
Classical and Atypical Scrapie in Sheep and Goats. Review on the Etiology, Genetic Factors, Pathogenesis, Diagnosis, and Control Measures of Both Diseases

 , ,
, ,
Abstract
:Simple Summary
Abstract
1. Introduction
Causal Agent
- Biological properties: prion strains result in specific phenotypes for different diseases, which can be identified by their incubation periods, clinical signs, histopathological lesions (lesion profile), distribution of PrPSc, and the tissue and cellular tropisms that are all studied in mice models.
- Biochemical properties: Each prion strain is associated with a specific group of biochemical characteristics, in order to highlight the stability against denaturing agents, glycosylation patterns, electrophoretic mobility after digestion with proteinase K, and resistance to proteolytic degradation. Research also reported that strains may differ in their binding affinity for copper [34].
- Conformal properties: Different strains can show similar patterns of resistance to protease but can be distinguished by their conformations. The differences in conformation can be revealed by sedimentation techniques [35], light scattering [36], transmission electron microscopy, and atomic force microscopy [37], through studies of structural change, and by circular dichroism [38], by binding staining [39], by site mapping of binding using a conformation-dependent immunoassay (CDI) [40], and, finally, by mass spectrometry [41].
2. Genetic Factors
2.1. Genotype of the PRNP Gene and Classical Scrapie in Sheep
- The VRQ haplotype is the most closely related to susceptibility to classical scrapie. Homozygous animals for this haplotype are those that present a higher risk. Heterozygotes with the resistant haplotypes (ARR and AHQ) have lower risk.
- The ancestral form ARQ was also associated with susceptibility to classical scrapie, although with a lower risk or less penetration than VRQ.
- The ARR and AHQ alleles were associated with resistance to this TSE, but only if they are present in homozygosis, ARR/ARR or AHQ/AHQ.
- R1: indicates a very low risk of developing the disease in an individual and a very low risk in the first-generation progeny.
- R2: indicates a low risk to an individual and progeny.
- R3: indicates an individual low risk, but that of the progeny may increase based on the genotype of the other parent.
- R4: indicates that scrapie can be found occasionally and that the progeny has greater risk.
- R5: indicates that this sheep has the highest risk of developing scrapie; protease-resistant prion protein (PrP) where the superscript is the haplotype with three polymorphic codons (136 A (Alanine)/V (Valine), 154 R (Arginine)/H (Histidine), and 171 Q (Glutamine)/R/H).
2.2. Genotype of the PRNP Gene and Atypical Sheep Scrapie
2.3. Genotype of the PRNP Gene and Classical Scrapie in Goats
3. Pathogenesis and Transmission of the Disease
3.1. Classical Scrapie
3.2. Atypical Scrapie
4. Diagnostic Methods
4.1. Clinical Diagnosis
4.1.1. Classical Scrapie
- Changes in the mental and behavioral status, such as separation from the flock, bruxism, and repeated licking of the lips.
- Postural and locomotion changes. Wide-based limb posture, hypermetric movements, and ataxia.
- Head tremors.
- Loss of body condition.
4.1.2. Atypical Scrapie
4.2. Laboratory Diagnosis
4.2.1. Histological Diagnosis: Spongiform Change
Classical Scrapie
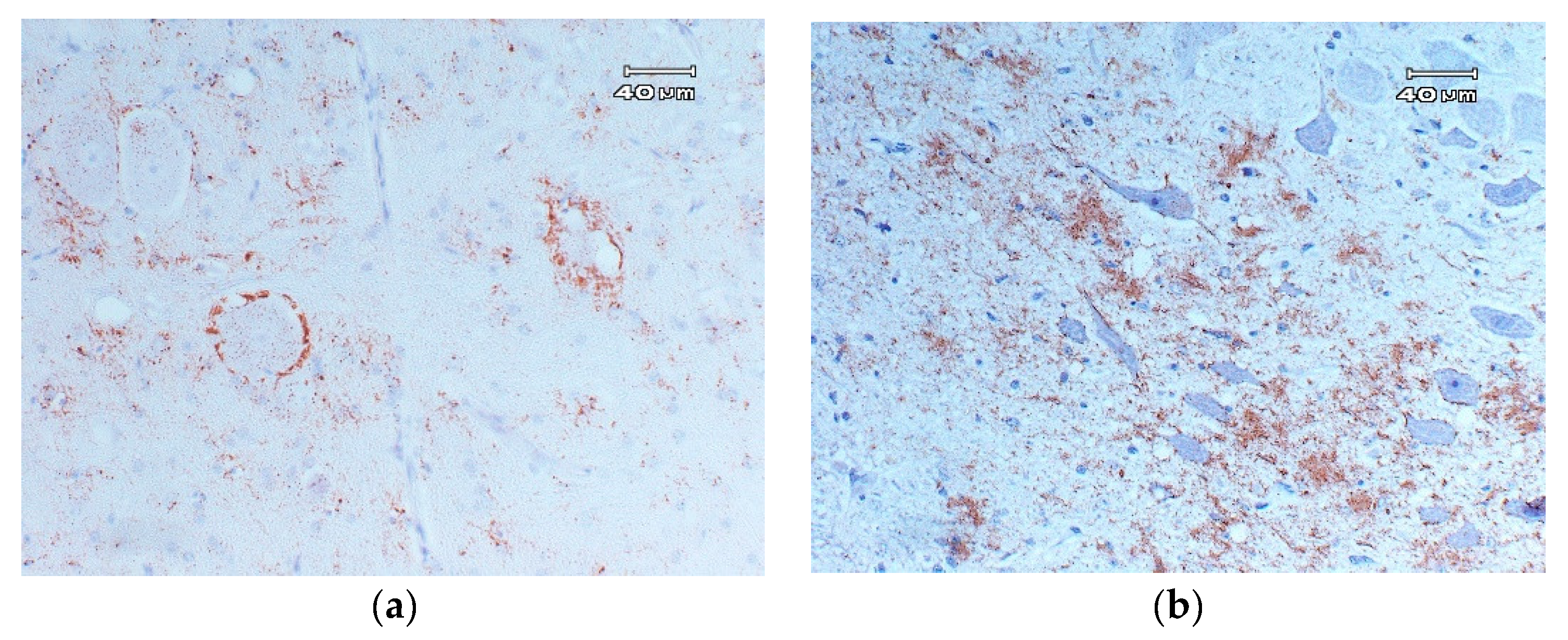
- Spongiform degeneration: Vacuolization, typically bilateral and symmetrical, of the neuronal perikaryon and neuropil gray matter (Figure 1) (spongiosis) located in specific neuroanatomic regions [18,124,125]. The main areas of the CNS where vacuolization is located are the following: In the spinal medulla, the dorsal horns. In the medulla oblongata, the nucleus of the solitary tract, the dorsal nucleus of the vagus nerve, the spinal tract of the trigeminal nerve, the vestibular nuclei, and the reticular formation. In the midbrain, the central gray substance. In the hypothalamus, the paraventricular area, and in the thalamus, the septal area [18,126,127]. The lesion profile can be affected by the strain of scrapie, the genotype of the PRNP gene, the pathway of infection, the age of the host, and the duration of the clinical phase [125,128].
- Gliosis. Gliosis is a common and nonspecific response of glial cells against various stimuli and is often present in prion diseases [129]. In scrapie, hypertrophic astrogliosis and an activation of the microglia, generally associated with PrPSc deposits, vacuolization, and neuronal degeneration, are seen [130,131,132,133]. Both astrocytes and microglia can accumulate PrPSc in natural and experimental cases of scrapie [95,134].
- Neuronal degeneration and loss. Other characteristic lesions of scrapie are neuronal degeneration and loss, which include disseminated necrotic neurons sometimes accompanied by neuronophagy, dystrophic neurites, and basophilic neurons [135]. Cerebral amyloidosis is also common in scrapie in sheep [127,135,136].
Atypical Scrapie
4.2.2. Detection of PrPSc Scrapie Using Immunohistochemistry Techniques
Classical Scrapie
- Intraneuronal: disseminated granular deposits in the neuronal pericarion.
- Intraglial: disseminated granular or ovoid deposits in the cytoplasm of glial cells.
- Glial-associated: radial deposits associated with glial cells.
- Subpial: multifocal or continuous accumulations, associated with glial cells below the pia mater.
- Perivascular: deposits associated with glia cells but localized around blood vessels.
- Subependymal: deposits, generally discontinuous, located in the glia cell layer located below the ependymal cells of the ventricular system.
- Ependymal: deposits on the apical edge of the ependymal cells.
- Linear: thick deposits in linear organization.
- Fine punctate: small granules in the neuropil.
- Coarse punctate: similar to the previous, but with larger deposits that are irregular in shape.
