High Symmetry Double Bubble Clusters as Secondary Building Units
We consider the 1-1 compounds that are predicted to have stable (lowest energy for a particular size) and metastable bubble, or fullerene like, structures. Perfect versions of these structures are composed of only three-coordinated atoms, sets of which create rings with an even number of sides that can be visualised as one of the faces of the bubble; an odd number of sides is unlikely as this would require at least one neighbouring pair of vertices of only cations or anions. Except for the smallest sized clusters, in which the curvature of the layer is important, cluster configurations containing one- or two-coordinated atoms are less stable than the perfect bubbles. Another characteristic of the stable bubbles is that, typically, the number of tetragonal rings is minimised (and, to a lesser extent, the distance between these should be maximised), while the number of hexagonal rings is maximised. A layer consisting of only hexagonal rings has no curvature, and therefore a perfect bubble of hexagons would require an infinite number of atoms (i.e., a 2D infinite hexagonal sheet). To obtain a perfect bubble with a finite number of atoms, the sheet requires the introduction of six tetragonal rings (Euler’s rule) as each tetragonal ring increases the curvature of the sheet. Increasing the number of tetragonal rings results in open (as opposed to closed) perfect bubbles, which contain much larger rings, e.g., octagonal, assuming the chemistry of the compound does not favour the bonding and coordination required for the formation of cuboids, i.e., cuts from rock salt.
The higher symmetry configurations of the perfect bubbles are typically found to be more stable. As high symmetry cluster structures are only possible for certain sizes, they are not only the stable state for their particular size, but usually have a greater stability than clusters of neighbouring sizes. In our previous studies [
6,
9,
10] we have, therefore, focused our attention on families of high symmetry structures, and, in particular, those with symmetry
Th,
Td and
T. These (MX)
n structures are found if
n = 4 (
Td), 12 (
Th), 16 (
Td), 28 (
T), 36 (
Td), 48 (
Th), 64 (
Td), … Larger
Th and
Td clusters include
n = 108 and 192 and
n =100, 144 and 196, respectively; examples are shown in
Figure 1. These clusters can be visualised as truncated octahedra, where there is one tetragonal ring of the bubble at each of the six truncated corners, and the hexagonal patchworks form the octahedron’s faces. In this morphology, the distance between all tetragonal rings is maximised for a given size
n, and the separation increases monotonically with
n.
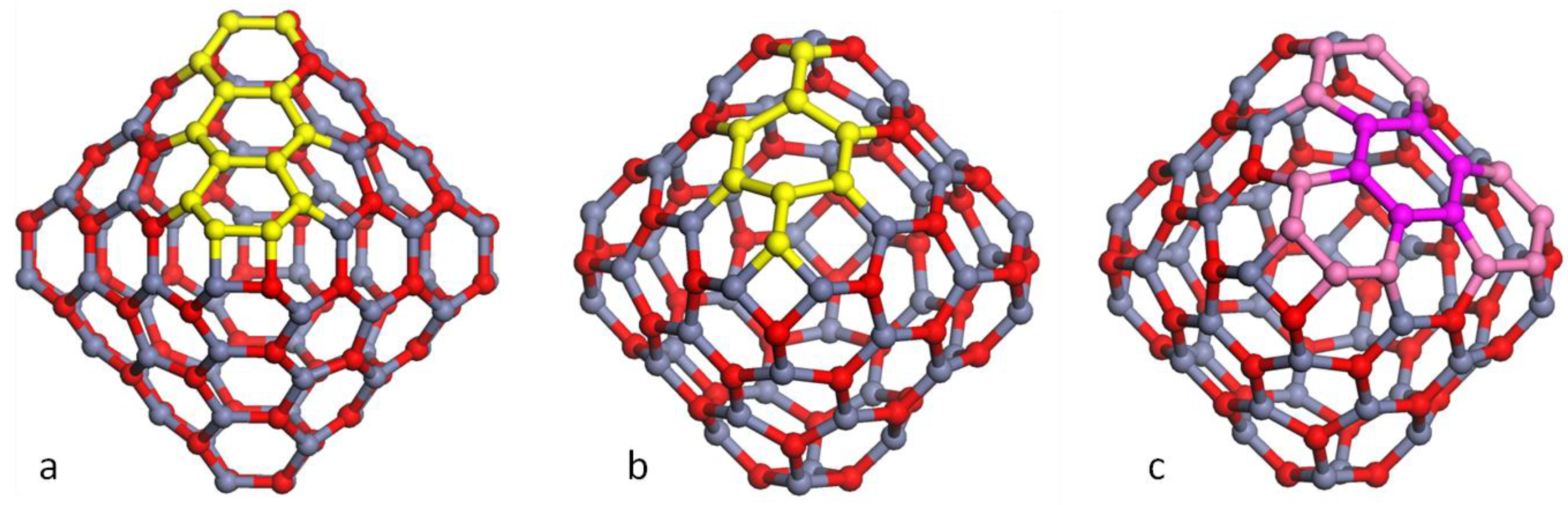
Figure 1.
