
Environmental and Lifestyle Risk Factors in the Carcinogenesis of Gallbladder Cancer
by
 ,
,
Pablo Pérez-Moreno
1,†, 
Ismael Riquelme
2,†,
Patricia García
1,
Priscilla Brebi
3 and
Juan Carlos Roa
1,* 1
Millennium Institute on Immunology and Immunotherapy, Department of Pathology, School of Medicine, Pontificia Universidad Católica de Chile, Santiago 8380000, Chile
2
Institute of Biomedical Sciences, Faculty of Health Sciences, Universidad Autónoma de Chile, Temuco 4810101, Chile
3
Millennium Institute on Immunology and Immunotherapy, Laboratory of Integrative Biology (LiBi), Centro de Excelencia en Medicina Translacional (CEMT), Scientific and Technological Bioresource Nucleus (BIOREN), Universidad de La Frontera, Temuco 4810296, Chile
*
Author to whom correspondence should be addressed.
†
These authors contributed equally to this work.
J. Pers. Med. 2022, 12(2), 234; https://doi.org/10.3390/jpm12020234
Submission received: 18 September 2021
/
Revised: 8 November 2021
/
Accepted: 23 December 2021
/
Published: 8 February 2022
(This article belongs to the Special Issue Personalized Prevention of Gallbladder Cancer)
{kind=link}
{kind=link}
{kind=link}
Abstract
:Gallbladder cancer (GBC) is an aggressive neoplasm that in an early stage is generally asymptomatic and, in most cases, is diagnosed in advanced stages with a very low life expectancy because there is no curative treatment. Therefore, understanding the early carcinogenic mechanisms of this pathology is crucial to proposing preventive strategies for this cancer. The main risk factor is the presence of gallstones, which are associated with some environmental factors such as a sedentary lifestyle and a high-fat diet. Other risk factors such as autoimmune disorders and bacterial, parasitic and fungal infections have also been described. All these factors can generate a long-term inflammatory state characterized by the persistent activation of the immune system, the frequent release of pro-inflammatory cytokines, and the constant production of reactive oxygen species that result in a chronic damage/repair cycle, subsequently inducing the loss of the normal architecture of the gallbladder mucosa that leads to the development of GBC. This review addresses how the different risk factors could promote a chronic inflammatory state essential to the development of gallbladder carcinogenesis, which will make it possible to define some strategies such as anti-inflammatory drugs or public health proposals in the prevention of GBC.
1. Introduction
Gallbladder cancer (GBC) is a neoplasm that causes around 84,695 deaths per year worldwide [1], affecting more commonly women than men with an age-standardized incidence rate of 1.4 and 0.89 per 100,000 people, respectively [1]. The onset of this disease is characterized by a slow and silent development, resulting in many GBC cases being frequently diagnosed at late stages when patients have a 5-year life expectancy of less than 5% [2]. For this reason, the understanding of how different risk factors influence the early development of gallbladder carcinogenic processes is crucial to improving the prevention strategies for this malignancy.
GBC has multifactorial causes that converge to form a new tumor. The most important risk factor described for GBC development is gallstone disease (GSD) due to its high correlation with GBC [3,4,5,6]. Gallstones can lead to a chronic inflammatory state of tissue damage/regeneration characterized by an imbalance between anti- and pro-inflammatory factors that induce carcinogenic processes in the gallbladder mucosa [6]. In fact, some environmental and lifestyle risk factors may be involved in the development of gallstones and the subsequent GBC formation, such as body mass index (BMI), geographical location, nutritional aspects (high-fat and high-sugar diets), bacterial infections (e.g., Salmonella typhi), chronic diseases (e.g., sclerosing cholangitis, type II diabetes, metabolic syndrome and/or dyslipidemias), parasitic infections, smoking, and alcohol consumption [2,7,8,9,10,11,12,13,14,15,16].
Therefore, this article reviews the relationship between environmental risk factors and the formation of chronic inflammation, and how this inflammatory process is involved in gallbladder carcinogenesis.
2. Search and Selection of Literature
A literature search was performed using different databases including Pubmed and Web of Science. The search terms were as follows: (gallbladder cancer OR gallbladder neoplasia OR gallbladder carcinoma) AND (inflammation OR environmental risk factors OR carcinogenesis OR lifestyle OR bacterial infections OR viral infections OR parasitic infections OR fungal infections OR immune diseases OR metabolic diseases OR alcohol OR smoking OR aflatoxins). The selection criteria of articles were as follows: (1) articles related to risk factors associated with gallbladder cancer and (2) articles related to inflammation in gallbladder cancer. Those studies that were not related to GBC were excluded from this review. In those cases of multiple publications regarding the same topic, we consider only the most relevant studies. We identified one retrospective [17], five prospective [15,16,18,19,20], nine meta-analyses [9,13,14,21,22,23,24,25,26], eleven case–control [3,4,27,28,29,30,31,32,33,34], and two Mendelian randomization [35,36] studies. Other additional sources of information consisted of experimental studies and review articles. The search diagram is represented in Figure 1.
The selection criteria of articles were: (i) articles related to risk factors associated with gallbladder cancer and (ii) articles related to inflammation in gallbladder cancer. The exclusion criteria used in the screening phase focused on those studies that were not related to GBC; meanwhile, in the eligibility phase, the exclusion criteria were focused on those studies regarding the same topic.
3. Inflammation in Gallbladder Carcinogenesis
Inflammation is a process that involves the activation of the innate and adaptive immune system to defend the host against pathogens, as well as to repair and remodel tissues to recover their homeostasis [37]. Thus, the main physiological role of inflammation is to be activated after foreign damage that occurs in a specific area of the body, eliminating the Noxa and allowing the restoration of homeostasis in the affected area [37,38]. Under normal conditions this process begins with vasodilation and hyperemia within the blood vessels that promote vascular permeability, mainly induced by the increase in nitric oxide (NO) and allowing the immune cells and inflammatory mediators (the inflammatory exudate) to exit towards the extravascular tissues. This induces many circulating leukocytes to be able to adhere to the endothelium through molecules, such as E-selectin and VCAM-1, located on the surface of activated endothelial cells. Then, the monocytes are transformed into macrophages that release many hydrolytic enzymes, pro-inflammatory cytokines such as IL-1 and TNF-α, and free radicals that result in the development of an acute inflammatory process in epithelial cells. Finally, when the injuring agent has disappeared, macrophages stimulate fibroblasts for collagen synthesis and endothelial cells to produce growth factors that stimulate tissue repair. Fortunately, under normal conditions, this process is strictly regulated [37,39]. However, in a pathological context such as cancer, this process occurs chronically, allowing a vicious cycle of damage/repair, which leads to chronic DNA damage, generating mutations in different genes, which can promote the development of cancer [37,39,40,41].
Unlike what occurs with a normal inflammatory response, in cancer there is a hyperproliferation of epithelial cells destined to increase the number of macrophages able to react against tumor cells, which acquire different protein expression patterns and phenotypical changes that lead to being recognized as foreign to the host [41,42]. In addition, after an oncogenic injury, the immune response will not be able to eliminate the Noxa (oncogenic injury); thus, an increase in the inflammatory reaction will be observed, resulting in a higher tumor growth rather than a restoration of tissue homeostasis [42]. Another difference between normal inflammation and cancer-inducing inflammation is that in a normal inflammation context against harmful agents (e.g., bacterial infections), the inflammatory response has an acute character by inducing the recruitment of macrophages from the nearby tissues and the diapedesis of monocytes from nearby blood vessels to be transformed into macrophages in the corresponding tissue. Conversely, due to chronic inflammation during the pre-carcinogenic stages, cancer is commonly characterized by a local proliferation of macrophages and migration of more macrophages from nearby sites to the affected area, which would indicate a weaker immune system activation. However, this effect takes longer and could increase as the tumor grows [42,43,44]. Interestingly, about 15% of cancers—including GBC—are preceded by chronic inflammation, which can be local (e.g., inflammation due to gallstones or infections) or systemic (e.g., metabolic syndrome) [45,46]. Therefore, the inflammatory component of GBC can come from different sources, producing an inflammatory state of a summative nature that provides the ideal niche for carcinogenic progress.
As mentioned above, chronic inflammation is widely described as a crucial early factor in the development of cancer within different organs [39,47]. One of the most studied cytokines that has been linked to cancer is tumor necrosis factor α (TNF-α) [48]. It has been shown that cancer cells release TNF-α, promoting cell transformation, tumorigenesis, and metastasis [48]. Surprisingly, TNF-α expression increases from hyperplasia to carcinoma, and also during TNM stages in GBC tissues [49], suggesting that TNF-α could participate in carcinogenic processes and may be a progression marker in GBC patients. Other cytokines are IL-2 and IL-8, which have been shown to promote COX-2 expression, which is overexpressed in chronic cholecystitis, increasing their expression in GBC tissues [50], suggesting that metabolites produced via cyclooxygenases (e.g., arachidonic acid) mediate the inflammatory response of the gallbladder [51]. Interestingly, the pain produced by gallstones is treated with non-steroidal anti-inflammatory drugs (NSAIDs), inhibiting the activity of COX-2 [52,53]. In addition, the COX-2 protein is intensively expressed in the hyperplastic mucosa of the gallbladder in patients with an anomalous arrangement of the pancreaticobiliary duct (AAPBD) [54], a congenital malformation in which the pancreatic and bile ducts join outside the wall of the duodenum, allowing pancreatic juices to chronically damage the bile duct causing an increased risk of developing GBC [55,56]. Interestingly, about 61% of patients with AAPBD have hyperplasia in the gallbladder mucosa [57,58], suggesting that the function and expression of COX-2 are increased from early stages in GBC carcinogenesis, participating in the malignant transformation of gallbladder mucosal cells. In addition, the prevalence of KRAS mutations is higher in AAPBD-related GBC [59], providing evidence that mutations in KRAS are involved in gallbladder carcinogenesis [60,61,62].
In 1986, Yamagiwa et al. proposed a GBC progression sequence through which a normal mucosa develops metaplasia, then dysplasia and ultimately a carcinoma [63]. It has been found that some common genetic markers such as KRAS [64,65], PIK3CA [64], and HER2/neu (c-Erbb-2) [66] are altered in different progression stages of this disease through loss of heterozygosity (LOH) and changes in the DNA methylation [67]. Accordingly, different genes have shown a deregulated expression in the early stages of GBC [68,69,70,71]. Recently, a study by Brägelmann et al. [72] showed different epigenetic changes that occur during the different stages of GBC carcinogenesis (i.e., GSD -> dysplasia -> GBC). For in-stance, epigenetic changes were observed to be accentuated in different genes as the tumor progression advanced, highlighting the hypermethylation of cytosine-guanine dinucleotide islands (CpG) and gene promoter regions. The results showed that methylation occurred in genes involved in the control of certain signaling pathways such as Wnt, an important pathway that is overactive during the carcinogenic process of different malignancies, including GBC. Among the differentially methylated regions were found the promoter regions of ZIC1, HHIP and PTCH1 (two negative regulators of Hedgehog signaling), WIF1, RUNX3, P73, RPRM, TWIST1, HBE1, and DCLK1, among other genes. In addition, gene promoters with hypomethylation were found in HMGA1, ERBB2, CDCA7, and RUNX1, among others. Furthermore, higher copy numbers were frequently found in some proto-oncogenes such as MDM2 and YEATS4, which suggest that these findings could have a real impact on GBC carcinogenesis and malignancy [72]. In addition, alterations in the TP53 gene have been observed in tissues from patients with chronic cholecystitis [69,73]. Another example is the case of the fragile histidine triad (FHIT) gene, which encodes an enzyme involved in purine metabolism that commonly sees a 55% loss of expression and a 46% allelic loss in dysplastic lesions in GBC patients [74]. In a study by Roa et al. [75], some somatic mutations in PI3KCA were found in the tissues of early GBC cases. Similarly, a study by Li et al. demonstrated that PI3K was overexpressed in gallbladder polyps and even higher in GBC tissues [76]. Interestingly, PI3K actively participates in the regulation of the immune response [77], suggesting that early alterations in PI3K could drastically alter the balance of the inflammatory response. Furthermore, Espinoza et al. also described Claudin-18 (CLDN-18) as a membrane marker of gallbladder metaplasia within the progression sequence towards GBC, where the CLDN-18 expression is also present in ~50% of gallbladder tumors [78].
A study by Mishra et al. [79] indicated that certain mutations in IDH1, IDH2, and KMT2C genes could generate a greater susceptibility to developing GBC. Additionally, Salazar et al. addressed the alteration of other important genes that could also be involved in gallbladder carcinogenesis, including Rb, VHL, EGFR, MSI, and hTERT [80]. Similarly, a study by Pandey et al. showed that some cholecystitis samples had alterations in the PIK3R2, CHD1, TP53, and CDKN2A genes. Interestingly, these alterations were also found in GBC samples, suggesting that these mutations are present from the early stages of gallbladder carcinogenesis towards the establishment of carcinoma. In addition, these authors identified other significantly mutated genes in GBC, including CCTNNB1, ELF3, ERBB2, ARID2, ERBB3, STK11, SMAD4, ARID1A, KRAS, EHF, PIK3CA, BRAF, ACVR2A, PSIP1, NFE2L2, CHRM3, ZNF107, SMARCA4, APC, NF1, KAT8, MAP2K4, and HIST1H2AG. In particular, mutations in the TP53, ELF3, CTNNB1, ERBB2, ARID1A, and CDKN2A genes resulted in the formation of different peptides as neoantigens. Among these genes, ELF3 had the highest number (n = 9) of predicted neoantigens capable of inducing T-cell activation and, therefore, these neoantigens may become candidates for GBC vaccines [81].
All these results suggest that the mutations and aberrant expression of these genes could be responsible for promoting gallbladder carcinogenesis [82].
Alterations in these genes have also been found in the carcinogenic sequences of other cancer types. For instance, different types of TP53 mutations have been described in the early preneoplastic stages of gastric, colorectal, and lung cancers [83,84,85,86], strongly suggesting that TP53 is altered in the initial stages of cancer probably induced by early and persistent inflammatory processes.
