Advancements in copy number variation screening in herbivorous livestock genomes and their association with phenotypic traits
 Xiaotong Liu
Xiaotong Liu  Wenting Chen
Wenting Chen  Bingjian Huang
Bingjian Huang  Xinrui Wang Yongdong Peng Xinhao Zhang
Xinrui Wang Yongdong Peng Xinhao Zhang  Wenqiong Chai
Wenqiong Chai  Muhammad Zahoor Khan* Changfa Wang*
Muhammad Zahoor Khan* Changfa Wang*- Liaocheng Research Institute of Donkey High-Efficiency Breeding, Liaocheng University, Liaocheng, China
Copy number variations (CNVs) have garnered increasing attention within the realm of genetics due to their prevalence in human, animal, and plant genomes. These structural genetic variations have demonstrated associations with a broad spectrum of phenotypic diversity, economic traits, environmental adaptations, epidemics, and other essential aspects of both plants and animals. Furthermore, CNVs exhibit extensive sequence variability and encompass a wide array of genomes. The advancement and maturity of microarray and sequencing technologies have catalyzed a surge in research endeavors pertaining to CNVs. This is particularly prominent in the context of livestock breeding, where molecular markers have gained prominence as a valuable tool in comparison to traditional breeding methods. In light of these developments, a contemporary and comprehensive review of existing studies on CNVs becomes imperative. This review serves the purpose of providing a brief elucidation of the fundamental concepts underlying CNVs, their mutational mechanisms, and the diverse array of detection methods employed to identify these structural variations within genomes. Furthermore, it seeks to systematically analyze the recent advancements and findings within the field of CNV research, specifically within the genomes of herbivorous livestock species, including cattle, sheep, horses, and donkeys. The review also highlighted the role of CNVs in shaping various phenotypic traits including growth traits, reproductive traits, pigmentation and disease resistance etc., in herbivorous livestock. The main goal of this review is to furnish readers with an up-to-date compilation of knowledge regarding CNVs in herbivorous livestock genomes. By integrating the latest research findings and insights, it is anticipated that this review will not only offer pertinent information but also stimulate future investigations into the realm of CNVs in livestock. In doing so, it endeavors to contribute to the enhancement of breeding strategies, genomic selection, and the overall improvement of herbivorous livestock production and resistance to diseases.
1 Introduction
China, renowned as one of the earliest nations to engage in livestock domestication (1), has a rich history of nurturing herbivorous livestock, including cattle, sheep, horses, and donkeys. This ancient practice has played a pivotal role in fulfilling diverse human needs, ranging from the procurement of essential animal-derived products such as meat, eggs, milk, and leather to harnessing domesticated animals for laborious tasks (2). Over time, the scope of domestication has expanded to encompass a multitude of applications. Throughout this evolutionary process, natural and artificial selection mechanisms have yielded an array of domestic animal breeds characterized by varying traits (3), including phenotypic attributes, economic characteristics, environmental adaptability, and resistance to diseases. Nonetheless, the intricate genetic underpinnings responsible for these disparities remain incompletely elucidated.
In recent years, the exploration of genomic variation has emerged as a central focus of scientific inquiry in the fields of animal production and health regulation, as evidenced by numerous studies (4–10). This emphasis on genetic variation holds significant significance in our quest to comprehend the intricate interplay between genetic diversity and a wide array of phenotypic and economic traits exhibited by animals (11–13). Furthermore, it serves as a robust theoretical foundation for elucidating genetic mechanisms and advancing the field of molecular breeding. Since the introduction of genomic selection, a range of livestock species, including sheep, goats, cattle, and horses, have undergone genotyping to assess their suitability for important economic traits, as demonstrated by previous studies (14–16). Up to this point, single nucleotide polymorphisms (SNPs) have been the primary focus of genomic research within the animal breeding community (17, 18). Significant strides have been made in establishing a solid genetic foundation for enhancing production and disease resistance in animals (18, 19). However, it is worth noting that despite these advancements, ~25% of the identified copy number variants (CNVs) exhibit no significant linkage disequilibrium with any SNP, leading to the conclusion that CNVs harbor genetic information that cannot be solely elucidated through SNP analysis (20).
CNVs are heritable chromosomal structural variations, characterized by deletions or insertions exceeding 50 base pairs (21). Notably, CNVs encompass a larger proportion of the genome compared to SNPs (22, 23). Consequently, CNVs are being proposed as an additional reservoir of information to elucidate the genetic variance underlying complex traits that may not be fully accounted for by SNPs alone (20). To date, several methodologies have been commonly employed for CNV detection, including comparative genome hybridization, extracting CNV data from SNP arrays, and whole-genome sequencing (WGS) approaches (24–26). Notably, recent research endeavors have delved into the investigation of the association between CNV in specific genes and a variety of phenotypic traits in animals, including growth characteristics in cattle (27, 28), goats (29–31), sheep (32) and horses (33–38). These studies have also extended their focus to examine the link between CNVs and other vital phenotypes, such as reproduction traits (39, 40) and disease resistance (7, 41). These studies have unveiled CNV as a key player linked to diverse facets of phenotypic diversity and economic traits in animal realms.
The confluence of two pivotal trends, the rising prevalence of molecular markers in livestock breeding and the maturation of microarray and sequencing technologies, necessitates a contemporary and comprehensive review of the burgeoning body of research on CNVs. This review paper seeks to illuminate the intrinsic value and biological ramifications of CNV in the landscape of genetic variation, with a particular focus on its potential as a potent molecular marker in the realm of livestock breeding. Through this approach, our review aims to introduce fresh viewpoints regarding genetic diversity and molecular breeding. Thus, in current review, we have focused on the progress of CNV screening methods in genomes of various herbivorous livestock including cattle, horses, donkeys, sheep and goat. In addition, we have briefly evaluated the association of CNVs in genes with different phenotypic traits including growth traits, reproductive traits, pigmentation, disease resistance, and environmental adaptability, etc., in cattle, horses, sheep and goat. The overall progress in screening CNVs within livestock genomes is summarized in Figure 1 (32–35, 37, 42–70).
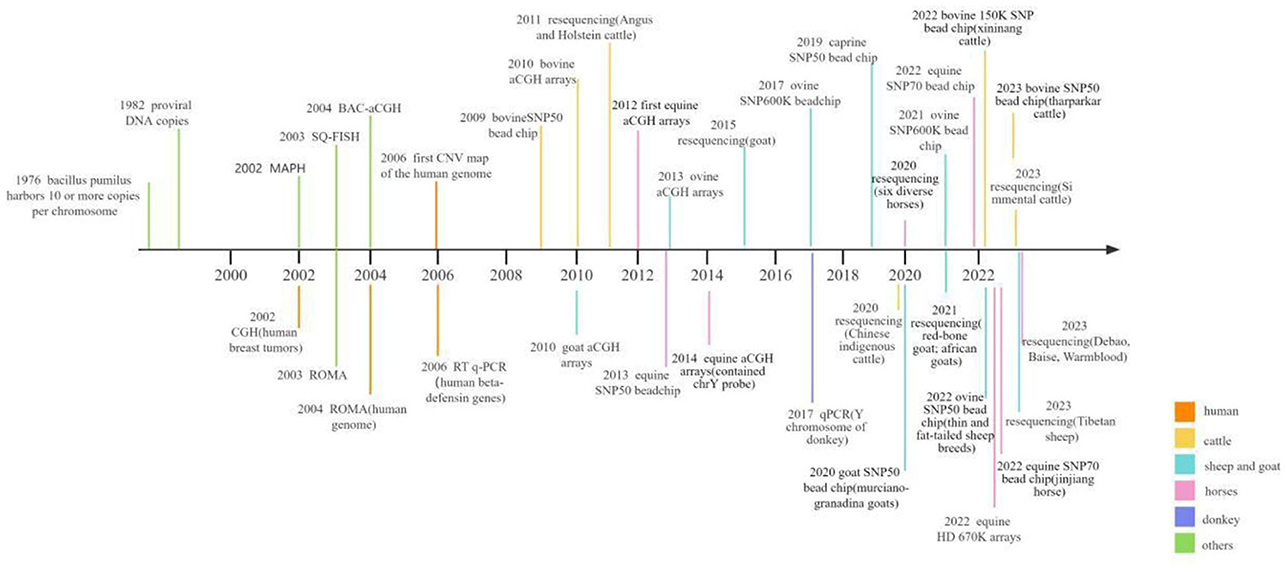
Figure 1. Graphical presentation of the research progress on CNV screening in herbivorous livestock and humans.
2 Overview of CNV biology
2.1 CNV definition
A CNV, typically resulting from genome rearrangement, refers to the amplification or reduction in the copy number of a large genome segment of 1 kb or greater in length, primarily demonstrated via sub-microscopic deletions and duplications (50, 71). The common variant forms of CNV are illustrated in Figure 2 (72).
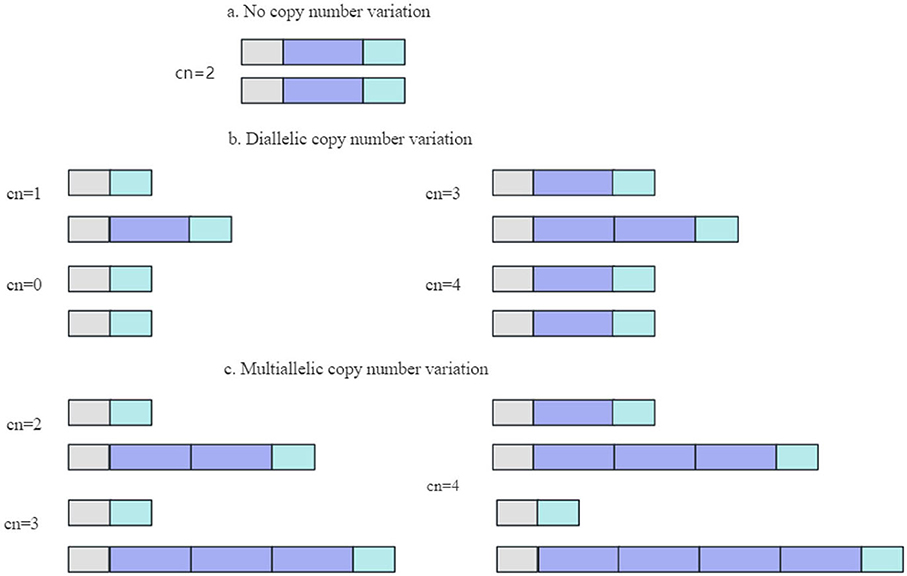
Figure 2. Common types of CNV.
Several nearby CNVs and partially overlapping CNVs in the same genomic region can be merged into a single CNV segment (73). CNVs play a vital role in genomic structural variation (SV) (74). The number of base pairs regulated by CNVs is over five times greater than the number regulated by single nucleotide variants (SNVs) in each individual. Each CNV is correlated more than three times with one genome-wide association signal and fifty times with expression quantitative trait loci (eQTL) compared to SNVs (75). Furthermore, CNVs have been recognized for its significant influence on the evolution of phenotypic diversity, disease resistance, and evolutionary processes in organisms (76, 77).
It is widely recognized that CNVs are common in the genomes of plants, animals, and humans (78), but there is no final conclusion on the mechanism of CNV formation. According to existing studies, three primary formation mechanisms may be involved: (1) Non-allelic homologous Recombination, NAHR: Rearrangements occurring between homologous chromosomes of an individual's genomic DNA during meiosis can lead to duplications, deletions, inversions, and translocations (79, 80). (2) Non-homologous end-joining, NHEJ: A mechanism of genomic rearrangement that occurs during the period of DNA double-strand break repair and that can result in numerous simple CNVs (79, 80). (3) Replication fork smiling and template Switching, FoSTeS: Refers to the stalling of DNA replication forks which causes the lagging strand to break away from the DNA template and switch to other replication forks to continue DNA replication synthesis. This can lead to DNA duplications or deletions and the emergence of a significant number of CNVs. Consequently, the formation of complex structural CNVs may be attributed to this phenomenon (80, 81).
2.2 CNV detection methods
2.2.1 Chip technology
Array comparative genomic hybridization (aCGH) has proven to be an efficient technique for the identification of CNVs at a genome-wide level (82–84). The fundamental principle of CGH involves labeling the DNA under analysis and control DNA with distinct fluorescent dyes. Subsequently, these treated DNAs are hybridized to standard chromosomes, and a digital fluorescence imaging system is employed to scan them, with a priority on fluorescence intensity ratios (85, 86). The aCGH is a microarray technology that utilizes two types of microarrays depending on the probes used during fabrication: Bacterial Artificial Chromosome CGH microarrays and Oligonucleotide Probe CGH microarrays. These aCGH microarray probes cover the entire genome and exhibit exceptional sensitivity, accuracy, and resolution, facilitating high-throughput screening. Previous research has provided robust support for the credibility of the experimental data generated by CGH (87, 88).