- Coalescent: deposits in the neuropil that are constituted by the fusion of accumulations of coarse particles of PrPSc, forming amorphous masses, or a mesh-like structure.
- Perineuronal: fine deposits around the neuronal perikaryon and neurites.
- Plaques: large accumulations of PrPSc with a fibrillary and radiated form, typically localized around blood vessels.
Atypical Scrapie
4.2.3. Detection of PrPSc Scrapie Using Western Blotting Techniques
Classical Scrapie
Atypical Scrapie
4.2.4. Diagnosis of Classical and Atypical Scrapie by Rapid Test
4.2.5. New Diagnostic Methods of Scrapie
5. Surveillance and Control Methods Established in the European Union
- Exclusion of specific risk materials (SRM) from the human food chain. Some studies on the pathogenesis of the disease indicate that infectivity is mainly localized in the CNS and, in the case of scrapie, infectivity is also distributed by the LRS [156]. The tissues with higher infectivity are defined as specific risk materials, and their exclusion from the food chain was one of the most important measures for human consumer protection [157,158,159]. During the bovine spongiform encephalopathy epidemic, sheep and goats were possibly less exposed to contaminated meat and bone meal, which does not exempt the transmission of the disease to these species [160]. In this way, it was shown that small ruminants could be experimentally infected with BSE [161,162,163,164,165], along with diagnosing the first goat BSE case in France by active surveillance [166]. In sheep and goats, the SRM are:
- ○
- The spleen and the ileum of sheep of all ages.
- ○
- The skull, including the brains and eyes, tonsils, and spinal cord from animals over 12 months or with a permanent incisor erupted.
- Surveillance programs. Surveillance programs of scrapie allow a reliable knowledge of the epidemiological situation in each member state. The annual monitoring program is based on active surveillance (testing without previous suspicion) and passive surveillance (testing of clinical suspects).
- ○
- Active surveillance. The active surveillance covers testing of three subpopulations of sheep and goats:
- ▪
- Animals over 18 months of age that are not slaughtered for human consumption, such as fallen stock, which have died or been killed, but not in the framework of an epidemic.
- ▪
- Animals culled in the framework of TSE eradication.
- ▪
- Healthy animals over 18 months of age slaughtered for human consumption. Only member states with major ovine or caprine populations are required to test an annual minimum sample size of such animals.
- ○
- Passive surveillance. Testing animals identified as TSE suspects as scrapie is a notifiable disease.
- Genetic selection in sheep. Member states may introduce breeding programs to select for resistance to TSE in their ovine populations. The breeding program shall concentrate on flocks of high genetic merit, as defined in point 3 of Annex I of Commission Decision 2002/1003/EC [167]. In brief, breeding programs are aimed to increase the frequency of the ARR allele; any male animal carrying the VRQ allele shall be slaughtered or castrated. The flocks are finally recognized at two levels:
- ○
- Level I flocks shall be flocks composed entirely of ovine animals of the ARR/ARR genotype.
- ○
- Level II flocks shall be flocks whose progeny has been sired exclusively by rams of the ARR/ARR genotype.
- Genetic selection in goats. Member states may introduce breeding programs to select for resistance to TSEs in their goat populations. The K222, D146, and S146 alleles confer genetic resistance against classical scrapie strains [57,58,59,168]. Breeding for resistance can be an effective tool for controlling classical scrapie in goats [65].
6. Infectivity of the Different Tissues
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- McGowan, J.P. Scrapie in sheep. Scott. J. Agric. 1922, 5, 365–375. [Google Scholar]
- Wells, G.; Scott, A.C.; Johnson, C.T.; Gunning, R.F.; Hancock, R.D.; Jeffrey, M.; Dawson, M.; Bradley, R. A novel progressive spongiform encephalopathy in cattle. Vet. Rec. 1987, 121, 419–420. [Google Scholar] [CrossRef] [PubMed]
- Bruce, M.E.; Will, R.G.; Ironside, J.W.; McConnell, I.; Drummond, D.; Suttie, A.; McCardle, L.; Chree, A.; Hope, J.; Birkett, C.; et al. Transmissions to mice indicate that «new variant » CJD is caused by the BSE agent. Nature 1997, 389, 498–501. [Google Scholar] [CrossRef] [PubMed]
- Casalone, C.; Zanusso, G.; Acutis, P.; Ferrari, S.; Capucci, L.; Tagliavini, F.; Monaco, S.; Caramelli, M. Identification of a second bovine amyloidotic spongiform encephalopathy: Molecular similarities with sporadic Creutzfeldt-Jakob disease. Proc. Natl. Acad. Sci. USA 2004, 101, 3065–3070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biacabe, A.; Laplanche, J.; Ryder, S.; Baron, T. Distinct molecular phenotypes in bovine prion diseases. EMBO Rep. 2004, 5, 110–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilesmith, J.W.; Wells, A.G.; Cranwell, M.P.; Ryan, J.B. Bovine spongiform encephalopathy: Epidemiological studies. Vet. Rec. 1988, 123, 638–644. [Google Scholar] [CrossRef]
- Baron, T.; Vulin, J.; Biacabe, A.-G.; Lakhdar, L.; Verchere, J.; Torres, J.-M.; Bencsik, A. Emergence of classical bse strain properties during serial passages of h-bse in wild-type mice. PLoS ONE 2011, 6, e15839. [Google Scholar] [CrossRef]
- Torres, J.-M.; Andréoletti, O.; Lacroux, C.; Prieto, I.; Lorenzo, P.; Larska, M.; Baron, T.; Espinosa, J.-C. Classical bovine spongiform encephalopathy by transmission of h-type prion in homologous prion protein context. Emerg. Infect. Dis. 2011, 17, 1636–1644. [Google Scholar] [CrossRef]
- Huor, A.; Espinosa, J.C.; Vidal, E.; Cassard, H.; Douet, J.Y.; Lugan, S.; Aron, N.; Marín-Moreno, A.; Lorenzo, P.; Aguilar-Calvo, P.; et al. The emergence of classical BSE from atypical/Nor98 scrapie. Proc. Natl. Acad. Sci. USA 2019, 116, 26853–26862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuillé, J.; Chelle, P.L. La maladie dite tremblante du mouton est-elle inoculable? C. R. Acad. Sci. Ser. D 1936, 203, 1552–1554. [Google Scholar]
- Chelle, P.L. Un cas de tremblante chez la chevre. Bull. Acad. Vét. Fr. 1942, 15, 294–295. [Google Scholar]
- Collinge, J.; Clarke, A.R. A general model of prion strains and their pathogenicity. Science 2007, 318, 930–936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Healy, A.M.; Weavers, E.; McElroy, M.; Gomez-Parada, M.; Collins, J.D.; O’Doherty, E.; Sweeney, T.; Doherty, M.L. The clinical neurology of scrapie in Irish sheep. J. Vet. Intern. Med. 2003, 17, 908–916. [Google Scholar] [CrossRef] [PubMed]
- Benestad, S.L.; Sarradin, P.; Thu, B.; Schönheit, J.; Tranulis, M.A.; Bratberg, B. Cases of scrapie with unusual features in Norway and designation of a new type, Nor98. Vet. Rec. 2003, 153, 202–208. [Google Scholar] [CrossRef]
- Mitchell, G.B.; O’Rourke, K.I.; Harrington, N.P.; Soutyrine, A.; Simmons, M.M.; Dudas, S.; Zhuang, D.; Laude, H.; Balachandran, A. Identification of atypical scrapie in Canadian sheep. J. Vet. Diagn. Investig. 2010, 22, 408–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kittelberger, R.; Chaplin, M.J.; Black, H.; Pigott, C.J.; O’Keefe, J.S.; Simmons, M.M.; Ramirez-Villaescusa, A.; McIntyre, L.; MacDiarmid, S.C.; Hannah, M.J.; et al. Atypical scrapie/Nor98 in a sheep from New Zealand. J. Vet. Diagn. Investig. 2010, 22, 863–875. [Google Scholar] [CrossRef] [Green Version]
- Matsuura, Y.; Miyazawa, K.; Imamura, M.; Yokoyama, T.; Iwamaru, Y. First case of atypical scrapie in a goat in Japan. J. Vet. Med. Sci. 2019, 81, 986–989. [Google Scholar] [CrossRef] [PubMed]
- Office International des Epizooties (OIE). Scrapie. 2016. Available online: https://www.oie.int/index.php?id=169&L=0&htmfile=chapitre_scrapie.htm. (accessed on 15 December 2020).
