
Differential Peripheral Blood Glycoprotein Profiles in Symptomatic and Asymptomatic COVID-19

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Biological Samples
2.2. Chemicals and Reagents
2.3. Preanalytical Sample Preparation
2.4. Liquid Chromatography–Mass Spectrometry (LC-MS) Analysis
2.5. Data Analysis
2.6. Pathway Analysis
3. Results
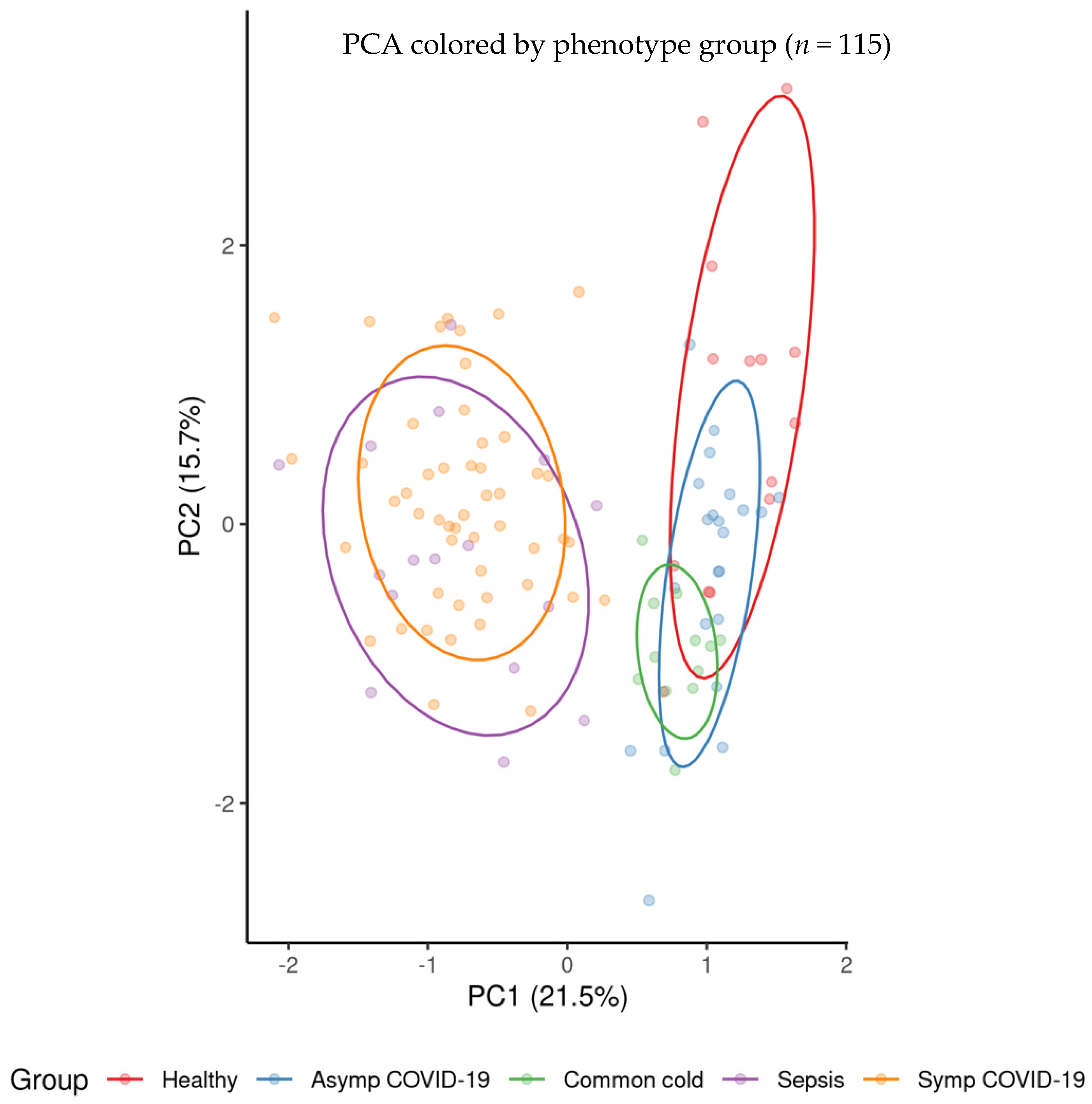
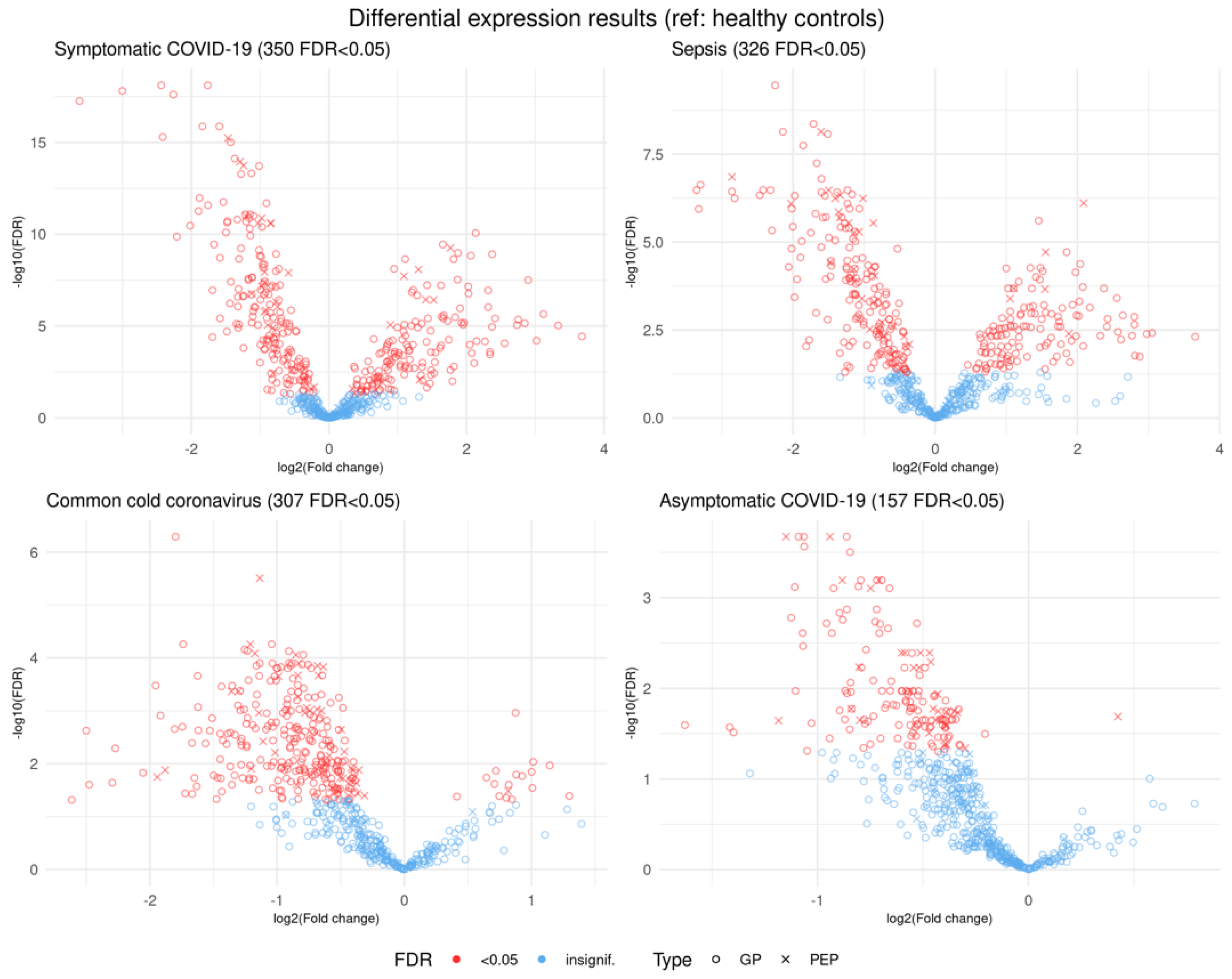
3.1. Logistic Regression Results Comparing Individual Phenotype Groups
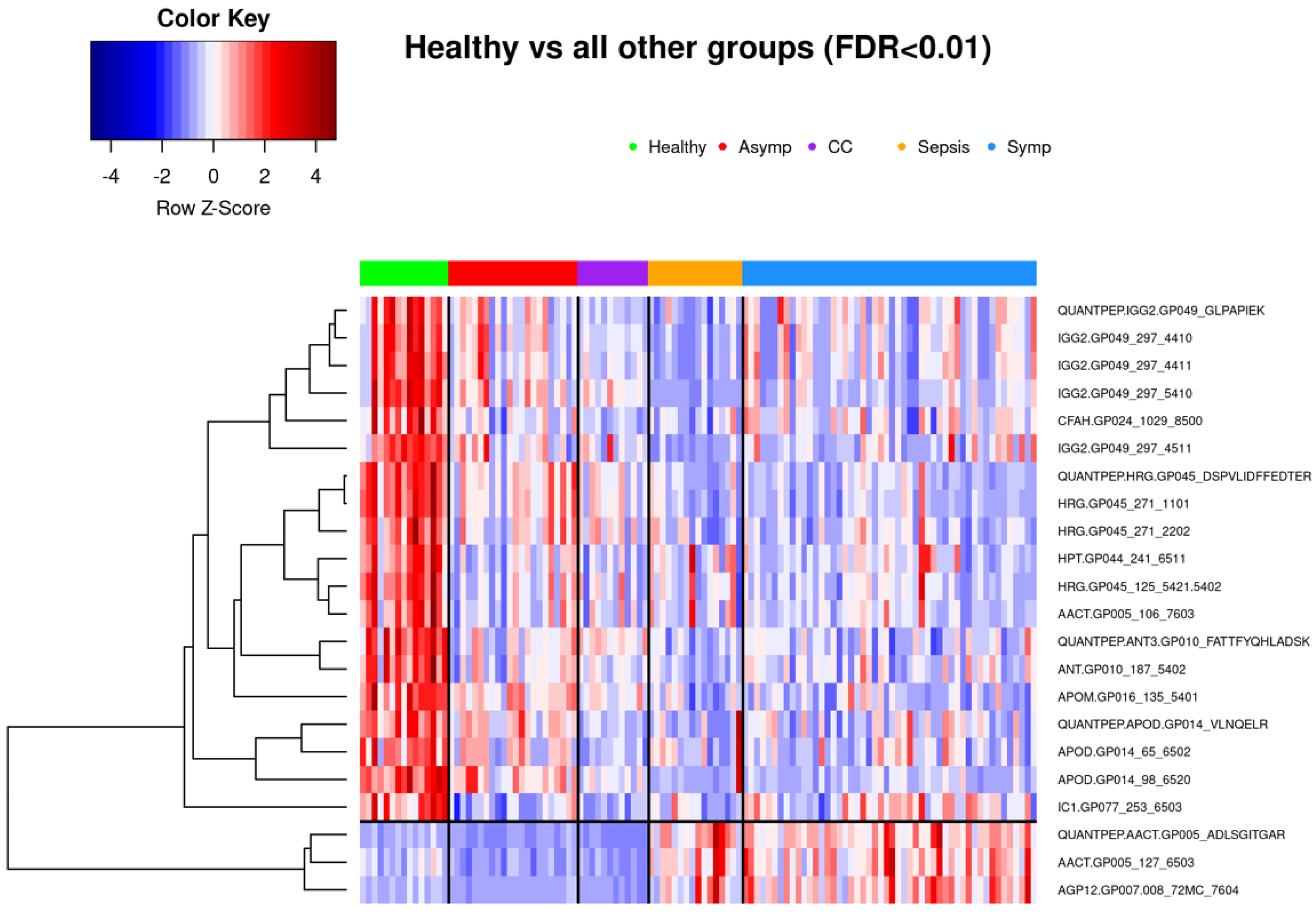
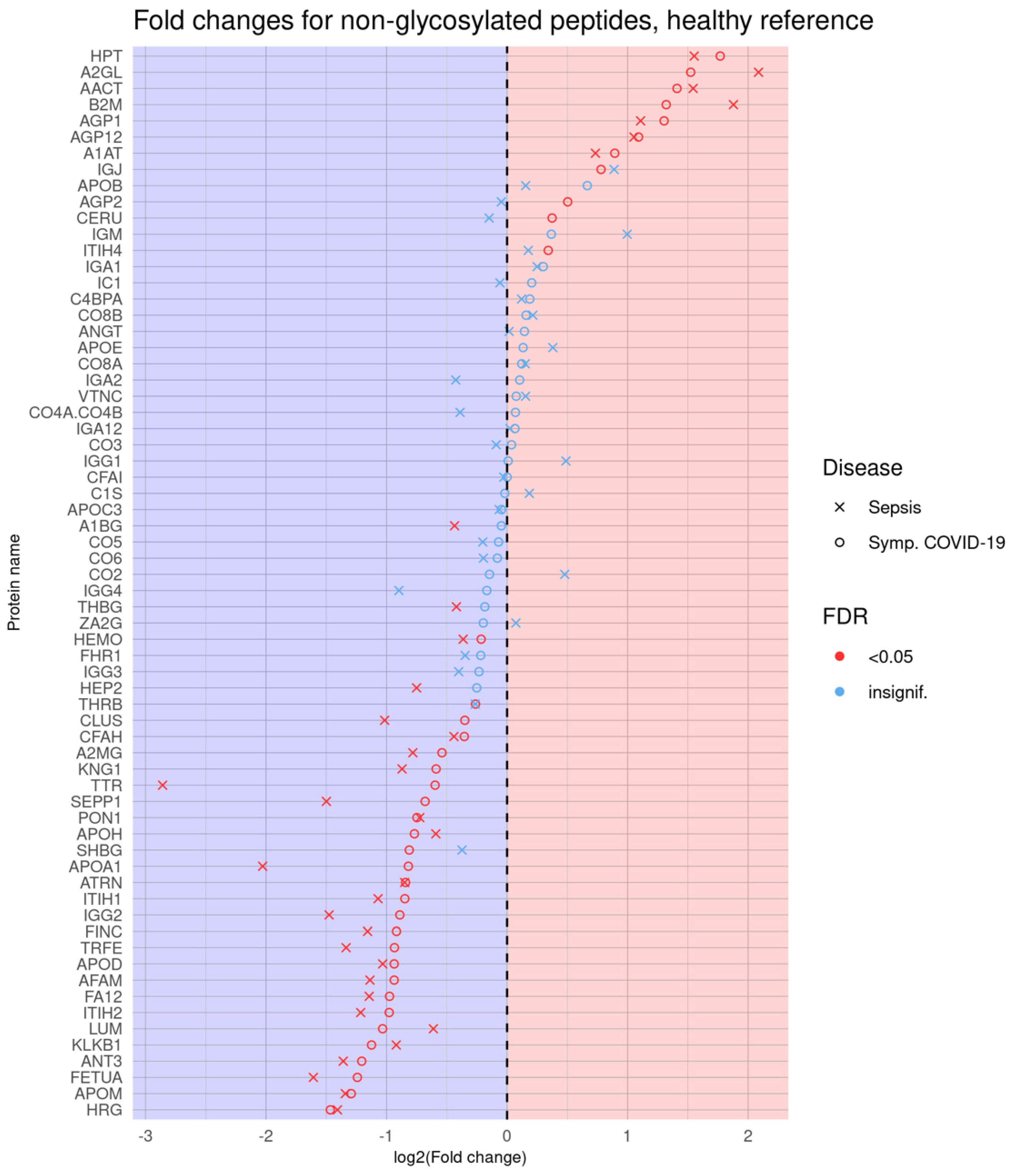
3.1.1. Comparison of Healthy Control Samples to Other Groups
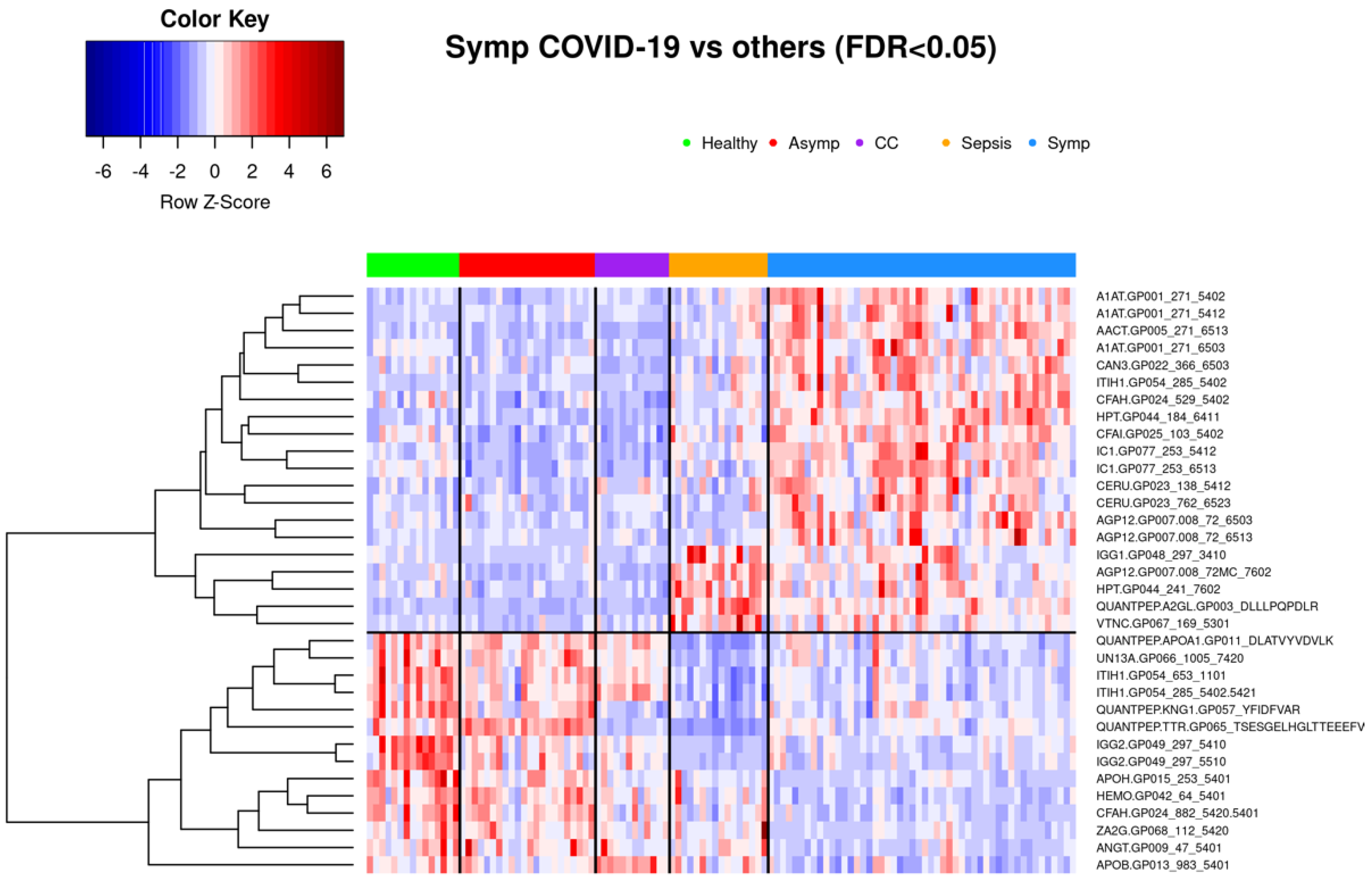
3.1.2. Comparison of Symptomatic and Asymptomatic COVID-19 Samples
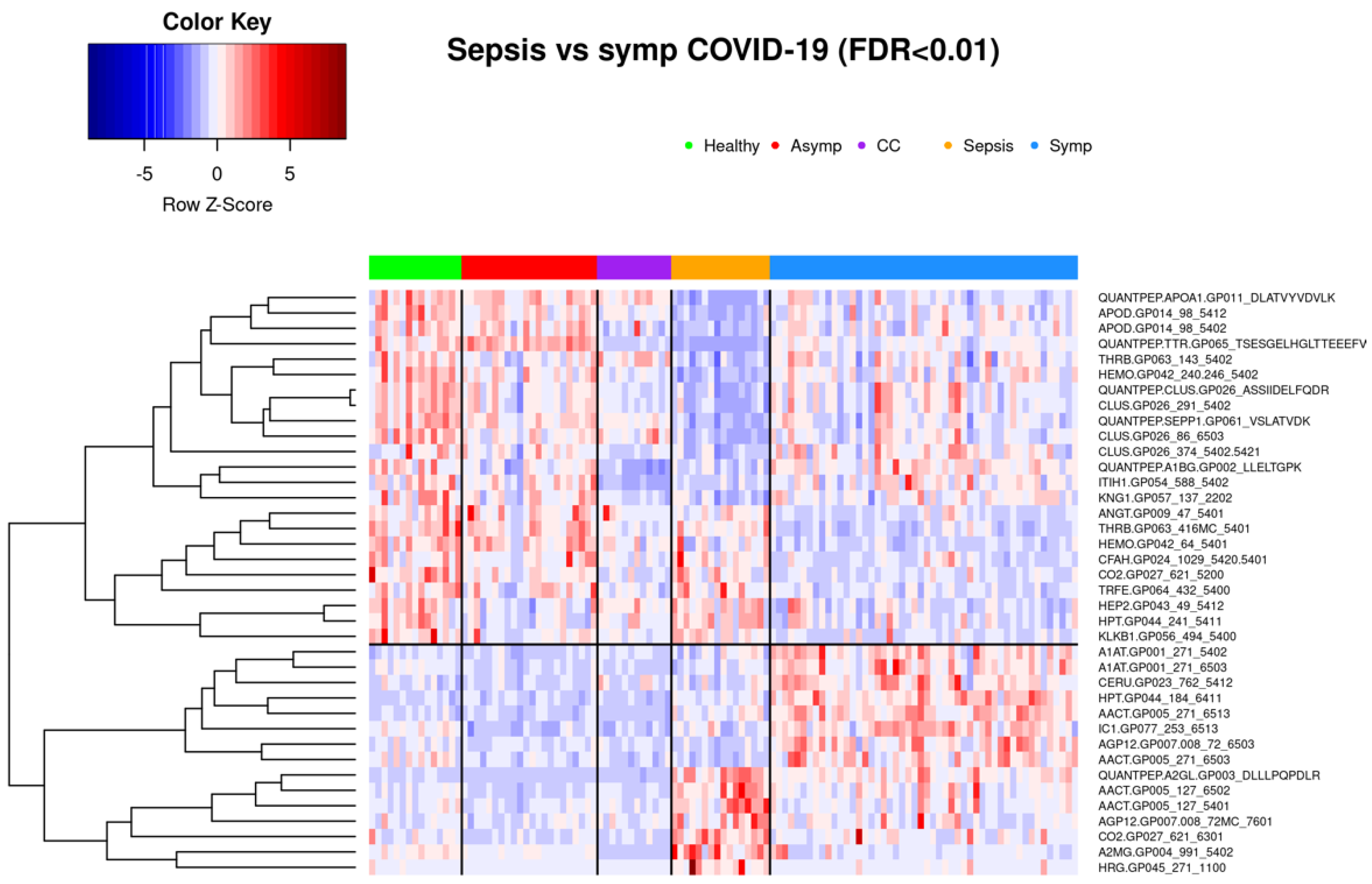
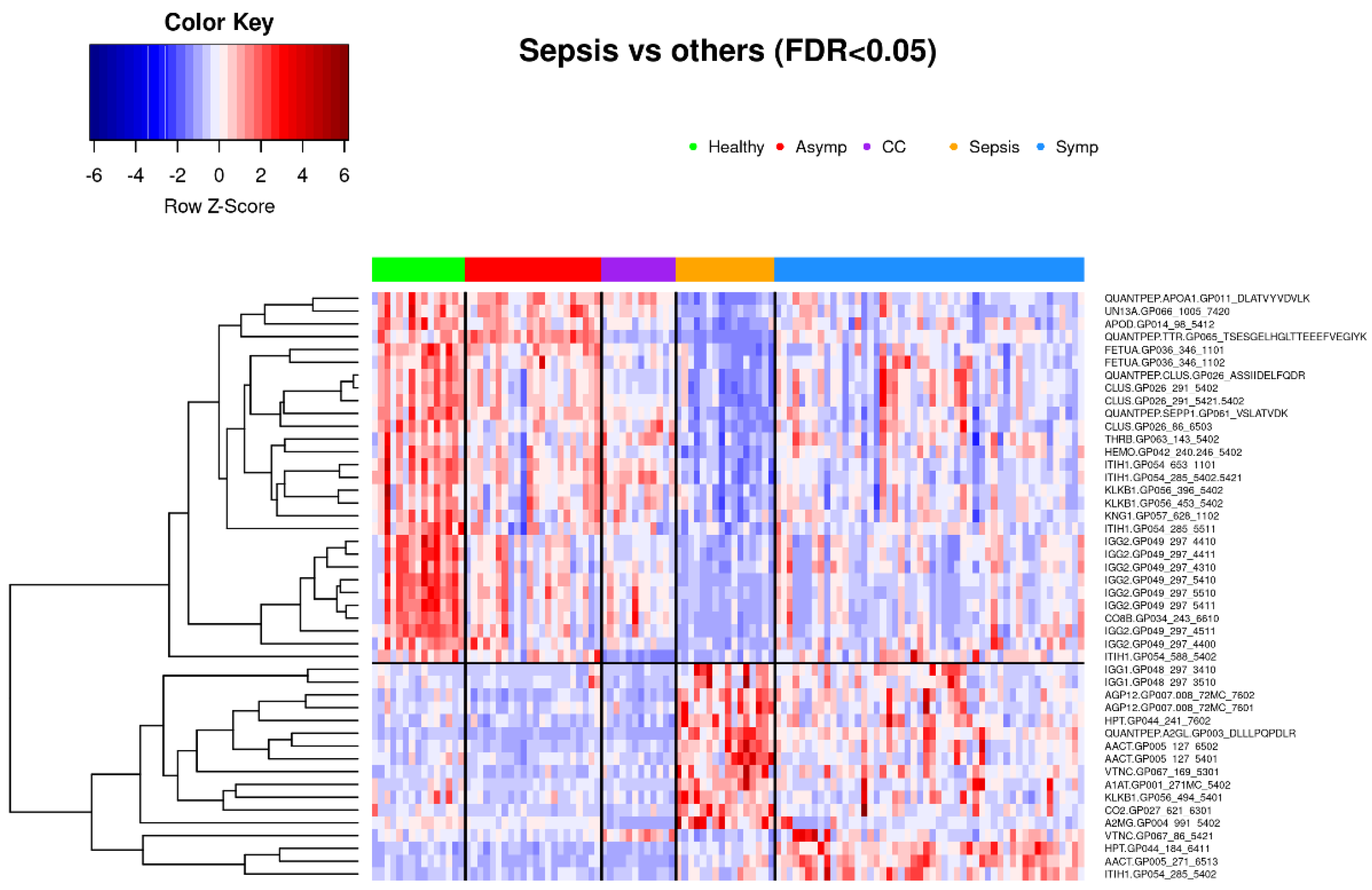