Single nucleotide polymorphism (SNP) microarray technology represents another effective method for detecting CNVs. SNP microarray technology requires only a single hybridization, thus obviating the need for the simultaneous double hybridization of two DNA samples with probes. It determines the genomic copy equivalents of each locus by comparing variations in signal intensities between the samples being tested (89). A SNP microarray demonstrates remarkable stability and high resolution, enabling the detection of diverse forms of CNVs, including sub-microscopic deletions, duplications, and more (90). In comparison to Comparative Genomic Hybridization, SNP microarrays offer the advantage of simultaneously detecting CNVs while determining their genotypes and revealing heterozygous deletions. This approach is not only more cost-effective but also facilitates large-scale CNV testing (91). Several software and programs are currently available for CNV detection using chip technology, including CNVPartition, PennCNV, and QuantiSNP. A study revealed through genome sequencing of horses that the optimal order of performance for the three assays was CNVPartition, PennCNV, and QuantiSNP (92). Furthermore, the combination of PennCNV and QuantiSNP exhibited improved accuracy in CNV detection.
2.2.2 Sequencing technology
As gene sequencing technology has matured, numerous tools and software have been developed to enhance the efficiency and precision of copy number variant detection. Next-generation sequencing (NGS) has emerged as the most commonly used method for detecting CNVs in recent years (93), with Illumina's Solexa/HiSeq technology being a prominent representative. Second-generation sequencing technology aims to synthesize and sequence DNA simultaneously. Fluorescent signals are excited by lasers and recorded using optical equipment. These recorded signals are then converted into bases using computer technology. Second-generation sequencing methods are not limited to target sequences hybridized with primer probes and can identify genome-wide CNVs, structural variants, and other variations (94). In the current landscape, four primary strategies and methods are employed for detecting variants in NGS data: paired-end mapping (PEM), split-read (SR), read depth (RD), and de novo assembly (AS), in addition to a combined approach based on the above four methods. Compared to microarray-based methods, next-generation sequencing technology offers advantages in terms of speed, resolution, cost-effectiveness, and reproducibility (95–97). Furthermore, widely used sequencing technologies for identifying CNVs, software including CNVnator, CNVpytor, CNVcaller, and ERDS have been documented.
3 Overview of screening methods for CNVs in herbivorous livestock genomes
3.1 Cattle genome
Cattle ranching represent a promising industry with substantial economic, ecological, and social implications. The diversity of cattle breeds, including Holstein for dairy production, Charolais for beef, and regionally adapted yaks and buffaloes, caters to a wide range of human needs. CNVs assume significant importance in the exploration of phenotypic traits and adaptation to various environments, shedding light on the domestication origins of these animals. As dietary preferences evolve toward beef with higher protein and lower fat content, copy number variation regions (CNVRs) have been found to be associated with cattle carcass traits, offering theoretical support for future breeding endeavors. In recent years, numerous investigations have identified CNVs within the bovine genome. Table 1 provides a comprehensive summary of some of these studies. Consistently, a study identified a total of 79 CNV loci across six distinct cattle breeds, employing the BovineSNP50 BeadChip (51). Notably, 10 CNVs were found to overlap with those previously identified through aCGH data. Similarly another study conducted an extensive genome-wide analysis of CNVs in 90 modern domesticated cattle, uncovering over 200 potential CNVRs (52). In addition, this high-quality bovine CNV map fills critical gaps in current genome-wide association and selection studies based on SNP genotyping. Previous studies generated genome-wide CNV maps for cattle using the BovineSNP50 BeadChip and aCGH arrays, respectively (22, 98). Similarly, Kumar et al. (69) pioneered CNV detection in Indian Tharparkar cattle, identifying a total of 8881 CNVs, which were subsequently filtered down to 693 CNVs and merged into 447 CNVRs, representing ~2.17% of the cattle genome. Utilizing a purebred Angus cow as a reference, a study documented 605 CNVRs through a genome-wide analysis of CNVs in comparative genomic crossbreeding arrays of 29 Chinese domestic bulls (100). Detailed distribution maps of these CNVRs were constructed on their respective genomes. In a consistent manner, Kooverjee et al. (103) conducted a comprehensive investigation, wherein they successfully identified a total of 355 CNVRs in a cohort of five crossbred cows. This identification was accomplished through the utilization of the Panelcn. MOPS software. Notably, these CNVRs exhibited an average length of 9318 base pairs, collectively representing ~0.15% coverage of the bovine genome. Previous studies employed different platforms for genotyping Holstein cattle, followed by CNV analysis, to investigate the impact of genotype array density on CNV detection, thereby contributing to our understanding of genetic variation in Holstein cattle (24, 26). Accordingly, Sun et al. (104) harnessed sequencing technology to sequence the entire genome of Simmental bulls, detecting 2944 CNVRs, which were subsequently subject to genetic analysis, revealing associations with reproduction, immunity, and fertility. These findings constitute a valuable molecular breeding resource for cattle. A study analyzed common CNV regions in Xinjiang brown cattle and compared differences between the ARS and UMD reference genomes, suggesting the ARS reference genome's superior effectiveness in CNV detection (102). Likewise, previously studies conducted on CNVs in the bovine genome utilizing the BovineHD Genotyping BeadChip (38, 102). Conversely, studies undertook an analysis of CNVs in the bovine genome through sequencing technologies (23, 63, 99, 105).
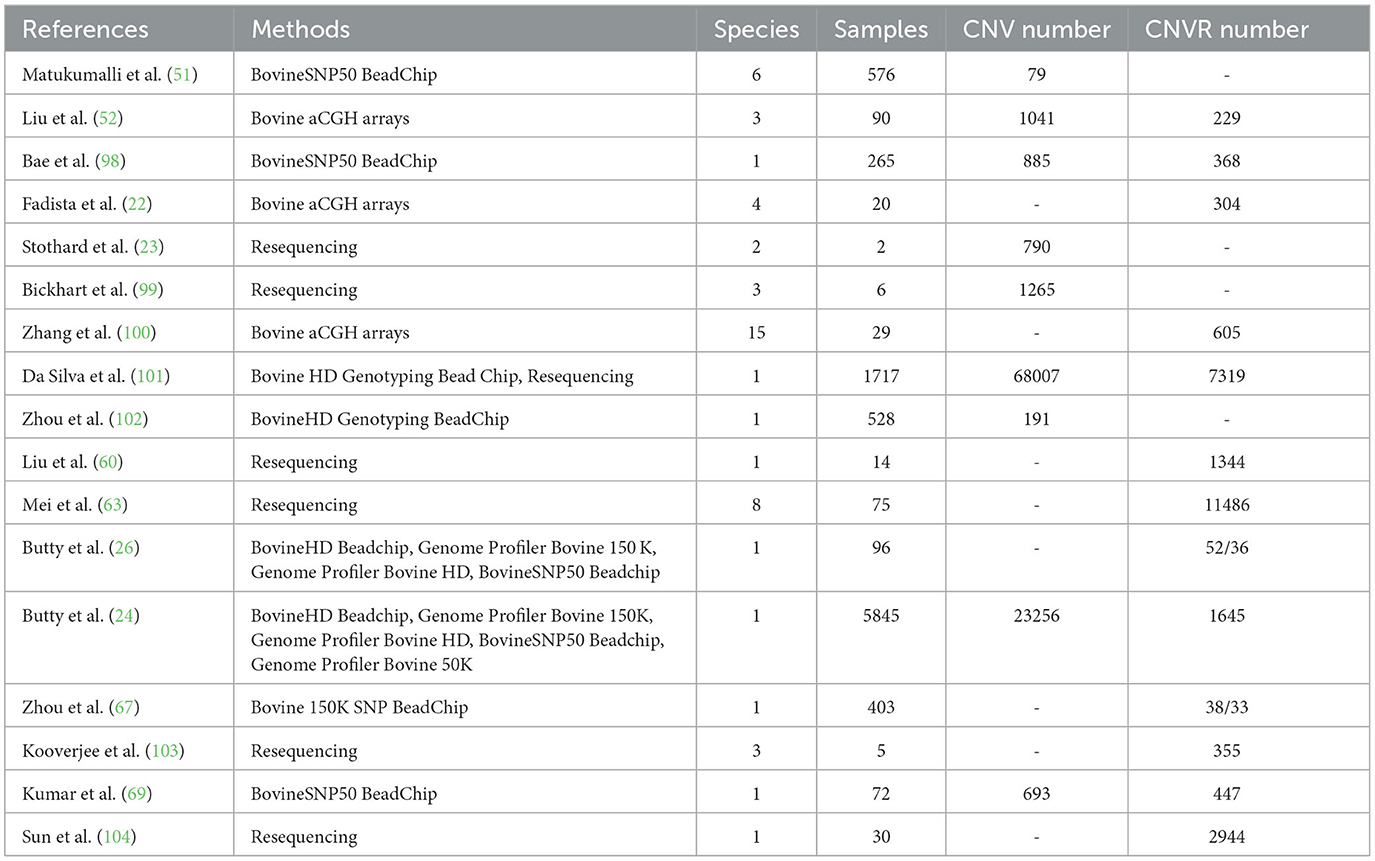
Table 1. Summary of CNVs and identification methods performed in cattle.
3.2 Sheep and goat genomes
Sheep products, encompassing mutton meat, milk, wool, and cashmere, are of considerable significance within the sheep industry. They not only enhance the quality of life for individuals but also contribute substantially to industrial development, augment the incomes of farmers and herdsmen, and provide high-quality fertilizers for farmland, among other advantages. Genomic selection technology has emerged as a pivotal approach in sheep breeding. Current research has unveiled associations between CNVs and various traits in sheep, including growth characteristics, wool color, cashmere quality, disease resistance, and reproduction.
Numerous studies focused on mapping CNVs within the sheep genome have significantly enriched our understanding of genomic variations in sheep. Table 2 provides a comprehensive summary of research pertaining to CNVs in the sheep genome. A study identified a total of 238 CNVRs and establishing a CNV map in the genomes of three distinct sheep breeds using the sheep SNP50K microarray (54). Consequently, Ma et al. (106) investigated CNVs in the genomes of eight sheep breeds using the sheep SNP50K microarray, identifying 111 CNV regions from 160 sheep and mapping the distribution of CNVRs across autosomal chromosomes. Consistently, total of 13,347 CNVs based on sequencing data from six domesticated goats and two wild goats were detected (56). While another experimental trial employed high-density sheep SNP microarrays to identify 371, 301, and 66 autosomal CNVRs within the genomes of big-tailed frigid sheep, Altai sheep, and Tibetan sheep, respectively (107). This endeavor resulted in the creation of the first high-resolution sheep CNV map, offering a valuable resource for comprehending genomic variation in sheep. In continuity, Yang et al. (108) identified 24,558 CNVs from 2,254 sheep across various geographic regions worldwide, culminating in 619 CNV regions with a combined length of 197 megabases (Mb). This encompasses 6.9% of the sheep genome and establishes a comprehensive CNV map that can assist in genome annotation for sheep. In addition, Igoshin et al. (111) detected 4,527 CNVs among 354 sheep representing 16 Russian indigenous breeds. Gene function enrichment analysis revealed significant impacts of CNVs on olfactory perception and immunity within 12 of the breeds. In line, Taghizadeh et al. (68) identified 328 and 187 CNVRs in fat-tailed and thin-tailed sheep breeds, respectively. These CNVRs were found to be located within or overlapping with 790 known sheep genes, covering ~73.85 Mb of the sheep genome. Previous report conducted a genome-wide analysis on 48 beach sheep, documented 1,296 CNV regions and constructed a CNVs map of the Tan sheep genome, thereby complemented the data on CNVs in the Chinese sheep genome (59). While another study identified 6,286 CNVs in a total of 1,023 sheep representing 50 different breeds worldwide, employing the pennCNV tool. The results unveiled differences in CNVs among populations across different geographic regions (60). Furthermore, a study reported 42 CNVs from 120 samples representing five dairy goat breeds and established significant associations between two CNVs (CNV5 and CNV25) and two milk production traits (109). Consequently, Guan et al. (62) detected 1,461 regions of CNV within the Spanish dairy goat genome, with an average length of 196.89 kilobases (kb). The total length of all CNV regions accounted for 3.9% of the autosomal genome, leading to the creation of a CNV map. Previous study has identified 127 CNVRs in four breeds of goats, covering ~11.39 Mb of the bovine genome, thereby establishing the first CNVR map for goats (70). In addition, a study reported 1,217 CNVRs in 67 sheep breeds worldwide (66). Furthermore, a study identified 4,769 high-quality CNVRs in 47 sheep breeds globally, subsequently generating CNV maps. Additionally, they investigated the influence of solar radiation on CNVs within sheep genomes (66). Consistently, a study employed resequencing to detect CNVs in two Tibetan sheep breeds, ultimately identifying 368 distinct CNVRs, which may contribute to determining population disparities (32). Yuan and co-authors were the first to utilize resequencing technology to establish a CNV map of Chinese fine-wool sheep and analyzed the overlap of CNVRs with several quantitative trait loci related to economic traits, providing vital insights for the future improvement of fine-wool sheep (110). Consistently previous studies employed resequencing to analyze the genomic CNVs of Mile red-boned goats and African goats, respectively (64, 65). These findings facilitated a deeper understanding of the genetic traits of these animals. Consequently, another study identified a total of 702 CNVs in 120 dairy goats, resulting in the creation of a CNVR map, which promises to be beneficial for further research on the association between CNVs and phenotypic variations (13).
