Mapping Adverse Outcome Pathways for Kidney Injury as a Basis for the Development of Mechanism-Based Animal-Sparing Approaches to Assessment of Nephrotoxicity
 Angela Mally
Angela Mally Sebastian Jarzina
Sebastian Jarzina- Department of Toxicology, University of Würzburg, Würzburg, Germany
In line with recent OECD activities on the use of AOPs in developing Integrated Approaches to Testing and Assessment (IATAs), it is expected that systematic mapping of AOPs leading to systemic toxicity may provide a mechanistic framework for the development and implementation of mechanism-based in vitro endpoints. These may form part of an integrated testing strategy to reduce the need for repeated dose toxicity studies. Focusing on kidney and in particular the proximal tubule epithelium as a key target site of chemical-induced injury, the overall aim of this work is to contribute to building a network of AOPs leading to nephrotoxicity. Current mechanistic understanding of kidney injury initiated by 1) inhibition of mitochondrial DNA polymerase γ (mtDNA Polγ), 2) receptor mediated endocytosis and lysosomal overload, and 3) covalent protein binding, which all present fairly well established, common mechanisms by which certain chemicals or drugs may cause nephrotoxicity, is presented and systematically captured in a formal description of AOPs in line with the OECD AOP development programme and in accordance with the harmonized terminology provided by the Collaborative Adverse Outcome Pathway Wiki. The relative level of confidence in the established AOPs is assessed based on evolved Bradford-Hill weight of evidence considerations of biological plausibility, essentiality and empirical support (temporal and dose-response concordance).
1 Introduction
There is general consensus that comprehensive understanding of how chemicals and drugs cause adverse effects is key to the development of human relevant, animal-sparing solutions for safety testing. Adverse Outcome Pathways (AOPs) represent a formal description of the mechanistic linkage between a molecular initiating event, a series of intermediate steps and key events at different levels of biological organization, and an adverse outcome. While the concept of mode-of-action (or AOPs) has been a key aspect of human cancer risk assessment for some time, it was recently adopted by the Organisation for Economic Co-operation and Development (OECD) as a pragmatic tool which may facilitate transition of chemical safety assessment from measurement of apical endpoints in animals to toxicity prediction based on mechanistic information (Vinken, 2013). Identification of key events and systematic mapping of AOPs for a given hazard endpoint can form the basis for the development of alternative tests (in vitro, lower organisms, refined in vivo) as part of a science-based integrated testing strategy to eventually replace conventional guideline studies (Tollefsen et al., 2014; Sakuratani et al., 2018). To promote implementation of this concept into chemical safety assessment, the OECD has published guidance documents for the development, assessment and reporting of AOPs as well as use of AOPs to support Integrated Approaches to Testing and Assessment (IATAs) (OECD, 2017b; OECD, 2017a).
The kidney and in particular the proximal tubule epithelium presents one of the key target sites of chemical-induced injury. A wide-range of structurally diverse chemicals, including heavy metals, fungal toxins and drugs are known to cause kidney injury. The particular susceptibility to toxic insult is due to a number of physiological factors that jointly lead to high renal exposure to xenobiotics present in the systemic circulation. The kidneys receive roughly 25% of the cardiac output and hence the rate of delivery of toxicants to the kidney is high. The kidneys’ ability to concentrate solutes further increases exposure of renal cells to xenobiotics (Pfaller and Gstraunthaler, 1998; Khan and Alden, 2002). Uptake of xenobiotics into kidney tubule cells and intrarenal bioactivation to toxic metabolites is facilitated by active transporters and drug metabolizing enzymes that are abundantly expressed particularly throughout the proximal tubule, which renders this segment of the nephron especially susceptible to toxicity (Pfaller and Gstraunthaler, 1998; Khan and Alden, 2002). Acute or chronic damage to proximal tubule cells can lead to kidney dysfunction and ultimately acute or chronic kidney failure. Moreover, chronic cytotoxicity and compensatory regenerative hyperplasia is a well-established mode-of-action by which some chemicals cause kidney tumor formation (Lock and Hard, 2004). Considering the varied chemical nature of nephrotoxic compounds (Pfaller and Gstraunthaler, 1998), it is perhaps not surprising that multiple mechanisms can lead to proximal tubule damage and loss of kidney function. Structurally diverse chemicals may interact with an equally diverse number of molecular targets, and these molecular initiating events (MIE) may each trigger a cascade of molecular and cellular events (Key events, KE) that ultimately result in kidney injury.
In line with recent OECD activities on the use of AOPs in developing Integrated Approaches to Testing and Assessment (IATAs), the overall aim of the present work was to contribute to building a network of AOPs leading to kidney injury through development and critical evaluation of AOPs. Here, we focused on three distinct mechanisms by which certain chemicals or drugs may cause nephrotoxicity and systematically captured the current mechanistic understanding in a formal description of AOPs in accordance with the harmonized terminology provided by the Collaborative Adverse Outcome Pathway Wiki (AOP Wiki; https://aopwiki.org/), a central repository for all AOPs developed as part of the OECD AOP Development Effort. The AOPs considered here are initiated by 1) inhibition of mitochondrial DNA polymerase γ (mtDNA Polγ), 2) receptor mediated endocytosis and lysosomal overload, and 3) covalent protein binding. Human and experimental data on selected chemical stressors for each AOP were identified via Pubmed literatur search and assembled to support the sequence of events leading to kidney injury. The relative level of confidence in the established AOPs was assessed based on evolved Bradford-Hill weight of evidence considerations of biological plausibility, essentiality and empirical support (temporal and dose-response concordance) provided by Becker et al. (2015) and OECD guidance documents for developing and assessing AOPs (OECD, 2017b; 2018) (Box 1).
BOX 1 |

Considering that implementation of the AOP conceptual framework for translation of mechanistic data into regulatory decisions requires quantitative understanding of the relationships between key events within an AOP (Box 1), information on quantitative relationship between two pairs of KEs - as far as available–is assembled and data gaps that need to be filled in order to move from qualitative descriptions of AOPs to quantitative AOPs are highlighted.
2 Inhibition of Mitochondrial Deoxyribonucleic Acid Polymerase γ Leading to Kidney Toxicity (AOP-256)
This Adverse Outcome Pathway describes the sequential key events that link inhibition of mitochondrial DNA polymerase γ (mtDNA Polγ) to kidney toxicity. Nucleoside and nucleotide (nucleos(t)ide) analogs, which are widely used as antiviral drugs for the effective treatment of viral infections, including human immunodeficiency virus (HIV) and chronic hepatitis B virus infections, may act as chemical stressors for this pathway. As structural analogs of substrate nucleotides, these drugs act as chain terminators of viral DNA synthesis via competitive inhibition of reverse transcriptase or viral DNA polymerases, thereby blocking virus replication. Besides targeting viral enzymes, nucleos(t)ide antiviral agents may also interact with human DNA polymerases, which may lead to moderate to life-threatening adverse drug reactions, including peripheral neuropathy, myopathy, lactic acidosis, and acute and chronic kidney injury (Lewis and Dalakas, 1995; Johnson et al., 2001; Fontana, 2009; Fung et al., 2014).
2.1 Nephrotoxicity Associated With Long-Term Intake of Acyclic Nucleoside Phosphonates
The acyclic nucleoside phosphonates (ANPs) adefovir, tenofovir and cidofovir (Figure 1) were introduced into drug therapy of viral infections 15–20 years ago. Compared to existing antiviral drugs, this new class of antivirals offered a broad-spectrum activity against DNA viruses and retroviruses and lower risk of resistance development. However, long-term therapy with ANPs was subsequently found to cause renal proximal tubulopathy and even acute kidney injury. Based on its in vivo antiretroviral potency (Balzarini et al., 1989), adefovir [9-(2-phosphonylmethoxyethyl)adenine; PMEA] and its prodrug adefovir dipivoxil were originally developed for the treatment of HIV infections and cytomegaly virus (CMV) disease (James, 1997). While initial clinical studies reported effective antiretroviral activity and safety of adefovir dipivoxil (125 mg/d) in patients with advanced HIV infections (Deeks et al., 1997), a subsequent multi-center, randomized, double-blind and placebo-controlled trial in adult patients infected with HIV revealed an increased incidence of nephrotoxic effects in patients receiving adefovir (120 mg/d), characterized primarily by elevations in serum creatinine or hypophosphatemia (Kahn et al., 1999). Considering the risk for serious kidney toxicity in long-term use, the U.S. Food and Drug Administration (FDA) denied approval of adefovir for the treatment of HIV (Highleyman, 1999). However, at lower doses, adefovir dipivoxil was subsequently approved by the FDA for the treatment of hepatitis B, although safety concerns due to nephrotoxicity remain. Tenofovir [9-(2-Phosphonyl-methoxypropyly)adenine; PMPA], respectively its prodrug tenofovir disoproxil fumarate (TDF), obtained FDA approval for the treatment of HIV-1 infections in combination with other antiretroviral medicines in 2002, and subsequently for the treatment of chronic hepatitis B in adults in 2008. While tenofovir is now widely used as a first-line therapy against HIV and hepatitis B virus (HBV) infections, long-term treatment with tenofovir is associated with renal toxicity manifested as proximal tubule dysfunction, renal Fanconi syndrome or even acute kidney injury (Woodward et al., 2009; Agarwala et al., 2010; Herlitz et al., 2010; Gara et al., 2012; Hall, 2013; Sise et al., 2015). Cidofovir [(S)-l-(3-hydroxy-2-phosphonylmethoxypropyl)cytosine, HPMPC] is an acyclic nucleotide analog of deoxycytidine that was approved in 1996 for i.v. treatment of acquired immunodeficiency syndrome (AIDS) associated CMVretinitis in adults. Dose-dependent nephrotoxicity was found to be the major dose-limiting toxicity related to cidofovir treatment, and cases of acute renal failure were reported after treatment with as few as one or two doses (Lalezari et al., 1995; Group, 1997; The Studies of Ocular Complications of AIDS Research Group in collaboration with the AIDS Clinical Trials Group, 2000).
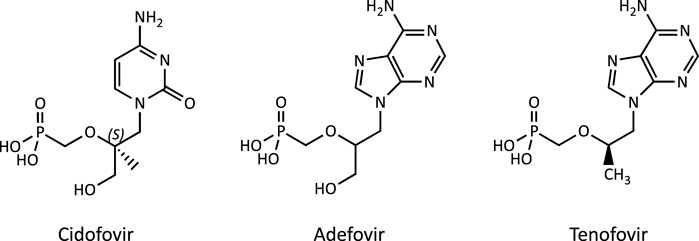
FIGURE 1. Chemical structure of the acyclic nucleoside phosphonates (ANPs) adefovir, tenofovir and cidofovir.
2.2 Mechanism of Acyclic Nucleoside Phosphonate Induced Nephrotoxicity
While the sequence of molecular events leading to ANP associated nephrotoxicity appears to present a universal mechanism by which nucleos(t)ide analogs may cause toxicity in a wide range of organs and tissues, including liver, heart, muscle and the nervous system (Lewis and Dalakas, 1995; Lewis et al., 2003; Fontana, 2009; Fung et al., 2014), the particular susceptibility of the kidney, respectively the proximal tubule, to ANP toxicity is a result of transporter-mediated uptake into proximal tubule cells, leading to intracellular accumulation of ANPs. Organic anion transporter 1 (OAT1) and to a lesser extent organic anion transporter 3 (OAT3) located at the basolateral membrane of proximal tubule cells are recognized as the major membrane carriers for uptake of ANPs (Figure 2) (Uwai et al., 2007; Zhang et al., 2015; Lash et al., 2018). In support of this, inhibition of basolateral membrane transporters has been shown to reduce ANP nephrotoxicity (Lalezari et al., 1995). In addition, recent data demonstrate ANP uptake into primary human kidney cells via apical carriers, potentially OAT4 or organic anion-transporting polypeptides (OATPs) (Lash et al., 2018). Thus, the tissue-specificity of ANP toxicity appears to be determined predominantly by toxicokinetics and renal handling of these drugs. Once taken up into kidney cells, the phosphonate analogs are transported across the mitochondrial membrane prior or subsequent to metabolic conversion into the active triphosphate form via nucleotide kinases present in mitochondria and the cytosol (Robbins et al., 1995; Lewis et al., 2003; Izzedine et al., 2005; Uwai et al., 2007; Kohler et al., 2011). While designed to inhibit viral reverse transcriptase and DNA polymerases with high efficiency, ANPs may also interact with human DNA polymerases, including mitochondrial DNA Pol γ, which is essential for mitochondrial DNA replication (Figure 2). Phosphorylated ANPs compete with endogenous deoxyribonucleotides for incorporation into DNA, thereby inhibiting mitochondrial DNA Pol γ and consequently mtDNA replication. As a result, mtDNA, which encodes 13 components of the electron transport chain essential to oxidative phosphorylation, is depleted. This leads to impaired mitochondrial function, i.e. reduced respiration, electron leakage and energy decline, and ultimately cell death (Figure 2) (Perazella, 2010; Fernandez-Fernandez et al., 2011). Although additional pathways are discussed as potential contributors to mitochondrial toxicity of nucleos(t)ide analogs (Apostolova et al., 2011), there is sufficient evidence from in vitro and in vivo studies in humans and rodents to support mitochondrial dysfunction as a consequence of inhibition of mt Pol γ dependent mtDNA replication as the primary mechanism of ANP induced proximal tubule injury.
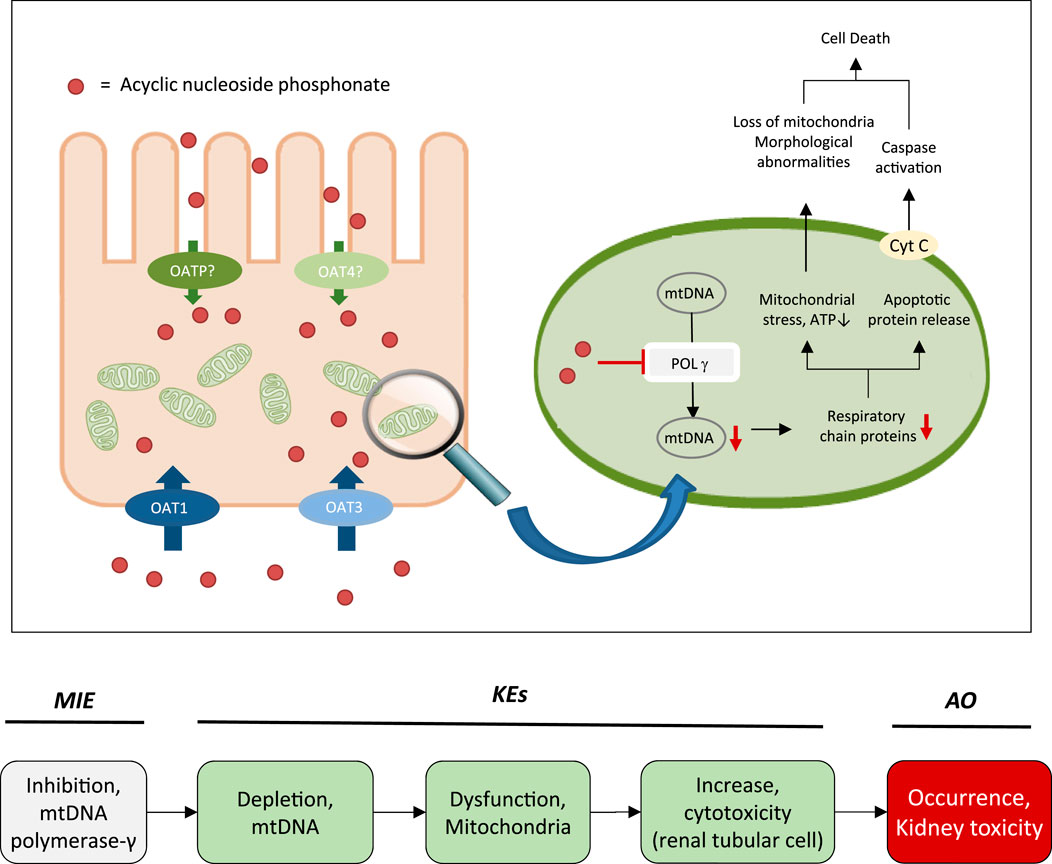
FIGURE 2. Active transport of acyclic nucleoside phosphonates into proximal tubule cells predominantly via basolateral influx carriers (OATs and OAT3) and subsequent disruption of proximal tubular mitochondrial function due to inhibition of mitochondrial DNA Pol γ (modified from (Fernandez-Fernandez et al., 2011)). Adverse outcome pathway of inhibition of mitochondrial DNA Pol γ leading to kidney toxicity.
2.3 The Adverse Outcome Pathway of Inhibition of mtDNA Polymerase γ Leading to Kidney Toxicity
Based on these mechanistic considerations, the sequence of key events (KE) leading to kidney injury as an adverse outcome can be described as inhibition of mt Pol γ as the molecular initiating event (MIE), leading to mitochondrial DNA (mtDNA) depletion (KE1), mitochondrial dysfunction (KE2) and proximal tubule cell toxicity (KE3) (Figure 2). Evidence for inhibition of mitochondrial DNA polymerase γ leading to kidney toxicity as an adverse outcome primarily comes from experimental in vitro and in vivo studies on tenofovir, adefovir and cidofovir that serve as chemical stressors for this pathway, as well as from clinical trials and reports of patients treated with ANPs (Tables 1A–C). Collectively, these studies show a strong association between mitochondrial toxicity and ANP induced nephrotoxicity (Tanji et al., 2001; Cote et al., 2006; Kohler et al., 2009; Lebrecht et al., 2009; Herlitz et al., 2010; Ramamoorthy et al., 2014), with some studies also demonstrating concomitant mtDNA depletion (Tanji et al., 2001; Kohler et al., 2009; Lebrecht et al., 2009; Kohler and Hosseini, 2011). Additional support for mtDNA depletion and mitochondrial dysfunction as down-stream events of mt Pol γ inhibition is derived from studies on nucleos(t)ide analogs that induce mitochondrial toxicity in other target organs via the same principle mechanism. Moreover, there is a wealth of data that link point mutations in the gene encoding for the catalytic subunit of Pol γ with a wide range of human mitochondrial disorders that typically affect tissues with high energy requirement with varying symptoms and severity (Nurminen et al., 2017). In the following sections, evidence supporting the KEs and KE relationships (KERs) in this AOP will be presented, followed by a critical assessment of the AOP in terms of temporal and dose-response concordance, essentiality of key events, biological plausibility, coherence, and consistency of the experimental evidence.
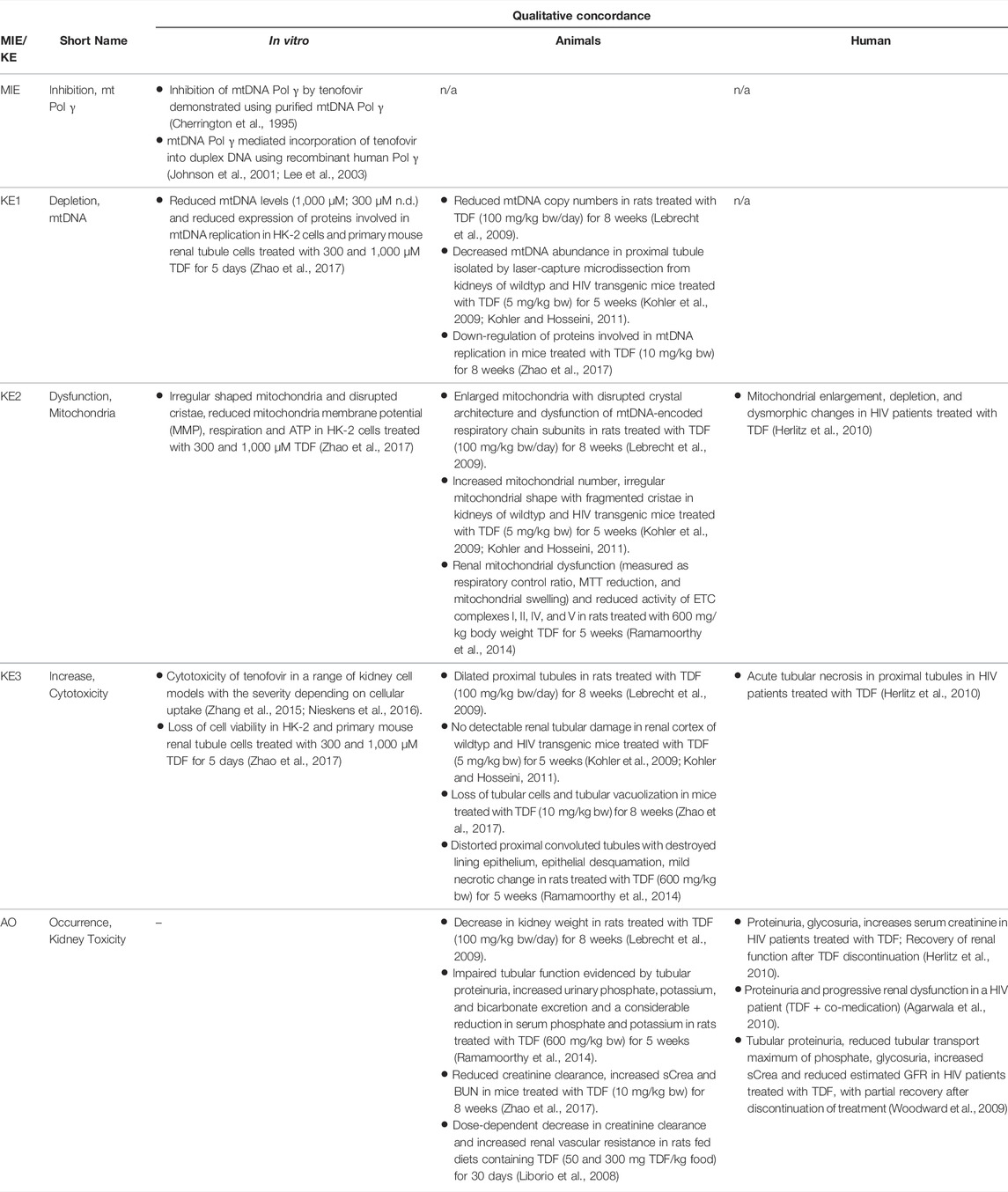
TABLE 1A. Evidence from human, animal and in vitro studies on tenofovir or its prodrug tenofovir disoproxil fumarate (TDF) supporting the key events and qualitative concordance of KEs within this AOP (n/a = no data available).
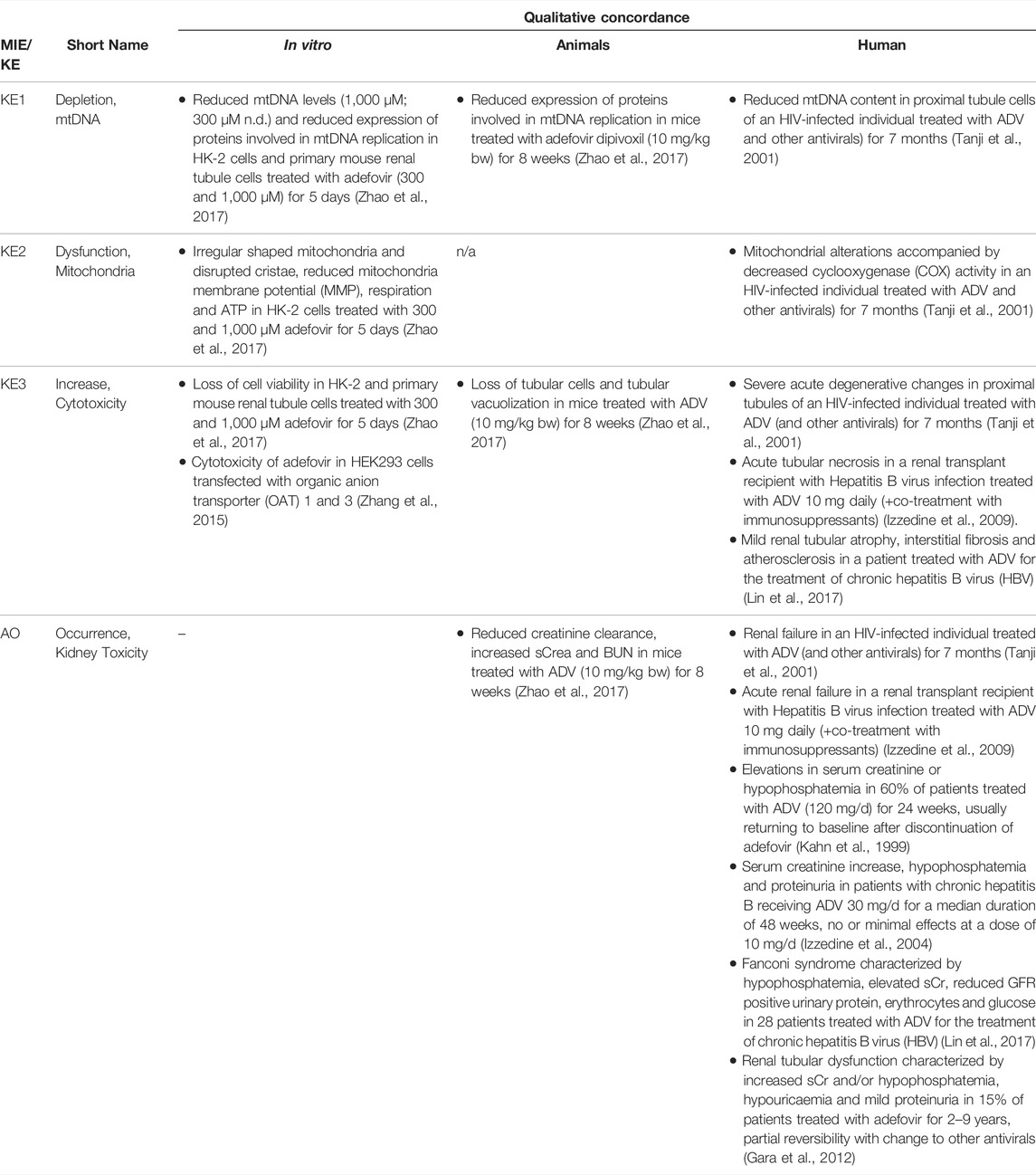
TABLE 1B. Evidence from human, animal and in vitro studies on adefovir or its prodrug adefovir dipivoxil (ADV) supporting the key events and qualitative concordance of KEs within this AOP (n/a = no data available).
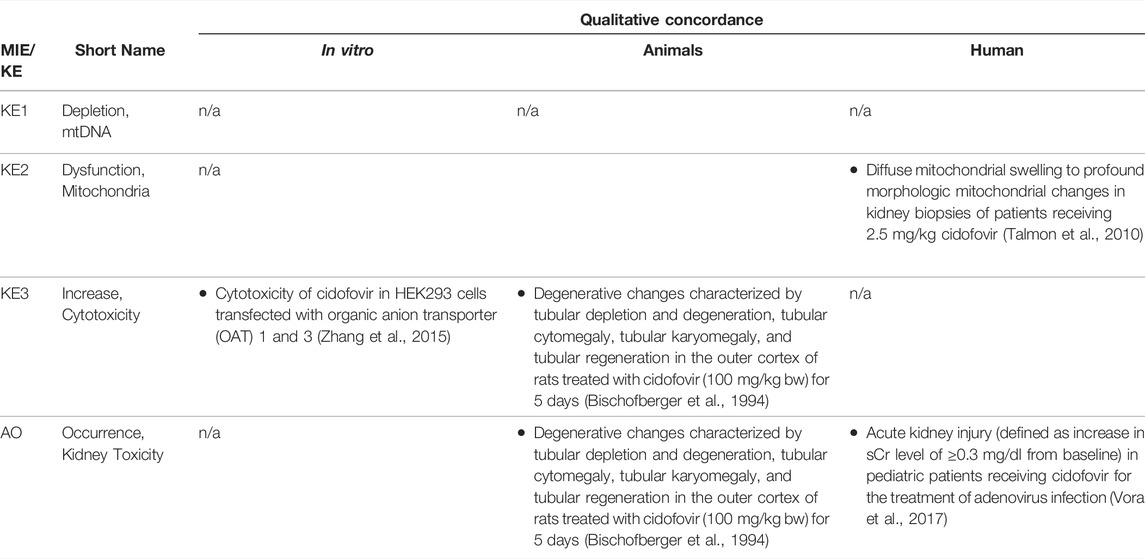
TABLE 1C. Evidence from human, animal and in vitro studies on cidofovir supporting the key events and qualitative concordance of KEs within this AOP (n/a = no data available).
Molecular Initiating Event: Inhibition of mtDNA Polymerase γ
As structural analogs of normal nucleotides that lack the 3′-OH group of the deoxyribose moiety, antiviral nucleos(t)ides were designed as alternative substrates for viral DNA polymerases that block virus replication by preventing chain-elongation. As an undesirable extension of their pharmacological action, antiviral nucleos(t)ides also interact with host DNA polymerases. Among the cellular replicative DNA polymerases, mitochondrial DNA Pol γ, which is responsible for maintenance of mtDNA, has been shown to be most sensitive to the inhibitory effects of these drugs, although nuclear DNA polymerases such as DNA polymerase α and β may also be affected. Numerous in vitro and in vivo studies document inhibitory effects of a wide range of nucleoside and nucleotide reverse transcriptase inhibitors on mtDNA Pol γ at concentrations achieved in vivo (reviewed in (Kakuda, 2000)). While there are significant differences in the ability of individual antiviral nucleos(t)ides to become incorporated into DNA by Pol γ, quantitative prediction of the overall inhibitory effect on mtDNA replication and subsequent mitochondrial toxicity is complicated by the 3′-5′exonuclease activity of Pol γ, which catalyses removal of incorporated nucleotides. This is exemplified by the case of zidovudine (3′-azido-3′-deoxythymidine), a drug that is a comparatively poor substrate for incorporation into mtDNA by Pol γ, which may still effectively block mtDNA replication due to inefficient excision of dideoxynucleotides and hence persistence in mtDNA (Lim and Copeland, 2001; Lim et al., 2003). In addition to the intrinsic 3′-5′exonuclease activity of Pol γ, a recent study also identified Pol β, previously thought to be exclusively located in the nucleus, as a major mtDNA repair enzyme (Prasad et al., 2017). Thus, the ability of nucleos(t)ide analogs to inhibit Pol β presents a further modifying events in this pathway, with progression to the next key event, i.e. mtDNA depletion, depending on the rate of nucleotide incorporation vs. the rate of removal by Pol γ (and presumably also by Pol β) relative to the time required to replicate mtDNA.