3.1.3. Comparison of Symptomatic COVID-19 and Sepsis Samples
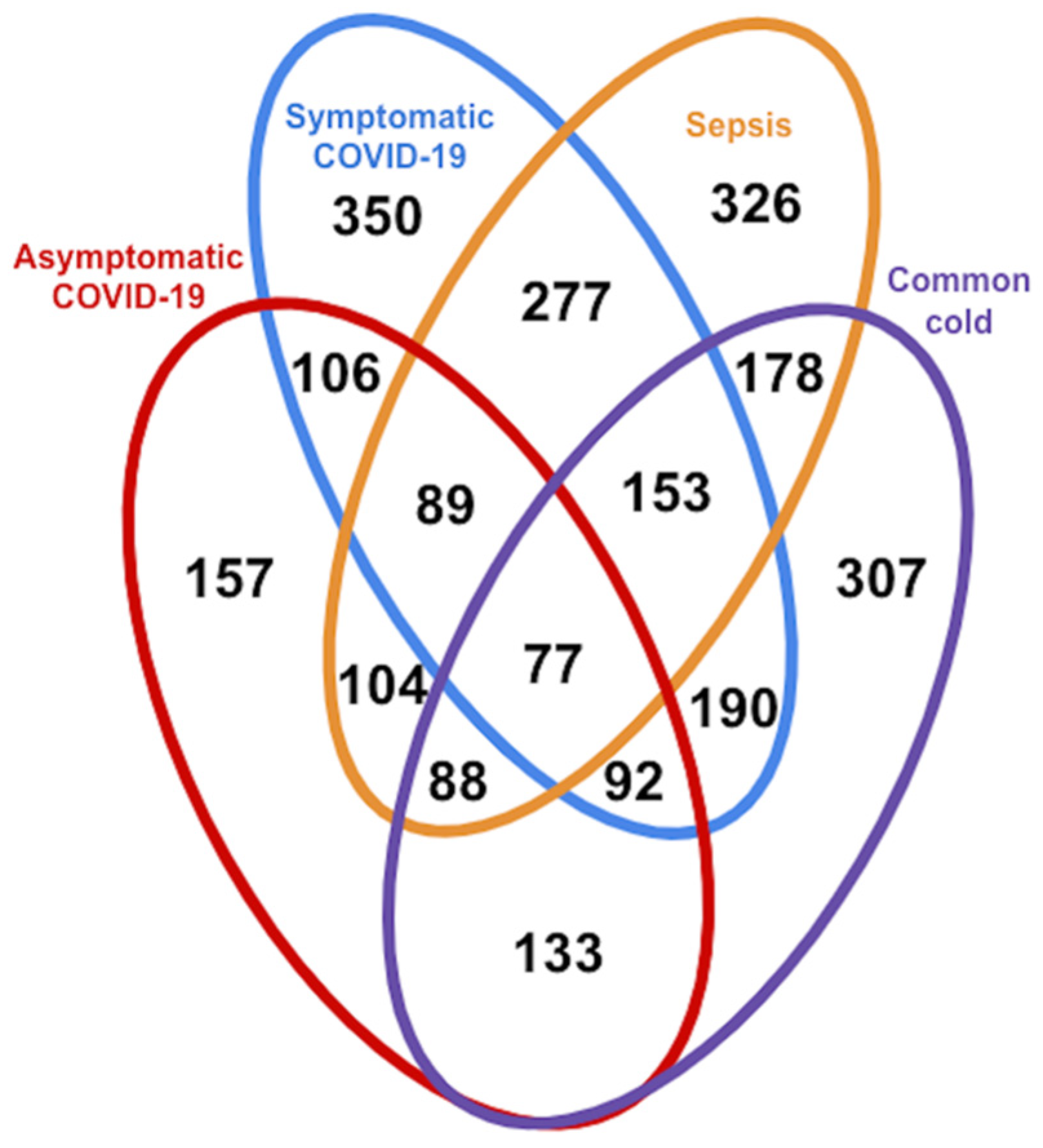
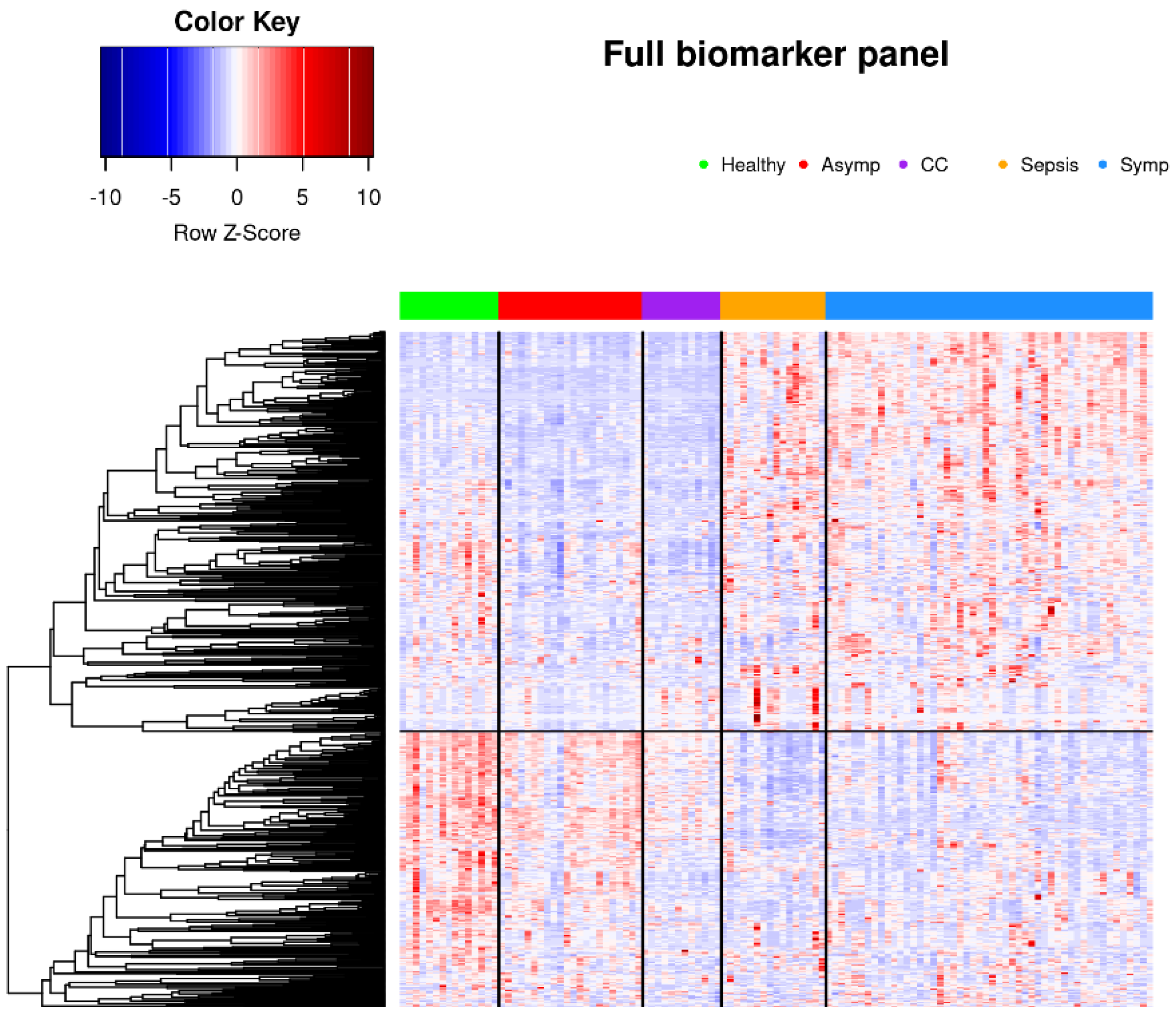
3.2. Glycoproteomic Signatures Predicting Symptomatic COVID-19 and Other Phenotype Status
3.2.1. Classification of Symptomatic COVID-19, Sepsis, and Other Samples Using K-Means Clustering
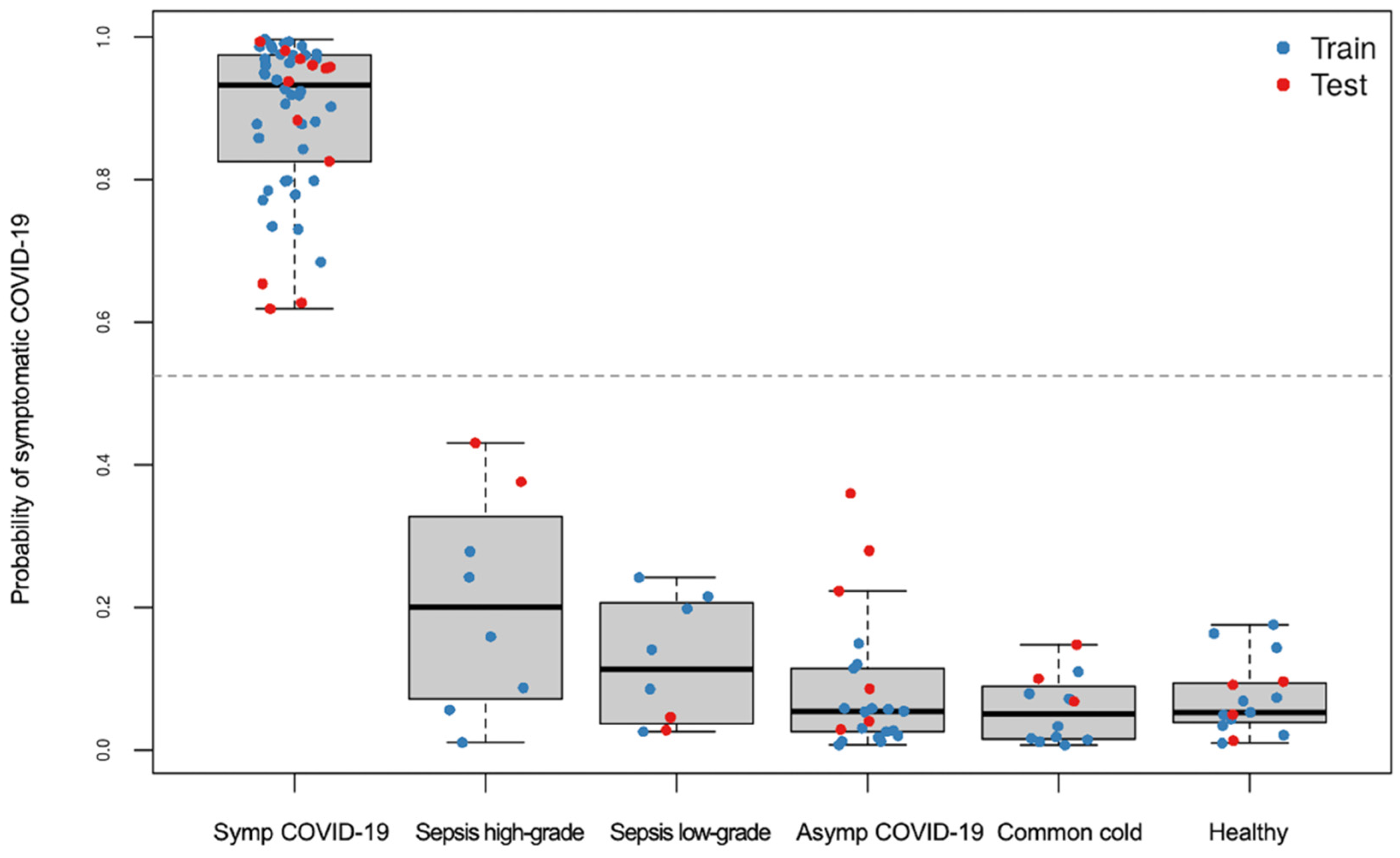
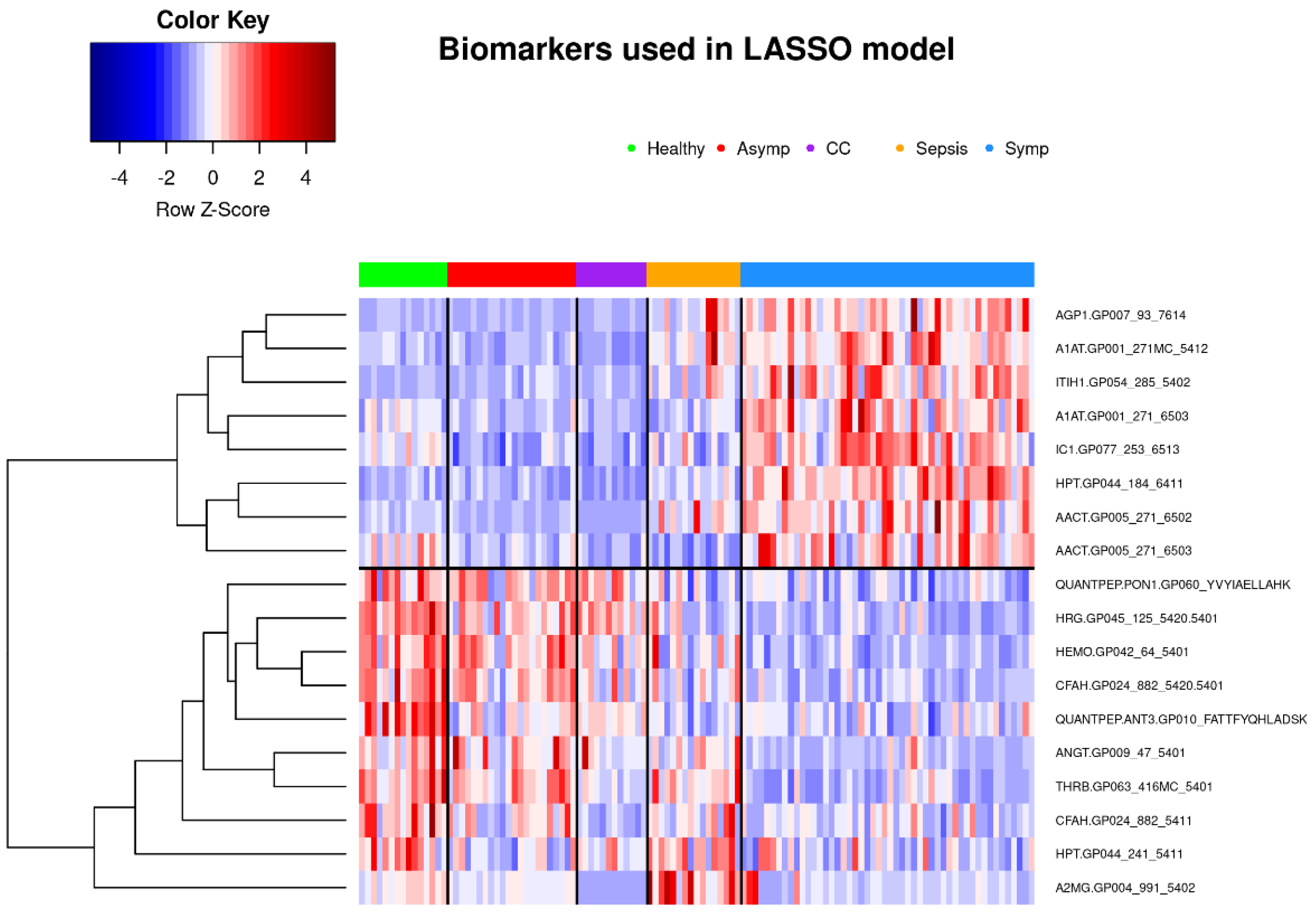
3.2.2. Classification of Symptomatic COVID-19 Using LASSO Regression
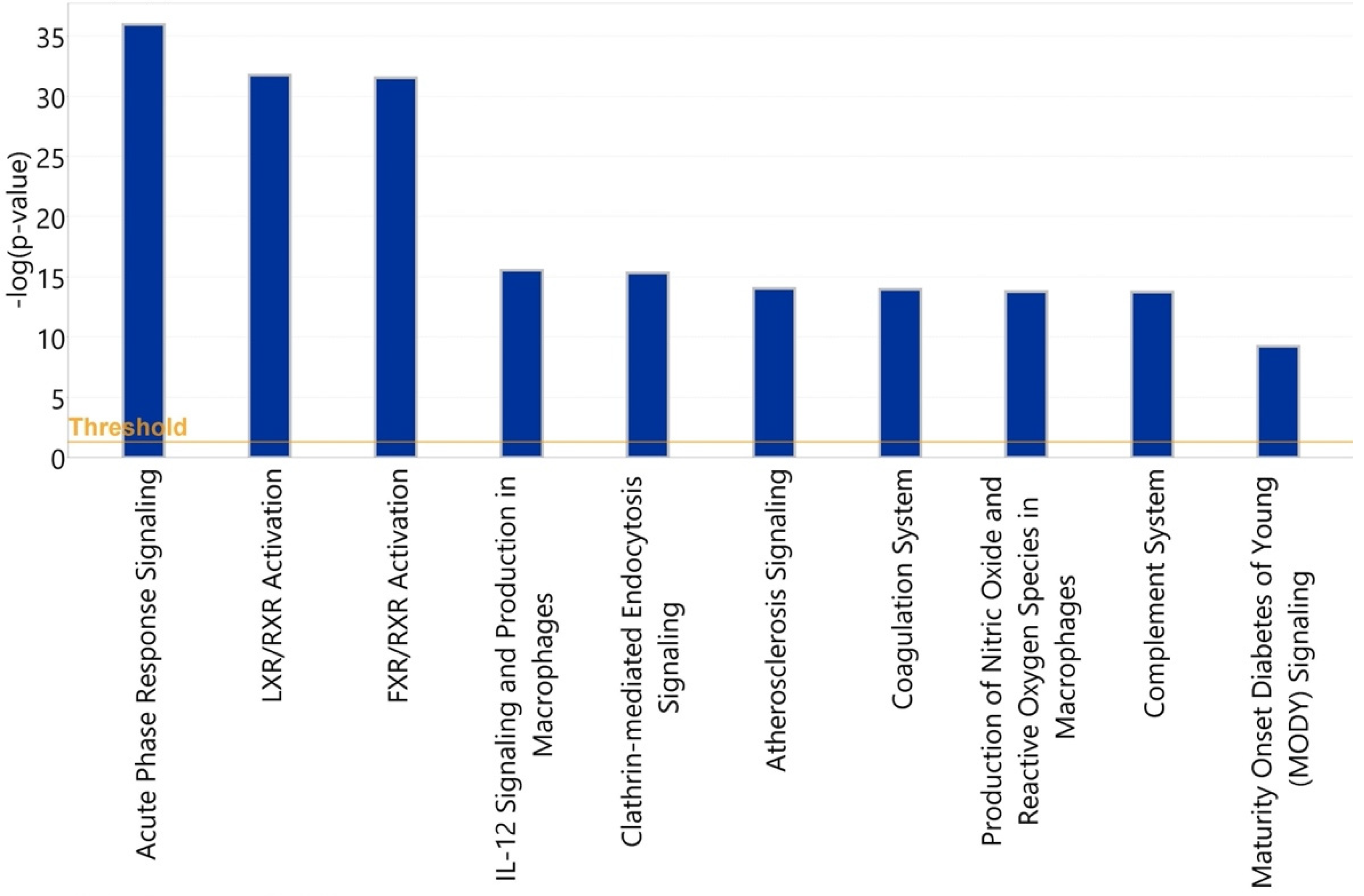
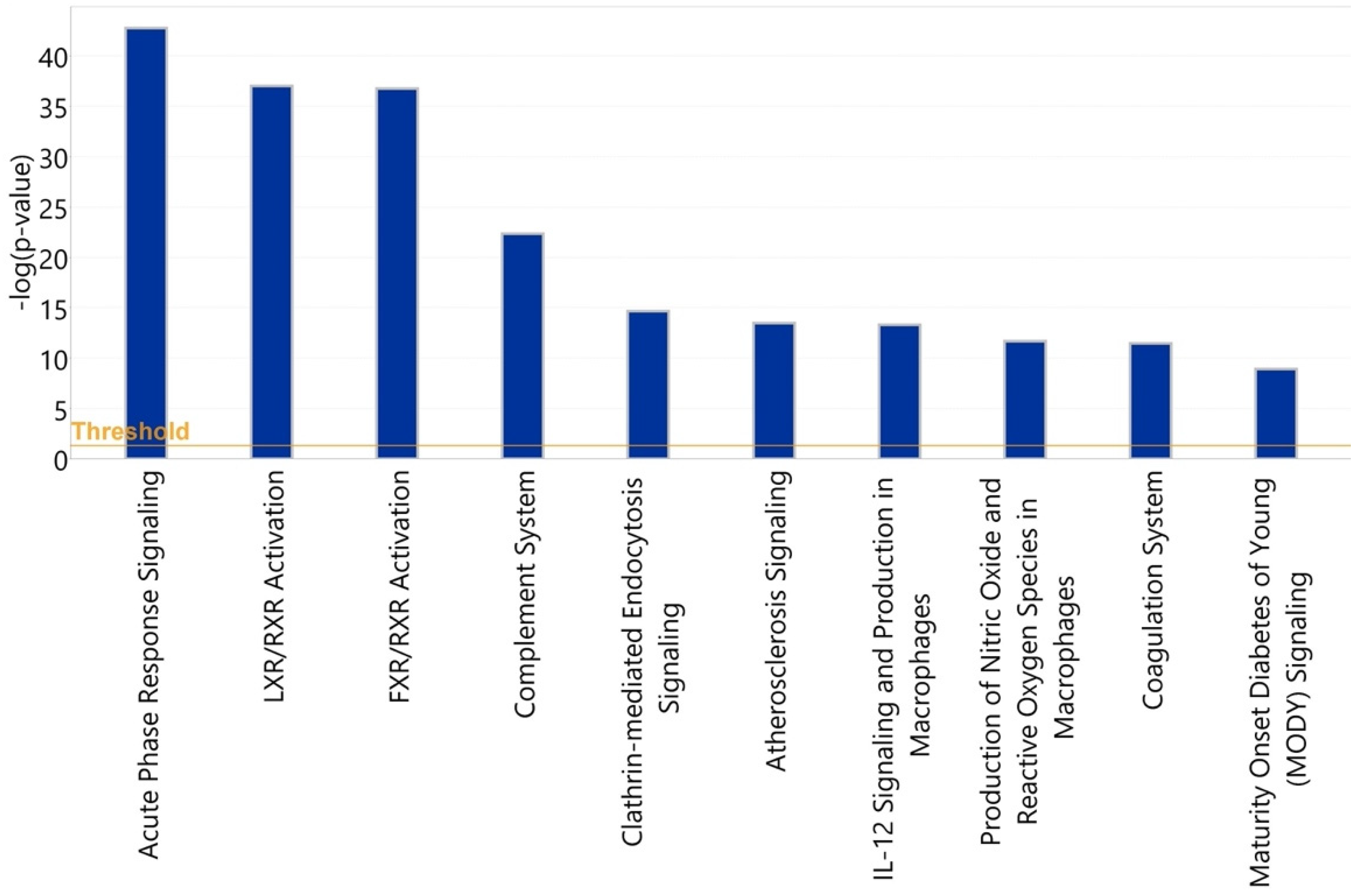
3.3. Bioinformatic Analysis of Observed Findings
3.3.1. Healthy vs. Symptomatic COVID-19
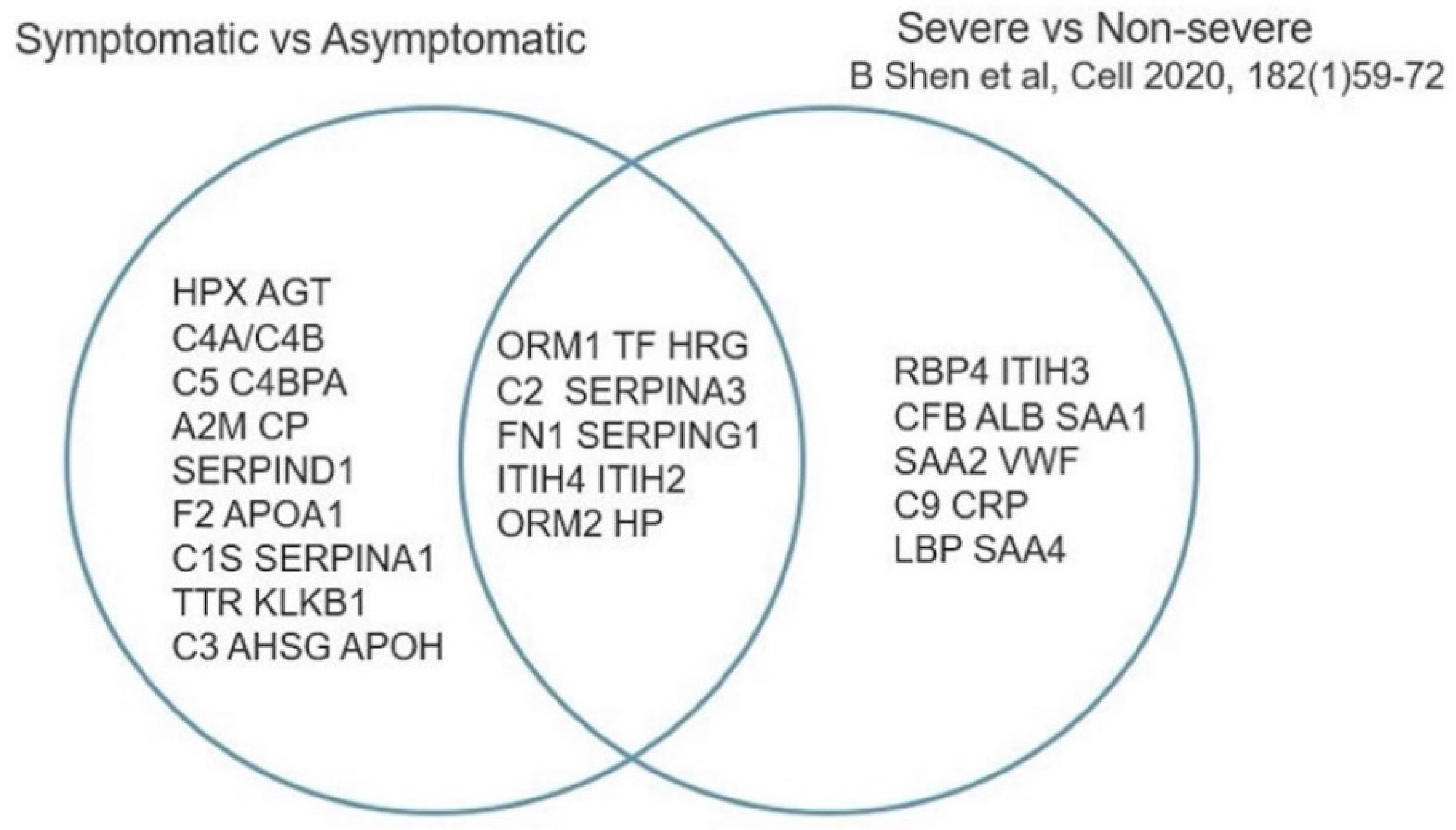
3.3.2. Asymptomatic vs. Symptomatic COVID-19
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Samprathi, M.; Jayashree, M. Biomarkers in COVID-19: An Up-To-Date Review. Front. Pediatr. 2020, 8, 607647. [Google Scholar] [CrossRef] [PubMed]