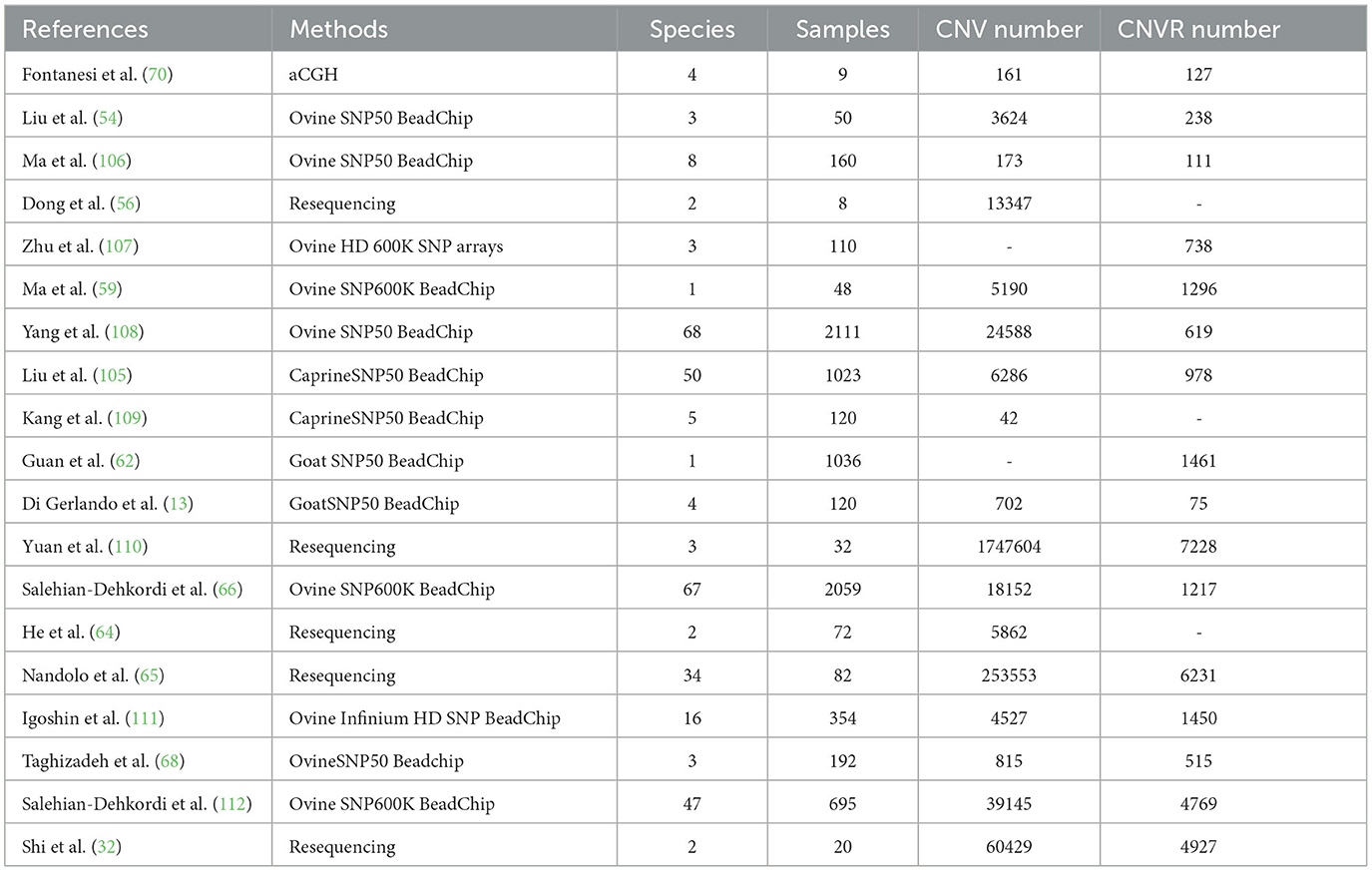
Table 2. Summary of CNVs and identification methods performed in sheep and goat.
3.3 Horses genome
China's horse industry has experienced remarkable growth owing to the country's rapid social and economic development, coupled with continuous improvements in living standards. Consequently, equestrian sports have gained significant popularity as leisure and recreational activities within the nation. The maturation of microarray and sequencing technologies has played a pivotal role in the identification of equine CNVs. These CNVs are of paramount importance for the study of equine trait variations, disease prevention and treatment, exploration of genetic diversity, tracing the origins of domestication, and the development of new equine breeds.
In recent years, the substantial progress in microarray and sequencing technologies has facilitated the detection of CNVs. Researchers from both domestic and international institutions have undertaken extensive investigations into equine genome CNV identification, with select research findings summarized in Table 3. Doan et al. (53) were the pioneers in reporting CNVs within equine genomes. They identified 775 CNV regions in 16 horses using an aCGH microarray, demonstrating the significant impact of CNVs on biological phenotypic diversity. Subsequently, in an effort to detect CNVs in both normal horses and Przewalski horses, Ghosh et al. (55) prepared an aCGH microarray that also included a Y chromosome probe. They successfully detected 258 CNVRs in autosomes, chrX, and chrUn, but none were found in chrY. Notably, the majority of these CNVRs were associated with genes related to sensory perception, the immune system, and reproduction (55). Previous study meticulously described the mapping of CNVs in Chinese horses using high-resolution array Comparative Genomic Hybridization (aCGH), a highly effective method for genome-wide CNV detection in animals (114). Consequently, Kader et al. (57) through whole-genome analysis of CNVs in 96 horses representing three Chinese breeds: Debo Shorthorn, Mongolian horse, and Yili horse. Their work identified a total of 287 CNVs, which were combined to form 122 CNV regions (CNVRs) with sizes ranging from 199 base pairs to 2,344 kilobases. Consistently, a study reported 15,041 CNVs and 5,350 CNVRs in 222 Friesian horses, creating a distribution map of CNVRs within the equine genome (115). Similarly, a recent study performed an analysis of CNV in 469 horses from four Korean breeds, uncovering 843 CNVRs that overlapped with 7.2% of the reference genome for horses (33). Furthermore, they constructed an autosomal map of CNVRs in horses. In addition, Laseca et al. (34) analyzed high-density SNP genotyping data from 654 horses, identifying a total of 19,902 CNV segments and 1,007 CNV regions, with CNVs covering 4.4% of the equine genome.
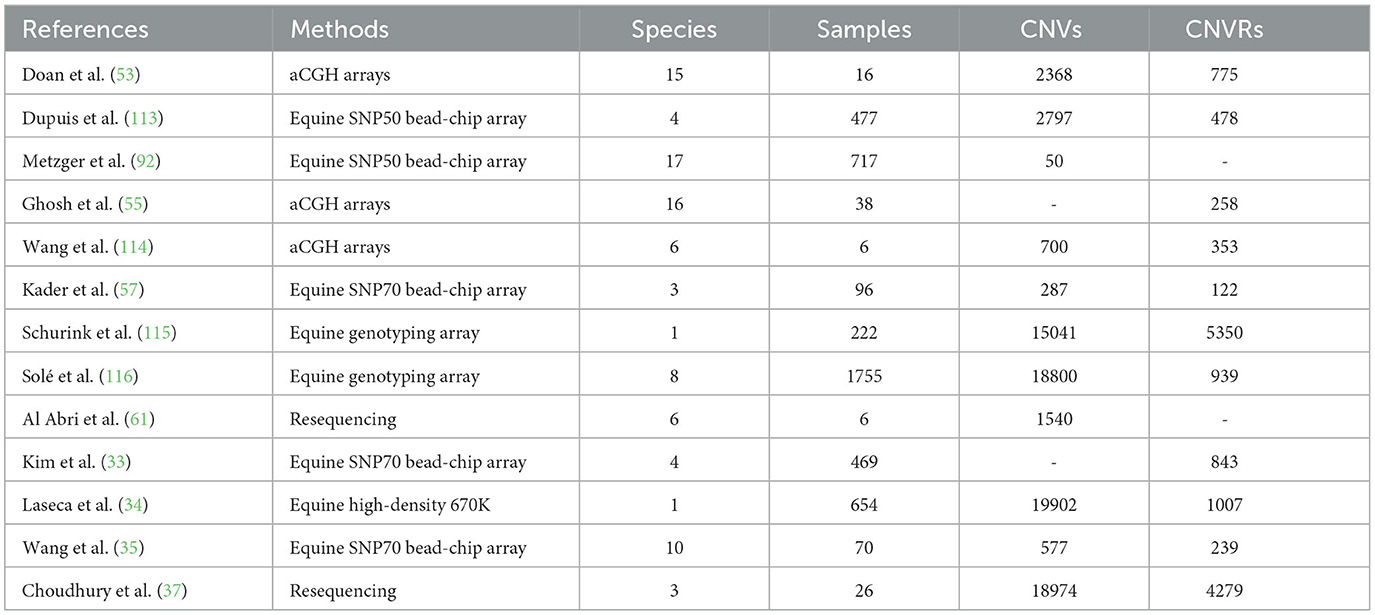
Table 3. Summary of CNVs and identification methods performed in horses.
A study utilized gene chips to identify CNVs in 1,755 horses representing eight breeds. Their findings revealed that the size of CNV regions varied from 1 kilobase to 21.3 megabases (116). Consequently a study revealed an average total of 1,540 CNVs per horse through whole-genome resequencing of six horses representing six different breeds. Their results suggested that a reduction in the number of LATH copies might be linked to the development of endurance in horses (61). While a recent study by Choudhury et al. (37) leveraged resequencing data for Debao (DB), Baise (BS), and Warmblood (WB) horses to identify CNVs and create a CNVR map of the equine genome. Their research indicated that differential CNVRs may influence the phenotypic characteristics of different breeds.
3.4 Donkey genome
The donkey industry has emerged as a significant contributor to the growth of the livestock sector in recent years. However, the detection of CNVs within the genetic variations of the donkey genome, both in China and internationally, remains a relatively understudied area. In a study conducted by Han, a cohort comprising 263 native Chinese donkeys representing 13 breeds from eight provinces and regions was employed to identify CNVs in five Y chromosome genes of donkeys (CUL4BY, ETSTY1, ETSTY4, ETSTY5, and SRY) through quantitative Polymerase Chain Reaction (qPCR) analysis (58). While studies related to Single Nucleotide Polymorphisms (SNPs) have been reported, studies focusing on CNVs in donkeys are limited. Consistently, another study detected ~7 million SNPs in 126 domestic donkeys and made a noteworthy discovery that black or chestnut coat color was attributed to a 1 base pair deletion downstream of the TBX3 gene (117). This deletion led to reduced gene expression and its inhibitory effect on pigmentation. In addition to SNPs, CNVs represent crucial genetic resources. They possess distinct advantages, including their ubiquitous presence in the genome, extensive coverage, and relative ease of detection compared to SNPs (50, 118). The continued advancement of sequencing technology promises to greatly facilitate the identification of CNVs within the donkey genome.
The subsequent section offers insights into potential reasons why CNVs have been relatively overlooked in donkey research:
1. Donkeys predominantly serve as working animals in many parts of the world. Consequently, researchers may prioritize the study of genetic markers associated with phenotype traits in species other than donkeys.
2. Initially, copy number variation was primarily detected using gene chips, which were less readily available for donkeys compared to other livestock species such as cattle, sheep, and horses. In recent years, the development of sequencing technology and the completion of whole-genome sequencing for donkeys have created favorable conditions for the detection of CNVs within the donkey genome.
3. Funding for research pertaining to donkeys may be comparatively constrained when compared to other livestock species like cattle and sheep. Consequently, researchers may face challenges in securing adequate resources for the study of CNVs in donkeys.
4. In many parts of the world, the commercial value of donkeys is primarily linked to their performance as working animals, rather than their utility for products such as milk or meat. As a result, there may be limited commercial interest in investigating CNVs in donkeys.
4 Gene ontology analysis for genes overlapping CNVRs
To identify genes that may be influenced by CNVRs within the genome, an annotation analysis of genes associated with CNVRs revealed that olfactory-related functions, specifically olfactory transduction and olfactory receptor activity, were frequently affected. This observation aligns with previous findings in various species, including cattle (119), sheep (62, 111), horses (34, 115, 120) and humans (121–123), where genes affected by copy number variation have shown enrichment for olfactory-related functions. In the case of humans, the sense of smell is considered a minor aspect of overall health and may not be closely linked with adaptation. Consequently, human olfactory receptor (OR) genes tend to evolve neutrally (121). However, in the animal kingdom, the sense of smell holds paramount importance as it plays a vital role in locating food, identifying harmful substances, avoiding predators, selecting mates, and ensuring long and healthy survival and reproduction (124). Consistently, a study proposed that olfactory receptors also play a role in appetite regulation and feeding efficiency in mammals. Hence, alterations in these receptors may lead to individual differences in feed intake, body weight, and body composition (125). Additionally, CNVRs have been found to intersect with Quantitative Trait Loci (QTL) associated with various factors such as morphology, disease resistance, and more. This intersection serves as a fundamental foundation for the examination of phenotypic diversity. Variations in CNVR frequency among different breeds have been identified in CNVR tests involving animals from various regions (52, 111, 126). These variations can be attributed to breed domestication and environmental adaptation.
