 eÅ−3 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Mori, M.; Tresoldi, A.; Villa, S.; Cazzaniga, G.; Bellinzoni, M.; Meneghetti, F. 5-(4-Nitrophenyl)furan-2-carboxylic Acid. Molbank 2022, 2022, M1515. https://doi.org/10.3390/M1515
AMA Style
Mori M, Tresoldi A, Villa S, Cazzaniga G, Bellinzoni M, Meneghetti F. 5-(4-Nitrophenyl)furan-2-carboxylic Acid. Molbank. 2022; 2022(4):M1515. https://doi.org/10.3390/M1515
Chicago/Turabian StyleMori, Matteo, Andrea Tresoldi, Stefania Villa, Giulia Cazzaniga, Marco Bellinzoni, and Fiorella Meneghetti. 2022. "5-(4-Nitrophenyl)furan-2-carboxylic Acid" Molbank 2022, no. 4: M1515. https://doi.org/10.3390/M1515
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.