
Gating Mechanism of Hv1 Studied by Molecular Dynamic Simulations †


1
Center for Advanced Chemistry, Institute of Research and Development, Duy Tan University, 03 Quang Trung, Hai Chau, Danang 550000, Vietnam
2
Faculty of Environmental and Chemical Engineering, Duy Tan University, 03 Quang Trung, Hai Chau, Danang 550000, Vietnam
†
Presented at the 2nd International Online-Conference on Nanomaterials, 15–30 November 2020; Available online: https://iocn2020.sciforum.net/ .
Mater. Proc. 2021, 4(1), 20; https://doi.org/10.3390/IOCN2020-07862
Published: 11 November 2020
(This article belongs to the Proceedings of The 2nd International Online-Conference on Nanomaterials)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:The voltage-gated proton channel (Hv1) plays the important role in proton extrusion, pH homeostasis, sperm motility, and cancer progression. The closed-state structure of Hv1 was recently revealed by X-ray crystallography. However, the opened-state structure has not been captured yet. To investigate the mechanism of proton transfer in Hv1, molecular dynamics (MD) simulations were performed with the closed-state structure of Hv1 under electric field and pH conditions. The residues arrangement on the closed-state structure revealed that the selectivity filter (Asp108) which is located in the hydrophobic layer (consists of two Phe residues 146 and 179) might prevent water penetration. In molecular dynamics simulations, we observed that the channel opened by moving 3 Arg up on the S4 helix and a continuous hydrogen-bonded chain of water molecules (a “water wire”) went through the channel when it opened. During simulations, the open channel allowed water molecules to pass through the channel but excluded other ions. This indicates the Hv1 channel is highly selective for protons. Our results clearly showed the Hv1 channel is voltage-and pH-gradient sensing.
1. Introduction
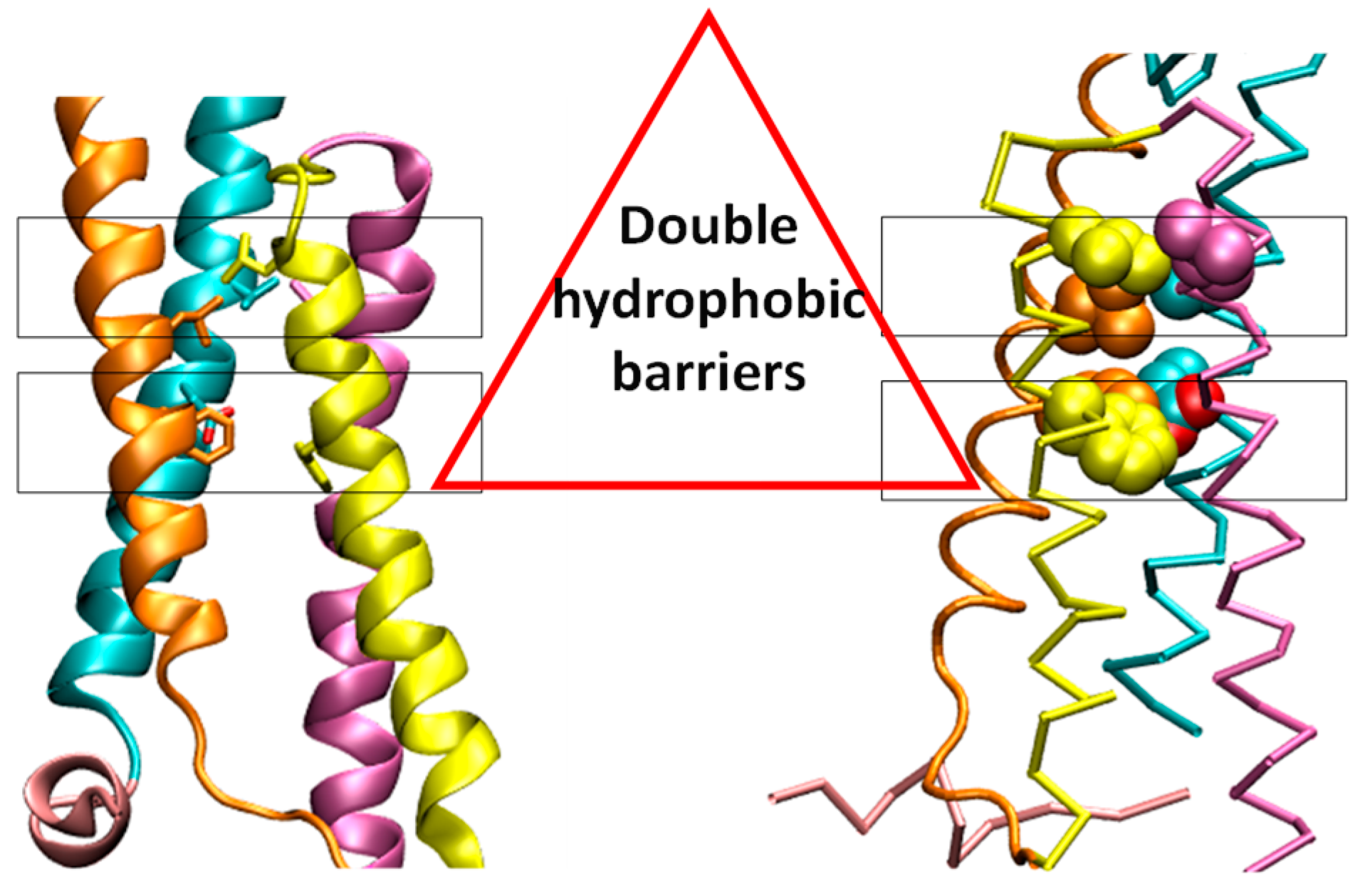
The voltage-gated proton channel (Hv1) consists of two functional domains: the votage sensing domains (VSDs) and the cytoplasmic coiled-coil domain. The crystal structure of mouse Hv1 chimera channel (mHv1cc) showed that the fourth transmembrane helix (S4) was directly connected to the cytoplasmic coiled-coil region to form a slightly bent, long helical structure [1]. The mHv1cc crystal structure showed two hydrophobic layers (Figure 1). In this model, the ASP108, which is critical for the selective proton permeation, is located in the hydrophobic layer. Two hydrophobic layers may work as a shield to prevent the permeation of water molecules.
The Grotthuss mechanism (hopping mechanism) is the mechanism by which an “excess” proton hops through the hydrogen bond network of water molecules or other hydrogen-bonded liquids through the formation or cleavage of covalent bonds [2]. Protons can hop through the single-file region of a water-filled pore without displacing the water, whereas ions must wait for the water in front to pass through, which takes a lot of time. The proton does not need to travel with an entourage of waters of hydration and can move quite well as a bare proton [3].
