From the Ground Up: Esophageal Atresia Types, Disease Severity Stratification and Survival Rates at a Single Institution
 Devon Michael Evanovich1,2
Devon Michael Evanovich1,2  Jue Teresa Wang1,3
Jue Teresa Wang1,3  Benjamin Zendejas3,4,5
Benjamin Zendejas3,4,5  Russell William Jennings3,4,5
Russell William Jennings3,4,5  Dusica Bajic1,3*
Dusica Bajic1,3*- 1Department of Anesthesiology, Critical Care and Pain Medicine, Boston Children's Hospital, Boston, MA, United States
- 2Tufts School of Medicine, Tufts University, Boston, MA, United States
- 3Harvard Medical School, Harvard University, Boston, MA, United States
- 4Department of Surgery, Boston Children's Hospital, Boston, MA, United States
- 5Esophageal and Airway Treatment Center, Boston Children's Hospital, Boston, MA, United States
Esophageal atresia (EA), although a rare congenital anomaly, represents one of the most common gastrointestinal birth defects. There is a gap in our knowledge regarding the impact of perioperative critical care in infants born with EA. This study addresses EA types, disease severity stratification, and mortality in a retrospective cohort at a single institution. Institutional Review Board approved our retrospective cross-sectional study of term-born (n = 53) and premature infants (28–37 weeks of gestation; n = 31) that underwent primary surgical repair of EA at a single institution from 2009–2020. Demographic and clinical data were obtained from the electronic medical record, Powerchart (Cerner, London, UK). Patients were categorized by (i) sex, (ii) gestational age at birth, (iii) types of EA (in relation to respiratory tract anomalies), (iv) co-occurring congenital anomalies, (v) severity of disease (viz. American Society of Anesthesiologists (ASA) and Pediatric Risk Assessment (PRAm) scores), (vi) type of surgical repair for EA (primary anastomosis vs. Foker process), and (vii) survival rate classification using Spitz and Waterston scores. Data were presented as numerical sums and percentages. The frequency of anatomical types of EA in our cohort parallels that of the literature: 9.5% (8/84) type A, 9.5% (8/84) type B, 80% (67/84) type C, and 1% (1/84) type D. Long-gap EA accounts for 88% (7/8) type A, 75% (6/8) type B, and 13% (9/67) type C in the cohort studied. Our novel results show a nearly equal distribution of sex per each EA type, and gestational age (term-born vs. premature) by anatomical EA type. PRAm scoring showed a wider range of disease severity (3–9) than ASA scores (III and IV). The survival rate in our EA cohort dramatically increased in comparison to the literature in previous decades. This retrospective analysis at a single institution shows incidence of EA per sex and gestational status for anatomical types (EA type A-D) and by surgical approach (primary anastomosis vs. Foker process for short-gap vs. long-gap EA, respectively). Despite its wider range, PRAm score was not more useful in predicting disease severity in comparison to ASA score. Increased survival rates over the last decade suggest a potential need to assess unique operative and perioperative risks in this unique population of patients. Presented findings also represent a foundation for future clinical studies of outcomes in infants born with EA.
Introduction
Esophageal atresia (EA), although a rare congenital anomaly with a stable world-wide prevalence (1) represents one of the most common gastrointestinal birth defects with reported incidence of 1 in 3,000 to 1 in 4,500 live births (2). If esophageal lumen interruption is left unrepaired, infants are prone to inadequate nutrition and growth, as well as infections such as pneumonia (3). EA is classified into 4 types (type A, B, C, and D) based on the anatomical description in relation to the airway structures (4) (Figure 1), that does not take into account the complexity of underlying disease.
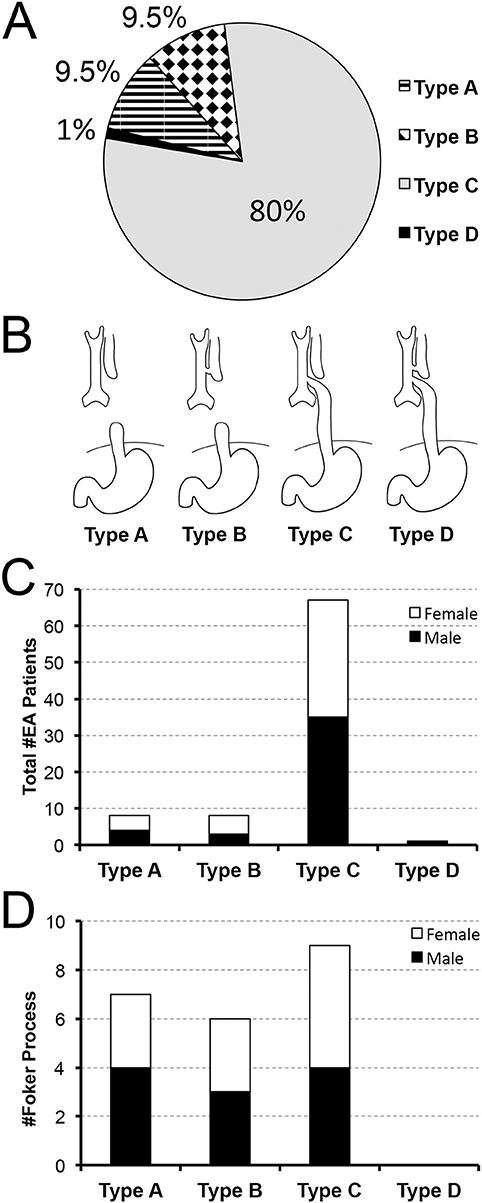
Figure 1. Retrospective Analysis of Incidence of Primary Esophageal Atresia Repair at a Single Institution. Data was retrospectively collected from 2009–2020 (n = 84). Pie chart in (A) summarizes the percent (%) incidence of esophageal atresia (EA) by anatomical classification that is schematically illustrated in (B). According to our retrospective data analysis, anatomical type C of EA is the most frequently encountered. Graph in (C) also illustrates nearly equal sex distribution per anatomical type with females found in 50% in type A, 62.5% in type B, and 48% in type C. Graph in (D) summarizes the incidence of long-gap EA patients (n = 22) that underwent Foker process (5–8). Incidence of long-gap EA follows the trend per anatomical type and sex, similar to the cohort as a whole (see Results for more detail).
In addition to associated malformation, the EA gap length dictates the complexity of perioperative care. If the gap of the esophagus is too large to be repaired by direct anastomosis (>3 cm or >2 vertebral bodies in length), EA is defined as long-gap EA, which is more commonly found in anatomical types A and B of EA (9). At our institution, the latter cases undergo a unique type of EA repair, the Foker process (5–8). Compared to short-gap esophageal atresia, long-gap EA is also more likely to be an isolated defect and associated with trisomy 21 (9). In contrast, short-gap EA is more commonly found with VACTERL anomalies (vertebral, anorectal, cardiac, tracheo-esophageal fistula and/or esophageal atresia, renal, and limb defects/ malformations) relative to long-gap EA (9). Last but not least, CHARGE syndrome (coloboma, heart defects, choanal atresia, growth retardation, genital abnormalities, and ear abnormalities) (10) can further complicate the care of infants born with EA.
The complexity of underlying disease with and without other congenital anomalies in the case of EA is undeniable. Although short-gap EA is repaired with direct anastomosis and requires shorter pain management, infants are vulnerable to post-operative feeding challenges (11) in addition to the impact of associated anomalies. In cases of long-gap EA, the revolutionary Foker process (5–8) encourages the natural growth and lengthening of infant's existing esophageal pouches, but it requires at least two separate thoracotomies/thoracoscopies with a subsequent prolonged postoperative intubation (12, 13) associated with development of physical dependence to drugs of sedation (13–15). The primary goal of this study addresses the severity of EA disease as a blueprint for a subsequent myriad of caregiving conditions that might impose challenges to developing infants born with EA. Specifically, we conducted a retrospective analysis at a single institution to analyze incidence of EA by severity of disease using American Society of Anesthesiologists (ASA) (16) and Pediatric Risk Assessment (PRAm) (17–19) scores in the context of (i) sex, (ii) gestational age (term-born and premature), (iii) anatomical classification of EA types, and (iv) the type of surgical repair (viz. direct anastomosis vs. Foker process with prolonged sedation). Our secondary clinical end-point measures looked into mortality risk in the context of other co-morbidities according to Spitz et al. (20) and Waterson et al. (21) classifications that take into account co-existing pneumonia and cardiac disease, respectively. This study was, in part, previously published as thesis (22).