5 CNVs and their association with phenotypic traits in herbivorous livestock
It has been well-established that genomic CNVs exert an impact on an organism's phenotype through various mechanisms, including changes in gene dosage, modulation of gene expression, modulation of gene transcriptional regulators, and positional effects (127). The association of CNVs in genes and their association with various phenotypic traits (growth traits, reproductive traits, pigmentation, and diseases resistance) in herbivorous livestock (cattle, sheep, goat, and horses) have been summarized in Table 4.
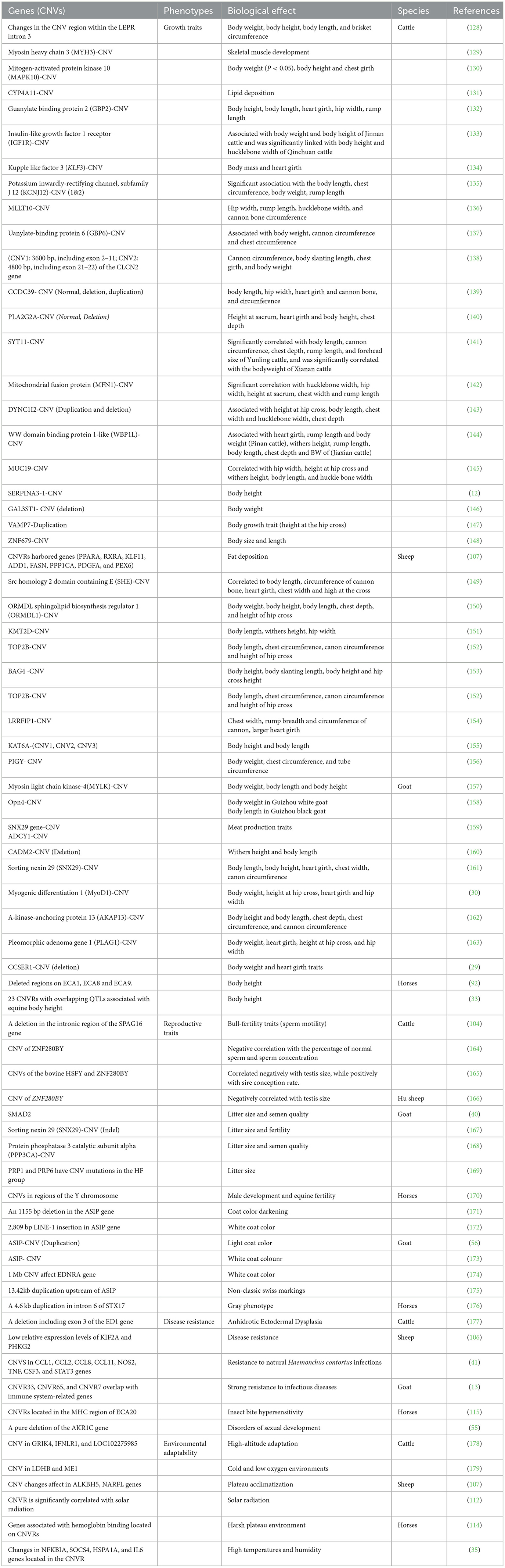
Table 4. Summary of CNVs in genes and their association with phenotypic traits in herbivorous livestock.
5.1 CNVs associated with growth and reproductive traits in herbivorous livestock
CNVs have been extensively studied in various livestock species, including cattle, sheep, donkey and horses, and its association with important growth, reproduction, and fertility traits has been documented. This comprehensive review discusses key findings and contributions from various research studies in these livestock species. In cattle, Yang et al. (131) identified CNV in the cytochrome P-450 4A11 (CYP4A11) gene, which was associated with increased growth. Multiple copies of CYP4A11 were found to promote the differentiation of 3T3-L1 cells into adipocytes, potentially leading to increased fat deposition. Previous studies investigated SERPINA3-1 and GAL3ST1 gene CNVs in different Chinese cattle breeds, revealed associations with growth traits such as body height, body weight, and rump width (146, 180). These genes hold promise as candidate genes for Chinese cattle breeding. Correspondingly, Hu et al. (139) analyzed CCDC39 gene CNVs and their impact on body length and hip width, noting significant effects, particularly in the Pinan (PN) breed. Additionally, Shi et al. (128) found a correlation between leptin gene CNV and various phenotypic traits, including body weight, body height, body length, and brisket circumference, in multiple cattle breeds.
In sheep, Zhu et al. (107) identified adiposity-related genes, including PPARA, RXRA, KLF11, ADD1, FASN, PPP1CA, and PDGFA, in CNV regions of fat-tailed sheep. These genes were associated with fat deposition, with individuals carrying copy number deletions exhibiting higher body weight. Similarly, a recent study by Wang et al. (155) documented a significant correlation between CNV in the KAT6A gene and body height and hip width in Hu sheep (HU). They also found that CNV3 duplicates were associated with higher body height and weight. Yang et al. (153) highlighted the BAG4 gene's role in regulating body height in sheep and its potential as a molecular marker for molecular breeding. Feng et al. (156) discovered that CNVs of the PIGY gene significantly impacted body weight, chest circumference, and tube circumference in sheep. Additionally, Xu et al. (29) found that CNV types of the CCSER1 gene were correlated with body weight and heart girth traits in Guizhou white goats (GZW).
Horses were also subject to CNV studies. Consistently, Metzger et al. (92) conducted a genome-wide analysis of CNVs and their association with equine body height traits. They identified deleted regions in ECA1, ECA8, and ECA9, which were significantly linked to equine body height. While Kim et al. (33) reported CNVRs with overlapping quantitative trait loci (QTLs) associated with equine body height in Jeju riding horses and Hanra horses. These findings provided valuable insights into the genetic factors influencing equine body height.
In the context of fertility, CNVs have also been explored. Consistently, a deletion in the intronic region of the SPAG16 gene has been identified in bulls with poor sperm motility (PSM), suggesting its potential role in bull fertility (104). A comprehensive analysis documented CNVs in Laoshan dairy goat populations with differing fertility levels and showed that CNV mutations in PRP1 and PRP6 genes, which affect mammalian fertility (169). A study investigated CNVs in Y chromosome-specific regions in male horses, identifying potential genes linked to stallion fertility and contributing to our understanding of equine male development and fertility (170). These studies collectively underscore the significance of CNVs in shaping various traits in cattle, sheep, and horses, from growth and fat deposition to fertility. The identification of specific genes and regions associated with these traits holds promise for selective breeding programs and further genetic research in these livestock species.
5.2 Pigmentation
The role of CNVs in determining coat color and disease resistance in cattle, horses, and sheep has been the subject of extensive scientific investigation. In this discussion, we will delve into various studies that have shed light on the influence of CNVs on these traits in these livestock species. A study reported two sequence translocations between chromosomes 6 and 29 in Belgian Blue and Swiss Brown cattle, affecting the KIT gene, leading to color-sidedness (181). Similarly another study reported that a deletion of 1155 bp within the ASIP gene in Nellore cattle results in dark hair color in specific regions by elevating melanin production (171). Furthermore, a 2,809 bp LINE-1 insertion in the ASIP gene icausing a white coat color phenotype by impeding melanin production has been identified in buffalo (172). Consequently a study postulated that a 4.6 kb duplication within intron 6 of the STX17 gene leads to a pronounced upregulation of both STX17 and NR4A3 gene expression (176). This heightened gene expression subsequently instigates the proliferation of melanocytes, ultimately culminating in the manifestation of a gray coat phenotype in affected horses. Furthermore, it is noteworthy that horses harboring this mutant phenotype exhibit a gradual transition in hair color from gray to white as they advance in age. In a similar vein, previous investigations encompassed genomic analyses of feral and domestic goat populations, which unveiled the intriguing revelation that 13 genes situated within CNV regions overlap with the comprehensive roster of cloned color genes provided by the European Society for Pigment Cell Research (ESPCR) (56). Moreover, these investigations substantiated the substantial impact of CNVs within the ASIP gene on the lightening of coat color in domestic goat breeds, employing rigorous resequencing analyses. A subsequent inquiry brought to light the potential influence of CNVs on the ASIP gene, which may, in turn, lead to the emergence of a white coat color phenotype in the Girgentana and Saanen goat breeds, as corroborated by Fontanesi et al. (173). A recent report presented compelling evidence demonstrating the significant association between a 13.42 kb repeat sequence located upstream of the ASIP gene and non-classic Swiss markings in goats, utilizing CNV assays and quantitative Polymerase Chain Reaction (qPCR) techniques (175). This finding further underscores the pivotal role of CNVs in shaping coat color patterns in goats. Furthermore, Menzi et al. (174) unraveled the involvement of the EDNRA gene, situated within a 1 Mb CNV region on chromosome 17, in potentially attenuating melanism among Boer goats. Notably, an elevation in the copy number within this CNV region could potentially lead to a reduction in skin pigmentation, thereby culminating in the manifestation of a white-spotted phenotype. In summary, these comprehensive investigations collectively shed light on the intricate interplay between CNVs and the genetic determinants of coat color diversity in horses and goats, providing valuable insights into the underlying genetic mechanisms governing these phenotypic variations.
5.3 Disease resistance
The CNVs hold significant potential to influence disease resistance in livestock species, including cattle, sheep, and horses. Notably, Liu et al. (52) conducted an exhaustive investigation in cattle, uncovering multiple CNVs that play pivotal roles in crucial biological processes such as drug detoxification, innate and adaptive immunity, as well as receptor and signal recognition. In the context of cattle, it is worth highlighting the association between CNVs and disease susceptibility. For instance, Drögemüller et al. (177) reported that a deletion encompassing exon 3 of the ED1 gene has been linked to anhidrotic ectodermal dysplasia, underscoring the critical role of CNVs in disease vulnerability. Furthermore, a study reported CNVs in 18 candidate genes (TERT, NOTCH1, SLC6A3, CLPTM1L, PPARα, BCL-2, ABO, VAV2, CACNA1S, TRAF2, RELA, ELF3, DBH, CDK5, NF2, FASN, EWSR1 and MAP3K11) which were associated with milk somatic cells count and mastitis resistance in dairy cattle (182). Consistently, another study reported that CNVs in ZNF496 and NLRP3 were significantly associated with resistance to gastrointestinal nematodes in Angus cattle (129).
Shifting our focus to small ruminants, Di Gerlando et al. (13) embarked on an exploration of Sicilian goat breeds. Their study unveiled intriguing findings, as CNVR33, CNVR65, and CNVR7 were found to overlap with genes closely associated with the immune system. This discovery offers a potential explanation for the remarkable resistance of these goat breeds against infectious diseases. In the case of Florida Native sheep, Estrada-Reyes et al. (41) delved into the genetic underpinnings of resistance against gastrointestinal nematodes. Their meticulous investigation revealed that 14 CNVs exhibited overlaps with QTLs associated with gastrointestinal nematode resistance. Moreover, these CNVs demonstrated significant correlations with fecal egg count (FEC), suggesting a potential influence of CNVs on parasite resistance in these sheep. Ma et al. (106) conducted an extensive study encompassing 160 Chinese sheep breeds, leading to the identification of 111 CNV regions. Their functional analysis highlighted an enrichment of CNV regions with genes closely linked to environmental responses. Notably, 17 candidates genes emerged from this analysis, primarily associated with specific diseases, metabolic processes, and development. Particularly intriguing was the observation of lower relative expression levels of KIF2A and PHKG2 in domestic sheep breeds compared to introduce sheep breeds, implying enhanced disease resistance within modern Gansu sheep breeding populations.
In the equine domain, Schurink et al. (115) undertook a thorough investigation into CNVs within Friesian horses. Their research unveiled a staggering 15,041 CNVs identified across 222 individuals. Importantly, this study integrated genome-wide association study (GWAS) leveraging both SNPs and CNVs data. A significant finding was the association between CNV regions situated in the major histocompatibility complex (MHC) region of ECA20 and insect bite hypersensitivity (IBH) in Friesian horses. Notably, approximately half of the horses included in the study were afflicted by this condition. Consistently, Ghosh et al. (55) conducted a comprehensive genomic analysis encompassing healthy horses representing 16 distinct breeds. Their investigation identified 258 CNV regions. Notably, the study extended its inquiry to horses exhibiting sexual developmental impairments, wherein they identified a pure deletion of the AKR1C gene in two male pseudohermaphrodites. This discovery suggests a potential association between this gene deletion and the observed abnormalities.
5.4 Environmental adaptability
A comparative examination of CNVs in herbivorous livestock originating from diverse regional breeds has illuminated the potential influence of CNVs on their environmental adaptability. A performed a comprehensive investigation into the differential distribution of CNVs within yak populations hailing from the Tibet and Gansu regions (178). Their study identified seven candidate CNVs, specifically annotating three genes (GRIK4, IFNLR1, and LOC102275985) enriched in five well-established signaling pathways that play pivotal roles in the animals' acclimatization to their environments and various physiological functions. Of particular note is the regulation of physiological processes in hypoxic environments. This research significantly contributes to our understanding of the molecular mechanisms underlying the high-altitude environmental adaptability of yaks. Qaidam cattle, known for their adaptation to cold and low-oxygen environments, underwent genomic analysis by Guo et al. (179). This study identified LDHB and ME1 as potential key genes influencing the cattle's remarkable adaptability to such harsh conditions.
