
Depleting Ly6G Positive Myeloid Cells Reduces Pancreatic Cancer-Induced Skeletal Muscle Atrophy

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Mice
2.2. Pancreatic Tumor Model
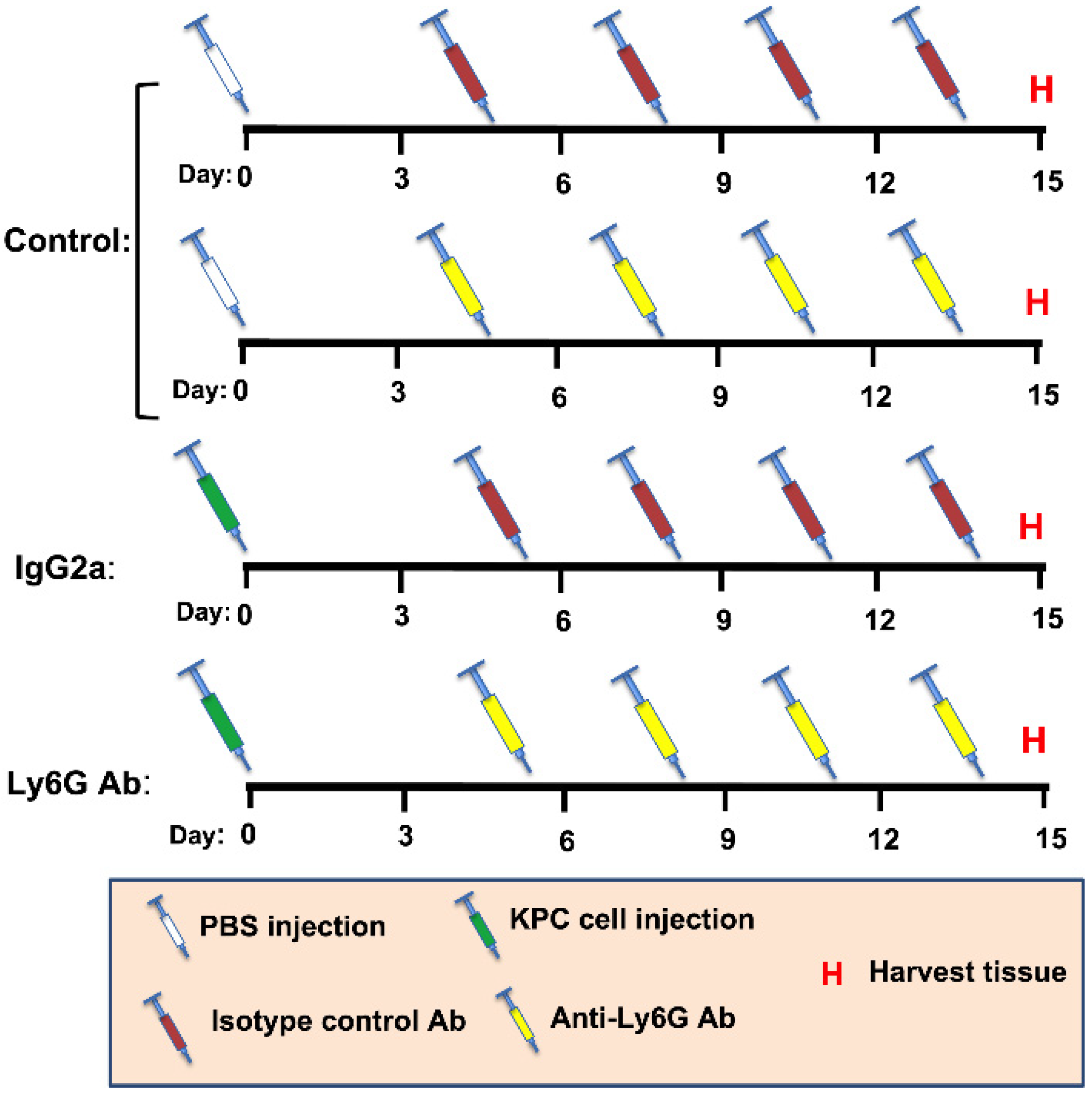
2.3. Ly6G Antibody-Mediated Cell Depletion
2.4. Tissue Specimen Collection and Storage
2.5. Histology
2.6. Micrograph Acquisition and Analysis
2.7. Atrophy-Related Gene Expression
2.8. Statistics and Data Presentation
3. Results
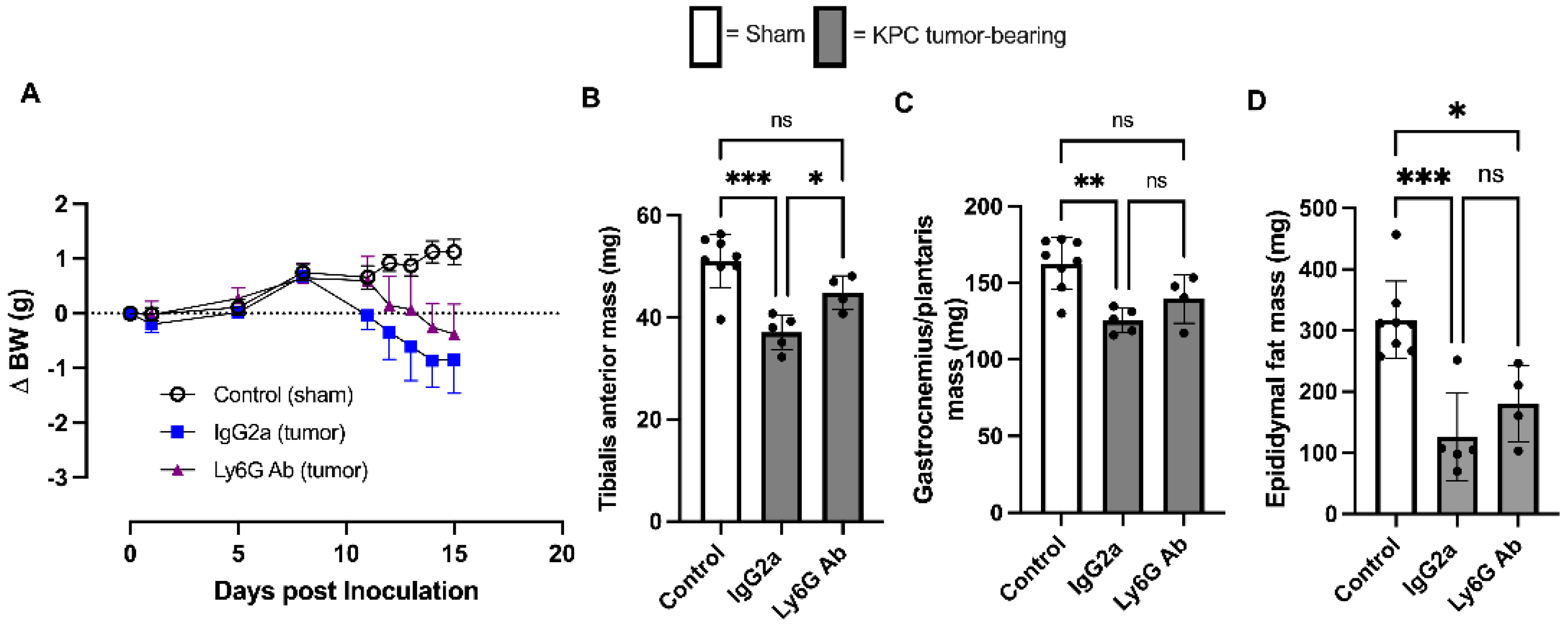
3.1. Anti-Ly6G Treatment Delays Body Wasting and Reduces Loss of Skeletal Muscle Mass
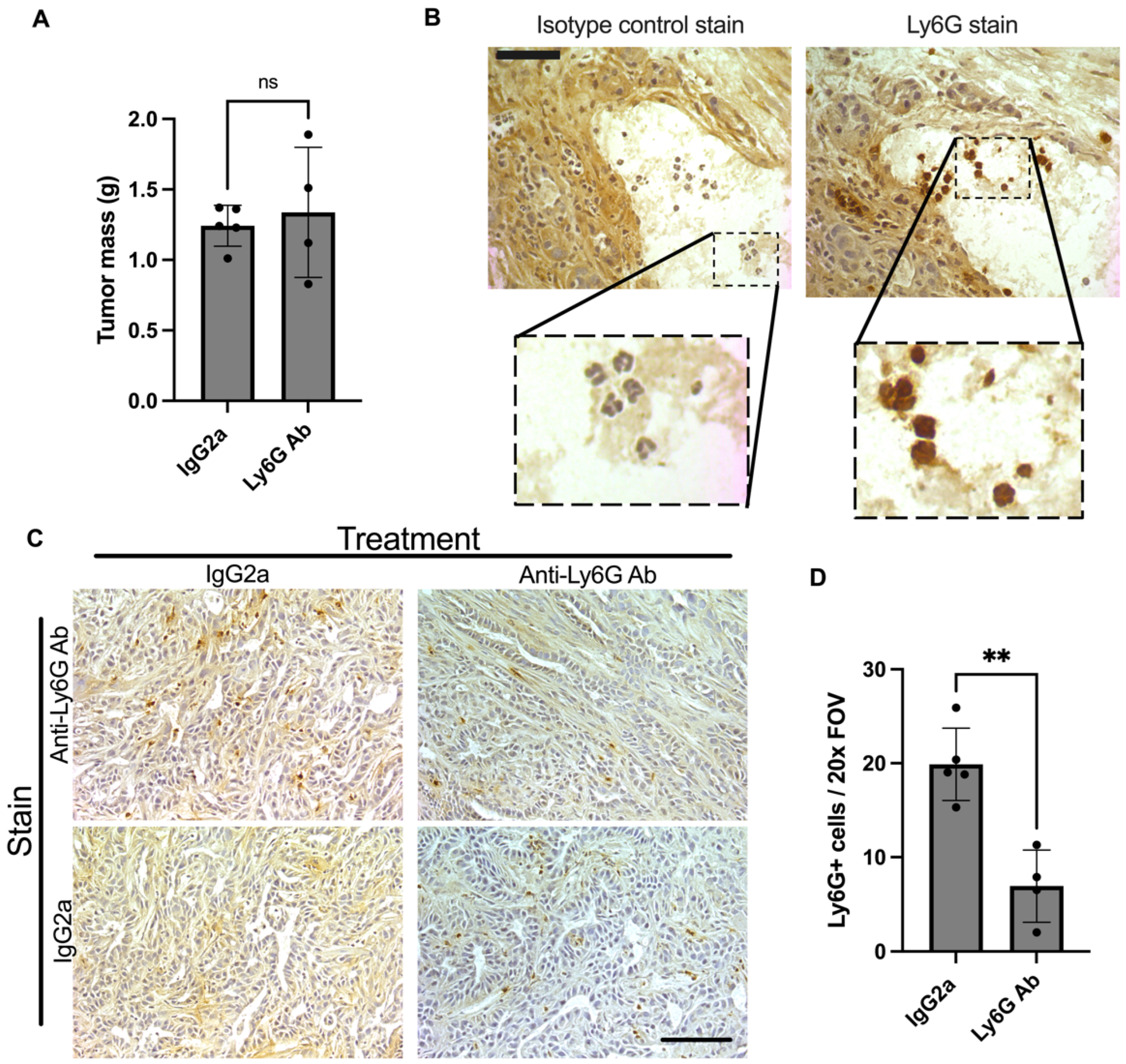
3.2. Anti-Ly6G Treatment Depletes Ly6G+ Cells in the Tumor and TA Muscle
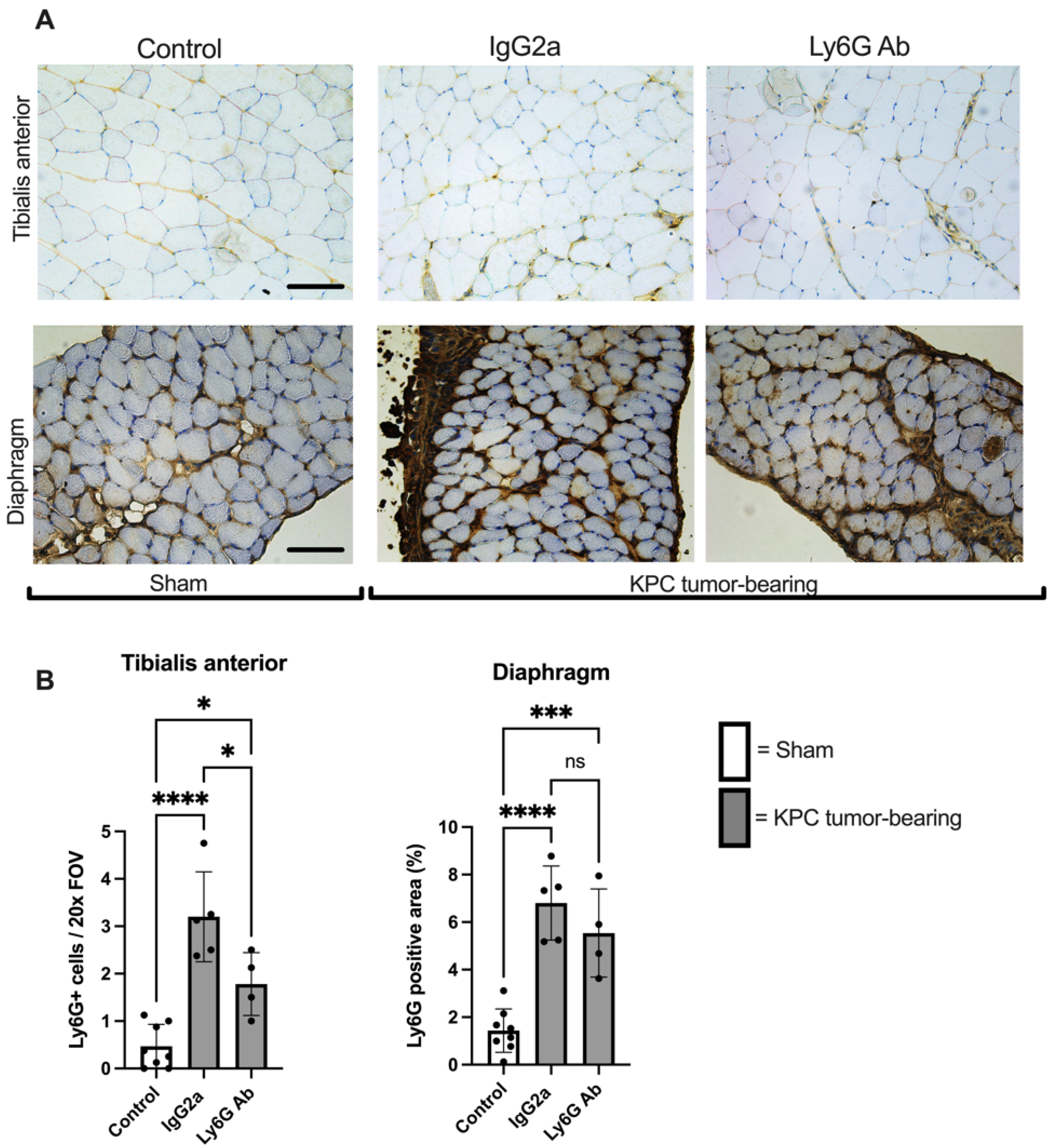
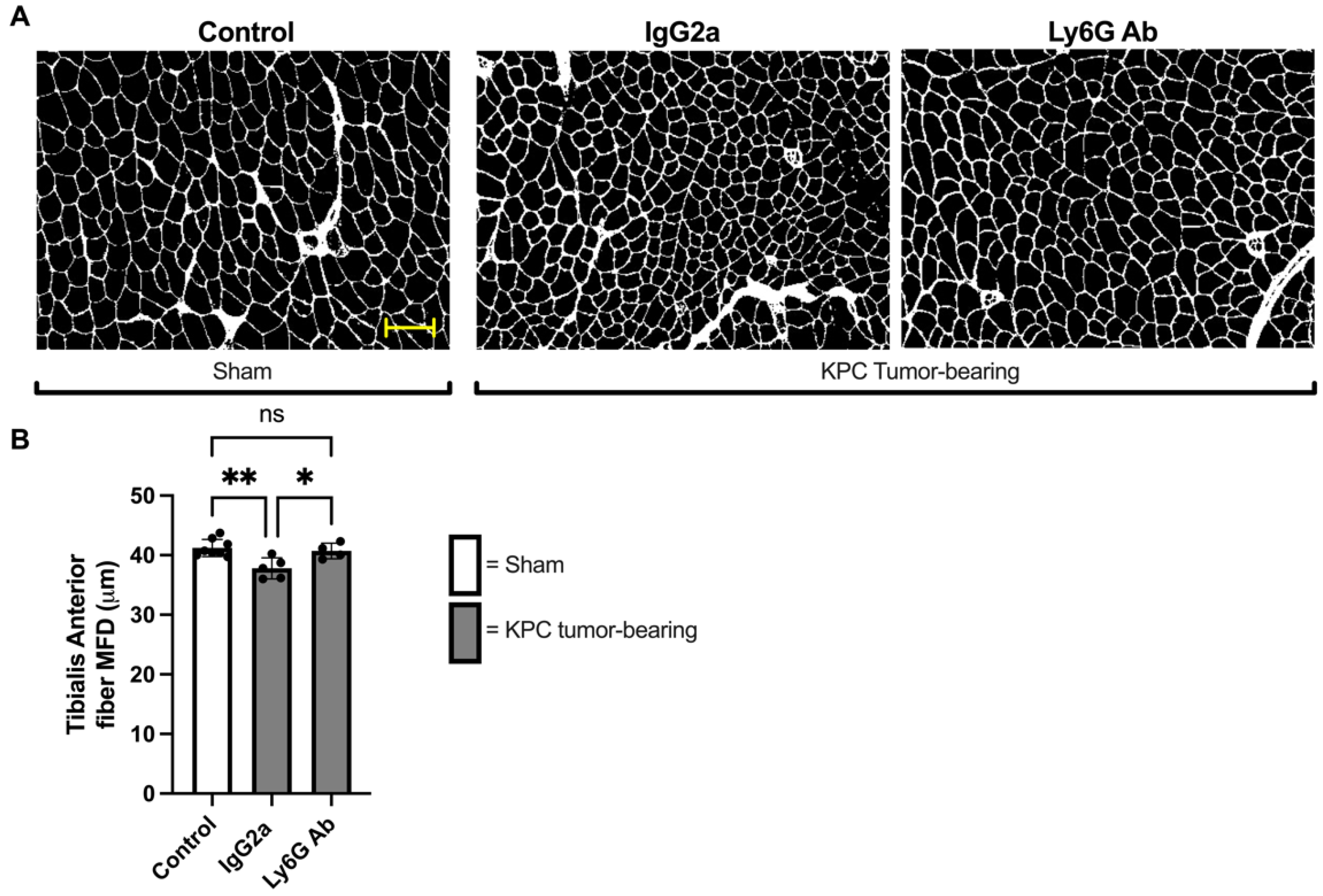
3.3. Anti-Ly6G Treatment Attenuates Muscle Fiber Atrophy
3.4. KPC Tumor Burden Causes a IIx/b to IIa Fiber Type Shift Independent of Ly6G Treatment
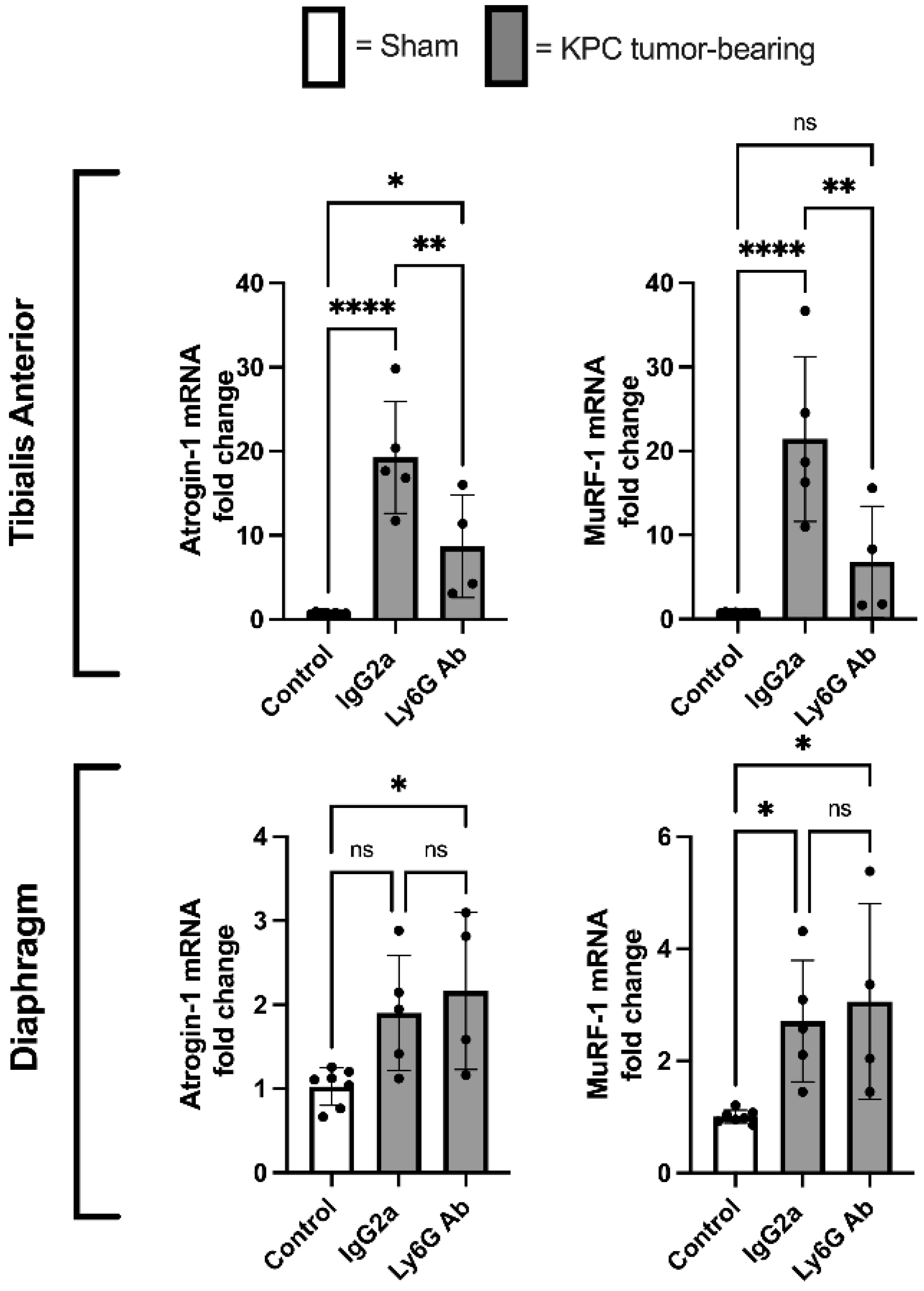
3.5. Anti-Ly6G Treatment Attenuates Atrophic Gene Expression in TA Muscle
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Finisguerra, V.; Di Conza, G.; Di Matteo, M.; Serneels, J.; Costa, S.; Thompson, A.A.; Wauters, E.; Walmsley, S.; Prenen, H.; Granot, Z.; et al. MET is required for the recruitment of anti-tumoural neutrophils. Nature 2015, 522, 349–353. [Google Scholar] [CrossRef] [PubMed]
- Roumenina, L.T.; Daugan, M.V.; Petitprez, F.; Sautes-Fridman, C.; Fridman, W.H. Context-dependent roles of complement in cancer. Nat. Rev. Cancer 2019, 19, 698–715. [Google Scholar] [CrossRef] [PubMed]
- Clemente, C.G.; Mihm, M.C., Jr.; Bufalino, R.; Zurrida, S.; Collini, P.; Cascinelli, N. Prognostic value of tumor infiltrating lymphocytes in the vertical growth phase of primary cutaneous melanoma. Cancer 1996, 77, 1303–1310. [Google Scholar] [CrossRef]