Methods
Study Design and Subjects Equations
Institutional Review Board at Boston Children's Hospital approved this retrospective cross-sectional research study (IRB-P000007855) of infants born with esophageal atresia (EA) that underwent primary surgical repair at a single institution. The study conformed to the standards set by the Declaration of Helsinki and Good Clinical Practice guidelines. The patient information was obtained from a prospectively maintained clinical database, The Esophageal and Airway Treatment Center REDCap database, established in 2009. Considering our institution is not a birthing center, all infants cared for at our institution are considered outborn. Eligibility criteria included: (1) term-born (defined as birth between 37 and 42 weeks of gestation) and premature infants (28–37 weeks of gestation) born with EA of any type, and (2) patients that received their primary surgical repair at Boston Children's Hospital. Cohort patients underwent surgery in the first month of life with exception of two patients that were born outside of the State and underwent primary surgical repair at our institution at 2 and 3 months of age. Exclusion criteria included: (1) extreme prematurity (<28 weeks of gestation), and (2) any surgical repair at other institutions (including but not limited to EA repair). Our retrospective study included a total of 84 patients (n = 53 term-born; n = 31 premature) over the period of 11 years (2009–2020).
Chart Review
Electronic medical record, Powerchart (Cerner, London, UK) was used to collect demographic data (viz. date of birth; gestational age at birth (weeks); birth weight (kg)) and clinical data. The latter included several end-point measures.
Esophageal Atresia Types
In addition to classification of EA into 4 anatomical types (type A, B, C, and D) based on the anatomical description in relation to the airway structures (4) – in particular to co-existence with tracheo-esophageal fistula (TEF), we also classified EA cases based on the length of EA gap into: short-gap (that was repaired by primary anastomosis) and long-gap that underwent repair by Foker process (5–8). Some of the patients with long-gap EA were managed with our newer minimally invasive Foker process which entails an internal adjustable traction system that is adjusted every 5–7 days via a thoracoscopy. As such, it leads to less postoperative muscle paralysis and sedation in comparison to the external traction Foker process via thoracotomy (8, 23, 24). For the purpose of this study, we identify short-gap EA with direct anastomosis repair, and long-gap EA with the Foker process repair - as these are the two main surgical approaches at our Institution.
Medical/Surgical Comorbidities
As EA often presents with other congenital anomalies and/or co-morbidities, we collected clinical data regarding any other genetic or chromosomal anomalies (e.g., Trisomy 18, 21 etc.), and any other associated congenital anomalies (e.g., vertebral, cardiac, anal anomalies etc.), some of which are a part of the complex congenital syndromes associated with EA such as VACTERL (25) or CHARGE syndrome (26). Co-existing cardiac anomalies were classified as minor (not requiring surgical intervention such as patent foramen ovale, patent ductus arteriosus, atrial septal defect, ventricular septal defect, dextrocardia), or complex (requiring surgical correction such as large ventricular septal defect, large atrial septal defect, coarctation of the aorta, large patent ductus arteriosus, and Tetralogy of Fallow). We quantified the incidence of other associated co-morbidities such as: (i) pneumonia treatment, and (ii) cardiac surgeries, which both served as a basis for mortality risk evaluation (see below). Due to the retrospective study design, characterization of associated co-morbidities was obtained from the medical records as part of the clinical diagnostics and treatment.
Disease Severity
Complexity of clinical status in the context of other comorbidities was assessed using two scoring systems: American Society of Anesthesiologists (ASA) (16) and Pediatric Risk Assessment (PRAm) (17–19) scores at the time of EA repair surgery. Assigning an ASA Physical Status classification level is a clinical decision based on several factors (16) and represents the most commonly used assessment of system level disease severity by anesthesiologists (Figure 3A–Table). ASA scores are based on several factors and range from ASA I (normal healthy patient) to ASA VI (a declared brain dead patient) (16). In contrast, PRAm scoring is a relatively novel measure introduced in 2017 (17–19) that involves 5 scoring points: urgency of surgical procedure (+1), presence of at least one comorbidity (+2), presence of at least one indication of critical illness (+3), age <12 months at surgery (+3), and co-existing malignancy (+4) for a range of scores from 0 to 13. PRAm scoring has been designed as a less subjective assessment of disease severity for the use specifically in pediatric populations (scores 0 – 13; Figure 3B–Table).
Mortality Risk Assessment
Considering our retrospective data collection spans period of last 11 years (2009-2020), it was used for comparison to previously published survival rates in infants born with EA as per two different scoring systems: (i) Waterston et al. (21), and (ii) Spitz et al. (20). As originally described by Waterston et al. (21), this scoring system takes into account weight of the patient, co-existence of other congenital anomalies, and pneumonia. Scoring is described as low risk (group A: birth weight >2.5 kg with no or co-existing congenital anomaly or pneumonia), moderate risk (group B: birth weight 1.8–2.5 kg with co-existing mild pneumonia and mild congenital anomaly), or high risk (group C: birth weight 1.8–2.5 kg with co-existing severe pneumonia and severe congenital anomaly). To improve clarity and transparency with scoring definitions, we modified the original scoring by Waterson et al. (21) for moderate and high risk groups. In this report, the moderate mortality risk group (group B) included infants with co-existing moderate pneumonia (defined as receiving antibiotics), and/or a moderate congenital anomaly (viz. limb anomalies, cleft lip or palate, atrial-septal defect or small patent ductus arteriosus). High mortality risk (group C) in this study referred to infants with severe pneumonia (defined as requiring mechanical ventilation) and/or a severe congenital anomaly (viz. defined as making survival difficult or impossible without surgical repair). The second scoring system, described by Spitz et al. (20), takes into account weight and co-existence of a severe cardiac congenital anomaly. The risk is described as low (I), moderate (II), and high (III) risk of mortality with major cardiac anomalies defined as one that required medical or surgical treatment.
Statistical Analysis
Data was presented as numerical sums and percentages for (i) sex and anatomical classification of EA, (ii) gestational age at birth, (iii) distribution of long-gap EA patients for sex and gestational age, (iv) distribution of congenital anomalies, (v) disease severity scores, and (vi) survival rate. PRAm scores were also presented as numerical sums and as boxplot distributions indicating median scores, first and third quartile ranges, and absolute values for minimum and maximum values.
Results
The retrospective chart review included infants that underwent EA repair at a single institution over a period of 11 years (2009–2020; n = 84): 53 term-born, and 31 premature (born between 28 and 37 weeks of gestation).
Demographic Information and Incidence of Esophageal Atresia Types
Anatomical Types
Figures 1A,B illustrate incidence of EA patients according to the anatomical types of EA (4): type A (isolated EA; 8/84, 9.5%), type B (TEF at the upper esophageal pouch; 8/84; 9.5%), type C (the most common type of EA with TEF at the lower esophageal pouch; 67/84; 80%), type D (the rarest type of EA with TEF at each esophageal pouch; 1/84; 1%). Our novel data implicate equal distribution of sex for infants born with EA (Figure 1C) with 49% (41/84) female and 51% (43/84) male patients of nearly equal distribution per anatomical types of EA: 50% (4/8) female with type A, 62.5% (5/8) female with type B, and 48% (32/67) female with type C.
Esophageal Gap: Short-Gap vs. Long-Gap
We also distinguished between short-gap and long-gap EA (see Method's section). The latter is equated to Foker process repair (5–8), that represented 26% of the cohort (22/84). Unlike the cohort as a whole (Figure 1C), the incidence of long-gap EA cases showed nearly equal distribution by anatomical types (Figure 1D): type C (9/22; 41%), type A (7/22; 32%), and type B (6/22; 27%). However, long-gap EA accounted for 88% (7/8) of patients in type A, 75% (6/8) in type B, and 13% (9/67) in type C (not graphically shown). Importantly, infants born with long-gap EA showed exactly equal distribution of sex (50%; 11/22 female) with nearly equal distribution per anatomical types of EA (Figure 1D): 42% (3/7) female with type A, 50% (3/6) female with type B, and 56% (5/9 female) with type C.