Using purified mtDNA Pol γ and activated calf thymus DNA as a primer template, adefovir, tenofovir and cidofovir were all shown to inhibit mammalian DNA polymerases α, β, and γ (Tables 1A–C). The kinetic inhibition constants (Ki) values of the diphosphates of the three nucleoside phosphonates against Pol γ were 0.97, 59.5 and 299 µM for adefovir, tenofovir and cidofovir, respectively, and with the exception of adefovir - significantly higher than Ki values of some of the other nucleos(t)ide analogs such as 2′,3′-dideoxycytidine (0.034 µM) or zidovudine (18.3 µM) (Cherrington et al., 1994; Cherrington et al., 1995). The kinetic inhibition constants against mammalian DNA polymerases β were 70.4, 81.7 and 520 µM for adefovir, tenofovir and cidofovir, respectively (Cherrington et al., 1994; Cherrington et al., 1995). The lower inhibitory activity of tenofovir and cidofovir against human DNA polymerases compared to adefovir and some of the other antiviral nucleos(t)ide analogs were considered to be in line with the relatively lower toxicity of tenofovir and cidofovir. Similarly, a toxicity index calculated based on single turnover kinetic studies using reconstituted human Pol γ holoenzyme to measure the rates of incorporation and exonuclease removal also suggested relatively low mitochondrial toxicity of tenofovir as compared to some other drugs, e.g., 2′,3′-dideoxycytidine (Johnson et al., 2001; Lee et al., 2003).
The inhibitory effects of antiviral drugs on Pol γ dependent mtDNA replication resemble mitochondrial genetic diseases associated with inactivating mutations in the gene encoding Pol γ. Pathogenic mutations in the catalytic subunit of Pol γ cluster into five distinct regions involving the active site, residues of the upstream DNA binding channel, and regions responsible for regulating polymerase vs. exonuclease activity and enzyme processivity (Nurminen et al., 2017). The clinical manifestations of Pol γ syndromes comprise a continuum of phenotypic abnormalities with varying degree of severity, age of onset and tissues affected. There is a close relationship between the age of onset and the severity of the symptoms, i.e. the earlier the onset, the more severe the condition. These range from prenatally-fatal to severe early childhood multi-system disorders such as Alpers-Huttenlocher syndrome (AHS), a progressive neurodegenerative disorder accompanied by disturbed hepatocellular function and tissue-specific DNA depletion (liver > skeletal muscle, heart) that progressively leads to psychomotor regression, epilepsy and liver failure, to adult-onset milder diseases such as progressive external ophthalmoplegia (Cohen et al., 1993; Nurminen et al., 2017). The latter initially presents with weakness of the eye muscles but may also involve other multisystemic features including generalized mitochondrial myopathy with ragged-red fibers, ataxia, axonal sensory-motor polyneuropathy, sensorineural hearing loss, depression, and lactic acidosis (Cohen et al., 1993). Childhood myocerebrohepatopathy spectrum (MCHS) is another Pol γ-related disorder that presents in the first few months of life with developmental delay, lactic acidosis, myopathy and further symptoms such as frequent vomiting, hearing loss, liver failure, pancreatitis and renal tubular acidosis (Cohen et al., 1993).
Key Event 1: mtDNA Depletion
As Pol γ is essential for mtDNA replication, a gradual decrease in mtDNA is an obvious and biologically plausible consequence of sustained inhibition of Pol γ. While there are no reports on the effect of cidofovir on mtDNA content (Table 1C), experimental in vitro and in vivo studies demonstrate reduced mtDNA copy numbers associated with decreased expression of proteins involved in mtDNA replication in kidney tubule cells in response to adefovir and tenofovir (Tables 1A, B). A reduction in the ratio of mitochondrial to nuclear DNA was also reported in proximal tubule cells of an HIV-infected individual maintained on highly active antiretroviral therapy that included adefovir dipivoxil for 7 months (Tanji et al., 2001). In contrast to these studies, Birkus et al. found no effect of tenofovir on mtDNA content in HepG2 cells, skeletal muscle cells and human renal proximal tubule epithelial cells (Birkus et al., 2002). It needs to be emphasized, however, that uptake of tenofovir into cells was not verified in this study, either by directly measuring intracellular levels of tenofovir or by characterization of cells with regard to expression of relevant drug transporters. Considering that tenofovir toxicity depends on transporters that mediate cellular uptake (Uwai et al., 2007; Zhang et al., 2015; Lash et al., 2018), it is questionable if sufficiently high intracellular concentrations to inhibit Pol γ and block mtDNA replication were achieved in this model.
The causal relationship between inhibition of Pol γ and loss of mtDNA is further supported by studies investigating the mechanism of toxicity of nucleos(t)ide analogs in other cells and tissues. For instance, a significant reduction in mtDNA was observed in muscle biopsies of zidovudine-treated HIV positive patients with myopathy as compared non-HIV-patient controls (Arnaudo et al., 1991). Inhibition of mtDNA synthesis and loss of cell number was also observed in a T-lymphoid leukemic cell line (Molt-4) treated with several anti-HIV and anti-HBV nucleoside analogs (d4T, 3′-deoxy-2′,3′-didehydrothymidine; FLT, 3′-fluoro-3′-deoxythynidine; ddC, 2′,3′-dideoxycytidine), which were also identified as potent inhibitors of Pol γ. Similar to the study on tenofovir by Birkus et al. (Birkus et al., 2002), a number of potent Pol γ inhibitors did not cause significant effects on mtDNA synthesis and cell viability (Martin et al., 1994). Based on these findings, the authors concluded that there was no clear quantitative or qualitative correlation between the inhibition of isolated Pol γ and inhibition of mitochondrial DNA synthesis in vitro, and moreover that these data are not predictive of in vivo toxicity (Martin et al., 1994). It is however important to stress that toxicokinetics, most notably cellular uptake of the tested antivirals, were not considered in this assessment. Thus, it is possible that some of the most potent inhibitors of Pol γ identified in a cell-free assay failed to induce mtDNA depletion and cytotoxicity in this cell model simply because of insufficient cellular uptake (Martin et al., 1994).
Experimental evidence for functional inhibition of mitochondrial Pol γ as the underlying cause of mtDNA depletion and associated mitochondriopathies also comes from animal studies. Functional knockout of Pol γ in mice leads to complete loss of mtDNA and embryonic lethality in mice (Hance et al., 2005). Similarly, Pol γ function was demonstrated to be essential for maintenance of mtDNA and development in Drosophila melanogaster (Iyengar et al., 2002). In contrast, polg−/− mutant zebrafish carrying mutations within the polymerase domain survived up to 4 weeks post-fertilization, but showed delayed growth and regenerative defects accompanied by a gradual decrease in mtDNA that correlated with impaired basal and maximal FCCP (carbonyl cyanide p-trifluoro-methoxyphenyl hydrazone)-uncoupled respiration (Rahn et al., 2015). This study also revealed tissue specific differences in the basal levels of mtDNA copy numbers per cell in wildtype animals, with the tail region of zebrafish containing higher levels of mtDNA compared to the region containing the gills, heart and internal organs, and yet lower levels in the central nervous system (CNS) region containing eyes and the brain. Moreover, the degree of mtDNA depletion upon Pol γ knockout was shown to differ between tissues, with the tissue most severely depleted being the organ fraction (mtDNA content in polg−/− 14% of wildtype), followed by CNS (38% of wildtype) and finally tail as the least affected region (52% of wildtype) (Rahn et al., 2015). Considering the finding that the organ fraction, which contains e.g. the liver, along with CNS were most affected by Pol γ knockout, it was suggested that the polg−/−zebrafish model closely resembles human Pol γ-associated mitochondrial diseases that typically present with first symptoms in organs with high energy demand, i.e., CNS and the liver.
Differences in the rate of mtDNA synthesis between tissues are also likely to be an important determinant of tissue-specific responses to Pol γ inhibition. In vivo mtDNA labeling with BrdU in adult wild-type mice showed that BrdU was more rapidly incorporated into mtDNA in the brain as compared to the liver (Fuke et al., 2014), suggesting more rapid mitochondrial biogenesis in the brain.
Key Event 2: Mitochondrial Dysfunction
Although the vast majority of proteins localized in mitochondria is encoded by nuclear DNA, the mitochondrial genome is essential for oxidative energy metabolism as all 13 polypeptides coded for by mitochondrial genes are subunits of complexes of the respiratory chain/oxidative phosphorylation system. It is therefore inevitable that depletion of mtDNA leads to mitochondrial dysfunction. Evidence for mitochondrial dysfunction as a key event in this AOP comes from human, animal and in vitro studies treated with ANPs as chemical stressors for this pathway (Tables 1A–C). Mitochondrial changes indicative of impaired mitochondrial function are typically described as mitochondrial enlargement with disrupted crystal architecture and reduced activity of mtDNA-encoded respiratory chain subunits. These alterations were frequently reported to occur concomitant with mtDNA depletion and cytotoxicity, further supporting a direct link between upstream and downstream key events (Kohler et al., 2009; Lebrecht et al., 2009; Kohler and Hosseini, 2011; Zhao et al., 2017). Likewise, studies in Pol γ deficient animal models demonstrate a close correlation between loss of mtDNA induced by Pol γ inactivation and altered mitochondrial function (e.g. impaired basal and maximal FCCP-uncoupled respiration (Rahn et al., 2015)). Although genetic knockdown of Pol γ typically affects different tissues than ANPs which specifically target the kidney in a transporter-dependent manner, these studies provide substantial evidence for a causal relationship between mtDNA depletion and mitochondrial dysfunction.
Key Event 3: Cytotoxicity
Mitochondria are not only critical for cellular metabolism and energy production that are fundamental to cell viability, they also act as signalling organelles and as such play a key role in cellular life-and-death decisions. Mitochondria participate in both the extrinsic and intrinsic pathway of apoptosis, the latter of which involves opening of the mitochondrial outer membrane and subsequent release of pro-apoptotic factors such as cytochrome C from mitochondria. Interference with the energy-producing function of mitochondria, e.g. through impairment of oxidative phosphorylation as a result of decreased mtDNA content, leads to adenosine triphosphate (ATP) depletion and consequently disturbed cellular function that culminates in necrosis.
While there are multiple mechanisms by which drugs and chemicals can target mitochondria and impair mitochondrial ATP synthesis (e.g. uncoupling of the mitochondrial respiratory chain, inhibition of ATP synthesis, damage to mtDNA, interference with mtDNA replication), mitochondrial toxicity is well established as a key cause of toxicity of a wide range of drugs and chemicals that affect different target organs, including the liver, heart, skeletal muscle, central nervous system, and the kidney. Similarly, mitochondrial dysfunction caused by inherited or sporadic mutations in mtDNA is considered to play a critical role in the pathogenesis of a range of diseases, including acute and chronic kidney injury that involve damage to the proximal tubule (Martin-Hernandez et al., 2005; Emma et al., 2012; Che et al., 2014; Hall and Schuh, 2016). Within the kidney, tubule cells and particularly those of the proximal tubules, are particularly vulnerable to mitochondrial dysfunction. Active transport of solutes in the proximal tubule requires large amounts of ATP generated predominantly via mitochondrial oxidative phosphorylation. To meet the high energy demand, proximal tubule cells contain numerous large mitochondria. While it is generally acknowledged that mitochondrial dysfunction may lead to activation of cell-death pathways, evidence for a mechanistic link between mitochondrial dysfunction caused by inhibition of mtDNA Pol γ and proximal tubule toxicity comes from in vitro and in vivo studies that demonstrate loss of cell viability, dilated proximal tubules and degenerative changes affecting proximal tubules in experimental animals and humans treated with ANPs (Tables 1A–C).
Adverse Outcome: Kidney Toxicity
Through excretion of metabolic wastes and regulation of acid-base balance, electrolyte concentrations and extracellular fluid volume, the kidney plays a key role in maintaining whole-body homeostasis. Functional integrity of the proximal tubule, which contributes to fluid, electrolyte, and nutrient homeostasis by reabsorbing approximately 60–80% of filtered solute and water as well as virtually all of the filtered nutrients (e.g., glucose and amino acids) and low-molecular-weight proteins, is critical for whole-kidney function. Consequently, injury to the proximal tubule will lead to a decline in kidney function, although minor proximal tubule changes may not cause significant effects on renal function due to the kidney’s functional reserve and capacity to regenerate. Numerous drugs and chemicals are known to cause nephrotoxicity primarily by killing proximal tubule cells. Depending on the nature and severity of the insult, altered tubule or whole kidney function may be evidenced by altered renal handling of electrolytes (e.g., sodium, phosphate, calcium, bicarbonate), an increase in urinary glucose, amino acids and low-molecular-weight proteins indicative of impaired tubular reabsorption, and a rise in blood urea nitrogen (BUN) and serum creatinine (sCr). Such changes are evident in experimental animals and patients treated with ANPs that act as chemical stressors for this AOP (Tables 1A–C). For instance, tubular proteinuria and increased urinary phosphate, potassium, and bicarbonate excretion accompanied by reduced serum phosphate and potassium were observed in rats treated with TDF at 600 mg/kg bw for 5 weeks (Ramamoorthy et al., 2014). In another study in rats, a dose-dependent decrease in creatinine clearance was observed (Liborio et al., 2008). Reduced creatinine clearance accompanied by increased sCr and BUN were also reported in mice treated with TDF and adefovir dipivoxil (Zhao et al., 2017). In humans, kidney toxicity associated with intake of ANPs is predominantly characterized by glucosuria and proteinuria, hypophosphatemia, and increased sCr (Kahn et al., 1999; Izzedine et al., 2004; Woodward et al., 2009; Agarwala et al., 2010; Herlitz et al., 2010; Gara et al., 2012; Lin et al., 2017; Vora et al., 2017).
2.4 Assessment of the Adverse Outcome Pathway of Inhibition of mtDNA Polymerase γ Leading to Kidney Toxicity
The relative level of confidence in the overall AOP was assessed based on evolved Bradford-Hill weight of evidence considerations provided by Becker et al. (2015) and OECD guidance documents for developing and assessing AOPs (OECD, 2017b; OECD, 2018) (Box 1).
Biological Plausibility
Mitochondrial DNA replication relies on Pol γ activity. As detailed in Section 2.3, sustained inhibition of Pol γ inevitably leads to reduced mtDNA synthesis and in consequence to a gradual decrease in mtDNA. Loss of mtDNA is thus an obvious and biologically plausible consequence of inhibition of Pol γ (Table 2). All 13 polypeptides encoded by mtDNA are subunits of complexes of the respiratory chain/oxidative phosphorylation system that are required for maintaining mitochondrial function. Biological plausibility for the KER between depletion of mtDNA and mitochondrial dysfunction is therefore considered high (Table 2). It is also well established that mitochondrial function is vital for cell survival, particularly in cells with a high energy demand such as proximal tubule cells. Finally, it is well established and supported by an extensive body of evidence that proximal tubule cell injury impairs kidney function. Thus, the level of confidence in the biological plausibility of all key event relationships (KERs) within the proposed AOP can be considered as high (Table 3).
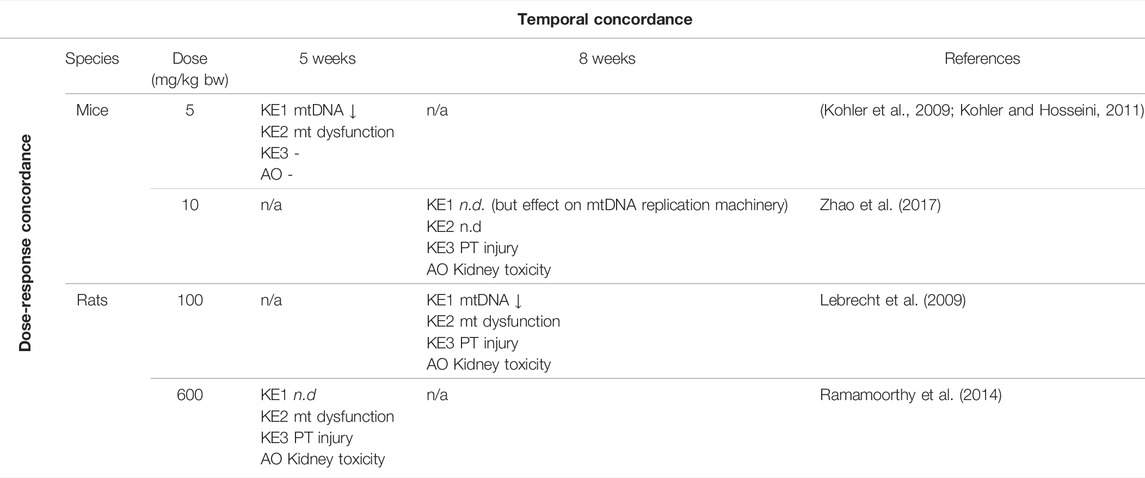
TABLE 2. Dose-Time Concordance of KEs based on rodent studies with tenofovir disoproxil fumarate (TDF) as a specific stressor for the adverse outcome pathway of inhibition of mitochondrial DNA polymerase γ leading to kidney toxicity (n.d. = not determined; n/a = no data available).
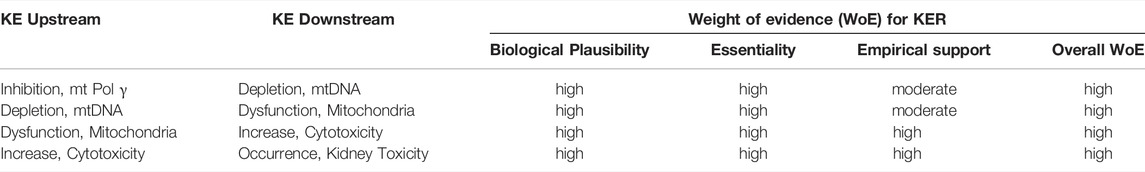
TABLE 3. Weight-of-evidence analysis of KERs in the adverse outcome pathway of inhibition of mitochondrial DNA polymerase γ leading to kidney toxicity.
Essentiality of Key Events
While there are numerous studies to demonstrate that blocking Pol γ function via pharmacological inhibition, genetic knock-out or mutational inactivation is detrimental to cells as it leads to mtDNA depletion and reduced mitochondrial function, there do not appear to be any experimental studies to investigate if restoration of Pol γ function (e.g., through overexpression of Pol γ) maintains mtDNA copy numbers and mitochondrial function. However, studies on arterial aging in mice show that restoring mtDNA copy numbers through overexpression of the mitochondrial helicase Twinkle (Tw+) preserves arterial mitochondrial respiration in aging mice (Foote et al., 2018). Similarly, in a mouse model of volume overload-induced heart failure, increased mtDNA copy numbers in transgenic mice overexpressing human transcription factor A of mitochondria (TFAM) or Twinkle helicase afforded cardioprotection through maintaining mitochondrial enzymatic activities (Ikeda et al., 2015). These data provide experimental support for the essentiality of mtDNA copy number for mitochondrial respiration. Maintaining mitochondrial function has been recognized as a promising therapeutic target for the treatment of acute kidney injury (Hall and Schuh, 2016). Strategies to increase mitochondrial biogenesis, e.g., through activation or overexpression of peroxisome proliferator-activated receptor-γ coactivator-1α (PGC-1α) that acts as a master regulator of mitochondrial biogenesis, have been demonstrated to restore mitochondrial activity and/or kidney function in ischemia-reperfusion induced renal injury or drug-induced acute kidney injury (Sivarajah et al., 2003; Rasbach and Schnellmann, 2007; Liborio et al., 2008; Funk and Schnellmann, 2013; Jesinkey et al., 2014; Jesse et al., 2014). Collectively, these studies provide good evidence that mitochondrial dysfunction and kidney toxicity as the adverse outcome can be prevented by maintaining mtDNA levels, which requires mitochondrial biogenesis and hence Pol γ mediated mtDNA replication. Moreover, recovery of kidney function was reported in patients after discontinuation of ANP treatment (Kahn et al., 1999; Woodward et al., 2009; Herlitz et al., 2010; Gara et al., 2012). Based on direct evidence illustrating essentiality for at least one of the important KEs, the level of confidence for essentiality of KEs in this AOP can thus be considered as high (Table 2).
Empirical Evidence: Dose-Response and Temporal Concordance
Overall, there is only very limited data on dose-related effects of ANPs in vitro and in vivo. In HK-2 cells and primary mouse renal tubule cells treated with TDF and ADV at 300 and 1,000 µM for 5 days, irregularly shaped mitochondria accompanied by reduced mitochondrial respiration and ATP production were observed at both concentrations (Zhao et al., 2017). While treatment with 1,000 µM TDF and ADV resulted significant inhibition of cell growth, decrease in cell viability and induction of apoptosis, treatment with 300 µM of each ANP did not significantly affect cell viability (Zhao et al., 2017). These data provide evidence that mitochondrial dysfunction (KE2) occurs at equal and lower concentrations of adefovir and tenofovir than cytotoxicity (KE3). Unfortunately, mtDNA copy number and expression of proteins involved in mtDNA replication were only investigated at 1,000 µM of TDF and ADV, at which they were significantly altered. Although these data show that all KE were impacted at the same concentration, it is not possible to conclude that KE1 (mtDNA depletion) occurs at lower concentration than the downstream KEs. There are no in vivo studies investigating dose-related effects of ANPs on the proposed KEs, i.e., all studies conducted in experimental animals so far are limited to a single dose group per study (Table 3). Cross-study comparison to establish dose-response concordance is not possible due to variations in experimental design, including species, strain, dose and treatment duration. In studies in mice given TDF at a dose of 5 mg/kg bw for 5 weeks, loss of mtDNA and mitochondrial dysfunction were observed in the absence of proximal tubule injury and impaired kidney function (Kohler et al., 2009; Kohler and Hosseini, 2011), suggesting that either higher doses or prolonged treatment may be required to trigger the final KE and the AO in this AOP (Table 3). In another study, effects on mtDNA replication machinery, proximal tubule injury and kidney toxicity were evident in mice given TDF at 10 mg/kg bw for 8 weeks, while mitochondrial function was not assessed (Table 3) (Zhao et al., 2017). In rats, all 3 KE and kidney toxicity as the adverse outcome were observed after treatment with TDF (100 mg/kg bw) for 8 weeks (Table 3) (Lebrecht et al., 2009). Mitochondrial dysfunction, proximal tubule injury and kidney toxicity were also evident in rats given TDF at 600 mg/kg bw for 5 weeks, but mtDNA copy number was not assessed in this study (Table 3) (Ramamoorthy et al., 2014). Collectively, the available in vitro and in vivo studies conducted using chemical stressors for this AOP generally demonstrate effects on KEs across the entire AOP at equal doses/concentrations of the stressor, with some evidence for upstream events occurring at lower concentrations than downstream KEs. There are no data that would disagree with the assumption of dose-response concordance.
As can be seen from Table 3, there are no in vivo time-course studies on tenofovir or other specific stressors for this AOP, and hence temporal concordance for the entire sequence of events cannot be demonstrated. However, there is a large body of evidence to demonstrate that changes in mitochondrial bioenergetics and dynamics precede proximal tubule damage in kidney injury induced by nephrotoxic drugs and chemicals (Lock et al., 1993) as well as in diabetic kidney disease (Coughlan et al., 2016).
Based on the criteria for assessing AOP (Box 1), evidence for dose-response concordance with relevant stressors but lack of time-course studies to demonstrate temporal concordance, the level of confidence for empirical support for the KERs in this AOP is considered moderate (Table 2).
Weight-Of-Evidence Analysis
Based on the high level of confidence in the biological plausibility of KERs, strong support for essentality of the KEs provided i.e. by experimental studies demonstrating mtDNA depletion and reduced mitochondrial function in response to genetic knock-out or mutational inactivation of Pol γ, and moderate empirical support for the KER in this AOP, the overall weight-of-evidence of this AOP can be considered as high (Table 2).
2.5 Quantitative and Temporal Understanding of Key Event Relationships
Based on the available literature, there is at present little or no quantitative information on the response-response relationship between two pairs of KEs in the AOP of inhibition of Pol γ leading to depletion of mtDNA, but experiments are underway within the Risk-IT project to define these.
KER1: Inhibition of Pol γ Leading to Depletion of mtDNA
As outlined above, the quantitative relationship between inhibition of Pol γ and mtDNA depletion is still poorly defined. Efforts to predict a compounds inhibitory effect on mtDNA replication based on its inhibitory activity against Pol γ did not yield satisfactory results. In establishing response-response relationships, several aspects that determine the KER need to be considered. Firstly, inhibitory effects on Pol γ are typically assessed in cell-free systems, whereas studying effects on mtDNA replication require intact cells. Nominal concentrations added to a cell culture system may not adequately reflect concentrations at the molecular target, e.g., due to active transport, drug metabolism, or binding to plastic. Thus, adequate understanding of the in vitro toxicokinetics of the chemical stressors is needed to extrapolate from a cell-free to a cell-based assay. Second, the overall effect of a chemical stressor on Pol γ mediated mtDNA replication as the downstream KEs depends on its persistence in mtDNA, which is a function not only of the stressors ability to bind to Pol γ and become incorporated into mtDNA (feed-forward loops), but also on the rate of excision of nucleotides by the intrinsic proofreading 3′-5′exonuclease activity of Pol γ (Johnson et al., 2001; Lim and Copeland, 2001; Lim et al., 2003), which presents a feed-back loop (Figure 3). Moreover, recent evidence suggest that Pol β also contributes to base excision repair in mammalian mitochondria (Prasad et al., 2017). Thus, the ability of a chemical stressor to inhibit Pol β is also likely to influence its persistence in mtDNA and thus its overall effect on mtDNA replication. Finally, the rate of mitochondrial biogenesis is a critical determinant. If the stressor is removed more quickly from mtDNA than is required for mtDNA to replicate, mtDNA copy number may not be affected. Similarly, exposure to a stressor for only a short period of time may not be sufficient to trigger mtDNA depletion and subsequent mitochondrial toxicity in this AOP. The maximum lifetime of mitochondria in the kidney cortex has been estimated to be 15 days (Pfeifer and Scheller, 1975). This may explain why effects on KEs downstream of Pol γ inhibition in kidneys of patients and experimental animals generally occur only after continuous exposure for several weeks. This temporal delay between the MIE and the first KE in this AOP is also important to consider when developing in vitro test related to KEs in this AOP.
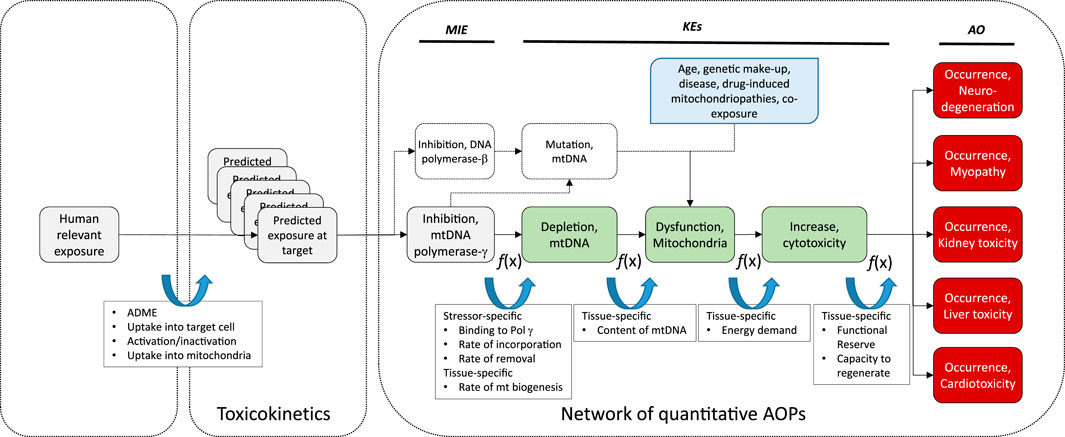
FIGURE 3. Schematic presentation of a universal AOP of Pol γ inhibition leading to adverse outcomes in kidney and extrarenal tissues, with predicted exposure at target site, feed-forward and feedback mechanisms, and potential modulating factors as determinants of the tissue-specific biological response to a chemical stressor.
While it is known that mtDNA content and rates of mitochondrial biogenesis differ between tissues, presumably due to tissue-specific energy demands, recent evidence suggests that there are also tissue-specific differences in the mode of mtDNA replication (Herbers et al., 2018). Since the sequence of KEs in this AOP is also relevant to nucleos(t)ide analog toxicity in extrarenal tissues including liver, heart, muscle and the nervous system, tissue-specific rates and modes of mitochondrial biogenesis may be important determinants of the tissue-specific downstream response to a particular nucleos(t)ide analog in addition to toxicokinetic factors as previously discussed (Figure 3).
Moreover, replication of mtDNA and mitochondrial biogenesis are complex processes that are regulated by a range of factors, including nitric oxide (NO), sirtuins, mitogen-activated protein kinase (p38 MAPK), AMP-activated protein kinase (AMPK) and calcium/calmodulin-dependent protein kinase IV (CaMKIV) (Jornayvaz and Shulman, 2010). This suggest that temporal or inter-individual variation in the activity of these pathways may act as modulating factors of the relationship between MIE and KE1. Estrogens are also known to be involved in the control of mitochondrial biogenesis, and thus sex-differences in the KER may exist (Klinge, 2017; Ventura-Clapier et al., 2017).
KER2: Depletion of mtDNA Leading to Mitochondrial Dysfunction
It is plausible to assume that the level of depletion of mtDNA required to cause mitochondrial toxicity may be cell- and tissue-specific, with metabolically active cells such as kidney tubule cells being most susceptible. There is a lack of quantitative information on the extent of mtDNA depletion required to induce a significant change in mitochondrial function.
While energy decline is thought to be an immediate consequence of mtDNA depletion, it may further increase mitochondrial stress through generation of oxidative stress that may cause mutations in the mitochondrial genome. It is also possible that an increased mtDNA mutation load due to impaired proofreading activity of Pol γ may contribute to mitochondrial dysfunction. Moreover, nucleos(t)ide analogs often also interact with Pol β, which plays a key role in mtDNA repair and maintenance of mitochondrial genome stability (Prasad et al., 2017). Thus, it needs to be considered that a further pathway initiated by inhibition of DNA Pol β by the very same chemical stressor, leading to increased mtDNA mutations and subsequently altered mitochondrial function, may combine with Pol γ inhibition to cause mitochondrial disturbance (Figure 3).