Hv1 has been shown to play important roles in proton extrusion, pH homeostasis, sperm motility, and cancer progression and has also been shown to be highly expressed in cell lines and tissue samples from patients with breast cancer. However, the understanding of the gating mechanisms of Hv1 is still unclear. We used the X-structure to perform molecular dynamics simulations with the electric field and pH conditions to investigate the gating mechanism of Hv1.
2. Methods
2.1. System Preparation
The missing loop of the X-ray structure of the murine Hv1 channel (mHv1cc, 3WKV) was completed by SWISS-MODEL [4]. An MD simulation with a prepared structure was run to get the stable closed conformation. Two histidines that make coordinates with zinc were assigned as HSD136, HSE190. Four simulations were run with four electric fields including 0 mV, 50 mV, 150 mV, 250 mV. To mimic pH conditions with pHo (external pH) and pHi (internal pH), histidines at the extracellular were assumed to be in the deprotonated state, and the intracellular histidine was assumed to be in the protonated state. The CHARMm force field with explicit TIP3 water molecules was used in all simulations. For systems were solvated in a truncated octahedral box with TIP3 water molecules, respectively. These systems were neutralized with NaCl (0.15 M). The purpose of solvation and neutralization was required to achieve an electrically neutral solvate system.
2.2. Molecular Dynamics Simulations
All simulations were performed by NAMD 2.10 [5] and CHARMm 36 [6] force fields under the periodic boundary condition. Initially, an energy minimization step was performed to remove bad contacts within the 1000 steps. Then, the MD simulations were started by gradually heating the systems from 10 to 298 K at a constant volume in 60 ps. The simulation systems were switched to a constant pressure and constant temperature (NPT) afterward. The simulation systems were equilibrated in the course of 200 ps. All heavy atoms in these steps of energy minimization, heating, and equilibration were restrained with a harmonic constant of 1 kcal/mol·A2. After that, the harmonic restraints were removed, allowing all atoms in the systems to relax. All bonds involving hydrogen atoms were constrained, allowing an integration time step of 2 fs, and the average pressure and temperature were maintained at 1 bar and 298 K. The nonbonded interactions were smoothly truncated from 10 Å to 12 Å cutoff, and the particle-mesh Ewald method [7] was used to treat long-range electrostatic interactions. The total lengths of the MD trajectories of each were 100 ns. Then, all conformational changes were observed by using the Visual Molecular Dynamics (VMD) program 1.9.1 [8]. All the results were analyzed after the equilibration state of the simulations.