Gestational Age
Taking into account exclusion of extreme prematurity, this retrospective cohort shows slightly higher frequency of term-born (53/84, 63%) than premature patients (31/84; 37%) with similar trend per anatomical classification of EA types: 75% (6/8) term-born with type A, 50% (4/8) term-born with type B, and 63% (42/67) term-born with type C (Figures 2A,B). There was only one term-born patient with type D EA. Similarly, infants that underwent Foker process for long-gap EA repair (Figure 2C) had a similar frequency of term-born and premature patients (11/22; 50%). However, term-born patients with long-gap EA were predominantly noted in type A (6/7; 86%) while premature infants with long-gap EA represented majority in type B (4/6; 67%) and type C (6/9; 67%) as illustrated in Figure 2C.
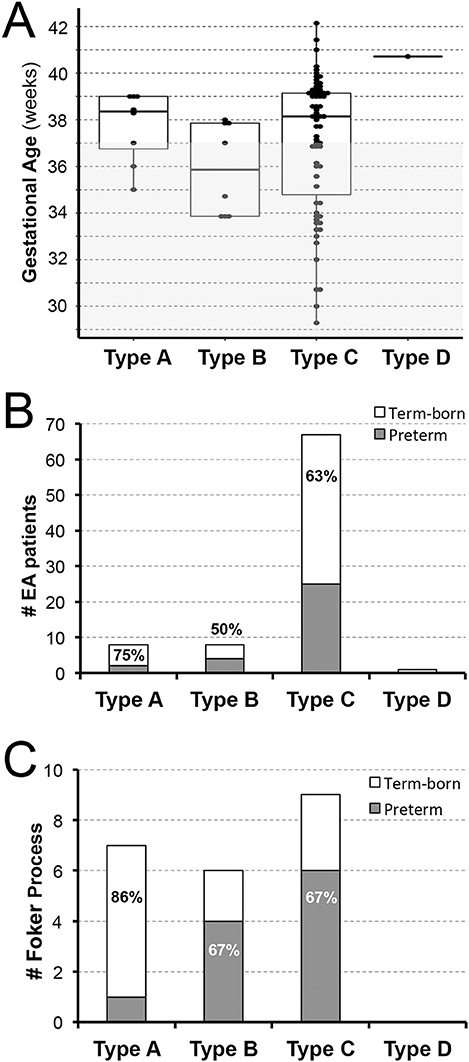
Figure 2. Retrospective Analysis of Esophageal Atresia Classification by Gestational Age at Birth. Retrospective data of infants born with esophageal atresia (EA) was collected from 2009–2020 (n = 84) and included infants born ≥28 weeks of gestation that were classified as term-born (37–42 weeks gestation) or premature (28–37 weeks of gestation). (A) Illustrates individual distribution of gestational age at birth per EA type (dots), while gray area schematically marks prematurity (<37 weeks of gestation). (B) Summarizes percent (%) incidence of EA per anatomical type and gestational age groups with either equal (type B) or predominant incidence of term-born patients (type A and C). In this cohort of infants with primary surgical repair of EA at our institution, we report only one term-born infant with type D EA. For illustration of anatomical EA types, please see Figure 1B. (C) Summarizes incidence of infants that underwent Foker process (5–8) for long-gap EA repair. We report equal incidence of term-born and preterm patients. However, term-born patients with long-gap EA were predominantly noted in type A (6/7; 86%) while premature infants with long-gap EA represented majority in type B (4/6; 67%) and type C (6/9; 67%) anatomical EA type.
Incidence of Co-existing Congenital Anomalies
Syndromes Associated With Esophageal Atresia
Table 1 summarizes the incidence of other co-existing anomalies with EA. About 42% (35/84) of the cohort patients had complex EA disease as part of a syndrome or known chromosomal abnormality (Table 1A). Of those, the most frequent was VACTERL syndrome (31/35, 89%), although we also report cases of CHARGE syndrome (2/35, 6%), trisomy 21 (Down's syndrome; 1/35) and trisomy 18 (Edwards syndrome; 1/35). We also report a similar incidence of VACTERL syndrome in those that underwent primary repair (viz. short-gap EA; 24/62; 39%) compared to infants that underwent the Foker process for the repair of long-gap EA (7/22; 32%).

Table 1. Incidence of esophageal atresia in the context of other congenital anomalies.
Esophageal Atresia in the Absence of Syndrome
For infants born with EA without associated syndrome (49/84; 58%), the majority had either 2 (17/49; 35%) or more than 2 (16/49; 33%) co-occurring congenital anomalies. Only a minority of patients had no co-existing congenital anomalies (6/49, 12%; Table 1B). Interestingly, a majority of patients – apart from syndromic patients (49/84; 58%) - had a cardiac anomaly (38/49; 78%) and no patients had a documented anorectal anomaly occurring outside of a syndrome (Table 1C).
Cardiac Co-anomalies
Of all the infants born with EA that had co-existing cardiac anomalies (72/84; 86%), only 18% (15/84) had congenital heart disease severe enough to require surgical repair (Table 2A). Of those that underwent cardiac surgery, 93% (14/15) had type C EA, and only one patient had type B EA. We report a similar pattern of co-existing congenital cardiac anomalies in infants with long-gap EA that underwent the Foker process repair (Table 2B): 86% (19/22) had co-existing cardiac anomalies, but only 14% (3/22) required cardiac surgery and were diagnosed with either type C EA (2/3) or type B EA (1/3). Table 2 summarizes severity of cardiac findings in this cohort.
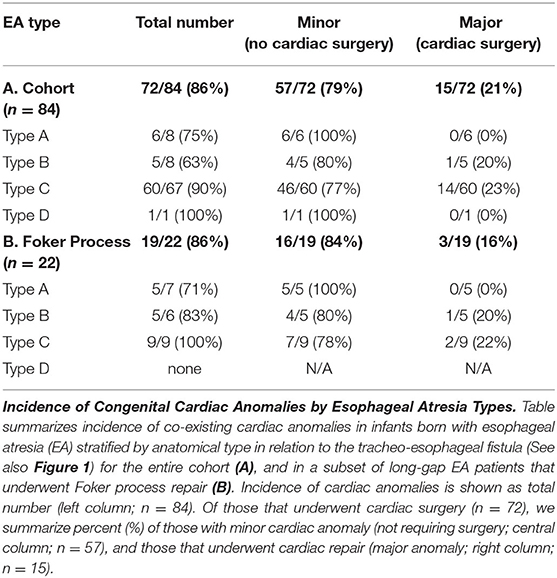
Table 2. Incidence of co-existing congenital cardiac anomalies.
Severity Stratification of Underlying Disease
Figure 3 illustrates 2 different disease severity scores by anatomical EA types (types A-D; Figure 1B) and gestational age (term-born vs. premature).
