
The Rabies Virus L Protein Catalyzes mRNA Capping with GDP Polyribonucleotidyltransferase Activity


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
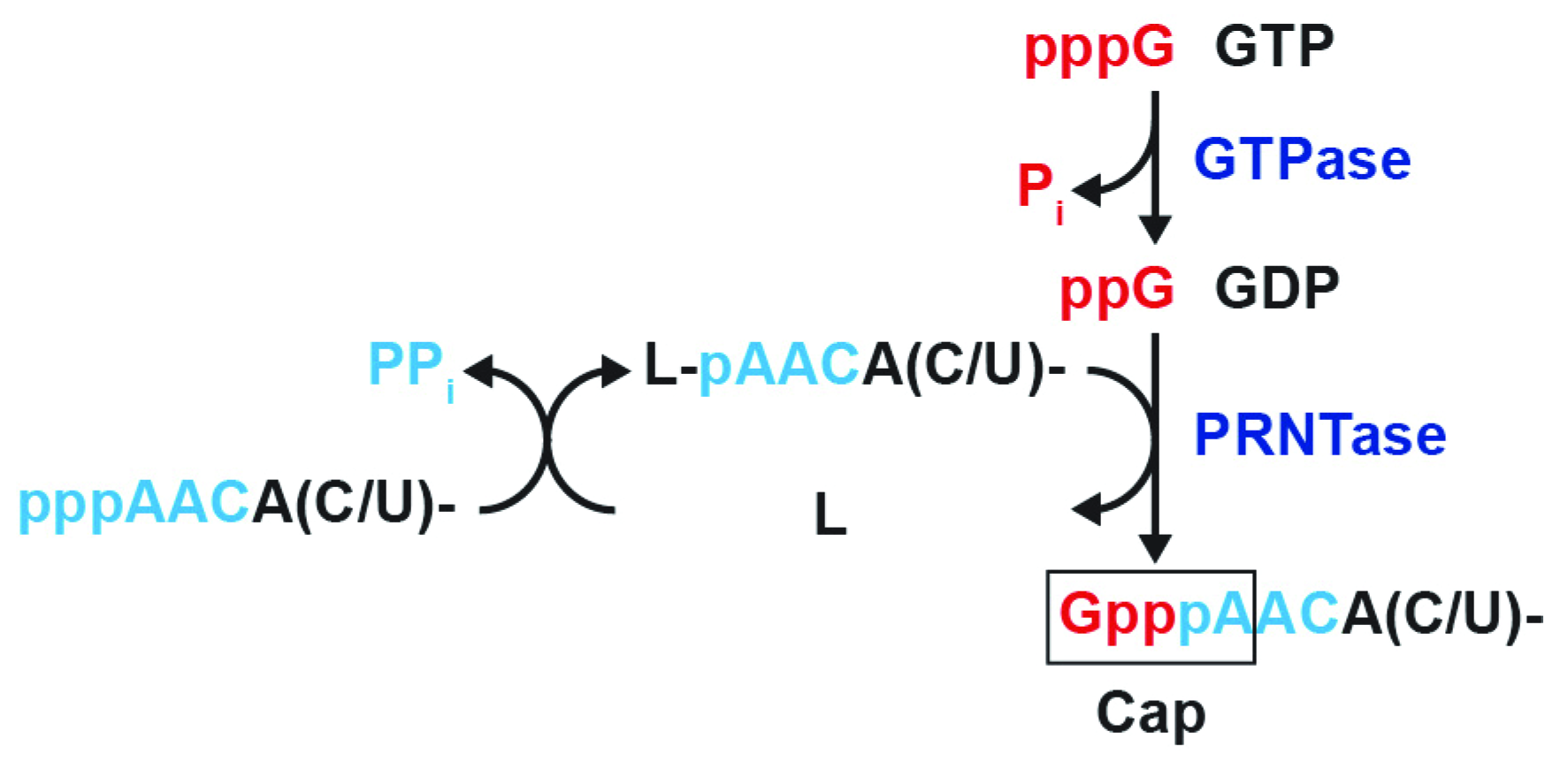
Abstract
:1. Introduction
2. Materials and Methods
2.1. Expression and Purification of the Recombinant RABV L Protein
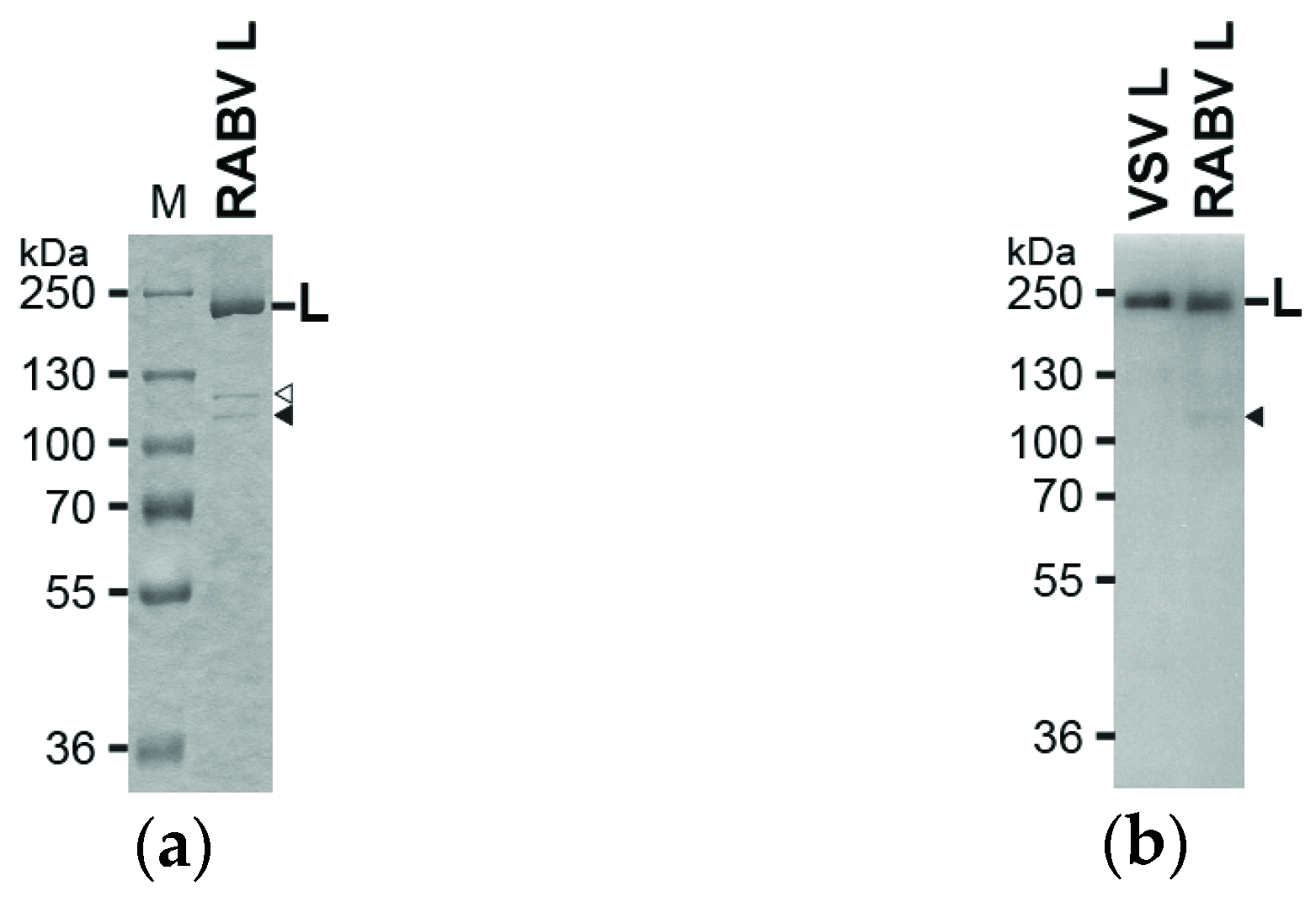
2.2. Sodium Dodecyl Sulfate Polyacrylamide Gel Electrophoresis (SDS-PAGE) and Western Blotting
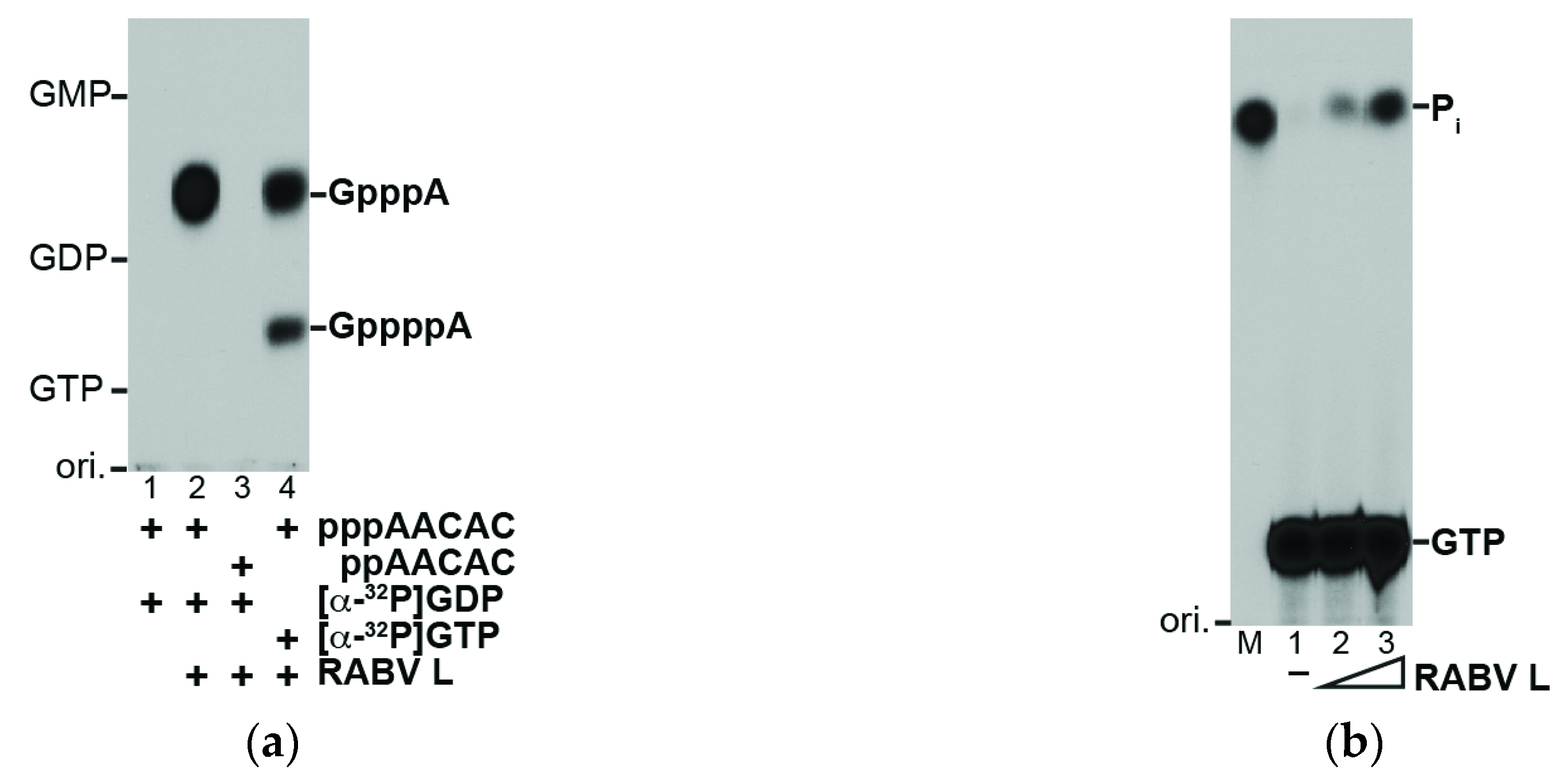
2.3. RNA Capping and GTP Hydrolysis Assays
3. Results
3.1. The RABV L Protein Catalyzes Unconventional RNA Capping
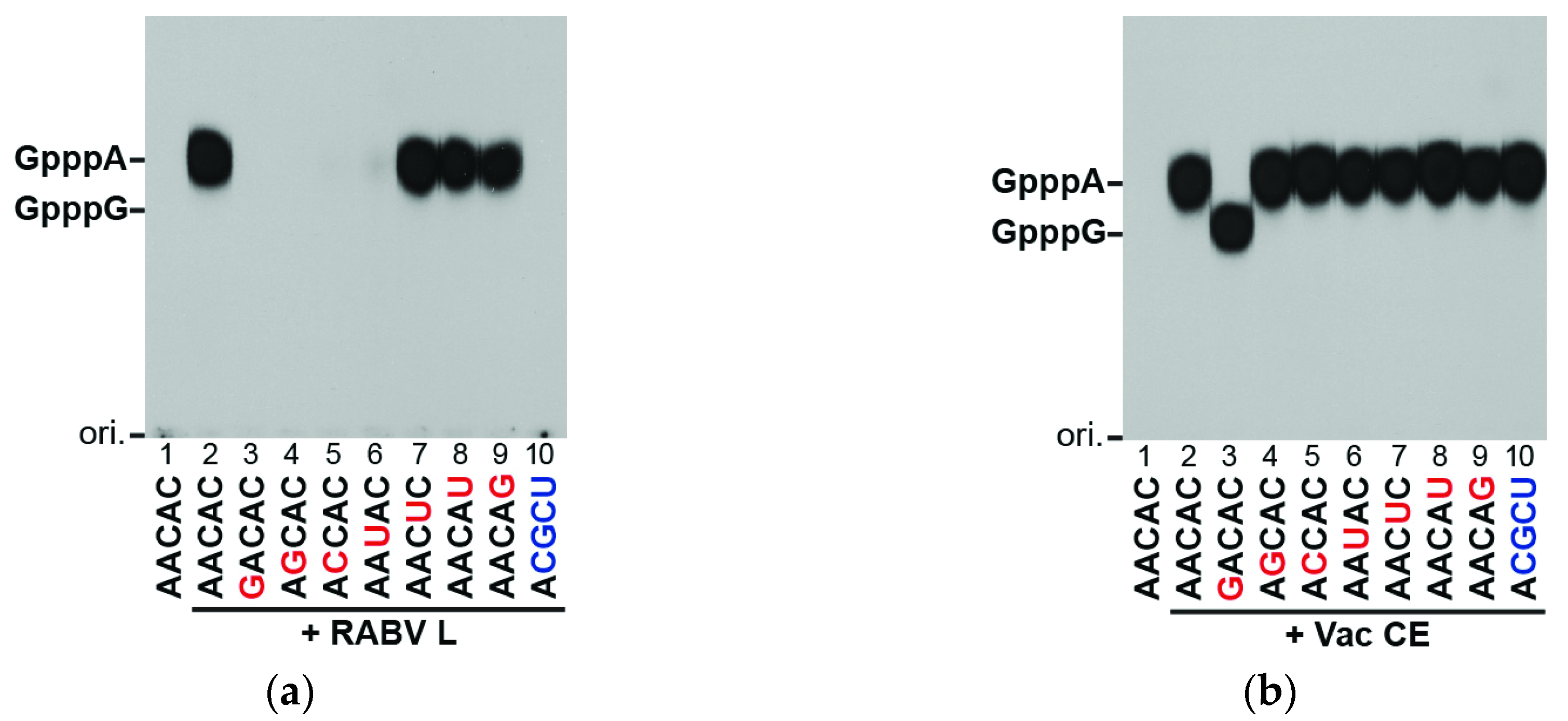
3.2. The RABV L Protein Specifically Caps the RABV mRNA-Start Sequence
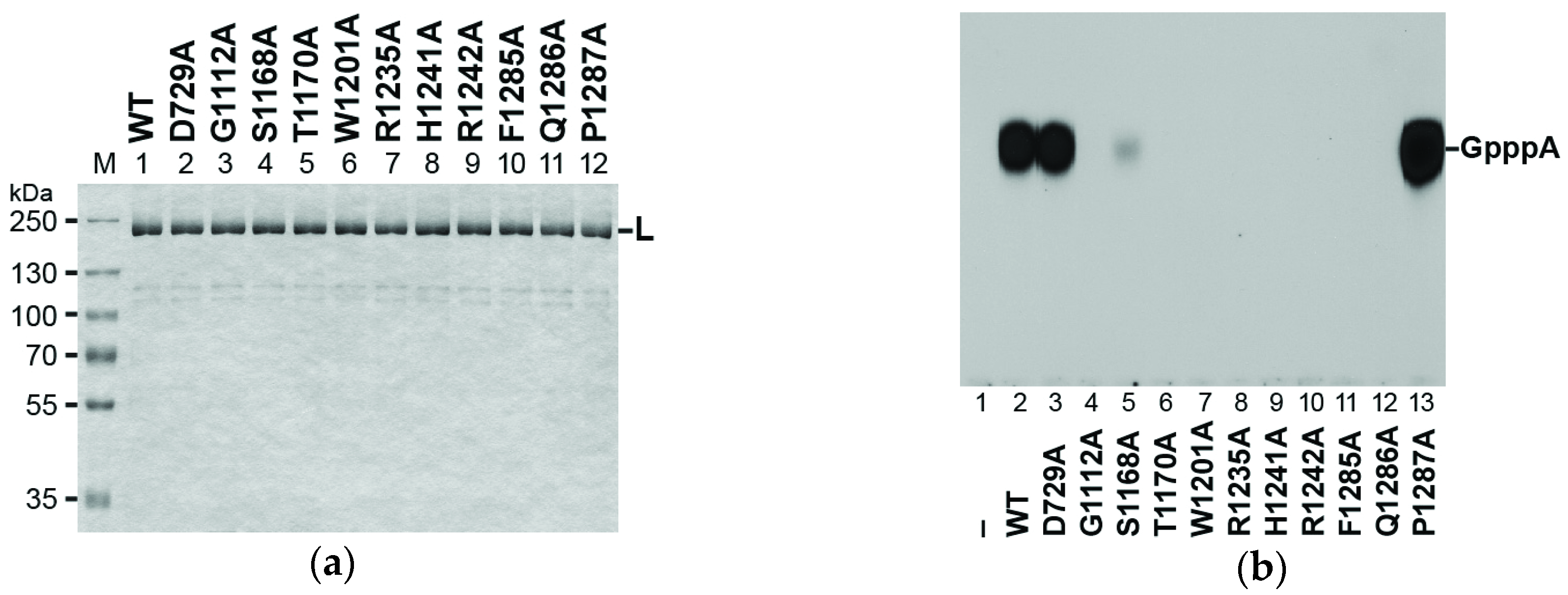
3.3. Conserved Amino Acid Residues in PRNTase Motifs A–E of the RABV L Protein are Required for mRNA Cap Formation
4. Discussion
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| RABV | rabies virus |
| VSV | vesicular stomatitis virus |
| CHPV | Chandipura virus |
| PRNTase | polyribonucleotidyltransferase |
| GTPase | guanosine 5′-triphosphatase |
| RdRp | RNA-dependent RNA polymerase |
| SDS-PAGE | sodium dodecyl sulfate polyacrylamide gel electrophoresis |