- Gavier-Widén, D.; Stack, M.J.; Baron, T.; Balachandran, A.; Simmons, M. Diagnosis of transmissible spongiform encephalopathies in animals: A review. J. Vet. Diagn. Investig. 2005, 17, 509–527. [Google Scholar] [CrossRef] [Green Version]
- Andréoletti, O.; Orge, L.; Corbière, F.; Costes, P.; Morel, N.; Schelcher, F.; Lacroux, C.; Benestad, S.L.; Beringue, V.; Litaise, C.; et al. Atypical/Nor98 scrapie infectivity in sheep peripheral tissues. PLoS Pathog. 2011, 7, e1001285. [Google Scholar] [CrossRef] [Green Version]
- Brown, P.; Bradley, R. 1755 and all that: A historical primer of transmissible spongiform encephalopathy. BMJ 1998, 317, 1688–1692. [Google Scholar] [CrossRef] [Green Version]
- Le Dur, A.; Béringue, V.; Andréoletti, O.; Reine, F.; Laï, T.L.; Baron, T.; Bratberg, B.; Vilotte, J.L.; Sarradin, P.; Benestad, S.L.; et al. A newly identified type of scrapie agent can naturally infect sheep with resistant PrP genotypes. Proc. Natl. Acad. Sci. USA 2005, 102, 16031–16036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simmons, M.M.; Konold, T.A.; Simmons, H.; Spencer, Y.I.; Lockey, R.; Spiropoulos, J.; Everitt, S.; Clifford, D. Experimental transmission of atypical scrapie to sheep. BMC Vet. Res. 2007, 3, 20. [Google Scholar] [CrossRef] [Green Version]
- Fediaevsky, A.; Tongue, S.C.; Nöremark, M.; Calavas, D.; Ru, G.; Hopp, P. A descriptive study of the prevalence of atypical and classical scrapie in sheep in 20 European countries. BMC Vet. Res. 2008, 4, 19. [Google Scholar] [CrossRef] [PubMed]
- Prusiner, S.B. The prion diseases. Sci. Am. 1995, 272, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Poser, C.M. Notes on the history of the prion diseases. Part I. Clin. Neurol. Neurosurg. 2002, 104, 1–9. [Google Scholar] [CrossRef]
- Baral, P.K.; Yin, J.; Aguzzi, A.; James, M.N.G. Transition of the prion protein from a structured cellular form (PrPC) to the infectious scrapie agent (PrPSc). Prot. Sci. 2019, 28, 2055–2063. [Google Scholar] [CrossRef] [PubMed]
- Martins, V.R.R.; Linden, M.; Prado, R.; Walz, A.C.; Sakamoto, I.; Izquierdo, R.R.; Brentani. Cellular prion protein: On the road for functions. FEBS Lett. 2002, 512, 25–28. [Google Scholar] [CrossRef] [Green Version]
- Westergard, L.; Christensen, H.M.; Harris, D.A. The cellular prion protein (PrP(C)): Its physiological function and role in disease. Biochim. Biophys. Acta 2007, 1772, 629–644. [Google Scholar] [CrossRef] [Green Version]
- Nishida, N.; Tremblay, P.; Sugimoto, T.; Shigematsu, K.; Shirabe, S.; Petromilli, C.; Erpel, S.P.; Nakaoke, R.; Atarashi, R.; Houtani, T.; et al. A mouse prion protein transgene rescues mice deficient for the prion protein gene from purkinje cell degeneration and demyelination. Lab. Investig. 1999, 79, 689–697. [Google Scholar] [PubMed]
- Benestad, S.L.; Austbø, L.; Tranulis, A.M.; Espenes, A.; Olsaker, I. Healthy goats naturally devoid of prion protein. BMC Vet. Res. 2012, 43, 87. [Google Scholar] [CrossRef] [Green Version]
- Skedsmo, F.S.; Malachin, G.; Våge, D.I.; Hammervold, M.M.; Salvesen, Ø.; Ersdal, C.; Ranheim, B.; Stafsnes, M.H.; Bartosova, Z.; Bruheim, P.; et al. Demyelinating polyneuropathy in goats lacking prion protein. FASEB J. 2019, 34, 2359–2375. [Google Scholar] [CrossRef]
- Requena, J.R. The protean prion protein. PLoS Biol. 2020, 18, e3000754. [Google Scholar] [CrossRef]
- Wadsworth, J.D.; Jackson, G.S.; Hill, A.F.; Collinge, J. Molecular biology of prion propagation. Curr. Opin. Genet. Dev. 1999, 9, 338–345. [Google Scholar] [CrossRef]
- Bessen, A.R.; Marsh, R.F. Biochemical and physical properties of the prion protein from two strains of the transmissible mink encephalopathy agent. J. Virol. 1992, 66, 2096–2101. [Google Scholar] [CrossRef] [Green Version]
- Scheibel, T.; Lindquist, S.L. The role of conformational flexibility in prion propagation and maintenance for Sup35p. Nat. Genet. 2001, 8, 958–962. [Google Scholar] [CrossRef]
- Serio, T.R.; Cashikar, A.G.; Kowal, A.S.; Sawicki, G.J.; Moslehi, J.J.; Serpell, L.; Arnsdorf, M.F.; Lindquist, S.L. Nucleated conformational conversion and the replication of conformational information by a prion determinant. Science 2000, 289, 1317–1321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Safar, J.; Wille, H.; Itri, V.; Groth, D.; Serban, H.; Torchia, M.; Cohen, F.E.; Prusiner, S.B. Eight prion strains have PrP(Sc) molecules with different conformations. Nat. Med. 1998, 4, 1157–1165. [Google Scholar] [CrossRef] [PubMed]
- Sigurdson, C.J.; Nilsson, K.P.R.; Hornemann, S.; Manco, G.; Polymenidou, M.; Schwarz, P.; Leclerc, M.; Hammarström, P.; Wüthrich, K.; Aguzzi, A. Prion strain discrimination using luminescent conjugated polymers. Nat. Chem. Biol. 2007, 4, 1023–1030. [Google Scholar] [CrossRef]
- Zou, W.-Q.; Capellari, S.; Parchi, P.; Sy, M.-S.; Gambetti, P.; Chen, S.G. Identification of novel proteinase k-resistant C-terminal fragments of prp in Creutzfeldt-Jakob disease. J. Biol. Chem. 2003, 278, 40429–40436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, A.R.; Faris, R.; Priola, A.S. Proteomics applications in prion biology and structure. Expert Rev. Proteom. 2015, 12, 171–184. [Google Scholar] [CrossRef] [PubMed]
- Hunter, N. PrP genetics in sheep and the application for scrapie and BSE. Trends Microbiol. 1997, 5, 331–334. [Google Scholar] [CrossRef]
- Bossers, A.; Belt, P.B.G.M.; Raymond, G.J.; Caughey, B.; De Vries, R.; Smits, M.A. Scrapie susceptibility-linked polymorphisms modulate the in vitro conversion of sheep prion protein to protease-resistant forms. Proc. Natl. Acad. Sci. USA 1997, 4, 4931–4936. [Google Scholar] [CrossRef] [Green Version]
- Houston, F.; Goldmann, W.; Foster, J.; González, L.; Jeffrey, M.; Hunter, N. Comparative susceptibility of sheep of different origins, breeds and prnp genotypes to challenge with bovine spongiform encephalopathy and scrapie. PLoS ONE 2015, 10, e0143251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thorgeirsdottir, S.; Sigurdarson, S.; Thorisson, H.M.; Georgsson, G.; Palsdottir, A. PrP gene polymorphism and natural scrapie in Icelandic sheep. J. Gen. Virol. 1999, 80, 2527–2534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dawson, M.; Hoinville, L.J.; Hosie, B.D.; Hunter, N. Guidance on the use of PrP genotyping as an aid to the control of clinical scrapie. Scrapie Information Group. Vet. Rec. 1998, 142, 623–625. [Google Scholar]
- Moum, T.; Olsaker, I.; Hopp, P.; Moldal, T.; Valheim, M.; Moum, T.; Benestad, S.L. Polymorphisms at codons 141 and 154 in the ovine prion protein gene are associated with scrapie Nor98 cases. J. Gen. Virol. 2005, 86, 231–235. [Google Scholar] [CrossRef]
- Goldmann, W.; Ryan, K.; Foster, J.; Stewart, P.; Parnham, D.; Xicohtencatl, R.; Fernandez, N.; Saunders, G.; Windl, O.; González, L.; et al. Caprine prion gene polymorphisms are associated with decreased incidence of classical scrapie in goat herds in the United Kingdom. Vet. Res. 2011, 42, 110. [Google Scholar] [CrossRef] [Green Version]