As suggested by the different studies reviewed above, alterations in the TP53 gene could be a crucial event for the beginning of a carcinogenic process. The main consequence of TP53 alterations is the loss of its function, decreasing its capacity as a tumor suppressor, causing cells to begin to lose regulation of the cell cycle, and perpetuating the successive mutations in DNA that translate into greater genomic instability [87]. In this regard, the frequency of TP53 abnormalities observed in early stages, such as chronic cholecystitis, can be subgrouped into 35% of LOH, 25% of mutations, and 11% of gene inactivation; meanwhile, in invasive GBC the frequency is 81%, 67 %, and 52%, respectively. This suggests that alterations in TP53 increase dramatically as disease progression increases [69]. From the point of view of the inflammatory process, TP53 abnormalities are significant since one of their functions is to repress the inflammatory response mainly through the NF-κb pathway [88]. The NF-κb pathway has been described as a key regulator of pro-inflammatory response in cancer progression [89,90] by promoting the expression of certain cytokines (e.g., IL-1, IL-2, IL-6, and TNF-α), some chemokines (e.g., CXCL1, CXCL10, and MCP-1), adhesion molecules (ICAM-1, VCAM-1, and ECAM-1), and antiapoptotic factors such as BCL-2, c-Flip, and survivin, allowing the recruitment and activation of leukocytes and tumor cell survival [91,92]. Therefore, all these previously described factors suggest that the loss of p53 function in GBC could be an important cause in the imbalance of the early inflammatory response and, as a consequence, in gallbladder carcinogenesis.
4. Nutritional and Lifestyle Aspects That Increase Susceptibility to Gallbladder Carcinogenesis
As previously mentioned, gallstone disease (GSD) is one of the most important predisposing factors to developing GBC. Thus, metabolic syndrome and some factors that increase the probability of forming gallstones, including sedentary lifestyle, the consumption of sugar-sweetened and artificially sweetened beverages, obesity, high-fat diet, hypercholesterolemia, and the consumption of red meat, can consequently promote carcinogenesis by inducing a chronic pro-inflammatory state in the gallbladder tissue [2,6,27,93]. Conversely, a physically active lifestyle and the consumption of some vegetables, such as radish and sweet potato, have been shown to reduce the risk of developing gallstones and GBC [27,94]. In this regard, an increased BMI constitutes an important risk factor for GBC and other types of cancer [21,95]. For instance, a study by Barahona et al. showed that BMI has a causal effect on gallstone disease, which subsequently increases the GBC risk. The meta-analysis performed by Tan et al. showed that the relative risk of GBC was 1.14 (95% CI, 1.04–1.25) for overweight people (BMI 25–30 kg/m2) and 1.56 (95% CI, 1.41–1.73) for obese individuals (BMI > 30 kg/m2), evidencing a higher GBC risk in women than men [22]. This difference between genders could be explained by the fact that estrogens promote a greater cholesterol storage in the bile, which is consistent with the higher obesity and BMI rates observed in women, and the increased risk of developing gallstones [96]. Interestingly, higher C-reactive protein (CRP) concentrations increased GBC risk in the European population, suggesting that the inflammatory status is crucial in developing GBC [36].
The matter of a sedentary lifestyle can be explained according to metabolic disorders inferred from the study by Skoumas et al., in which physically active women had significantly lower levels of total serum cholesterol, LDL-c, oxidized LDL cholesterol, and triglycerides compared to sedentary women [94]. Other studies have also observed that a more sedentary lifestyle is directly correlated with the risk of developing metabolic syndrome and elevated plasma levels of triglycerides and cholesterol [97,98]. On the other hand, the consumption of sugar-sweetened and artificially sweetened beverages can significantly increase (double) the risk of GBC when individuals consume two or more servings per day (200 mL/serving) of sweetened beverages compared to those without consumption (hazard ratio = 2.24; 95% CI = 1.02 to 4.89) [18]. A probable explanation for this is that the increased sugar intake has been associated with increased body weight and diabetes mellitus, predisposing to gallstone disease-dependent GBC [19]. In fact, diabetic patients have shown a higher risk of GBC compared to non-diabetic individuals [13]. This phenomenon is probably due to the fact that hyperinsulinemia caused by sugar ingestion induces the expression of the insulin-like growth factor receptor (IGFR-1), which is able to induce malignant properties in several types of cancer by increasing cell proliferation, tumorigenic capacity, and apoptosis resistance through the activation of PI3K and MAPK pathways [99].
In general, these data suggest that a sedentary lifestyle and sugar consumption—powered by high-fat food—can promote gallstone formation and subsequently GBC.
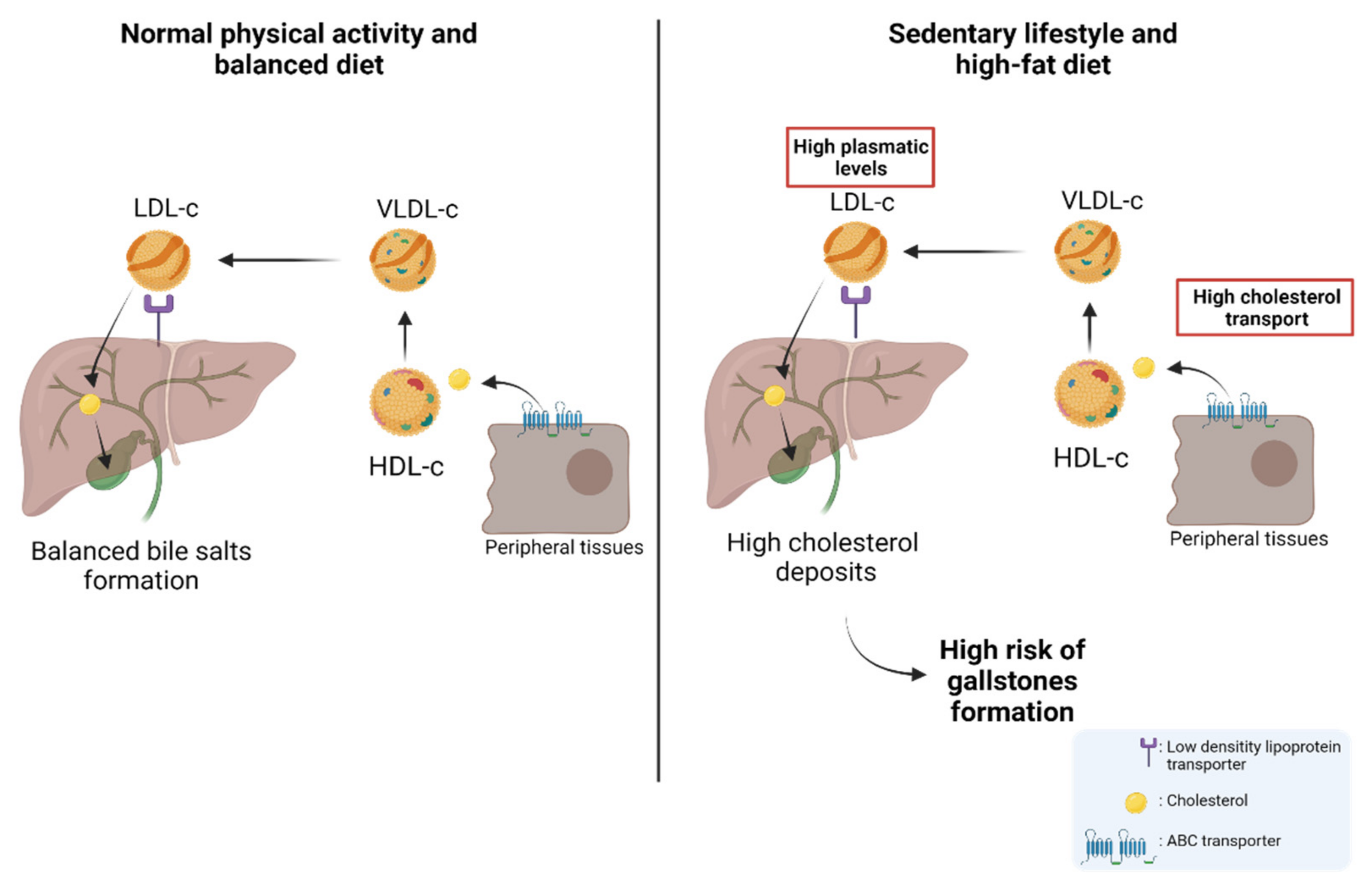
About 80% of gallstones are comprised of cholesterol; hence, patients with permanently high levels of plasma cholesterol are candidates for gallstones [100,101,102,103]. Physiologically, cholesterol is transported through different lipoproteins such as high-density lipoproteins (HDL-c) and low-density lipoproteins (LDL-c). HDL-c takes the cholesterol from peripheral tissues through ABC transporters (e.g., ABCA1) and esterifies it through lecithin cholesterol acyltransferase (LCAT) [104]. Subsequently, HDL-c can be recognized by the scavenger receptor class B type I (SR-BI), which facilitates the uptake of high-density lipoprotein cholesterol esters in the liver. In addition, cholesterol can be transported to very-low-density lipoproteins (VLDL-c) via the cholesteryl ester transfer protein (CETP) [105,106]. Then, VLDL-c is transformed into LDL-c and is recognized by the low-density lipoprotein receptor (LDLr) and LDL-related protein (LRP) to induce the internalization of cholesterol into the hepatocytes in the liver [105,107]. Finally, the cholesterol within the hepatocytes can be used in bile formation to be transported to the bile canaliculus and stored in the gallbladder [105,108]. Interestingly, a study by Selcuk Atamanalp et al. showed that high cholesterol and LDL-c levels in plasma were closely correlated with a higher presence of cholesterol gallstones. Conversely, low serum HDL levels did not affect the occurrence of cholesterol gallstones [109]. In addition, Wang et al. showed that patients with increased plasma levels of cholesterol, triglycerides, HDL-c, LDL-c, and apolipoprotein B (APOB), also showed a significantly higher recurrence of gallstones compared to control patients [110], which was similar to those results found by Hayat et al., who showed that patients with gallstones had significantly higher plasma levels of triglycerides and HDL-c than the control patients [20]. All these data reaffirm the idea that high plasma levels of cholesterol and other lipids induce gallstone formation and, therefore, are heavily involved in the subsequent development of GBC. The cholesterol metabolism and its implication in the risk of developing GSD are represented in Figure 2.
There are different pathophysiological processes that promote gallstone formation, including cholesterol crystallization/nucleation, alteration in mucin secretion, changes in biliary motility, alteration in intestinal cholesterol transport, and intestinal motility [93]. Normally, cholesterol is found in unilamellar vesicles that bind bile salts to allow cholesterol solubilization. However, when there is an increase in cholesterol deposits, the solubilizing capacity of the bile is saturated promoting the solidification and subsequent nucleation of cholesterol/bile acids, allowing the formation of gallstones [111]. The mechanism by which cholelithiasis predisposes to GBC has not yet been established; however, it has been reported that the DNA mutation rate is higher in the inflammation microenvironment induced by gallstones than in normal tissues, increasing the risk of developing GBC [112]. This inflammatory state is generated in different ways, including the direct damage by gallstones due to continuous friction with the mucosa, which leads to a continuous damage/repair loop in the gallbladder epithelium and, on the other hand, the increase in gallstone size which can obstruct the bile duct, which also increases the susceptibility to infections that give rise to the inflammation process [93,113].
Different animal models have been used to study gallstone formation and its effect on the development of preneoplastic lesions [114,115,116,117]. Our research group established an animal model of gallbladder preneoplasia through a lithogenic diet. The results showed that after administration of a high-cholesterol diet (lithogenic diet) for 9 months, early gallstone formation was induced within the gallbladder. Furthermore, animals fed a lithogenic diet evidenced fatty liver and higher plasma cholesterol levels than animals fed a normal diet in a similar manner to that observed in humans. More interestingly, those animals treated with a lithogenic diet had metaplastic and dysplastic architecture in gallbladder tissues, with no invasive features, but with a well-defined inflammatory component showing a predominance of lymphocytes and polymorphonuclear cells [116]. A similar study showed that mice treated with a lithogenic diet had epithelial hyperplasia along with the occurrence of acute and chronic inflammation characterized by the presence of eosinophils, neutrophils, and lymphocytes within the lamina propria [117]. Recently, Kato et al. described an animal model generated by the orthotopic implantation of gallbladder organoids containing mutant loss of KRAS and TP53 genes developed in vitro using lentiviral Cre transduction and CRISPR/Cas9 gene editing, respectively. The data showed that the tumor transcriptomic profiles are similar to that found in human tissues, as well as the immune cell infiltration observed during tumor formation, suggesting that this model could be an interesting approach to study carcinogenesis in GBC [118].
Different inflammatory components have been described as participants in the pathophysiology of inflammation including some inflammatory cytokines such as IL-6, IL-10, IL-12, and visfatin. Interestingly, visfatin is considered a key molecule in the activation of human leukocytes and the production of pro-inflammatory cytokines; therefore, it seems to be associated with the risk of developing gallstones [119,120]. In this regard, patients with acute cholecystitis often present a higher expression of visfatin in peripheral blood mononuclear cells (PBMCs), serum, and in grossly inflamed gallbladder tissues. Moreover, the gene overexpression of visfatin observed on in vitro models of acute cholecystitis has been frequently accompanied by an increased expression of other pro-inflammatory mediators including IL-10, TNF-α, IL-6, ICAM-1, and VCAM-1 [121]. Similarly, Nien Wang et al. showed that serum visfatin levels were markedly higher in subjects that presented pigment and cholesterol gallstones than in healthy controls. Additionally, serum levels of cholesterol, triglycerides, AST, ALT, leukocyte count, and fasting glucose were significantly higher in those individuals with gallstones. Interestingly, high AST levels and the increased white blood cell count were considered significant predictors of gallbladder lithiasis, while the elevated values of visfatin in serum were also suggested as a significant risk factor for gallstone formation [120]. These results suggested that visfatin could be a predictive marker of inflammation and predisposition to gallbladder lithiasis.