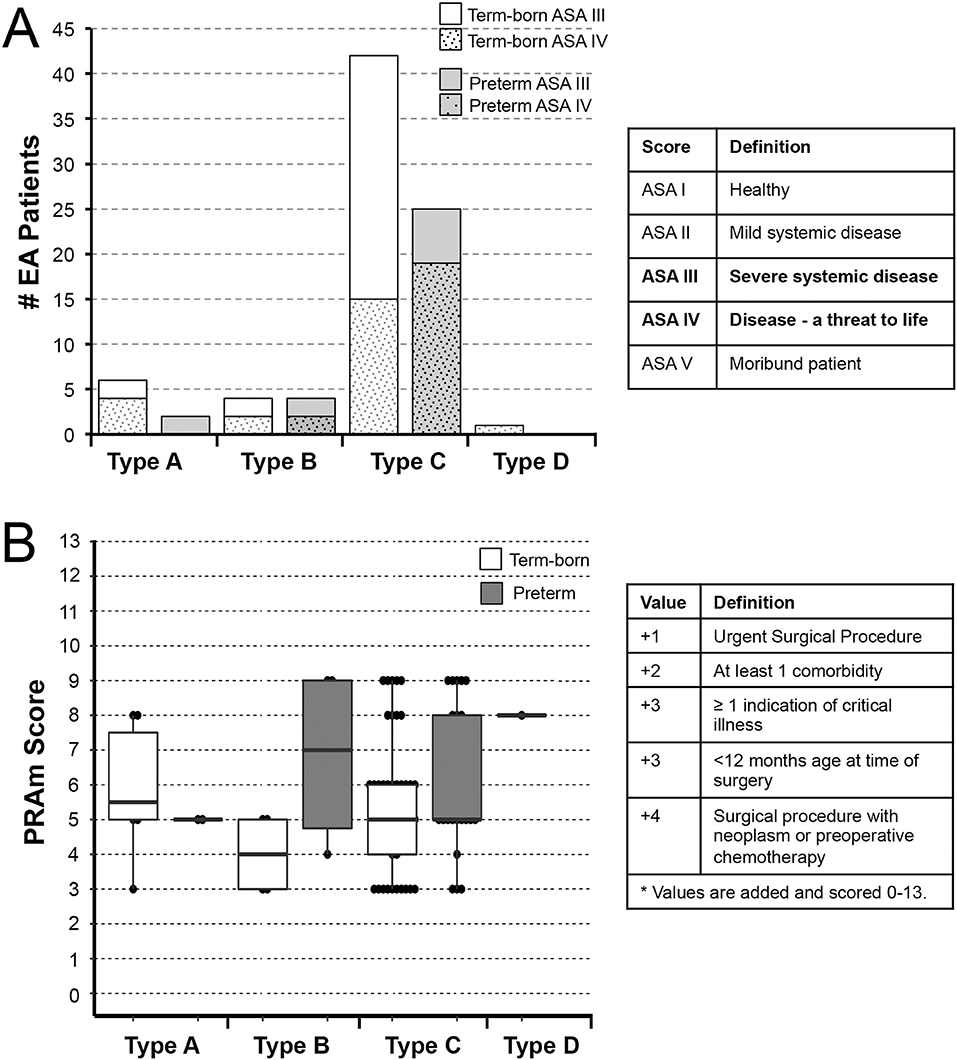
Figure 3. Underlying Disease Severity Stratification in Esophageal Atresia Cohort. Graphs illustrate incidence of American Society of Anesthesiologists (ASA) Physical Status classification (A) and Pediatric Risk Assessment (PRAm) severity scores (B) in infants born with esophageal atresia (EA) that underwent primary repair at a single institution from 2009-2020 (n = 84). Definition of scoring is shown in summary tables on the right for both the ASA Physical Status (16) and PRAm scoring (17–19). Graphs show stratification by anatomical types of EA (type A-D) and gestational age (term-born and premature; see Methods section). Specifically, all patients in this study were rated as either ASA Physical Status III or IV (A). For the most common type C EA, the majority of term-born patients had ASA III status (27/42; 64%), while the majority of premature infants were assigned ASA IV status (19/25; 76%) implicating premature infants were more critically ill in the most common type of EA, type C. Graph in (B) illustrates distribution of PRAm scores per anatomical type and gestational age. Considering all infants had surgery when <12 months of age, the minimal score was 3. Since none of the infants had co-existing malignancy, the highest score was 9. From the graph in (B), one can infer that premature infants had higher median score for type B EA, but lower median score for type C (thick horizontal line). Individual values are represented as dots, boxes span the interquartile range (IQR) (first and third quartile), and whiskers represent maximum and minimum values.
American Society of Anesthesiologists Physical Status Classification
Half of the patients of this cohort were rated either ASA Physical Status III (49% (41/84) with severe systemic disease; 76% (31/41) term-born; 24% (10/41) premature), or ASA Physical Status IV (51% (43/84) with severe systemic disease that is a constant threat to life; 51% (22/43) term-born; 49% (21/43) premature). Figure 3A shows ASA Physical Status classification of the cohort infants per anatomical types of EA. For the most common type of EA – type C, the majority of term-born patients received ASA III status (27/42; 64%), while the majority of premature infants received ASA IV status (19/25; 76%).
Pediatric Risk Assessment Scores
Considering all patients in this retrospective cohort underwent surgical repair in infancy, and none had any associated malignancy, the PRAm score ranged from 3 to 9 across all anatomical types and gestational age groups (Figure 3B). For the cohort as a whole, we report Median PRAm score of 5 for both term-born (interquartile range of 4–6) and premature infants (interquartile range of 5–8). When classified according to anatomical EA types, premature infants had a higher median score for type B EA (Median 7; interquartile range of 4.75–9), but a lower median score for most frequent type C (Median 5; interquartile range 5–8) in comparison to term-born infants (Median 4; interquartile range of 3–5 for type B; Median 5; interquartile range of 4–6 for type C). While type B shows higher PRAm scores for premature infants, there is a limited number of patients in this group (n = 8) compared to type C (n = 67). Furthermore, we report a wide PRAm score classification (Figure 4A) for term-born patients with propensity for lower PRAm scores (25% (13/53) PRAm 3; 9% (5/53) PRAm 9), and premature infants with propensity for higher PRAm scores (10% (3/31) PRAm 3; 23% (7/31) PRAm 9).
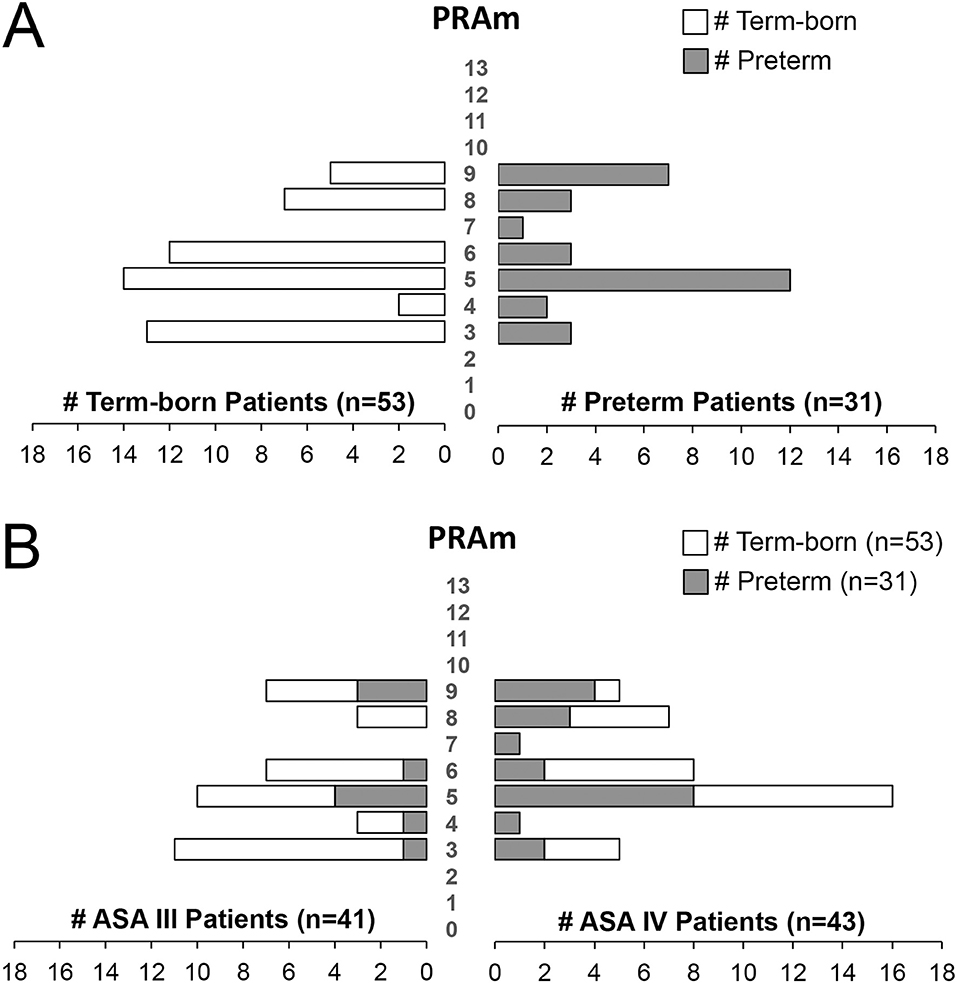
Figure 4. Pediatric Risk Assessment (PRAm) Scores in a Retrospective Cohort of Infants Born with Esophageal Atresia. (A) Shows PRAm scores of term-born (left) and preterm patients (right) illustrating a wide range of PRAm score across gestational age of infants born with esophageal atresia (EA; n = 84). Note a subtle tendency of term-born patients for lower, and premature patients for higher PRAm scores. (B) Plots PRAm scores in relation to American Society of Anesthesiologists (ASA) Physical Status Classification. Despite more premature infants having had higher ASA IV classification (21/31; 68%) in comparison to term-born (22/53; 41%; see also Figure 3A), PRAm score shows wide distribution of scores between 3 and 9. Such outlining is in support of ASA and not PRAm scoring in assessing disease severity when gestational age is the primary factor.