- Shkurnikov, M.; Nersisyan, S.; Jankevic, T.; Galatenko, A.; Gordeev, I.; Vechorko, V.; Tonevitsky, A. Association of HLA Class I Genotypes With Severity of Coronavirus Disease-19. Front. Immunol 2021, 12, 641900. [Google Scholar] [CrossRef] [PubMed]
- Beyerstedt, S.; Casaro, E.B.; Rangel, E.B. COVID-19: Angiotensin-converting enzyme 2 (ACE2) expression and tissue susceptibility to SARS-CoV-2 infection. Eur. J. Clin. Microbiol. Infect. Dis. 2021, 40, 905–919. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Kruger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280. [Google Scholar] [CrossRef]
- Fricke-Galindo, I.; Falfan-Valencia, R. Genetics Insight for COVID-19 Susceptibility and Severity: A Review. Front. Immunol. 2021, 12, 622176. [Google Scholar] [CrossRef]
- Kaiser, J. DNA test to predict odds of severe COVID-19 draws scrutiny. Science 2021, 372, 1139. [Google Scholar] [CrossRef]
- Park, J.; Kim, H.; Kim, S.Y.; Kim, Y.; Lee, J.S.; Dan, K.; Seong, M.W.; Han, D. In-depth blood proteome profiling analysis revealed distinct functional characteristics of plasma proteins between severe and non-severe COVID-19 patients. Sci. Rep. 2020, 10, 22418. [Google Scholar] [CrossRef]
- Stukalov, A.; Girault, V.; Grass, V.; Karayel, O.; Bergant, V.; Urban, C.; Haas, D.A.; Huang, Y.; Oubraham, L.; Wang, A.; et al. Multilevel proteomics reveals host perturbations by SARS-CoV-2 and SARS-CoV. Nature 2021, 594, 246–252. [Google Scholar] [CrossRef]
- Shu, T.; Ning, W.; Wu, D.; Xu, J.; Han, Q.; Huang, M.; Zou, X.; Yang, Q.; Yuan, Y.; Bie, Y.; et al. Plasma Proteomics Identify Biomarkers and Pathogenesis of COVID-19. Immunity 2020, 53, 1108–1122. [Google Scholar] [CrossRef]