Models of high symmetry (MX)n bubbles with: (a) n = 12 with symmetry Th; (b) n = 48 with symmetry Th; (c) n = 28, 36 and 48 with octahedra superimposed.
Figure 1.
Models of high symmetry (MX)n bubbles with: (a) n = 12 with symmetry Th; (b) n = 48 with symmetry Th; (c) n = 28, 36 and 48 with octahedra superimposed.
Smaller bubble clusters can be readily generated using an appropriate global optimiser (e.g., one based on Monte Carlo basin hopping [
17] or genetic algorithm [
18] routines), and once one has determined the relationship between them, the larger bubble clusters can be constructed by simply increasing the number of rows of hexagonal rings in each face or edge of the octahedron. For example, each octahedron’s edge of a
Td bubble links a side of two tetragonal rings via a “ladder” of
m hexagonal rings (one hexagon wide)—see the highlighted ladder in
Figure 2a—with the remaining hexagonal rings completing the faces of the octahedron. Note that the line of the octahedron edge bisects the rings of this ladder and that the tetragonal ring is out of phase with the tetragonal face created by truncating the octahedron. Constructions with
m = 0, 1, 2, 3, 4 and 5 corresponds to perfect bubbles at
n = 4, 16, 36, 64, 100, 144 and 196, respectively. In contrast, each octahedron edge in a
Th bubble links the corner of two tetragonal rings via
m + 1 M–X sticks that are separated by
m hexagonal rings, forming an alternating linear pattern. Each stick is actually a shared side of two hexagonal rings, with each ring part of a hexagonal patchwork that covers a face of the octahedron. These sticks form a line segment of the octahedron edge (see highlighting in
Figure 2b), and this line bisects opposite angles of the hexagonal rings, rather than opposite sides, and the tetragonal rings are in-phase with the tetragonal face created by truncating the octahedron. The smallest
Th bubble,
m = 0, or
n = 12, has just one stick between two neighbouring tetragonal rings. The next smallest size
Th bubble,
m = 1, or
n = 48, is constructed using two sticks and one hexagonal ring; then, for
m = 2, or
n = 108, there are three sticks and two hexagonal rings. Comparing the growth of the octahedron edges, it is evident why there are more bubbles with
Td rather than
Th symmetry.
Figure 2.
Models of high symmetry (MX)n bubbles with: (a) n = 64, symmetry Td and the ladder of hexagonal rings, highlighted in yellow, that corresponds to one of the twelve edges of an octahedron; (b) n = 48, symmetry Th with a fragment that corresponds to one of twelve edges highlighted in yellow; (c) n = 48, symmetry Th with one of twelve patchworks that correspond to the octahedron side highlighted in purple.
Figure 2.
Models of high symmetry (MX)n bubbles with: (a) n = 64, symmetry Td and the ladder of hexagonal rings, highlighted in yellow, that corresponds to one of the twelve edges of an octahedron; (b) n = 48, symmetry Th with a fragment that corresponds to one of twelve edges highlighted in yellow; (c) n = 48, symmetry Th with one of twelve patchworks that correspond to the octahedron side highlighted in purple.
As discussed in the Introduction, framework structures with an increased density are typically more stable [
19,
20]. We therefore choose to investigate double bubbles that are composed of high symmetry perfect bubbles as these are more dense. Two bubbles are combined by inserting the smaller bubble inside the larger; aligned with the same centre of mass and identical direction of orthogonal axes, with each axis passing through the centre of mass and the centre of opposite truncated corners, or tetragonal faces. The rotation of these tetragonal faces about the octahedral axes is dependent upon the symmetry of the cluster. When clusters have
Th and
Td symmetry this rotation is 45° out-of-phase, and, if one of the bubbles has T symmetry, between 0° and 45°. For stability, the best match is obtained when the inner and outer bubbles are taken from the set of
Th bubbles and the highest density obtained by combining the smallest two of these:
n = 12 (a sodalite cage) and
n = 48 [
10].