| PEI-cellulose TLC | thin layer chromatography on a polyethyleneimine-cellulose plate |
| WT | wild-type |
References
- Albertini, A.A.; Ruigrok, R.W.; Blondel, D. Rabies virus transcription and replication. Adv. Virus Res. 2011, 79, 1–22. [Google Scholar] [PubMed]
- Davis, B.M.; Rall, G.F.; Schnell, M.J. Everything you always wanted to know about rabies virus (but were afraid to ask). Annu. Rev. Virol. 2015, 2, 451–471. [Google Scholar] [CrossRef] [PubMed]
- Lankester, F.; Hampson, K.; Lembo, T.; Palmer, G.; Taylor, L.; Cleaveland, S. Infectious Disease. Implementing Pasteur’s vision for rabies elimination. Science 2014, 345, 1562–1564. [Google Scholar] [CrossRef] [PubMed]
- Johnson, N.; Vos, A.; Freuling, C.; Tordo, N.; Fooks, A.R.; Muller, T. Human rabies due to lyssavirus infection of bat origin. Vet. Microbiol. 2010, 142, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Villarreal, L.P.; Holland, J.J. Transcribing complexes in cells infected by vesicular stomatitis virus and rabies virus. J. Virol. 1974, 14, 441–450. [Google Scholar] [PubMed]
- Kawai, A. Transcriptase activity associated with rabies virion. J. Virol. 1977, 24, 826–835. [Google Scholar] [PubMed]