There are also numerous factors independent of chemical stressors of this AOP (e.g., age, genetic make-up, disease, drug-induced mitochondriopathies, co-exposure) that may affect mitochondrial function and increase the susceptibility of mitochondria to mitotoxicity induced by mtDNA depletion. These modulating factors are depicted in Figure 3.
KER3: Mitochondrial Dysfunction Leading to Cytotoxicity
Mitochondrial dysfunction is characterized by a reduced efficiency of oxidative phosphorylation and reduced synthesis of high-energy molecules, such as adenosine-5′-triphosphate (ATP). Expression of toxicity in response to a decline in mitochondrial function may be influenced by the cellular dependence on mitochondrial function, which is known to vary between tissues. Clearly, proximal tubule cells depend on cellular respiration and mitochondrial ATP production to fuel active transport of solutes. However, there is no systematic assessment as to how much decline in mitochondrial function or ATP depletion may be tolerated by a proximal tubule cell before it commits to apoptosis or necrosis. Rather, assays determining mitochondrial activity such as the [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] (MTT) assay (succinate dehydrogenase activity) or ATP content (e.g., Cell Titer-Glo®) are widely used as cytotoxicity assays based on the assumption that mitochondrial activity is related to the number of viable cells. On the other hand, there are numerous studies employing more than one cytotoxicity assay that show poor correlation between cell viability assays measuring mitochondrial activity vs. other end-points such as compromised plasma membrane integrity (e.g., lactate dehydrogenase (LDH) release), with mitochondrial activity assays generally reported to be more sensitive indicators of cytotoxicity than other endpoints (Fotakis and Timbrell, 2006; Xu et al., 2008). This is consistent with mitochondrial dysfunction being a distinct KE that generally precedes cell demise (Vinken and Blaauboer, 2017). It is important to point out that antivirals as chemical stressors for this AOP may have more than one target and thus several independent pathways may contribute to the overall outcome. Integration into nuclear DNA, telomere shortening, nuclear DNA hypermethylation, interference with ATP synthesis and transport of nucleotides into cellular compartments have all been suggested as possible mechanisms unrelated to mitochondrial dysfunction due to mtDNA Pol γ inhibition (Ho et al., 2000; Moyle, 2000). Biotransformation of cidofovir has been shown to give rise to cidofovir-phosphocholine, which may interfere with synthesis or degradation of membrane phospholipids based on its structural similarity with arabinofuranosyl-cytosine 5′-diphosphocholine (Eisenberg et al., 1998; Ho et al., 2000).
KER4: Cytotoxicity (Proximal Tubule Cell) Leading to Kidney Toxicity (Impaired Kidney Function).
Due to the functional reserve of the kidney, homeostasis may be maintained even in the presence of severe kidney damage. It is generally accepted that 70–80% of the renal epithelial mass must be lost before significant changes in serum creatinine (sCr) and blood urea nitrogen (BUN) occur (Pfaller and Gstraunthaler, 1998; Amin et al., 2004). Moreover, even though sCr and BUN are widely used as indicators of renal function in the clinic and in preclinical safety assessment, they are recognized as relatively insensitive markers that only start to rise when renal function is significantly impaired (approximately 50%) (Pfaller and Gstraunthaler, 1998; Amin et al., 2004). Markers related to renal handling of electrolytes, glucose and proteins, including urinary low-molecular weight proteins such as β2-microglobulin, cystatin C and neutrophil gelatinase-associated lipocalin (NGAL) may be more sensitive indicators of proximal tubule function, but despite numerous in vivo studies investigating nephrotoxic effects of drugs and chemicals, there does not appear to be a systematic quantitative assessment as to the extent of proximal tubule injury required to cause significant changes in these markers. However, a multiscale mathematical model was recently developed and applied to prediction of gentamicin-induced kidney injury based on urinary excretion of kidney injury molecule-1 (Kim-1) (Gebremichael et al., 2018) (see Section 3.5). The authors suggest that the developed model should be generalizable to proximal tubule injury induced by nephrotoxins irrespective of their primary mechanism.
3 Receptor-Mediated Endocytosis and Lysosomal Overload Leading to Kidney Toxicity (AOP-257)
This Adverse Outcome Pathway describes the sequence key events that link receptor mediated endocytosis and lysosomal overload to kidney toxicity. Polybasic drugs and compounds with peptidic structure (e.g., aminoglycosides, glycopeptides, polymyxins) (Figure 4), as well as urinary proteins that act as ligands for multiligand, endocytic receptors (megalin, cubilin) expressed at the brush-boarder of renal tubule cells are efficiently taken up into proximal tubule cell via receptor-mediated endocytosis (Moestrup et al., 1995; Khan and Alden, 2002; Verroust et al., 2002; Thevenod, 2003; Schnellmann, 2013; Liu W. J. et al., 2015). Due to low lysosomal pH, endocytosed compounds may be trapped within lysosomes and accumulate in this organelle, leading to disruption of lysosomal function and eventually permeabilization of lysosomal membranes with release of reactive oxygen species and cytotoxic lysosomal enzymes (Figure 5) (Khan and Alden, 2002; Schnellmann, 2013; Liu D. et al., 2015).
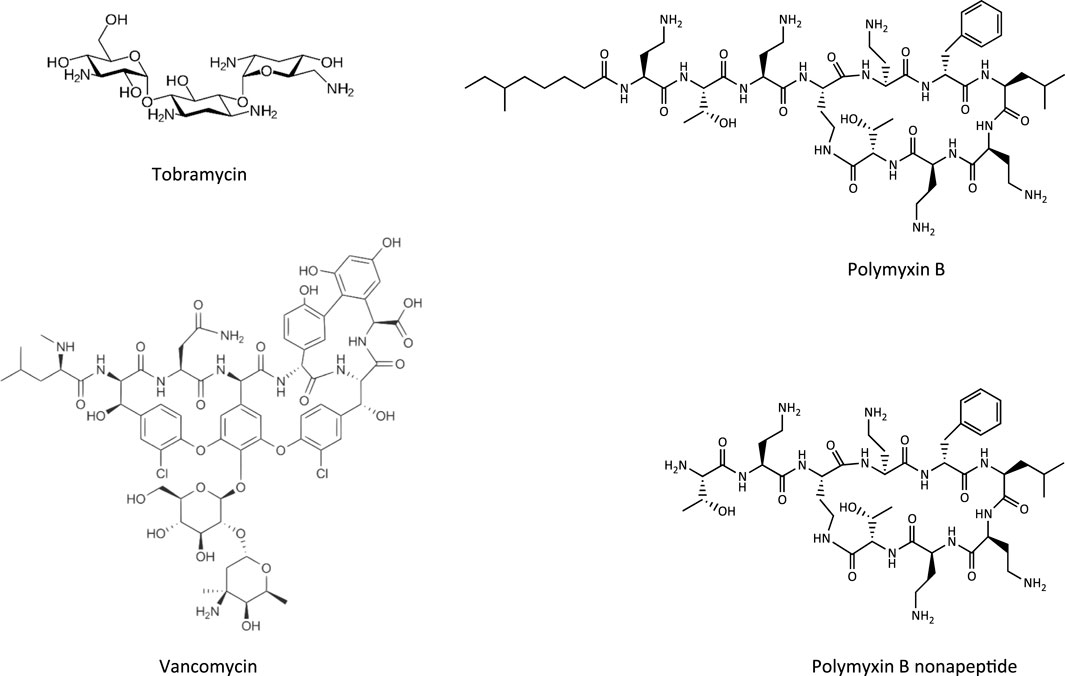
FIGURE 4. Chemical structures of tobramycin as an example of an aminoglycoside, vancomycin as a glycopeptide, and polymyxin B and its less toxic analogue polymyxin B nonapeptide.
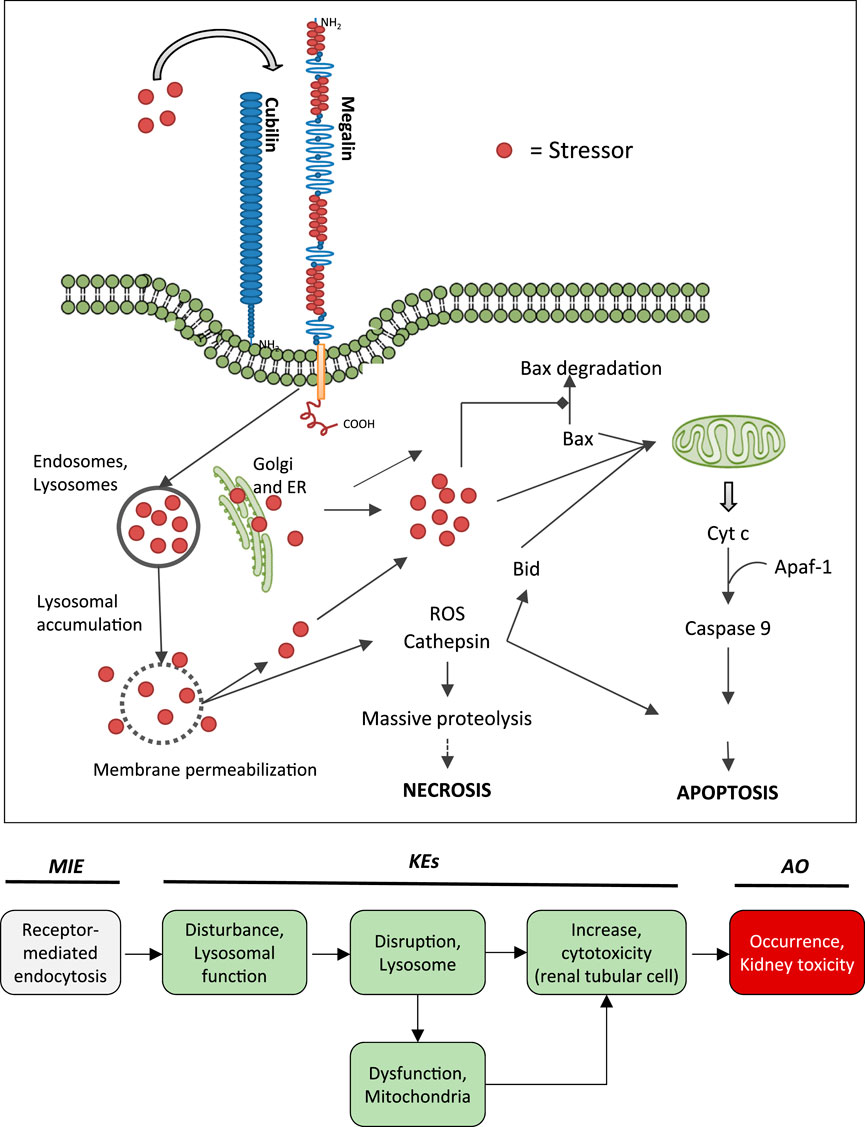
FIGURE 5. Uptake of polybasic drugs and compounds with peptidic structure via multiligand, endocytic receptors (megalin, cubilin) expressed at the brush-boarder of renal tubule cells. Endocytosed compounds may accumulate within lysosomes, leading to lysosomal swelling and disruption of lysosomes. Release of reactive oxygen species, cytotoxic lysosomal enzymes and possibly also endocytosed compounds may trigger cell death via necrosis or apoptosis. This may occur as a direct consequence (e.g., via massive proteolysis) or secondary to mitochondrial dysfunction. Adverse outcome pathway of receptor mediated endocytosis and lysosomal overload leading to kidney toxicity.
3.1 Nephrotoxicity Associated With Lysosomal Accumulation of Ligands of Endocytic Receptors Expressed at the Brush-Boarder of Proximal Tubule Cells
A pivotal function of the renal tubule epithelium is to reabsorb and recycle filtered proteins, carrier-bound vitamins and trace elements from the glomerular ultrafiltrate in order to retrieve nutrients (Eshbach and Weisz, 2017). Cells lining the proximal convoluted tubule are highly specialized for apical endocytosis due to abundant expression of the multiligand endocytic receptors megalin and cubilin at their brush-boarder. Megalin and cubilin ligands include vitamin carrier proteins (e.g., Vitamin D-binding protein, retinol binding protein, transcobalamin), other carrier proteins (e.g., albumin, hemoglobin, liver-type fatty acid-binding protein, metallothionein, transferrin), lipoproteins, enzymes and enzyme inhibitors, immunoglobulin light chains, as well as a number of drugs, including aminoglycosides and polymyxins (Eshbach and Weisz, 2017). Under physiological conditions, endocytosed physiological substrates are efficiently degraded by lysosomal proteases. However, lysosomal swelling and rupture leading to damage of proximal tubule cells may occur if lysosomal degradation is blocked or overwhelmed by substrate overload (Figure 5).
Overproduction of proteins or glomerular injury resulting in increased glomerular permeability and leakage of plasma proteins into urine leads to excessive protein reabsorption and overload of proximal tubule cells. Sustained proteinuria is thus recognized not only as a marker of renal dysfunction but also as a key mediator of tubular injury that contributes to progression of a range of human kidney diseases (Zoja et al., 2004; Erkan et al., 2005). Direct evidence for tubulotoxic effects of excess urinary protein comes from experimental models of albumin or light chain (LC) protein overload nephropathy (Eddy, 1989; Liu et al., 2008; Fang et al., 2018) as well as from in vitro studies with isolated kidney tubule cells (Erkan et al., 2001; Li et al., 2008).
Glycopeptide antibiotics, aminoglycosides and polymyxins (Figure 4) are drugs that exhibit bactericidal activity against Gram-positive and/or Gram-negative bacteria. Due to their polybasic structure, these drugs mimic endogenous ligands of endocytic receptors and highjack the endocytic system to enter proximal tubule cells (Avedissian et al., 2019). Polymyxins were first discovered in 1947 as antimicrobial agents with high activity against Gram-negative bacteria, including Pseudomonas aeruginosa, Klebsiella pneumoniae and Haemophilus influenzae (Stansly et al., 1947) based on their ability to disrupt the outer and inner membranes of Gram-negative bacteria after binding to lipopolysaccharide (LPS). Clinical signs of nephrotoxicity such as albuminuria and increased blood non-protein nitrogen were soon after reported in patients administered polymyxin B (Stansly, 1949), limiting their clinical use by the early 1970s. Despite the high risk of nephrotoxicity with conventional doses, polymyxins play an increasingly important role today as salvage therapy of life-threatening multidrug-resistant bacterial infections (Wertheim et al., 2013). The incidence of acute kidney injury following systemic polymyxin therapy with polymyxin B or colistin is reported to range beween 18 and 61% (Justo and Bosso, 2015).
In contrast to polymyxins, the glycopeptide vancomycin and aminoglycosides are mainstream therapy of serious bacterial infections. However, drug-induced kidney injury is a common and dose-limiting adverse effect of vancomycin and aminoglycoside antibiotic treatment. The incidence of acute kidney injury in patients treated with aminoglycoside antibiotics is reported to range between 10–33%, with the wide variation reflecting the characteristics of the population (Lopez-Novoa et al., 2011; McWilliam et al., 2017). The incidence of vancomycin-associated nephrotoxicity is reported to range up to 43%, depending on the target population (Jeffres, 2017).
3.2 Mechanism of Proximal Tubule Injury Induced by Aminoglycosides, Polymyxins, Vancomycin and Protein Overload
The key mechanism of kidney injury induced by aminoglycosides is fairly well established (Tulkens, 1989; Lopez-Novoa et al., 2011). Aminoglycosides are rapidly eliminated via urinary excretion (80% within 24 h). Following glomerular filtration, however, 5–10% of the dose are reabsorbed and accumulate within the renal cortex (Antoine et al., 2010). The proximal tubule, and specifically the proximal convoluted tubule, presents the critical target of aminoglycoside nephrotoxicity. The site-specificity of renal toxicity is consistent with the abundant expression of megalin and cubilin, which facilitate efficient uptake into epithelial cells of this nephron segment. Following receptor-mediated endocytosis, aminoglycosides accumulate within the endosomal compartment, particularly within lysosomes (Silverblatt and Kuehn, 1979). Due to their cationic structure, aminoglycosides bind to membrane phospholipids, e.g. within lysosomes, and alter their function (e.g., inhibition of A1, A2, C1 phospholipases). Accumulation of aminoglycosides within lysosomes eventually leads to permeabilization of lysosomal membranes and release of lysosomal content and free aminoglycosides into the cytosol. While cytosolic aminoglycosides have been suggested to directly interfere with the mitochondrial electron transport chain and mitochondrial energy production, and to activate the intrinsic apoptotic pathway, release of cathepsin proteases and reactive oxygen species from lysosomes is on its own detrimental to cells (Lopez-Novoa et al., 2011). Depending on their concentration, cathepsins can induce cell death by either apoptosis through cleavage of caspases and activation of Bid or necrosis through massive proteolysis. Besides lysosomal overload as the major pathway involved in the mechanism aminoglycoside toxicity, it has also been suggested that accumulation of aminoglycosides within the endoplasmic reticulum (ER) may induce ER stress by interfering with protein synthesis and protein folding (Lopez-Novoa et al., 2011).
Following glomerular filtration of polymyxins, renal tubule cells reabsorb 90% of the drug and thus polymyxins accumulate substantially within proximal tubule cells located within the renal cortex (Azad et al., 2019). Although there is some evidence to suggest that polypeptide transporters (PEPT1 and PEPT2) contribute to cellular uptake of polymyxins, megalin-mediated reabsorption is considered to play a key role in accumulation of polymyxins within proximal tubule cells (Yun et al., 2015; Azad et al., 2019). While there is limited data on intracellular distribution of polymyxins in kidney cells (Azad et al., 2019), recent studies in human alveolar epithelial cells demonstrate co-localization of polymyxin B with early endosomes, lysosomes, and mitochondria (Ahmed et al., 2019). Similar to aminoglycosides, polymyxin B was shown to affect release of the hydrolytic lysosomal enzyme N-acetyl-β-glucosaminidase from lysosomes in vitro (Powell and Reidenberg, 1983). Although there are as yet no mechanistic studies investigating the causal relationship between polymyxin mediated disruption of lysosomal function and death, the evident similarities in renal handling and lysosomal localization suggests that polymyxins act at least in part via the same mechanism as aminoglycosides. Besides lysosomal toxicity, polymyxins have been suggested to cause oxidative stress and apoptosis via mitochondrial, death receptor, and endoplasmic reticulum pathways (Dai et al., 2014; Azad et al., 2019), yet the interlinkage between these effects remains unclear.
Compared to aminoglycosides and polymyxins, the mechanism of vancomycin induced nephrotoxicity is less well studied. However, based on vancomycin being a ligand for megalin and lyososomal accumulation of vancomycin within the S1 and S2 segment of the proximal tubule (Beauchamp et al., 1992; Fujiwara et al., 2012), a similar mechanism can be assumed.
The mechanism of proximal tubule injury induced by protein overload initiated by receptor-mediated endocytosis of urinary proteins induced is also linked to lysosomal dysfunction and membrane permeabilization. Protein overload leads to increased lysosomal number and volume, impaired lysosome-mediated proteolytic degradation as a result of defective lysosomal acidification, and finally lysosomal membrane permeabilization (Liu W. J. et al., 2015). Activation of NF-κB in tubular epithelial cells is thought to play an important role in the progression of tubulointerstitial injury by promoting initerstitial infiltration of mononuclear cells, interstitial edema, and fibrosis (Zoja et al., 2004).
3.3 The Adverse Outcome Pathway of Receptor Mediated Endocytosis and Lysosomal Overload Leading to Kidney Toxicity
Binding to multiligand, endocytic receptors expressed at the brush-boarder of renal tubule cells, resulting in proximal tubule cell uptake via receptor-mediated endocytosis can be defined as the molecular initiating event (MIE) in this AOP. Although toxicokinetics are typically not considered as part of an AOP, the molecular interaction between ligand and receptor appears to be essential for the lysosomal accumulation of chemical stressors and subsequent disturbance of lysosomal function (KE1), disruption of lysosomes (KE2) and proximal tubule cell toxicity (KE3) (Figure 5).
Evidence for receptor-mediated endocytosis and lysosomal overload leading to kidney toxicity as an adverse outcome primarily comes from experimental in vitro and in vivo studies on aminoglycosides, polymyxins, and low molecular weight urinary proteins that serve as chemical stressors for this pathway (Tables 4A–C). In the following sections, evidence supporting the key events and key event relationships in this AOP is presented, followed by a critical assessment of the AOP in terms of temporal and dose-response concordance, essentiality of key events, biological plausibility, coherence, and consistency of the experimental evidence.

TABLE 4A. Evidence from human, animal and in vitro studies on aminoglycosides supporting the key events and qualitative concordance of KEs within this AOP (n/a = not data available).
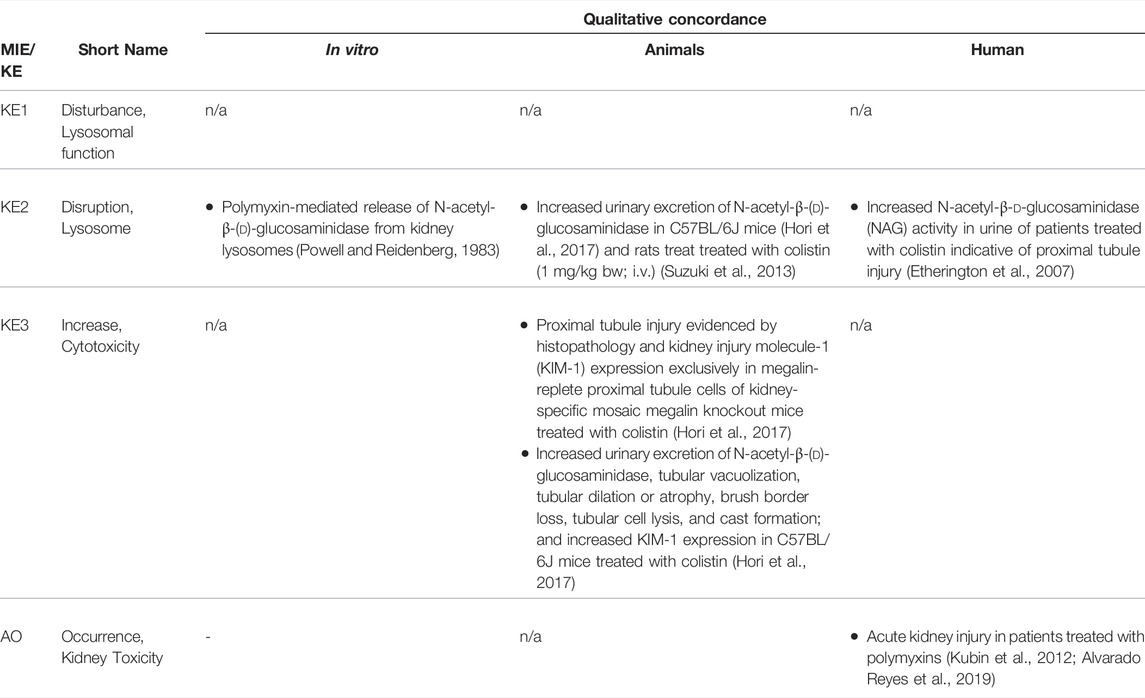
TABLE 4B. Evidence from human, animal and in vitro studies on polymyxins supporting the key events and qualitative concordance of KEs within this AOP (n/a = not data available).
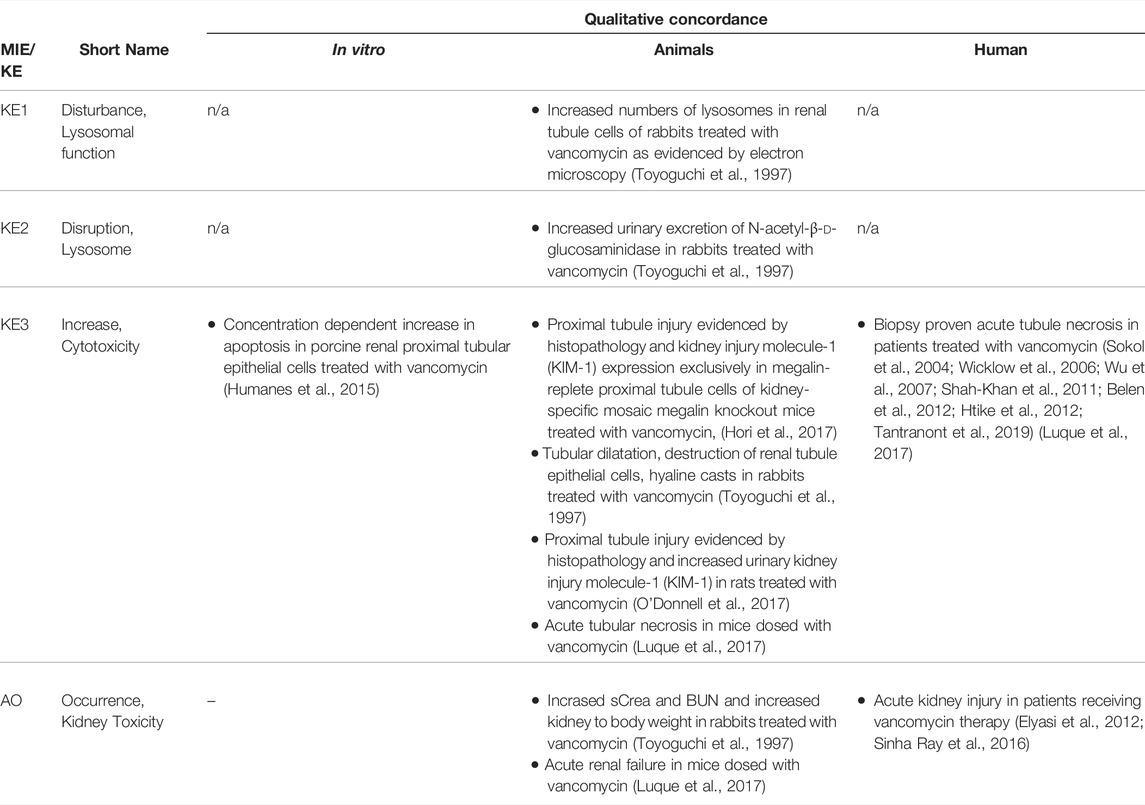
TABLE 4C. Evidence from human, animal and in vitro studies on vancomycin supporting the key events and qualitative concordance of KEs within this AOP (n/a = not data available).
Molecular Initiating Event: Receptor-Mediated Endocytosis
Receptor-mediated endocytosis via binding to the multi-ligand receptor system megalin/cubilin constitutes the principle pathway of cellular uptake of polybasic drugs and low molecular weight proteins (LMWP) from the glomerular filtrate. The interaction of polybasic drugs and LMWPs with the receptor located at the brush border is facilitated by the negative charge of acidic membrane phospholipids and interaction of basic residues of the ligand with negatively charged receptor domains (Moestrup et al., 1995). The site-specific toxicity of polybasic drugs and LMWPs to the proximal tubule, the proximal convoluted tubule, corresponds with the abundant expression of megalin and high endocytic capacity within this nephron segment (Schuh et al., 2018). Receptor-mediated endocytosis is primarily responsible for delivery of polybasic drugs and LMWPs to lysosomes and subsequent disturbance of lysosomal function that ultimately leads to kidney toxicity. There are several lines of in vitro and in vivo evidence that support receptor-mediated endocytosis as the key initiating event: 1) stressors for this AOP are ligands of megalin, 2) uptake and subsequent toxicity of stressors for this AOP can be prevented by competitive inhibitors and indirectly by statins, which block post-translational prenylation of guanosine-5′-triphosphate (GTP)-binding proteins are required for megalin-mediated endocytosis (Antoine et al., 2010; McWilliam et al., 2018). 3) Loss of megalin, e.g., through megalin knockout, protects from accumulation and toxicity of stressors. Experimental evidence from in vitro and in vivo studies on aminoglycosides, polymyxins and vancomycin as chemical stressors of this AOP are summarized in Tables 4A–C. In addition to these chemical stressors, silencing of megalin and cubulin has been shown to inhibit myeloma light chain endocytosis and reduce the toxicity of myeloma light chains (Li et al., 2008), which are exessively produced in multiple myeloma and may cause proximal tubule alterations through overload of the endocytic process (Batuman, 2007). Similarly, siRNA mediated knockdown of megalin and cubilin was shown to block albumin-induced tubular injury (Liu D. et al., 2015). Although there are as yet no data related to the MIE in humans, it is noted that a phase IIa randomized controlled clinical trial investigating prevention of aminoglycoside-induced kidney injury by rosuvastatin in children with cystic fibrosis has been initiated (McWilliam et al., 2018).
Key Event 1: Disturbance, Lysosomal Function
There is substantial evidence from in vitro and in vivo studies that accumulation of endocytosed drugs and LMWPs within lysosomes leads to ultrastructural changes, including increased number and size of lysosomes in renal proximal convoluted cells (Houghton et al., 1978b; Kaloyanides and Pastoriza-Munoz, 1980; Feldman et al., 1982; Mandal and Bennett, 1988; Toyoguchi et al., 1997). These ultastructural changes are considered to occur as a consequence of substrate overload or reduced lysosomal proteolytic capacity due to compound binding to lysosomal phospholipid membrane and inhibition of phospholipases. Interference with phospholipid metabolism results in accumulation of phospholipids with formation of lysosomal myeloid bodies, i.e. concentric multilaminar phospholipid membrane whorls. Experimental in vitro and in vivo studies demonstrating inhibition of phospholipases, phospholipidosis and myeloid body formation by chemical stressors for this AOP as well as detection of myeloid bodies in renal biopsies and urinary sediment of patients treated with aminoglycosides support disturbed lysosomal function as a key event in this AOP (Tables 4A–C). In addition to interference with lysosomal phospholipid metabolism, lysosomal protein overload through excessive exposure of proximal tubule cells to ligands of the endocytic receptor may also lead to altered lysosomal function that expresses itself in hyaline droplet formation. Recent data suggest that changes in the tertiary structure of albumin may interfere with lysosomal proteolysis (Medina-Navarro et al., 2019). This is reminiscent of stabilization of α2u-globulin through binding of chemicals to α2u-globulin, resulting in resistance of the α2u-globulin–chemical complexes to lysosomal degradation and subsequent protein droplet formation in α2u-nephropathy (Lehman-McKeeman et al., 1990).