Tibetan sheep, native to high-altitude plateaus, were investigated by Zhu et al. (107), who identified 66 CNVRs associated with their plateau acclimatization. Notably, α-ketoglutarate-dependent dioxygenase alkB homolog 5 (ALKBH5) and nuclear prelamin A recognition factor-like (NARFL) were found to be associated with plateau adaptation within the identified CNVRs. A study performed by Salehian-Dehkordi et al. (112) documented CNVs in 47 sheep breeds. Their research revealed 155 CNVs highly significantly correlated with various environmental parameters, with 35 CNVRs showing significant correlations with solar radiation. Moreover, genes overlapping with CNVs, such as B3GNTL1, UBE2L3, TRAF2, GTF2F1, and IGFALS, were significantly correlated with climatic variables, further emphasizing the role of CNVs in environmental adaptation.
In the equine domain, Wang et al. (114) utilized aCGH to identify CNVs in six horse breeds, uncovering a total of 700 CNVs. These CNVs were classified into 353 CNVRs, and their genetic examination revealed specific genes associated with hemoglobin binding, suggesting a potential influence on horses' adaptability to the challenging plateau environment. The Jinjiang horse, an indigenous breed exclusive to the southeastern coast of China and adapted to high temperatures and humidity, was the focus of a study by Wang et al. (35). They identified 229 genes that overlapped with CNVRs, with four specific candidate genes (NFKBIA, SOCS4, HSPA1A, and IL6) highlighted due to their strong correlation with cellular thermal acclimatization.
6 Conclusions
Common herbivorous livestock, like cattle and sheep, are vital to human daily life due to their significant contributions to meat and dairy production, playing crucial roles in animal husbandry. Recently, China has seen remarkable growth in its equine and donkey industries, driven by advancements in science, technology, and societal progress, establishing themselves as emerging specialties within the livestock sector. Therefore, investigating CNVs within the genomes of these animals holds profound importance. Advancements in microarray and sequencing technologies, along with decreasing sequencing costs, have provided a robust foundation for identifying and studying CNVs. These CNVs, characterized by extended mutant fragment lengths and their substantial impact on genes, represent a formidable genetic resource for exploring genetic variations in livestock and poultry. Extensive research focusing on CNVs in herbivore genomes has unequivocally demonstrated the pivotal role CNVs play in shaping phenotypic diversity, influencing economic traits, enhancing disease resistance, and facilitating environmental adaptation. Researchers worldwide have dedicated their efforts to elucidating the connection between CNVs and phenotypic differences as well as diseases in livestock and poultry. These findings offer compelling support for exploring the potential applications of CNVs as genetic markers in regulation of various productive and disease resistance traits. Consequently, CNVs emerge as a promising avenue for augmenting genetic diversity and expediting molecular breeding strategies in common herbivorous livestock, including cattle, sheep, horses, and donkeys. In conclusion, CNVs represent a valuable and dynamic field of study poised to make a lasting impact on the genetic improvement of herbivorous livestock species, ultimately benefiting both human society and the global livestock industry.
Author contributions
XL: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Software, Writing—original draft, Writing—review & editing. WChe: Methodology, Writing—review & editing, Data curation, Formal analysis. BH: Writing—review & editing, Investigation, Software. XW: Investigation, Software, Writing—review & editing, Data curation, Formal analysis. YP: Data curation, Software, Writing—review & editing, Conceptualization. XZ: Data curation, Software, Writing—review & editing, Investigation. WCha: Software, Writing—review & editing, Formal analysis, Methodology. MK: Methodology, Writing—review & editing, Conceptualization, Project administration, Supervision, Writing—original draft. CW: Conceptualization, Funding acquisition, Investigation, Methodology, Project administration, Resources, Supervision, Validation, Visualization, Writing—original draft, Writing—review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This research was funded by the National Key R&D Program of China (grant number 2022YFD1600103), The Shandong Province Modern Agricultural Technology System Donkey Industrial Innovation Team (grant no. SDAIT-27), Livestock and Poultry Breeding Industry Project of the Ministry of Agriculture and Rural Affairs (grant number 19211162), The National Natural Science Foundation of China (grant no. 31671287), The Open Project of Liaocheng University Animal Husbandry Discipline (grant no. 319312101-14), The Open Project of Shandong Collaborative Innovation Center for Donkey Industry Technology (grant no. 3193308), Research on Donkey Pregnancy Improvement (grant no. K20LC0901), and Liaocheng University scientific research fund (grant no. 318052025).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Hancock JF. Origins of World Crops and Livestock, World Agriculture Before and After 1492: Legacy of the Columbian Exchange. Cham: Springer (2022), 5–18.
2. Caldwell CD. Domestication in agricultural systems In CD Caldwell, S Wang, editors. Introduction to Agroecology. Singapore: Springer Singapore, 157–67.
3. Andersson L, Purugganan M. Molecular genetic variation of animals and plants under domestication. Proc Nat Acad Sci. (2022) 119:e2122150119. doi: 10.1073/pnas.2122150119
4. Verbiest M, Maksimov M, Jin Y, Anisimova M, Gymrek M, Bilgin Sonay T, et al. Mutation and selection processes regulating short tandem repeats give rise to genetic and phenotypic diversity across species. J Evol Biol. (2023) 36:321–36. doi: 10.1111/jeb.14106
5. Lee YL, Bosse M, Takeda H, Moreira GC, Karim L, Druet T, et al. High-resolution structural variants catalogue in a large-scale whole genome sequenced bovine family cohort data. BMC Genomics. (2023) 24:1–7. doi: 10.1186/s12864-023-09259-8
6. Khan MZ, Wang J, Ma Y, Chen T, Ma M, Ullah Q, et al. Genetic polymorphisms in immune-and inflammation-associated genes and their association with bovine mastitis resistance/susceptibility. Front Immunol. (2023) 14:1082144. doi: 10.3389/fimmu.2023.1082144
7. Khan IM, Khan A, Liu H, Khan MZ. Genetic markers identification for animal production and disease resistance. Front Genet. (2023) 14:1243793. doi: 10.3389/fgene.2023.1243793
8. Wang T, Wang X, Liu Z, Shi X, Ren W, Huang B, et al. Genotypes and haplotype combination of DCAF7 gene sequence variants are associated with number of thoracolumbar vertebrae and carcass traits in Dezhou donkey. J Appl Anim Res. (2023) 51:31–9. doi: 10.1080/09712119.2022.2149538
9. Khan MZ, Ma Y, Ma J, Xiao J, Liu Y, Liu S, et al. Association of DGAT1 with cattle, buffalo, goat, and sheep milk and meat production traits. Frontiers in Veterinary Science. (2021) 8:712470. doi: 10.3389/fvets.2021.712470
10. Weischenfeldt J, Symmons O, Spitz F, Korbel JO. Phenotypic impact of genomic structural variation: insights from and for human disease. Nat Rev Genet. (2013) 14:125–38. doi: 10.1038/nrg3373
11. Begna T. Role and economic importance of crop genetic diversity in food security. Int J Agric Sci Food Technol. (2021) 7:164–9. doi: 10.17352/2455-815X.000104
12. Huang B, Khan MZ, Chai W, Ullah Q, Wang C. Exploring genetic markers: mitochondrial dna and genomic screening for biodiversity and production traits in donkeys. Animals. (2023) 13:2725. doi: 10.3390/ani13172725
13. Di Gerlando R, Mastrangelo S, Moscarelli A, Tolone M, Sutera AM, Portolano B, et al. Genomic structural diversity in local goats: analysis of copy-number variations. Animals. (2020) 10:1040. doi: 10.3390/ani10061040
14. Haque MA, Alam MZ, Iqbal A, Lee YM, Dang CG, Kim JJ, et al. Genome-wide association studies for body conformation traits in korean holstein population. Animals. (2023) 13:2964. doi: 10.3390/ani13182964
15. Wang P, Li X, Zhu Y, Wei J, Zhang C, Kong Q, et al. Genome-wide association analysis of milk production, somatic cell score, and body conformation traits in Holstein cows. Front Vet Sci. (2022) 9:932034. doi: 10.3389/fvets.2022.932034
16. Yang P, Wang G, Jiang S, Chen M, Zeng J, Pang Q, et al. Comparative analysis of genome-wide copy number variations between Tibetan sheep and White Suffolk sheep. Anim Biotechnol. (2023) 34:986–93. doi: 10.1080/10495398.2021.2007937
17. Sölzer N, May K, Yin T, König S. Genomic analyses of claw disorders in Holstein cows: genetic parameters, trait associations, and genome-wide associations considering interactions of SNP and heat stress. J Dairy Sci. (2022) 105:8218–36. doi: 10.3168/jds.2022-22087
18. Junior LP, Pinto LF, Cruz VA, Junior GA, Oliveira HR, Chud TS, et al. Genome-wide association and functional genomic analyses for various hoof health traits in North American Holstein cattle. J Dairy Sci. (2023).
19. Schneider H, Segelke D, Tetens J, Thaller G, Bennewitz J. A genomic assessment of the correlation between milk production traits and claw and udder health traits in Holstein dairy cattle. J Dairy Sci. (2023) 106:1190–205. doi: 10.3168/jds.2022-22312
20. Hay EH, Utsunomiya YT, Xu L, Zhou Y, Neves HH, Carvalheiro R, et al. Genomic predictions combining SNP markers and copy number variations in Nellore cattle. BMC Genomics. (2018) 19:1–8. doi: 10.1186/s12864-018-4787-6
21. Sudmant PH, Rausch T, Gardner EJ, Handsaker RE, Abyzov A, Huddleston J, et al. An integrated map of structural variation in 2,504 human genomes. Nature. (2015) 526:75–81. doi: 10.1038/nature15394
22. Fadista J, Thomsen B, Holm L, Bendixen C. Copy number variation in the bovine genome. BMC Genomics. (2010) 11:284. doi: 10.1186/1471-2164-11-284
23. Stothard P, Choi J, Basu U, Sumner-Thomson JM, Meng Y, Liao X, et al. Whole genome resequencing of black angus and holstein cattle for snp and cnv discovery. BMC Genomics. (2011) 12:559. doi: 10.1186/1471-2164-12-559
24. Butty AM, Chud TCS, Cardoso DF, Lopes LSF, Miglior F, Schenkel FS, et al. Genome-wide association study between copy number variants and hoof health traits in holstein dairy cattle. J Dairy Sci. (2021) 104:8050–61. doi: 10.3168/jds.2020-19879
25. Alkan C, Coe BP, Eichler EE. Genome structural variation discovery and genotyping. Nat Rev Genet. (2011) 12:363–76. doi: 10.1038/nrg2958
26. Butty AM, Chud TCS, Miglior F, Schenkel FS, Kommadath A, Krivushin K, et al. High confidence copy number variants identified in holstein dairy cattle from whole genome sequence and genotype array data. Sci Rep. (2020) 10:8044. doi: 10.1038/s41598-020-64680-3
27. Braga LG, Chud TC, Watanabe RN, Savegnago RP, Sena TM. Identification of copy number variations in the genome of Dairy Gir cattle. PLoS ONE. (2023) 18:e0284085. doi: 10.1371/journal.pone.0284085