- Tosolini, M.; Kirilovsky, A.; Mlecnik, B.; Fredriksen, T.; Mauger, S.; Bindea, G.; Berger, A.; Bruneval, P.; Fridman, W.H.; Pages, F.; et al. Clinical impact of different classes of infiltrating T cytotoxic and helper cells (Th1, th2, treg, th17) in patients with colorectal cancer. Cancer Res. 2011, 71, 1263–1271. [Google Scholar] [CrossRef] [PubMed]
- Vesalainen, S.; Lipponen, P.; Talja, M.; Syrjanen, K. Histological grade, perineural infiltration, tumour-infiltrating lymphocytes and apoptosis as determinants of long-term prognosis in prostatic adenocarcinoma. Eur. J. Cancer 1994, 30, 1797–1803. [Google Scholar] [CrossRef]
- Balachandran, V.P.; Luksza, M.; Zhao, J.N.; Makarov, V.; Moral, J.A.; Remark, R.; Herbst, B.; Askan, G.; Bhanot, U.; Sendabaoglu, Y.; et al. Identification of unique neoantigen qualities in long-term survivors of pancreatic cancer. Nature 2017, 551, 512–516. [Google Scholar] [CrossRef]
- Li, J.; Byrne, K.T.; Yan, F.; Yamazoe, T.; Chen, Z.; Baslan, T.; Richman, L.P.; Lin, J.H.; Sun, Y.H.; Rech, A.J.; et al. Tumor Cell-Intrinsic Factors Underlie Heterogeneity of Immune Cell Infiltration and Response to Immunotherapy. Immunity 2018, 49, 178–193.e7. [Google Scholar] [CrossRef]
- Balachandran, V.P.; Beatty, G.L.; Dougan, S.K. Broadening the Impact of Immunotherapy to Pancreatic Cancer: Challenges and Opportunities. Gastroenterology 2019, 156, 2056–2072. [Google Scholar] [CrossRef]
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F.; et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 2014, 371, 1507–1517. [Google Scholar] [CrossRef]
- Brahmer, J.R.; Tykodi, S.S.; Chow, L.Q.; Hwu, W.J.; Topalian, S.L.; Hwu, P.; Drake, C.G.; Camacho, L.H.; Kauh, J.; Odunsi, K.; et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N. Engl. J. Med. 2012, 366, 2455–2465. [Google Scholar] [CrossRef]
- Kim, R.; Emi, M.; Tanabe, K. Cancer immunosuppression and autoimmune disease: Beyond immunosuppressive networks for tumour immunity. Immunology 2006, 119, 254–264. [Google Scholar] [CrossRef] [PubMed]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef] [PubMed]