In the MX bulk phase considered here, the atoms are four-coordinated tetrahedra, so the stability of the double bubbles will improve if M–X linkages between layers are found. These linkages can be expected to be located between aligned pairs of hexagonal patchworks that form the faces of the octahedra rather than between the truncated corners. For the
n = 60 double bubble, the inner
n = 12 bubble has one hexagonal ring on each face, whereas the outer
n = 48 bubble is composed of a patchwork of five and a half hexagonal rings; a central hexagonal ring that is linked via one hexagonal ring to each of three nearest tetragonal rings and three hexagonal rings that are shared with neighbouring faces of the octahedron (see
Figure 2c). Importantly, the central hexagonal ring can bond with the hexagonal ring of the inner bubble; see
Figure 3. Analogous to our structures, experimentally observed cages of boron nitride (BN), [
15,
16] and molybdenum sulphide (MoS
2) [
21,
22] have been reported to be constructed from four and six (hexagonal) membered ring building units. CdSe cage structures have been experimentally observed to be stable and formed from truncated-octahedra [
23]. DFT calculations on cage structures of CdSe that are similar to our structures have also been reported [
24].
Figure 3.
Models of the n = 60 Th double bubble, with inter-layer links between the inner n = 12 sodalite cage and the eight hexagonal rings that are in the centres of the octahedron faces of the outer n = 48 bubble, highlighted using ball-and-sticks rather than line representation for: (a) no bridging links; (b) four bridging links; and (c) all eight bridging links.
Figure 3.
Models of the n = 60 Th double bubble, with inter-layer links between the inner n = 12 sodalite cage and the eight hexagonal rings that are in the centres of the octahedron faces of the outer n = 48 bubble, highlighted using ball-and-sticks rather than line representation for: (a) no bridging links; (b) four bridging links; and (c) all eight bridging links.
The first framework is constructed from
Th bubble (sodalite cage) secondary building units (SBUs) of (ZnO)
12 and (GaN)
12, see
Figure 1a. As the typical Zn–O and Ga–N bond lengths are similar (1.98 Å and 1.95 Å in their ground state wurtzite form), their respective SBUs are also similar in size. Consider each SBU as an octahedron. By corner sharing the octahedra, and assuming an equal number of SBUs for each compound, we construct an fcc, rock-salt like lattice, as shown in
Figure 4c. The second framework is constructed from the
n = 60
Th double bubbles; see
Figure 1c and
Figure 4b. Again, imagining each SBU as an octahedron, but rather than corner sharing they are now stacked so that they share edges, each double bubble is surrounded by twelve others (see
Figure 4d), and each edge of the outer bubble is one bond length from an edge of a neighbouring bubble forming an
n = 6 double ring (a drum) and two
n = 2 rings. Each tetragonal ring of an outer
n = 48 bubble combines with five others to form an
n = 12
Th bubble,
i.e., the void is a sodalite cage. The inner sodalite cage of each double bubble is formed from (i) the same compound and (ii) two compounds, which we alternate.