28. Liu Y, Mu Y, Wang W, Ahmed Z, Wei X, Lei C, et al. Analysis of genomic copy number variations through whole-genome scan in Chinese Qaidam cattle. Front Vet Sci. (2023) 10:1148070. doi: 10.3389/fvets.2023.1148070
29. Xu Z, Wang X, Song X, An Q, Wang D, Zhang Z, et al. Association between the copy number variation of ccser1 gene and growth traits in chinese capra hircus (goat) populations. Anim Biotechnol. (2023) 34:1377–83. doi: 10.1080/10495398.2022.2025818
30. Ren H, Wei Z, Li X, Wang Q, Chen H, Lan X, et al. Goat MyoD1: mRNA expression, InDel and CNV detection and their associations with growth traits. Gene. (2023) 866:147348. doi: 10.1016/j.gene.2023.147348
31. Yao Z, Zhang S, Wang X, Guo Y, Xin X, Zhang Z, et al. Genetic diversity and signatures of selection in BoHuai goat revealed by whole-genome sequencing. BMC Genomics. (2023) 24:116. doi: 10.1186/s12864-023-09204-9
32. Shi H, Li T, Su M, Wang H, Li Q, Lang X, et al. Identification of copy number variation in tibetan sheep using whole genome resequencing reveals evidence of genomic selection. BMC Genomics. (2023) 24:555. doi: 10.1186/s12864-023-09672-z
33. Kim Y, Ha S, Seong H, Choi J, Baek H, Yang B, et al. Identification of copy number variations in four horse breed populations in south korea. Animals. (2022) 12. doi: 10.3390/ani12243501
34. Laseca N, Molina A, Valera M, Antonini A, Demyda-Peyrás S. Copy number variation (cnv): a new genomic insight in horses. Animals. (2022) 12:1435. doi: 10.3390/ani12111435
35. Wang M, Liu Y, Bi X, Ma H, Zeng G, Guo J, et al. Genome-wide detection of copy number variants in chinese indigenous horse breeds and verification of cnv-overlapped genes related to heat adaptation of the jinjiang horse. Genes. (2022) 13:603. doi: 10.3390/genes13040603
36. Zandi MB, Salek Ardestani S, Vahedi SM, Mahboudi H, Mahboudi F, Meskoob A, et al. Detection of common copy number of variants underlying selection pressure in middle eastern horse breeds using whole-genome sequence data. J Heredity. (2022) 113:421–30. doi: 10.1093/jhered/esac027
37. Choudhury MP, Wang Z, Zhu M, Teng S, Yan J, Cao S, et al. Genome-wide detection of copy number variations associated with miniature features in horses. Genes. (2023) 14:1934. doi: 10.3390/genes14101934
38. Castaneda C, Radović L, Felkel S, Juras R, Davis BW, Cothran EG, Wallner B, Raudsepp T. Copy number variation of horse Y chromosome genes in normal equine populations and in horses with abnormal sex development and subfertility: relationship of copy number variations with Y haplogroups. G3 J. (2022) 12:jkac278. doi: 10.1093/g3journal/jkac278
39. Sun X, Jiang J, Wang G, Zhou P, Li J, Chen C, et al. Genome-wide association analysis of nine reproduction and morphological traits in three goat breeds from Southern China. Anim Biosci. (2023) 36:191. doi: 10.5713/ab.21.0577
40. Wijayanti D, Luo Y, Bai Y, Pan C, Qu L, Guo Z, et al. New insight into copy number variations of goat SMAD2 gene and their associations with litter size and semen quality. Theriogenology. (2023) 206:114–22. doi: 10.1016/j.theriogenology.2023.05.012
41. Estrada-Reyes ZM, Ogunade IM, Pech-Cervantes AA, Terrill TH. Copy number variant-based genome wide association study reveals immune-related genes associated with parasite resistance in a heritage sheep breed from the united states. Parasite Immunol. (2022) 44:e12943. doi: 10.1111/pim.12943
42. Lovett PS, Duvall EJ, Keggins KM. Bacillus pumilus plasmid ppl10: properties and insertion into bacillus subtilis 168 by transformation. J Bacteriol. (1976) 127:817–28. doi: 10.1128/jb.127.2.817-828.1976
43. Buckler CE, Staal SP, Rowe WP, Martin MA. Variation in the number of copies and in the genomic organization of ecotropic murine leukemia virus proviral sequences in sublines of akr mice. J Virol. (1982) 43:629–40. doi: 10.1128/jvi.43.2.629-640.1982
44. Hollox EJ, Armour JAL, Barber JCK. Extensive normal copy number variation of a β-defensin antimicrobial-gene cluster. The Am J Hum Genetics. (2003) 73:591–600. doi: 10.1086/378157
45. Pollack JR, Sørlie T, Perou CM, Rees CA, Jeffrey SS, Lonning PE, et al. Microarray analysis reveals a major direct role of dna copy number alteration in the transcriptional program of human breast tumors. Proc Nat Acad Sci. (2002) 99:12963–8. doi: 10.1073/pnas.162471999
46. Lucito R, Healy J, Alexander J, Reiner A, Esposito D, Chi M, et al. Representational oligonucleotide microarray analysis: a high-resolution method to detect genome copy number variation. Genome Res. (2003) 13:2291–305. doi: 10.1101/gr.1349003
47. Guillaud-Bataille M, Valent A, Soularue P, Perot C, Inda M, Receveur A, et al. Detecting single dna copy number variations in complex genomes using one nanogram of starting dna and bac-array cgh. Nucleic Acids Res. (2004) 32:e112. doi: 10.1093/nar/gnh108
48. Sebat J, Lakshmi B, Troge J, Alexander J, Young J, Lundin P, et al. Large-scale copy number polymorphism in the human genome. Science. (2004) 305:525–8. doi: 10.1126/science.1098918
49. Chen Q, Book M, Fang X, Hoeft A, Stuber F. Screening of copy number polymorphisms in human β-defensin genes using modified real-time quantitative pcr. J Immunol Methods. (2006) 308:231–40. doi: 10.1016/j.jim.2005.11.001
50. Redon R, Ishikawa S, Fitch KR, Feuk L, Perry GH, Andrews TD, et al. Global variation in copy number in the human genome. Nature. (2006) 444:444–54. doi: 10.1038/nature05329
51. Matukumalli LK, Lawley CT, Schnabel RD, Taylor JF, Allan MF, Heaton MP, et al. Development and characterization of a high density snp genotyping assay for cattle. PLoS ONE. (2009) 4:e5350. doi: 10.1371/journal.pone.0005350
52. Liu GE, Hou Y, Zhu B, Cardone MF, Jiang L, Cellamare A, et al. Analysis of copy number variations among diverse cattle breeds. Genome Res. (2010) 20:693–703. doi: 10.1101/gr.105403.110
53. Doan R, Cohen N, Harrington J, Veazy K, Juras R, Cothran G, et al. Identification of copy number variants in horses. Genome Res. (2012) 22:899–907. doi: 10.1101/gr.128991.111
54. Liu J, Zhang L, Xu L, Ren H, Lu J, Zhang X, et al. Analysis of copy number variations in the sheep genome using 50k snp beadchip array. BMC Genomics. (2013) 14:229. doi: 10.1186/1471-2164-14-229
55. Ghosh S, Qu Z, Das PJ, Fang E, Juras R, Cothran EG, et al. Copy number variation in the horse genome. PLoS Genet. (2014) 10:e1004712. doi: 10.1371/journal.pgen.1004712
56. Dong Y, Zhang X, Xie M, Arefnezhad B, Wang Z, Wang W, et al. Reference genome of wild goat (Capra aegagrus) and sequencing of goat breeds provide insight into genic basis of goat domestication. BMC Genomics. (2015) 16:431. doi: 10.1186/s12864-015-1606-1
57. Kader A, Liu X, Dong K, Song S, Pan J, Yang M, et al. Identification of copy number variations in three chinese horse breeds using 70k single nucleotide polymorphism beadchip array. Anim Genet. (2016) 47:560–9. doi: 10.1111/age.12451
58. Han H, Zhao X, Xia X, Chen H, Lei C, Dang R, et al. Copy number variations of five y chromosome genes in donkeys. Arch Anim Breed. (2017) 60:391–7. doi: 10.5194/aab-60-391-2017
59. Ma Q, Liu X, Pan J, Ma L, Ma Y, He X, et al. Genome-wide detection of copy number variation in chinese indigenous sheep using an ovine high-density 600 k SNP array. Sci Rep. (2017) 7:912. doi: 10.1038/s41598-017-00847-9
60. Liu M, Zhou Y, Rosen BD, Van Tassell CP, Stella A, Tosser-Klopp G, et al. Diversity of copy number variation in the worldwide goat population. Heredity. (2019) 122:636–46. doi: 10.1038/s41437-018-0150-6
61. Al Abri MA, Holl HM, Kalla SE, Sutter NB, Brooks SA. Whole genome detection of sequence and structural polymorphism in six diverse horses. PLoS ONE. (2020) 15:e230899. doi: 10.1371/journal.pone.0230899
62. Guan D, Martínez A, Castelló A, Landi V, Luigi-Sierra MG, Fernández-Álvarez J, et al. genome-wide analysis of copy number variation in Murciano-Granadina goats. Genetics Select Evol. (2020) 52:1–0. doi: 10.1186/s12711-020-00564-4
63. Mei C, Junjvlieke Z, Raza SHA, Wang H, Cheng G, Zhao C, et al. Copy number variation detection in chinese indigenous cattle by whole genome sequencing. Genomics. (2020) 112:831–6. doi: 10.1016/j.ygeno.2019.05.023
64. He Y, Hong Q, Zhou D, Wang S, Yang B, Yuan Y, et al. Genome-wide selective detection of mile red-bone goat using next-generation sequencing technology. Ecol E11. (2021) 14805–12. doi: 10.1002/ece3.8165
65. Nandolo W, Mészáros G, Wurzinger M, Banda LJ, Gondwe TN, Mulindwa HA, et al. Detection of copy number variants in african goats using whole genome sequence data. BMC Genomics. (2021) 22:398. doi: 10.1186/s12864-021-07703-1
66. Salehian-Dehkordi H, Xu Y, Xu S, Li X, Luo L, Liu Y, et al. Genome-wide detection of copy number variations and their association with distinct phenotypes in the world's sheep. Front Genet. (2021) 12:670582. doi: 10.3389/fgene.2021.670582
67. Zhou J, Liu L, Reynolds E, Huang X, Garrick D, Shi Y, et al. Discovering copy number variation in dual-purpose xinjiang brown cattle. Front Genet. (2022) 12. doi: 10.3389/fgene.2021.747431
68. Taghizadeh S, Gholizadeh M, Rahimi-Mianji G, Moradi MH, Costilla R, Moore S, et al. Genome-wide identification of copy number variation and association with fat deposition in thin and fat-tailed sheep breeds. Sci Rep. (2022) 12:8834. doi: 10.1038/s41598-022-12778-1
69. Kumar H, Panigrahi M, Saravanan KA, Rajawat D, Parida S, Bhushan B, et al. Genome-wide detection of copy number variations in tharparkar cattle. Anim Biotechnol. (2023) 34:448–55. doi: 10.1080/10495398.2021.1942027
70. Fontanesi L, Martelli PL, Beretti F, Riggio V, Dall'Olio S, Colombo M, et al. An initial comparative map of copy number variations in the goat (Capra hircus) genome. BMC genomics. (2010) 11:1–5. doi: 10.1186/1471-2164-11-639
71. Feuk L, Carson AR, Scherer SW. Structural variation in the human genome. Nat Rev Genet. (2006) 7:85–97. doi: 10.1038/nrg1767
72. Mei-lin J, Zeng-kui L, Qing L, Xiao-juan F, Ming-xing C, Cai-hongW. Research progress on copy number variation of livestock and poultry. J Agric Biotechnol. (2019) 10:1840–48.
73. Fernandes AC, da Silva VH, Goes CP, Moreira GC, Godoy TF, Ibelli AM, et al. Genome-wide detection of CNVs and their association with performance traits in broilers. BMC Genomics. (2021) 22:1–8. doi: 10.1186/s12864-021-07676-1
74. Pokrovac I, Pezer Ž. Recent advances and current challenges in population genomics of structural variation in animals and plants. Front Genet. (2022) 13:1060898. doi: 10.3389/fgene.2022.1060898
75. Hsieh P, Vollger MR, Dang V, Porubsky D, Baker C, Cantsilieris S, et al. Adaptive archaic introgression of copy number variants and the discovery of previously unknown human genes. Science. (2019) 366: aax2083. doi: 10.1126/science.aax2083
76. Freeman JL, Perry GH, Feuk L, Redon R, McCarroll SA, Altshuler DM, et al. Copy number variation: new insights in genome diversity. Genome Res. (2006) 16:949–61. doi: 10.1101/gr.3677206
77. Pös O, Radvanszky J, Buglyó G, Pös Z, Rusnakova D, Nagy B, et al. copy number variation: main characteristics, evolutionary significance, and pathological aspects. Biomed J. (2021) 44:548–59.