As previously mentioned, evidence accumulated over many years indicates that gallstones can induce an inflammatory microenvironment that increases the risk of developing GBC. However, this risk can be fostered by a greater genetic predisposition [122], for example, the genetic variability present in genes that encode the different ATP-binding cassette (ABC) transporters in the hepatocanalicular membrane, which are involved in the different processes of the exportation of bile salts in biliary tracts, including the transportation of ABCB11, the transport of phosphatidylcholine (ABCB4), and secretion of cholesterol and phytosterols into bile (heterodimer ABCG5/8) [123]. In this regard, a genetic variant in the ABCG8 gene (variant rs11887534 or D19H) has been associated with a higher gallstone development through cholesterol hypersecretion and cholesterol supersaturation in the bile [122,124]. In addition, other variants (rs1558375, rs17209837, and rs4148808) have been determined in the 7q21.12 region harboring both the ABCB1 and ABCB4 genes, which showed a higher risk of developing GBC [28]. A study by Bustos et al. showed that in the Chilean Mapuche ancestry population, variants in ABCG8 (rs11887534) and TRAF3 (rs12882491) were associated with GSD. In addition, it was shown that TRAF3 levels were lower in individuals affected by GSD, suggesting that these variants could be used as risk markers for GBC [125,126]. Other mutations occur in the ABCB4 gene and are classified as nonsense mutations (class I), missense mutations affecting maturation (class II), activity (class III), or protein stability (class IV), and mutations with no identifiable effect (class V) [127]. It has been shown that mutations in this gene increase the risk of developing gallstones in subjects under 40 years, mainly by inducing an ABCB4 deficiency that results in low biliary phosphatidylcholine concentrations, which is consistent with the spontaneous occurrence of cholecystolithiasis [128,129]. In fact, the homozygous ABCB4 mutations lead to the complete absence of the phospholipid transporter and no secretion of phospholipids into bile, which finally causes a decrease in the solubility of bile, and consequently, a greater predisposition to bile crystallization and gallstone formation [130]. Other mutations and alterations have been described in different genes and proteins potentially involved in a higher risk of developing gallstones, such as ABCB11 [130], cholesterol 7a-hydroxylase (CYPA1) [131,132], APOB gene [133], and cholecystokinin A receptor (CCKAR) [134,135]. In addition, certain alterations in genes related to the immune system, inflammation, and oxidative stress have also been implicated in a greater risk of developing GBC, including mutations in TLR2, TLR4 [136], IL1RN, IL1B [137], IL10, IL8, IL8RB, RNASEL, VEGF [138], and CCR5 [139], as well as rs7504990 variant in the DCC [140]. These data show that the predisposition to developing cholecystolithiasis and GBC also have an important genetic background that needs to be considered in GBC carcinogenesis.
Finally, another risk factor associated with GBC is the appearance of bluish and brittle calcifications in the inner gallbladder wall named "porcelain gallbladder" [141]. Porcelain gallbladder has an incidence of less than 1% in patients with gallbladder disease, being more prevalent in women [141]. This rare condition is considered a risk factor for GBC because approximately 60% to 90% of these cases show gallstones [141,142]. Despite the pathophysiology of "porcelain gallbladder" not being clear, this condition could be a consequence of a previous chronic inflammatory process or could be the result of an obstruction produced by gallstones that induce the accumulation and precipitation of calcium in the mucosal layer of the gallbladder wall [143]. Whatever the origin of this disease, it is also unknown whether calcium levels play a role in the “porcelain gallbladder” pathogenesis. Recently, Berger et al. found significantly higher calcium and parathormone (PTH) levels in the plasma of individuals with porcelain gallbladder compared to controls [144], which suggests that individuals with diseases that induce persistent hypercalcemia (e.g., primary hyperparathyroidism) also have a higher risk of developing porcelain gallbladder. In this regard, we venture to propose that persistent hypercalcemia could be an initiating factor of porcelain gallbladder, which would eventually trigger the formation of a chronic inflammatory state in the inner layer of the gallbladder, increasing in this manner the risk of developing GBC. However, more studies are still needed to demonstrate this hypothesis.
5. Infections and Chronic Inflammatory Diseases
5.1. Infections Related to Gallbladder Carcinogenesis
Despite gallstone development and genetic abnormalities being important risk factors in inducing GBC, chronic infections also seem to be a factor to consider in gallbladder carcinogenesis [145] since certain pathogens, such as bacteria, viruses, and parasites, are capable of generating direct tissue damage that leads to the activation of an acute or chronic inflammatory response by increasing the levels of different pro-inflammatory cytokines and inducing the reaction of neutrophils and lymphocytes [146].
In the case of bacteria, different bacterial genera have been found in the gallbladder of patients with cholecystitis and cholelithiasis, including Salmonella spp., Escherichia spp., Klebsiella spp., and Helicobacter spp. [145]. Another study showed that most bile samples from cholecystectomy patients had E. coli, Salmonella spp., and Klebsiella spp.; however, a few samples also evidenced the presence of Pseudomonas sp., Acinetobacter sp., Enterobacter spp., Citrobacter freundii, Vibrio spp., and Serratia marcescens [147]. In addition, the genetic material of other bacterial species, including Collibacillus spp., Bacteroides fragilis, Klebsiella spp., Clostridium perfringens, and Clostridium spp., has been found in GBC tissues suggesting that aerobic and anaerobic bacteria can colonize the gallbladder during this malignancy [147].
From these bacterial agents, the most implicated species in gallbladder carcinogenesis are those belonging to the Salmonella genus; a large number of Salmonella spp. sequences have been found in GBC samples, suggesting a possible role of the infection by this bacterial genus in GBC carcinogenesis through an inflammatory process [148]. Interestingly, those subjects whose gallbladders had Salmonella also showed a higher number of cholecystitis, empyema, and neutrophil infiltration, which indicates the activation of the immune system and inflammatory process [6]. In particular, chronic carriers of Salmonella typhi have shown to be implicated in greater development of gallstones [149] and GBC [29,150]. In fact, this risk of acquiring GBC increases about 12-fold in subjects with a history of typhoid fever [29]. This strong likelihood of producing GBC has been associated with the presence of the Vi polysaccharide from Salmonella typhi in northern Indian subjects with biliary disease [151]. Evidence has shown that not only chronic infection with Salmonella typhi, but also Salmonella parathypi presents a high risk of advancing to GBC [150]. On the other hand, a study by Scanu et al. showed that Salmonella enterica can induce cell transformation in organoids derived from pre-transformed murine gallbladders from mice deficient in the Ink4b-Arf-Ink4a locus that implies an inactive TP53, suggesting that alterations in this gene are necessary to malignant transformation [152].
Helicobacter has been widely described as inducing different pathologies, specifically Helicobacter pilori, which is involved in gastric carcinogenesis [148]. The presence of Helicobacter species, such as Helicobacter bilis and Helicobacter pullorum, has also been demonstrated in bile samples and gallbladder tissues from patients with chronic cholecystitis [153]. In particular, the presence of Helicobacter bilis in bile and in biliary tract neoplasms has been linked to a high risk of bile duct carcinoma and gallstones [131,132,133]. Therefore, both Helicobacter bilis and Helicobacter pullorum have been associated in some way with the risk of developing GBC [23,153,154,155].
Regarding parasitic infections, no direct associations with GBC have been described; however, the presence of liver flukes such as Clonorchis sinensis, Schistosomiasis japonica and Opisthorchis viverrine have been associated with an increased risk of cholangiocarcinoma [156]. The mechanisms by which these parasites damage the epithelium of the bile ducts are not clear yet, but some authors suggest that trematode eggs cause intense and persistent local inflammation by increasing ROS formation and activating the immune system response [156]. For example, Clonorchis sinensis can cause a partial obstruction of the bile ducts, increasing biliary pressure and provoking a repetitive ulceration/inflammation cycle that finally leads to DNA damage and the risk of developing cholangiocarcinoma [157].
Regarding viral infections, some studies performed in Taiwan and China have established that infection with hepatitis B (HBV) and hepatitis C (HCV) viruses increases the risk of extrahepatic bile duct cancer [158,159]. In fact, HBV infection has also been associated with cholangiocarcinoma, cholecystolithiasis, and choledocholithiasis [30]. Interestingly, a study on the Korean population showed a positive association in patients with metabolic syndrome infected with HBV with the risk of bile duct cancer [160]. A meta-analysis by Chen et al. [24] showed that the Epstein–Barr virus (EBV) could have a positive association with the risk of developing hepatobiliary cancers; however, many studies are still needed to determine its effect on the risk of developing GBC.
The development of biliary pathologies from fungal infections is uncommon since these types of infections mainly occur in immunosuppressed patients [161]. An article by Szvalb et al. reported only 42 cases of cholecystitis between 1976 and 2019 where Candida infection was involved. About 86% of these cholecystitis cases had Candida species as a single microbial agent isolated from gallbladder tissue or fluids, whereas the remaining 14% of cases had one or more co-infecting bacteria. The Candida species found were Candida albicans (67%), Candida glabrata (14%), Candida parapsilosis (12%), and Candida tropicalis (7%) [162]. Interestingly, a particular fungi genus known as Fusarium was also isolated from cholecystitis samples in a neutropenic patient with leukemia who was previously diagnosed with disseminated fusariosis [162]. In addition, a retrospective study that analyzed the bile of patients between 2014 and 2019 who underwent a percutaneous transhepatic cholangiogram (PTC) for obstructive jaundice found that out of 71 patients, only 5 had a positive Candida culture (two cases of cholangiocarcinoma and only one of GBC) [17].
All these data indicate that infections could be recognized as important carcinogens that can have a great impact on the development of GBC, since they induce a significant increase in the inflammatory response due to different pathophysiological mechanisms that promote the activation of the immune system, which is considered a determining risk factor in GBC carcinogenesis.
5.2. Immune Inflammatory Diseases Involved in Gallbladder Carcinogenesis
Evidence has stated that autoimmune diseases can increase the predisposition to the development of cancer because the recognition made by the immune system of itself as foreign triggers a chronic inflammatory process that leads to carcinogenesis [163,164]. Different autoimmune diseases have been associated with the development of GBC, including celiac disease and Crohn’s disease; however, the most studied GBC-related autoimmune disease is primary sclerosing cholangitis (PSC) [165,166]. PSC is a chronic inflammatory disease characterized by the destruction of the intrahepatic and extrahepatic bile ducts with the consequent fibrosis, which has been described as a high-risk disease for developing cholangiocarcinoma and GBC [167,168,169,170]. Lewis et al. performed an anatomopathological study in 72 gallbladder tissues from PSC patients and found that 69 samples contained at least focal pyloric metaplasia, 27 samples evidenced dysplasia (15 cases with high-grade dysplasia and 12 cases with low-grade dysplasia), and 35 samples showed moderate or advanced diffuse chronic lymphoplasmacytic cholecystitis. In addition, this study also found a significant correlation between dysplasia and the risk of GBC [171]. In another study by Hebillas et al., they showed that of 102 gallbladder cholecystectomy samples from patients with PSC, 8 of them had adenocarcinoma of which 57% were associated with dysplasia [172]. Another study showed that the frequency of gallbladder polyps (GBPs) in patients with PSC was 10.6%, of which 10 had malignant/premalignant lesions, and 4 had high-grade dysplasia [31]. Similarly, a study in Karolinska University Hospital between 1985 and 2006 showed that dysplasia and carcinoma were found in 30% of cases with PSC and a great presence of inflammation and fibrosis [173]. In addition, another study by Castro et al. showed that among 30 autoimmune pathologies, Crohn’s disease and systemic lupus erythematosus (SLE), as well as pernicious anemia and psoriasis, had a higher risk of developing GBC. In the case of SLE, this disease also showed a greater risk of developing extrahepatic bile duct cancer [165]. Therefore, these data suggested that a certain type of autoimmune diseases, such as SLE, Crohn´s disease, and PSC, which are characterized by an abnormal immune reaction to self-antigens, could be an important inflammatory source for the carcinogenic development in GBC. However, further research is required to clarify the mechanisms by which these autoantibodies-released throughout the pathogenesis of these autoimmune diseases-could induce gallbladder neoplasia.
6. Consumption of Alcohol and Tobacco as a Risk Factor for Gallbladder Carcinogenesis
Alcohol consumption and smoking are widely known risk factors for some types of malignancies such as colon and lung cancer, respectively [25,32]. However, the role of alcohol and smoking in the development of GBC has not yet been established. For instance, O’Keeffe et al. showed that smoking is associated with a greater GBC risk in both genders; however, alcohol consumption increases the risk of GBC only in men [25], mainly because men consume more alcohol than women [174,175,176]. Alcohol is a well-known inducer of alcoholic fatty liver [177], which is characterized by an increment of cholesterol transportation through the bile canaliculi into the gallbladder, inducing cholesterol crystallization and thus gallstone formation. In addition, greater alcohol consumption can lead to liver cirrhosis, which is directly linked to a higher incidence of gallstones [178,179]. Another study by McGee et al. analyzed 26 prospective studies in which the risk of bile duct cancer was evaluated in people who consumed alcohol and smoked. Results showed that smoking increased the risk of developing intrahepatic bile duct, extrahepatic bile duct, and ampulla of Vater cancers, but not GBC. In addition, alcohol consumption was also associated with a higher risk for developing intrahepatic bile duct cancer, but not for GBC [16]. An article by Wenbin et al. reviewed 10 case–control studies and one prospective study, showing that smoking was able to augment the risk of developing GBC, being an independent factor from alcohol consumption and gallstone formation for GBC development [14].
7. Exposure to Aflatoxins and Other Elements and Compounds as Risk Factors for Gallbladder Carcinogenesis
Aflatoxins are compounds derived from some fungi species, such as Aspergillus flavus and Aspergillus parasiticus, which can be found in certain foods such as corn or peanuts [180]. Aflatoxins form adducts with albumin from the blood, binding in lysine residues [181]. Some authors have estimated that the consumption of aflatoxins could increase the risk of GBC and liver cancer [182,183] since this compound could be accumulated in the bile ducts [184,185]. A study carried out in Chile evaluated the effect of the weekly consumption of ají rojo (a type of hot dried pepper) in patients with GBC, finding that 64% of these cases had AFB1 adducts in plasma [186]. Another study by Koshiol et al. showed that the plasma aflatoxin B1 (AFB1) was present in 32% of GBC patients. Interestingly, none of the 54 sequenced tumors had the R249S mutation in the TP53 gene, which has been associated with aflatoxin exposure [187], suggesting that there could be other mechanisms involved in gallbladder carcinogenesis which are different from those observed in the development of other cancer types.