Key Event 2: Disruption, Lysosome
While reduced release of N-acetyl-β-d-glucosaminidase suggestive of lysosomal membrane stabilization may be an initial response to chemical stressors of this AOP (Powell and Reidenberg, 1982; Powell and Reidenberg, 1983), swelling of lysosomes due to intralysosomal accumulation of chemical stressors and macromolecules (protein, phospholipids) ultimately leads to lysosomal membrane permeabilization or lysosome rupture. It has been suggested that impaired phospholipid metabolism may increase the hydrophobicity of the lysosomal membrane, thereby interfering with transport of water-soluble products across the lysosomal membrane and subsequent osmotic disruption of lysosomes (Powell and Reidenberg, 1982; Powell and Reidenberg, 1983). In addition to the evidence for chemical stressors presented in Tables 4A–C, proximal tubule toxicity induced by albumin and urinary proteins has been shown to involve lysosomal membrane permeabilization and lysosome rupture (Liu D. et al., 2015; Liu W. J. et al., 2015). As a result of lysosomal disruption, lysosomal enzymes such as cathepsins are released into the cytosol (Lopez-Novoa et al., 2011), which may be evident by reduced lysosomal cathepsin activity or immunoreactivity (Li et al., 2000).
Key Event 3: Increase, Cytotoxicity (Renal Tubular Cell)
The link between disruption of lysosomes and cell death is well established (Bursch, 2001; Turk et al., 2002; Guicciardi et al., 2004). Leakage of lysosomal proteases such as cathepsins may trigger apoptosis directly through activation of pro-caspases or indirectly via promoting release of cytochrome C from mitochondria, whereas extensive lysosomal rupture results in necrosis. Evidence for this comes from studies demonstrating that controlled lysosomal rupture induced by a lysosomoropic detergent causes cathepsin release prior to apoptosis (Li et al., 2000). This study also shows that changes in mitochondrial membrane potential occur seondary to lysomal rupture (Li et al., 2000). There is ample evidence from in vitro experiments and studies in animals and humans that demonstrate proximal tubule toxicity of stressors of this AOP (Tables 4A–C). On a cautionary note, establishment of stable cell lines often involves use of aminoglycosides as selection antibiotics and thus renal cell lines generated via this protocol may be resistant to aminoglycoside toxicity.
Adverse Outcome: Kidney Toxicity
The link between proximal tubule injury and impaired kidney function has already been described in Section 2.3. Since receptor-mediated endocytosis occurs primarily within the S1 segment of the proximal tubule, which is also the primary site of glucose reabsorption, increased urinary glucose is often one of the earliest signs of proximal tubule injury induced by stressors of this AOP. Increased urinary excretion of (low-molecular-weight) proteins that are normally endocytosed and degraded is also frequently observed as an early response to stressors of this AOP, although it is not entirely clear if such changes necessarily always reflect impaired tubular reabsorption as a result of tubule damage or rather competitive inhibition of receptor-mediated endocytosis by the chemical stressor of this AOP. With increasing severity of tubule damage, nephrotoxicity induced by stressors of this AOP may progress to changes in blood urea nitrogen (BUN) and serum creatinine (sCrea), reduced glomerular filtration, and oligo-anuric renal failure. Such changes are evident in experimental animals treated with aminoglycosides and polymyxins as well as in patients receiving aminoglycoside, glycopeptide and polymyxin antibiotics (Tables 4A–C).
3.4 Assessment of the Adverse Outcome Pathway of Receptor Mediated Endocytosis and Lysosomal Overload Leading to Kidney Toxicity
Biological Plausibility
The mechanistic basis for a causal relationship between the KEs in this AOP is detailed in Section 3.3. Considering the high endocytic activity of convoluted proximal tubule cells, the physiological role of lysosomes in the degradation of endocytosed material, the proteolytic function of lysosomal enzymes and toxicity of highly reactive oxygen species that leak into the cytosol upon lysosomal membrane permeabilization subsequent to lysosomal overload, and the critical role of the proximal tubule for kidney function, the level of confidence in the biological plausibility of key event relationships (KERs) within the proposed AOP can be considered as high (Table 5).
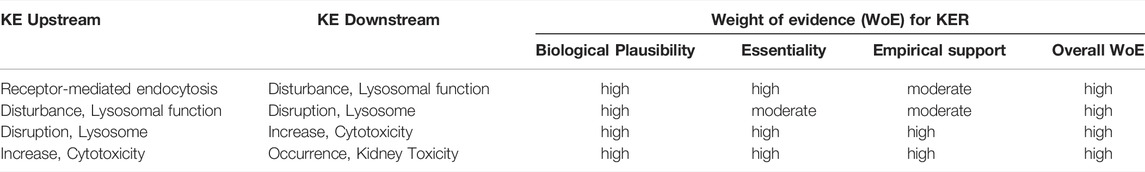
TABLE 5. Weight-of-evidence analysis of KERs in the adverse outcome pathway of receptor-mediated endocytosis and lysosomal overload leading to kidney toxicity.
Essentiality of Key Events
Essentiality of the MIE in this AOP is clearly supported by a range of in vitro and in vivo studies in experimental animals that demonstrate reduced cellular uptake, accumulation and cytotoxicity of model stressors for this pathway in megalin deficient kidney cells or in the presence of competitive inhibitors of receptor mediated endocytosis, e.g., (Moestrup et al., 1995; Schmitz et al., 2002; Watanabe et al., 2004; Takamoto et al., 2005a; Takamoto et al., 2005b; Wolff et al., 2006; Wolff et al., 2008; Raggi et al., 2011; Onodera et al., 2012; Suzuki et al., 2013; Liu W. J. et al., 2015; Hori et al., 2017) (Tables 4A–D) and can thus be considered high (Table 5). Pharmacological inhibition of cathepsins has been shown to ameliorate protein overload-triggered tubule cell apoptosis (Liu W. J. et al., 2015), providing evidence that lysosomal membrane permeabilization and associated cathepsin release is an essential trigger for cell death in this pathway. Similarly, Song et al. (2017) showed that caspase-3 activation and apoptosis caused by lead-induced lysosomal membrane permeabilization in primary rat proximal tubular cells is significantly reduced by cathepsin B and D inhibitors (Song et al., 2017). Based on direct evidence for essentiality of the MIE and an important KE upstream of cytotoxicity, the level of confidence for essentiality of KEs in this AOP can thus be considered as high (Table 5).
Empirical Evidence: Dose-Response and Temporal Concordance
There are numerous studies that provide dose-response data on aminoglycoside nephrotoxicity in experimental animals through comparative analysis of histopathological changes, clinical chemistry parameters indicative of renal function, and novel biomarkers of kidney injury. These studies frequently report proximal tubule injury (KE3) at doses lower than those required to induce a significant decline in kidney function (Table 6). In contrast, there are only few studies that considered early upstream KEs in this AOP, i.e. lysosomal alterations (Table 6). While it is not possible from the available data to conclude that aminoglycoside-mediated effects on lysosomes occur at lower doses compared to those required to induce proximal tubule injury and kidney failure, it is evident that these lysosomal changes are recorded at an equal dose.
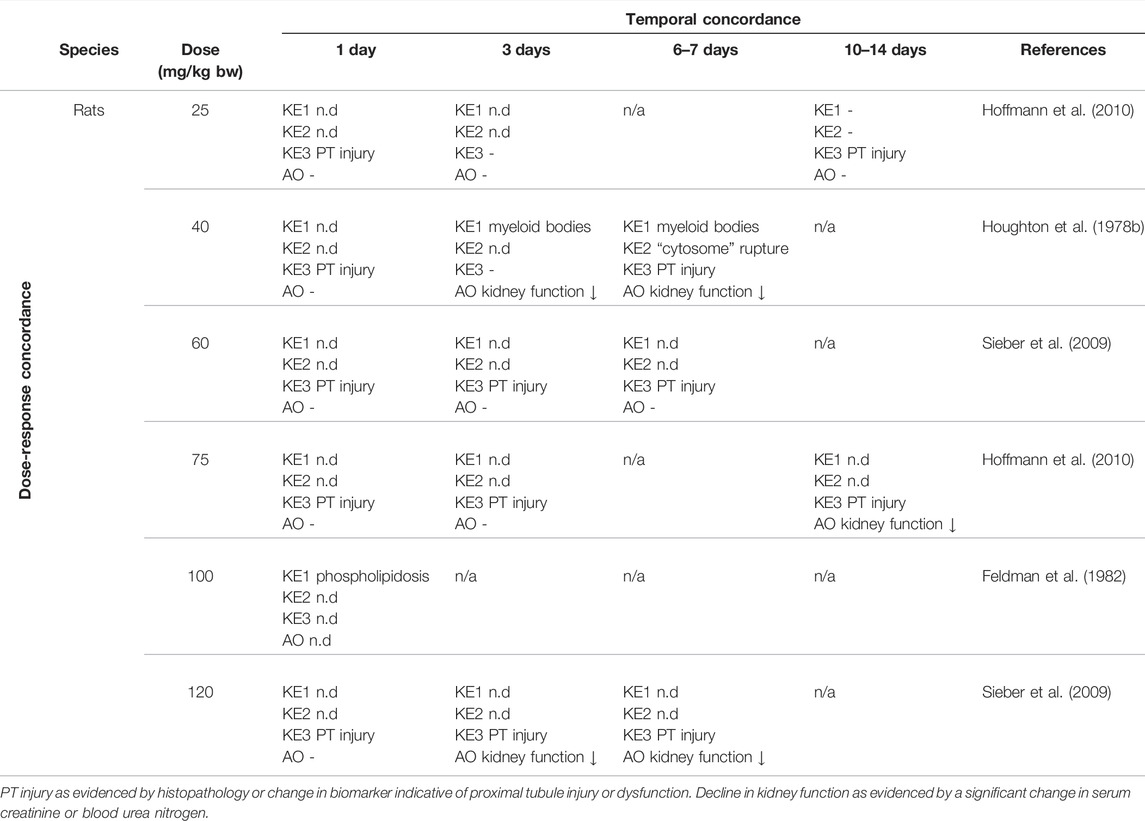
TABLE 6. Dose-Time Concordance of KEs based on rodent studies with gentamicin as a specific stressor for the adverse outcome pathway of receptor-mediated endocytosis and lysosomal overload leading to kidney toxicity (n.d. = not determined; n/a = not data available)).
Collectively, the available in vitro and in vivo studies conducted using chemical stressors for this AOP generally demonstrate effects on KEs across the entire AOP at equal doses/concentrations of the stressor, with some evidence for upstream events occurring at lower concentrations than downstream KEs. There are no data that would disagree with the assumption of dose-response concordance. Based on the criteria for assessing AOP (Box 1), the level of confidence for concordance of dose-response can thus be regarded as high.
There are some studies investigating the time-course of aminoglycoside nephrotoxicity in rats that collectively support the temporal sequence of KEs in this AOP (Table 6). While many of these studies focused on analysing the time-course of aminoglycoside-induced histopathological changes and impact on kidney function, there are also some studies that demonstrate that injury to the convoluted proximal tubule occurs subsequent to lysosomal changes. In Fischer F344 rats treated with either gentamicin or tobramycin, ultrastructural changes, most prominently vacuolar structures containing myeloid bodies (referred to as cytosomes by the authors) were recorded in proximal tubules prior to any other evidence of injury (Houghton et al., 1978b). In this study, increased numbers of “cytosomes” and “cytosomal” rupture were observed concomitant with tubule cell injury (Houghton et al., 1978b). In a further study in rats, phospholipidosis was detected in the renal cortex within 24 h of a single dose of gentamicin or netilmicin, leading the authors to conclude that alterations in phosholipid metabolism are an early event in the pathogenesis of aminoglycoside toxicity that precedes signs of tubule injury (Feldman et al., 1982). In vitro, lysosomal membrane permeabilization in gentamicin-treated renal LLC-PK1 cells was shown to precede mitochondrial changes and apoptosis (Servais et al., 2005; Denamur et al., 2011). Similarly, recent time-resolved analyses conducted within the frame of the Risk-IT project showed that cytotoxicity induced by polymyxin B was preceded by a decrease in lysosomal number, thus supporting the temporal sequence of events within this AOP (Jarzina et al., 2022).
Further support for the sequence of KEs comes from studies in patients receiving aminoglycoside therapy. A retrospective analysis of renal biopsies obtained from patients that received gentamicin within 6 weeks of biopsy reported ultrastructural changes in renal proximal tubule lysosomes in the absence of clinical signs of nephrotoxicity (Houghton et al., 1978a). Similarly, early lysosomal changes were observed in proximal tubular cells of patients receiving therapeutic doses of aminoglycosides for four consecutive days prior to nephrectomy (De Broe et al., 1984). Although no additional histopathological or clinical chemistry data were reported, the authors suggested that these lysosomal alterations occur before the onset of excretory failure (De Broe et al., 1984). In urine samples obtained from 20 patients receiving aminoglycoside therapy for 3–26 days, myeloid bodies were found in urinary sediment irrespective of whether or not the patient developed acute renal failure, although the number of myeloid bodies was increased in patients with acute renal failure (Mandal et al., 1987). In contrast, both the incidence of the appearance of renal tubule cells in the urinary sediment and their number was significantly increased in patients with aminoglycoside-induced renal failure as compared to the non-renal failure group, thus supporting the temporal sequence of events leading from lysosomal alterations to proximal tubule dysfunction and necrosis and ultimately renal excretory failure (Mandal et al., 1987).
Increased activity of the lysosomal enzyme N-acetyl-β-d-glucosaminidase (NAG) was observed in urine of patients treated with tobramycin or colistin in the absence of changes in sCrea and BUN (Etherington et al., 2007).
Weight-Of-Evidence Analysis
Based on biological plausibility and empirical support, the overall weight-of-evidence of KERs in this AOP can be considered as high (Table 5).
3.5 Quantitative and Temporal Understanding of Key Event Relationships
Based on the available literature, there is at present little or no quantitative information on the response-response relationship between two pairs of KEs in this AOP.
KER1: Receptor-Mediated Endocytosis Leading to Disturbance of Lysosomal Function
Numerous studies demonstrate that inhibition of ligand binding and receptor-mediated endocytosis reduces toxicity of the chemical stressor. However, there are no data to describe the quantitative relationship between receptor-mediated endocytosis and disturbance of lysosomal function. While being an essential and thus indispensable component in this AOP, the MIE receptor-mediated endocytosis directly links toxicokinetics to molecular and cellular responses. As such, the relationship between the MIE and KE1 may no longer be chemical-agnostic when moving from qualitative descriptions to quantitiative AOPs. Rather, determination of intralysosomal or intracellular accumulation of the stressor may present the best quantifiable measure of receptor-mediated endocytosis, whereby the intracellular stressor concentration necessary to impair lysosomal function may vary between stressors. Not suprisingly, within a group of structurally related compounds such as polymyxin antibiotics, there is a positive correlation between biological responses and affinity to endocytic receptors at the brush boarder membrane or renal accumulation (Vaara et al., 2008; Keirstead et al., 2014; Jarzina et al., 2022).
KER2: Disturbance of Lysosomal Function Leading to Disruption of Lysosomes
There is as yet little information as to the degree of disturbance of lysosomal function necessary to cause permeabilization of lysosomal membranes and release of lysosomal content. Within the Risk-IT project, the response-response relationship betweeen lyososmal membrane associated proteins (LAMP-1/2), reflecting disturbed lysosomal function (KE1), and release of cathepsin D from lysosomes as an endpoint reflecting lysosomal disruption (KE2) was established from experimental data on polymyxin B and successfully employed to predict the downstreamt KE of structural analogs based on experimental KE1 data (Jarzina et al., 2022).
KER3: Disruption of Lysosomes Leading to Increased Proximal Tubule Cytotoxicity
While a response-response relationship between lysosomal permeabilization and proximal tubule cytotoxicity has not yet been established, a study investigating controlled lysosomal rupture by the synthetic lysosomotropic detergent O-methyl-serine dodecylamide hydrochloride (MSDH) in a murine macrophage cell line provides important quantitative and temporal information on this KER (Li et al., 2000). At low concentrations of the lysosomotropic detergent, lysosomal membrane destabilization was observed by reduced acridine orange fluoresence intensity and granularity scoring of cathepsin D immunoreactivity. Lysosomal leakage preceded morphological signs of apoptosis, activation of caspase-3-like proteases and mitochondrial changes, indicating that cell death occurred secondary to partial lysosomal rupture, presumably due to the apoptotic role of lysosomal proteases (Li et al., 2000; Turk et al., 2002). In contrast, extensive lysosomal rupture, e.g., induced by high concentrations of MSDH, results in necrosis as the predominant type of cell death (Li et al., 2000; Turk et al., 2002). In an attempt to define the quantitative relationship between disruption of lysosomes and proximal tubule cytotoxicity, the response-response between cathepsin D release from lysosomes and cytotoxicity was established from experimental data on polymyxin B. Although there was some concern regarding the reliability of the in vitro cathepsin assay, the response-response relationship was successfully employed for prediction of cytotoxicity of structural analogs (Jarzina et al., 2022).
KER4: Proximal Tubule Cytotoxicity Leading to Kidney Toxicity
Considering that proximal tubule epithelial cell injury is a common key event involved in various AOPs that lead to acute and/or chronic kidney injury, Gebremichael et al. (2018) developed a multiscale quantitative systems pharmacology model to relate drug induced proximal tubule cell injury (i.e., a celluar event) to renal dysfuction (i.e., an adverse outcome at organ level) (Gebremichael et al., 2018). The model is based on the assumption that “the relationships between cell injury and death and subsequent effects on tubular dysfunction, biomarker expression, and organ-level dysfunction should be independent of the injury mechanism” (Gebremichael et al., 2018) and thus independent of the nephrotoxic agent (i.e., chemical agnostic). Thus, the authors consider that their mathematical model should be applicable to prediction of drug-induced changes in kidney function based on the extent of cell injury and cell death inferred from urinary biomarker responses. The model, which consists of a cellular injury submodel and a systems renal physiology model, was developed using histopathology and biomarker data obtained from a single dose cisplatin study in rats. Model parameters were fitted to the experimentally established time-course of urinary biomarker responses (Kim-1, albumin, glucose, αGST) to determine the fractions of functional, injured and dead proximal tubule cells, and subsequently to simulate serum creatinine levels as a readout for alterations in kidney function. The model was successfully applied not only to predict the serum creatinine time course in response to repeated cisplatin administration but also in response to gentamicin as a structurally unrelated drug based on urinary Kim-1 data. Thus, the developed model holds great promise for translation of time-resolved urinary biomarker data as proxy of proximal tubule cell injury and death into the time-course and severity of proximal tubule injury and organ-level dysfunction. Moreover, combined with quantitative in vitro to in vivo extrapolation, the model may help to bridge the gap between in vitro and in vivo responses by facilitating prediction of kidney injury and dysfunction based on in vitro cytotoxicity data obtained in proximal tubule cells.
4 Renal Protein Alkylation Leading to Kidney Toxicity (AOP-258)
This Adverse Outcome Pathway describes the sequential key events that link protein alkylation to kidney toxicity. It is well established that bioactivation of xenobiotics to reactive intermediates that covalently bind to proteins presents a major mechanism by which xenobiotics may cause proximal tubule injury. Examples for compounds that form covalent protein adducts in proximal tubule cells include haloalkenes (e.g., trichloroethylene, tetrachloroethylene, hexachloro-1,3-butadiene, chloroform), quinones (derived from e.g. hydroquinone, bromobenzene, 4-aminophenol), cephalosporins, and N-(3,5-dichlorophenyl)succinimide (Birner et al., 1994; Lau, 1995; Tune, 1997; Griffin and Harvison, 1998; Kleiner et al., 1998; Pahler et al., 1998) (Figure 6). Covalent interaction of a chemical or a metabolite with cellular proteins represents the molecular initiating event (MIE) that triggers perturbation of cellular functions, of which mitochondrial dysfunction leading to ATP depletion appears to be most critical for proximal tubule cell death by apoptosis and/or necrosis (Figure 7) (Aleo et al., 1991; Groves et al., 1991; Hill et al., 1992; Tune, 1997; Chen et al., 2001). Alternative events that may contribute to toxicity include endoplasmic reticulum (ER) stress, glutathione depletion and oxidative stress (van de Water et al., 1996). Tubular obstruction and inflammatory responses to proximal tubule injury including activation of complement may cause secondary toxicity and thus amplify kidney injury, resulting in a progressive decline in kidney function (evidenced by e.g. rise in sCrea and BUN).
FIGURE 6. Chemical structures of nephrotoxic haloalkenes (A) and phenols and heteroatom-substituted benzene derivatives that are bioactivated to nephrotoxic quinones (B).
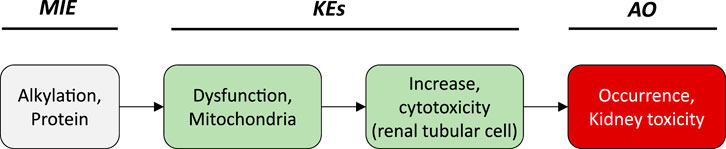
FIGURE 7. Adverse outcome pathway of renal protein alkylation leading to kidney toxicity.
4.1 Nephrotoxicity and Mechanism of Kidney Injury Induced by Agents That Covalently Bind to Proteins
Ever since the pioneering work of the Millers on the role of liver protein alkylation in the carcinogenicity of aminoazo dyes (Miller and Miller, 1947; Miller and Miller, 1966) and subsequent work by Brodie and co-workers demonstrating that covalent protein binding of acetaminophen precedes hepatic necrosis (Jollow et al., 1973), covalent modification of critical target proteins has been established as an important mechanism of toxicity mediated by reactive intermediates. While intial work in this field focussed on liver toxicity and/or carcinogenicity, the concept of protein alkylation leading to toxicity is universal and not restricted to a particular target tissue. Rather, it is often the site of bioactivation to a chemically reactive intermediate that determines the site of toxicity. Selective nephrotoxicity of several haloalkenes, such as hexachlorobutadiene (HCB), trichloroethene (TRI) and perchloroethene (PER), and hydroquinones and aminophenols frequently involves formation of glutathione-S-conjugates (Dekant, 2001). Although conjugation with glutathione (GSH) occurs largely in the liver, it serves to direct xenobiotics to the kidneys. By the sequential action of γ-glutamyltransferase (GGT) and aminopeptidases/dipeptidases that are abundantly expressed by renal tubule cells, glutathione S-conjugates delivered to the kidneys are cleaved to the corresponding cysteine S-conjugates. Following active uptake into kidney epithelial cells via organic-anion transporters, the cysteine S-conjugates of haloalkenes may undergo cysteine conjugate β-lyase-mediated cleavage to a reactive intermediate capable of covalent binding to cellular proteins. In contrast, the nephrotoxicity of aminophenol and hydroquinone S-conjugates does not depend on β-lyases but appears to be linked to oxidation to a reactive quinone (Dekant, 2001).
Hexachloro-1,3-butadiene (HCBD) is a persistent organic pollutant and by-product in the production of various chlorinated hydrocarbons that was previously used as a pesticide and component of transformer, hydraulic and heat-transfer liquids. Renal toxicity and carcinogenicity of HCBD observed in experimental animals (Kociba et al., 1977a; Kociba et al., 1977b; NTP, 1991) has been linked to GSH-mediated biotransformation in the liver, yielding 1-(glutathion-S-yl)-1,2,3,4,4-pentachlorobutadiene (PCBG), subsequent translocation of the GSH-conjugate to the kidneys and processing by GGT and dipeptidases to the corresponding cysteine S-conjugate, S-(1,2,3,4,4-pentachloro-1:3-butadienyl)-l-cysteine (PCBC) (Dekant et al., 1990). Uptake of PCBC into proximal tubule cells and renal cysteine-conjugate β-lyase mediated cleavage gives rise to reactive intermediates that may bind to tissue nucleophiles (Dekant et al., 1990).
Trichloroethylene (TRI) and tetrachloroethylene (PER) are haloalkenes that are widely used as industrial solvents, e.g., for degreasing metals and dry-cleaning fabrics. TRI is nephrotoxic and carcinogenic based on experimental evidence in laboratory animals and epidemiological human data, while PER is nephrotoxic and considered likely to be a human carcinogen (NTP, 1986, 1988; NTP, 1990; IARC, 2014). Similar to HCBD, TRI and PER are bioactivated to reactive intermediates via glutathione-S-transferase-mediated GSH conjugation in liver to form S-(1,2-dichlorovinyl)-glutathione (DCVG) and S-(1,2,2-trichlorovinyl)-glutathione (TCVG), respectively. Following transport to the kidney and renal processing of the GSH conjugates to S-(1,2-dichlorovinyl)-l-cysteine (DCVC) and S-(1,2,2-trichlorovinyl)-l-cysteine (TCVC), respectively, DCVC and TCVC are taken up into proximal tubule cells and bioactivated to a reactive thioketene via cysteine-conjugate β-lyases.
Hydroquinone (HQ) is an intermediate used in the chemical industry that has been identified as a nephrotoxin and renal carcinogen in rodents (NTP, 1989). It is metabolized in the liver by cytochrome P450 enzymes to 1,4-benzoquinone (1,4-BQ), which reacts with GSH to form 2-(glutathion-S-yl)HQ. Further oxidation and conjugation reactions with GSH addition lead to formation of 2,3,5-tris-(glutathion-S-yl)HQ (TGHQ), a potent nephrotoxic metabolite of HQ. It is thought that site-specific toxicity of TGHQ to S3 proximal tubule cells is linked to uptake in form of its cysteine conjugate, intracellular redox cycling and protein adduction (Labenski et al., 2011).
The industrial chemical bromobenzene produces hepatotoxicity and nephrotoxicity. Bromobenzene is bioactivated via cytochrome P450 enzymes to bromophenol and subsequently to 2-bromohydroquinone (BHQ), which reacts with GSH to form three positional isomers of 2-bromo-(glutathione-S-yl)hydroquinone as well as a nephrotoxic bisglutathione conjugate, 2-bromo-bis(glutathione-S-yl)hydroquinone. The organ-specific toxicity of bromobenzene in the kidney has been linked to transport of the GSH-conjugates to the kidney and conversion of the corresponding cystein conjugates. Covalent binding of BHQ and quinol-thiother-derived covalent protein adducts have been found in kidney tubule cells (Schnellmann et al., 1989; Rodeheaver and Schnellmann, 1991) and/or kidneys of rats exposed to BHQ (Kleiner et al., 1998).
4-Aminophenol, used as a photographic developer, for dyeing textiles, hair and furs, and an intermediate in the manufacture of pharmaceuticals and azo dyes, was shown to cause site-specific toxicity to the proximal tubule epithelium (Green et al., 1969). 4-Aminophenol is metabolized in the liver via GSH-dependent pathways, giving rise to toxic GSH conjugates, including 4-amino-3-(glutathion-S-yl)phenol, 4-amino-2,5-bis(glutathion-S-yl)phenol, and 4-amino-2,3,5(or6)-tris(glutathion-S-yl)phenol) (Klos et al., 1992). Delivery of the GSH-conjugates to the kidneys, processing by GGT and dipeptidases, uptake into S3 proximal tubule cells as cysteine conjugates, and intrarenal oxidation to electrophilic quinone imines that covalently bind to tissue nucleophils are considered to be responsible for 4-aminophenol nephrotoxicity (Klos et al., 1992; Dekant, 2001).
4.2 The Adverse Outcome Pathway of Renal Protein Alkylation Leading to Kidney Toxicity
Molecular Initiating Event: Protein Alkylation
In vitro and in vivo studies using radiolabeled compounds or immunochemical approaches provide clear evidence for covalent binding of chemically reactive intermediates of chemical stressors of this AOP to kidney proteins (Tables 7A–E). Covalent binding to proteins of the proximal tubule is consistent with the formation of reactive metabolites of the chemical stressors for this AOP, and corresponds to the site of uptake and/or bioactivation. Inhibition of metabolic pathways that lead to reactive metabolite formation has been shown to block covalent binding to renal proteins, as exemplified by inhibition of covalent protein binding by aminooxyacetic acid, which blocks β-lyase mediated cleavage of S-(pentachlorbutadienyl)-cysteine and S-(1,2-dichlorovinyl)-cysteine (Hayden and Stevens, 1990; van de Water et al., 1995). There is also evidence from studies on bromobenzene that scavenging of reactive metabolites by GSH inhibits covalent protein binding and protects from mitochondrial toxicity (Lau and Monks, 1987; Schnellmann et al., 1989), further supporting adduction of renal proteins as an initiating event required for nephrotoxicity induced by these compounds. Analyses of the subcellular localization of covalently bound proteins provide evidence that both cytosolic and mitochondrial proteins are targeted by reactive metabolites (Hayden and Stevens, 1990; Birner et al., 1994), consistent with the presence of both cytosolic and mitochondrial cysteine-conjugate β-lyases.