- Goldmann, W.; Martin, T.; Foster, J.; Hughes, S.; Smith, G.; Hughes, K.; Dawson, M. Novel polymorphisms in the caprine PrP gene: A codon 142 mutation associated with scrapie incubation period. J. Gen. Virol. 1996, 77, 2885–2891. [Google Scholar] [CrossRef]
- Gonzalez, L.; Martin, S.; Hawkins, S.A.; Goldmann, W.; Jeffrey, M.; Siso, S. Pathogenesis of natural goat scrapie: Modulation by host PRNP genotype and effect of co-existent conditions. Vet. Res. 2010, 41, 48. [Google Scholar] [CrossRef] [Green Version]
- Goldmann, W.; Hunter, N.; Chong, A.; Foster, J.; Hope, J. The shortest known prion protein gene allele occurs in goats, has only three octapeptide repeats and is non-pathogenic. J. Gen. Virol. 1998, 79, 3173–3176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Billinis, C.; Panagiotidis, C.H.; Psychas, V.; Argyroudis, S.; Nicolaou, A.; Leontides, S.; Papadopoulos, O.; Sklaviadis, T. Prion protein gene polymorphisms in natural goat scrapie. J. Gen. Virol. 2002, 83, 713–721. [Google Scholar] [CrossRef]
- Barillet, F.; Mariat, D.; Amigues, Y.; Faugeras, R.; Caillat, H.; Moazami-Goudarzi, K.; Rupp, R.; Babilliot, J.M.; Lacroux, C.; Lugan, S.; et al. Identification of seven haplotypes of the caprine PrP gene at codons 127, 142, 154, 211, 222 and 240 in French Alpine and Saanen breeds and their association with classical scrapie. J. Gen. Virol. 2009, 90, 769–776. [Google Scholar] [CrossRef] [PubMed]
- Bouzalas, I.G.; Dovas, C.I.; Banos, G.; Papanastasopoulou, M.; Kritas, S.; Oevermann, A.; Papakostaki, D.; Evangelia, C.; Papadopoulos, O.; Seuberlich, T.; et al. Caprine PRNP polymorphisms at codons 171, 211, 222 and 240 in a Greek herd and their association with classical scrapie. J. Gen. Virol. 2010, 91, 1629–1634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colussi, S.; Vaccari, G.; Maurella, C.; Bona, C.; Lorenzetti, R.; Troiano, P.; Casalinuovo, F.; Di Sarno, A.; Maniaci, M.G.; Zuccon, F.; et al. Histidine at codon 154 of the prion protein gene is a risk factor for Nor98 scrapie in goats. J. Gen. Virol. 2008, 89, 3173–3176. [Google Scholar] [CrossRef] [PubMed]
- Vaccari, G.; Panagiotidis, C.H.; Acin, C.; Peletto, S.; Barillet, F.; Acutis, P.; Bossers, A.; Langeveld, J.; Van Keulen, L.; Sklaviadis, T.; et al. State-of-the-art review of goat TSE in the European Union, with special emphasis onPRNPgenetics and epidemiology. Vet. Res. 2009, 40, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Papasavva-Stylianou, P.; Windl, O.; Saunders, G.; Mavrikiou, P.; Toumazos, P.; Kakoyiannis, C. PrP gene polymorphisms in Cyprus goats and their association with resistance or susceptibility to natural scrapie. Vet. J. 2011, 187, 245–250. [Google Scholar] [CrossRef] [PubMed]
- Acutis, P.L.; Bossers, A.; Priem, J.; Riina, M.V.; Peletto, S.; Mazza, M.; Casalone, C.; Forloni, G.; Ru, G.; Caramelli, M. Identification of prion protein gene polymorphisms in goats from Italian scrapie outbreaks. J. Gen. Virol. 2006, 87, 1029–1033. [Google Scholar] [CrossRef]
- Vaccari, G.; Di Bari, M.A.; Morelli, L.; Nonno, R.; Chiappini, B.; Antonucci, G.; Marcon, S.; Esposito, E.; Fazzi, P.; Palazzini, N.; et al. Identification of an allelic variant of the goat PrP gene associated with resistance to scrapie. J. Gen. Virol. 2006, 87, 1395–1402. [Google Scholar] [CrossRef]
- Acutis, P.L.; Martucci, F.; Zuccon, F.; Corona, C.; Martinelli, N.; Casalone, C.; Caramelli, M.; Lombardi, G.; D’Angelo, A.; Peletto, S.; et al. Resistance to classical scrapie in experimentally challenged goats carrying mutation K222 of the prion protein gene. Vet. Res. 2012, 43, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corbière, F.; Perrin-Chauvineau, C.; Lacroux, C.; Costes, P.; Thomas, M.; Brémaud, I.; Martin, S.; Lugan, S.; Chartier, C.; Schelcher, F.; et al. PrP-associated resistance to scrapie in five highly infected goat herds. J. Gen. Virol. 2013, 94, 241–245. [Google Scholar] [CrossRef]
- Aguilar-Calvo, P.; Espinosa, J.C.; Pintado, B.; Gutierrez-Adan, A.; Alamillo, E.; Miranda, A.; Prieto, I.; Bossers, A.; Andreoletti, O.; Torres, J.M. Role of the goat K222-PrP(C) polymorphic variant in prion infection resistance. J. Virol. 2014, 88, 2670–2676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lacroux, C.; Perrin-Chauvineau, C.; Barillet, F.; Andréoletti, O.; Caughey, B.W.; Corbière, F.; Aron, N.; Aguilar-Calvo, P.; Torres, J.M.; Costes, P.; et al. Genetic resistance to scrapie infection in experimentally challenged goats. J. Virol. 2013, 88, 2406–2413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dassanayake, R.P.; White, S.N.; Madsen-Bouterse, S.A.; Schneider, D.A.; O’Rourke, K.I. Role of the PRNP S127 allele in experimental infection of goats with classical caprine scrapie. Anim. Genet. 2015, 46, 341. [Google Scholar] [CrossRef]
- EFSA Panel on Biological Hazards (BIOHAZ); Ricci, A.; Allende, A.; Bolton, D.; Chemaly, M.; Davies, R.; Fernández Escámez, P.S.; Gironés, R.; Herman, L.; Koutsoumanis, K.; et al. Genetic resistance to transmissible spongiform encephalopathies (TSE) in goats. EFSA J. 2017, 15, e04962. [Google Scholar]
- Konold, T.; Moore, S.J.; Bellworthy, S.J.; Simmons, A.H. Evidence of scrapie transmission via milk. BMC Vet. Res. 2008, 4, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foster, J.D.; Goldmann, W.; Hunter, N. Evidence in sheep for pre-natal transmission of scrapie to lambs from infected mothers. PLoS ONE 2013, 8, e79433. [Google Scholar] [CrossRef] [Green Version]
- Garza, M.C.; Eraña, H.; Castilla, J.; Acín, C.; Vargas, A.; Badiola, J.J.; Monleón, E. Protein misfolding cyclic amplification corroborates the absence of PrPSc accumulation in placenta from foetuses with the ARR/ARQ genotype in natural scrapie. Vet. Microbiol. 2017, 203, 294–300. [Google Scholar] [CrossRef]
- Mohan, J.; Brown, K.L.; Farquhar, C.F.; Bruce, E.M.; Mabbott, A.N. Scrapie transmission following exposure through the skin is dependent on follicular dendritic cells in lymphoid tissues. J. Dermatol. Sci. 2004, 35, 101–111. [Google Scholar] [CrossRef]
- Mohan, J.; Bruce, M.E.; Mabbott, N.A. Neuroinvasion by scrapie following inoculation via the skin is independent of migratory langerhans cells. J. Virol. 2005, 79, 1888–1897. [Google Scholar] [CrossRef] [Green Version]
- Glaysher, B.R.; Mabbott, N.A. Role of the draining lymph node in scrapie agent transmission from the skin. Immunol. Lett. 2007, 109, 64–71. [Google Scholar] [CrossRef]
- Haybaeck, J.; Heikenwalder, M.; Stitz, L.; Aguzzi, A.; Klevenz, B.; Schwarz, P.; Margalith, I.; Bridel, C.; Mertz, K.; Zirdum, E.; et al. Aerosols transmit prions to immunocompetent and immunodeficient mice. PLoS Pathog. 2011, 7, e1001257. [Google Scholar] [CrossRef] [Green Version]