- Poch, O.; Blumberg, B.M.; Bougueleret, L.; Tordo, N. Sequence comparison of five polymerases (L proteins) of unsegmented negative-strand RNA viruses: Theoretical assignment of functional domains. J. Gen. Virol. 1990, 71, 1153–1162. [Google Scholar] [CrossRef] [PubMed]
- Bujnicki, J.M.; Rychlewski, L. In silico identification, structure prediction and phylogenetic analysis of the 2′-O-ribose (cap 1) methyltransferase domain in the large structural protein of ssRNA negative-strand viruses. Protein Eng. 2002, 15, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Ogino, T.; Banerjee, A.K. An unconventional pathway of mRNA cap formation by vesiculoviruses. Virus Res. 2011, 162, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Abraham, G.; Banerjee, A.K. Sequential transcription of the genes of vesicular stomatitis virus. Proc. Natl. Acad. Sci. USA 1976, 73, 1504–1508. [Google Scholar] [CrossRef] [PubMed]
- Ball, L.A.; White, C.N. Order of transcription of genes of vesicular stomatitis virus. Proc. Natl. Acad. Sci. USA 1976, 73, 442–446. [Google Scholar] [CrossRef] [PubMed]
- Testa, D.; Chanda, P.K.; Banerjee, A.K. Unique mode of transcription in vitro by Vesicular stomatitis virus. Cell 1980, 21, 267–275. [Google Scholar] [CrossRef]