- Al Sayed, M.F.; Amrein, M.A.; Buhrer, E.D.; Huguenin, A.L.; Radpour, R.; Riether, C.; Ochsenbein, A.F. T-cell-Secreted TNFalpha Induces Emergency Myelopoiesis and Myeloid-Derived Suppressor Cell Differentiation in Cancer. Cancer Res. 2019, 79, 346–359. [Google Scholar] [CrossRef] [PubMed]
- Sica, A.; Guarneri, V.; Gennari, A. Myelopoiesis, metabolism and therapy: A crucial crossroads in cancer progression. Cell Stress 2019, 3, 284–294. [Google Scholar] [CrossRef] [PubMed]
- Moore, F.A.; Phillips, S.M.; McClain, C.J.; Patel, J.J.; Martindale, R.G. Nutrition Support for Persistent Inflammation, Immunosuppression, and Catabolism Syndrome. Nutr. Clin. Pract. 2017, 32, 121S–127S. [Google Scholar] [CrossRef]
- Wu, W.C.; Sun, H.W.; Chen, H.T.; Liang, J.; Yu, X.J.; Wu, C.; Wang, Z.; Zheng, L. Circulating hematopoietic stem and progenitor cells are myeloid-biased in cancer patients. Proc. Natl. Acad. Sci. USA 2014, 111, 4221–4226. [Google Scholar] [CrossRef]
- Youn, J.I.; Nagaraj, S.; Collazo, M.; Gabrilovich, D.I. Subsets of myeloid-derived suppressor cells in tumor-bearing mice. J. Immunol. 2008, 181, 5791–5802. [Google Scholar] [CrossRef]
- Cassetta, L.; Bruderek, K.; Skrzeczynska-Moncznik, J.; Osiecka, O.; Hu, X.; Rundgren, I.M.; Lin, A.; Santegoets, K.; Horzum, U.; Godinho-Santos, A.; et al. Differential expansion of circulating human MDSC subsets in patients with cancer, infection and inflammation. J. Immunother. Cancer 2020, 8, e001223. [Google Scholar] [CrossRef]
- Chao, T.; Furth, E.E.; Vonderheide, R.H. CXCR2-Dependent Accumulation of Tumor-Associated Neutrophils Regulates T-cell Immunity in Pancreatic Ductal Adenocarcinoma. Cancer Immunol. Res. 2016, 4, 968–982. [Google Scholar] [CrossRef]
- Steele, C.W.; Karim, S.A.; Leach, J.D.G.; Bailey, P.; Upstill-Goddard, R.; Rishi, L.; Foth, M.; Bryson, S.; McDaid, K.; Wilson, Z.; et al. CXCR2 Inhibition Profoundly Suppresses Metastases and Augments Immunotherapy in Pancreatic Ductal Adenocarcinoma. Cancer Cell 2016, 29, 832–845. [Google Scholar] [CrossRef]