Relationship Between ASA Physical Status and PRAm Scores
To better gauge individual relationship of two different scores, Figure 4B illustrates relationship between ASA physical status and PRAm scores. Despite wide distribution of PRAm scores irrespective of the gestational age (Figure 4A), patients with assigned ASA IV classification had about equal distribution per gestational age groups: premature (21/43; 49%) and term-born patients (22/43; 51%). However, we do not show that premature infants with ASA IV physical classification (n = 21/43) align with higher PRAm score distribution (Figure 4B).
Disease Severity With Respect to Type of EA Surgical Repair
We also report a wide distribution of PRAm scores (scores 3-9) irrespective of the type of surgical repair (Figure 5A). However, infants born with short-gap EA undergoing direct anastomosis repair (n = 62) have a propensity for lower PRAm scores in term-born patients (62.5% (10/16) PRAm 3; 42% (5/12) PRAm 9) and higher PRAm scores in premature patients (12.5% (2/16) PRAm 3; 17% (2/12) PRAm 9) as illustrated in Figure 5B. Similarly, infants born with long-gap EA undergoing Foker process repair (n = 22) have a propensity for lower PRAm scores in term-born patients (19% (3/16) PRAm 3; 0% (0/12) PRAm 9) and higher PRAm scores in premature patients (6% (1/16) PRAm 3; 42% (5/12) PRAm 9) as illustrated in Figure 5B. Considering more premature patients with EA have ASA IV classification status (Figure 3A), prematurity should be considered a confounding factor for increased underlying disease severity. As such, other important aspects of prematurity, such as intra-uterine growth retardation and prematurity associated sequelae (e.g., respiratory distress syndrome) should be considered as potential indirect markers of prematurity in assessing outcomes following EA repair. Finally, when PRAm scores (with score range from 3–9) are graphed in relation to ASA physical status (Figure 5C), more infants with long-gap EA are scored as ASA IV classification (73%; 16/22) compared to short-gap EA patients (44%; 27/62). Future goals should include unique scoring system design that would include other potential confounders unique for EA repair, such as length of post-operative mechanical ventilation and antibiotic treatment as indirect markers of postoperative sedation and infections, respectively.
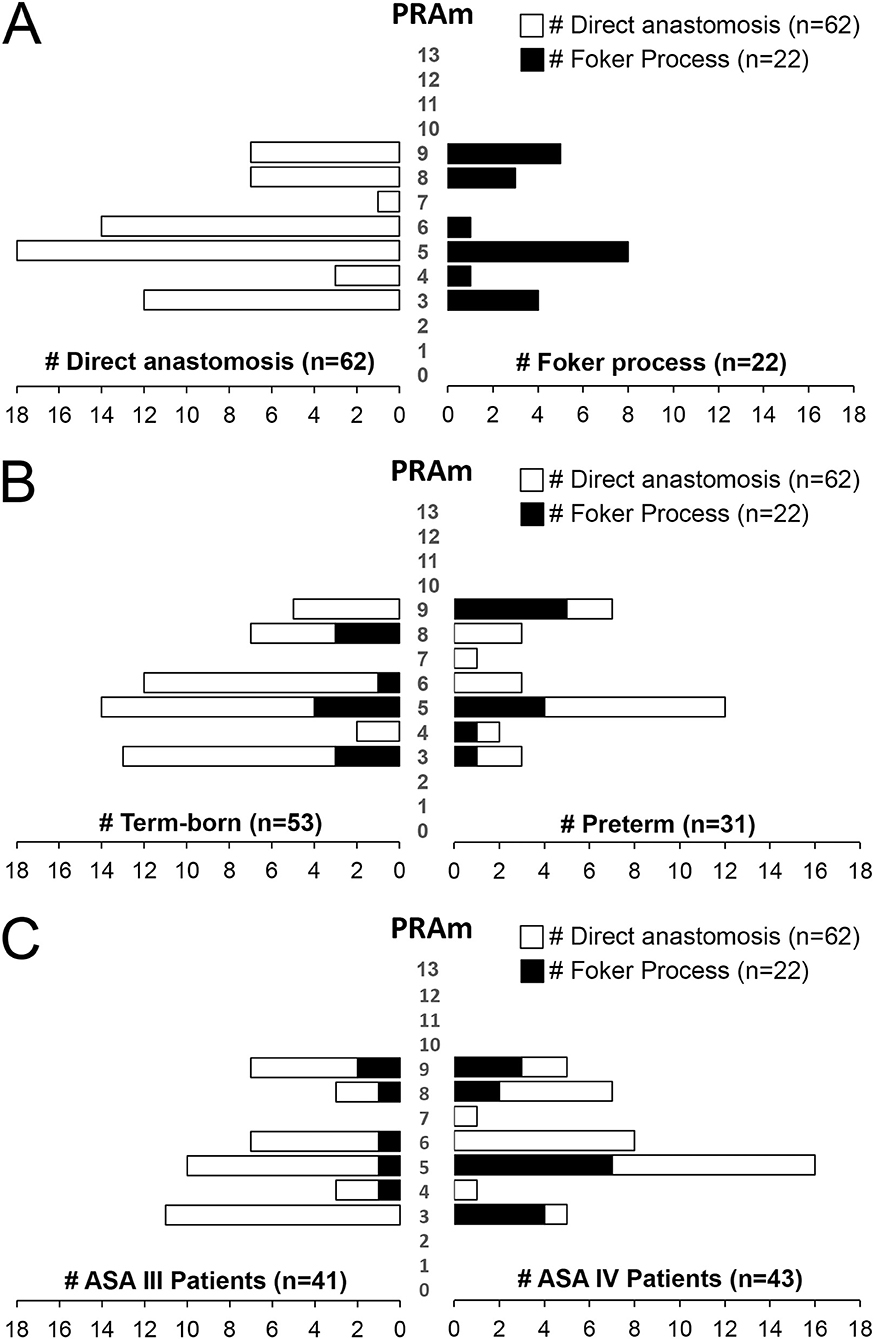
Figure 5. Disease Severity Stratification of Infants Born with Esophageal Atresia by Type of Surgical Repair. (A) Illustrates wide distribution of Pediatric Risk Assessment (PRAm) scores (from scores 3–9) by the type of surgical repair: (i) direct anastomosis for short-gap esophageal atresia (EA) repair (left), and (ii) Foker process (5–8) for long-gap EA repair (right). (B) Summarizes severity of disease per type of surgical repair in relation to the gestation age at birth. Similar to data in Figure 4A, note the subtle tendency of term-born patients having had lower, and premature patients higher PRAm scores. When PRAm scores are graphed in relation to American Society of Anesthesiologists (ASA) physical status (C), we report more infants with long-gap EA with an ASA IV classification (73%; 16/22).