- Emerson, S.U. Reconstitution studies detect a single polymerase entry site on the vesicular stomatitis virus genome. Cell 1982, 31, 635–642. [Google Scholar] [CrossRef]
- Banerjee, A.K.; Rhodes, D.P. In vitro synthesis of RNA that contains polyadenylate by virion-associated RNA polymerase of vesicular stomatitis virus. Proc. Natl. Acad. Sci. USA 1973, 70, 3566–3570. [Google Scholar] [CrossRef] [PubMed]
- Abraham, G.; Rhodes, D.P.; Banerjee, A.K. The 5′ terminal structure of the methylated mRNA synthesized in vitro by vesicular stomatitis virus. Cell 1975, 5, 51–58. [Google Scholar] [CrossRef]
- Ogino, T.; Banerjee, A.K. Unconventional mechanism of mRNA capping by the RNA-dependent RNA polymerase of vesicular stomatitis virus. Mol. Cell 2007, 25, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Ogino, T.; Banerjee, A.K. Formation of guanosine(5′)tetraphospho(5′)adenosine cap structure by an unconventional mRNA capping enzyme of vesicular stomatitis virus. J. Virol. 2008, 82, 7729–7734. [Google Scholar] [CrossRef] [PubMed]
- Ogino, T.; Banerjee, A.K. The HR motif in the RNA-dependent RNA polymerase L protein of Chandipura virus is required for unconventional mRNA-capping activity. J. Gen. Virol. 2010, 91, 1311–1134. [Google Scholar] [CrossRef] [PubMed]