- Pylayeva-Gupta, Y.; Lee, K.E.; Hajdu, C.H.; Miller, G.; Bar-Sagi, D. Oncogenic Kras-induced GM-CSF production promotes the development of pancreatic neoplasia. Cancer Cell 2012, 21, 836–847. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Ma, X.; Zhu, C.; Liu, L.; Wang, G.; Yuan, X. The Role of Myeloid-Derived Suppressor Cells in Patients with Solid Tumors: A Meta-Analysis. PLoS ONE 2016, 11, e0164514. [Google Scholar] [CrossRef] [PubMed]
- Stromnes, I.M.; Brockenbrough, J.S.; Izeradjene, K.; Carlson, M.A.; Cuevas, C.; Simmons, R.M.; Greenberg, P.D.; Hingorani, S.R. Targeted depletion of an MDSC subset unmasks pancreatic ductal adenocarcinoma to adaptive immunity. Gut 2014, 63, 1769–1781. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Kim, H.S.; Shi, J. Neutrophil in the Pancreatic Tumor Microenvironment. Biomolecules 2021, 11, 1170. [Google Scholar] [CrossRef] [PubMed]
- Barker, T.; Fulde, G.; Moulton, B.; Nadauld, L.D.; Rhodes, T. An elevated neutrophil-to-lymphocyte ratio associates with weight loss and cachexia in cancer. Sci. Rep. 2020, 10, 7535. [Google Scholar] [CrossRef] [PubMed]
- Penafuerte, C.A.; Gagnon, B.; Sirois, J.; Murphy, J.; MacDonald, N.; Tremblay, M.L. Identification of neutrophil-derived proteases and angiotensin II as biomarkers of cancer cachexia. Br. J. Cancer 2016, 114, 680–687. [Google Scholar] [CrossRef]
- Xu, C.; Yuan, J.; Du, W.; Wu, J.; Fang, Q.; Zhang, X.; Li, H. Significance of the Neutrophil-to-Lymphocyte Ratio in p16-Negative Squamous Cell Carcinoma of Unknown Primary in Head and Neck. Front. Oncol. 2020, 10, 39. [Google Scholar] [CrossRef]
- Fearon, K.; Arends, J.; Baracos, V. Understanding the mechanisms and treatment options in cancer cachexia. Nat. Rev. Clin. Oncol. 2013, 10, 90–99. [Google Scholar] [CrossRef]