78. Huang Y, Li Y, Wang X, Yu J, Cai Y, Zheng Z, et al. An atlas of cnv maps in cattle, goat and sheep. Sci China Life Sci. (2021) 64:1747–64. doi: 10.1007/s11427-020-1850-x
79. Lupski JR, Stankiewicz P. Genomic disorders: molecular mechanisms for rearrangements and conveyed phenotypes. PLoS Genet. (2005) 1:e49. doi: 10.1371/journal.pgen.0010049
80. Gu W, Zhang F, Lupski JR. Mechanisms for human genomic rearrangements. Pathogenetics. (2008) 1:4. doi: 10.1186/1755-8417-1-4
81. Lee JA, Carvalho CMB, Lupski JR. A dna replication mechanism for generating nonrecurrent rearrangements associated with genomic disorders. Cell. (2007) 131:1235–47. doi: 10.1016/j.cell.2007.11.037
82. Pinkel D, Albertson DG. Comparative genomic hybridization. Annu Rev Genomics Hum Genet. (2005) 6:331–54. doi: 10.1146/annurev.genom.6.080604.162140
83. Chehbani F, Tomaiuolo P, Picinelli C, Baccarin M, Castronovo P, Scattoni ML, et al. Yield of array-cgh analysis in tunisian children with autism spectrum disorder. Mol Genet Genom Med. (2022) 10:e1939. doi: 10.1002/mgg3.1939
84. Kowalczyk K, Smyk M, Bartnik-Głaska M, Plaskota I, Wiśniowiecka-Kowalnik B, Bernaciak J, et al. Application of array comparative genomic hybridization (ACGH) for identification of chromosomal aberrations in the recurrent pregnancy loss. J Assist Reprod Genet. (2022) 39:357–67. doi: 10.1007/s10815-022-02400-8
85. Oostlander AE, Meijer GA, Ylstra B. Microarray-based comparative genomic hybridization and its applications in human genetics. Clin Genet. (2004) 66:488–95. doi: 10.1111/j.1399-0004.2004.00322.x
86. De Ravel D, Devriendt TJL, Fryns K, Vermeesch JJR. What's new in karyotyping? The move towards array comparative genomic hybridisation (CGH). Eur J Pediatr. (2007) 166:637–43. doi: 10.1007/s00431-007-0463-6
87. Vissers LELM, de Vries BBA, Osoegawa K, Janssen IM, Feuth T, Choy CO, et al. Array-based comparative genomic hybridization for the genomewide detection of submicroscopic chromosomal abnormalities. The Am J Hum Genetics. (2003) 73:1261–70. doi: 10.1086/379977
88. Weiss MM, Snijders AM, Kuipers EJ, Ylstra B, Pinkel D, Meuwissen SG, et al. Determination of amplicon boundaries at 20q13. 2 in tissue samples of human gastric adenocarcinomas by high-resolution microarray comparative genomic hybridization. The J Pathol Soc Great Britain Ireland. (2003) 200:320–6. doi: 10.1002/path.1359
89. LaFramboise T. Single nucleotide polymorphism arrays: a decade of biological, computational and technological advances. Nucleic Acids Res. (2009) 37:4181–93. doi: 10.1093/nar/gkp552
90. Nowak D, Hofmann W, Koeffler HP. Genome-wide mapping of copy number variations using snp arrays. Transfus Med Hemother. (2009) 36:246–51. doi: 10.1159/000225372
91. Walker BA, Morgan GJ. Use of single nucleotide polymorphism–based mapping arrays to detect copy number changes and loss of heterozygosity in multiple myeloma. Clin Lymphoma Myeloma. (2006) 7:186–92. doi: 10.3816/CLM.2006.n.057
92. Metzger J, Philipp U, Lopes MS, da Camara Machado A, Felicetti M, Silvestrelli M, et al. Analysis of copy number variants by three detection algorithms and their association with body size in horses. BMC Genomics. (2013) 14:1–5. doi: 10.1186/1471-2164-14-487
93. Zhao M, Wang Q, Wang Q, Jia P, Zhao Z. Computational tools for copy number variation (cnv) detection using next-generation sequencing data: features and perspectives. BMC Bioinf. (2013) 14:S1. doi: 10.1186/1471-2105-14-S11-S1
94. Liu GE, Bickhart DM. Copy number variation in the cattle genome. Funct Integr Genomics. (2012) 12:609–24. doi: 10.1007/s10142-012-0289-9
95. Liu L, Li Y, Li S, Hu N, He Y, Pong R, et al. Comparison of next-generation sequencing systems. J Biomed Biotechnol. (2012) 2012:251364. doi: 10.1155/2012/251364
96. Grada A, Weinbrecht K. Next-generation sequencing: methodology and application. J Invest Dermatol. (2013) 133:1–4. doi: 10.1038/jid.2013.248
97. Chen D, Xu Y, Ding C, Wang Y, Fu Y, Cai B, et al. The inconsistency between two major aneuploidy-screening platforms—single-nucleotide polymorphism array and next-generation sequencing—in the detection of embryo mosaicism. BMC Genomics. (2022) 23:62. doi: 10.1186/s12864-022-08294-1
98. Bae JS, Cheong HS, Kim LH, NamGung S, Park TJ, Chun JY, et al. Identification of copy number variations and common deletion polymorphisms in cattle. BMC Genomics. (2010) 11:1–10. doi: 10.1186/1471-2164-11-232
99. Bickhart DM, Hou Y, Schroeder SG, Alkan C, Cardone MF, Matukumalli LK, et al. Copy number variation of individual cattle genomes using next-generation sequencing. Genome Res. (2012) 22:778–90. doi: 10.1101/gr.133967.111
100. Zhang L, Jia S, Yang M, Xu Y, Li C, Sun J, et al. Detection of copy number variations and their effects in chinese bulls. BMC Genomics. (2014) 15:480. doi: 10.1186/1471-2164-15-480
101. Da Silva D, Giachetto JM, Da Silva PF, Cintra LO, Paiva LC, Yamagishi SR, et al. Genome-wide copy number variation (CNV) detection in nelore cattle reveals highly frequent variants in genome regions harboring QTLS affecting production traits. BMC Genomics. (2016) 17:454. doi: 10.1186/s12864-016-2752-9
102. Zhou Y, Connor EE, Wiggans GR, Lu Y, Tempelman RJ, Schroeder SG, et al. Genome-wide copy number variant analysis reveals variants associated with 10 diverse production traits in holstein cattle. BMC Genomics. (2018) 19:314. doi: 10.1186/s12864-018-4699-5
103. Kooverjee BB, Soma P, van der Nest MA, Scholtz MM, Neser FW. Copy number variation discovery in South African Nguni-Sired and Bonsmara-Sired crossbred cattle. Animals. (2023) 13:2513. doi: 10.3390/ani13152513
104. Sun T, Pei S, Liu Y, Hanif Q, Xu H, Chen N, et al. Whole genome sequencing of simmental cattle for snp and cnv discovery. BMC Genomics. (2023) 24:179. doi: 10.1186/s12864-023-09248-x
105. Liu S, Kang X, Catacchio CR, Liu M, Fang L, Schroeder SG, et al. Computational detection and experimental validation of segmental duplications and associated copy number variations in water buffalo (Bubalus bubalis). Funct Integr Genomics. (2019) 19:409–19. doi: 10.1007/s10142-019-00657-4
106. Ma Y, Zhang Q, Lu Z, Zhao X, Zhang Y. Analysis of copy number variations by snp50 beadchip array in chinese sheep. Genomics. (2015) 106:295–300. doi: 10.1016/j.ygeno.2015.08.001
107. Zhu C, Fan H, Yuan Z, Hu S, Ma X, Xuan J, et al. Genome-wide detection of cnvs in chinese indigenous sheep with different types of tails using ovine high-density 600k snp arrays. Sci Rep. (2016) 6:27822. doi: 10.1038/srep27822
108. Yang L, Xu L, Zhou Y, Liu M, Wang L, Kijas JW, et al. Diversity of copy number variation in a worldwide population of sheep. Genomics. (2018) 110:143–8. doi: 10.1016/j.ygeno.2017.09.005
109. Kang X, Li M, Liu M, Liu S, Pan MG, Wiggans GR, et al. Copy number variation analysis reveals variants associated with milk production traits in dairy goats. Genomics. (2020) 112:4934–7. doi: 10.1016/j.ygeno.2020.09.007
110. Yuan C, Lu Z, Guo T, Yue Y, Wang X, Wang T, et al. A global analysis of cnvs in chinese indigenous fine-wool sheep populations using whole-genome resequencing. BMC Genomics. (2021) 22:78. doi: 10.1186/s12864-021-07387-7
111. Igoshin AV, Deniskova TE, Yurchenko AA, Yudin NS, Dotsev AV, Selionova MI, et al. Copy number variants in genomes of local sheep breeds from russia. Anim Genet. (2022) 53:119–32. doi: 10.1111/age.13163
112. Salehian-Dehkordi H, Huang J, Pirany N, Mehrban H, Lv X, Sun W, et al. Genomic landscape of copy number variations and their associations with climatic variables in the worldandrsquo;s sheep. Genes. (2023) 14:1256. doi: 10.3390/genes14061256
113. Dupuis MC, Zhang Z, Durkin K, Charlier C, Lekeux P, Georges M, et al. Detection of copy number variants in the horse genome and examination of their association with recurrent laryngeal neuropathy. Anim Genet. (2013) 44:206–8. doi: 10.1111/j.1365-2052.2012.02373.x
114. Wang W, Wang S, Hou C, Xing Y, Cao J, Wu K, et al. Genome-wide detection of copy number variations among diverse horse breeds by array cgh. PLoS ONE. (2014) 9:e86860. doi: 10.1371/journal.pone.0086860
115. Schurink A, Da Silva D, Velie VH, Dibbits BD, Crooijmans BW. Copy number variations in friesian horses and genetic risk factors for insect bite hypersensitivity. BMC Genet. (2018) 19:49. doi: 10.1186/s12863-018-0657-0
116. Solé M, Ablondi M, Binzer-Panchal A, Velie BD, Hollfelder N, Buys N, et al. Inter-and intra-breed genome-wide copy number diversity in a large cohort of European equine breeds. BMC Genomics. (2019) 20:1–2. doi: 10.1186/s12864-019-6141-z
117. Wang C, Li H, Guo Y, Huang J, Sun Y, Min J, et al. Donkey genomes provide new insights into domestication and selection for coat color. Nat Commun. (2020) 11:6014. doi: 10.1038/s41467-020-19813-7
118. Huang Y, Xu T, Li L, Li Z. Jun-yong W, Hong C, Zhao LC. Research progress of copy number variation of cattle and sheep china cattle. Science. (2018) 44:55–60. doi: 10.3969/j.issn.1001-9111.2018.04.015
119. Keel BN, Lindholm-Perry AK, Snelling WM. Evolutionary and functional features of copy number variation in the cattle genome. Front Genet. (2016) 7:207. doi: 10.3389/fgene.2016.00207
120. Corbi-Botto CM, Morales-Durand H, Zappa ME, Sadaba SA, Peral-García P, Giovambattista G, et al. Genomic structural diversity in criollo argentino horses: analysis of copy number variations. Gene. (2019) 695:26–31. doi: 10.1016/j.gene.2018.12.067
121. Nozawa M, Kawahara Y, Nei M. Genomic drift and copy number variation of sensory receptor genes in humans. Proc Nat Acad Sci. (2007) 104:20421–6. doi: 10.1073/pnas.0709956104
122. Young JM, Endicott RM, Parghi SS, Walker M, Kidd JM, Trask BJ, et al. Extensive copy-number variation of the human olfactory receptor gene family. The Am J Hum Genetics. (2008) 83:228–42. doi: 10.1016/j.ajhg.2008.07.005
123. Veerappa AM, Vishweswaraiah S, Lingaiah K, Murthy M, Manjegowda DS, Nayaka R, et al. Unravelling the complexity of human olfactory receptor repertoire by copy number analysis across population using high resolution arrays. PLoS ONE. (2013) 8:e66843. doi: 10.1371/journal.pone.0066843
124. Spehr M, Munger SD. Olfactory receptors: g protein-coupled receptors and beyond. J Neurochem. (2009) 109:1570–83. doi: 10.1111/j.1471-4159.2009.06085.x
125. Palouzier-Paulignan B, Lacroix MC, Aime P, Baly C, Caillol M, Congar P, et al. Olfaction under metabolic influences. Chem Senses. (2012) 37:769–97. doi: 10.1093/chemse/bjs059
126. Ghosh S, Das PJ, McQueen CM, Gerber V, Swiderski CE, Lavoie JP, et al. Analysis of genomic copy number variation in equine recurrent airway obstruction (heaves). Anim Genet. (2016) 47:334–44. doi: 10.1111/age.12426
127. Zhang F, Gu W, Hurles ME, Lupski JR. Copy number variation in human health, disease, and evolution. Annu Rev Genomics Hum Genet. (2009) 10:451–81. doi: 10.1146/annurev.genom.9.081307.164217
128. Shi T, Xu Y, Yang M, Huang Y, Lan X, Lei C, et al. Copy number variations at lepr gene locus associated with gene expression and phenotypic traits in chinese cattle. Anim Sci J. (2016) 87:336–43. doi: 10.1111/asj.12531
129. Xu L, Hou Y, Bickhart DM, Song J, Van Tassell CP, Sonstegard TS, et al. genome-wide survey reveals a deletion polymorphism associated with resistance to gastrointestinal nematodes in Angus cattle. Funct Integ Genomics. (2014) 14:333–9. doi: 10.1007/s10142-014-0371-6
130. Liu M, Li B, Huang Y, Yang M, Lan X, Lei C, et al. Copy number variation of bovine MAPK10 modulates the transcriptional activity and affects growth traits. Livest Sci. (2016) 194:44–50. doi: 10.1016/j.livsci.2016.09.014
131. Yang M, Lv J, Zhang L, Li M, Zhou Y, Lan X, et al. Association study and expression analysis of cyp4a11 gene copy number variation in chinese cattle. Sci Rep. (2017) 7:46599. doi: 10.1038/srep46599
132. Zhang GM, Zheng L, He H, Song CC, Zhang ZJ, Cao XK, et al. Associations of GBP2 gene copy number variations with growth traits and transcriptional expression in Chinese cattle. Gene. (2018) 647:101–6. doi: 10.1016/j.gene.2018.01.004