Mortality Risk Assessment of Infants Born With Esophageal Atresia
Mortality Risk Assessment I
Table 3 summarizes the mortality risk assessment as originally described by Waterston et al. (21) in EA patients according to the (i) birth weight, (ii) co-existing congenital anomalies (Tables 1 and 2), and (iii) co-morbidity with pneumonia into: low (group A), moderate (group B), and high mortality risk (group C). Indeed, our retrospective study is of similar population size of EA patients (n = 84) as in the original report (n = 113) (21). However, our cohort had smaller numbers of infants in low risk group and much higher number of infants in high mortality group, group C (Table 4). With that in mind, we also show striking survival rates especially in the moderate risk (group B; 100% (42/42) vs. 68% (29/43 in the Waterson's study) and high risk group [group C; 95% (38/40) vs. 6% (2/32) in the original study (21)].
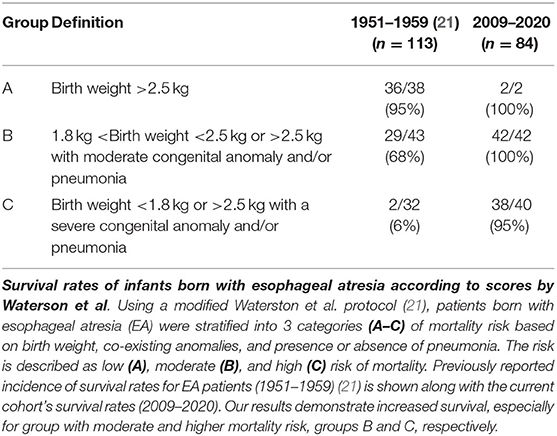
Table 3. Mortality risk assessment I.
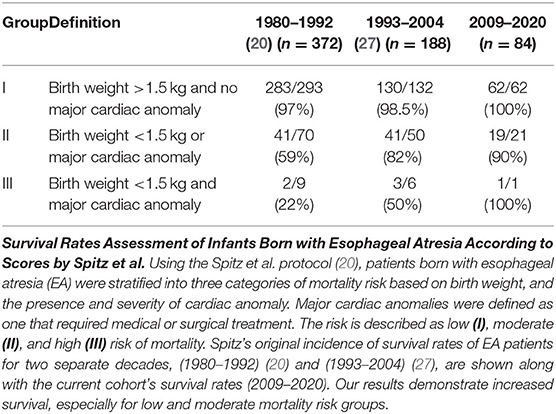
Table 4. Mortality risk assessment II.
Mortality Risk Assessment II
The second scoring system by Spitz et al. (20) takes into account (i) birth weight and (ii) co-existence and (iii) severity of cardiac congenital anomalies. The risk is described as low (I), moderate (II), and high (III) risk of mortality. Although our retrospective cohort has lower power (n = 84) in comparison to previous decades' reports: 1980–1992 (n = 372) (20) and 1993–2004 (n = 188) (27), we report improved survival rates (Table 4), especially for low risk group (I; 100%; 62/62) and moderate risk group (II) of 90% (19/21). This data are in stark contrast to moderate risk group (II) survival rates of 59% (41/70) during 1980s (1980–1992) (20) and 82% (41/50) survival rate in the following decade (1993–2004) (27). Since our cohort only had only one patient that met criteria for group III - in part due to the exclusion criteria of extremely premature patients (<28 weeks), future clinical studies are needed to evaluate group III survival rates at our institution.
Dicussion
Our novel results using retrospective approach from a single institution show near equal distribution of sex and gestational age (term-born vs. premature) by anatomical type of EA (types A–D) and by type of surgical repair (direct anastomosis vs. Foker process). We also share the incidence of co-occurring congenital anomalies with EA, with special emphasis on cardiac anomalies that have been shown to be a major mortality risk factor for infants born with EA (20). Although PRAm score showed a wider range of disease severity (3–9), ASA scores (III and IV) are more useful in predicting disease severity. We also report increased survival rate in our EA cohort in comparison to the literature in previous decades.
Limitations of the Retrospective Chart Review
In keeping with the retrospective study design (28), data collected were originally intended for reasons other than research (29, 30). Due to the dependence of patient information stored for clinical practice, retrospective analysis may represent incomplete or missing documentation, poorly recorded or absent chart information, as well as difficult identification of desired patient data [e.g., attainment of PRAm scores (17) for cases of EA repair prior to 2017].
Study Size
Despite the exclusion of cases with extreme prematurity, and surgical repair at an outside institution, this study retained a moderate sample size with enough power to evaluate EA disease characteristics. The main challenges imposed with the exclusion criteria are that of generalizability since our institution represents a highly specialized level of care.
High-Risk Mortality Scores
Mortality risk assessment scores were quantified according to the scores previously described in the literature by Waterston et al. (21), and Spitz et al. (20). For both types of scoring, the high-risk groups used very low birth weights as surrogates for extreme prematurity. For this study, we defined Waterston risk assessment for the highest risk group (group C) as either (i) a low birth weight (associated with prematurity) or (ii) a standard birthweight with severe congenital anomaly or severe pneumonia. Therefore, analysis of Waterston et al. (21) group C was likely not significantly impacted by the exclusion of extreme prematurity. In contrast, the Spitz risk assessment required a birth weight <1.5 kg for the highest risk group (group III) for which only one patient from our retrospective cohort met criteria. Therefore, risk assessment of group III of the Spitz et al. (20) classification is not powered in our retrospective study.
Characteristics of Esophageal Atresia Cohort
Anatomical Types of Esophageal Atresia
We report similar distribution of EA patients according to the anatomical classification (Figure 1A) compared to the literature (4, 31, 32) although one should keep in mind that definition of long-gap EA might differ. Our slightly increased incidence for type A and type B EA could be explained by the fact that data was obtained from a single institution, which pioneered the Foker process (5–8) for repair of long-gap EA (9) and receives patients locally, nationally and internationally. Indeed, we report higher incidence of long-gap EA in more rare types of EA: types A and type B, despite type C having the highest number of long-gap EA patients in this cohort (Figure 1D). The definitions for long-gap EA have not been agreed upon and can confound the comparison of data within the literature. Currently, a common method of classification is to simply define types A and B as long-gap EA and all others as short-gap EA (33, 34). The presence of long-gap EA patients with type C EA (Figure 1D) is in direct contrast to those studies that classify long-gap EA by purely anatomical classification. However, Ure et al. in 1995 (35) reported a total of 9 long-gap EA patients and a majority of them having type C EA. Study by Donoso et al. in 2016 (34) reported that a single patient with type C EA underwent long-gap EA repair despite being classified as short-gap in their study. Due to the inconsistency in the literature, we defined long-gap EA by surgical procedure [viz. Foker process (5–8)] implicating complex perioperative critical care instead of anatomical definitions based on the location of the EA gap.
Sex Distribution
We report nearly equal sex distribution for the entire cohort (n = 84), which is consistent with previous large retrospective reports (36–39). Since sex distribution for EA patients was only reported for the entire cohort (34, 39–41), we indicate – for the first time – that there is nearly equal sex distribution in EA patients by anatomical types (Figure 1C). We also report equal sex distribution for long-gap EA patients, which is in accordance with previous report in a larger cohort (9). Findings of equal sex distribution indicate that there is possibly no sex preference in infants born with EA – the subject of interest that continues to be investigated.
Distribution as Per Gestational Age
Our study found a slightly higher incidence of term-born patients in comparison to premature infants (28–37 weeks of gestation) with EA (Figures 2A,B). This finding stands in contrast to a large national cohort study of EA that included all gestational ages and found a higher prevalence of EA in premature patients (39). This discrepancy could be explained, in part, by our exclusion criteria that eliminated extremely premature patients from the cohort. However, our findings of higher incidence of term-born patients are consistent with the national studies in France (41) and Italy (40) reporting similar results. While the incidence of gestational age in long-gap EA patients are reported in the literature (6, 42), our novel results outline incidence of long-gap EA by anatomical types (Figure 1B). Discrepancy among gestational age within EA studies and novel findings of gestational age distribution in long-gap EA patients (with exclusion of extreme prematurity) suggests the need for future analysis to discern demographics of prematurity, as it represents an important risk factor in hospital mortality (43).
Incidence of Associated Congenital Anomalies and Comorbidities With Esophageal Atresia
Congenital co-anomalies with EA can present as a wide spectrum across multiple organ systems (26), and can pose a challenge for care of infants with EA with increased risk of mortality and morbidity (44).