- Dewys, W.D.; Begg, C.; Lavin, P.T.; Band, P.R.; Bennett, J.M.; Bertino, J.R.; Cohen, M.H.; Douglass, H.O., Jr.; Engstrom, P.F.; Ezdinli, E.Z.; et al. Prognostic effect of weight loss prior to chemotherapy in cancer patients. Am. J. Med. 1980, 69, 491–497. [Google Scholar] [CrossRef]
- Bachmann, J.; Heiligensetzer, M.; Krakowski-Roosen, H.; Buchler, M.W.; Friess, H.; Martignoni, M.E. Cachexia worsens prognosis in patients with resectable pancreatic cancer. J. Gastrointest. Surg. 2008, 12, 1193–1201. [Google Scholar] [CrossRef]
- Mendes, M.C.; Pimentel, G.D.; Costa, F.O.; Carvalheira, J.B. Molecular and neuroendocrine mechanisms of cancer cachexia. J. Endocrinol. 2015, 226, R29–R43. [Google Scholar] [CrossRef] [PubMed]
- Marceca, G.P.; Londhe, P.; Calore, F. Management of Cancer Cachexia: Attempting to Develop New Pharmacological Agents for New Effective Therapeutic Options. Front. Oncol. 2020, 10, 298. [Google Scholar] [CrossRef] [PubMed]
- Judge, S.M.; Deyhle, M.R.; Neyroud, D.; Nosacka, R.L.; D’Lugos, A.C.; Cameron, M.E.; Vohra, R.S.; Smuder, A.J.; Roberts, B.M.; Callaway, C.S.; et al. MEF2c-dependent downregulation of Myocilin mediates cancer-induced muscle wasting and associates with cachexia in cancer patients. Cancer Res. 2020, 80, 1861–1874. [Google Scholar] [CrossRef]
- Jamieson, T.; Clarke, M.; Steele, C.W.; Samuel, M.S.; Neumann, J.; Jung, A.; Huels, D.; Olson, M.F.; Das, S.; Nibbs, R.J.; et al. Inhibition of CXCR2 profoundly suppresses inflammation-driven and spontaneous tumorigenesis. J. Clin. Investig. 2012, 122, 3127–3144. [Google Scholar] [CrossRef] [PubMed]
- Daley, J.M.; Thomay, A.A.; Connolly, M.D.; Reichner, J.S.; Albina, J.E. Use of Ly6G-specific monoclonal antibody to deplete neutrophils in mice. J. Leukoc. Biol. 2008, 83, 64–70. [Google Scholar] [CrossRef]
- Roberts, B.M.; Ahn, B.; Smuder, A.J.; Al-Rajhi, M.; Gill, L.C.; Beharry, A.W.; Powers, S.K.; Fuller, D.D.; Ferreira, L.F.; Judge, A.R. Diaphragm and ventilatory dysfunction during cancer cachexia. FASEB J. 2013, 27, 2600–2610. [Google Scholar] [CrossRef] [PubMed]