- Prucha, M.; Bellingan, G.; Zazula, R. Sepsis biomarkers. Clin. Chim. Acta 2015, 440, 97–103. [Google Scholar] [CrossRef]
- Kim, M.H.; Choi, J.H. An Update on Sepsis Biomarkers. Infect. Chemother. 2020, 52, 1–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caval, T.; Lin, Y.H.; Varkila, M.; Reiding, K.R.; Bonten, M.J.M.; Cremer, O.L.; Franc, V.; Heck, A.J.R. Glycoproteoform Profiles of Individual Patients’ Plasma Alpha-1-Antichymotrypsin are Unique and Extensively Remodeled Following a Septic Episode. Front. Immunol. 2020, 11, 608466. [Google Scholar] [CrossRef] [PubMed]
- Novokmet, M.; Lukic, E.; Vuckovic, F.; Ethuric, Z.; Keser, T.; Rajsl, K.; Remondini, D.; Castellani, G.; Gasparovic, H.; Gornik, O.; et al. Changes in IgG and total plasma protein glycomes in acute systemic inflammation. Sci. Rep. 2014, 4, 4347. [Google Scholar] [CrossRef] [Green Version]
- Joenvaara, S.; Saraswat, M.; Kuusela, P.; Saraswat, S.; Agarwal, R.; Kaartinen, J.; Jarvinen, A.; Renkonen, R. Quantitative N-glycoproteomics reveals altered glycosylation levels of various plasma proteins in bloodstream infected patients. PLoS ONE 2018, 13, e0195006. [Google Scholar] [CrossRef] [PubMed]
- Sorrentino, J.T.; Toledo, A.G.; Golden, G.; Diaz-Peña, R.; Campos, A.R.; Nizet, V.; Malmstrom, J.; Smith, J.W.; Lewis, N.E.; Esko, J.D. Decoding Glycoproteome Remodeling in Sepsis through Integrative Multi-Omics Analysis and Parts-Based Data Representation. FASEB J. 2020, 34, 1. [Google Scholar] [CrossRef]
- De Coux, A.; Tian, Y.; De Leon-Pennell, K.Y.; Nguyen, N.T.; de Castro Bras, L.E.; Flynn, E.R.; Cannon, P.L.; Griswold, M.E.; Jin, Y.F.; Puskarich, M.A.; et al. Plasma Glycoproteomics Reveals Sepsis Outcomes Linked to Distinct Proteins in Common Pathways. Crit. Care Med. 2015, 43, 2049–2058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larsen, M.D.; de Graaf, E.L.; Sonneveld, M.E.; Plomp, H.R.; Nouta, J.; Hoepel, W.; Chen, H.J.; Linty, F.; Visser, R.; Brinkhaus, M.; et al. Afucosylated IgG characterizes enveloped viral responses and correlates with COVID-19 severity. Science 2021, 371, eabc8378. [Google Scholar] [CrossRef]
- Chakraborty, S.; Gonzalez, J.; Edwards, K.; Mallajosyula, V.; Buzzanco, A.S.; Sherwood, R.; Buffone, C.; Kathale, N.; Providenza, S.; Xie, M.M.; et al. Proinflammatory IgG Fc structures in patients with severe COVID-19. Nat. Immunol. 2021, 22, 67–73. [Google Scholar] [CrossRef]
- Available online: https://scikit-learn.org/stable (accessed on 25 February 2022).