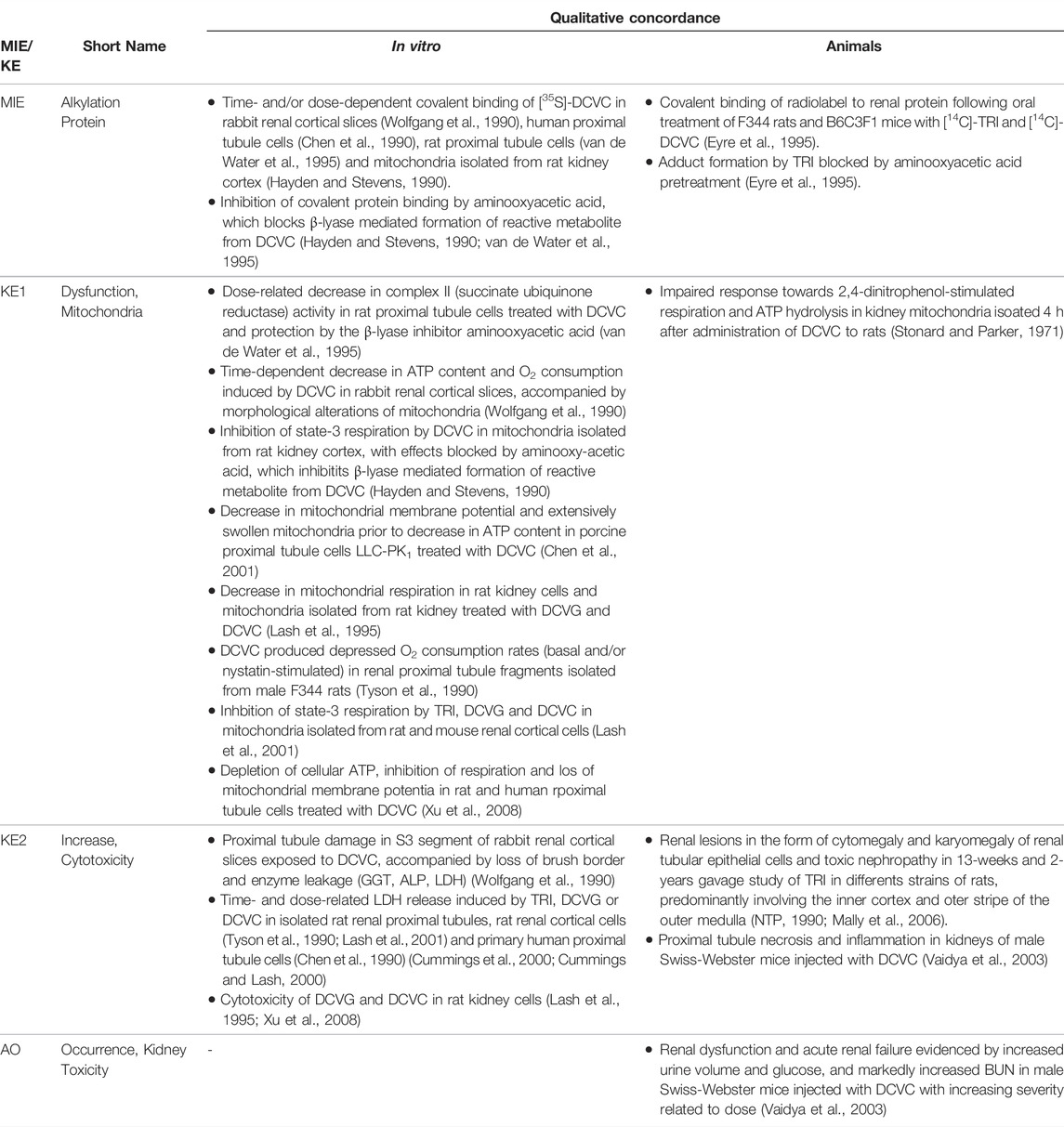
TABLE 7A. Evidence from in vitro and in vivo studies on trichloroethylene (TRI) and its metabolites S-(1,2-dichlorovinyl)-glutathione (DCVG) and S-(1,2-dichlorovinyl)-cysteine (DCVC) supporting the key events and qualitative concordance of KEs within this AOP (n/a = no data available).

TABLE 7B. Evidence from human, animal and in vitro studies on perchloroethylene (PER) and its and its glutathione- and cysteine-S-conjugates TCVG and TCVC supporting the key events and qualitative concordance of KEs within this AOP (n/a = no data available).
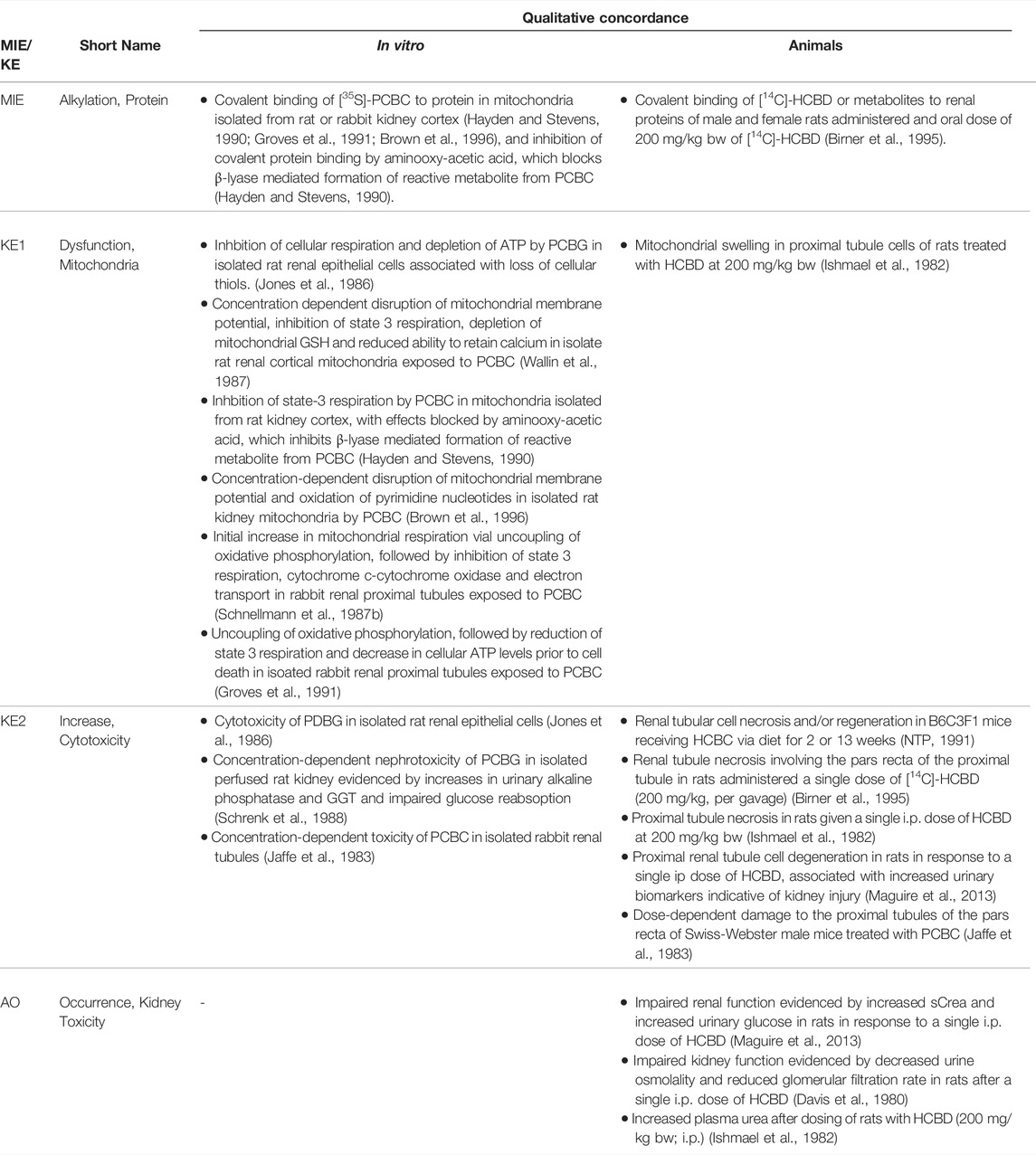
TABLE 7C. Evidence from human, animal and in vitro studies on hexachloro-1,3-butadiene (HCBD) and its glutathione- and cysteine-S-conjugates (S-(pentachlorbutadienyl)glutathione (PCBG) and S-(pentachlorbutadienyl)-cysteine (PCBC)) supporting the key events and qualitative concordance of KEs within this AOP.
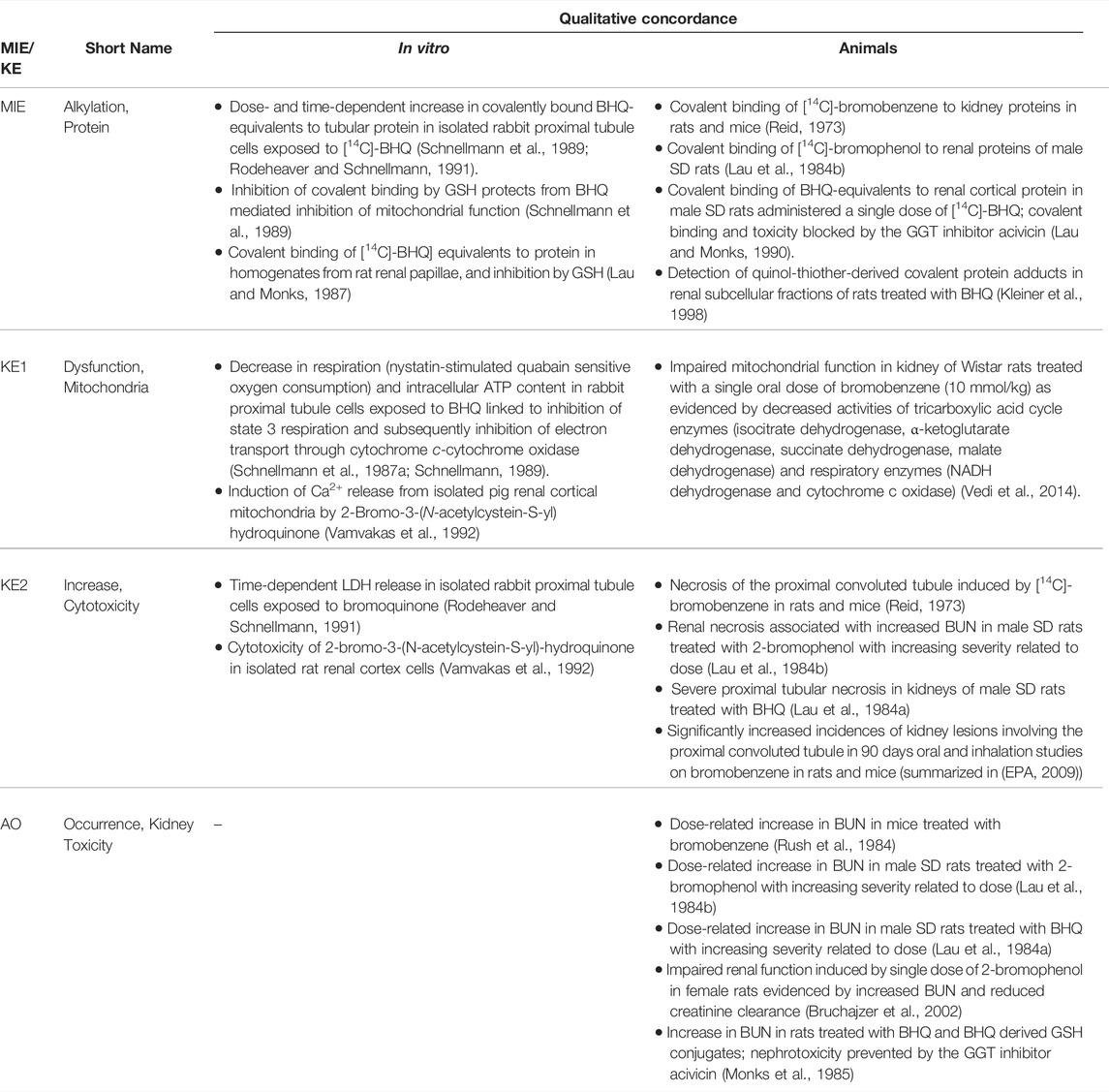
TABLE 7D. Evidence from animal and in vitro studies on bromobenzene and its metabolites 2-bromophenol and 2-bromohydroquinone (BHQ) supporting the key events and qualitative concordance of KEs within this AOP. (Data on human toxicity of bromobenzene are not available (EPA, 2009).).
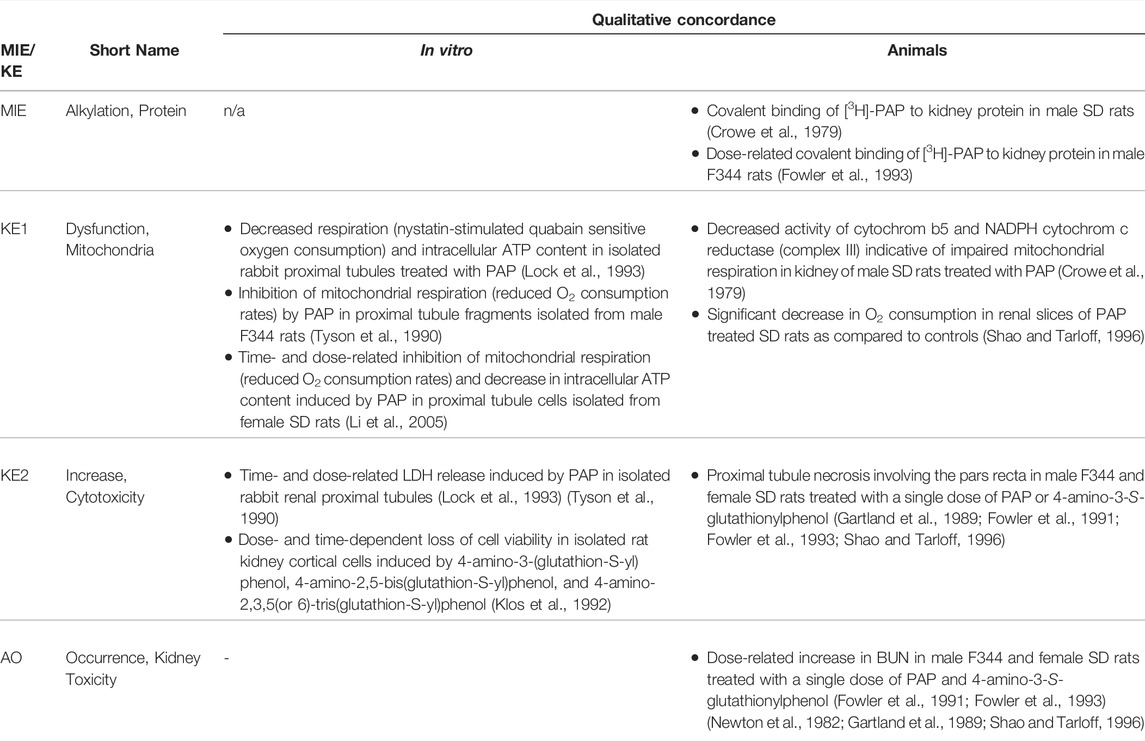
TABLE 7E. Evidence from animal and in vitro studies on 4-aminophenol (PAP) and its nephrotoxic GSH conjugates supporting the key events and qualitative concordance of KEs within this AOP. Human toxicity data on PAP are not available (EPA, 2005). (n/a = no data available).
Studies on tetrafluoroethene and its metabolite S-(1,1,2,2-tetrafluoroethyl)-l-cysteine (TFEC) in rats demonstrated acylation of renal proteins (Harris et al., 1992), with a high specificity for covalent binding to mitochondrial proteins (Hayden et al., 1991). Purification and NH2-terminal sequence analysis identified mitochondrial HSP60/P1-protein and HSP70-like protein (Mortalin) as major targets of TFEC (Bruschi et al., 1993), although it is still unclear if adduction of these proteins contributes to toxicity. As some compounds, such as the nontoxic acetaminophen analogue 3-hydroxyacetanilide, cause covalent binding in the absence of toxicity, it is now recognized that the binding pattern to certain cellular targets rather than the absolute level of binding may encode the biological response (Myers et al., 1995). Considering that different electrophiles preferentially attack different amino acid residues, it is also clear that different reactive metabolites will give rise to differential patterns of target protein modifications and cellular responses. For instance, bromobenzene and its hydroquinone metabolites appear to preferentially alkylate cysteine residues (Slaughter and Hanzlik, 1991), whereas the thioketenes formed by bioactivation of TRI and PER target both cysteine and lysine residues. Overall, specific proteins critical for subsequent cellular responses leading to proximal tubule toxicity remain to be identified. Moreover, the expected differential alkylation of proteins by various electrophiles highlights the necessity to further refine the AOP of renal protein alkylation leading to kidney toxicity based on future understanding of the contribution of different target proteins to toxic outcome.
Key Event 1: Mitochondrial Dysfunction
Evidence for mitochondrial dysfunction as a key event in this AOP comes primarily from a wide range of in vitro studies, consistently demonstrating inhibition of cellular respiration, depletion of ATP and disruption of mitochondrial membrane potential by chemical stressors of this AOP or their nephrotoxic metabolites (Tables 7A–E). These in vitro findings are supported by data from a limited number of in vivo studies, reporting mitochondrial swelling and/or decreased O2 consumption, decreased activity of tricarboxylic acid cycle enzymes and respiratory enzymes indicative of impaired mitochondrial respiration in kidneys of rats treated with a chemical stressor for this AOP (Crowe et al., 1979; Ishmael et al., 1982; Shao and Tarloff, 1996; Vedi et al., 2014). Importantly, the β-lyase inhibitor aminooxyacetic acid, which prevents β-lyase mediated cleavage of cysteine-S-conjugates to reactive intermediates, was shown to block both covalent binding and mitochondrial effects of S-(pentachlorbutadienyl)-cysteine and S-(1,2-dichlorovinyl)-cysteine (Hayden and Stevens, 1990; van de Water et al., 1995), supporting a causal link between reactive metabolite formation, protein adduction and mitochondrial toxicity.
Key Event 2: Increase, Cytotoxicity (Renal Tubular Cell)
It is generally accepted that interference with mitochondrial energy production may lead to cell death via apoptosis or necrosis. Mitochondrial toxicity is recognized as a critical event in drug-induced kidney injury induced by a wide range of chemicals (Gai et al., 2020). Proximal tubule cells highly depend on mitochondria to ensure an adequate ATP supply for active transporters expressed on the basolateral and brush border membrane of proximal tubular cells to facilitate tubular secretion and reabsorption. Inhibition of the triarboxylic acid cycle and the electron transport chain ultimately results in ATP depletion. In addition, opening of the mitochondrial permeability transition pore leads to mitochondrial dysfunction via mitochondrial depolarization, ATP depletion, release of Ca2+ from mitochondria, and inhibition of respiration–mitochondrial changes typically observed in response to chemical stressors of this AOP. There is also ample evidence from in vitro and in vivo studies in rodents for proximal tubule cell toxicity induced by the chemical stressors for this AOP and their respective nephrotoxic metabolites (Tables 7A–E).
Adverse Outcome: Kidney Toxicity
The link between proximal tubule injury and impaired kidney function has already been described in Section 2.3. Renal dysfunction and renal failure evidenced by increased urine volume, BUN or sCrea have been reported in experimental animals and humans exposed to chemical stressors of this AOP (Tables 7A–E).
4.3 Assessment of the Adverse Outcome Pathway of Renal Protein Alkylation Leading to Kidney Toxicity
Biological Plausibility
The covalent binding hypothesis of chemical toxicity, which goes back to the early 1970s, is a well-established principle in toxicology. The sequence of events leading from bioactivation of a xenobiotic to a reactive electrophile, which covalently binds to proteins and alters protein function, to toxicity and cell death is experimentally well supported. There is ample evidence that covalent binding to renal proteins is causally linked to the nephrotoxicity of a range of chemicals. Although there is yet a paucity of information on specific target proteins and their link to impaired mitochondrial function and cell death, the level of confidence in the biological plausibility of key event relationships (KERs) within the proposed AOP can be considered as high (Table 8).
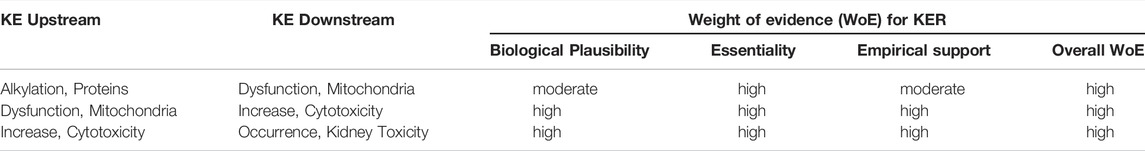
TABLE 8. Weight-of-evidence analysis of KERs in the adverse outcome pathway of renal protein alkylation leading to kidney toxicity.
Essentiality of Key Events
Essentiality of the MIE in this AOP is supported by a number of in vitro and in vivo studies, which demonstrate that inhibition of covalent binding of reactive metabolites to cellular proteins (via enzyme inhibitors that block reactive metabolite formation or scavenging of electrophiles by GSH) protects from mitochondrial toxicity and/or toxicity of chemical stressors for this AOP (Monks et al., 1985; Schnellmann et al., 1989; Hayden and Stevens, 1990; Lau and Monks, 1990; van de Water et al., 1995). As the entire sequence of KE and the AO can be blocked by inhbition of the MIE, there is clear evidence that covalent binding to proteins is essential (Table 8). Using inhibitors of specific mitochondrial processes, Xu et al. (2008) showed that mitochondrial dysfunction is an essential step in cell injury induced by DCVC in human proximal tubule cells (Xu et al., 2008). While these experimental data support the biologically plausible link between adduction of mitochondrial proteins, mitochondrial dysfunction and toxicity, it is important to recognize that covalent protein binding may well affect other organelles and thereby cause toxicity by mechanisms other than mitochondrial dysfunction, e.g. through induction of ER stress. This, however, does not contest the causal relationship between covalent protein binding, mitochondrial dysfunction and toxicity and thus the validity of mitochondrial dysfunction as a KE, but rather suggests that several KEs may branch out of the MIE (covalent protein binding) and combine to cause nephrotoxicity as the AO. The relative contribution of such branches to the overall outcome may differ between chemical stressors and depend on several factors, including the chemical reactivity of the stressor or its metabolite, the dose over time and consequently the target proteins affected by adduction, and their essentiality for cell homeostasis. These considerations are vital when it comes to application of AOPs for toxicity prediction, as the quantitative relationships between the measurable and essential, but yet mechanistically poorly defined MIE and downstream KEs including the AO may not be universal for all stressors that trigger the MIE, as exemplified by the poor correlation between covalent protein binding and hepatotoxicity of the acetaminophen analogue 3-hydroxyacetanilide.
Empirical Evidence: Dose-Response and Temporal Concordance
There is ample experimental evidence to support the temporal sequence of events in this AOP. In kidneys of rodents treated with a single i.p. dose of bromobenzene, covalent protein binding preceded the onset of histopathological lesions (Reid, 1973). Similarly, in isolated rabbit proximal tubules treated with bromohydroquinone, covalent binding to tubular protein and mitochondrial changes occurred rapidly within 15 min and preceded loss of cell viability (Schnellmann et al., 1987a; Schnellmann et al., 1989). Inhibition of mitochondrial respiration and loss of ATP was also shown to occur prior to cell death in rabbit proximal tubule cells treated with 4-aminophenol (Lock et al., 1993).
In a 12 h time-course study of DCVC toxicity in rabbit renal cortical slices, a time-dependent increase in covalent binding of [35S]DCVC was observed between 5 and 120 min. These effects were followed by a decline in mitochondrial function, oxygen consumption and ATP content, which manifested at 4–8h, and histological evidence of S3 proximal tubule injury after 8 h exposure (Wolfgang et al., 1990). Moreover, pulsed versus continuous exposure to DCVC demonstrated that 30 min exposure to DCVC, in which substantial covalent binding occured, was sufficient to trigger proximal tubule toxicity (Wolfgang et al., 1990). In a similar study on the time-course of DCVC toxicity in porcine proximal tubule cells, a decrease in mitochondrial membrane potential was evident at 4h, whereas biochemical changes associated with apoptosis (cytochrome C release, caspase-3 activity, DNA fragmentation) and decreases in cellular ATP manifested at 6–8 h (Chen et al., 2001). Support for mitochondrial dysfunction as an early event that precedes proximal tubule injury also comes from studies on the hexachorobutadiene metabolite S-pentachloro-1,3-butadienyl)-l-cysteine (PCBC) in rabbit renal proximal tubules. Here, mitochondrial changes were recorded within 15 min of exposure, while a decrease in cell viability was evident after 60 min of exposure to PDBC (Schnellmann et al., 1987b).
Although no detailed in vivo dose-response studies are available, covalent binding and proximal tubule damage, or mitochondrial changes, proximal tubule cell necrosis and impaired function were all observed in rats following a single oral or i.p. dose of HCBD at 200 mg/kg bw (Ishmael et al., 1982; Birner et al., 1995). Similarly, covalent binding of [14C]-bromobenzene or [14C]-2-bromophenol to mouse or rat kidney proteins in vivo was recorded at the same dose that resulted in histological evidence of kidney injury (Reid, 1973) or impaired kidney function (Lau et al., 1984a). In vitro studies on DCVC indicate a high concordance between concentrations that cause covalent protein binding, mitochondrial effects and cytotoxicity (van de Water et al., 1995).
Collectively, the available in vitro and in vivo studies conducted using chemical stressors for this AOP support the temporal sequence of KE and demonstrate effects on KEs across the entire AOP at equal doses/concentrations of the stressor. Based on the criteria for assessing AOP (Box 1), the level of confidence for temporal and dose-response concordance can thus be regarded as high (Table 8).
Weight-Of-Evidence Analysis
Based on biological plausibility and empirical support, the overall weight-of-evidence of KERs in this AOP can be considered as high (Table 8).
4.5 Quantitative and Temporal Understanding of Key Event Relationships
Based on the available literature, there is at present little or no quantitative information on the response-response relationship between two pairs of KEs in this AOP.
KER1: Protein Alkylation Leading to Mitochondrial Dysfunction
There is as yet no information regarding the quantitiative response-response relationship between covalent protein binding and mitochondrial dysfunction or toxicity in general. This stems from an insufficient mechanistic understanding of which specific target proteins are critical for toxicity. It is widely appreciated that total covalent protein binding cannot be utilized as a good predictor of cytotoxic potential, but rather that selective binding to critical cellular targets may drive the outcome (Cohen et al., 1997).
KER2: Mitochondrial Dysfunction Leading to Proximal Tubule Cytotoxicity
As outlined in Section 2.5, it is evident that proximal tubule cells depend on cellular respiration and mitochondrial ATP production to provide energy for active transport of solutes. There is, however, no systematic assessment as to how much decline in mitochondrial function or ATP depletion and for how long may be tolerated by a proximal tubule cell before it commits to apoptosis or necrosis.
KER3: Proximal Tubule Cytotoxicity Leading to Kidney Toxicity
Proximal tubule cytotoxicity is a common KE across all three AOPs discussed here. The link between proximal tubule toxicity and impaired kidney function is well established, although it is less clear how much cell killing over time is needed to cause functional impairment. For further considerations, the reader is referred to Sections 2.5 and 3.5.
5 Towards a Network of Adverse Outcome Pathways for Nephrotoxicity and Considerations for Implementation of Adverse Outcome Pathways for Safety Assessment
Herein, we describe a set of AOPs for kidney injury that are triggered by different MIEs but involve proximal tubule toxicity as a common KE. Through shared KEs, our AOPs tie in with a previously established AOP “Alpha2u-microglobulin cytotoxicity leading to renal tubular adenomas and carcinomas (in male rat)” (https://aopwiki.org/aops/105) to build a first network of kidney related AOs (Figure 8), which is expected to be expanded progressively as further AOPs are being developed and validated. Eventually, a comprehensive AOP network may then provide a unique basis for the identification of a battery of in vitro and/or in vivo endpoints that cover the entire mechanistic landscape of chemically-induced kidney injury and can be integrated with endpoints relevant to other target organs into an integrated testing strategy to collectively address repeated dose toxicity. While the AOP on skin sensitisation (OECD, 2014) convincingly demonstrates how mechanistic information systematically captured in form of AOPs can be translated into new test guidelines for a specific hazard endpoint, it is evident that identification and characterization of potential health harzards that arise from repeated exposure and that may affect a broad range of targets of toxicity is increasingly more complex and is still in its infancy. The AOPs developed and evaluated here in view of identification of mechanistically relevant endpoints for renal safety assessment highlight a number of open issues that need to be discussed and addressed by the scientific community on the way to implementation of AOP based testing strategies for assessment of repeated dose toxicity, particularly for regulatory decision making beyond hazard identification.
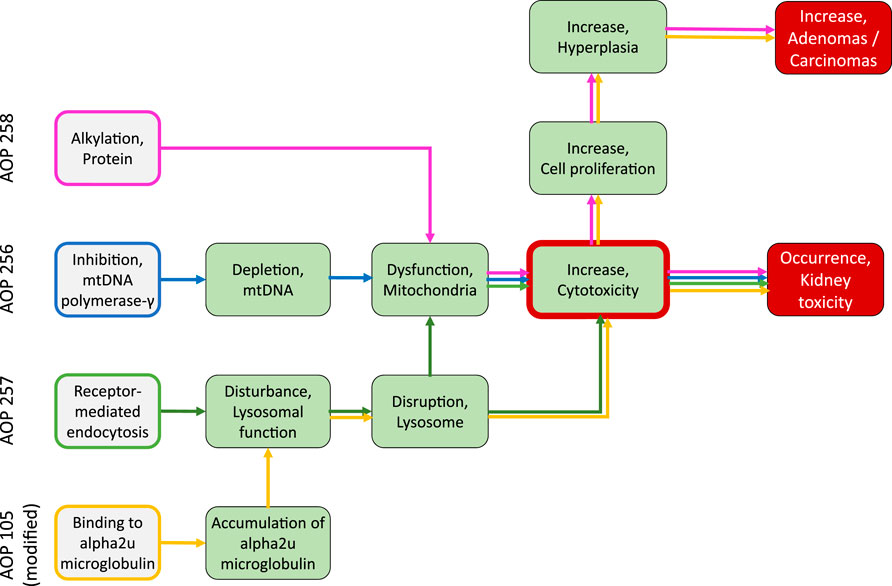
FIGURE 8. An initial network of AOPs leading to kidney toxicity, with mitochondrial dysfunction and renal tubular cell cytotoxicity as common KEs. Sustained renal tubular cell cytotoxicity is also recognized as an important trigger for regenerative cell proliferation, resulting in hyperplasia and ultimately renal tumors. While this has been proposed as an AOP in itself with cytotoxicity irrespective of the primary insult as the MIE, this AOP can be initiated by chemicals that bind to alpha2u macroglobulin in serum in male rats (leading to accumulation of alpha2u macroglobulin in lysosomes of renal tubular cells, associated with disturbance of lysosomal function and ultimately disruption of lysosomes) but also by chemicals that cause sustained cytotoxicity through protein alkylation. The AOP “Alpha2u-microglobulin cytotoxicity leading to renal tubular adenomas and carcinomas (in male rat)” developed by Charles Wood (US EPA) and published in AOPWiki (https://aopwiki.org/aops/105) was slightly extended to highlight overlapping KEs between alpha2u nephropathy and the AOP of receptor-mediated endocytosis leading to kidney toxicity. The colored arrows indicate the pathways that lead from a specific MIE through shared KEs to the respective AO in kidney. While cytotoxicity is central KE in all presented AOPs, it is the extent of cytotoxicity over time that is expected to determine the AO. Note that - with the exemption of “Binding to alpha2u macroglobulin” which occurs in serum–all MIEs and KEs shown relate to renal tubule cells or renal tubules.