- Salminen, A.; Kaarniranta, K.; Kauppinen, A. The role of myeloid-derived suppressor cells (MDSC) in the inflammaging process. Ageing Res Rev 2018, 48, 1–10. [Google Scholar] [CrossRef]
- Onesti, J.K.; Guttridge, D.C. Inflammation based regulation of cancer cachexia. Biomed. Res. Int. 2014, 2014, 168407. [Google Scholar] [CrossRef]
- Winfield, R.D.; Delano, M.J.; Pande, K.; Scumpia, P.O.; Laface, D.; Moldawer, L.L. Myeloid-derived suppressor cells in cancer cachexia syndrome: A new explanation for an old problem. J. Parenter. Enter. Nutr. 2008, 32, 651–655. [Google Scholar] [CrossRef]
- Buijs, N.; Luttikhold, J.; Houdijk, A.P.; van Leeuwen, P.A. The role of a disturbed arginine/NO metabolism in the onset of cancer cachexia: A working hypothesis. Curr. Med. Chem. 2012, 19, 5278–5286. [Google Scholar] [CrossRef]
- Lee, P.Y.; Wang, J.X.; Parisini, E.; Dascher, C.C.; Nigrovic, P.A. Ly6 family proteins in neutrophil biology. J. Leukoc. Biol. 2013, 94, 585–594. [Google Scholar] [CrossRef] [PubMed]
- Cuenca, A.G.; Cuenca, A.L.; Winfield, R.D.; Joiner, D.N.; Gentile, L.; Delano, M.J.; Kelly-Scumpia, K.M.; Scumpia, P.O.; Matheny, M.K.; Scarpace, P.J.; et al. Novel role for tumor-induced expansion of myeloid-derived cells in cancer cachexia. J. Immunol. 2014, 192, 6111–6119. [Google Scholar] [CrossRef] [PubMed]
- Stephens, N.A.; Skipworth, R.J.; Fearon, K.C. Cachexia, survival and the acute phase response. Curr Opin Support Palliat Care 2008, 2, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Derman, B.A.; Macklis, J.N.; Azeem, M.S.; Sayidine, S.; Basu, S.; Batus, M.; Esmail, F.; Borgia, J.A.; Bonomi, P.; Fidler, M.J. Relationships between longitudinal neutrophil to lymphocyte ratios, body weight changes, and overall survival in patients with non-small cell lung cancer. BMC Cancer 2017, 17, 141. [Google Scholar] [CrossRef] [PubMed]
- VanderVeen, B.N.; Murphy, E.A.; Carson, J.A. The Impact of Immune Cells on the Skeletal Muscle Microenvironment During Cancer Cachexia. Front. Physiol. 2020, 11, 1037. [Google Scholar] [CrossRef]