- Denkers, N.D.; Hayes-Klug, J.; Hoover, E.A.; Anderson, K.R.; Seelig, D.M.; Haley, N.J.; Dahmes, S.J.; Osborn, D.A.; Miller, K.V.; Warren, R.J.; et al. Aerosol transmission of chronic wasting disease in white-tailed deer. J. Virol. 2013, 87, 1890–1892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nichols, T.A.; Spraker, T.R.; O’Rourke, K.; Telling, G.C.; Bowen, R.; Zabel, M.D.; Vercauteren, K.C.; Rigg, T.D.; Meyerett-Reid, C.; Hoover, C.; et al. Intranasal inoculation of white-tailed deer (Odocoileus virginianus) With lyophilized chronic wasting disease prion particulate complexed to montmorillonite clay. PLoS ONE 2013, 8, e62455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruce, E.M. TSE strain variation. Br. Med. Bull. 2003, 66, 99–108. [Google Scholar] [CrossRef]
- Grassi, J. Pre-clinical diagnosis of transmissible spongiform encephalopathies using rapid tests. Transfus. Clin. Biol. 2003, 10, 19–22. [Google Scholar] [CrossRef]
- Unterberger, U.; Budka, H. Pathogenesis of prion diseases. Acta Neuropathol. 2005, 109, 32–48. [Google Scholar] [CrossRef] [PubMed]
- Taylor, D.M.; McConnell, I.; Fraser, H. Scrapie infection can be established readily through skin scarification in immunocompetent but not immunodeficient mice. J. Gen. Virol. 1996, 77, 1595–1599. [Google Scholar] [CrossRef] [PubMed]
- Maignien, T.; Zas, C.I.L.; Beringue, V.; Dormont, D.; Deslys, J.-P. Pathogenesis of the oral route of infection of mice with scrapie and bovine spongiform encephalopathy agents. J. Gen. Virol. 1999, 80, 3035–3042. [Google Scholar] [CrossRef]
- Detwiler, L.; Baylis, M. The epidemiology of scrapie. Rev. Sci. Tech. l’OIE 2003, 22, 121–143. [Google Scholar] [CrossRef]
- Hamir, A.N.; Kunkle, R.A.; Bulgin, M.S.; Rohwer, R.G.; Gregori, L.; Richt, J.A. Experimental transmission of scrapie agent to susceptible sheep by intralingual or intracerebral inoculation. Can. J. Vet. Res. 2008, 72, 63–67. [Google Scholar]
- Rose, S.G.S.; Hunter, N.; Matthews, L.; Foster, J.D.; Chase-Topping, E.M.; Kruuk, L.E.B.; Shaw, D.J.; Rhind, S.M.; Will, R.G.; Woolhouse, M.E.J. Comparative evidence for a link between Peyer’s patch development and susceptibility to transmissible spongiform encephalopathies. BMC Infect. Dis. 2006, 6, 5. [Google Scholar] [CrossRef] [Green Version]
- Mabbott, N.A.; Bruce, M.E. The immunobiology of TSE diseases. J. Gen. Virol. 2001, 82, 2307–2318. [Google Scholar] [CrossRef] [Green Version]
- Van Keulen, L.J.M.; Vromans, M.E.W.; Van Zijderveld, F.G. Early and late pathogenesis of natural scrapie infection in sheep. APMIS 2002, 110, 23–32. [Google Scholar] [CrossRef] [Green Version]
- Beekes, M.; McBride, A.P. Early accumulation of pathological PrP in the enteric nervous system and gut-associated lymphoid tissue of hamsters orally infected with scrapie. Neurosci. Lett. 2000, 278, 181–184. [Google Scholar] [CrossRef]
- Hunter, N.; Foster, J.; Chong, A.; McCutcheon, S.; Parnham, D.; Eaton, S.; MacKenzie, C.; Houston, F. Transmission of prion diseases by blood transfusion. J. Gen. Virol. 2002, 83, 2897–2905. [Google Scholar] [CrossRef]
- Aguzzi, A.; Heikenwalder, M. Pathogenesis of prion diseases: Current status and future outlook. Nat. Rev. Genet. 2006, 4, 765–775. [Google Scholar] [CrossRef]
- Sisó, S.; Jeffrey, M.; González, L. Neuroinvasion in sheep transmissible spongiform encephalopathies: The role of the haematogenous route. Neuropathol. Appl. Neurobiol. 2009, 35, 232–246. [Google Scholar] [CrossRef] [PubMed]
- Begara-McGorun, I.; Clark, A.M.; Martin, S.; Jeffrey, M. Prevalence of vacuolar lesions consistent with scrapie in the brains of healthy cull sheep of the Shetland Islands. Vet. Rec. 2000, 147, 439–441. [Google Scholar] [CrossRef] [PubMed]
- Ryder, S.J.; Spencer, Y.I.; Bellerby, P.J.; March, S.A. Immunohistochemical detection of PrP in the medulla oblongata of sheep: The spectrum of staining in normal and scrapie-affected sheep. Vet. Rec. 2001, 148, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Jeffrey, M.; Martin, S.; Barr, J.; Chong, A.; Fraser, J.R. Onset of accumulation of PrPRes in murine ME7 scrapie in relation to pathological and PrP immunohistochemical changes. J. Comp. Pathol. 2001, 124, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Heggebø, R.; Press, C.M.; Gunnes, G.; González, L.; Jeffrey, M. Distribution and accumulation of PrP in gut-associated and peripheral lymphoid tissue of scrapie-affected Suffolk sheep. J. Gen. Virol. 2002, 83, 479–489. [Google Scholar] [CrossRef]
- Hoinville, L.J. A review of the epidemiology of scrapie in sheep. Rev. Sci. Tech. 1996, 15, 827–852. [Google Scholar] [CrossRef] [PubMed]
- Onodera, T.; Ikeda, T.; Muramatsu, Y.; Shinagawa, M. Isolation of scrapie agent from the placenta of sheep with natural scrapie in Japan. Microbiol. Immunol. 1993, 37, 311–316. [Google Scholar] [CrossRef] [PubMed]
- Andreoletti, O.; Lacroux, C.; Chabert, A.; Monnereau, L.; Tabouret, G.; Lantier, F.; Berthon, P.; Eychenne, F.; Lafond-Benestad, S.J.; Elsen, M.; et al. PrP(Sc) accumulation in placentas of ewes exposed to natural scrapie: Influence of foetal PrP genotype and effect on ewe-to-lamb transmission. J. Gen. Virol. 2002, 83, 2607–2616. [Google Scholar] [CrossRef]
- Tuo, W.; O’Rourke, K.I.; Zhuang, D.; Cheevers, W.P.; Spraker, T.R.; Knowles, D.P. Pregnancy status and fetal prion genetics determine PrPSc accumulation in placentomes of scrapie-infected sheep. Proc. Natl. Acad. Sci. USA 2002, 99, 6310–6315. [Google Scholar] [CrossRef] [Green Version]
- Terry, A.L.; Howells, L.; Bishop, K.; Baker, A.C.; Everest, S.; Thorne, L.; Maddison, B.C.; Gough, K.C. Detection of prions in the faeces of sheep naturally infected with classical scrapie. Vet. Res. 2011, 42, 65. [Google Scholar] [CrossRef] [Green Version]
- Miller, M.W.; Williams, E.S.; Hobbs, N.T.; Wolfe, L.L. Environmental sources of prion transmission in mule deer. Emerg. Infect. Dis. 2004, 10, 1003–1006. [Google Scholar] [CrossRef]
- Madsen-Bouterse, S.A.; Highland, M.A.; Dassanayake, R.P.; Zhuang, D.; Schneider, D.A. Low-volume goat milk transmission of classical scrapie to lambs and goat kids. PLoS ONE 2018, 13, e0204281. [Google Scholar] [CrossRef]
- Lacroux, C.; Simon, S.; Weisbecker, J.-L.; Moldal, T.; Simmons, H.; Lantier, F.; Tarisse, C.F.; Morel, N.; Schelcher, F.; Grassi, J.; et al. Prions in milk from ewes incubating natural scrapie. PLoS Pathog. 2008, 4, e1000238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ligios, C.; Cancedda, G.M.; Margalith, I.; Santucciu, C.; Madau, L.; Maestrale, C.; Basagni, M.; Saba, M.; Heikenwalder, M. Intraepithelial and interstitial deposition of pathological prion protein in kidneys of scrapie-affected sheep. PLoS ONE 2007, 2, e859. [Google Scholar] [CrossRef]
- Vascellari, M.; Nonno, R.; De Grossi, L.; Rosone, F.; Giordani, F.; Agrimi, U.; Mutinelli, F.; Bigolaro, M.; Di Bari, M.A.; Melchiotti, E.; et al. Prpsc in salivary glands of scrapie-affected sheep. J. Virol. 2007, 81, 4872–4876. [Google Scholar] [CrossRef] [Green Version]