The AOPs discussed here cover fairly well established mechanisms by which certain chemicals or drugs are thought to cause nephrotoxicity. Compared to other mechanisms or mode of actions that are less well defined, there is thus a wealth of data derived from multiple stressors to support the MIEs, KEs and KERs. While the overall confidence in these AOPs can be considered as high, there are still data gaps, as exemplified by the as yet insufficient mechanistic understanding of the causal link between alkylation of specific target proteins of reactive metabolites and mitochondrial dysfunction/toxicity. For AOPs to serve as a mechanistic framework to derive suitable endpoints for hazard identification, it may however not be necessary to fully understand and describe in detail all the molecular and cellular events and their causal relationships. Rather, a simple representation of the AOP by a few essential, generalized KEs using harmonized KE umbrella terms may present a pragmatic approach. Thus, even less well defined adverse outcome pathways may be integrated into an AOP network to ensure full coverage of the mechanistic landscape of an adverse outcome, which is essential for future testing approaches. Omitting pathways relevant to a particular health hazard because they are mechanistically poorly understood may otherwise hold the risk of creating critical gaps in future test strategies, which may leave some chemical hazards undetected. It thus appears equally important to integrate as yet ill-defined mechanisms with a low level of confidence as well as well-established mechanisms to obtain a wholistic network view of pathways leading to an adverse outcome. It may, however, be helpful to indicate the level of confidence or uncertainties in the graphical representations of AOPs, including the network view.
On the other hand, development of quantitative AOPs for toxicity prediction may require more specified KE terms rather than KE umbrella terms and also consider potential modulating events. Again, this is exemplified by the poor predictivity of total protein alkylation for toxicity. Understanding which specific protein targets contribute to perturbation of downstream events (in this case mitochondrial dysfunction, cytotoxicity, kidney injury), and to which extent, would be required to allow quantitative predictions. It is conceivable that the overall adverse effect initiated by protein alkylation may involve several pathways in parallel (e.g., covalent binding to ATP synthase leading to ATP depletion; covalent binding to protein thiols involved in redox regulation leading to impaired antioxidant defense). There should be consensus on whether such parallel pathways, that are likely to be affected by all chemical stressors but perhaps to a varying degree, should all form individual AOPs or rather an AOP family tree subsummized under a more generalized MIE term.
Similarly, there needs to be consensus on how to define and represent AOPs that operate in different target organs, such as protein alkylation, which may also cause hepato- and nephrotoxicity, or inhbition of mtPol γ, which has been linked to neurodegeneration, myopathy, cardiotoxicity and hepatotoxicity in addition to nephrotoxicity (Figure 3). Particularly when it comes to quantitative description of KERs, it needs to be considered that the tissue-specific response to a molecular initiating event may depend on the biological context of the cell and organ affected. For instance, the response to inhibition of mtDNA polymerase γ may depend on the rate of mitochondrial biogenesis, mtDNA content and energy demand on the cellular level, as well as on the functional reserve and regenerative capacity of the organ, all of which are tissues-specific (Figure 3). Despite a similar effect on mtPol γ, mitochondrial dysfunction and subsequent events are expected to have more detrimental effects in tissues with a high rate of mitochondrial biogenesis, a high energy demand, low functional reserve and capacity to regenerate. In addition, sex, age, and other susceptibility factors are likely to influence the quantitative KERs. Similarly, the temporal scale of effects may vary between tissues, and potential temporal delays between the MIE and the first KE in this AOP are important to consider when developing KE related endpoints for toxicity testing.
In our AOP-257 we designated receptor-mediated endocytosis as the MIE. As endocytosis facilitates uptake of stressors into the cell, it may rather be seen as part of the toxicokinetics of the stressors rather than an element of an AOP. However, as receptor-mediated endocytosis directs ligands to the lysosomal compartment, it is considered essential for down-stream lysosomal events to occur. However, this illustrates that strict dissociation of toxicokinetics and MIEs at the chemical-biological interface may not always be straightforward.
Finally, while AOPs are by definition chemically agnostic, it is important to realize that empirical support comes primarily from (presumed) chemical stressors of the AOP. Similarly, quantitative KERs are likely to be derived through use of chemical stressors, which poses a source of uncertainty as chemicals often act by more than one mechanism or pathway. An example is hydroquinone, which may covalenty bind to proteins but may also cause oxidative tress through redox cycling. As the relative contribution of multiple pathway to the overall outcome is rarely known even for toxicologically well-characterized chemicals, prediction of the toxicity of chemicals that act by more than one AOP will present a major scientific challenge.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Author Contributions
Conceptualization, AM; methodology, formal analysis and investigation AM; writing—original draft preparation AM; writing—review and editing, AM, SJ; visualization, AM, SJ; acquisition of funding, AM. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded through the “Innovative systems toxicology for alternatives to animal testing” joint funding scheme by ZonMw (Project InnosysTox - Risk-IT) and the German Federal Ministry of Education and Research (BMBF) (Project InnosysTox - Risk-IT: 031L0019A). This publication was supported by the Open Access Publication Fund of the University of Wuerzburg.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors thank all members and external advisors of the Risk-IT project for excellent collaboration and valuable scientific advice.
Abbreviations
AOP, Adverse Outcome Pathway; AO, Adverse Outcome; BHQ, 2-Bromohydroquinone; BUN, Blood Urea Nitrogen; DCVC, S-(1,2-Dichlorovinyl)-cysteine; DCVG, S-(1,2-Dichlorovinyl)-glutathione; HCBD, Hexachloro-1,3-butadiene; KE, Key Event;KER, Key Event Relationship; MIE, Molecular Initiating Event;PAP, 4-Aminophenol; PCBC, S-(Pentachlorbutadienyl)-cysteine;PCBG, S-(Pentachlorbutadienyl)-glutathione; PER,Perchloroethylene; sCrea, Serum Creatinine; TDF, tenofovirDipivoxil Fumarate; TRI, trichloroethylene; TCVC, S-(1,2,2-Trichlorovinyl)-cysteine; TCVG, S-(1,2,2-Trichlorovinyl)- glutathione.
References
Agarwala, R., Mohan, S., Herlitz, L. C., and Cheng, J.-T. (2010). The Case ∣ 41-Year-Old HIV Patient with Proteinuria and Progressive Renal Dysfunction. Kidney Int. 77 (5), 475–476. doi:10.1038/ki.2009.486
Ahmed, M. U., Velkov, T., Zhou, Q. T., Fulcher, A. J., Callaghan, J., Zhou, F., et al. (2019). Intracellular Localization of Polymyxins in Human Alveolar Epithelial Cells. J. Antimicrob. Chemother. 74 (1), 48–57. doi:10.1093/jac/dky409
Aleo, M. D., Rankin, G. O., Cross, T. J., and Schnellmann, R. G. (1991). Toxicity of N-(3,5-dichlorophenyl)succinimide and Metabolites to Rat Renal Proximal Tubules and Mitochondria. Chemico-Biological Interactions 78 (1), 109–121. doi:10.1016/0009-2797(91)90107-i
Alvarado Reyes, Y., Cruz, R., Gonzalez, J., Perez, Y., and Wolowich, W. R. (2019). Incidence of Acute Kidney Injury in Intermittent versus Continuous Infusion of Polymyxin B in Hospitalized Patients. Ann. Pharmacother. 53 (9), 886–893. doi:10.1177/1060028019841898
Amin, R. P., Vickers, A. E., Sistare, F., Thompson, K. L., Roman, R. J., Lawton, M., et al. (2004). Identification of Putative Gene Based Markers of Renal Toxicity. Environ. Health Perspect. 112 (4), 465–479. doi:10.1289/ehp.6683
Antoine, D. J., Srivastava, A., Pirmohamed, M., and Park, B. K. (2010). Statins Inhibit Aminoglycoside Accumulation and Cytotoxicity to Renal Proximal Tubule Cells. Biochem. Pharmacol. 79 (4), 647–654. doi:10.1016/j.bcp.2009.09.021
Apostolova, N., Blas-García, A., and Esplugues, J. V. (2011). Mitochondrial Interference by Anti-HIV Drugs: Mechanisms beyond Pol-γ Inhibition. Trends Pharmacol. Sci. 32 (12), 715–725. doi:10.1016/j.tips.2011.07.007
Arnaudo, E., Shanske, S., DiMauro, S., Schon, E. A., Moraes, C. T., Schon, E. A., et al. (1991). Depletion of Muscle Mitochondrial DNA in AIDS Patients with Zidovudine-Induced Myopathy. The Lancet 337 (8740), 508–510. doi:10.1016/0140-6736(91)91294-5
Aubert-Tulkens, G., Van Hoof, F., and Tulkens, P. (1979). Gentamicin-induced Lysosomal Phospholipidosis in Cultured Rat Fibroblasts. Quantitative Ultrastructural and Biochemical Study. Lab. Invest. 40 (4), 481–491.
Avedissian, S. N., Liu, J., Rhodes, N. J., Lee, A., Pais, G. M., Hauser, A. R., et al. (2019). A Review of the Clinical Pharmacokinetics of Polymyxin B. Antibiotics 8 (1), 31. doi:10.3390/antibiotics8010031
Azad, M. A. K., Nation, R. L., Velkov, T., and Li, J. (2019). Mechanisms of Polymyxin-Induced Nephrotoxicity. Adv. Exp. Med. Biol. 1145, 305–319. doi:10.1007/978-3-030-16373-0_18
Balzarini, J., Naesens, L., Herdewijn, P., Rosenberg, I., Holy, A., Pauwels, R., et al. (1989). Marked In Vivo Antiretrovirus Activity of 9-(2-phosphonylmethoxyethyl)adenine, a Selective Anti-human Immunodeficiency Virus Agent. Proc. Natl. Acad. Sci. U.S.A. 86 (1), 332–336. doi:10.1073/pnas.86.1.332
Batuman, V. (2006). Proximal Tubular Injury in Myeloma. Contrib. Nephrol. 153, 87–104. doi:10.1159/000096762
Beauchamp, D., Gourde, P., Simard, M., and Bergeron, M. G. (1992). Subcellular Localization of Tobramycin and Vancomycin Given Alone and in Combination in Proximal Tubular Cells, Determined by Immunogold Labeling. Antimicrob. Agents Chemother. 36 (10), 2204–2210. doi:10.1128/aac.36.10.2204
Becker, R. A., Ankley, G. T., Edwards, S. W., Kennedy, S. W., Linkov, I., Meek, B., et al. (2015). Increasing Scientific Confidence in Adverse Outcome Pathways: Application of Tailored Bradford-Hill Considerations for Evaluating Weight of Evidence. Regul. Toxicol. Pharmacol. 72 (3), 514–537. doi:10.1016/j.yrtph.2015.04.004
Belen, C., Budhiraja, P., Bracamonte, E., and Popovtzer, M. (2012). Biopsy-proven Acute Tubular Necrosis Associated with Vancomycin in an Adult Patient. Ren. Fail. 34 (4), 502–505. doi:10.3109/0886022X.2012.655683
Bertino, J. S., Booker, L. A., Franck, P. A., Jenkins, P. L., Franck, K. R., and Nafziger, A. N. (1993). Incidence of and Significant Risk Factors for Aminoglycoside-Associated Nephrotoxicity in Patients Dosed by Using Individualized Pharmacokinetic Monitoring. J. Infect. Dis. 167 (1), 173–179. doi:10.1093/infdis/167.1.173
Birkus, G., Hitchcock, M. J. M., and Cihlar, T. (2002). Assessment of Mitochondrial Toxicity in Human Cells Treated with Tenofovir: Comparison with Other Nucleoside Reverse Transcriptase Inhibitors. Antimicrob. Agents Chemother. 46 (3), 716–723. doi:10.1128/aac.46.3.716-723.2002
Birner, G., Richling, C., Henschler, D., Anders, M. W., and Dekant, W. (1994). Metabolism of Tetrachloroethene in Rats: Identification of N.epsilon.-(Dichloroacetyl)-L-Lysine and N.epsilon.-(Trichloroacetyl)-L-Lysine as Protein Adducts. Chem. Res. Toxicol. 7 (6), 724–732. doi:10.1021/tx00042a003
Birner, G., Werner, M., Ott, M. M., and Dekant, W. (1995). Sex Differences in Hexachlorobutadiene Biotransformation and Nephrotoxicity. Toxicol. Appl. Pharmacol. 132 (2), 203–212. doi:10.1006/taap.1995.1100
Bischofberger, N., Hitchcock, M. J., Chen, M. S., Barkhimer, D. B., Cundy, K. C., Kent, K. M., et al. (1994). 1-[((S)-2-hydroxy-2-oxo-1,4,2-dioxaphosphorinan-5-yl)methyl] Cytosine, an Intracellular Prodrug for (S)-1-(3-hydroxy-2-phosphonylmethoxypropyl)cytosine with Improved Therapeutic index In Vivo. Antimicrob. Agents Chemother. 38 (10), 2387–2391. doi:10.1128/aac.38.10.2387
Brown, P. C., Sokolove, P. M., McCann, D. J., Stevens, J. L., and Jones, T. W. (1996). Induction of a Permeability Transition in Rat Kidney Mitochondria by Pentachlorobutadienyl Cysteine: A β-Lyase-Independent Process. Arch. Biochem. Biophys. 331 (2), 225–231. doi:10.1006/abbi.1996.0302
Bruchajzer, E., Szymanska, J. A., and Piotrowski, J. K. (2002). Acute and Subacute Nephrotoxicity of 2-bromophenol in Rats. Toxicol. Lett. 134(1-3), 245–2a52. doi:10.1016/s0378-4274(02)00172-8
Bruschi, S. A., West, K. A., Crabb, J. W., Gupta, R. S., and Stevens, J. L. (1993). Mitochondrial HSP60 (P1 Protein) and a HSP70-like Protein (Mortalin) Are Major Targets for Modification during S-(1,1,2,2-tetrafluoroethyl)-L-cysteine-induced Nephrotoxicity. J. Biol. Chem. 268 (31), 23157–23161. doi:10.1016/s0021-9258(19)49440-4
Bursch, W. (2001). The Autophagosomal-Lysosomal Compartment in Programmed Cell Death. Cell Death Differ 8 (6), 569–581. doi:10.1038/sj.cdd.4400852
Calvert, G. M., Ruder, A. M., and Petersen, M. R. (2011). Mortality and End-Stage Renal Disease Incidence Among Dry Cleaning Workers. Occup. Environ. Med. 68 (10), 709–716. doi:10.1136/oem.2010.060665
Che, R., Yuan, Y., Huang, S., and Zhang, A. (2014). Mitochondrial Dysfunction in the Pathophysiology of Renal Diseases. Am. J. Physiology-Renal Physiol. 306 (4), F367–F378. doi:10.1152/ajprenal.00571.2013
Chen, J. C., Stevens, J. L., Trifillis, A. L., and Jones, T. W. (1990). Renal Cysteine Conjugate β-lyase-mediated Toxicity Studied with Primary Cultures of Human Proximal Tubular Cells. Toxicol. Appl. Pharmacol. 103 (3), 463–473. doi:10.1016/0041-008x(90)90319-p
Chen, Y., Cai, J., Anders, M. W., Stevens, J. L., and Jones, D. P. (2001). Role of Mitochondrial Dysfunction in S-(1,2-dichlorovinyl)-l-cysteine-induced Apoptosis. Toxicol. Appl. Pharmacol. 170 (3), 172–180. doi:10.1006/taap.2000.9107
Cherrington, J. M., Allen, S. J., McKee, B. H., and Chen, M. S. (1994). Kinetic Analysis of the Interaction between the Diphosphate of (S)-1-(3-hydroxy-2-phosphonylemthoxypropyl)cytosine, ddCTP, AZTTP, and FIAUTP with Human DNA Polymerases β and γ. Biochem. Pharmacol. 48 (10), 1986–1988. doi:10.1016/0006-2952(94)90600-9
Cherrington, J. M., Allen, S. J. W., Bischofberger, N., and Chen, M. S. (1995). Kinetic Interaction of the Diphosphates of 9-(2-phosphonylmethoxyethyl)adenine and Other Anti-HIV Active Purine Congeners with HIV Reverse Transcriptase and Human DNA Polymerases α, β and γ. Antivir. Chem. Chemother. 6 (4), 217–221. doi:10.1177/095632029500600403
Choi, Y. H., Kim, N., Seo, Y. S., Choi, S. J., Yang, J. O., Lee, E.-Y., et al. (2003). ARF Requiring Hemodialysis after Accidental Perchloroethylene Ingestion. Am. J. Kidney Dis. 41 (3), 1–e11. doi:10.1053/ajkd.2003.50138
Cohen, B. H., Chinnery, P. F., and Copeland, W. C. (1993). "POLG-related Disorders," in GeneReviews((R)), eds. M. P. Adam, H. H. Ardinger, R. A. Pagon, S. E. Wallace, L. J. H. Bean, and K. Stephens.(Seattle (WA)).
Cohen, S. D., Pumford, N. R., Khairallah, E. A., Boekelheide, K., Pohl, L. R., Amouzadeh, H. R., et al. (1997). Selective Protein Covalent Binding and Target Organ Toxicity. Toxicol. Appl. Pharmacol. 143 (1), 1–12. doi:10.1006/taap.1996.8074
Côté, H. C., Magil, A. B., Harris, M., Scarth, B. J., Gadawski, I., Wang, N., et al. (2006). Exploring Mitochondrial Nephrotoxicity as a Potential Mechanism of Kidney Dysfunction Among HIV-Infected Patients on Highly Active Antiretroviral Therapy. Antivir. Ther. 11 (1), 79–86.
Coughlan, M. T., Nguyen, T.-V., Penfold, S. A., Higgins, G. C., Thallas-Bonke, V., Tan, S. M., et al. (2016). Mapping Time-Course Mitochondrial Adaptations in the Kidney in Experimental Diabetes. Clin. Sci. (Lond) 130 (9), 711–720. doi:10.1042/CS20150838
Crowe, C. A., Yong, A. C., Calder, I. C., Ham, K. N., and Tange, J. D. (1979). The Nephrotoxicity of P-Aminophenol. I. The Effect on Microsomal Cytochromes, Glutathione and Covalent Binding in Kidney and Liver. Chem. Biol. Interact 27(2-3), 235–2a43. doi:10.1016/0009-2797(79)90128-5
Cummings, B. S., Zangar, R. C., Novak, R. F., and Lash, L. H. (2000). Cytotoxicity of Trichloroethylene and S-(1, 2-Dichlorovinyl)-L-Cysteine in Primary Cultures of Rat Renal Proximal Tubular and Distal Tubular Cells. Toxicology 150 (1-3), 83–98. doi:10.1016/s0300-483x(00)00252-3
Cummings, B. S., and Lash, L. H. (2000). Metabolism and Toxicity of Trichloroethylene and S-(1,2-dichlorovinyl)-L-cysteine in Freshly Isolated Human Proximal Tubular Cells. Toxicol. Sci. 53 (2), 458–466. doi:10.1093/toxsci/53.2.458
Dagil, R., O'Shea, C., Nykjær, A., Bonvin, A. M. J. J., and Kragelund, B. B. (2013). Gentamicin Binds to the Megalin Receptor as a Competitive Inhibitor Using the Common Ligand Binding Motif of Complement Type Repeats. J. Biol. Chem. 288 (6), 4424–4435. doi:10.1074/jbc.M112.434159
Dai, C., Li, J., Tang, S., Li, J., and Xiao, X. (2014). Colistin-induced Nephrotoxicity in Mice Involves the Mitochondrial, Death Receptor, and Endoplasmic Reticulum Pathways. Antimicrob. Agents Chemother. 58 (7), 4075–4085. doi:10.1128/AAC.00070-14
Davis, M. E., Berndt, W. O., and Mehendale, H. M. (1980). Disposition and Nephrotoxicity of Hexachloro-1,3-Butadiene. Toxicology 16 (3), 179–191. doi:10.1016/0300-483x(80)90115-8
De Broe, M. E., Paulus, G. J., Verpooten, G. A., Roels, F., Buyssens, N., Wedeen, R., et al. (1984). Early Effects of Gentamicin, Tobramycin, and Amikacin on the Human Kidney. Kidney Int. 25 (4), 643–652. doi:10.1038/ki.1984.69
Deeks, S. G., Collier, A., Lalezari, J., Pavia, A., Rodrigue, D., Drew, W. L., et al. (1997). The Safety and Efficacy of Adefovir Dipivoxil, a Novel Anti‐Human Immunodeficiency Virus (HIV) Therapy, in HIV‐Infected Adults: A Randomized, Double‐Blind, Placebo‐Controlled Trial. J. Infect. Dis. 176 (6), 1517–1523. doi:10.1086/514150
Dekant, W. (2001). Chemical-induced Nephrotoxicity Mediated by Glutathione S-Conjugate Formation. Toxicol. Lett. 124 (1-3), 21–36. doi:10.1016/s0378-4274(00)00285-x
Dekant, W., Vamvakas, S., and Anders, M. W. (1990). Bioactivation of Hexachlorobutadiene by Glutathione Conjugation. Food Chem. Toxicol. 28 (4), 285–293. doi:10.1016/0278-6915(90)90041-k
Denamur, S., Tyteca, D., Marchand-Brynaert, J., Van Bambeke, F., Tulkens, P. M., Courtoy, P. J., et al. (2011). Role of Oxidative Stress in Lysosomal Membrane Permeabilization and Apoptosis Induced by Gentamicin, an Aminoglycoside Antibiotic. Free Radic. Biol. Med. 51 (9), 1656–1665. doi:10.1016/j.freeradbiomed.2011.07.015
Eddy, A. A. (1989). Interstitial Nephritis Induced by Protein-Overload Proteinuria. Am. J. Pathol. 135 (4), 719–733.
Eisenberg, E. J., Lynch, G. R., Bidgood, A. M., Krishnamurty, K., and Cundy, K. C. (1998). Isolation and Identification of a Metabolite of Cidofovir from Rat Kidney. J. Pharm. Biomed. Anal. 16 (8), 1349–1356. doi:10.1016/s0731-7085(97)00162-3
Elyasi, S., Khalili, H., Dashti-Khavidaki, S., and Mohammadpour, A. (2012). Vancomycin-induced Nephrotoxicity: Mechanism, Incidence, Risk Factors and Special Populations. A Literature Review. Eur. J. Clin. Pharmacol. 68 (9), 1243–1255. doi:10.1007/s00228-012-1259-9
Emma, F., Bertini, E., Salviati, L., and Montini, G. (2012). Renal Involvement in Mitochondrial Cytopathies. Pediatr. Nephrol. 27 (4), 539–550. doi:10.1007/s00467-011-1926-6
EPA (2005). in Provisional Peer Reviewed Toxicity Values for P-Aminophenol (CASRN 123-30-8)National Center for Environmental Assessment, Superfund Health Risk Technical Support Center, Office of Research and Development (Cincinnati, OH: U.S. Environmental Protection Agency). Superfund Health Risk Technical Support Center.
EPA (2009). Toxicological Review of Bromobenzene (CAS No. 108-86-1) in Support of Summary Information on the Integrated Risk Information System (IRIS). U.S.E.P. Agency. Washington, DC: U.S. Environmental Protection Agency.
Erkan, E., De Leon, M., and Devarajan, P. (2001). Albumin Overload Induces Apoptosis in LLC-PK1cells. Am. J. Physiology-Renal Physiol. 280 (6), F1107–F1114. doi:10.1152/ajprenal.2001.280.6.F1107
Erkan, E., Garcia, C. D., Patterson, L. T., Mishra, J., Mitsnefes, M. M., Kaskel, F. J., et al. (2005). Induction of Renal Tubular Cell Apoptosis in Focal Segmental Glomerulosclerosis: Roles of Proteinuria and Fas-dependent Pathways. Jasn 16 (2), 398–407. doi:10.1681/ASN.2003100861
Eshbach, M. L., and Weisz, O. A. (2017). Receptor-Mediated Endocytosis in the Proximal Tubule. Annu. Rev. Physiol. 79, 425–448. doi:10.1146/annurev-physiol-022516-034234
Etherington, C., Bosomworth, M., Clifton, I., Peckham, D. G., and Conway, S. P. (2007). Measurement of Urinary N-Acetyl-B-D-Glucosaminidase in Adult Patients with Cystic Fibrosis: before, during and after Treatment with Intravenous Antibiotics. J. Cystic Fibrosis 6 (1), 67–73. doi:10.1016/j.jcf.2006.05.013
Eyre, R. J., Stevens, D. K., Parker, J. C., and Bull, R. J. (1995). Acid‐labile Adducts to Protein Can Be Used as Indicators of the cysteineS‐conjugate Pathway of Trichloroethene Metabolism. J. Toxicol. Environ. Health 46 (4), 443–464. doi:10.1080/15287399509532048
Fang, H., Deng, M., Zhang, L., Lu, A., Su, J., Xu, C., et al. (2018). Role of (Pro)renin Receptor in Albumin Overload-Induced Nephropathy in Rats. Am. J. Physiology-Renal Physiol. 315 (6), F1759–F1768. doi:10.1152/ajprenal.00071.2018
Feldman, S., Wang, M. Y., and Kaloyanides, G. J. (1982). Aminoglycosides Induce a Phospholipidosis in the Renal Cortex of the Rat: an Early Manifestation of Nephrotoxicity. J. Pharmacol. Exp. Ther. 220 (3), 514–520.
Fernandez-Fernandez, B., Montoya-Ferrer, A., Sanz, A. B., Sanchez-Niño, M. D., Izquierdo, M. C., Poveda, J., et al. (2011). Tenofovir Nephrotoxicity: 2011 Update. AIDS Res. Treat. 2011, 1–11. doi:10.1155/2011/354908
Fontana, R. J. (2009). Side Effects of Long-Term Oral Antiviral Therapy for Hepatitis B. Hepatology 49 (5 Suppl. l), S185–S195. doi:10.1002/hep.22885
Foote, K., Reinhold, J., Yu, E. P. K., Figg, N. L., Finigan, A., Murphy, M. P., et al. (2018). Restoring Mitochondrial DNA Copy Number Preserves Mitochondrial Function and Delays Vascular Aging in Mice. Aging Cell 17, e12773. doi:10.1111/acel.12773
Fotakis, G., and Timbrell, J. A. (2006). In Vitro cytotoxicity Assays: Comparison of LDH, Neutral Red, MTT and Protein Assay in Hepatoma Cell Lines Following Exposure to Cadmium Chloride. Toxicol. Lett. 160 (2), 171–177. doi:10.1016/j.toxlet.2005.07.001
Fowler, L. M., Foster, J. R., and Lock, E. A. (1993). Effect of Ascorbic Acid, Acivicin and Probenecid on the Nephrotoxicity of 4-aminophenol in the Fischer 344 Rat. Arch. Toxicol. 67 (9), 613–621. doi:10.1007/BF01974068
Fowler, L. M., Moore, R. B., Foster, J. R., and Lock, E. A. (1991). Nephrotoxicity of 4-aminophenol Glutathione Conjugate. Hum. Exp. Toxicol. 10 (6), 451–459. doi:10.1177/096032719101000615
Fujiwara, K., Yoshizaki, Y., Shin, M., Miyazaki, T., Saita, T., and Nagata, S. (2012). Immunocytochemistry for Vancomycin Using a Monoclonal Antibody that Reveals Accumulation of the Drug in Rat Kidney and Liver. Antimicrob. Agents Chemother. 56 (11), 5883–5891. doi:10.1128/aac.01267-12
Fuke, S., Kametani, M., Yamada, K., Kasahara, T., Kubota‐Sakashita, M., Kujoth, G. C., et al. (2014). Heterozygous Polg Mutation Causes Motor Dysfunction Due to Mt DNA Deletions. Ann. Clin. Transl Neurol. 1 (11), 909–920. doi:10.1002/acn3.133
Fung, J., Seto, W.-K., Lai, C.-L., and Yuen, M.-F. (2014). Extrahepatic Effects of Nucleoside and Nucleotide Analogues in Chronic Hepatitis B Treatment. J. Gastroenterol. Hepatol. 29 (3), 428–434. doi:10.1111/jgh.12499
Funk, J. A., and Schnellmann, R. G. (2013). Accelerated Recovery of Renal Mitochondrial and Tubule Homeostasis with SIRT1/PGC-1α Activation Following Ischemia-Reperfusion Injury. Toxicol. Appl. Pharmacol. 273 (2), 345–354. doi:10.1016/j.taap.2013.09.026
Gai, Z., Gui, T., Kullak-Ublick, G. A., Li, Y., and Visentin, M. (2020). The Role of Mitochondria in Drug-Induced Kidney Injury. Front. Physiol. 11, 1079. doi:10.3389/fphys.2020.01079
Gara, N., Zhao, X., Collins, M. T., Chong, W. H., Kleiner, D. E., Jake Liang, T., et al. (2012). Renal Tubular Dysfunction during Long-Term Adefovir or Tenofovir Therapy in Chronic Hepatitis B. Aliment. Pharmacol. Ther. 35 (11), 1317–1325. doi:10.1111/j.1365-2036.2012.05093.x
Gartland, K. P. R., Bonner, F. W., Timbrell, J. A., and Nicholson, J. K. (1989). Biochemical Characterisation of Para-Aminophenol-Induced Nephrotoxic Lesions in the F344 Rat. Arch. Toxicol. 63 (2), 97–106. doi:10.1007/bf00316430
Gebremichael, Y., Lu, J., Shankaran, H., Helmlinger, G., Hallow, K. M., and au, K. M. (2018). Multiscale Mathematical Model of Drug-Induced Proximal Tubule Injury: Linking Urinary Biomarkers to Epithelial Cell Injury and Renal Dysfunction. Toxicol. Sci. 162 (1), 200–211. doi:10.1093/toxsci/kfx239
Glass, S., Plant, N. D., and Spencer, D. A. (2005). The Effects of Intravenous Tobramycin on Renal Tubular Function in Children with Cystic Fibrosis. J. Cystic Fibrosis 4 (4), 221–225. doi:10.1016/j.jcf.2005.09.003
Green, C. R., Ham, K. N., and Tange, J. D. (1969). Kidney Lesions Induced in Rats by P-Aminophenol. Bmj 1 (5637), 162–164. doi:10.1136/bmj.1.5637.162
Griffin, R. J., and Harvison, P. J. (1998). In Vivo metabolism and Disposition of the Nephrotoxicant N-(3, 5-dichlorophenyl)succinimide in Fischer 344 Rats. Drug Metab. Dispos 26 (9), 907–913.