2.3. Water Dynamics Analyze
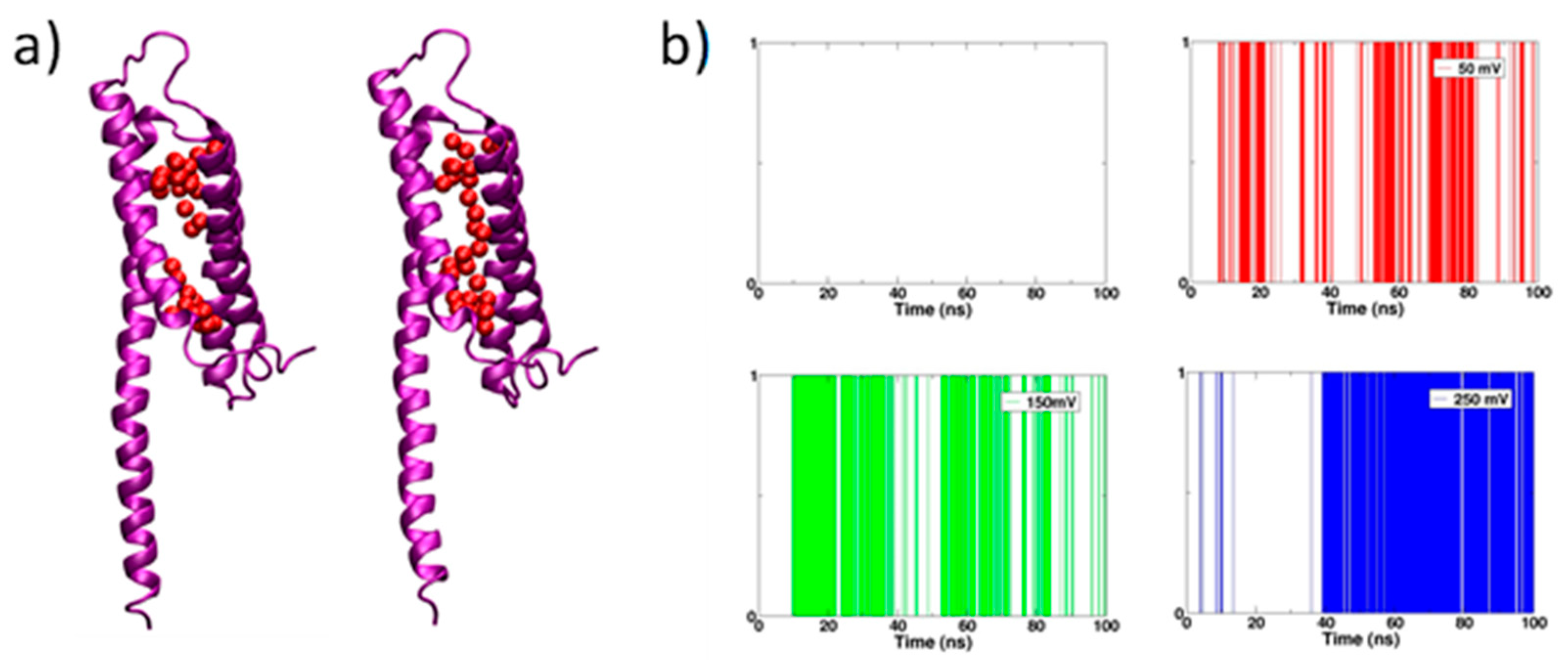
We defined the water wire by the distance between two adjacent water molecules. The distance of two oxygen of two water molecules is less than 3.4 Å. Water wire formation along each simulation trajectory was modeled as a binary process, yes or no.