- Maddison, B.C.; Baker, C.A.; Terry, L.A.; Bellworthy, S.J.; Thorne, L.; Rees, H.C.; Gough, K.C. Environmental sources of scrapie prions. J. Virol. 2010, 84, 11560–11562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gough, K.C.; Baker, C.A.; Rees, H.C.; Terry, L.A.; Spiropoulos, J.; Thorne, L.; Maddison, B.C. The oral secretion of infectious scrapie prions occurs in preclinical sheep with a range of prnp genotypes. J. Virol. 2011, 86, 566–571. [Google Scholar] [CrossRef] [Green Version]
- Rubenstein, R.; Bulgin, M.S.; Chang, B.; Sorensen-Melson, S.; Petersen, R.B.; LaFauci, G. PrP(Sc) detection and infectivity in semen from scrapie-infected sheep. J. Gen. Virol. 2012, 93, 1375–1383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Race, R.; Jenny, A.; Sutton, D. Scrapie infectivity and proteinase k-resistant prion protein in sheep placenta, brain, spleen, and lymph node: Implications for transmission and antemortem diagnosis. J. Infect. Dis. 1998, 178, 949–953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spiropoulos, J.; Hawkins, S.A.C.; Simmons, M.M.; Bellworthy, S.J. Evidence of in utero transmission of classical scrapie in sheep. J. Virol. 2014, 88, 4591–4594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benestad, S.L.; Arsac, J.-N.; Goldmann, W.; Nöremark, M. Atypical/Nor98 scrapie: Properties of the agent, genetics, and epidemiology. Vet. Res. 2008, 39, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cassmann, E.D.; Mammadova, N.; Moore, S.J.; Benestad, S.; Greenlee, J.J. Transmission of the atypical/Nor98 scrapie agent to Suffolk sheep with VRQ/ARQ, ARQ/ARQ, and ARQ/ARR genotypes. PLoS ONE 2021, 16, e0246503. [Google Scholar] [CrossRef]
- Fediaevsky, A.; Morignat, E.; Ducrot, C.; Calavas, D. A case-control study on the origin of atypical scrapie in sheep, France. Emerg. Infect. Dis. 2009, 15, 710–718. [Google Scholar] [CrossRef]
- Ortiz-Peláez, A.; Arnold, M.E.; Vidal-Diez, A. Epidemiological investigations on the potential transmissibility of a rare disease: The case of atypical scrapie in Great Britain. Epidemiol. Infect. 2016, 144, 2107–2116. [Google Scholar] [CrossRef]
- Clark, A.M.; Moar, A.J. Scrapie: A clinical assessment. Vet. Rec. 1992, 130, 377–378. [Google Scholar] [CrossRef] [PubMed]
- EU TSE Reference Laboratory. Clinical Signs of Transmissible Spongiform Encephalopathies in Sheep. Animal and Plant Health Agency (APHA). 2017. Available online: https://science.vla.gov.uk/tseglobalnet/documents/clinical-signs-tse-sheep-stills.pdf (accessed on 15 December 2020).
- Jeffrey, M.; González, L. Classical sheep transmissible spongiform encephalopathies: pathogenesis, pathological phenotypes and clinical disease. Neuropathol. Appl. Neurobiol. 2007, 33, 373–394. [Google Scholar] [CrossRef] [PubMed]
- Vargas, F.; Lujan, L.; Bolea, R.; Monleón, E.; Martín-Burriel, I.; Fernández, A.; De Blas, I.; Badiola, J.J. Detection and clinical evolution of scrapie in sheep by 3rd eyelid biopsy. J. Vet. Intern. Med. 2006, 20, 187–193. [Google Scholar] [CrossRef]
- Barnett, K.C.; Palmer, A.C. Retinopathy in sheep affected with natural scrapie. Res. Vet. Sci. 1971, 12, 383–385. [Google Scholar] [CrossRef]
- Vargas, F.; Bolea, R.; Monleón, E.; Acín, C.; Vargas, A.; De Blas, I.; Luján, L.; Badiola, J.J. Clinical characterisation of natural scrapie in a native Spanish breed of sheep. Vet. Rec. 2005, 156, 318–319. [Google Scholar] [CrossRef]
- Sharp, M.W.; Collings, D.F. Ovine abomasal enlargement and scrapie. Vet. Rec. 1987, 120, 215. [Google Scholar] [CrossRef]
- Capucchio, M.T.; Guarda, F.; Pozzato, N.; Coppolino, S.; Caracappa, S.; Di Marco, V. Clinical signs and diagnosis of scrapie in Italy: A comparative study in sheep and goats. J. Vet. Med. Ser. A 2001, 48, 23–31. [Google Scholar] [CrossRef]
- Konold, T.; Davis, A.; Bone, G.; Bracegirdle, J.; Everitt, S.; Chaplin, M.; Saunders, G.C.; Cawthraw, S.; Simmons, M.M. Clinical findings in two cases of atypical scrapie in sheep: A case report. BMC Vet. Res. 2007, 3, 2. [Google Scholar] [CrossRef] [Green Version]
- Espenes, A.; Press, C.; Landsverk, T.; Tranulis, M.; Aleksandersen, M.; Gunnes, G.; Benestad, S.; Fuglestveit, R.; Ulvund, M. Detection of prpsc in rectal biopsy and necropsy samples from sheep with experimental scrapie. J. Comp. Pathol. 2006, 134, 115–125. [Google Scholar] [CrossRef] [PubMed]
- O’Rourke, K.I.; Baszler, T.V.; Jenny, A.; Knowles, D.P.; Besser, T.E.; Miller, J.M.; Cutlip, R.C.; Wells, G.A.H.; Ryder, S.J.; Parish, S.M.; et al. Preclinical diagnosis of scrapie by immunohistochemistry of third eyelid lymphoid tissue. J. Clin. Microbiol. 2000, 38, 3254–3259. [Google Scholar] [CrossRef]
- Schreuder, B.E.C.; van Keulen, L.J.M.; Vromans, M.E.W.; Langeveld, J.P.M.; Smits, M.A. Tonsillar biopsy and PrPSc detection in the preclinical diagnosis of scrapie. Vet. Rec. 1998, 142, 564–568. [Google Scholar] [CrossRef] [Green Version]
- Wells, G.; McGill, I. Recently described scrapie-like encephalopathies of animals: Case definitions. Res. Vet. Sci. 1992, 53, 1–10. [Google Scholar] [CrossRef]
- Ligios, C.; Jeffrey, M.; Ryder, S.; Bellworthy, S.; Simmons, M. Distinction of scrapie phenotypes in sheep by lesion profiling. J. Comp. Pathol. 2002, 127, 45–57. [Google Scholar] [CrossRef]
- Detwiler, L. Scrapie. Rev. Sci. Tech. l’OIE 1992, 11, 491–537. [Google Scholar] [CrossRef] [Green Version]
- Wood, J.L.N.; McGill, I.S.; Done, S.H.; Bradley, R. Neuropathology of scrapie: A study of the distribution patterns of brain lesions in 222 cases of natural scrapie in sheep, 1982-1991. Vet. Rec. 1997, 140, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Begara-McGorum, I.; González, L.; Simmons, M.; Hunter, N.; Houston, F.; Jeffrey, M. Vacuolar lesion profile in sheep scrapie: Factors influencing its variation and relationship to disease-specific PRP accumulation. J. Comp. Pathol. 2002, 127, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Summers, R.J.; Papaioannou, M.; Harris, S.; Evans, B.A. Expression of beta 3-adrenoceptor mRNA in rat brain. Br. J. Pharmacol. 1995, 116, 2547–2548. [Google Scholar] [CrossRef]
- Wells, G.A.H.; Wilesmith, J.W.; McGill, I.S. Bovine spongiform encephalopathy: A neuropathological perspective. Brain Pathol. 1991, 1, 69–78. [Google Scholar] [CrossRef]
- Lazarini, F.; Boussin, F.; Deslys, J.; Tardy, M.; Dormont, D. Astrocyte gene expression in experimental mouse scrapie. J. Comp. Pathol. 1994, 111, 87–98. [Google Scholar] [CrossRef]
- Rezaie, P.; Lantos, P.L. Microglia and the pathogenesis of spongiform encephalopathies. Brain Res. Brain Res. Rev. 2001, 35, 55–72. [Google Scholar] [CrossRef]
- Titeux, M.; Galou, M.; Gomes, F.C.A.; Dormont, D.; Neto, V.M.; Paulin, D. Differences in the activation of the GFAP gene promoter by prion and viral infections. Mol. Brain Res. 2002, 109, 119–127. [Google Scholar] [CrossRef]