- Ogino, T.; Yadav, S.P.; Banerjee, A.K. Histidine-mediated RNA transfer to GDP for unique mRNA capping by vesicular stomatitis virus RNA polymerase. Proc. Natl. Acad. Sci. USA 2010, 107, 3463–3468. [Google Scholar] [CrossRef] [PubMed]
- Shuman, S. Structure, mechanism, and evolution of the mRNA capping apparatus. Prog. Nucleic Acid Res. Mol. Biol. 2001, 66, 1–40. [Google Scholar] [PubMed]
- Ogino, T. Capping of vesicular stomatitis virus pre-mRNA is required for accurate selection of transcription stop-start sites and virus propagation. Nucleic Acids Res. 2014, 42, 12112–12125. [Google Scholar] [CrossRef] [PubMed]
- Neubauer, J.; Ogino, M.; Green, T.J.; Ogino, T. Signature motifs of GDP polyribonucleotidyltransferase, a non-segmented negative strand RNA viral mRNA capping enzyme, domain in the L protein are required for covalent enzyme-pRNA intermediate formation. Nucleic Acids Res. 2016, 44, 330–341. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Rahmeh, A.; Morelli, M.; Whelan, S.P. A conserved motif in region v of the large polymerase proteins of nonsegmented negative-sense RNA viruses that is essential for mRNA capping. J. Virol. 2008, 82, 775–784. [Google Scholar] [CrossRef] [PubMed]