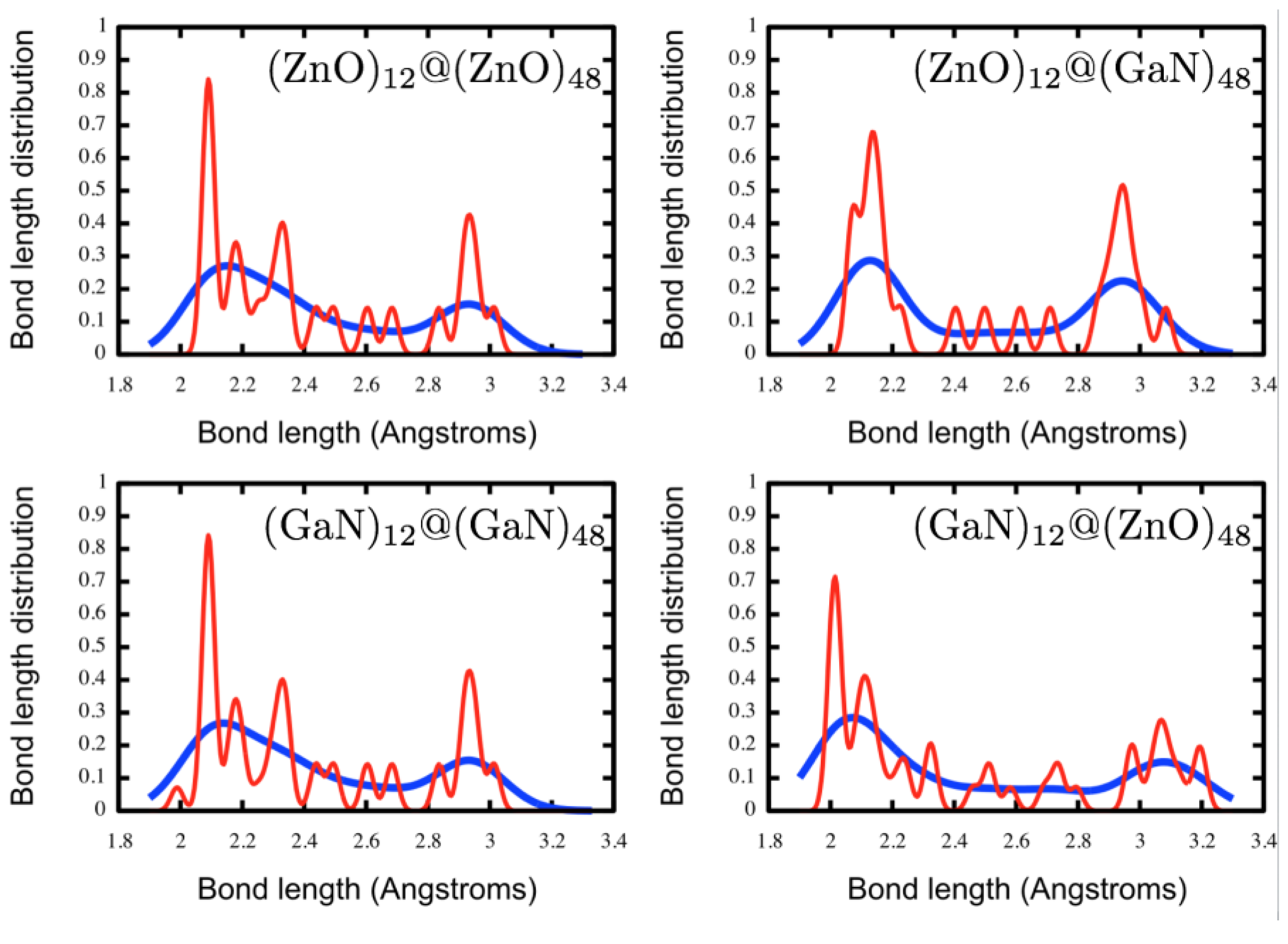
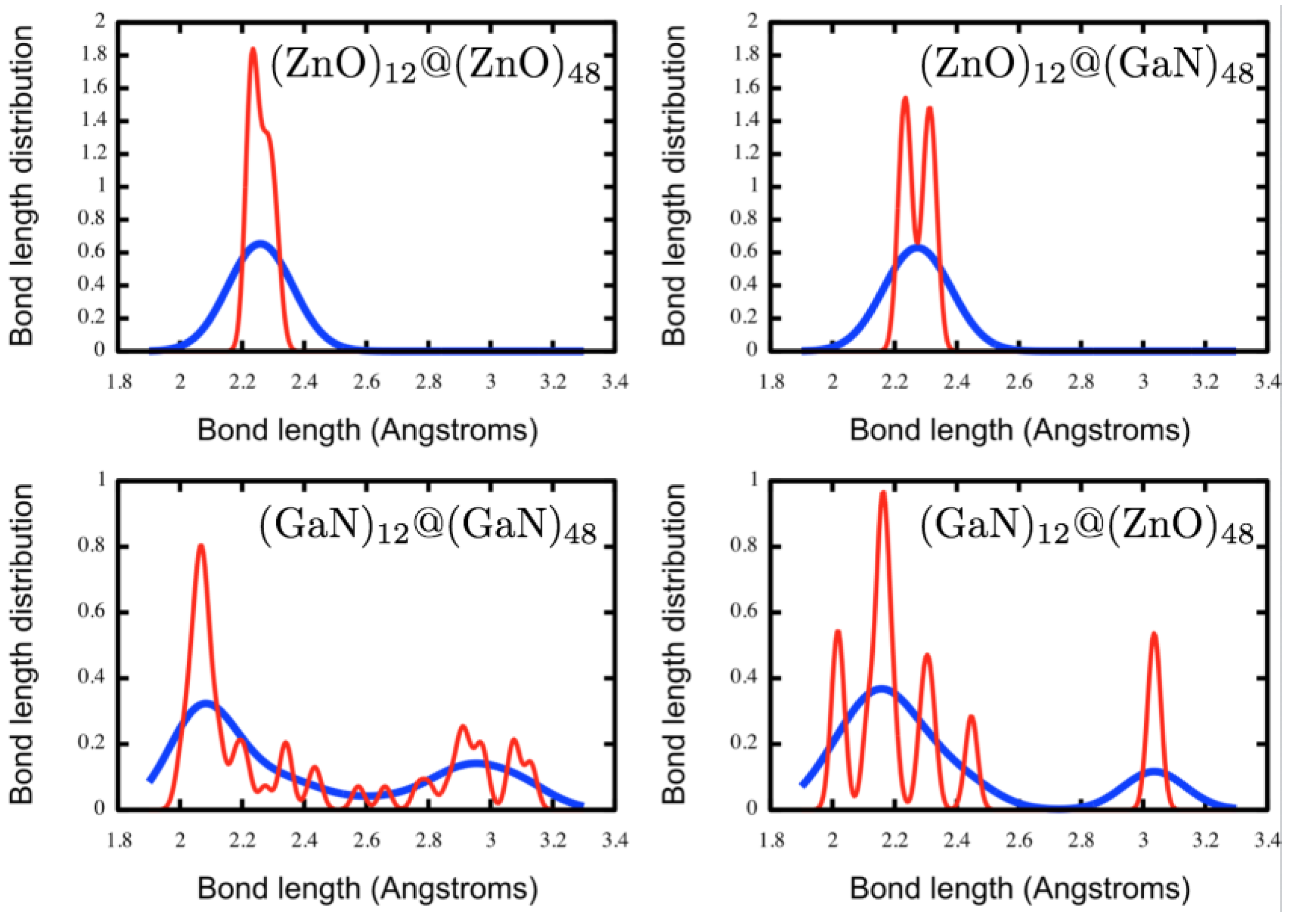
We start the double bubble construction from two relaxed single bubbles. If the distance between each inner hexagonal ring and its corresponding central hexagonal ring of the outer bubble is approximately a typical M–X bond length, then we shall refer to this as an ideal match, and the relaxed double bubble is expected to maintain
Th symmetry. Whether there is an ideal match depends on the composition: if the two layers are of the same composition and there is not an ideal match then the inner bubble is too small. The outer eight planes of hexagonal patchworks, or octahedron faces, have more flexibility than the corners. During a geometry relaxation of the double bubble, the central hexagonal ring of each outer patchwork can move inward, maintaining the
Th symmetry or, due to the repulsion between neighbouring patchworks, only the central hexagons from alternate patchworks,
i.e., four of the eight, move inwards reducing the symmetry to
T; see
Figure 3.
For all ZnO/GaN compositions investigated here, n = 60 double bubble structures of high symmetry (Th and T) were constructed and then geometry optimised. Low symmetry structural distortions were allowed in the optimisation process in order to find the lowest energy double bubble configuration.