Group (1997). Parenteral Cidofovir for Cytomegalovirus Retinitis in Patients with AIDS: the HPMPC Peripheral Cytomegalovirus Retinitis Trial. A Randomized, Controlled Trial. Studies of Ocular Complications of AIDS Research Group in Collaboration with the AIDS Clinical Trials Group. Ann. Intern. Med. 126(4), 264–2a74.doi:10.7326/0003-4819-126-4-199702150-00002
Groves, C. E., Schnellmann, R. G., Sokol, P. P., Steffens, T. G., and Lock, E. A. (1991). Pentachlorobutadienyl-L-cysteine (PCBC) Toxicity: the Importance of Mitochondrial Dysfunction. J. Biochem. Toxicol. 6 (4), 253–260. doi:10.1002/jbt.2570060404
Guicciardi, M. E., Leist, M., and Gores, G. J. (2004). Lysosomes in Cell Death. Oncogene 23 (16), 2881–2890. doi:10.1038/sj.onc.1207512
Hall, A. M., and Schuh, C. D. (2016). Mitochondria as Therapeutic Targets in Acute Kidney Injury. Curr. Opin. Nephrol. Hypertens. 25 (4), 355–362. doi:10.1097/MNH.0000000000000228
Hall, A. M. (2013). Update on Tenofovir Toxicity in the Kidney. Pediatr. Nephrol. 28 (7), 1011–1023. doi:10.1007/s00467-012-2269-7
Hance, N., Ekstrand, M. I., and Trifunovic, A. (2005). Mitochondrial DNA Polymerase Gamma Is Essential for Mammalian Embryogenesis. Hum. Mol. Genet. 14 (13), 1775–1783. doi:10.1093/hmg/ddi184
Harris, J. W., Dekant, W., and Anders, M. W. (1992). In Vivo detection and Characterization of Protein Adducts Resulting from Bioactivation of Haloethene Cysteine S-Conjugates by Fluorine-19 NMR: Chlorotrifluoroethene and Tetrafluoroethene. Chem. Res. Toxicol. 5 (1), 34–41. doi:10.1021/tx00025a007
Hayden, P. J., and Stevens, J. L. (1990). Cysteine Conjugate Toxicity, Metabolism, and Binding to Macromolecules in Isolated Rat Kidney Mitochondria. Mol. Pharmacol. 37 (3), 468–476.
Hayden, P. J., Ichimura, T., McCann, D. J., Pohl, L. R., and Stevens, J. L. (1991). Detection of Cysteine Conjugate Metabolite Adduct Formation with Specific Mitochondrial Proteins Using Antibodies Raised against Halothane Metabolite Adducts. J. Biol. Chem. 266 (28), 18415–18418. doi:10.1016/s0021-9258(18)55074-2
Herbers, E., Kekäläinen, N. J., Hangas, A., Pohjoismäki, J. L., and Goffart, S. (2019). Tissue Specific Differences in Mitochondrial DNA Maintenance and Expression. Mitochondrion 44, 85–92. doi:10.1016/j.mito.2018.01.004
Herlitz, L. C., Mohan, S., Stokes, M. B., Radhakrishnan, J., D'Agati, V. D., and Markowitz, G. S. (2010). Tenofovir Nephrotoxicity: Acute Tubular Necrosis with Distinctive Clinical, Pathological, and Mitochondrial Abnormalities. Kidney Int. 78 (11), 1171–1177. doi:10.1038/ki.2010.318
Highleyman, L. (1999). FDA Panel Fails to Recommend Adefovir Approval. Food and Drug Administration. BETA 12 (4), 4.
Hill, B. A., Monks, T. J., and Lau, S. S. (1992). The Effects of 2,3,5-(triglutathion-S-Yl)hydroquinone on Renal Mitochondrial Respiratory Function In Vivo and In Vitro: Possible Role in Cytotoxicity. Toxicol. Appl. Pharmacol. 117 (2), 165–171. doi:10.1016/0041-008x(92)90233-i
Ho, E. S., Lin, D. C., Mendel, D. B., and Cihlar, T. (2000). Cytotoxicity of Antiviral Nucleotides Adefovir and Cidofovir Is Induced by the Expression of Human Renal Organic Anion Transporter 1. Jasn 11 (3), 383–393. doi:10.1681/asn.v113383
Hoffmann, D., Adler, M., Vaidya, V. S., Rached, E., Mulrane, L., Gallagher, W. M., et al. (2010). Performance of Novel Kidney Biomarkers in Preclinical Toxicity Studies. Toxicol. Sci. 116 (1), 8–22. doi:10.1093/toxsci/kfq029
Hori, Y., Aoki, N., Kuwahara, S., Hosojima, M., Kaseda, R., Goto, S., et al. (2017). Megalin Blockade with Cilastatin Suppresses Drug-Induced Nephrotoxicity. Jasn 28 (6), 1783–1791. doi:10.1681/ASN.2016060606
Houghton, D. C., Campbell-Boswell, M. V., Bennett, W. M., Porter, G. A., and Brooks, R. E. (1978a). Myeloid Bodies in the Renal Tubules of Humans: Relationship to Gentamicin Therapy. Clin. Nephrol. 10 (4), 140–145.
Houghton, D. C., Plamp, C. E., DeFehr, J. M., Bennett, W. M., Porter, G., and Gilbert, D. (1978b). Gentamicin and Tobramycin Nephrotoxicity. A Morphologic and Functional Comparison in the Rat. Am. J. Pathol. 93 (1), 137–152.
Htike, N. L., Santoro, J., Gilbert, B., Elfenbein, I. B., and Teehan, G. (2012). Biopsy-proven Vancomycin-Associated Interstitial Nephritis and Acute Tubular Necrosis. Clin. Exp. Nephrol. 16 (2), 320–324. doi:10.1007/s10157-011-0559-1
Humanes, B., Jado, J. C., Camaño, S., López-Parra, V., Torres, A. M., Álvarez-Sala, L. A., et al. (2015). Protective Effects of Cilastatin against Vancomycin-Induced Nephrotoxicity. Biomed. Res. Int. 2015, 1–12. doi:10.1155/2015/704382
IARC (2014). Trichloroethylene, Tetrachloroethylene, and Some Other Chlorinated Agents. IARC Monogr. Eval. Carcinogenic Risks Humans 106, 1–525.
Ikeda, M., Ide, T., Fujino, T., Arai, S., Saku, K., Kakino, T., et al. (2015). Overexpression of TFAM or Twinkle Increases mtDNA Copy Number and Facilitates Cardioprotection Associated with Limited Mitochondrial Oxidative Stress. PLoS One 10 (3), e0119687. doi:10.1371/journal.pone.0119687
Ishmael, J., Pratt, I., and Lock, E. A. (1982). Necrosis of the Pars Recta (S3 Segment) of the Rat Kidney Produced by Hexachloro 1:3 Butadiene. J. Pathol. 138 (2), 99–113. doi:10.1002/path.1711380202
Iyengar, B., Luo, N., Farr, C. L., Kaguni, L. S., and Campos, A. R. (2002). The Accessory Subunit of DNA Polymerase γ Is Essential for Mitochondrial DNA Maintenance and Development in Drosophila melanogaster. Proc. Natl. Acad. Sci. U.S.A. 99 (7), 4483–4488. doi:10.1073/pnas.072664899
Izzedine, H., Hulot, J. S., Launay-Vacher, V., Marcellini, P., Hadziyannis, S. J., Currie, G., et al. (2004). Renal Safety of Adefovir Dipivoxil in Patients with Chronic Hepatitis B: Two Double-Blind, Randomized, Placebo-Controlled Studies. Kidney Int. 66 (3), 1153–1158. doi:10.1111/j.1523-1755.2004.00866.x
Izzedine, H., Kheder-Elfekih, R., Housset, P., Sarkozy, C., Brocheriou, I., and Deray, G. (2009). Adefovir Dipivoxil-Induced Acute Tubular Necrosis and Fanconi Syndrome in a Renal Transplant Patient. AIDS 23 (4), 544–545. doi:10.1097/QAD.0b013e32832407f7
Izzedine, H., Launay-Vacher, V., and Deray, G. (2005). Antiviral Drug-Induced Nephrotoxicity. Am. J. Kidney Dis. 45 (5), 804–817. doi:10.1053/j.ajkd.2005.02.010
Jado, J. C., Humanes, B., Lopez-Parra, V., Camano, S., Lara, J. M., Cercenado, E., et al. (2014). Effects of Cilastatin on Gentamicin-Induced Renal Damage. In Vitro and In Vivo Evidence. Nephrol. Dial. Transplant. 29, 90–90.
Jaffe, D. R., Hassall, C. D., Brendel, K., and Gandolfi, A. J. (1983). In Vivoandin Vitronephrotoxicity of the Cysteine Conjugate of Hexachlorobutadiene. J. Toxicol. Environ. Health 11 (4-6), 857–867. doi:10.1080/15287398309530389
James, J. S. (1997). GS 840 (Adefovir Dipivoxil): Broad-Spectrum Antiviral Trial, CD4 Count under 100. AIDS Treat. News (264), 4–5. (No
Jarzina, S., Di Fiore, S., Ellinger, B., Reiser, P., Frank, S., Glaser, M., et al. (2022). Application of the Adverse Outcome Pathway (AOP) Concept to in Vitro Nephrotoxicity Assessment: Kidney Injury Due to Receptor-Mediated Endocytosis and Lysosomal Overload as a Case Study. Front. Toxicol. 4, 864441. doi:10.3389/ftox.2022.864441
Jeffres, M. N. (2017). The Whole Price of Vancomycin: Toxicities, Troughs, and Time. Drugs 77 (11), 1143–1154. doi:10.1007/s40265-017-0764-7
Jesinkey, S. R., Funk, J. A., Stallons, L. J., Wills, L. P., Megyesi, J. K., Beeson, C. C., et al. (2014). Formoterol Restores Mitochondrial and Renal Function after Ischemia-Reperfusion Injury. Jasn 25 (6), 1157–1162. doi:10.1681/ASN.2013090952
Jesse, C. R., Bortolatto, C. F., Wilhelm, E. A., Roman, S. S., Prigol, M., and Nogueira, C. W. (2014). The Peroxisome Proliferator-Activated Receptor-γ Agonist Pioglitazone Protects against Cisplatin-Induced Renal Damage in Mice. J. Appl. Toxicol. 34 (1), 25–32. doi:10.1002/jat.2818
Johnson, A. A., Ray, A. S., Hanes, J., Suo, Z., Colacino, J. M., Anderson, K. S., et al. (2001). Toxicity of Antiviral Nucleoside Analogs and the Human Mitochondrial DNA Polymerase. J. Biol. Chem. 276 (44), 40847–40857. doi:10.1074/jbc.M106743200
Jollow, D. J., Mitchell, J. R., Potter, W. Z., Davis, D. C., Gillette, J. R., and Brodie, B. B. (1973). Acetaminophen-induced Hepatic Necrosis. II. Role of Covalent Binding In Vivo. J. Pharmacol. Exp. Ther. 187 (1), 195–202.
Jones, T. W., Wallin, A., Thor, H., Gerdes, R. G., Ormstad, K., and Orrenius, S. (1986). The Mechanism of Pentachlorobutadienyl-Glutathione Nephrotoxicity Studied with Isolated Rat Renal Epithelial Cells. Arch. Biochem. Biophys. 251 (2), 504–513. doi:10.1016/0003-9861(86)90358-9
Jornayvaz, F. R., and Shulman, G. I. (2010). Regulation of Mitochondrial Biogenesis. Essays Biochem. 47, 69–84. doi:10.1042/bse0470069
Justo, J. A., and Bosso, J. A. (2015). Adverse Reactions Associated with Systemic Polymyxin Therapy. Pharmacotherapy 35 (1), 28–33. doi:10.1002/phar.1493
Kahn, J., Lagakos, S., Wulfsohn, M., Cherng, D., Miller, M., Cherrington, J., et al. (1999). Efficacy and Safety of Adefovir Dipivoxil with Antiretroviral TherapyA Randomized Controlled Trial. JAMA 282 (24), 2305–2312. doi:10.1001/jama.282.24.2305
Kakuda, T. N. (2000). Pharmacology of Nucleoside and Nucleotide Reverse Transcriptase Inhibitor-Induced Mitochondrial Toxicity. Clin. Ther. 22 (6), 685–708. doi:10.1016/S0149-2918(00)90004-3
Kaloyanides, G. J., and Pastoriza-Munoz, E. (1980). Aminoglycoside Nephrotoxicity. Kidney Int. 18 (5), 571–582. doi:10.1038/ki.1980.175
Keirstead, N. D., Wagoner, M. P., Bentley, P., Blais, M., Brown, C., Cheatham, L., et al. (2014). Early Prediction of Polymyxin-Induced Nephrotoxicity with Next-Generation Urinary Kidney Injury Biomarkers. Toxicol. Sci. 137 (2), 278–291. doi:10.1093/toxsci/kft247
Khan, K., and Alden, C. (2002). “Kidney,” in Handbook of Toxicologic Pathology. Editors C. G. R. W. M. Haschek, and M. A. Wallig (San Diego: Elsevier Science), 255–336. doi:10.1016/b978-012330215-1/50034-x
Kleiner, H. E., Rivera, M. I., Pumford, N. R., Monks, T. J., and Lau, S. S. (1998). Immunochemical Detection of Quinol−Thioether-Derived Protein Adducts. Chem. Res. Toxicol. 11 (11), 1283–1290. doi:10.1021/tx980134e
Klinge, C. M. (2017). Estrogens Regulate Life and Death in Mitochondria. J. Bioenerg. Biomembr 49 (4), 307–324. doi:10.1007/s10863-017-9704-1
Klos, C., Koob, M., Kramer, C., and Dekant, W. (1992). p-Aminophenol Nephrotoxicity: Biosynthesis of Toxic Glutathione Conjugates. Toxicol. Appl. Pharmacol. 115 (1), 98–106. doi:10.1016/0041-008x(92)90372-y
Kociba, R. J., Schwetz, B. A., Keyes, D. G., Jersey, G. C., Ballard, J. J., Dittenber, D. A., et al. (1977b). Chronic Toxicity and Reproduction Studies of Hexachlorobutadiene in Rats. Environ. Health Perspect. 21, 49–53. doi:10.1289/ehp.772149
Kohler, J. J., Hosseini, S. H., Green, E., Abuin, A., Ludaway, T., Russ, R., et al. (2011). Tenofovir Renal Proximal Tubular Toxicity Is Regulated by OAT1 and MRP4 Transporters. Lab. Invest. 91 (6), 852–858. doi:10.1038/labinvest.2011.48
Kohler, J. J., Hosseini, S. H., Hoying-Brandt, A., Green, E., Johnson, D. M., Russ, R., et al. (2009). Tenofovir Renal Toxicity Targets Mitochondria of Renal Proximal Tubules. Lab. Invest. 89 (5), 513–519. doi:10.1038/labinvest.2009.14
Kohler, J. J., and Hosseini, S. H. (2011). Subcellular Renal Proximal Tubular Mitochondrial Toxicity with Tenofovir Treatment. Methods Mol. Biol. 755, 267–277. doi:10.1007/978-1-61779-163-5_22
Kqciba, R. J., Keyes, D. G., Jersey, G. C., Ballard, J. J., Dittenber, D. A., Quast, J. F., et al. (1977a). Results of a Two Year Chronic Toxicity Study with Hexachlorobutadiene in Rats. Am. Ind. Hyg. Assoc. J. 38 (11), 589–602. doi:10.1080/00028897708984403
Kubin, C. J., Ellman, T. M., Phadke, V., Haynes, L. J., Calfee, D. P., and Yin, M. T. (2012). Incidence and Predictors of Acute Kidney Injury Associated with Intravenous Polymyxin B Therapy. J. Infect. 65 (1), 80–87. doi:10.1016/j.jinf.2012.01.015
Labenski, M. T., Fisher, A. A., Monks, T. J., and Lau, S. S. (2011). One-dimensional Western Blotting Coupled to LC-MS/MS Analysis to Identify Chemical-Adducted Proteins in Rat Urine. Methods Mol. Biol. 691, 327–338. doi:10.1007/978-1-60761-849-2_20
Lalezari, J. P., Drew, W. L., Glutzer, E., James, C., Miner, D., Flaherty, J., et al. (1995). (S)-1-[3-hydroxy-2-(phosphonylmethoxy)propyl]cytosine (Cidofovir): Results of a Phase I/II Study of a Novel Antiviral Nucleotide Analogue. J. Infect. Dis. 171 (4), 788–796. doi:10.1093/infdis/171.4.788
Lash, L. H., Qian, W., Putt, D. A., Hueni, S. E., Elfarra, A. A., Krause, R. J., et al. (2001). Renal and Hepatic Toxicity of Trichloroethylene and its Glutathione-Derived Metabolites in Rats and Mice: Sex-, Species-, and Tissue-dependent Differences. J. Pharmacol. Exp. Ther. 297 (1), 155–164.
Lash, L. H., Xu, Y., Elfarra, A. A., Duescher, R. J., and Parker, J. C. (1995). Glutathione-dependent Metabolism of Trichloroethylene in Isolated Liver and Kidney Cells of Rats and its Role in Mitochondrial and Cellular Toxicity. Drug Metab. Dispos 23 (8), 846–853.
Lash, L. H., Lee, C. A., Wilker, C., and Shah, V. (2018). Transporter-dependent Cytotoxicity of Antiviral Drugs in Primary Cultures of Human Proximal Tubular Cells. Toxicology 404-405, 10–24. doi:10.1016/j.tox.2018.05.002
Lash, L. H., Qian, W., Putt, D. A., Hueni, S. E., Elfarra, A. A., Sicuri, A. R., et al. (2002). Renal Toxicity of Perchloroethylene and S-(1,2,2-trichlorovinyl)glutathione in Rats and Mice: Sex- and Species-dependent Differences. Toxicol. Appl. Pharmacol. 179 (3), 163–171. doi:10.1006/taap.2001.9358
Lau, S. S., and Monks, T. J. (1987). Co-oxidation of 2-bromohydroquinone by Renal Prostaglandin Synthase. Modulation of Prostaglandin Synthesis by 2-bromohydroquinone and Glutathione. Drug Metab. Dispos 15 (6), 801–807.
Lau, S. S., Monks, T. J., and Gillette, J. R. (1984a). Identification of 2-bromohydroquinone as a Metabolite of Bromobenzene and O-Bromophenol: Implications for Bromobenzene-Induced Nephrotoxicity. J. Pharmacol. Exp. Ther. 230 (2), 360–366.
Lau, S. S., Monks, T. J., Greene, K. E., and Gillette, J. R. (1984b). The Role of Ortho-Bromophenol in the Nephrotoxicity of Bromobenzene in Rats. Toxicol. Appl. Pharmacol. 72 (3), 539–549. doi:10.1016/0041-008x(84)90131-5
Lau, S. S., and Monks, T. J. (1990). The In Vivo Disposition of 2-bromo-[14C]hydroquinone and the Effect of γ-glutamyl Transpeptidase Inhibition. Toxicol. Appl. Pharmacol. 103 (1), 121–132. doi:10.1016/0041-008x(90)90268-y
Lau, S. S. (1995). Quinone-thioether-mediated Nephrotoxicity. Drug Metab. Rev. 27 (1-2), 125–141. doi:10.3109/03602539509029819
Lebrecht, D., Venhoff, A. C., Kirschner, J., Wiech, T., Venhoff, N., and Walker, U. A. (2009). Mitochondrial Tubulopathy in Tenofovir Disoproxil Fumarate-Treated Rats. Jaids-Journal of Acquired Immune Deficiency Syndromes 51 (3), 258–263. doi:10.1097/qai.0b013e3181a666eb
Lee, H., Hanes, J., and Johnson, K. A. (2003). Toxicity of Nucleoside Analogues Used to Treat AIDS and the Selectivity of the Mitochondrial DNA Polymerase. Biochemistry 42 (50), 14711–14719. doi:10.1021/bi035596s
Lehman-McKeeman, L. D., Rivera-Torres, M. I., and Caudill, D. (1990). Lysosomal Degradation of α2u-globulin and α2u-globulin-xenobiotic Conjugates. Toxicol. Appl. Pharmacol. 103 (3), 539–548. doi:10.1016/0041-008x(90)90326-p
Lewis, W., and Dalakas, M. C. (1995). Mitochondrial Toxicity of Antiviral Drugs. Nat. Med. 1 (5), 417–422. doi:10.1038/nm0595-417
Lewis, W., Day, B. J., and Copeland, W. C. (2003). Mitochondrial Toxicity of NRTI Antiviral Drugs: an Integrated Cellular Perspective. Nat. Rev. Drug Discov. 2 (10), 812–822. doi:10.1038/nrd1201
Li, M., Balamuthusamy, S., Simon, E. E., and Batuman, V. (2008). Silencing Megalin and Cubilin Genes Inhibits Myeloma Light Chain Endocytosis and Ameliorates Toxicity in Human Renal Proximal Tubule Epithelial Cells. Am. J. Physiology-Renal Physiol. 295 (1), F82–F90. doi:10.1152/ajprenal.00091.2008
Li, W., Yuan, X., Nordgren, G., Dalen, H., Dubowchik, G. M., Firestone, R. A., et al. (2000). Induction of Cell Death by the Lysosomotropic Detergent MSDH. FEBS Lett. 470 (1), 35–39. doi:10.1016/s0014-5793(00)01286-2
Li, Y., Bentzley, C. M., and Tarloff, J. B. (2005). Comparison of Para-Aminophenol Cytotoxicity in Rat Renal Epithelial Cells and Hepatocytes. Toxicology 209 (1), 69–76. doi:10.1016/j.tox.2004.12.008
Libório, A. B., Andrade, L., Pereira, L. V. B., Sanches, T. R. C., Shimizu, M. H., and Seguro, A. C. (2008). Rosiglitazone Reverses Tenofovir-Induced Nephrotoxicity. Kidney Int. 74 (7), 910–918. doi:10.1038/ki.2008.252
Lim, S. E., and Copeland, W. C. (2001). Differential Incorporation and Removal of Antiviral Deoxynucleotides by Human DNA Polymerase γ. J. Biol. Chem. 276 (26), 23616–23623. doi:10.1074/jbc.M101114200
Lim, S. E., Ponamarev, M. V., Longley, M. J., and Copeland, W. C. (2003). Structural Determinants in Human DNA Polymerase γ Account for Mitochondrial Toxicity from Nucleoside Analogs. J. Mol. Biol. 329 (1), 45–57. doi:10.1016/s0022-2836(03)00405-4
Lin, Y., Pan, F., Wang, Y., Chen, Z., Lin, C., Yao, L., et al. (2017). Adefovir Dipivoxil-Induced Fanconi Syndrome and its Predictive Factors: A Study of 28 Cases. Oncol. Lett. 13 (1), 307–314. doi:10.3892/ol.2016.5393
Lipsky, J. J., and Lietman, P. S. (1982). Aminoglycoside Inhibition of a Renal Phosphatidylinositol Phospholipase C. J. Pharmacol. Exp. Ther. 220(2), 287–2a92.
Liu, D., Wen, Y., Tang, T.-T., Lv, L.-L., Tang, R.-N., Liu, H., et al. (2015). Megalin/Cubulin-Lysosome-mediated Albumin Reabsorption Is Involved in the Tubular Cell Activation of NLRP3 Inflammasome and Tubulointerstitial Inflammation. J. Biol. Chem. 290 (29), 18018–18028. doi:10.1074/jbc.m115.662064
Liu, F. Y., Li, Y., Peng, Y. M., Ye, K., Li, J., Liu, Y.-H., et al. (2008). Norcantharidin Ameliorates Proteinuria, Associated Tubulointerstitial Inflammation and Fibrosis in Protein Overload Nephropathy. Am. J. Nephrol. 28 (3), 465–477. doi:10.1159/000112850
Liu, W. J., Xu, B.-H., Ye, L., Liang, D., Wu, H.-L., Zheng, Y.-Y., et al. (2015). Urinary Proteins Induce Lysosomal Membrane Permeabilization and Lysosomal Dysfunction in Renal Tubular Epithelial Cells. Am. J. Physiology-Renal Physiol. 308 (6), F639–F649. doi:10.1152/ajprenal.00383.2014
Lock, E. A., Cross, T. J., and Schnellmann, R. G. (1993). Studies on the Mechanism of 4-Aminophenol-Induced Toxicity to Renal Proximal Tubules. Hum. Exp. Toxicol. 12 (5), 383–388. doi:10.1177/096032719301200507
Lock, E. A., and Hard, G. C. (2004). Chemically Induced Renal Tubule Tumors in the Laboratory Rat and Mouse: Review of the NCI/NTP Database and Categorization of Renal Carcinogens Based on Mechanistic Information. Crit. Rev. Toxicol. 34 (3), 211–299. doi:10.1080/10408440490265210
Lopez-Novoa, J. M., Quiros, Y., Vicente, L., Morales, A. I., and Lopez-Hernandez, F. J. (2011). New Insights into the Mechanism of Aminoglycoside Nephrotoxicity: an Integrative point of View. Kidney Int. 79 (1), 33–45. doi:10.1038/ki.2010.337
Luque, Y., Louis, K., Jouanneau, C., Placier, S., Esteve, E., Bazin, D., et al. (2017). Vancomycin-Associated Cast Nephropathy. Jasn 28 (6), 1723–1728. doi:10.1681/ASN.2016080867
Maguire, D. P., Turton, J. A., Scudamore, C. L., Swain, A. J., McClure, F. J., Smyth, R., et al. (2013). Correlation of Histopathology, Urinary Biomarkers, and Gene Expression Responses Following Hexachloro-1:3-Butadiene-Induced Acute Nephrotoxicity in Male Hanover Wistar Rats. Toxicol. Pathol. 41 (5), 779–794. doi:10.1177/0192623312464306
Mally, A., Walker, C. L., Everitt, J. I., Dekant, W., and Vamvakas, S. (2006). Analysis of Renal Cell Transformation Following Exposure to Trichloroethene In Vivo and its Metabolite S-(dichlorovinyl)-L-cysteine In Vitro. Toxicology 224 (1-2), 108–118. doi:10.1016/j.tox.2006.04.036
Mandal, A. K., and Bennett, W. M. (1988). Transmission Electron Microscopy of Urinary Sediment in the Assessment of Aminoglycoside Nephrotoxicity in the Rat. Nephron 49 (1), 67–73. doi:10.1159/000184989
Mandal, A. K., Mize, G. N., and Birnbaum, D. B. (1987). Transmission Electron Microscopy of Urinary Sediment in Aminoglycoside Nephrotoxicity. Ren. Fail. 10 (2), 63–81. doi:10.3109/08860228709056320
Martin, J. L., Brown, C. E., Matthews-Davis, N., and Reardon, J. E. (1994). Effects of Antiviral Nucleoside Analogs on Human DNA Polymerases and Mitochondrial DNA Synthesis. Antimicrob. Agents Chemother. 38 (12), 2743–2749. doi:10.1128/aac.38.12.2743
Martín-Hernández, E., García-Silva, M. T., Vara, J., Campos, Y., Cabello, A., Muley, R., et al. (2005). Renal Pathology in Children with Mitochondrial Diseases. Pediatr. Nephrol. 20 (9), 1299–1305. doi:10.1007/s00467-005-1948-z
McWilliam, S. J., Antoine, D. J., and Pirmohamed, M. (2018). Repurposing Statins for Renal Protection: Is it a Class Effect? Clin. Translational Sci. 11 (2), 100–102. doi:10.1111/cts.12521
McWilliam, S. J., Antoine, D. J., Smyth, R. L., and Pirmohamed, M. (2017). Aminoglycoside-induced Nephrotoxicity in Children. Pediatr. Nephrol. 32 (11), 2015–2025. doi:10.1007/s00467-016-3533-z
Medina-Navarro, R., Torres-Ramos, Y. D., Guzmán-Grenfell, A. M., Díaz-Flores, M., León-Reyes, G., and Hicks G., J. J. (2019). Lysosomal Dysfunction Induced by Changes in Albumin's Tertiary Structure: Potential Key Factor in Protein Toxicity during Diabetic Nephropathy. Life Sci. 230, 197–207. doi:10.1016/j.lfs.2019.05.069
Miller, E. C., and Miller, J. A. (1966). Mechanisms of Chemical Carcinogenesis: Nature of Proximate Carcinogens and Interactions with Macromolecules. Pharmacol. Rev. 18 (1), 805–838.
Miller, E. C., and Miller, J. A. (1947). The Presence and Significance of Bound Aminoazo Dyes in the Livers of Rats Fed P-Dimethylaminoazobenzene. Cancer Res. 7 (7), 468–480.
Moestrup, S. K., Cui, S., Vorum, H., Bregengård, C., Bjørn, S. E., Norris, K., et al. (1995). Evidence that Epithelial Glycoprotein 330/megalin Mediates Uptake of Polybasic Drugs. J. Clin. Invest. 96 (3), 1404–1413. doi:10.1172/JCI118176
Monks, T. J., Lau, S. S., Highet, R. J., and Gillette, J. R. (1985). Glutathione Conjugates of 2-bromohydroquinone Are Nephrotoxic. Drug Metab. Dispos 13 (5), 553–559.