- Ye, X.; Scallet, A.C.; Kascsak, R.J.; Carp, I.R. Astrocytosis and amyloid deposition in scrapie-infected hamsters. Brain Res. 1998, 809, 277–287. [Google Scholar] [CrossRef]
- Wood, J.L.; Done, S.H. Natural scrapie in goats: Neuropathology. Vet. Rec. 1992, 131, 93–96. [Google Scholar] [CrossRef] [PubMed]
- Jeffrey, M.; Goodsir, C.M.; Holliman, A.; Higgins, R.J.; Bruce, M.E.; McBride, P.A.; Fraser, J.R. Determination of the frequency and distribution of vascular and parenchymal amyloid with polyclonal and N-terminal-specific PrP antibodies in scrapie-affected sheep and mice. Vet. Rec. 1998, 142, 534–537. [Google Scholar] [CrossRef]
- Simmons, M.M.; Konold, T.; Thurston, L.; Bellworthy, S.J.; Chaplin, M.J.; Moore, S.J. The natural atypical scrapie phenotype is preserved on experimental transmission and sub-passage in PRNP homologous sheep. BMC Vet. Res. 2010, 6, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- González, L.; Martin, S.; Jeffrey, M. Distinct profiles of PrPd immunoreactivity in the brain of scrapie- and BSE-infected sheep: Implications for differential cell targeting and PrP processing. J. Gen. Virol. 2003, 84, 1339–1350. [Google Scholar] [CrossRef]
- Miller, J.M.; Jenny, A.L.; Taylor, W.D.; Marsh, R.F.; Rubenstein, R.; Race, R.E. Immunohistochemical detection of prion protein in sheep with scrapie. J. Vet. Diagn. Investig. 1993, 5, 309–316. [Google Scholar] [CrossRef] [PubMed]
- Debeer, S.O.; Baron, T.G.; Bencsik, A.A. Immunohistochemistry of PRpSC within bovine spongiform encephalopathy brain samples with graded autolysis. J. Histochem. Cytochem. 2001, 49, 1519–1524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaplin, M.; Barlow, N.; Ryder, S.; Simmons, M.; Spencer, Y.; Hughes, R.; Stack, M. Evaluation of the effects of controlled autolysis on the immunodetection of PrPSc by immunoblotting and immunohistochemistry from natural cases of scrapie and bse. Res. Vet. Sci. 2002, 72, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Monleón, E.; Monzón, M.; Hortells, P.; Vargas, A.; Badiola, J.J. Detection of PrP(sc) in samples presenting a very advanced degree of autolysis (BSE liquid state) by immunocytochemistry. J. Histochem. Cytochem. 2003, 51, 15–18. [Google Scholar] [CrossRef] [Green Version]
- Hardt, M.; Baron, T.; Groschup, M. A comparative study of immunohistochemical methods for detecting abnormal prion protein with monoclonal and polyclonal antibodies. J. Comp. Pathol. 2000, 122, 43–53. [Google Scholar] [CrossRef]
- O’Rourke, K.I.; Baszler, T.V.; Miller, J.M.; Spraker, T.R.; Sadler-Riggleman, I.; Knowles, D.P. Monoclonal antibody F89/160.1.5 defines a conserved epitope on the ruminant prion protein. J. Clin. Microbiol. 1998, 36, 1750–1755. [Google Scholar] [CrossRef] [Green Version]
- González, L.; Martin, S.; Begara-McGorum, I.; Hunter, N.; Houston, F.; Simmons, M.; Jeffrey, M. Effects of Agent Strain and Host Genotype on PrP Accumulation in the Brain of Sheep Naturally and Experimentally Affected with Scrapie. J. Comp. Pathol. 2002, 126, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Deslys, J.-P.; Lasmézas, C.; Comoy, E.; Domont, D. Diagnosis of bovine spongiform encephalopathy. Vet. J. 2001, 161, 1–4. [Google Scholar] [CrossRef]
- Meyer, R.K.; McKinley, M.P.; Bowman, K.A.; Braunfeld, M.B.; Barry, R.A.; Prusiner, S.B. Separation and properties of cellular and scrapie prion proteins. Proc. Natl. Acad. Sci. USA 1986, 83, 2310–2314. [Google Scholar] [CrossRef] [Green Version]
- Oesch, B.; Westaway, D.; Wälchli, M.; McKinley, M.P.; Kent, S.B.; Aebersold, R.; Barry, R.A.; Tempst, P.; Teplow, D.B.; Hood, L.E.; et al. A cellular gene encodes scrapie PrP 27-30 protein. Cell 1985, 40, 735–746. [Google Scholar] [CrossRef]
- Madec, J.-Y.; Groschup, M.; Buschmann, A.; Belli, P.; Calavas, D.; Baron, T. Sensitivity of the Western blot detection of prion protein PrPres in natural sheep scrapie. J. Virol. Methods 1998, 75, 169–177. [Google Scholar] [CrossRef]
- Moynagh, J.; Schimmel, H. Tests for BSE evaluated. Bovine spongiform encephalopathy. Nature 1999, 400, 105. [Google Scholar]
- International Reference Laboratory for TSE. Available online: https://science.vla.gov.uk/tseglobalnet/ (accessed on 15 December 2020).
- Grassi, J.; Maillet, S.; Simon, S.; Morel, N. Progress and limits of TSE diagnostic tools. Vet. Res. 2008, 39, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Wilham, J.M.; Orrú, C.D.; Bessen, R.A.; Atarashi, R.; Sano, K.; Race, B.; Meade-White, K.D.; Taubner, L.M.; Timmes, A.; Caughey, B. Rapid end-point quantitation of prion seeding activity with sensitivity comparable to bioassays. PLoS Pathog. 2010, 6, e1001217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mammadova, N.; Greenlee, M.H.W.; Moore, S.J.; Hwang, S.; Lehmkuhl, A.D.; Nicholson, E.M.; Greenlee, J.J. Evaluation of antemortem diagnostic techniques in goats naturally infected with scrapie. Front. Vet. Sci. 2020, 7. [Google Scholar] [CrossRef]
- European Commission. Communication from the Commission to the European Parliament and the Council—The TSE Road Map 2. OIB; European Commission: Brussel, Belgium, 2010. [Google Scholar]
- WHO. Tables on Tissue Infectivity Distribution in Transmissible Spongiform Encephalopathies. 2010. WHO/EMP/QSM/2010.1; WHO Press, World Health Organization: Geneva, Switzerland, 2010. [Google Scholar]
- Bradley, R. Bovine spongiform encephalopathy. Update. Acta Neurobiol. Exp. 2002, 62, 183–195. [Google Scholar]
- Dormont, D. Prion diseases: Pathogenesis and public health concerns. FEBS Lett. 2002, 529, 17–21. [Google Scholar] [CrossRef] [Green Version]
- Heim, D.; Kihm, U. Risk management of transmissible spongiform encephalopathies in Europe. Rev. Sci. Tech. l’OIE 2003, 22, 179–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baylis, M.; Houston, F.; Kao, R.R.; McLean, A.R.; Hunter, N.; Gravenor, M.B. BSE-a wolf in sheep’s clothing? Trends Microbiol. 2002, 10, 563–570. [Google Scholar] [CrossRef]
- Foster, J.D.; Hope, J.; Fraser, H. Transmission of bovine spongiform encephalopathy to sheep and goats. Vet. Rec. 1993, 133, 339–341. [Google Scholar] [CrossRef] [PubMed]
- Houston, E.F.; Gravenor, M.B. Clinical signs in sheep experimentally infected with scrapie and BSE. Vet. Rec. 2003, 152, 333–334. [Google Scholar] [CrossRef]
- Bellworthy, S.J.; Dexter, G.; Stack, M.; Chaplin, M.; Hawkins, S.A.C.; Simmons, M.M.; Jeffrey, M.; Martin, S.; Gonzalez, L.; Hill, P. Natural transmission of BSE between sheep within an experimental flock. Vet. Rec. 2005, 157, 206. [Google Scholar] [CrossRef]
- Bellworthy, S.J.; Dexter, G.; Stack, M.; Chaplin, M.; Hawkins, S.A.C.; Simmons, M.M.; Jeffrey, M.; Martin, S.; Gonzalez, L.; Hill, P. Oral transmission of BSE to VRQ/VRQ sheep in an experimental flock. Vet. Rec. 2008, 162, 130–131. [Google Scholar] [CrossRef]