- Judge, S.M.; Nosacka, R.L.; Delitto, D.; Gerber, M.H.; Cameron, M.E.; Trevino, J.G.; Judge, A.R. Skeletal Muscle Fibrosis in Pancreatic Cancer Patients with Respect to Survival. JNCI Cancer Spectr. 2018, 2, pky043. [Google Scholar] [CrossRef] [PubMed]
- Kusmartsev, S.; Gabrilovich, D.I. Role of immature myeloid cells in mechanisms of immune evasion in cancer. Cancer Immunol. Immunother. 2006, 55, 237–245. [Google Scholar] [CrossRef]
- Li, Y.P.; Reid, M.B. NF-kappaB mediates the protein loss induced by TNF-alpha in differentiated skeletal muscle myotubes. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2000, 279, R1165–R1170. [Google Scholar] [CrossRef]
- Sartori, R.; Milan, G.; Patron, M.; Mammucari, C.; Blaauw, B.; Abraham, R.; Sandri, M. Smad2 and 3 transcription factors control muscle mass in adulthood. Am. J. Physiol. Cell Physiol. 2009, 296, C1248–C1257. [Google Scholar] [CrossRef]
- Tidball, J.G. Mechanisms of muscle injury, repair, and regeneration. Compr Physiol 2011, 1, 2029–2062. [Google Scholar] [CrossRef]
- Zhang, Y.; Chandra, V.; Sanchez, E.R.; Dutta, P.; Quesada, P.R.; Rakoski, A.; Zoltan, M.; Arora, N.; Baydogan, S.; Horne, W.; et al. Interleukin-17-induced neutrophil extracellular traps mediate resistance to checkpoint blockade in pancreatic cancer. J. Exp. Med. 2020, 217. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Mendoza, M.G.; Inman, D.R.; Ponik, S.M.; Jeffery, J.J.; Sheerar, D.S.; Van Doorn, R.R.; Keely, P.J. Neutrophils drive accelerated tumor progression in the collagen-dense mammary tumor microenvironment. Breast Cancer Res. 2016, 18, 49. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Wang, M.; Yin, T.; Zhao, Y.; Wei, X. Myeloid-Derived Suppressor Cells Promote Metastasis in Breast Cancer After the Stress of Operative Removal of the Primary Cancer. Front. Oncol. 2019, 9, 855. [Google Scholar] [CrossRef] [PubMed]