- Ogino, T.; Banerjee, A.K. mRNA capping by vesicular stomatitis virus and other related viruses. In Negative Strand RNA Virus; Luo, M., Ed.; World Scientific: Singapore, 2011; pp. 79–94. [Google Scholar]
- Liang, B.; Li, Z.; Jenni, S.; Rahmeh, A.A.; Morin, B.M.; Grant, T.; Grigorieff, N.; Harrison, S.C.; Whelan, S.P. Structure of the L protein of vesicular stomatitis virus from electron cryomicroscopy. Cell 2015, 162, 314–327. [Google Scholar] [CrossRef] [PubMed]
- Ito, N.; Takayama, M.; Yamada, K.; Sugiyama, M.; Minamoto, N. Rescue of rabies virus from cloned cDNA and identification of the pathogenicity-related gene: Glycoprotein gene is associated with virulence for adult mice. J. Virol. 2001, 75, 9121–9128. [Google Scholar] [CrossRef] [PubMed]
- Ogino, T. In vitro capping and transcription of rhabdoviruses. Methods 2013, 59, 188–198. [Google Scholar] [CrossRef] [PubMed]
- Bourhy, H.; Kissi, B.; Tordo, N. Molecular diversity of the Lyssavirus genus. Virology 1993, 194, 70–81. [Google Scholar] [CrossRef] [PubMed]
- Schnell, M.J.; Conzelmann, K.K. Polymerase activity of in vitro mutated rabies virus L protein. Virology 1995, 214, 522–530. [Google Scholar] [CrossRef] [PubMed]