GBC may also be induced by exposure to certain toxic elements and compounds. For instance, arsenic (As) is a highly toxic semi-metallic element that can induce disease in humans by different sources of exposure, mainly via contaminated groundwater, residues from textile manufacturing, pesticides, mine dust and others, representing one of the greatest threats to public health in different regions of the world [188]. It has been described that the mechanisms by which arsenic can induce cancer are different, including inducing ROS formation and modulating epigenetic changes through histone modification and DNA methylation [189]. This element can enter the body predominantly by consuming contaminated water used for drinking, for food preparation, and for vegetable irrigation, as well as by exposure to pesticides and other chemicals products [188]. Exposure to the trivalent inorganic form (iAs) (III) and its mono- and dimethylated derivatives MMA(III) and DMA(III), respectively, are associated with cancers of the skin, lung, bladder, kidney, and liver [189,190]. Interestingly, in certain locations a significant correlation has been observed between the frequency of GBC and the presence of arsenic in waters destined for drinking or food preparation, suggesting a potential carcinogenic role in gallbladder carcinogenesis [191]. Surprisingly, some studies have suggested arsenic’s protective effect on GBC [192]. In this regard, Barahona et al. found a protective effect of iAs% on the risk of GBC (OR = 0.80, p = 0.03), showing that a poor metabolizing capacity, marked by the highest percentage of MMA, showed a protective effect (OR = 0.85, p = 0.08). On the other hand, the highest percentage of DMA, a marker of efficient arsenic metabolism, showed a non-protective effect on the risk of GBC (OR=1.10, p = 0.06). Interestingly, the presence of some variants of AS3MT (rs9527) and FTCD (rs61735836) were shown to be associated with MMA, iAS, and a low risk of GBC. While the rs11191527 variant of the AS3MT gene showed discrepant results [35].
Other elements and compounds such as nickel (Ni), cadmium (Cd), chromium (Cr) [33,193], molybdenum (Mo) [194], copper (Cu) [195], asbestos [34], coal or wood dust [196], and radon (Rn) have been associated with GBC or bile duct cancer [197]. For example, serum Mo levels are higher in patients with gallstones and GBC [194], suggesting that exposure to this metal could be a risk factor for developing GBC. By contrast, lower serum levels of selenium (Se), zinc (Zn), manganese (Mn), vitamin E, and vitamin C, elements that have been described as antioxidant agents and modulators of the immune system [198], were found in GBC when compared to cholelithiasis and healthy controls. In fact, copper (Cu) levels and the Cu/Zn ratio also showed a significant increase in the serum, bile, and gallbladder tissues of subjects with GBC compared to the other elements [195], indicating that the deficiency of Se, Zn, Mn, vitamin E, and vitamin C could be a risk for developing GBC, while the Cu/Zn ratio could be another biological parameter to also determine the risk for GBC.
Different articles have addressed gallbladder carcinogenesis in different ways, focusing mainly on epidemiological [67,80,199,200] and clinical aspects [201]. This review comprehensively addresses the cellular and/or molecular aspects induced by risk factors that lead to gallbladder carcinogenesis. These aspects include immune system activation, chronic inflammation, nutritional features, infections, genomic alterations/genetic variability, signaling pathway activation, and lipid metabolism. Many of these mechanisms result in gallstone formation and/or chronic inflammation that subsequently leads to GBC. Moreover, we address recent topics regarding neoantigens that could be useful for developing tumor immunotherapies against GBC and the implications of aflatoxin consumption from food that initiate an inflammatory state before gallbladder carcinogenesis. In summary, this article emphasizes GBC from the many aspects involving an inflammation response that could help to better understand this devastating disease.
Figure 3 shows the relationship between the risk factors described above and their involvement in GBC inflammation and carcinogenesis.
8. Conclusions
The cumulative evidence described in this review shows that GBC is a chronic inflammatory disease promoted by different risk factors including gallstone disease (GSD), sedentary lifestyle, smoking, alcohol consumption, metabolic disorders, high-fat diet, hypercholesterolemia, and some types of infections. In addition, gallbladder carcinogenesis has been strongly associated with specific populations (e.g., Mapuche ancestry), possibly because these populations present some genetic background (e.g., ABCG8 and TRAF3 gene variants) that predispose them to being more susceptible to gallstone formation and hence a greater predisposition to the development GBC, which could explain the high incidence of GBC in this population.
On the other hand, there are certain genetic alterations that could induce or maintain a gallbladder carcinogenic state. For instance, TP53 mutations are usually described as regulators of the immune response through the NF-κB pathway, suggesting that alterations in TP53 could be considered a pivotal step to gallbladder carcinogenesis. In addition, mutations in other genes (e.g., FHIT, IL10, and IL8), as well as the overexpression of many inflammatory proteins (e.g., COX-2 and TNF-α), are usually considered crucial in the maintenance of the chronic inflammatory state characterized by a continuous damage/regeneration loop that results in the carcinogenic process. Therefore, regarding the above-mentioned topic, that is, whether populations that already have a greater inherited genetic susceptibility (e.g., Mapuche ancestry) are able to develop additional genetic alterations, the incidence of GBC might be eventually maintained or increased; thus, the search for new therapies that seek to avoid chronic inflammation and continuous carcinogenic development in the affected population would have a great impact in decreasing the incidence of this malignancy.
Infections by different pathogens (e.g., bacteria) can also generate an exacerbated immune response due to the repetitive damage produced in the gallbladder mucosa through the different molecular mechanisms that these pathogens use to survive in the host. This review has shown that the presence of different bacterial genera (e.g., Salmonella spp and Helicobacter species) in the bile of patients with cholecystitis is strongly associated with gallbladder carcinogenesis, suggesting that infections by different pathogens could be a feasible cause of a chronic inflammation state to promote GBC [26,202,203].
Similarly, autoimmune diseases also predispose to a chronic inflammatory state in the affected tissues. One of these pathologies is PSC, which is characterized mainly by the destruction of the intrahepatic and extrahepatic bile ducts, which induces a greater presence of inflammatory components, suggesting that PSC and other inflammatory diseases could be significant to GBC development in these patients. The treatments for such diseases are mainly focused on reducing the inflammatory effect and the activity of the immune system in order to slow down the pathological consequences in patients, but not on completely eliminating the chronicity of the tissue damage. Furthermore, if inherited and acquired mutational heterogeneity is added to the previous variables, the scenario is very favorable to promoting a persistent carcinogenic development.
Smoking and alcohol consumption are widely known risk factors for several cancer types; however, these factors have not been well defined for GBC. This article reviewed some studies that propose smoking could be a risk factor for the development of GBC. Regarding the involvement of alcohol in GBC, the evidence is not conclusive because studies have not found a reliable correlation between GBC and the amount of alcohol consumed by individuals. Interestingly, some studies have found that men have a higher risk of developing GBC than women. This could be explained by the fact that alcohol consumption is frequently higher in men than in women. However, in recent years a considerable increase in smoking and alcohol consumption has been evidenced among young people; therefore, it would not be unusual to observe an increase in the GBC incidence due to these causes in the coming years. Finally, there is worldwide concern about human exposure to harmful elements and compounds (e.g., arsenic) commonly found in water sources and foods. Unfortunately, this problem is more frequent in lower-income countries where health policies may be insufficient compared to developed countries. Although some of these elements have not been directly associated with GBC yet, they could be an added value in the predisposition to the development of GBC. An example of this is arsenic, an element widely described as dangerous to humans. Regarding this, some studies have shown arsenic as a protective element and others as a risk factor for GBC; therefore, considering this problem, the "protective" concept is still inconclusive, requiring even more studies to demonstrate this hypothesis.
In summary, gallbladder carcinogenesis is induced by several risk factors that regardless of their origin trigger a chronic inflammatory state. Therefore, one of the challenges for the scientific community is the search for preventive treatments that can reduce the effect of chronic inflammation in the gallbladder, and hence, slow the onset of GBC. Finally, it is necessary that government authorities along with researchers commit to promoting massive sanitary and health prevention strategies in the global population, especially in poor and developing nations.
Author Contributions
Conceptualization, P.P.-M.; writing—original draft preparation, P.P.-M.; writing—review and editing, I.R.; supervision, J.C.R., P.G., and P.B.; funding acquisition, J.C.R. All authors have read and agreed to the published version of the manuscript.
Funding
This research received funding from the National Fund for Scientific and Technological Development (FONDECYT) [No. 3210237 to P.P.-M.; No. 1210440 to P.B.; No. 1221345 to J.C.R.], the Research Direction from Universidad Autónoma de Chile (DIUA) [No. 226-2021 to I.R.], the Re-search Direction from Universidad de La Frontera (DIUFRO) [No. DI20-0128 to P.B.], FONDEF Idea [No. ID21I10027 to P.B.], the Millennium Institute on Immunology and Immunotherapy (IMII) [No. ICN2021_045 to P.P.-M., P.G., J.C.R. and P.B.], and the European Research Council (ERC) under the European Union’s Horizon 2020 Research and Innovation Programme [No. 825510 to J.C.R.].
Conflicts of Interest
The authors declare no conflict of interest.
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Hundal, R.; Shaffer, E.A. Gallbladder cancer: Epidemiology and outcome. Clin Epidemiol 2014, 6, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Alvi, A.R.; Siddiqui, N.A.; Zafar, H. Risk factors of gallbladder cancer in Karachi-a case-control study. World J. Surg. Oncol. 2011, 9, 164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roa, I.; Ibacache, G.; Roa, J.; Araya, J.; de Aretxabala, X.; Muñoz, S. Gallstones and gallbladder cancer-volume and weight of gallstones are associated with gallbladder cancer: A case-control study. J. Surg. Oncol. 2006, 93, 624–628. [Google Scholar] [CrossRef] [PubMed]
- Hsing, A.W.; Gao, Y.-T.; Han, T.-Q.; Rashid, A.; Sakoda, L.C.; Wang, B.-S.; Shen, M.-C.; Zhang, B.-H.; Niwa, S.; Chen, J.; et al. Gallstones and the risk of biliary tract cancer: A population-based study in China. Br. J. Cancer 2007, 97, 1577–1582. [Google Scholar] [CrossRef] [PubMed]
- Espinoza, J.A.; Bizama, C.; García, P.; Ferreccio, C.; Javle, M.; Miquel, J.F.; Koshiol, J.; Roa, J.C. The inflammatory inception of gallbladder cancer. Biochim. Et Biophys. Acta (BBA)-Bioenerg. 2016, 1865, 245–254. [Google Scholar] [CrossRef]
- Stokes, C.S.; Krawczyk, M.; Lammert, F. Gallstones: Environment, Lifestyle and Genes. Dig. Dis. 2011, 29, 191–201. [Google Scholar] [CrossRef]
- Bouvard, V.; Baan, R.; Straif, K.; Grosse, Y.; Secretan, B.; El Ghissassi, F.; Benbrahim-Tallaa, L.; Guha, N.; Freeman, C.; Galichet, L.; et al. A review of human carcinogens—Part B: Biological agents. Lancet Oncol. 2009, 10, 321–322. [Google Scholar] [CrossRef]
- Nagaraja, V.; Eslick, G.D. Systematic review with meta-analysis: The relationship between chronicSalmonella typhicarrier status and gall-bladder cancer. Aliment. Pharmacol. Ther. 2014, 39, 745–750. [Google Scholar] [CrossRef]