Moyle, G. (2000). Toxicity of Antiretroviral Nucleoside and Nucleotide Analogues Drug Saf. 23(6), 467–481. doi:10.2165/00002018-200023060-00001
Myers, T. G., Dietz, E. C., Anderson, N. L., Khairallah, E. A., Cohen, S. D., and Nelson, S. D. (1995). A Comparative Study of Mouse Liver Proteins Arylated by Reactive Metabolites of Acetaminophen and its Nonhepatotoxic Regioisomer, 3'-hydroxyacetanilide. Chem. Res. Toxicol. 8 (3), 403–413. doi:10.1021/tx00045a012
Newton, J. F., Kuo, C.-H., Gemborys, M. W., Mudge, G. H., and Hook, J. B. (1982). Nephrotoxicity of P-Aminophenol, a Metabolite of Acetaminophen, in the Fischer 344 Rat. Toxicol. Appl. Pharmacol. 65 (2), 336–344. doi:10.1016/0041-008x(82)90017-5
Nieskens, T. T. G., Peters, J. G. P., Schreurs, M. J., Smits, N., Woestenenk, R., Jansen, K., et al. (2016). A Human Renal Proximal Tubule Cell Line with Stable Organic Anion Transporter 1 and 3 Expression Predictive for Antiviral-Induced Toxicity. AAPS J. 18 (2), 465–475. doi:10.1208/s12248-016-9871-8
NTP (1990). NTP Carcinogenesis Studies of Trichloroethylene (Without Epichlorohydrin) (CAS No. 79-01-6) in F344/N Rats and B6C3F1 Mice (Gavage Studies). Natl. Toxicol. Program Tech. Rep. Ser. 243, 1–174.
NTP (1986). NTP Toxicology and Carcinogenesis Studies of Tetrachloroethylene (Perchloroethylene) (CAS No. 127-18-4) in F344/N Rats and B6C3F1 Mice (Inhalation Studies). Natl. Toxicol. Program Tech. Rep. Ser. 311, 1–197.
NTP (1988). NTP Toxicology and Carcinogenesis Studies of Trichloroethylene (CAS No. 79-01-6) in Four Strains of Rats (ACI, August, Marshall, Osborne-Mendel) (Gavage Studies). Natl. Toxicol. Program Tech. Rep. Ser. 273, 1–299.
NTP (1991). Toxicity Studies of Hexachloro-1,3-Butadiene in B6C3F1 Mice (Feed Studies). NIH Publication No. 91-3120.).
NTP (1989). Toxicology and Carcinogenesis Studies of Hydroquinone (CAS No. 123-31-9) in F344/N Rats and B6C3F1 Mice (Gavage Studies). Natl. Toxicol. Program Tech. Rep. Ser. 366, 1–248.
Nurminen, A., Farnum, G. A., and Kaguni, L. S. (2017). Pathogenicity in POLG Syndromes: DNA Polymerase Gamma Pathogenicity Prediction Server and Database. BBA Clin. 7, 147–156. doi:10.1016/j.bbacli.2017.04.001
O'Donnell, J. N., Rhodes, N. J., Lodise, T. P., Prozialeck, W. C., Miglis, C. M., Joshi, M. D., et al. (2017). 24-Hour Pharmacokinetic Relationships for Vancomycin and Novel Urinary Biomarkers of Acute Kidney Injury. Antimicrob. Agents Chemother. 61 (11). doi:10.1128/AAC.00416-17
OECD (2017a). “Guidance Document for the Use of Adverse Outcome Pathways in Developing Integrated Approaches to Testing and Assessment (IATA),” in Series on Testing & Assessment No. 260 (Paris: Environment, Health and Safety, Environment Directorate, OECD).
OECD (2017b). “Revised Guidance Document on Developing and Assessing Adverse Outcome Pathways,” in Series on Testing and Assessment No. 184 Revised (Paris: Environment, Health and Safety, Environment Directorate, OECD).
OECD (2014). The Adverse Outcome Pathway for Skin Sensitisation Initiated by Covalent Binding to Proteins.
OECD (2018). “Users´ Handbook Supplement to the Guidance Document for Developing and Assessing AOPs,” in Series on Testing & Assessment No. 233, Series on Adverse Outcome Pathways No. 1 (Paris: Environment, Health and Safety, Environment Directorate, OECD).
Oliveira, J. F. P., Silva, C. A., Barbieri, C. D., Oliveira, G. M., Zanetta, D. M. T., and Burdmann, E. A. (2009). Prevalence and Risk Factors for Aminoglycoside Nephrotoxicity in Intensive Care Units. Antimicrob. Agents Chemother. 53 (7), 2887–2891. doi:10.1128/AAC.01430-08
Onodera, A., Tani, M., Michigami, T., Yamagata, M., Min, K.-S., Tanaka, K., et al. (2012). Role of Megalin and the Soluble Form of its Ligand RAP in Cd-Metallothionein Endocytosis and Cd-Metallothionein-Induced Nephrotoxicity In Vivo. Toxicol. Lett. 212 (2), 91–96. doi:10.1016/j.toxlet.2012.05.012
Pähler, A., Birner, G., Parker, J., and Dekant, W. (1998). Generation of Antibodies to Di- and Trichloroacetylated Proteins and Immunochemical Detection of Protein Adducts in Rats Treated with Perchloroethene. Chem. Res. Toxicol. 11 (9), 995–1004. doi:10.1021/tx9800102
Pahler, A., Parker, J., and Dekant, W. (1999). Dose-dependent Protein Adduct Formation in Kidney, Liver, and Blood of Rats and in Human Blood after Perchloroethene Inhalation. Toxicol. Sci. 48 (1), 5–13. doi:10.1093/toxsci/48.1.5
Perazella, M. A. (2010). Tenofovir-induced Kidney Disease: an Acquired Renal Tubular Mitochondriopathy. Kidney Int. 78 (11), 1060–1063. doi:10.1038/ki.2010.344
Pfaller, W., and Gstraunthaler, G. (1998). Nephrotoxicity Testing In Vitro: What We Know and what We Need to Know. Environ. Health Perspect. 106 (Suppl. 2), 559–569. doi:10.2307/3433806
Pfeifer, U., and Scheller, H. (1975). A Morphometric Study of Cellular Autophagy Including Diurnal Variations in Kidney Tubules of normal Rats. J. Cel Biol 64 (3), 608–621. doi:10.1083/jcb.64.3.608
Powell, J. H., and M. Reidenberg, M. (1982). In Vitro response of Rat and Human Kidney Lysosomes to Aminoglycosides. Biochem. Pharmacol. 31 (21), 3447–3453. doi:10.1016/0006-2952(82)90625-6
Powell, J. H., and Reidenberg, M. M. (1983). Further Studies of the Response of Kidney Lysosomes to Aminoglycosides and Other Cations. Biochem. Pharmacol. 32 (21), 3213–3220. doi:10.1016/0006-2952(83)90206-x
Prasad, R., Çağlayan, M., Dai, D.-P., Nadalutti, C. A., Zhao, M.-L., Gassman, N. R., et al. (2017). DNA Polymerase β: A Missing Link of the Base Excision Repair Machinery in Mammalian Mitochondria. DNA Repair 60, 77–88. doi:10.1016/j.dnarep.2017.10.011
Raggi, C., Fujiwara, K., Leal, T., Jouret, F., Devuyst, O., and Terryn, S. (2011). Decreased Renal Accumulation of Aminoglycoside Reflects Defective Receptor-Mediated Endocytosis in Cystic Fibrosis and Dent's Disease. Pflugers Arch. - Eur. J. Physiol. 462 (6), 851–860. doi:10.1007/s00424-011-1026-2
Rahn, J. J., Bestman, J. E., Stackley, K. D., and Chan, S. S. L. (2015). Zebrafish Lacking Functional DNA Polymerase Gamma Survive to Juvenile Stage, Despite Rapid and Sustained Mitochondrial DNA Depletion, Altered Energetics and Growth. Nucleic Acids Res. 43 (21), gkv1139–10352. doi:10.1093/nar/gkv1139
Ramamoorthy, H., Abraham, P., and Isaac, B. (2014). Mitochondrial Dysfunction and Electron Transport Chain Complex Defect in a Rat Model of Tenofovir Disoproxil Fumarate Nephrotoxicity. J. Biochem. Mol. Toxicol. 28 (6), 246–255. doi:10.1002/jbt.21560
Rasbach, K. A., and Schnellmann, R. G. (2007). PGC-1α Over-expression Promotes Recovery from Mitochondrial Dysfunction and Cell Injury. Biochem. Biophysical Res. Commun. 355 (3), 734–739. doi:10.1016/j.bbrc.2007.02.023
Reid, W. D. (1973). Mechanism of Renal Necrosis Induced by Bromobenzene. Exp. Mol. Pathol. 19 (2), 197–214. doi:10.1016/0014-4800(73)90079-8
Robbins, B. L., Greenhaw, J., Connelly, M. C., and Fridland, A. (1995). Metabolic Pathways for Activation of the Antiviral Agent 9-(2-phosphonylmethoxyethyl)adenine in Human Lymphoid Cells. Antimicrob. Agents Chemother. 39 (10), 2304–2308. doi:10.1128/aac.39.10.2304
Rodeheaver, D. P., and Schnellmann, R. G. (1991). Mechanism of pH Amelioration of 2-Bromohydroquinone-Induced Toxicity to Rabbit Renal Proximal Tubules. J. Pharmacol. Exp. Ther. 256 (3), 917–921.
Roels, F., Paulus, G., and De Broe, M. E. (1984). Lysosomal Modifications in Human Proximal Tubule Induced by Aminoglycoside Treatment: Visualisation by Light Microscopical Cytochemistry. Pathol. - Res. Pract. 179 (2), 230–234. doi:10.1016/S0344-0338(84)80135-1
Rush, G. F., Kuo, C.-H., and Hook, J. B. (1984). Nephrotoxicity of Bromobenzene in Mice. Toxicol. Lett. 20 (1), 23–32. doi:10.1016/0378-4274(84)90178-4
Sakuratani, Y., Horie, M., and Leinala, E. (2018). Integrated Approaches to Testing and Assessment: OECD Activities on the Development and Use of Adverse Outcome Pathways and Case Studies. Basic Clin. Pharmacol. Toxicol. 123, 20–28. doi:10.1111/bcpt.12955
Schmitz, C., Hilpert, J., Jacobsen, C., Boensch, C., Christensen, E. I., Luft, F. C., et al. (2002). Megalin Deficiency Offers protection from Renal Aminoglycoside Accumulation. J. Biol. Chem. 277 (1), 618–622. doi:10.1074/jbc.M109959200
Schnellmann, R. G. (1989). 2-Bromohydroquinone-induced Toxicity to Rabbit Renal Proximal Tubules: Evidence against Oxidative Stress. Toxicol. Appl. Pharmacol. 99 (1), 11–18. doi:10.1016/0041-008x(89)90106-3
Schnellmann, R. G., Ewell, F. P. Q., Sgambati, M., and Mandel, L. J. (1987a). Mitochondrial Toxicity of 2-bromohydroquinone in Rabbit Renal Proximal Tubules. Toxicol. Appl. Pharmacol. 90 (3), 420–426. doi:10.1016/0041-008x(87)90134-7
Schnellmann, R. G., Lock, E. A., and Mandel, L. J. (1987b). A Mechanism of S-(1,2,3,4,4-pentachloro-1,3-butadienyl)-L-cysteine Toxicity to Rabbit Renal Proximal Tubules. Toxicol. Appl. Pharmacol. 90 (3), 513–521. doi:10.1016/0041-008x(87)90143-8
Schnellmann, R. G., Monks, T. J., Mandel, L. J., and Lau, S. S. (1989). 2-Bromohydroquinone-induced Toxicity to Rabbit Renal Proximal Tubules: the Role of Biotransformation, Glutathione, and Covalent Binding. Toxicol. Appl. Pharmacol. 99 (1), 19–27. doi:10.1016/0041-008x(89)90107-5
Schnellmann, R. G. (2013). “Toxic Responses of the Kidney,” in Casarett and Doull´s Toxicology. The Basic Science of Poisons. Editors C. D. Klaassen. 8th Edition (Kansas City: Mcgraw-Hill Education Ltd).
Schrenk, D., Dekant, W., Wünsch, P. H., and Henschler, D. (1988). Role of Metabolic Activation in the Toxicity of S-(pentachlorobutadienyl)glutathione and in the Isolated Perfused Rat Kidney. Toxicol. Vitro 2 (4), 283–290. doi:10.1016/0887-2333(88)90047-1
Schuh, C. D., Polesel, M., Platonova, E., Haenni, D., Gassama, A., Tokonami, N., et al. (2018). Combined Structural and Functional Imaging of the Kidney Reveals Major Axial Differences in Proximal Tubule Endocytosis. Jasn 29 (11), 2696–2712. doi:10.1681/ASN.2018050522
Servais, H., Van Der Smissen, P., Thirion, G., Van der Essen, G., Van Bambeke, F., Tulkens, P. M., et al. (2005). Gentamicin-induced Apoptosis in LLC-PK1 Cells: Involvement of Lysosomes and Mitochondria. Toxicol. Appl. Pharmacol. 206 (3), 321–333. doi:10.1016/j.taap.2004.11.024
Shah-Khan, F., Scheetz, M. H., and Ghossein, C. (2011). Biopsy-Proven Acute Tubular Necrosis Due to Vancomycin Toxicity. Int. J. Nephrol. 2011, 1–4. doi:10.4061/2011/436856
Shao, R., and Tarloff, J. B. (1996). Lack of Correlation between Para-Aminophenol Toxicity In Vivo and In Vitro in Female Sprague-Dawley Rats. Toxicol. Sci. 31 (2), 268–278. doi:10.1093/toxsci/31.2.268
Sieber, M., Hoffmann, D., Adler, M., Vaidya, V. S., Clement, M., Bonventre, J. V., et al. (2009). Comparative Analysis of Novel Noninvasive Renal Biomarkers and Metabonomic Changes in a Rat Model of Gentamicin Nephrotoxicity. Toxicol. Sci. 109 (2), 336–349. doi:10.1093/toxsci/kfp070
Silverblatt, F. J., and Kuehn, C. (1979). Autoradiography of Gentamicin Uptake by the Rat Proximal Tubule Cell. Kidney Int. 15 (4), 335–345. doi:10.1038/ki.1979.45
Sinha Ray, A., Haikal, A., Hammoud, K. A., and Yu, A. S. L. (2016). Vancomycin and the Risk of AKI: A Systematic Review and Meta-Analysis. Cjasn 11 (12), 2132–2140. doi:10.2215/CJN.05920616
Sise, M. E., Hirsch, J. S., Canetta, P. A., Herlitz, L., and Mohan, S. (2015). Nonalbumin Proteinuria Predominates in Biopsy-Proven Tenofovir Nephrotoxicity. AIDS 29 (8), 941–946. doi:10.1097/QAD.0000000000000628
Sivarajah, A., Chatterjee, P. K., Patel, N. S. A., Todorovic, Z., Hattori, Y., Brown, P. A. J., et al. (2003). Agonists of Peroxisome-Proliferator Activated Receptor-Gamma Reduce Renal Ischemia/reperfusion Injury. Am. J. Nephrol. 23 (4), 267–276. doi:10.1159/000072088
Slaughter, D. E., and Hanzlik, R. P. (1991). Identification of Epoxide- and Quinone-Derived Bromobenzene Adducts to Protein Sulfur Nucleophiles. Chem. Res. Toxicol. 4 (3), 349–359. doi:10.1021/tx00021a015
Sokol, H., Vigneau, C., Maury, E., Guidet, B., and Offenstadt, G. (2004). Biopsy-proven Anuric Acute Tubular Necrosis Associated with Vancomycin and One Dose of Aminoside. Nephrol. Dial. Transplant. 19 (7), 1921–1922. doi:10.1093/ndt/gfh170
Song, X.-B., Liu, G., Liu, F., Yan, Z.-G., Wang, Z.-Y., Liu, Z.-P., et al. (2017). Autophagy Blockade and Lysosomal Membrane Permeabilization Contribute to lead-induced Nephrotoxicity in Primary Rat Proximal Tubular Cells. Cell Death Dis 8 (6), e2863. doi:10.1038/cddis.2017.262
Stansly, P. G., Shepherd, R. G., and White, H. J. (1947). Polymyxin: a New Chemotherapeutic Agent. Bull. Johns Hopkins Hosp. 81 (1), 43–54.
Stonard, M. D., and Parker, V. H. (1971). 2-oxoacid Dehydrogenases of Rat Liver Mitochondria as the Site of Action of S-(1,2 Dichlorovinyl)-L-Cysteine and S-(1,2 Dichlorovinyl)-3-Mercaptopropionic Acid. Biochem. Pharmacol. 20 (9), 2417–2427. doi:10.1016/0006-2952(71)90242-5
Suzuki, T., Yamaguchi, H., Ogura, J., Kobayashi, M., Yamada, T., and Iseki, K. (2013). Megalin Contributes to Kidney Accumulation and Nephrotoxicity of Colistin. Antimicrob. Agents Chemother. 57 (12), 6319–6324. doi:10.1128/AAC.00254-13
Takamoto, K., Kawada, M., and Ikeda, D. (2005a). Prevention of Neomycin-Induced Nephrotoxic Event in Pig Proximal Tubular Epithelial Cell Line by Apolipoprotein E3. J. Antibiot. 58 (5), 353–355. doi:10.1038/ja.2005.45
Takamoto, K., Kawada, M., Ikeda, D., and Yoshida, M. (2005b). Apolipoprotein E3 (apoE3) Safeguards Pig Proximal Tubular LLC-PK1 Cells against Reduction in SGLT1 Activity Induced by Gentamicin C. Biochim. Biophys. Acta (Bba) - Gen. Subjects 1722 (3), 247–253. doi:10.1016/j.bbagen.2004.12.006
Talmon, G., Cornell, L. D., and Lager, D. J. (2010). Mitochondrial Changes in Cidofovir Therapy for BK Virus Nephropathy. Transplant. Proc. 42 (5), 1713–1715. doi:10.1016/j.transproceed.2009.11.039
Tanji, N., Tanji, K., Kambham, N., Markowitz, G. S., Bell, A., and D'Agati, V. D. (2001). Adefovir Nephrotoxicity: Possible Role of Mitochondrial DNA Depletion. Hum. Pathol. 32 (7), 734–740. doi:10.1053/hupa.2001.25586
Tantranont, N., Obi, C., Luque, Y., and Truong, L. D. (2019). Vancomycin Nephrotoxicity: Vancomycin Tubular Casts with Characteristic Electron Microscopic Findings. Cncs 7, 66–72. doi:10.5414/CNCS109817
The Studies of Ocular Complications of AIDS Research Group in collaboration with the AIDS Clinical Trials Group (2000). Long-term Follow-Up of Patients with AIDS Treated with Parenteral Cidofovir for Cytomegalovirus Retinitis: the HPMPC Peripheral Cytomegalovirus Retinitis Trial. The Studies of Ocular Complications of AIDS Research Group in Collaboration with the AIDS Clinical Trials Group. AIDS 14 (11), 1571–1581.
Thévenod, F. (2003). Nephrotoxicity and the Proximal Tubule. Nephron Physiol. 93 (4), p87–p93. doi:10.1159/000070241
Tollefsen, K. E., Scholz, S., Cronin, M. T., Edwards, S. W., de Knecht, J., Crofton, K., et al. (2014). Applying Adverse Outcome Pathways (AOPs) to Support Integrated Approaches to Testing and Assessment (IATA). Regul. Toxicol. Pharmacol. 70 (3), 629–640. doi:10.1016/j.yrtph.2014.09.009
Toyoguchi, T., Takahashi, S., Hosoya, J., Nakagawa, Y., and Watanabe, H. (1997). Nephrotoxicity of Vancomycin and Drug Interaction Study with Cilastatin in Rabbits. Antimicrob. Agents Chemother. 41 (9), 1985–1990. doi:10.1128/aac.41.9.1985
Tulkens, P. M. (1989). Nephrotoxicity of Aminoglycoside Antibiotics. Toxicol. Lett. 46 (1-3), 107–123. doi:10.1016/0378-4274(89)90121-5
Tulkens, P., and Van Hoof, F. (1980). Comparative Toxicity of Aminoglycoside Antibiotics towards the Lysosomes in a Cell Culture Model. Toxicology 17 (2), 195–199. doi:10.1016/0300-483x(80)90094-3
Tune, B. M. (1997). Nephrotoxicity of Beta-Lactam Antibiotics: Mechanisms and Strategies for Prevention. Pediatr. Nephrol. 11 (6), 768–772. doi:10.1007/s004670050386
Turk, B., Stoka, V., Rozman-Pungercar, J., Cirman, T., Droga-Mazovec, G., Oreic, K., et al. (2002). Apoptotic Pathways: Involvement of Lysosomal Proteases. Biol. Chem. 383 (7-8), 1035–1044. doi:10.1515/BC.2002.112
Tyson, C. A., Dabbs, J. E., Cohen, P. M., Green, C. E., and Melnick, R. L. (1990). Studies of Nephrotoxic Agents in an Improved Renal Proximal Tubule System. Toxicol. Vitro 4 (4-5), 403–408. doi:10.1016/0887-2333(90)90090-g
Uwai, Y., Ida, H., Tsuji, Y., Katsura, T., and Inui, K.-i. (2007). Renal Transport of Adefovir, Cidofovir, and Tenofovir by SLC22A Family Members (hOAT1, hOAT3, and hOCT2). Pharm. Res. 24 (4), 811–815. doi:10.1007/s11095-006-9196-x
Vaara, M., Fox, J., Loidl, G., Siikanen, O., Apajalahti, J., Hansen, F., et al. (2008). Novel Polymyxin Derivatives Carrying Only Three Positive Charges Are Effective Antibacterial Agents. Antimicrob. Agents Chemother. 52 (9), 3229–3236. doi:10.1128/AAC.00405-08
Vaidya, V. S., Shankar, K., Lock, E. A., Bucci, T. J., and Mehendale, H. M. (2003). Renal Injury and Repair Following S-1, 2 Dichlorovinyl-L-Cysteine Administration to mice☆☆Presented in Part at the 39th Annual Meeting of the Society of Toxicology, March 19-23, 2000. Toxicol. Appl. Pharmacol. 188 (2), 110–121. doi:10.1016/s0041-008x(02)00080-7
Vamvakas, S., Bittner, D., Koob, M., Glück, S., and Dekant, W. (1992). Glutathione Depletion, Lipid Peroxidation, DNA Double-Strand Breaks and the Cytotoxicity of 2-Bromo-3-(n-Acetylcystein-S-Yl)hydroquinone in Rat Renal Cortical Cells. Chemico-Biological Interactions 83 (2), 183–199. doi:10.1016/0009-2797(92)90045-m
van de Water, B., Zoeteweij, J. P., de Bont, H. J., and Nagelkerke, J. F. (1995). Inhibition of Succinate:ubiquinone Reductase and Decrease of Ubiquinol in Nephrotoxic Cysteine S-Conjugate-Induced Oxidative Cell Injury. Mol. Pharmacol. 48 (5), 928–937.
van de Water, B., Zoeteweij, J. P., and Nagelkerke, J. F. (1996). Alkylation-induced Oxidative Cell Injury of Renal Proximal Tubular Cells: Involvement of Glutathione Redox-Cycle Inhibition. Arch. Biochem. Biophys. 327 (1), 71–80. doi:10.1006/abbi.1996.0094
Vedi, M., Rasool, M., and Sabina, E. P. (2014). Protective Effect of Administration ofWithania Somiferaagainst Bromobenzene Induced Nephrotoxicity and Mitochondrial Oxidative Stress in Rats. Ren. Fail. 36 (7), 1095–1103. doi:10.3109/0886022x.2014.918812
Ventura-Clapier, R., Moulin, M., Piquereau, J., Lemaire, C., Mericskay, M., Veksler, V., et al. (2017). Mitochondria: a central Target for Sex Differences in Pathologies. Clin. Sci. (Lond) 131 (9), 803–822. doi:10.1042/CS20160485
Verroust, P. J., Birn, H., Nielsen, R., Kozyraki, R., and Christensen, E. I. (2002). The Tandem Endocytic Receptors Megalin and Cubilin Are Important Proteins in Renal Pathology. Kidney Int. 62 (3), 745–756. doi:10.1046/j.1523-1755.2002.00501.x
Vinken, M., and Blaauboer, B. J. (2017). In Vitro testing of Basal Cytotoxicity: Establishment of an Adverse Outcome Pathway from Chemical Insult to Cell Death. Toxicol. Vitro 39, 104–110. doi:10.1016/j.tiv.2016.12.004
Vinken, M. (2013). The Adverse Outcome Pathway Concept: a Pragmatic Tool in Toxicology. Toxicology 312, 158–165. doi:10.1016/j.tox.2013.08.011
Vora, S. B., Brothers, A. W., and Englund, J. A. (2017). Renal Toxicity in Pediatric Patients Receiving Cidofovir for the Treatment of Adenovirus Infection. J. Pediatr. Infect Dis Soc 6, 399–402. doi:10.1093/jpids/pix011
Wallin, A., Jones, T. W., Vercesi, A. E., Cotgreave, I., Ormstad, K., and Orrenius, S. (1987). Toxicity of S-Pentachlorobutadienyl-L-Cysteine Studied with Isolated Rat Renal Cortical Mitochondria. Arch. Biochem. Biophys. 258 (2), 365–372. doi:10.1016/0003-9861(87)90357-2
Watanabe, A., Nagai, J., Adachi, Y., Katsube, T., Kitahara, Y., Murakami, T., et al. (2004). Targeted Prevention of Renal Accumulation and Toxicity of Gentamicin by Aminoglycoside Binding Receptor Antagonists. J. Controlled Release 95 (3), 423–433. doi:10.1016/j.jconrel.2003.12.005
Wertheim, H., Van Nguyen, K., Hara, G. L., Gelband, H., Laxminarayan, R., Mouton, J., et al. (2013). Global Survey of Polymyxin Use: A Call for International Guidelines. J. Glob. Antimicrob. Resist. 1 (3), 131–134. doi:10.1016/j.jgar.2013.03.012
Wicklow, B. A., Ogborn, M. R., Gibson, I. W., and Blydt-Hansen, T. D. (2006). Biopsy-proven Acute Tubular Necrosis in a Child Attributed to Vancomycin Intoxication. Pediatr. Nephrol. 21 (8), 1194–1196. doi:10.1007/s00467-006-0152-0
Wiland, P., and Szechciński, J. (2003). Proximal Tubule Damage in Patients Treated with Gentamicin or Amikacin. Pol. J. Pharmacol. 55 (4), 631–637.
Wolff, N. A., Abouhamed, M., Verroust, P. J., and Thévenod, F. (2006). Megalin-dependent Internalization of Cadmium-Metallothionein and Cytotoxicity in Cultured Renal Proximal Tubule Cells. J. Pharmacol. Exp. Ther. 318 (2), 782–791. doi:10.1124/jpet.106.102574
Wolff, N. A., Lee, W.-K., Abouhamed, M., and Thévenod, F. (2008). Role of ARF6 in Internalization of Metal-Binding Proteins, Metallothionein and Transferrin, and Cadmium-Metallothionein Toxicity in Kidney Proximal Tubule Cells. Toxicol. Appl. Pharmacol. 230 (1), 78–85. doi:10.1016/j.taap.2008.02.008
Wolfgang, G. H. I., Gandolfi, A. J., Nagle, R. B., Brendel, K., and Stevens, J. L. (1990). Assessment of S-(1,2-dichlorovinyl)-L-cysteine Induced Toxic Events in Rabbit Renal Cortical Slices. Biochemical and Histological Evaluation of Uptake, Covalent Binding, and Toxicity. Chemico-Biological Interactions 75 (2), 153–170. doi:10.1016/0009-2797(90)90115-4
Woodward, C., Hall, A., Williams, I., Madge, S., Copas, A., Nair, D., et al. (2009). Tenofovir-associated Renal and Bone Toxicity. Hiv Med. 10 (8), 482–487. doi:10.1111/j.1468-1293.2009.00716.x
Wu, C.-Y., Wang, J.-S., Chiou, Y.-H., Chen, C.-Y., and Su, Y.-T. (2007). Biopsy Proven Acute Tubular Necrosis Associated with Vancomycin in a Child: Case Report and Literature Review. Ren. Fail. 29 (8), 1059–1061. doi:10.1080/08860220701643773
Xu, F., Papanayotou, I., Putt, D. A., Wang, J., and Lash, L. H. (2008). Role of Mitochondrial Dysfunction in Cellular Responses to S-(1,2-dichlorovinyl)-L-cysteine in Primary Cultures of Human Proximal Tubular Cells. Biochem. Pharmacol. 76 (4), 552–567. doi:10.1016/j.bcp.2008.05.016
Yun, B., Azad, M. A. K., Wang, J., Nation, R. L., Thompson, P. E., Roberts, K. D., et al. (2015). Imaging the Distribution of Polymyxins in the Kidney. J. Antimicrob. Chemother. 70 (3), 827–829. doi:10.1093/jac/dku441
Zappitelli, M., Moffett, B. S., Hyder, A., and Goldstein, S. L. (2011). Acute Kidney Injury in Non-critically Ill Children Treated with Aminoglycoside Antibiotics in a Tertiary Healthcare centre: a Retrospective Cohort Study. Nephrol. Dial. Transplant. 26 (1), 144–150. doi:10.1093/ndt/gfq375
Zhang, X., Wang, R., Piotrowski, M., Zhang, H., and Leach, K. L. (2015). Intracellular Concentrations Determine the Cytotoxicity of Adefovir, Cidofovir and Tenofovir. Toxicol. Vitro 29 (1), 251–258. doi:10.1016/j.tiv.2014.10.019
Zhao, X., Sun, K., Lan, Z., Song, W., Cheng, L., Chi, W., et al. (2017). Tenofovir and Adefovir Down-Regulate Mitochondrial Chaperone TRAP1 and Succinate Dehydrogenase Subunit B to Metabolically Reprogram Glucose Metabolism and Induce Nephrotoxicity. Sci. Rep. 7, 46344. doi:10.1038/srep46344
Keywords: adverse outcome pathway, nephrotoxicity, protein alkylation, lysosomal disruption, mitochondrial DNA polymerase γ
Citation: Mally A and Jarzina S (2022) Mapping Adverse Outcome Pathways for Kidney Injury as a Basis for the Development of Mechanism-Based Animal-Sparing Approaches to Assessment of Nephrotoxicity. Front. Toxicology 4:863643. doi: 10.3389/ftox.2022.863643
Received: 27 January 2022; Accepted: 11 March 2022;
Published: 15 June 2022.
Edited by:
Catherine Willett, Humane Society International, United KingdomCopyright © 2022 Mally and Jarzina. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Angela Mally, mally@toxi.uni-wuerzburg.de