3. Results and Discussions
3.1. Water Wire Forms in the mHv1cc Channel
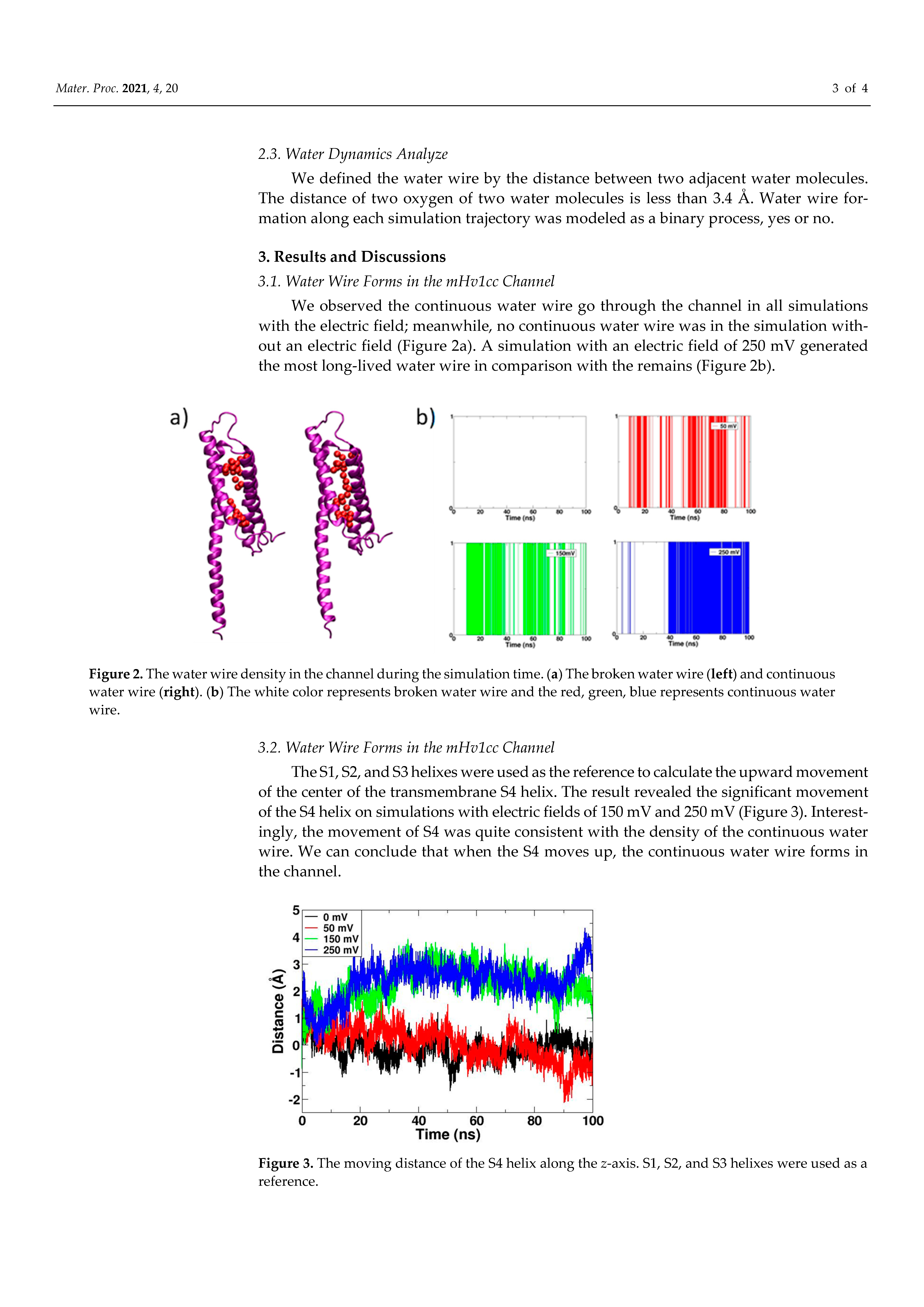
We observed the continuous water wire go through the channel in all simulations with the electric field; meanwhile, no continuous water wire was in the simulation without an electric field (Figure 2a). A simulation with an electric field of 250 mV generated the most long-lived water wire in comparison with the remains (Figure 2b).