- Zhu, Q.; Sun, X.; Ji, X.; Zhu, L.; Xu, J.; Wang, C.; Zhang, C.; Xue, F.; Liu, Y. The association between gallstones and metabolic syndrome in urban Han Chinese: A longitudinal cohort study. Sci. Rep. 2016, 6, 29937. [Google Scholar] [CrossRef] [Green Version]
- van Erp, L.W.; Cunningham, M.; Narasimman, M.; Ali, H.A.; Jhaveri, K.; Drenth, J.P.H.; Janssen, H.L.A.; Levy, C.; Hirschfield, G.M.; Hansen, B.E.; et al. Risk of gallbladder cancer in patients with primary sclerosing cholangitis and radiographically detected gallbladder polyps. Liver Int. 2020, 40, 382–392. [Google Scholar] [CrossRef] [PubMed]
- Deng, Z.; Xuan, Y.; Li, X.; Crawford, W.J.; Yuan, Z.; Chen, Z.; Brooks, A.; Song, Y.; Wang, H.; Liang, X.; et al. Effect of metabolic syndrome components on the risk of malignancy in patients with gallbladder lesions. J. Cancer 2021, 12, 1531–1537. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Yan, S.; Wang, B.; Shen, F.; Cao, H.; Fan, J.; Wang, Y. Type 2 diabetes mellitus and risk of gallbladder cancer: A systematic review and meta-analysis of observational studies. Diabetes/Metab. Res. Rev. 2015, 32, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Wenbin, D.; Zhuo, C.; Zhibing, M.; Chen, Z.; Ruifan, Y.; Jie, J.; Cheng, Q.; Zhenming, G. The effect of smoking on the risk of gallbladder cancer: A meta-analysis of observational studies. Eur. J. Gastroenterol. Hepatol. 2013, 25, 373–379. [Google Scholar] [CrossRef]
- Yagyu, K.; Kikuchi, S.; Obata, Y.; Lin, Y.; Ishibashi, T.; Kurosawa, M.; Inaba, Y.; Tamakoshi, A.; JACC Study Group. Cigarette smoking, alcohol drinking and the risk of gallbladder cancer death: A prospective cohort study in Japan. Int. J. Cancer 2008, 122, 924–929. [Google Scholar] [CrossRef] [PubMed]
- McGee, E.E.; Jackson, S.S.; Petrick, J.L.; Van Dyke, A.L.; Adami, H.-O.; Albanes, D.; Andreotti, G.; Beane-Freeman, L.E.; De Gonzalez, A.B.; Buring, J.E.; et al. Smoking, Alcohol, and Biliary Tract Cancer Risk: A Pooling Project of 26 Prospective Studies. J. Natl. Cancer Inst. 2019, 111, 1263–1278. [Google Scholar] [CrossRef] [PubMed]
- Al Manasra, A.R.A.; Jadallah, K.; Aleshawi, A.; Al-Omari, M.; Elheis, M.; Reyad, A.; Fataftah, J.; Al-Domaidat, H. Intractable Biliary Candidiasis in Patients with Obstructive Jaundice and Regional Malignancy: A Retrospective Case Series. Clin. Exp. Gastroenterol. 2021, 14, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Larsson, S.C.; Giovannucci, E.L.; Wolk, A. Sweetened Beverage Consumption and Risk of Biliary Tract and Gallbladder Cancer in a Prospective Study. J. Natl. Cancer Inst. 2016, 108. [Google Scholar] [CrossRef] [Green Version]
- Borena, W.; Edlinger, M.; Bjørge, T.; Häggström, C.; Lindkvist, B.; Nagel, G.; Engeland, A.; Stocks, T.; Strohmaier, S.; Manjer, J.; et al. A prospective study on metabolic risk factors and gallbladder cancer in the metabolic syndrome and cancer (Me-Can) collaborative study. PLoS ONE 2014, 9, e89368. [Google Scholar] [CrossRef] [Green Version]
- Hayat, S.; Hassan, Z.; Changazi, S.H.; Zahra, A.; Noman, M.; Zain Ul Abdin, M.; Javed, H.; Ans, A.H. Comparative analysis of serum lipid profiles in patients with and without gallstones: A prospective cross-sectional study. Ann. Med. Surg. 2019, 42, 11–13. [Google Scholar] [CrossRef]
- Renehan, A.G.; Tyson, M.; Egger, M.; Heller, R.F.; Zwahlen, M. Body-mass index and incidence of cancer: A systematic review and meta-analysis of prospective observational studies. Lancet 2008, 371, 569–578. [Google Scholar] [CrossRef]
- Tan, W.; Gao, M.; Liu, N.; Zhang, G.; Xu, T.; Cui, W. Body Mass Index and Risk of Gallbladder Cancer: Systematic Review and Meta-Analysis of Observational Studies. Nutrients 2015, 7, 8321–8334. [Google Scholar] [CrossRef] [Green Version]
- Xiao, M.; Gao, Y.; Wang, Y. Helicobacter species infection may be associated with cholangiocarcinoma: A meta-analysis. Int J Clin. Pract. 2014, 68, 262–270. [Google Scholar] [CrossRef]
- Chen, Z.X.; Peng, X.T.; Tan, L.; Zhai, G.Q.; Chen, G.; Gan, T.Q.; Li, J.J. EBV as a potential risk factor for hepatobiliary system cancer: A meta-analysis with 918 cases. Pathol. Res. Pract. 2019, 215, 278–285. [Google Scholar] [CrossRef]
- O’Keeffe, L.M.; Taylor, G.; Huxley, R.R.; Mitchell, P.; Woodward, M.; Peters, S.A.E. Smoking as a risk factor for lung cancer in women and men: A systematic review and meta-analysis. BMJ Open 2018, 8, e021611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Browne, A.J.; Kashef Hamadani, B.H.; Kumaran, E.A.P.; Rao, P.; Longbottom, J.; Harriss, E.; Moore, C.E.; Dunachie, S.; Basnyat, B.; Baker, S.; et al. Drug-resistant enteric fever worldwide, 1990 to 2018: A systematic review and meta-analysis. BMC Med. 2020, 18, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pandey, M.; Shukla, V.K. Diet and gallbladder cancer: A case-control study. Eur. J. Cancer Prev. 2002, 11, 365–368. [Google Scholar] [CrossRef]
- Mhatre, S.; Wang, Z.; Nagrani, R.; Badwe, R.; Chiplunkar, S.; Mittal, B.; Yadav, S.; Zhang, H.; Chung, C.C.; Patil, P.; et al. Common genetic variation and risk of gallbladder cancer in India: A case-control genome-wide association study. Lancet Oncol. 2017, 18, 535–544. [Google Scholar] [CrossRef]
- Strom, B.L.; Soloway, R.D.; Rios-Dalenz, J.L.; Rodriguez-Martinez, H.A.; West, S.L.; Kinman, J.L.; Polansky, M.; Berlin, J.A. Risk factors for gallbladder cancer. An international collaborative case-control study. Cancer 1995, 76, 1747–1756. [Google Scholar] [CrossRef]
- Lee, B.S.; Park, E.C.; Park, S.W.; Nam, C.M.; Roh, J. Hepatitis B virus infection, diabetes mellitus, and their synergism for cholangiocarcinoma development: A case-control study in Korea. World J. Gastroenterol. 2015, 21, 502–510. [Google Scholar] [CrossRef]
- Torabi Sagvand, B.; Edwards, K.; Shen, B. Frequency, Risk Factors, and Outcome of Gallbladder Polyps in Patients With Primary Sclerosing Cholangitis: A Case-Control Study. Hepatol. Commun. 2018, 2, 1440–1445. [Google Scholar] [CrossRef] [Green Version]
- Lin, T.C.; Chien, W.C.; Hu, J.M.; Tzeng, N.S.; Chung, C.H.; Pu, T.W.; Hsiao, C.W.; Chen, C.Y. Risk of colorectal cancer in patients with alcoholism: A nationwide, population-based nested case-control study. PLoS ONE 2020, 15, e0232740. [Google Scholar] [CrossRef] [PubMed]
- Shukla, V.K.; Prakash, A.; Tripathi, B.D.; Reddy, D.C.; Singh, S. Biliary heavy metal concentrations in carcinoma of the gall bladder: Case-control study. BMJ 1998, 317, 1288–1289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farioli, A.; Straif, K.; Brandi, G.; Curti, S.; Kjaerheim, K.; Martinsen, J.I.; Sparen, P.; Tryggvadottir, L.; Weiderpass, E.; Biasco, G.; et al. Occupational exposure to asbestos and risk of cholangiocarcinoma: A population-based case-control study in four Nordic countries. Occup. Environ. Med. 2018, 75, 191–198. [Google Scholar] [CrossRef] [Green Version]
- Barahona Ponce, C.; Scherer, D.; Boekstegers, F.; Garate-Calderon, V.; Jenab, M.; Aleksandrova, K.; Katzke, V.; Weiderpass, E.; Bonet, C.; Moradi, T.; et al. Arsenic and gallbladder cancer risk: Mendelian randomization analysis of European prospective data. Int. J. Cancer 2020, 146, 2648–2650. [Google Scholar] [CrossRef] [PubMed]
- Barahona Ponce, C.; Scherer, D.; Brinster, R.; Boekstegers, F.; Marcelain, K.; Gárate-Calderón, V.; Müller, B.; de Toro, G.; Retamales, J.; Barajas, O.; et al. Gallstones, Body Mass Index, C-Reactive Protein, and Gallbladder Cancer: Mendelian Randomization Analysis of Chilean and European Genotype Data. Hepatology 2021, 73, 1783–1796. [Google Scholar] [CrossRef]
- Medzhitov, R. Origin and physiological roles of inflammation. Nature 2008, 454, 428–435. [Google Scholar] [CrossRef]
- Weiss, U. Inflammation. Nature 2008, 454, 427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef]
- Ohshima, H.; Tatemichi, M.; Sawa, T. Chemical basis of inflammation-induced carcinogenesis. Arch. Biochem. Biophys 2003, 417, 3–11. [Google Scholar] [CrossRef]
- Okabe, Y.; Medzhitov, R. Tissue biology perspective on macrophages. Nat. Immunol. 2016, 17, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Greten, F.R.; Grivennikov, S.I. Inflammation and Cancer: Triggers, Mechanisms, and Consequences. Immunity 2019, 51, 27–41. [Google Scholar] [CrossRef]
- Loyher, P.L.; Hamon, P.; Laviron, M.; Meghraoui-Kheddar, A.; Goncalves, E.; Deng, Z.; Torstensson, S.; Bercovici, N.; Baudesson de Chanville, C.; Combadière, B.; et al. Macrophages of distinct origins contribute to tumor development in the lung. J. Exp. Med. 2018, 215, 2536–2553. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Herndon, J.M.; Sojka, D.K.; Kim, K.W.; Knolhoff, B.L.; Zuo, C.; Cullinan, D.R.; Luo, J.; Bearden, A.R.; Lavine, K.J.; et al. Tissue-Resident Macrophages in Pancreatic Ductal Adenocarcinoma Originate from Embryonic Hematopoiesis and Promote Tumor Progression. Immunity 2017, 47, 323–338.e326. [Google Scholar] [CrossRef]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, inflammation, and cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef] [Green Version]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef]
- Hussain, S.P.; Harris, C.C. Inflammation and cancer: An ancient link with novel potentials. Int. J. Cancer 2007, 121, 2373–2380. [Google Scholar] [CrossRef]
- Balkwill, F. Tumour necrosis factor and cancer. Nat. Rev. Cancer 2009, 9, 361–371. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.S.; Zhou, L.S.; Han, Y.; Zhu, A.J.; Sun, X.J.; Yang, Y.J. Expression of tumor necrosis factor and its receptor in gallstone and gallbladder carcinoma tissue. Hepatobiliary Pancreat Dis. Int. 2004, 3, 448–452. [Google Scholar]
- Nilsson, B.; Delbro, D.; Hedin, L.; Friman, S.; Andius, S.; Svanvik, J. Role of cyclooxygenase-2 for fluid secretion by the inflamed gallbladder mucosa. J. Gastrointest. Surg. 1998, 2, 269–277. [Google Scholar] [CrossRef]
- Longo, W.E.; Panesar, N.; Mazuski, J.E.; Kaminski, D. Synthetic pathways of gallbladder mucosal prostanoids: The role of cyclooxygenase-1 and 2. Prostaglandins Leukot. Essent. Fat. Acids 1999, 60, 77–85. [Google Scholar] [CrossRef]
- Thornell, E.; Jansson, R.; Kral, J.G.; Svanvik, J. Inhibition of prostaglandin synthesis as a treatment for biliary pain. Lancet 1979, 1, 584. [Google Scholar] [CrossRef]
- Babb, R.R. Managing gallbladder disease with prostaglandin inhibitors. Postgrad. Med. 1993, 94, 127–130. [Google Scholar] [CrossRef]
- Fumino, S.; Tokiwa, K.; Ono, S.; Iwai, N. Cyclooxygenase-2 expression in the gallbladder of patients with anomalous arrangement of the pancreaticobiliary duct. J. Pediatr. Surg. 2003, 38, 585–589. [Google Scholar] [CrossRef] [PubMed]
- Fumino, S.; Tokiwa, K.; Katoh, T.; Ono, S.; Iwai, N. New insight into bile flow dynamics in anomalous arrangement of the pancreaticobiliary duct. Br. J. Surg. 2002, 89, 865–869. [Google Scholar] [CrossRef]
- Tokiwa, K.; Iwai, N. Early mucosal changes of the gallbladder in patients with anomalous arrangement of the pancreaticobiliary duct. Gastroenterology 1996, 110, 1614–1618. [Google Scholar] [CrossRef]
- Sasatomi, E.; Tokunaga, O.; Miyazaki, K. Precancerous conditions of gallbladder carcinoma: Overview of histopathologic characteristics and molecular genetic findings. J. Hepatobiliary Pancreat. Surg. 2000, 7, 556–567. [Google Scholar] [CrossRef] [PubMed]
- Seretis, C.; Lagoudianakis, E.; Gemenetzis, G.; Seretis, F.; Pappas, A.; Gourgiotis, S. Metaplastic changes in chronic cholecystitis: Implications for early diagnosis and surgical intervention to prevent the gallbladder metaplasia-dysplasia-carcinoma sequence. J. Clin. Med. Res. 2014, 6, 26–29. [Google Scholar] [CrossRef] [Green Version]
- Hanada, K.; Tsuchida, A.; Iwao, T.; Eguchi, N.; Sasaki, T.; Morinaka, K.; Matsubara, K.; Kawasaki, Y.; Yamamoto, S.; Kajiyama, G. Gene mutations of K-ras in gallbladder mucosae and gallbladder carcinoma with an anomalous junction of the pancreaticobiliary duct. Am. J. Gastroenterol. 1999, 94, 1638–1642. [Google Scholar] [CrossRef]
- Jones, S.; Zhang, X.; Parsons, D.W.; Lin, J.C.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Kamiyama, H.; Jimeno, A.; et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science 2008, 321, 1801–1806. [Google Scholar] [CrossRef] [Green Version]
- Javle, M.; Rashid, A.; Churi, C.; Kar, S.; Zuo, M.; Eterovic, A.K.; Nogueras-Gonzalez, G.M.; Janku, F.; Shroff, R.T.; Aloia, T.A.; et al. Molecular characterization of gallbladder cancer using somatic mutation profiling. Hum. Pathol. 2014, 45, 701–708. [Google Scholar] [CrossRef] [Green Version]