- Venkatesan, S.; Moss, B. Donor and acceptor specificities of HeLa cell mRNA guanylyltransferase. J. Biol. Chem. 1980, 255, 2835–2842. [Google Scholar] [PubMed]
- Martin, S.A.; Moss, B. mRNA guanylyltransferase and mRNA (guanine-7-)-methyltransferase from vaccinia virions. Donor and acceptor substrate specificites. J. Biol. Chem. 1976, 251, 7313–7321. [Google Scholar] [PubMed]
- Furuichi, Y.; Muthukrishnan, S.; Tomasz, J.; Shatkin, A.J. Mechanism of formation of reovirus mRNA 5′-terminal blocked and methylated sequence, m7GpppGmpC. J. Biol. Chem. 1976, 251, 5043–5053. [Google Scholar] [PubMed]






© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ogino, M.; Ito, N.; Sugiyama, M.; Ogino, T. The Rabies Virus L Protein Catalyzes mRNA Capping with GDP Polyribonucleotidyltransferase Activity. Viruses 2016, 8, 144. https://doi.org/10.3390/v8050144
Ogino M, Ito N, Sugiyama M, Ogino T. The Rabies Virus L Protein Catalyzes mRNA Capping with GDP Polyribonucleotidyltransferase Activity. Viruses. 2016; 8(5):144. https://doi.org/10.3390/v8050144
Chicago/Turabian StyleOgino, Minako, Naoto Ito, Makoto Sugiyama, and Tomoaki Ogino. 2016. "The Rabies Virus L Protein Catalyzes mRNA Capping with GDP Polyribonucleotidyltransferase Activity" Viruses 8, no. 5: 144. https://doi.org/10.3390/v8050144