- Stack, M.; González, L.; Jeffrey, M.; Martin, S.; Macaldowie, C.; Chaplin, M.; Thorne, J.; Sayers, R.; Davis, L.; Bramwell, J.; et al. Three serial passages of bovine spongiform encephalopathy in sheep do not significantly affect discriminatory test results. J. Gen. Virol. 2009, 90, 764–768. [Google Scholar] [CrossRef] [PubMed]
- Eloit, M.; Adjou, K.; Biacabe, A.G.; Beringue, V.; Laude, H.; Le Dur, A.; Vilotte, J.L.; Comoy, E.; Deslys, J.P.; Grassi, J.; et al. BSE agent signatures in a goat. Vet. Rec. 2005, 156, 523–524. [Google Scholar] [CrossRef]
- European Comission. Commission Decision 2002/1003/EC. Commission Decision of 18 December 2002 Laying Down Minimum Requirements for a Survey of Prion Protein Genotypes of Sheep Breeds. Off. J. Eur. Union 2002, 349, 105–107. [Google Scholar]
- Papasavva-Stylianou, P.; Simmons, M.M.; Ortiz-Pelaez, A.; Windl, O.; Spiropoulos, J.; Georgiadou, S. Effect of Polymorphisms at Codon 146 of the Goat PRNP Gene on Susceptibility to Challenge with Classical Scrapie by Different Routes. J. Virol. 2017, 91, e01142-17. [Google Scholar] [CrossRef] [Green Version]
- Langeveld, J.P.M.; Jacobs, J.G.; Erkens, J.H.F.; Bossers, A.; Van Zijderveld, F.G.; Van Keulen, L.J.M. Rapid and discriminatory diagnosis of scrapie and BSE in retro-pharyngeal lymph nodes of sheep. BMC Vet. Res. 2006, 2, 19. [Google Scholar] [CrossRef] [Green Version]
- Houston, F.; Foster, J.; Chong, A.; Hunter, N.; Bostock, C. Transmission of BSE by blood transfusion in sheep. Lancet 2000, 356, 999–1000. [Google Scholar] [CrossRef]
- Houston, F.; McCutcheon, S.; Goldmann, W.; Chong, A.; Foster, J.; Sisó, S.; González, L.; Jeffrey, M.; Hunter, N. Prion diseases are efficiently transmitted by blood transfusion in sheep. Blood 2008, 112, 4739–4745. [Google Scholar] [CrossRef] [Green Version]
- Sisó, S.; Jeffrey, M.; Steele, P.; McGovern, G.; Martín, S.; Finlayson, J.; Chianini, F.; González, L. Occurrence and cellular localization of PrPd in kidneys of scrapie-affected sheep in the absence of inflammation. J. Pathol. 2008, 215, 126–134. [Google Scholar] [CrossRef] [PubMed]
- Murayama, Y.; Yoshioka, M.; Okada, H.; Takata, M.; Yokoyama, T.; Mohri, S. Urinary excretion and blood level of prions in scrapie-infected hamsters. J. Gen. Virol. 2007, 88, 2890–2898. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Romero, D.; Barria, M.A.; Leon, P.; Morales, R.; Soto, C. Detection of infectious prions in urine. FEBS Lett. 2008, 582, 3161–3166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bosque, P.J.; Ryou, C.; Telling, G.; Peretz, D.; Legname, G.; DeArmond, S.J.; Prusiner, S.B. Prions in skeletal muscle. Proc. Natl. Acad. Sci. USA 2002, 99, 3812–3817. [Google Scholar] [CrossRef] [Green Version]
- Thomzig, A.; Schulz-Schaeffer, W.; Kratzel, C.; Mai, J.; Beekes, M. Preclinical deposition of pathological prion protein PrPSc in muscles of hamsters orally exposed to scrapie. J. Clin. Investig. 2004, 113, 1465–1472. [Google Scholar] [CrossRef] [Green Version]
- Andreoletti, O.; Simon, S.; Lacroux, C.; Morel, N.; Tabouret, G.; Chabert, A.; Lugan, S.; Corbière, F.; Ferre, Ṕ.; Foucras, G.; et al. PrPSc accumulation in myocytes from sheep incubating natural scrapie. Nat. Med. 2004, 10, 591–593. [Google Scholar]
- Lacroux, C.; Corbière, F.; Tabouret, G.; Lugan, S.; Costes, P.; Mathey, J.; Delmas, J.M.; Weisbecker, J.L.; Foucras, G.; Cassard, H.; et al. Dynamics and genetics of PrPSc placental accumulation in sheep. J. Gen. Virol. 2007, 88, 1056–1061. [Google Scholar] [CrossRef] [PubMed]
- Mathiason, C.K.; Powers, J.G.; Wild, A.M.; Wolfe, L.L.; Spraker, T.R.; Miller, M.W.; Sigurdson, C.J.; Telling, G.C.; Hoover, A.E.; Dahmes, S.J.; et al. Infectious prions in the saliva and blood of deer with chronic wasting disease. Science 2006, 314, 133–136. [Google Scholar] [CrossRef] [Green Version]
- Mathiason, C.K.; Hays, S.A.; Hoover, E.A.; Powers, J.; Hayes-Klug, J.; Langenberg, J.; Dahmes, S.J.; Osborn, D.A.; Miller, K.V.; Warren, R.J.; et al. Infectious prions in pre-clinical deer and transmission of chronic wasting disease solely by environmental exposure. PLoS ONE 2009, 4, e5916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ligios, C.; Sigurdson, C.J.; Santucciu, C.; Carcassola, G.; Manco, G.; Basagni, M.; Maestrale, C.; Cancedda, M.G.; Madau, L.; Aguzzi, A. PrPSc in mammary glands of sheep affected by scrapie and mastitis. Nat. Med. 2005, 11, 1137–1138. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Animals | ||
| Disease | Host | Origin |
| Classical Scrapie | Sheep and Goat | Infectious |
| Atypical Scrapie | Sheep and Goat | Sporadic/spontaneous |
| Bovine Spongiform Encephalopathy (BSE) | Bovine | Infectious |
| Atypical BSE L or H | Bovine | Sporadic/spontaneous |
| Chronic Wasting Disease (CWD) | Deer | Infectious |
| Feline Spongiform Encephalopathy (FSE) | Feline | Infectious |
| Transmissible Mink Encephalopathy (TME) | Mink | Infectious |
| Exotic Ungulate Encephalopathy (EUE) | Antelope | Infectious |
| TSE in Non-Human Primates | Lemur | Infectious |
| Humans | ||
| Disease | Host | Origin |
| Iatrogenic Creutzfeldt–Jakob Disease (iCJD) | Human | Infectious |
| Sporadic Creutzfeldt–Jakob Disease (sCJD) | Human | Spontaneous |
| Familiar Creutzfeldt–Jakob Disease (fCJD) | Human | Hereditary |
| Variant Creutzfeldt–Jakob Disease (vCJD) | Human | Infectious |
| Gerstmann–Sträussler–Scheinker (GSS) | Human | Hereditary |
| Fatal Familiar Insomnia (FFI) | Human | Hereditary |
| Fatal Sporadic Insomnia (FSI) | Human | Spontaneous |
| Kuru | Human | Cannibalism |
| Variably Protease-Sensitive Prionopathy (VPSPr) | Human | Spontaneous |
| Genotype | Risk |
|---|---|
| PrPARR/PrPARR | R1 |
| PrPARR/PrPAHQ PrPAHQ/PrPAHQ | R2 |
| PrPARR/PrPARQ PrPARR/PrPARH PrPARQ/PrPAHQ PrPAHQ/PrPARH | R3 |
| PrPARH/PrPARH PrPARQ/PrPARH PrPARQ/PrPARQ PrPARR/PrPVRQ PrPAHQ/PrPVRQ | R4 |
| PrPARQ/PrPVRQ PrPARH/PrPVRQ PrPVRQ/PrPVRQ | R5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Acín, C.; Bolea, R.; Monzón, M.; Monleón, E.; Moreno, B.; Filali, H.; Marín, B.; Sola, D.; Betancor, M.; Guijarro, I.M.; et al. Classical and Atypical Scrapie in Sheep and Goats. Review on the Etiology, Genetic Factors, Pathogenesis, Diagnosis, and Control Measures of Both Diseases. Animals 2021, 11, 691. https://doi.org/10.3390/ani11030691
Acín C, Bolea R, Monzón M, Monleón E, Moreno B, Filali H, Marín B, Sola D, Betancor M, Guijarro IM, et al. Classical and Atypical Scrapie in Sheep and Goats. Review on the Etiology, Genetic Factors, Pathogenesis, Diagnosis, and Control Measures of Both Diseases. Animals. 2021; 11(3):691. https://doi.org/10.3390/ani11030691
Chicago/Turabian StyleAcín, Cristina, Rosa Bolea, Marta Monzón, Eva Monleón, Bernardino Moreno, Hicham Filali, Belén Marín, Diego Sola, Marina Betancor, Isabel M. Guijarro, and et al. 2021. "Classical and Atypical Scrapie in Sheep and Goats. Review on the Etiology, Genetic Factors, Pathogenesis, Diagnosis, and Control Measures of Both Diseases" Animals 11, no. 3: 691. https://doi.org/10.3390/ani11030691