- Wu, Z.; Serie, D.; Xu, G.; Zou, J. PB-Net: Automatic peak integration by sequential deep learning for multiple reaction monitoring. J. Proteom. 2020, 223, 103820. [Google Scholar] [CrossRef]
- Shen, B.; Yi, X.; Sun, Y.; Bi, X.; Du, J.; Zhang, C.; Quan, S.; Zhang, F.; Sun, R.; Qian, L.; et al. Proteomic and Metabolomic Characterization of COVID-19 Patient Sera. Cell 2020, 182, 59–72. [Google Scholar] [CrossRef]
- Pinho, S.S.; Reis, C.A. Glycosylation in cancer: Mechanisms and clinical implications. Nat. Rev. Cancer 2015, 15, 540–555. [Google Scholar] [CrossRef] [PubMed]
- Pietrobono, S.; Stecca, B. Aberrant Sialylation in Cancer: Biomarker and Potential Target for Therapeutic Intervention? Cancers 2021, 13, 2014. [Google Scholar] [CrossRef] [PubMed]
- Keeley, T.S.; Yang, S.; Lau, E. The Diverse Contributions of Fucose Linkages in Cancer. Cancers 2019, 11, 1241. [Google Scholar] [CrossRef] [Green Version]
- Wong, A.H.; Fukami, Y.; Sudo, M.; Kokubun, N.; Hamada, S.; Yuki, N. Sialylated IgG-Fc: A novel biomarker of chronic inflammatory demyelinating polyneuropathy. J. Neurol. Neurosurg. Psychiatry 2016, 87, 275–279. [Google Scholar] [CrossRef]
- Bohm, S.; Schwab, I.; Lux, A.; Nimmerjahn, F. The role of sialic acid as a modulator of the anti-inflammatory activity of IgG. In Seminars in Immunopathology; Springer: Berlin/Heidelberg, Germany, 2012; pp. 443–453. [Google Scholar]
- Li, D.; Lou, Y.; Zhang, Y.; Liu, S.; Li, J.; Tao, J. Sialylated immunoglobulin G: A promising diagnostic and therapeutic strategy for autoimmune diseases. Theranostics 2021, 11, 5430–5446. [Google Scholar] [CrossRef] [PubMed]
- Winzler, R.J. Plasma Proteins in Cancer. In Advances in Cancer Research; Academic Press: Cambridge, MA, USA, 1953; pp. 503–548. [Google Scholar]
- Gralinski, L.E.; Sheahan, T.P.; Morrison, T.E.; Menachery, V.D.; Jensen, K.; Leist, S.R.; Whitmore, A.; Heise, M.T.; Baric, R.S. Complement Activation Contributes to Severe Acute Respiratory Syndrome Coronavirus Pathogenesis. mBio 2018, 9, e01753-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, T.; Hu, M.; Zhang, X.; Li, H.; Zhu, L.; Liu, H.; Dong, Q.; Zhang, Z.; Wang, Z.; Hu, Y.; et al. Highly pathogenic coronavirus N protein aggravates lung injury by MASP-2-mediated complement over-activation. medRxiv 2020. [Google Scholar] [CrossRef]


















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Phenotype | Source | N * | Serum | Plasma | Train Set | Test Set | Male | Female | Med. Age (IQR) |
|---|---|---|---|---|---|---|---|---|---|
| Symptomatic COVID-19+ | Kaiser Permanente | 50 | 39 | 11 | 38 | 12 | 25 | 15 ** | 55.5 (50.5, 67.3) |
| Bacterial sepsis | U of Florida, Jacksonville | 16 | 0 | 16 | 12 | 4 | 11 | 5 | 60.5 (57, 73.3) |
| Common cold coronavirus | Stanford Blood Bank | 12 | 0 | 12 | 9 | 3 | n/a | n/a | n/a |
| Asymptomatic COVID-19+ | Stanford Blood Bank | 22 | 22 | 0 | 16 | 6 | 10 | 12 | 49 (40.3, 61) |
| Healthy control | Stanford Blood Bank | 15 | 15 | 0 | 11 | 4 | n/a | n/a | n/a |
| Phenotype | Predicted Cluster | ||
|---|---|---|---|
| True phenotype * | 1 | 2 | 3 |
| Symptomatic COVID-19 | 47 | 0 | 3 |
| Bacterial sepsis | 2 | 0 | 14 |
| Other phenotype | 0 | 49 | 0 |
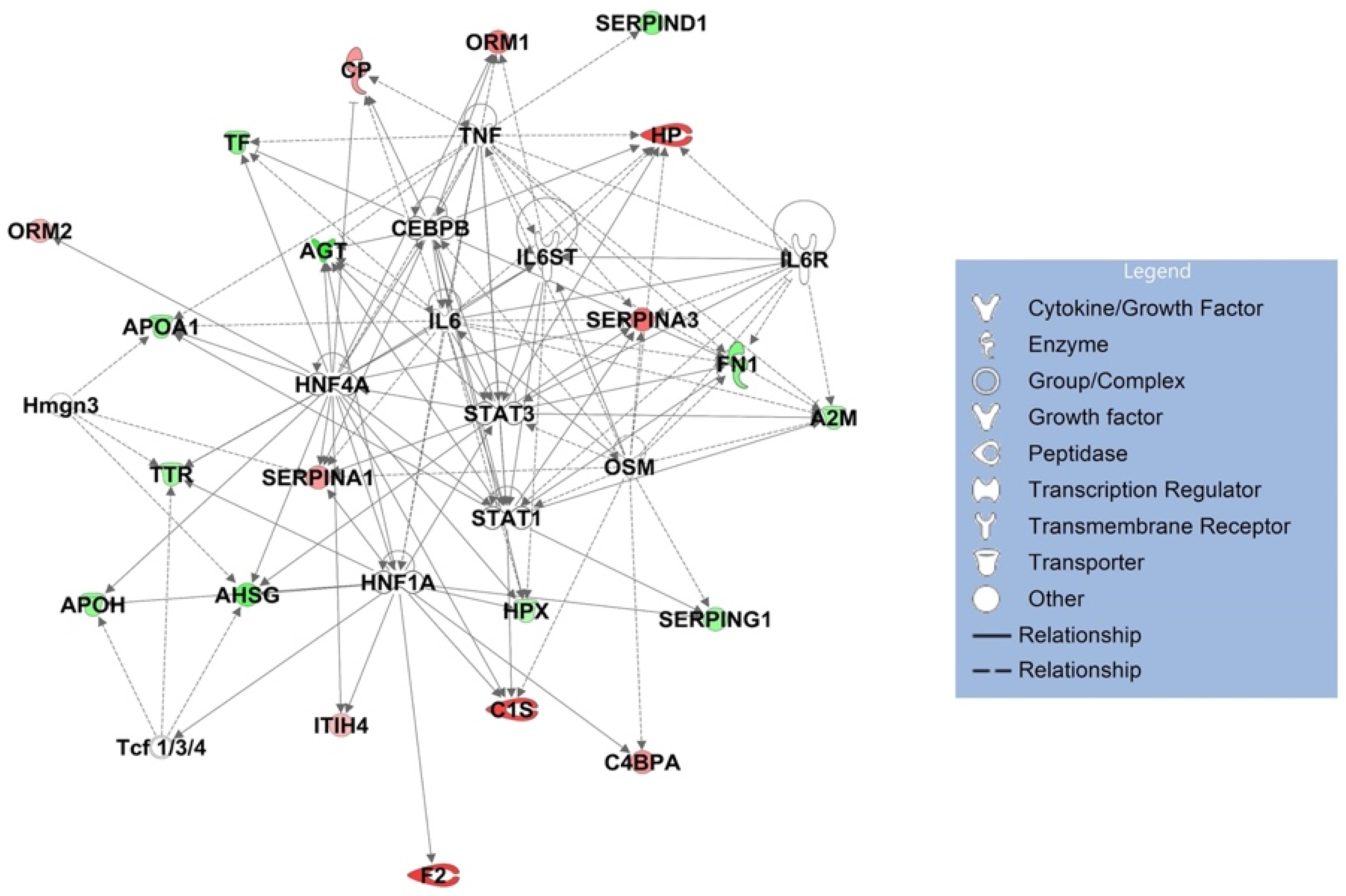
| Upstream Regulator | Molecule Type | p-Value of Target Molecules in Dataset | Target Molecules in Dataset |
|---|---|---|---|
| HNF1A | transcription regulator | 1.05 × 10−14 | AGT, AHSG, APOH, ATP, C1S, C4BPA, F2, HPX, ITIH4, SERPINA1, SERPING1, TTR |
| IL6 | cytokine | 1.24 × 10−13 | A2M, AGT, APOA1, ATP, CP, FN1, HP, HPX, ORM1, SERPINA1, SERPINA3, TF, TTR |
| HNF4A | transcription regulator | 1.1 × 10−10 | AGT, AHSG, APOA1, APOH, ATP, C1S, CP, HPX, ITIH4, ORM1, ORM2, SERPINA1, SERPINA3, TF, TTR |
| Tcf 1/3/4 | group | 1.66 × 10−8 | AHSG, APOH, TTR |
| Hmgn3 | other | 3.26 × 10−8 | AHSG, APOA1, SERPINA1, TTR |
| OSM | cytokine | 4.43 × 10−8 | A2M, C1S, C4BPA, FN1, HP, SERPINA1, SERPINA3, SERPING1 |
| CEBPB | transcription regulator | 8.56 × 10−8 | AGT, CP, FN1, HP, HPX, ORM1, SERPINA1, TF |
| STAT1 | transcription regulator | 1.21 × 10−7 | A2M, AGT, APOA1, C1S, FN1, SERPINA3, SERPING1 |
| STAT3 | transcription regulator | 1.66 × 10−7 | A2M, AGT, AHSG, ATP, FN1, HP, SERPINA1, SERPINA3 |
| IL6ST | transmembrane receptor | 6.29 × 10−7 | A2M, HP, HPX, ORM1 |
| TNF | cytokine | 1.13 × 10−6 | A2M, AGT, APOA1, ATP, CP, FN1, HP, ORM1, SERPINA3, SERPIND1, TF |
| IL6R | transmembrane receptor | 1.58 × 10−6 | A2M, FN1, HP, SERPINA3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pickering, C.; Zhou, B.; Xu, G.; Rice, R.; Ramachandran, P.; Huang, H.; Pham, T.D.; Schapiro, J.M.; Cong, X.; Chakraborty, S.; et al. Differential Peripheral Blood Glycoprotein Profiles in Symptomatic and Asymptomatic COVID-19. Viruses 2022, 14, 553. https://doi.org/10.3390/v14030553
Pickering C, Zhou B, Xu G, Rice R, Ramachandran P, Huang H, Pham TD, Schapiro JM, Cong X, Chakraborty S, et al. Differential Peripheral Blood Glycoprotein Profiles in Symptomatic and Asymptomatic COVID-19. Viruses. 2022; 14(3):553. https://doi.org/10.3390/v14030553
Chicago/Turabian StylePickering, Chad, Bo Zhou, Gege Xu, Rachel Rice, Prasanna Ramachandran, Hector Huang, Tho D. Pham, Jeffrey M. Schapiro, Xin Cong, Saborni Chakraborty, and et al. 2022. "Differential Peripheral Blood Glycoprotein Profiles in Symptomatic and Asymptomatic COVID-19" Viruses 14, no. 3: 553. https://doi.org/10.3390/v14030553