- Loberg, R.D.; Bradley, D.A.; Tomlins, S.A.; Chinnaiyan, A.M.; Pienta, K.J. The lethal phenotype of cancer: The molecular basis of death due to malignancy. CA Cancer J. Clin. 2007, 57, 225–241. [Google Scholar] [CrossRef] [PubMed]
- Burfeind, K.G.; Zhu, X.; Norgard, M.A.; Levasseur, P.R.; Huisman, C.; Buenafe, A.C.; Olson, B.; Michaelis, K.A.; Torres, E.R.; Jeng, S.; et al. Circulating myeloid cells invade the central nervous system to mediate cachexia during pancreatic cancer. Elife 2020, 9, e54095. [Google Scholar] [CrossRef] [PubMed]
- Boivin, G.; Faget, J.; Ancey, P.B.; Gkasti, A.; Mussard, J.; Engblom, C.; Pfirschke, C.; Contat, C.; Pascual, J.; Vazquez, J.; et al. Durable and controlled depletion of neutrophils in mice. Nat. Commun. 2020, 11, 2762. [Google Scholar] [CrossRef]
- Wang, Y.; Pessin, J.E. Mechanisms for fiber-type specificity of skeletal muscle atrophy. Curr. Opin. Clin. Nutr. Metab. Care 2013, 16, 243–250. [Google Scholar] [CrossRef]
- Burkholder, T.J.; Fingado, B.; Baron, S.; Lieber, R.L. Relationship between muscle fiber types and sizes and muscle architectural properties in the mouse hindlimb. J. Morphol. 1994, 221, 177–190. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Deyhle, M.R.; Callaway, C.S.; Neyroud, D.; D’Lugos, A.C.; Judge, S.M.; Judge, A.R. Depleting Ly6G Positive Myeloid Cells Reduces Pancreatic Cancer-Induced Skeletal Muscle Atrophy. Cells 2022, 11, 1893. https://doi.org/10.3390/cells11121893
Deyhle MR, Callaway CS, Neyroud D, D’Lugos AC, Judge SM, Judge AR. Depleting Ly6G Positive Myeloid Cells Reduces Pancreatic Cancer-Induced Skeletal Muscle Atrophy. Cells. 2022; 11(12):1893. https://doi.org/10.3390/cells11121893
Chicago/Turabian StyleDeyhle, Michael R., Chandler S. Callaway, Daria Neyroud, Andrew C. D’Lugos, Sarah M. Judge, and Andrew R. Judge. 2022. "Depleting Ly6G Positive Myeloid Cells Reduces Pancreatic Cancer-Induced Skeletal Muscle Atrophy" Cells 11, no. 12: 1893. https://doi.org/10.3390/cells11121893