VACTERL Syndrome
The incidence of VACTERL association in this cohort was high at 37% compared to the literature report at around 10% (44) (Table 1A). Higher incidences of VACTERL syndrome in our report has previously been recognized in other studies (9) and is likely due to recognized differences in VACTERL diagnostics (45). Our cohort was comprised of sicker infants due to the very low incidence of isolated EA at 12% (Table 1B) compared to very large cross-hospital findings of isolated EA at 45% (36), 57.3% (37), and 38.7% (38) at other institutions. Findings of increased incidence of VACTERL and other comorbidities in this study possibly reflects institutional reputation as a national and international referral center for infants born with EA. Our report of higher incidence of VACTERL patients in short-gap EA are in accordance with previous report from our institution (9). This is in contrast to the study from Tabriz Children's Hospital and Tehran Mofid Hospital in Iran that reported no difference in incidence of VACTERL spectrum defects irrespective of the type of surgical repair required (42). The analysis of etiology of VACTERL syndrome is outside the scope of this study but is described well in the literature (25, 45, 46) and continues to be investigated.
Congenital Cardiac Anomalies
It is well known that co-occurring cardiac anomalies can impact the length and complexity of care for EA patients (41). We identified that most EA patients had co-existing cardiac anomalies: 86% for the entire cohort (Table 2A) and 79% in the non-syndromic cases (Table 1C). This is consistent with literature report from the Children's Hospital of Chongqing Medical University, China (71.2%) (47) and a large multicenter study of EA patients across 43 hospitals (70%) (43). We and others (48) report that a great majority of co-existing cardiac anomalies were defined as simple. We also report, for the first time, that only about a fifth of EA patients with co-existing cardiac anomalies had undergone cardiac repair – of which majority were type C EA cases (Table 2A). Our novel data in infants with long-gap EA show having nearly equal incidence of cardiac co-anomalies compared to the entire cohort with a similar incidence of patients requiring cardiac surgery (Table 2B). Our results confirm that (i) non-syndromic patients with EA may present with additional and potentially life-threatening congenital anomalies and that (ii) infants with non-syndromic long-gap EA can have cardiac anomalies imposing additional risk to their care (20, 41).
Perioperative Risk Assessment
Underlying Disease Severity
Despite wider PRAm score variability (score 3-9; Figure 4A) and the same PRAm median score of 5 irrespective of the gestational age (Figure 3B), ASA Physical Status classification remains a golden standard in assessing underlying disease severity. We only noted a trend in term-born patients toward propensity for lower PRAm scores irrespective of the surgical type, while premature patients with long-gap EA showed propensity for higher PRAm scores (Figure 5B). Latter trends are in alignment with the seminal report in literature of a large cohort of infants undergoing non-cardiac surgery, validating PRAm scoring in predicting perioperative risk (17). Future work should also analyze unique risk factors related to surgical type of EA repair (viz. direct anastomosis vs. Foker process; open vs. laparoscopic approach) to expand on previous risk stratification of patients born with EA (49, 50). Morbidity risk assessment should also possibly include assessment of the neurological findings as our recent pilot study of infants with long-gap EA reported incidental brain findings for not only premature but term-born infants following Foker process repair (n = 13/group) (12, 15, 51, 52).
Mortality Risk Assessment
We report increased survival rates in this cohort as per two different mortality risk assessment scores. Despite our modification to the scoring schema by Waterston et al. (21), we report the total survival rates have vastly improved in the last decade for each of the described Waterston risk groups (Table 3). In addition, Spitz et al. (20) extended Waterston's mortality risk score in 1994 by including co-existing cardiac anomalies with EA. Indeed, the latter mortality risk score represents the most widely used mortality risk stratification that continues to be used for assessing risk in EA patients (27, 34). According to the most recent study of mortality predictors, major congenital heart disease was a significant predictor, while birth weight <1.5 kg was not (53). Indeed, we report increased survival rates in the most recent decade (2009–2020; Table 4) when compared to 1980s (20) and 1990s (27, 34) as previously published by Spitz et al. (20, 27). Such data are in support of great improvements in perioperative critical care of EA patients and treatment of their comorbidities, which may account for our reporting of improved survival. As per literature recommendation, the preferred clinical management of infants born with EA should be aided by highly specialized multidisciplinary team at expert centers (54) to help increase survival and decrease the incidence of morbidities (55). Therefore, improved outcomes in this report may be explained – in part, by the highly specialized nature of The Esophageal and Airway Treatment Center at our institution. Increased survival rates over the last decade suggest a potential need to assess unique operative and perioperative risks in this unique population of patients, as well as non-survival metrics such as functional status and quality of life.
Last, but not least, previous reports also suggests that extremely low birth weight infants with EA patients are at a higher risk of mortality (56), while a recent study showed potentially improved outcomes for extremely low birth-weight infants that underwent a staged repair for EA (57). Analysis of survival rates of extremely low birth weight infants with EA, especially those that underwent the Foker process for long-gap EA repair is needed to validate and potentially expand on these and our findings.
Conclusions
We present a comprehensive analysis of EA patient classification by anatomical types (type A-D) and by surgical repair type (direct anastomosis vs. Foker process for short-gap vs. long-gap EA, respectively). Despite a wider PRAm score distribution in infants born with EA, ASA scores remain the gold standard in assessing underlying disease severity stratification. With increase in survival rates over the last decade, future studies should be directed toward assessing unique aspects of EA group in the context of (i) severity of underlying disease with and without comorbidities, (ii) unique complexities of perioperative critical care (with and without prolonged sedation, repeated procedures, and infection/sepsis assessment), and (iii) survival metrics such as functional status (e.g., neurobehavioral outcomes and quality of life). It is our hope that this retrospective study will be of service to research community when designing future clinical studies of risks and outcomes in this uniquely vulnerable population of infant patients.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics Statement
The Institutional Review Board at Boston Children's Hospital approved this retrospective cross-sectional research study (IRB-P000007855) of infants born with esophageal atresia (EA) that underwent primary surgical repair at a single institution.
Author Contributions
Authorship credit was based on substantial contribution to (1) the conception and manuscript design (DE, JW, and DB), (2) acquisition (DE, JW, RJ, and DB), analysis (DE and DB), or interpretation of data (all authors), drafting the article (DE and DB) or critical revision for important intellectual content (all authors), (3) final approval of the version to be published (all authors), and (4) are accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved (all authors).
Funding
The Boston Children's Hospital 2019 OFD/BTREC/CTREC Faculty Career Development Fellowship, and 2017 Trailblazer Award from the Department of Anesthesiology, Critical Care and Pain Medicine, Boston Children's Hospital supported this work (DB). The content of this article is solely the responsibility of the authors and does not necessarily represent the official views of the funders.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors gratefully acknowledge (1) Quality Improvement Consultants, John J. Bennett, BS, MHA, and Heather B. Riley, MS, for their help with accessing The Esophageal and Airway Treatment Center REDCap data base; (2) Mark Breibart, Director of Anesthesia Information Technology from Department of Anesthesiology, Critical Care and Pain Medicine, Boston Children's Hospital for his help in obtaining PRAm scores; (3) Mackenzie S. Kagan, BS for help with graphs editing.
Abbreviations
ASA, American Society of Anesthesiologist; EA, Esophageal atresia; CHARGE, Coloboma, Heart defects, choanal Atresia, growth Retardation, Genital abnormalities, and Ear abnormalities; PRAm, Pediatric Risk Assessment; TEF, Tracheo-esophageal fistula; VACTERL, Vertebral, Anorectal, Cardiac, Tracheo-esophageal fistula and/or Esophageal atresia, Renal, and Limb defects/malformations.