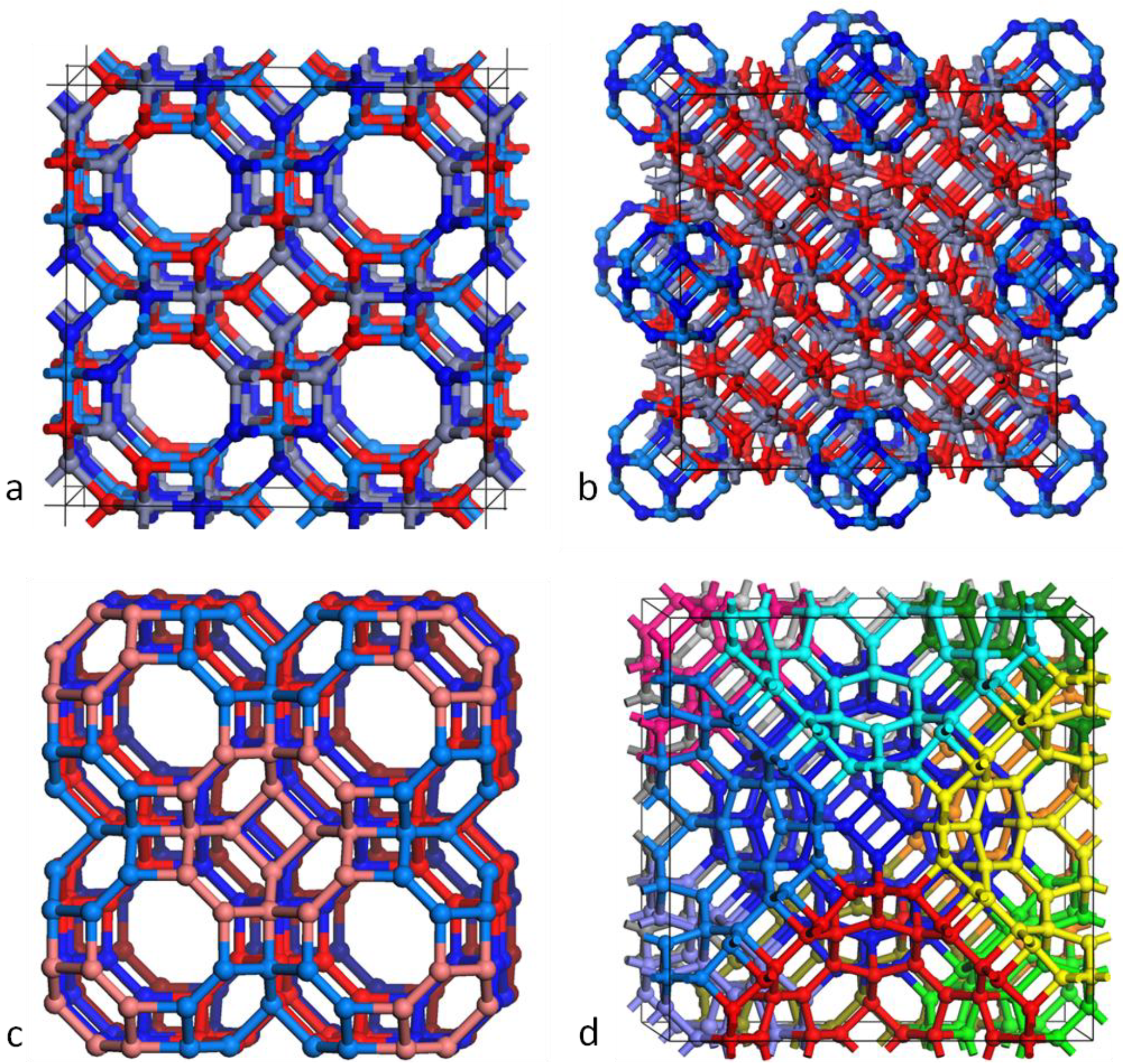
Figure 4.
Ball and stick models of two framework structures. (a) Constructed from Th bubbles of (GaN)12 and (ZnO)12; (b) Constructed from Th double bubbles of (ZnO)48 and (GaN)12; (c) the same structure as (a) but with each (GaN)12 coloured red and each (ZnO)12 coloured blue (lighter/darker shades used in the front/back row); (d) the same structure as (b) but with each (GaN)12 hidden and each (ZnO)48 uniquely coloured.
Figure 4.
Ball and stick models of two framework structures. (a) Constructed from Th bubbles of (GaN)12 and (ZnO)12; (b) Constructed from Th double bubbles of (ZnO)48 and (GaN)12; (c) the same structure as (a) but with each (GaN)12 coloured red and each (ZnO)12 coloured blue (lighter/darker shades used in the front/back row); (d) the same structure as (b) but with each (GaN)12 hidden and each (ZnO)48 uniquely coloured.








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}