- Matsubara, T.; Tsuji, T.; Miyama, A.; Yamaguchi, H.; Funabiki, T. Mutagenicity of bile and pancreatic juice from patients with pancreatico-biliary maljunction. Hepatogastroenterology 1995, 42, 113–116. [Google Scholar]
- Yamagiwa, H.; Tomiyama, H. Intestinal metaplasia-dysplasia-carcinoma sequence of the gallbladder. Pathol. Int. 1986, 36, 989–997. [Google Scholar] [CrossRef]
- Sharma, A.; Kumar, A.; Kumari, N.; Krishnani, N.; Rastogi, N. Mutational frequency of. Ecancermedicalscience 2017, 11, 757. [Google Scholar] [CrossRef] [Green Version]
- Singh, M.K.; Chetri, K.; Pandey, U.B.; Kapoor, V.K.; Mittal, B.; Choudhuri, G. Mutational spectrum of K-ras oncogene among Indian patients with gallbladder cancer. J. Gastroenterol. Hepatol. 2004, 19, 916–921. [Google Scholar] [CrossRef] [PubMed]
- Kiguchi, K.; Carbajal, S.; Chan, K.; Beltrán, L.; Ruffino, L.; Shen, J.; Matsumoto, T.; Yoshimi, N.; DiGiovanni, J. Constitutive expression of ErbB-2 in gallbladder epithelium results in development of adenocarcinoma. Cancer Res. 2001, 61, 6971–6976. [Google Scholar]
- Sharma, A.; Sharma, K.L.; Gupta, A.; Yadav, A.; Kumar, A. Gallbladder cancer epidemiology, pathogenesis and molecular genetics: Recent update. World J. Gastroenterol. 2017, 23, 3978–3998. [Google Scholar] [CrossRef] [PubMed]
- Narayan, R.R.; Creasy, J.M.; Goldman, D.A.; Gönen, M.; Kandoth, C.; Kundra, R.; Solit, D.B.; Askan, G.; Klimstra, D.S.; Basturk, O.; et al. Regional differences in gallbladder cancer pathogenesis: Insights from a multi-institutional comparison of tumor mutations. Cancer 2019, 125, 575–585. [Google Scholar] [CrossRef] [Green Version]
- Moreno, M.; Pimentel, F.; Gazdar, A.F.; Wistuba, I.I.; Miquel, J.F. TP53 abnormalities are frequent and early events in the sequential pathogenesis of gallbladder carcinoma. Ann. Hepatol. 2005, 4, 192–199. [Google Scholar] [CrossRef]
- Koda, M.; Yashima, K.; Kawaguchi, K.; Andachi, H.; Hosoda, A.; Shiota, G.; Ito, H.; Murawaki, Y. Expression of Fhit, Mlh1, and P53 protein in human gallbladder carcinoma. Cancer Lett. 2003, 199, 131–138. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, H.; Arai, Y.; Totoki, Y.; Shirota, T.; Elzawahry, A.; Kato, M.; Hama, N.; Hosoda, F.; Urushidate, T.; Ohashi, S.; et al. Genomic spectra of biliary tract cancer. Nat. Genet. 2015, 47, 1003–1010. [Google Scholar] [CrossRef] [PubMed]
- Brägelmann, J.; Barahona Ponce, C.; Marcelain, K.; Roessler, S.; Goeppert, B.; Gallegos, I.; Colombo, A.; Sanhueza, V.; Morales, E.; Rivera, M.T.; et al. Epigenome-Wide Analysis of Methylation Changes in the Sequence of Gallstone Disease, Dysplasia, and Gallbladder Cancer. Hepatology 2021, 73, 2293–2310. [Google Scholar] [CrossRef]
- Jain, K.; Mohapatra, T.; Das, P.; Misra, M.C.; Gupta, S.D.; Ghosh, M.; Kabra, M.; Bansal, V.K.; Kumar, S.; Sreenivas, V.; et al. Sequential occurrence of preneoplastic lesions and accumulation of loss of heterozygosity in patients with gallbladder stones suggest causal association with gallbladder cancer. Ann. Surg. 2014, 260, 1073–1080. [Google Scholar] [CrossRef] [PubMed]
- Wistuba, I.I.; Ashfaq, R.; Maitra, A.; Alvarez, H.; Riquelme, E.; Gazdar, A.F. Fragile histidine triad gene abnormalities in the pathogenesis of gallbladder carcinoma. Am. J. Pathol. 2002, 160, 2073–2079. [Google Scholar] [CrossRef] [Green Version]
- Roa, I.; Garcia, H.; Game, A.; de Toro, G.; de Aretxabala, X.; Javle, M. Somatic Mutations of PI3K in Early and Advanced Gallbladder Cancer: Additional Options for an Orphan Cancer. J. Mol. Diagn. 2016, 18, 388–394. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Yang, Z. Expression of phospho-ERK1/2 and PI3-K in benign and malignant gallbladder lesions and its clinical and pathological correlations. J. Exp. Clin. Cancer Res. 2009, 28, 65. [Google Scholar] [CrossRef] [Green Version]
- Hawkins, P.T.; Stephens, L.R. PI3K signalling in inflammation. Biochim. Biophys Acta 2015, 1851, 882–897. [Google Scholar] [CrossRef] [Green Version]
- Espinoza, J.A.; Riquelme, I.; Sagredo, E.A.; Rosa, L.; García, P.; Bizama, C.; Apud-Bell, M.; Leal, P.; Weber, H.; Benavente, F.; et al. Mucin 5B, carbonic anhydrase 9 and claudin 18 are potential theranostic markers of gallbladder carcinoma. Histopathology 2019, 74, 597–607. [Google Scholar] [CrossRef]
- Mishra, S.K.; Kumari, N.; Krishnani, N. Molecular pathogenesis of gallbladder cancer: An update. Mutat. Res. 2019, 816–818, 111674. [Google Scholar] [CrossRef]
- Salazar, M.; Ituarte, C.; Abriata, M.G.; Santoro, F.; Arroyo, G. Gallbladder cancer in South America: Epidemiology and prevention. Chin. Clin. Oncol. 2019, 8, 32. [Google Scholar] [CrossRef]
- Pandey, A.; Stawiski, E.W.; Durinck, S.; Gowda, H.; Goldstein, L.D.; Barbhuiya, M.A.; Schröder, M.S.; Sreenivasamurthy, S.K.; Kim, S.W.; Phalke, S.; et al. Integrated genomic analysis reveals mutated ELF3 as a potential gallbladder cancer vaccine candidate. Nat. Commun. 2020, 11, 4225. [Google Scholar] [CrossRef]
- Peng, M.; Mo, Y.; Wang, Y.; Wu, P.; Zhang, Y.; Xiong, F.; Guo, C.; Wu, X.; Li, Y.; Li, X.; et al. Neoantigen vaccine: An emerging tumor immunotherapy. Mol. Cancer 2019, 18, 128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ochiai, A.; Yamauchi, Y.; Hirohashi, S. p53 mutations in the non-neoplastic mucosa of the human stomach showing intestinal metaplasia. Int. J. Cancer 1996, 69, 28–33. [Google Scholar] [CrossRef]
- Shiao, Y.H.; Rugge, M.; Correa, P.; Lehmann, H.P.; Scheer, W.D. p53 alteration in gastric precancerous lesions. Am. J. Pathol. 1994, 144, 511–517. [Google Scholar] [PubMed]
- Hussain, S.P.; Amstad, P.; Raja, K.; Ambs, S.; Nagashima, M.; Bennett, W.P.; Shields, P.G.; Ham, A.J.; Swenberg, J.A.; Marrogi, A.J.; et al. Increased p53 mutation load in noncancerous colon tissue from ulcerative colitis: A cancer-prone chronic inflammatory disease. Cancer Res. 2000, 60, 3333–3337. [Google Scholar] [PubMed]
- Sozzi, G.; Miozzo, M.; Donghi, R.; Pilotti, S.; Cariani, C.T.; Pastorino, U.; Della Porta, G.; Pierotti, M.A. Deletions of 17p and p53 mutations in preneoplastic lesions of the lung. Cancer Res. 1992, 52, 6079–6082. [Google Scholar]
- Baugh, E.H.; Ke, H.; Levine, A.J.; Bonneau, R.A.; Chan, C.S. Why are there hotspot mutations in the TP53 gene in human cancers? Cell Death Differ. 2018, 25, 154–160. [Google Scholar] [CrossRef]
- Gudkov, A.V.; Gurova, K.V.; Komarova, E.A. Inflammation and p53: A Tale of Two Stresses. Genes Cancer 2011, 2, 503–516. [Google Scholar] [CrossRef]
- Karin, M.; Greten, F.R. NF-kappaB: Linking inflammation and immunity to cancer development and progression. Nat. Rev. Immunol. 2005, 5, 749–759. [Google Scholar] [CrossRef]
- Yu, H.; Pardoll, D.; Jove, R. STATs in cancer inflammation and immunity: A leading role for STAT3. Nat. Rev. Cancer 2009, 9, 798–809. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-κB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2. [Google Scholar] [CrossRef] [Green Version]
- Blagih, J.; Buck, M.D.; Vousden, K.H. p53, cancer and the immune response. J. Cell Sci. 2020, 133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Ciaula, A.; Portincasa, P. Recent advances in understanding and managing cholesterol gallstones. F1000Res 2018, 7. [Google Scholar] [CrossRef]
- Skoumas, J.; Pitsavos, C.; Panagiotakos, D.B.; Chrysohoou, C.; Zeimbekis, A.; Papaioannou, I.; Toutouza, M.; Toutouzas, P.; Stefanadis, C. Physical activity, high density lipoprotein cholesterol and other lipids levels, in men and women from the ATTICA study. Lipids Health Dis. 2003, 2, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsing, A.W.; Sakoda, L.C.; Rashid, A.; Chen, J.; Shen, M.C.; Han, T.Q.; Wang, B.S.; Gao, Y.T. Body size and the risk of biliary tract cancer: A population-based study in China. Br. J. Cancer 2008, 99, 811–815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Everson, G.T.; McKinley, C.; Kern, F. Mechanisms of gallstone formation in women. Effects of exogenous estrogen (Premarin) and dietary cholesterol on hepatic lipid metabolism. J. Clin. Invest. 1991, 87, 237–246. [Google Scholar] [CrossRef]
- Pinto Pereira, S.M.; Ki, M.; Power, C. Sedentary behaviour and biomarkers for cardiovascular disease and diabetes in mid-life: The role of television-viewing and sitting at work. PLoS ONE 2012, 7, e31132. [Google Scholar] [CrossRef]
- Crichton, G.E.; Alkerwi, A. Physical activity, sedentary behavior time and lipid levels in the Observation of Cardiovascular Risk Factors in Luxembourg study. Lipids Health Dis. 2015, 14, 87. [Google Scholar] [CrossRef] [Green Version]
- Arcidiacono, B.; Iiritano, S.; Nocera, A.; Possidente, K.; Nevolo, M.T.; Ventura, V.; Foti, D.; Chiefari, E.; Brunetti, A. Insulin resistance and cancer risk: An overview of the pathogenetic mechanisms. Exp. Diabetes Res. 2012, 2012, 789174. [Google Scholar] [CrossRef] [Green Version]
- Van Erpecum, K.J. Pathogenesis of cholesterol and pigment gallstones: An update. Clin. Res. Hepatol. Gastroenterol. 2011, 35, 281–287. [Google Scholar] [CrossRef]
- Rebholz, C.; Krawczyk, M.; Lammert, F. Genetics of gallstone disease. Eur. J. Clin. Invest. 2018, 48, e12935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryu, S.; Chang, Y.; Yun, K.E.; Jung, H.S.; Shin, J.H.; Shin, H. Gallstones and the Risk of Gallbladder Cancer Mortality: A Cohort Study. Am. J. Gastroenterol. 2016, 111, 1476–1487. [Google Scholar] [CrossRef] [PubMed]
- Stinton, L.M.; Shaffer, E.A. Epidemiology of gallbladder disease: Cholelithiasis and cancer. Gut Liver 2012, 6, 172–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rousset, X.; Vaisman, B.; Amar, M.; Sethi, A.A.; Remaley, A.T. Lecithin: Cholesterol acyltransferase--from biochemistry to role in cardiovascular disease. Curr. Opin. Endocrinol. Diabetes Obes. 2009, 16, 163–171. [Google Scholar] [CrossRef] [Green Version]
- Masson, D.; Jiang, X.C.; Lagrost, L.; Tall, A.R. The role of plasma lipid transfer proteins in lipoprotein metabolism and atherogenesis. J. Lipid Res. 2009, 50, S201–S206. [Google Scholar] [CrossRef] [Green Version]
- Acton, S.; Rigotti, A.; Landschulz, K.T.; Xu, S.; Hobbs, H.H.; Krieger, M. Identification of scavenger receptor SR-BI as a high density lipoprotein receptor. Science 1996, 271, 518–520. [Google Scholar] [CrossRef]
- Barter, P.J.; Brewer, H.B.; Chapman, M.J.; Hennekens, C.H.; Rader, D.J.; Tall, A.R. Cholesteryl ester transfer protein: A novel target for raising HDL and inhibiting atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 160–167. [Google Scholar] [CrossRef]
- Rader, D.J. Regulation of reverse cholesterol transport and clinical implications. Am. J. Cardiol. 2003, 92, 42J–49J. [Google Scholar] [CrossRef]
- Atamanalp, S.S.; Keles, M.S.; Atamanalp, R.S.; Acemoglu, H.; Laloglu, E. The effects of serum cholesterol, LDL, and HDL levels on gallstone cholesterol concentration. Pak. J. Med. Sci. 2013, 29, 187–190. [Google Scholar] [CrossRef]
- Wang, J.; Shen, S.; Wang, B.; Ni, X.; Liu, H.; Yu, R.; Suo, T. Serum lipid levels are the risk factors of gallbladder stones: A population-based study in China. Lipids Health Dis. 2020, 19, 50. [Google Scholar] [CrossRef] [Green Version]
- Reshetnyak, V.I. Concept of the pathogenesis and treatment of cholelithiasis. World J. Hepatol. 2012, 4, 18–34. [Google Scholar] [CrossRef]
- Shrikhande, S.V.; Barreto, S.G.; Singh, S.; Udwadia, T.E.; Agarwal, A.K. Cholelithiasis in gallbladder cancer: Coincidence, cofactor, or cause! Eur. J. Surg. Oncol. 2010, 36, 514–519. [Google Scholar] [CrossRef] [PubMed]
- Kuraishy, A.; Karin, M.; Grivennikov, S.I. Tumor promotion via injury- and death-induced inflammation. Immunity 2011, 35, 467–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jang, S.I.; Fang, S.; Kim, K.P.; Ko, Y.; Kim, H.; Oh, J.; Hong, G.Y.; Lee, S.Y.; Kim, J.M.; Noh, I.; et al. Combination treatment with n-3 polyunsaturated fatty acids and ursodeoxycholic acid dissolves cholesterol gallstones in mice. Sci. Rep. 2019, 9, 12740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maurer, K.J.; Rao, V.P.; Ge, Z.; Rogers, A.B.; Oura, T.J.; Carey, M.C.; Fox, J.G. T-cell function is critical for murine cholesterol gallstone formation. Gastroenterology 2007, 133, 1304–1315. [Google Scholar] [CrossRef] [Green Version]
- Rosa, L.; Lobos-González, L.; Muñoz-Durango, N.; García, P.; Bizama, C.; Gómez, N.; González, X.; Wichmann, I.A.; Saavedra, N.; Guevara, F.; et al. Evaluation of the chemopreventive potentials of ezetimibe and aspirin in a novel mouse model of gallbladder preneoplasia. Mol. Oncol. 2020, 14, 2834–2852. [Google Scholar] [CrossRef] [PubMed]