133. Ma YL, Wen YF, Cao XK, Cheng J, Huang YZ, Ma Y, et al. Copy number variation (CNV) in the IGF1R gene across four cattle breeds and its association with economic traits. Archives Anim Breeding. (2019) 62:171–9. doi: 10.5194/aab-62-171-2019
134. Xu JW, Zheng L, Li LJ, Yao YF, Hua H, Yang SZ, et al. Novel copy number variation of the KLF3 gene is associated with growth traits in beef cattle. Gene. (2019) 680:99–104. doi: 10.1016/j.gene.2018.08.040
135. Zheng L, Xu JW Li JC, Wang DH, An QM, Xu LN, et al. Distribution and association study in copy number variation of KCNJ12 gene across four Chinese cattle populations. Gene. (2019) 689:90–6. doi: 10.1016/j.gene.2018.12.019
136. Yang P, Zhang Z, Xu J, Qu K, Lyv S, Wang X, et al. The association of the copy number variation of the MLLT10 gene with growth traits of Chinese cattle. Animals. (2020) 10:250. doi: 10.3390/ani10020250
137. Hao D, Wang X, Thomsen B, Kadarmideen HN, Wang X, Lan X, et al. Copy number variations and expression levels of guanylate-binding protein 6 gene associated with growth traits of Chinese cattle. Animals. (2020) 10:566. doi: 10.3390/ani10040566
138. Tang J, Shen X, Yang Y, Yang H, Qi A, Yang S, et al. Two different copy number variations of the clcn2 gene in chinese cattle and their association with growth traits. Animals. (2021) 12:41. doi: 10.3390/ani12010041
139. Hu L, Yu J, Huang R, Yang P, Zhang Z, Chai Y, et al. Copy number variation of the CCDC39 gene is associated with growth traits in Chinese cattle. Vet Med Sci. (2022) 8:917–24. doi: 10.1002/vms3.712
140. Yang P, Cai C, Niu M, Liu X, Wang H, Liang H, et al. of copy number variation of PLA2G2A gene to growth traits in Chinese cattle. Gene. (2022) 809:146014. doi: 10.1016/j.gene.2021.146014
141. Yang H, Yue B, Yang Y, Tang J, Yang S, Qi A, et al. Distribution of copy number variation in syt11 gene and its association with growth conformation traits in Chinese cattle. Biology. (2022) 11:223. doi: 10.3390/biology11020223
142. Yao Z, Li J, Zhang Z, Chai Y, Liu X, Li J, et al. The relationship between MFN1 copy number variation and growth traits of beef cattle. Gene. (2022) 811:146071. doi: 10.1016/j.gene.2021.146071
143. Li X, Ding X, Liu L, Yang P, Yao Z, Lei C, et al. Copy number variation of bovine DYNC1I2 gene is associated with body conformation traits in chinese beef cattle. Gene. (2022) 810:146060. doi: 10.1016/j.gene.2021.146060
144. Zhang J, Zhang Z, Liu X, Chai Y, Yang P, Li J, et al. number variation of WBP1L gene revealed its association with growth traits across chinese cattle populations. J Agric Sci. (2022) 160:528–34. doi: 10.1017/S0021859622000387
145. Chen Y, Peng W, Zhang Z, Liu X, Yang P, Fu C, et al. The relationship between MUC19 copy number variation and growth traits of Chinese cattle. Gene. (2023) 851:147010. doi: 10.1016/j.gene.2022.147010
146. Liang J, Liu X, Yang P, Yao Z, Qu K, Wang H, et al. Copy number variation of gal3st1 gene is associated with growth traits of chinese cattle. Anim Biotechnol. (2023) 34:672–8. doi: 10.1080/10495398.2021.1996385
147. Liu X, Yang P, Sun H, Zhang Z, Cai C, Xu J, et al. analysis of VAMP7 gene reveals variation associated with growth traits in Chinese cattle. Anim Biotechnol. (2023) 34:1095–101. doi: 10.1080/10495398.2021.2011741
148. Song X, Li X, Liu X, Zhang Z, Ding X, Chai Y, et al. Copy number variation of the ZNF679 gene in cattle and its association analysis with growth traits. Anim Biotechnol. (2023) 21:1–7. doi: 10.1080/10495398.2023.2185628
149. Jiang R, Cheng J, Cao XK, Ma YL, Chaogetu B, Huang YZ, et al. Copy number variation of the SHE gene in sheep and its association with economic traits. Animals. (2019) 9:531. doi: 10.3390/ani9080531
150. Wang X, Cao X, Wen Y, Ma Y, Elnour IE, Huang Y, et al. Associations of ORMDL1 gene copy number variations with growth traits in four Chinese sheep breeds. Arch Anim Breeding. (2019) 62:571–8. doi: 10.5194/aab-62-571-2019
151. Cheng J, Jiang R, Yang Y, Cao X, Huang Y, Lan X, et al. Association analysis of KMT2D copy number variation as a positional candidate for growth traits. Gene. (2020) 753:144799. doi: 10.1016/j.gene.2020.144799
152. Toremurat Z, Ibrahim EE, Huang YZ, Lan X, Pi L, Chaogetu B, et al. Copy number variations of TOP2B gene are associated with growth traits in Chinese sheep breeds. Anim Biotechnol. (2022) 33:85–9. doi: 10.1080/10495398.2020.1773490
153. Yang Z, Cao X, Ma Y, Cheng J, Song C, Jiang R, et al. Novel copy number variation of the bag4 gene is associated with growth traits in three chinese sheep populations. Anim Biotechnol. (2021) 32:461–9. doi: 10.1080/10495398.2020.1719124
154. Wen Y, Wang E, Wang X, Qing S, Chaogetu B, Wang C, et al. Copy number variations of LRRFIP1 gene and the relationship with growth traits in four Chinese sheep. Anim Biotechnol. (2023) 34:3008–15. doi: 10.1080/10495398.2022.2126981
155. Wang X, Wang Y, Cao X, Huang Y, Li P, Lan X, et al. Copy number variations of the kat6a gene are associated with body measurements of chinese sheep breeds. Anim Biotechnol. (2023) 34:947–54. doi: 10.1080/10495398.2021.2005616
156. Feng Z, Li X, Cheng J, Jiang R, Huang R, Wang D, et al. Copy number variation of the pigy gene in sheep and its association analysis with growth traits. Animals. (2020) 10:688. doi: 10.3390/ani10040688
157. Shi SY Li LJ, Zhang ZJ, Wang EY, Wang J, Xu JW, et al. Copy number variation of MYLK4 gene and its growth traits of Capra hircus (goat). Anim Biotechnol. (2020) 31:532–7. doi: 10.1080/10495398.2019.1635137
158. Li L, Yang P, Shi S, Zhang Z, Shi Q, Xu J, et al. Association analysis to copy number variation (CNV) of Opn4 gene with growth traits of goats. Animals. (2020) 10:441. doi: 10.3390/ani10030441
159. Liu M, Woodward-Greene J, Kang X, Pan MG, Rosen B, Van Tassell CP, et al. Genome-wide CNV analysis revealed variants associated with growth traits in African indigenous goats. Genomics. (2020) 112:1477–80. doi: 10.1016/j.ygeno.2019.08.018
160. Xu Z, Wang X, Zhang Z, An Q, Wen Y, Wang D, et al. Copy number variation of CADM2 gene revealed its association with growth traits across Chinese Capra hircus (goat) populations. Gene. (2020) 741:144519. doi: 10.1016/j.gene.2020.144519
161. Wang Q, Wei Z, Zhu H, Pan C, Akhatayeva Z, Song X, et al. Goat pleomorphic adenoma gene 1 (PLAG1): mRNA expression, CNV detection and associations with growth traits. Animals. (2023) 13:2023. doi: 10.3390/ani13122023
162. Song X, Bai Y, Yuan R, Zhu H, Lan X, Qu L, et al. InDel and CNV within the AKAP13 gene revealing strong associations with growth traits in goat. Animals. (2023) 13:2746. doi: 10.3390/ani13172746
163. Wang Q, Song X, Bi Y, Zhu H, Wu X, Guo Z, et al. Detection distribution of CNVs of SNX29 in three goat breeds and their associations with growth traits. Front Vet Sci. (2023) 10:1132833. doi: 10.3389/fvets.2023.1132833
164. Pei SW, Qin F, Li WH, Li FD, Yue XP. Copy number variation of ZNF280AY across 21 cattle breeds and its association with the reproductive traits of Holstein and Simmental bulls. J Dairy Sci. (2019) 102:7226–36. doi: 10.3168/jds.2018-16063
165. Yue XP, Dechow C, Chang TC, DeJarnette JM, Marshall CE, Lei CZ, et al. Copy number variations of the extensively amplified Y-linked genes, HSFY and ZNF280BY, in cattle and their association with male reproductive traits in Holstein bulls. BMC Genomics. (2014) 15:1–2. doi: 10.1186/1471-2164-15-113
166. Pei S, Xu H, Wang L, Li F, Li W, Yue X. Copy number variation of ZNF280BY across eight sheep breeds and its association with testicular size of Hu sheep. J Anim Sci. (2022) 100:skac232. doi: 10.1093/jas/skac232
167. Wang Q, Bi Y, Wang Z, Zhu H, Liu M, Wu X, et al. Goat SNX29: mRNA expression, InDel and CNV detection, and their associations with litter size. Front Vet Sci. (2022) 9:981315. doi: 10.3389/fvets.2022.981315
168. Bai Y, Zhang T, Liu N, Wang C, Guo Z, Pan C, et al. Investigation of copy number variations (CNVs) of the goat PPP3CA gene and their effect on litter size and semen quality. Animals. (2022) 12:445. doi: 10.3390/ani12040445
169. Zhang R, Wang J, Zhang T, Zhai H, Shen W. Copy-number variation in goat genome sequence: a comparative analysis of the different litter size trait groups. Gene. (2019) 696:40–6. doi: 10.1016/j.gene.2019.02.027
170. Janečka JE, Davis BW, Ghosh S, Paria N, Das PJ, Orlando L, et al. Horse y chromosome assembly displays unique evolutionary features and putative stallion fertility genes. Nat Commun. (2018) 9:2945. doi: 10.1038/s41467-018-05290-6
171. Trigo BB, Utsunomiya ATH, Fortunato AAAD, Milanesi M, Torrecilha RBP, Lamb H, et al. Variants at the asip locus contribute to coat color darkening in nellore cattle. Genet Sel E. (2021) 53:40. doi: 10.1186/s12711-021-00633-2
172. Liang D, Zhao P, Si J, Fang L, Pairo-Castineira E, Hu X, et al. Genomic analysis revealed a convergent evolution of line-1 in coat color: a case study in water buffaloes (bubalus bubalis). Mol Biol E38. (2021) 1122–36. doi: 10.1093/molbev/msaa279
173. Fontanesi L, Beretti F, Riggio V, Gómez González E, Dall Olio S, Davoli R, et al. Copy number variation and missense mutations of the agouti signaling protein (asip) gene in goat breeds with different coat colors. Cytogenet Genome Res. (2010) 126:333–47. doi: 10.1159/000268089
174. Menzi F, Keller I, Reber I, Beck J, Brenig B, Schütz E, et al. Genomic amplification of the caprine ednra locus might lead to a dose dependent loss of pigmentation. Sci Rep. (2016) 6:28438. doi: 10.1038/srep28438
175. Guo J, Sun X, Mao A, Liu H, Zhan S, Li L, et al. 13 42-kb tandem duplication at the ASIP locus is strongly associated with the depigmentation phenotype of non-classic Swiss markings in goats. BMC Genomics. (2022) 23:437. doi: 10.1186/s12864-022-08672-9
176. Rosengren Pielberg G, Golovko A, Sundström E, Curik I, Lennartsson J, Seltenhammer MH, et al. A cis-acting regulatory mutation causes premature hair graying and susceptibility to melanoma in the horse. Nature Genet. (2008) 40:1004–9. doi: 10.1038/ng.185
177. Drögemüller C, Distl O, Leeb T. Partial deletion of the bovine ed1 gene causes anhidrotic ectodermal dysplasia in cattle. Genome Res. (2001) 11:1699–705. doi: 10.1101/gr.182501
178. Guang-Xin E, Yang BG, Zhu YB, Duang XH, Basang WD, Luo XL, et al. Genome-wide selective sweep analysis of the high-altitude adaptability of yaks by using the copy number variant. Biotech. (2020) 10:1–6. doi: 10.1007/s13205-020-02254-w
179. Guo S, Wu X, Pei J, Wang X, Bao P, Xiong L, et al. Genome-wide cnv analysis reveals variants associated with high-altitude adaptation and meat traits in qaidam cattle. Electron J Biotechnol. (2021) 54:8–16. doi: 10.1016/j.ejbt.2021.07.006
180. Huang Y, Shi QT, Shi S, Yang P, Zhang Z, Lyu S, et al. Association between copy number variation of serpina3-1 gene and growth traits in chinese cattle. Anim Biotechnol. (2023) 34:1524–31. doi: 10.1080/10495398.2022.2038183
181. Durkin K, Coppieters W, Drögemüller C, Ahariz N, Cambisano N, Druet T, et al. Serial translocation by means of circular intermediates underlies colour sidedness in cattle. Nature. (2012) 482:81–4. doi: 10.1038/nature10757
Keywords: copy number variation, herbivorous livestock, phenotypes, genome, molecular markers
Citation: Liu X, Chen W, Huang B, Wang X, Peng Y, Zhang X, Chai W, Khan MZ and Wang C (2024) Advancements in copy number variation screening in herbivorous livestock genomes and their association with phenotypic traits. Front. Vet. Sci. 10:1334434. doi: 10.3389/fvets.2023.1334434
Received: 07 November 2023; Accepted: 27 December 2023;
Published: 11 January 2024.
Edited by:
Lucas Lima Verardo, Universidade Federal dos Vales do Jequitinhonha e Mucuri (UFVJM), BrazilReviewed by:
Yongzhen Huang, Northwest A&F University, ChinaSharmila Ghosh, University of California, Davis, United States
Copyright © 2024 Liu, Chen, Huang, Wang, Peng, Zhang, Chai, Khan and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Muhammad Zahoor Khan, zahoorkhattak91@yahoo.com; Changfa Wang, wangchangfa@lcu.edu.cn