3.2. Water Wire Forms in the mHv1cc Channel
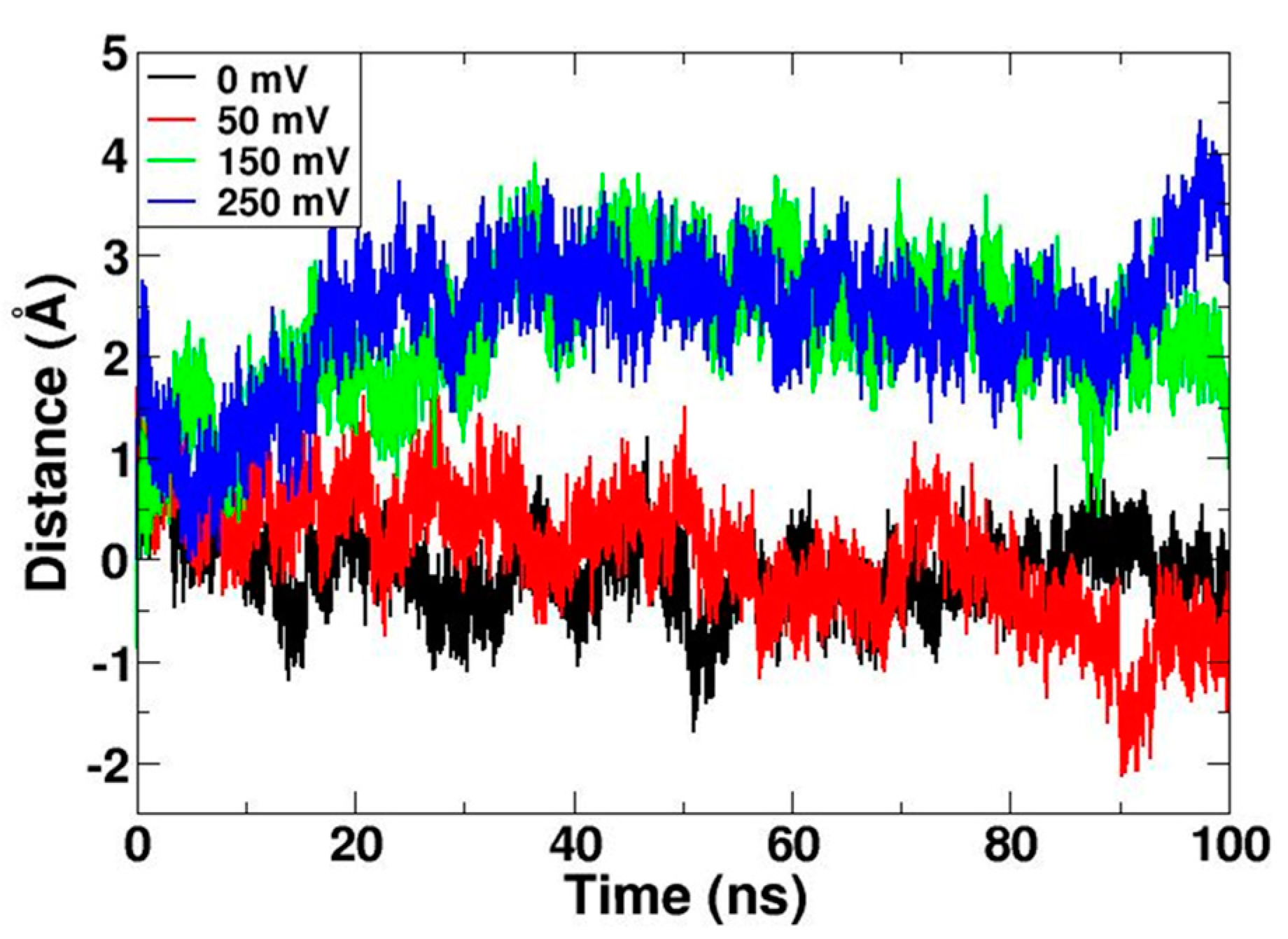
The S1, S2, and S3 helixes were used as the reference to calculate the upward movement of the center of the transmembrane S4 helix. The result revealed the significant movement of the S4 helix on simulations with electric fields of 150 mV and 250 mV (Figure 3). Interestingly, the movement of S4 was quite consistent with the density of the continuous water wire. We can conclude that when the S4 moves up, the continuous water wire forms in the channel.
3.3. The Opening of the Gating
In the closed state, D108 forms the salt bridges to both R202, R205 (belonging to the S4 helix). When the S4 helix moved up, D182 formed hydrogen bonds with both R202 and R205. The change of the salt bridges network is one piece of evidence for the upward movement of the S4 helix (Figure 4).
4. Conclusions
We observed the “open state” of the mHv1cc channel using the “closed state” X-ray crystal structure mHv1cc to set up MD simulations with the electric fields and pH conditions. The upward motion of the S4 helix of mHv1cc makes the channel transfer from “closed state” to “open state”. We have shown that mHv1cc exhibits water wire connectivity between the intracellular and extracellular sides when the channel is in an “open state”.