- Lavoie, B.; Nausch, B.; Zane, E.A.; Leonard, M.R.; Balemba, O.B.; Bartoo, A.C.; Wilcox, R.; Nelson, M.T.; Carey, M.C.; Mawe, G.M. Disruption of gallbladder smooth muscle function is an early feature in the development of cholesterol gallstone disease. Neurogastroenterol. Motil. 2012, 24, e313–e324. [Google Scholar] [CrossRef] [Green Version]
- Kato, S.; Fushimi, K.; Yabuki, Y.; Maru, Y.; Hasegawa, S.; Matsuura, T.; Kurotaki, D.; Suzuki, A.; Kobayashi, N.; Yoneda, M.; et al. Precision modeling of gall bladder cancer patients in mice based on orthotopic implantation of organoid-derived tumor buds. Oncogenesis 2021, 10, 33. [Google Scholar] [CrossRef]
- Liu, Z.; Kemp, T.J.; Gao, Y.T.; Corbel, A.; McGee, E.E.; Wang, B.; Shen, M.C.; Rashid, A.; Hsing, A.W.; Hildesheim, A.; et al. Association of circulating inflammation proteins and gallstone disease. J. Gastroenterol. Hepatol. 2018, 33, 1920–1924. [Google Scholar] [CrossRef]
- Moschen, A.R.; Kaser, A.; Enrich, B.; Mosheimer, B.; Theurl, M.; Niederegger, H.; Tilg, H. Visfatin, an adipocytokine with proinflammatory and immunomodulating properties. J. Immunol. 2007, 178, 1748–1758. [Google Scholar] [CrossRef] [Green Version]
- Park, J.W.; Kim, O.H.; Lee, S.C.; Kim, K.H.; Hong, H.E.; Seo, H.; Choi, H.J.; Kim, S.J. Serum level of visfatin can reflect the severity of inflammation in patients with acute cholecystitis. Ann. Surg. Treat. Res. 2020, 99, 26–36. [Google Scholar] [CrossRef] [PubMed]
- Grünhage, F.; Acalovschi, M.; Tirziu, S.; Walier, M.; Wienker, T.F.; Ciocan, A.; Mosteanu, O.; Sauerbruch, T.; Lammert, F. Increased gallstone risk in humans conferred by common variant of hepatic ATP-binding cassette transporter for cholesterol. Hepatology 2007, 46, 793–801. [Google Scholar] [CrossRef]
- Small, D.M. Role of ABC transporters in secretion of cholesterol from liver into bile. Proc Natl Acad Sci USA 2003, 100, 4–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Von Kampen, O.; Buch, S.; Nothnagel, M.; Azocar, L.; Molina, H.; Brosch, M.; Erhart, W.; von Schönfels, W.; Egberts, J.; Seeger, M.; et al. Genetic and functional identification of the likely causative variant for cholesterol gallstone disease at the ABCG5/8 lithogenic locus. Hepatology 2013, 57, 2407–2417. [Google Scholar] [CrossRef]
- Bustos, B.I.; Pérez-Palma, E.; Buch, S.; Azócar, L.; Riveras, E.; Ugarte, G.D.; Toliat, M.; Nürnberg, P.; Lieb, W.; Franke, A.; et al. Variants in ABCG8 and TRAF3 genes confer risk for gallstone disease in admixed Latinos with Mapuche Native American ancestry. Sci. Rep. 2019, 9, 772. [Google Scholar] [CrossRef]
- Jackson, S.S.; Van De Wyngard, V.; Pfeiffer, R.M.; Cook, P.; Hildesheim, A.; Pinto, L.A.; Jackson, S.H.; Choi, K.; Verdugo, R.A.; Cuevas, M.; et al. Inflammatory profiles in Chilean Mapuche and non-Mapuche women with gallstones at risk of developing gallbladder cancer. Sci. Rep. 2021, 11, 3686. [Google Scholar] [CrossRef]
- Delaunay, J.L.; Durand-Schneider, A.M.; Dossier, C.; Falguières, T.; Gautherot, J.; Davit-Spraul, A.; Aït-Slimane, T.; Housset, C.; Jacquemin, E.; Maurice, M. A functional classification of ABCB4 variations causing progressive familial intrahepatic cholestasis type 3. Hepatology 2016, 63, 1620–1631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakeeb, A.; Comuzzie, A.G.; Martin, L.; Sonnenberg, G.E.; Swartz-Basile, D.; Kissebah, A.H.; Pitt, H.A. Gallstones: Genetics versus environment. Ann. Surg. 2002, 235, 842–849. [Google Scholar] [CrossRef]
- Rosmorduc, O.; Hermelin, B.; Boelle, P.Y.; Parc, R.; Taboury, J.; Poupon, R. ABCB4 gene mutation-associated cholelithiasis in adults. Gastroenterology 2003, 125, 452–459. [Google Scholar] [CrossRef]
- Jansen, P.L.; Strautnieks, S.S.; Jacquemin, E.; Hadchouel, M.; Sokal, E.M.; Hooiveld, G.J.; Koning, J.H.; De Jager-Krikken, A.; Kuipers, F.; Stellaard, F.; et al. Hepatocanalicular bile salt export pump deficiency in patients with progressive familial intrahepatic cholestasis. Gastroenterology 1999, 117, 1370–1379. [Google Scholar] [CrossRef]
- Pullinger, C.R.; Eng, C.; Salen, G.; Shefer, S.; Batta, A.K.; Erickson, S.K.; Verhagen, A.; Rivera, C.R.; Mulvihill, S.J.; Malloy, M.J.; et al. Human cholesterol 7alpha-hydroxylase (CYP7A1) deficiency has a hypercholesterolemic phenotype. J. Clin. Invest. 2002, 110, 109–117. [Google Scholar] [CrossRef]
- Miyake, J.H.; Duong-Polk, X.T.; Taylor, J.M.; Du, E.Z.; Castellani, L.W.; Lusis, A.J.; Davis, R.A. Transgenic expression of cholesterol-7-alpha-hydroxylase prevents atherosclerosis in C57BL/6J mice. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 121–126. [Google Scholar] [CrossRef] [Green Version]
- Lancellotti, S.; Zaffanello, M.; Di Leo, E.; Costa, L.; Lonardo, A.; Tarugi, P. Pediatric gallstone disease in familial hypobetalipoproteinemia. J. Hepatol. 2005, 43, 188–191. [Google Scholar] [CrossRef] [PubMed]
- Miller, L.J.; Holicky, E.L.; Ulrich, C.D.; Wieben, E.D. Abnormal processing of the human cholecystokinin receptor gene in association with gallstones and obesity. Gastroenterology 1995, 109, 1375–1380. [Google Scholar] [CrossRef]
- Wang, D.Q.; Schmitz, F.; Kopin, A.S.; Carey, M.C. Targeted disruption of the murine cholecystokinin-1 receptor promotes intestinal cholesterol absorption and susceptibility to cholesterol cholelithiasis. J. Clin. Invest. 2004, 114, 521–528. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, K.; Srivastava, A.; Kumar, A.; Mittal, B. Significant association between toll-like receptor gene polymorphisms and gallbladder cancer. Liver Int. 2010, 30, 1067–1072. [Google Scholar] [CrossRef] [PubMed]
- Vishnoi, M.; Pandey, S.N.; Choudhuri, G.; Mittal, B. IL-1 gene polymorphisms and genetic susceptibility of gallbladder cancer in a north Indian population. Cancer Genet. Cytogenet. 2008, 186, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Hsing, A.W.; Sakoda, L.C.; Rashid, A.; Andreotti, G.; Chen, J.; Wang, B.S.; Shen, M.C.; Chen, B.E.; Rosenberg, P.S.; Zhang, M.; et al. Variants in inflammation genes and the risk of biliary tract cancers and stones: A population-based study in China. Cancer Res. 2008, 68, 6442–6452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srivastava, A.; Pandey, S.N.; Choudhuri, G.; Mittal, B. CCR5 Delta32 polymorphism: Associated with gallbladder cancer susceptibility. Scand. J. Immunol. 2008, 67, 516–522. [Google Scholar] [CrossRef]
- Cha, P.C.; Zembutsu, H.; Takahashi, A.; Kubo, M.; Kamatani, N.; Nakamura, Y. A genome-wide association study identifies SNP in DCC is associated with gallbladder cancer in the Japanese population. J. Hum. Genet. 2012, 57, 235–237. [Google Scholar] [CrossRef] [PubMed]
- Stephen, A.E.; Berger, D.L. Carcinoma in the porcelain gallbladder: A relationship revisited. Surgery 2001, 129, 699–703. [Google Scholar] [CrossRef]
- Morimoto, M.; Matsuo, T.; Mori, N. Management of Porcelain Gallbladder, Its Risk Factors, and Complications: A Review. Diagnostics 2021, 11, 1073. [Google Scholar] [CrossRef] [PubMed]
- Lam, R.; Zakko, A.; Petrov, J.C.; Kumar, P.; Duffy, A.J.; Muniraj, T. Gallbladder Disorders: A Comprehensive Review. Dis. Mon. 2021, 67, 101130. [Google Scholar] [CrossRef]
- Berger, M.; Dy, B.; McKenzie, T.; Thompson, G.; Wermers, R.; Lyden, M. Rates of hypercalcemia and hyperparathyroidism among patients with porcelain gallbladder. Am. J. Surg. 2020, 220, 127–131. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S. Infection as a risk factor for gallbladder cancer. J. Surg. Oncol. 2006, 93, 633–639. [Google Scholar] [CrossRef]
- Mogensen, T.H. Pathogen recognition and inflammatory signaling in innate immune defenses. Clin. Microbiol. Rev. 2009, 22, 240–273, Table of Contents. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Zhang, B.Y.; Shi, J.S.; Wu, L.Q. Expression of the bacterial gene in gallbladder carcinoma tissue and bile. Hepatobiliary Pancreat. Dis. Int. 2004, 3, 133–135. [Google Scholar] [PubMed]
- Iyer, P.; Barreto, S.G.; Sahoo, B.; Chandrani, P.; Ramadwar, M.R.; Shrikhande, S.V.; Dutt, A. Non-typhoidal Salmonella DNA traces in gallbladder cancer. Infect. Agents Cancer 2016, 11, 12. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Escobedo, G.; Marshall, J.M.; Gunn, J.S. Chronic and acute infection of the gall bladder by Salmonella Typhi: Understanding the carrier state. Nat. Rev. Microbiol. 2011, 9, 9–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caygill, C.P.; Hill, M.J.; Braddick, M.; Sharp, J.C. Cancer mortality in chronic typhoid and paratyphoid carriers. Lancet 1994, 343, 83–84. [Google Scholar] [CrossRef]
- Shukla, V.K.; Singh, H.; Pandey, M.; Upadhyay, S.K.; Nath, G. Carcinoma of the gallbladder--is it a sequel of typhoid? Dig. Dis. Sci. 2000, 45, 900–903. [Google Scholar] [CrossRef] [PubMed]
- Scanu, T.; Spaapen, R.M.; Bakker, J.M.; Pratap, C.B.; Wu, L.E.; Hofland, I.; Broeks, A.; Shukla, V.K.; Kumar, M.; Janssen, H.; et al. Salmonella Manipulation of Host Signaling Pathways Provokes Cellular Transformation Associated with Gallbladder Carcinoma. Cell Host Microbe 2015, 17, 763–774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fox, J.G.; Dewhirst, F.E.; Shen, Z.; Feng, Y.; Taylor, N.S.; Paster, B.J.; Ericson, R.L.; Lau, C.N.; Correa, P.; Araya, J.C.; et al. Hepatic Helicobacter species identified in bile and gallbladder tissue from Chileans with chronic cholecystitis. Gastroenterology 1998, 114, 755–763. [Google Scholar] [CrossRef]
- Matsukura, N.; Yokomuro, S.; Yamada, S.; Tajiri, T.; Sundo, T.; Hadama, T.; Kamiya, S.; Naito, Z.; Fox, J.G. Association between Helicobacter bilis in bile and biliary tract malignancies: H. bilis in bile from Japanese and Thai patients with benign and malignant diseases in the biliary tract. Jpn. J. Cancer Res. 2002, 93, 842–847. [Google Scholar] [CrossRef]
- Murata, H.; Tsuji, S.; Tsujii, M.; Fu, H.Y.; Tanimura, H.; Tsujimoto, M.; Matsuura, N.; Kawano, S.; Hori, M. Helicobacter bilis infection in biliary tract cancer. Aliment. Pharmacol. Ther. 2004, 20 (Suppl. 1), 90–94. [Google Scholar] [CrossRef]
- Prueksapanich, P.; Piyachaturawat, P.; Aumpansub, P.; Ridtitid, W.; Chaiteerakij, R.; Rerknimitr, R. Liver Fluke-Associated Biliary Tract Cancer. Gut Liver 2018, 12, 236–245. [Google Scholar] [CrossRef] [Green Version]
- Zheng, S.; Zhu, Y.; Zhao, Z.; Wu, Z.; Okanurak, K.; Lv, Z. Liver fluke infection and cholangiocarcinoma: A review. Parasitol. Res. 2017, 116, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Kamiza, A.B.; Su, F.H.; Wang, W.C.; Sung, F.C.; Chang, S.N.; Yeh, C.C. Chronic hepatitis infection is associated with extrahepatic cancer development: A nationwide population-based study in Taiwan. BMC Cancer 2016, 16, 861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsing, A.W.; Zhang, M.; Rashid, A.; McGlynn, K.A.; Wang, B.S.; Niwa, S.; Ortiz-Conde, B.A.; Goedert, J.J.; Fraumeni, J.F.; O’Brien, T.R.; et al. Hepatitis B and C virus infection and the risk of biliary tract cancer: A population-based study in China. Int. J. Cancer 2008, 122, 1849–1853. [Google Scholar] [CrossRef]
- Choe, J.W.; Hyun, J.J.; Kim, B.; Han, K.D. Influence of Metabolic Syndrome on Cancer Risk in HBV Carriers: A Nationwide Population Based Study Using the National Health Insurance Service Database. J. Clin. Med. 2021, 10. [Google Scholar] [CrossRef] [PubMed]
- Barnes, R.A. Early diagnosis of fungal infection in immunocompromised patients. J. Antimicrob. Chemother. 2008, 61 (Suppl. S1), i3–i6. [Google Scholar] [CrossRef]
- Szvalb, A.D.; Kontoyiannis, D.P. Acute acalculous cholecystitis due to Fusarium species and review of the literature on fungal cholecystitis. Mycoses 2019, 62, 847–853. [Google Scholar] [CrossRef]
- Wang, L.; Wang, F.S.; Gershwin, M.E. Human autoimmune diseases: A comprehensive update. J. Intern. Med. 2015, 278, 369–395. [Google Scholar] [CrossRef]
- Giat, E.; Ehrenfeld, M.; Shoenfeld, Y. Cancer and autoimmune diseases. Autoimmun. Rev. 2017, 16, 1049–1057. [Google Scholar] [CrossRef]
- Castro, F.A.; Liu, X.; Försti, A.; Ji, J.; Sundquist, J.; Sundquist, K.; Koshiol, J.; Hemminki, K. Increased risk of hepatobiliary cancers after hospitalization for autoimmune disease. Clin. Gastroenterol. Hepatol. 2014, 12, 1038–1045. [Google Scholar] [CrossRef]
- Horsley-Silva, J.L.; Rodriguez, E.A.; Franco, D.L.; Lindor, K.D. An update on cancer risk and surveillance in primary sclerosing cholangitis. Liver Int. 2017, 37, 1103–1109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burak, K.; Angulo, P.; Pasha, T.M.; Egan, K.; Petz, J.; Lindor, K.D. Incidence and risk factors for cholangiocarcinoma in primary sclerosing cholangitis. Am. J. Gastroenterol. 2004, 99, 523–526. [Google Scholar] [CrossRef] [PubMed]
- de Valle, M.B.; Björnsson, E.; Lindkvist, B. Mortality and cancer risk related to primary sclerosing cholangitis in a Swedish population-based cohort. Liver Int. 2012, 32, 441–448. [Google Scholar] [CrossRef] [PubMed]
- Boonstra, K.; Weersma, R.K.; van Erpecum, K.J.; Rauws, E.A.; Spanier, B.W.; Poen, A.C.; van Nieuwkerk, K.M.; Drenth, J.P.; Witteman, B.J.; Tuynman, H.A.; et al. Population-based epidemiology, malignancy risk, and outcome of primary sclerosing cholangitis. Hepatology 2013, 58, 2045–2055. [Google Scholar] [CrossRef]
- Barner-Rasmussen, N.; Pukkala, E.; Jussila, A.; Färkkilä, M. Epidemiology, risk of malignancy and patient survival in primary sclerosing cholangitis: A population-based study in Finland. Scand. J. Gastroenterol. 2020, 55, 74–81. [Google Scholar] [CrossRef]
- Lewis, J.T.; Talwalkar, J.A.; Rosen, C.B.; Smyrk, T.C.; Abraham, S.C. Prevalence and risk factors for gallbladder neoplasia in patients with primary sclerosing cholangitis: Evidence for a metaplasia-dysplasia-carcinoma sequence. Am. J. Surg. Pathol. 2007, 31, 907–913. [Google Scholar] [CrossRef]
- Buckles, D.C.; Lindor, K.D.; Larusso, N.F.; Petrovic, L.M.; Gores, G.J. In primary sclerosing cholangitis, gallbladder polyps are frequently malignant. Am. J. Gastroenterol. 2002, 97, 1138–1142. [Google Scholar] [CrossRef]
- Said, K.; Glaumann, H.; Björnstedt, M.; Bergquist, A. The value of thioredoxin family proteins and proliferation markers in dysplastic and malignant gallbladders in patients with primary sclerosing cholangitis. Dig. Dis. Sci. 2012, 57, 1163–1170. [Google Scholar] [CrossRef]
- Wilsnack, R.W.; Wilsnack, S.C.; Kristjanson, A.F.; Vogeltanz-Holm, N.D.; Gmel, G. Gender and alcohol consumption: Patterns from the multinational GENACIS project. Addiction 2009, 104, 1487–1500. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.K.; Neighbors, C.; Gilson, M.; Larimer, M.E.; Alan Marlatt, G. Epidemiological trends in drinking by age and gender: Providing normative feedback to adults. Addict. Behav. 2007, 32, 967–976. [Google Scholar] [CrossRef] [PubMed]
- Delker, E.; Brown, Q.; Hasin, D.S. Alcohol Consumption in Demographic Subpopulations: An Epidemiologic Overview. Alcohol Res. 2016, 38, 7–15. [Google Scholar] [PubMed]
- Lieber, C.S. Alcoholic fatty liver: Its pathogenesis and mechanism of progression to inflammation and fibrosis. Alcohol 2004, 34, 9–19. [Google Scholar] [CrossRef]
- Acalovschi, M. Gallstones in patients with liver cirrhosis: Incidence, etiology, clinical and therapeutical aspects. World J. Gastroenterol. 2014, 20, 7277–7285. [Google Scholar] [CrossRef]
- Del Olmo, J.A.; García, F.; Serra, M.A.; Maldonado, L.; Rodrigo, J.M. Prevalence and incidence of gallstones in liver cirrhosis. Scand. J. Gastroenterol. 1997, 32, 1061–1065. [Google Scholar] [CrossRef] [PubMed]
- Wilson, D.M. Analytical methods for aflatoxins in corn and peanuts. Arch. Environ. Contam. Toxicol. 1989, 18, 308–314. [Google Scholar] [CrossRef]
- Scholl, P.F.; Groopman, J.D. Long-term stability of human aflatoxin B1 albumin adducts assessed by isotope dilution mass spectrometry and high-performance liquid chromatography-fluorescence. Cancer Epidemiol. Biomark. Prev. 2008, 17, 1436–1439. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Wu, F. Global burden of aflatoxin-induced hepatocellular carcinoma: A risk assessment. Environ. Health Perspect. 2010, 118, 818–824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foerster, C.; Koshiol, J.; Guerrero, A.R.; Kogan, M.J.; Ferreccio, C. The case for aflatoxins in the causal chain of gallbladder cancer. Med. Hypotheses 2016, 86, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Emerole, G.O. Excretion of aflatoxin B1 as a glutathione conjugate. Eur. J. Drug Metab. Pharm. 1981, 6, 265–268. [Google Scholar] [CrossRef]
- Harland, E.C.; Cardeilhac, P.T. Excretion of carbon-14-labeled aflatoxin B1 via bile, urine, and intestinal contents of the chicken. Am. J. Vet. Res. 1975, 36, 909–912. [Google Scholar] [PubMed]
- Nogueira, L.; Foerster, C.; Groopman, J.; Egner, P.; Koshiol, J.; Ferreccio, C.; Group, G.C.C.W. Association of aflatoxin with gallbladder cancer in Chile. JAMA 2015, 313, 2075–2077. [Google Scholar] [CrossRef] [Green Version]
- Koshiol, J.; Gao, Y.T.; Dean, M.; Egner, P.; Nepal, C.; Jones, K.; Wang, B.; Rashid, A.; Luo, W.; Van Dyke, A.L.; et al. Association of Aflatoxin and Gallbladder Cancer. Gastroenterology 2017, 153, 488–494.e481. [Google Scholar] [CrossRef] [PubMed]
- Raju, N.J. Arsenic in the geo-environment: A review of sources, geochemical processes, toxicity and removal technologies. Environ. Res. 2021, 203, 111782. [Google Scholar] [CrossRef]
- Martinez, V.D.; Vucic, E.A.; Becker-Santos, D.D.; Gil, L.; Lam, W.L. Arsenic exposure and the induction of human cancers. J. Toxicol. 2011, 2011, 431287. [Google Scholar] [CrossRef] [Green Version]
- Tapio, S.; Grosche, B. Arsenic in the aetiology of cancer. Mutat. Res. 2006, 612, 215–246. [Google Scholar] [CrossRef]
- Ganesan, N.; Bambino, K.; Boffetta, P.; Labgaa, I. Exploring the potential carcinogenic role of arsenic in gallbladder cancer. Eur. J. Cancer Prev. 2020, 29, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.H.; Gao, Y.T.; Huang, Y.H.; McGee, E.E.; Lam, T.; Wang, B.; Shen, M.C.; Rashid, A.; Pfeiffer, R.M.; Hsing, A.W.; et al. A Metallomic Approach to Assess Associations of Serum Metal Levels With Gallstones and Gallbladder Cancer. Hepatology 2020, 71, 917–928. [Google Scholar] [CrossRef]
- Pandey, M. Environmental pollutants in gallbladder carcinogenesis. J. Surg. Oncol. 2006, 93, 640–643. [Google Scholar] [CrossRef]
- Versieck, J.; Hoste, J.; Vanballenberghe, L.; Barbier, F.; Cornelis, R.; Waelput, I. Serum molybdenum in diseases of the liver and biliary system. J. Lab. Clin. Med. 1981, 97, 535–544. [Google Scholar] [PubMed]
- Shukla, V.K.; Adukia, T.K.; Singh, S.P.; Mishra, C.P.; Mishra, R.N. Micronutrients, antioxidants, and carcinoma of the gallbladder. J. Surg. Oncol. 2003, 84, 31–35. [Google Scholar] [CrossRef] [PubMed]
- Jain, K.; Sreenivas, V.; Velpandian, T.; Kapil, U.; Garg, P.K. Risk factors for gallbladder cancer: A case-control study. Int. J. Cancer 2013, 132, 1660–1666. [Google Scholar] [CrossRef] [PubMed]
- Tomásek, L.; Darby, S.C.; Swerdlow, A.J.; Placek, V.; Kunz, E. Radon exposure and cancers other than lung cancer among uranium miners in West Bohemia. Lancet 1993, 341, 919–923. [Google Scholar] [CrossRef]
- Gröber, U.; Holzhauer, P.; Kisters, K.; Holick, M.F.; Adamietz, I.A. Micronutrients in Oncological Intervention. Nutrients 2016, 8, 163. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, M.A.; Marcano-Bonilla, L.; Roberts, L.R. Gallbladder cancer: Epidemiology and genetic risk associations. Chin. Clin. Oncol. 2019, 8, 31. [Google Scholar] [CrossRef]
- Rawla, P.; Sunkara, T.; Thandra, K.C.; Barsouk, A. Epidemiology of gallbladder cancer. Clin. Exp. Hepatol. 2019, 5, 93–102. [Google Scholar] [CrossRef]
- Valle, J.W.; Kelley, R.K.; Nervi, B.; Oh, D.Y.; Zhu, A.X. Biliary tract cancer. Lancet 2021, 397, 428–444. [Google Scholar] [CrossRef]
- Aljindan, R.Y.; Alkharsah, K.R. Pattern of increased antimicrobial resistance of Salmonella isolates in the Eastern Province of KSA. J. Taibah Univ. Med. Sci. 2020, 15, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Medalla, F.; Gu, W.; Friedman, C.R.; Judd, M.; Folster, J.; Griffin, P.M.; Hoekstra, R.M. Increased Incidence of Antimicrobial-Resistant Nontyphoidal Salmonella Infections, United States, 2004-2016. Emerg. Infect. Dis. 2021, 27, 1662–1672. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Diagram used in the systematic review.

Figure 2.
The cholesterol metabolism and the risk of developing gallstones. Left: Under normal conditions, cholesterol stored in peripheral tissues is transported to HDL-c via ABC transporters. The circulating HDL-c in the blood transports cholesterol to VLDL-c which is then transformed into LDL-c. Then, LDL-c transports cholesterol to the hepatocytes through a low-density lipoprotein receptor (LDLr). Cholesterol can be used in bile formation and stored in the gallbladder. Right: As a result of a high-fat diet and sedentary lifestyle, the concentration of plasma cholesterol increases, provoking a greater presence of cholesterol in peripheral tissues. This causes increased transportation of cholesterol from peripheral tissues to the liver via the different lipoproteins (HDL-c, VLDL-c, and LDL-c). Finally, this induces an increase in the storage of cholesterol in the liver and subsequently a greater release of cholesterol from the liver to the gallbladder, which leads to a high risk of gallstone formation.
Figure 2.
The cholesterol metabolism and the risk of developing gallstones. Left: Under normal conditions, cholesterol stored in peripheral tissues is transported to HDL-c via ABC transporters. The circulating HDL-c in the blood transports cholesterol to VLDL-c which is then transformed into LDL-c. Then, LDL-c transports cholesterol to the hepatocytes through a low-density lipoprotein receptor (LDLr). Cholesterol can be used in bile formation and stored in the gallbladder. Right: As a result of a high-fat diet and sedentary lifestyle, the concentration of plasma cholesterol increases, provoking a greater presence of cholesterol in peripheral tissues. This causes increased transportation of cholesterol from peripheral tissues to the liver via the different lipoproteins (HDL-c, VLDL-c, and LDL-c). Finally, this induces an increase in the storage of cholesterol in the liver and subsequently a greater release of cholesterol from the liver to the gallbladder, which leads to a high risk of gallstone formation.

Figure 3.
Carcinogenesis process in gallbladder cancer. The damage caused by the presence of risk factors such as gallstones, infections, lithogenic bile, alcohol, smoking, and genetic predisposition can induce continuous damage in the mucosa of the gallbladder, which is characterized by a chronic inflammatory state mainly highlighted by the activation of macrophages and lymphocytes that leads to the release of pro-inflammatory cytokines (TNF-α, IL-6, IL-1) and ROS stimulating the carcinogenic metaplasia/hyperplasia–dysplasia–carcinoma transition. This process can be marked by different gene alterations and protein expressions such as TP53 and FHIT mutations and COX-2, TNF-α, and CLDN-18 overexpression, respectively. BMI: Body Mass Index; TNF-α: Tumor necrosis factor-alpha; ROS: Reactive oxygen species; IL-1: Interleukin-1; IL-6; Interleukin-6; CLDN-18: Claudin 18; COX-2: Cyclooxygenase 2; TP53: Tumor protein 53; FHIT: Fragile Histidine Triad Diadenosine Triphosphatase. The risk factors in bold mean strong evidence. The risk factor in italics and underlined means weak evidence.
Figure 3.
Carcinogenesis process in gallbladder cancer. The damage caused by the presence of risk factors such as gallstones, infections, lithogenic bile, alcohol, smoking, and genetic predisposition can induce continuous damage in the mucosa of the gallbladder, which is characterized by a chronic inflammatory state mainly highlighted by the activation of macrophages and lymphocytes that leads to the release of pro-inflammatory cytokines (TNF-α, IL-6, IL-1) and ROS stimulating the carcinogenic metaplasia/hyperplasia–dysplasia–carcinoma transition. This process can be marked by different gene alterations and protein expressions such as TP53 and FHIT mutations and COX-2, TNF-α, and CLDN-18 overexpression, respectively. BMI: Body Mass Index; TNF-α: Tumor necrosis factor-alpha; ROS: Reactive oxygen species; IL-1: Interleukin-1; IL-6; Interleukin-6; CLDN-18: Claudin 18; COX-2: Cyclooxygenase 2; TP53: Tumor protein 53; FHIT: Fragile Histidine Triad Diadenosine Triphosphatase. The risk factors in bold mean strong evidence. The risk factor in italics and underlined means weak evidence.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Pérez-Moreno, P.; Riquelme, I.; García, P.; Brebi, P.; Roa, J.C. Environmental and Lifestyle Risk Factors in the Carcinogenesis of Gallbladder Cancer. J. Pers. Med. 2022, 12, 234. https://doi.org/10.3390/jpm12020234
AMA Style
Pérez-Moreno P, Riquelme I, García P, Brebi P, Roa JC. Environmental and Lifestyle Risk Factors in the Carcinogenesis of Gallbladder Cancer. Journal of Personalized Medicine. 2022; 12(2):234. https://doi.org/10.3390/jpm12020234
Chicago/Turabian StylePérez-Moreno, Pablo, Ismael Riquelme, Patricia García, Priscilla Brebi, and Juan Carlos Roa. 2022. "Environmental and Lifestyle Risk Factors in the Carcinogenesis of Gallbladder Cancer" Journal of Personalized Medicine 12, no. 2: 234. https://doi.org/10.3390/jpm12020234
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.