Data Availability Statement
Data is contained within the article.
References
- Takeshita, K.; Sakata, S.; Yamashita, E.; Fujiwara, Y.; Kawanabe, A.; Kurokawa, T.; Okochi, Y.; Matsuda, M.; Narita, H.; Okamura, Y.; et al. X-ray crystal structure of voltage-gated proton channel. Nat. Struct. Mol. Biol. 2014, 21, 352–357. [Google Scholar] [CrossRef] [PubMed]
- Agmon, N. The Grotthuss mechanism. Chem. Phys. Lett. 1995, 244, 456–462. [Google Scholar] [CrossRef]
- DeCoursey, T.E. The Voltage-Gated Proton Channel: A Riddle, Wrapped in a Mystery, inside an Enigma. Biochemistry 2015, 54, 3250–3268. [Google Scholar] [CrossRef] [PubMed]
- Guex, N.; Peitsch, M.C.; Schwede, T. Automated comparative protein structure modeling with SWISS-MODEL and Swiss-PdbViewer: A historical perspective. Electrophoresis 2009, 30, S162–S173. [Google Scholar] [CrossRef] [PubMed]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kale, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [PubMed]
- MacKerell, A.D.; Bashford, D.; Bellott, M.; Dunbrack, R.L.; Evanseck, J.D.; Field, M.J.; Fischer, S.; Gao, J.; Guo, H.; Ha, S.; et al. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B 1998, 102, 3586–3616. [Google Scholar] [CrossRef] [PubMed]
- Toukmaji, A.; Sagui, C.; Board, J.; Darden, T. Efficient particle-mesh Ewald based approach to fixed and induced dipolar interactions. J. Chem. Phys. 2000, 113, 10913–10927. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
Figure 1.
Double hydrophobic layers of the mHv1cc. The two hydrophobic residues, which are shown by a stick model. The upper layer at the extracellular consisted of four highly conserved hydrophobic residues (VAL112 (S1), LEU143 (S2), LEU186 (S3), LEU197 (S4)). The lower layer at the cytoplasmic side included PHE146 (S2) and PHE179 (S3). The four transmembrane segment S1 (cyan), S2 (orange), S3 (yellow), S4 (mauve), and S0 (pink) are shown.
Figure 1.
Double hydrophobic layers of the mHv1cc. The two hydrophobic residues, which are shown by a stick model. The upper layer at the extracellular consisted of four highly conserved hydrophobic residues (VAL112 (S1), LEU143 (S2), LEU186 (S3), LEU197 (S4)). The lower layer at the cytoplasmic side included PHE146 (S2) and PHE179 (S3). The four transmembrane segment S1 (cyan), S2 (orange), S3 (yellow), S4 (mauve), and S0 (pink) are shown.

Figure 2.
The water wire density in the channel during the simulation time. (a) The broken water wire (left) and continuous water wire (right). (b) The white color represents broken water wire and the red, green, blue represents continuous water wire.
Figure 2.
The water wire density in the channel during the simulation time. (a) The broken water wire (left) and continuous water wire (right). (b) The white color represents broken water wire and the red, green, blue represents continuous water wire.

Figure 3.
The moving distance of the S4 helix along the z-axis. S1, S2, and S3 helixes were used as a reference.
Figure 3.
The moving distance of the S4 helix along the z-axis. S1, S2, and S3 helixes were used as a reference.

Figure 4.
The position of residues forming the salt bridge before and after the applied electric field. The cartoon model of closed state (left) and the open state (right). The backline represents the hydrogen bond.
Figure 4.
The position of residues forming the salt bridge before and after the applied electric field. The cartoon model of closed state (left) and the open state (right). The backline represents the hydrogen bond.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Phan, T.T.V. Gating Mechanism of Hv1 Studied by Molecular Dynamic Simulations. Mater. Proc. 2021, 4, 20. https://doi.org/10.3390/IOCN2020-07862
AMA Style
Phan TTV. Gating Mechanism of Hv1 Studied by Molecular Dynamic Simulations. Materials Proceedings. 2021; 4(1):20. https://doi.org/10.3390/IOCN2020-07862
Chicago/Turabian StylePhan, Thi Tuong Vy. 2021. "Gating Mechanism of Hv1 Studied by Molecular Dynamic Simulations" Materials Proceedings 4, no. 1: 20. https://doi.org/10.3390/IOCN2020-07862