References
1. Sfeir R, Michaud L, Salleron J, Gottrand F. Epidemiology of esophageal atresia. Dis Esophagus. (2013) 26:354–5. doi: 10.1111/dote.12051
2. Moore KL, Persaud TVN, Torchia MG. Alimentary System. In Moore KL, Persaud TVN, Torchia MG, editors. The Developing Human: Clinically Oriented Embryology. London: Elsevier India (2012), p. 193–221. Available online at: https://www.elsevier.com/books/the-developing-human-clinically-oriented-embryology-9e/moore/978-81-312-3505-8
3. Pinheiro PF. Simões e Silva AC, Pereira RM. Current knowledge on esophageal atresia. World J Gastroenterol. (2012) 18:3662–72. doi: 10.3748/wjg.v18.i28.3662
4. Gross RE. The Surgery of Infancy and Childhood: its principles and techniques. Philadelphia, London: WB Saunders (1953).
5. Foker JE, Linden BC, Boyle EM Jr. Marquardt C. Development of a true primary repair for the full spectrum of esophageal atresia. Ann Surg. (1997) 226:533–41; discussion 41-3. doi: 10.1097/00000658-199710000-00014
6. Bairdain S, Hamilton TE, Smithers CJ, Manfredi M, Ngo P, Gallagher D, et al. Foker process for the correction of long gap esophageal atresia: primary treatment versus secondary treatment after prior esophageal surgery. J Pediatr Surg. (2015) 50:933–7. doi: 10.1016/j.jpedsurg.2015.03.010
7. Kunisaki SM, Foker JE. Surgical advances in the fetus and neonate: esophageal atresia. Clin Perinatol. (2012) 39:349–61. doi: 10.1016/j.clp.2012.04.007
8. Foker JE, Kendall Krosch TC, Catton K, Munro F, Khan KM. Long-gap esophageal atresia treated by growth induction: the biological potential and early follow-up results. Semin Pediatr Surg. (2009) 18:23–9. doi: 10.1053/j.sempedsurg.2008.10.005
9. Bairdain S, Zurakowski D, Vargas SO, Stenquist N, McDonald M, Towne MC, et al. Long-gap esophageal atresia is a unique entity within the esophageal atresia defect spectrum. Neonatology. (2017) 111:140–4. doi: 10.1159/000449241
10. Holland AJ, Fitzgerald DA. Oesophageal atresia and tracheo-oesophageal fistula: current management strategies and complications. Paediatr Respir Rev. (2010) 11:100–6; quiz 6–7. doi: 10.1016/j.prrv.2010.01.007
11. Harrington AW, Riebold J, Hernandez K, Staffa SJ, Svetanoff WJ, Zurakowski D, et al. Nutrition delivery and growth outcomes in infants with long-gap esophageal atresia who undergo the Foker process. J Pediatr Surg. (2021) 56:2133–39. doi: 10.1016/j.jpedsurg.2021.07.014
12. Mongerson CRL, Wilcox SL, Goins SM, Pier DB, Zurakowski D, Jennings RW, et al. Infant Brain Structural MRI Analysis in the Context of Thoracic Non-cardiac Surgery and Critical Care. Front Pediatrics. (2019) 7:315. doi: 10.3389/fped.2019.00315
13. Hodkinson DJ, Mongerson CRL, Jennings RW, Bajic D. Neonatal functional brain maturation in the context of perioperative critical care and pain management: a case report. Heliyon. (2019) 5:e02350-e. doi: 10.1016/j.heliyon.2019.e02350
14. Solodiuk JC, Jennings RW, Bajic D. Evaluation of postnatal sedation in full-term infants. Brain Sci. (2019) 9:114. doi: 10.3390/brainsci9050114
15. Rudisill SS, Wang JT, Jaimes C, Mongerson CRL, Hansen AR, Jennings RW, et al. Neurologic injury and brain growth in the setting of long-gap esophageal atresia perioperative critical care: a pilot study. Brain Sci. (2019) 9:383. doi: 10.3390/brainsci9120383
16. Abouleish AE, Leib ML, Cohen NH. ASA Provides Examples to Each ASA Physical Status Class. ASA Newsl. (2015) 79:38–49. Available online at: https://www.asahq.org/standards-and-guidelines/asa-physical-status-classification-system
17. Nasr VG, DiNardo JA, Faraoni D. Development of a pediatric risk assessment score to predict perioperative mortality in children undergoing noncardiac surgery. Anesth Analg. (2017) 124:1514–9. doi: 10.1213/ANE.0000000000001541
18. Valencia E, Staffa SJ, Faraoni D, DiNardo JA, Nasr VG. Prospective external validation of the pediatric risk assessment score in predicting perioperative mortality in children undergoing noncardiac surgery. Anesth Analg. (2019) 129:1014–20. doi: 10.1213/ANE.0000000000004197
19. Nasr VG, Valencia E, Staffa SJ, Faraoni D, DiNardo JA, Berry JG, et al. Comprehensive risk assessment of morbidity in pediatric patients undergoing noncardiac surgery: an institutional experience. Anesth Analg. (2020) 131:1607–15. doi: 10.1213/ANE.0000000000005157
20. Spitz L, Kiely EM, Morecroft JA, Drake DP. Oesophageal atresia: at-risk groups for the 1990s. J Pediatr Surg. (1994) 29:723–5. doi: 10.1016/0022-3468(94)90354-9
21. Waterston DJ, Carter RE, Aberdeen E. Oesophageal atresia: tracheo-oesophageal fistula. A study of survival in 218 infants. Lancet. (1962) 1:819–22. doi: 10.1016/S0140-6736(62)91837-8
22. Evanovich DM. Retrospective Analysis of Disease Severity Stratification in Infants Born with Esophageal Atresia at a Single Institution: [Masters in Biomedical Science Thesis]. Boston (MA): Tufts University School of Medicine (2021).
23. Till H, Muensterer OJ, Rolle U, Foker J. Staged esophageal lengthening with internal and subsequent external traction sutures leads to primary repair of an ultralong gap esophageal atresia with upper pouch tracheoesophagel fistula. J Pediatr Surg. (2008) 43:E33–5. doi: 10.1016/j.jpedsurg.2008.02.009
24. Svetanoff WJ, Zendejas B, Hernandez K, Davidson K, Ngo P, Manfredi M, et al. Contemporary outcomes of the Foker process and evolution of treatment algorithms for long-gap Esophageal Atresia. J Pediatr Surg. (2021) 56:2180–91. doi: 10.1016/j.jpedsurg.2021.02.054
25. Shaw-Smith C. Oesophageal atresia, tracheo-oesophageal fistula, and the VACTERL association: review of genetics and epidemiology. J Med Genet. (2006) 43:545–54. doi: 10.1136/jmg.2005.038158
26. Brown AK, Roddam AW, Spitz L, Ward SJ. Oesophageal atresia, related malformations, and medical problems: a family study. Am J Med Genet. (1999) 85:31–7. doi: 10.1002/(sici)1096-8628(19990702)85:1<31::aid-ajmg7>3.0.co
27. Lopez PJ, Keys C, Pierro A, Drake DP, Kiely EM, Curry JI, et al. Oesophageal atresia: improved outcome in high-risk groups? J Pediatr Surg. (2006) 41:331–4. doi: 10.1016/j.jpedsurg.2005.11.009
28. Gearing RE, Mian IA, Barber J, Ickowicz A. A methodology for conducting retrospective chart review research in child and adolescent psychiatry. J Can Acad Child Adolesc Psychiatry. (2006) 15:126–34.
30. Jansen AC, van Aalst-Cohen ES, Hutten BA, Büller HR, Kastelein JJ, Prins MH. Guidelines were developed for data collection from medical records for use in retrospective analyses. J Clin Epidemiol. (2005) 58:269–74. doi: 10.1016/j.jclinepi.2004.07.006
33. Deurloo JA, Ekkelkamp S, Hartman EE, Sprangers MA, Aronson DC. Quality of life in adult survivors of correction of esophageal atresia. Arch Surg. (2005) 140:976–80. doi: 10.1001/archsurg.140.10.976
34. Donoso F, Kassa AM, Gustafson E, Meurling S, Lilja HE. Outcome and management in infants with esophageal atresia - a single centre observational study. J Pediatr Surg. (2016) 51:1421–5. doi: 10.1016/j.jpedsurg.2016.03.010
35. Ure BM, Slany E, Eypasch EP, Gharib M, Holschneider AM, Troidl H. Long-term functional results and quality of life after colon interposition for long-gap oesophageal atresia. Eur J Pediatr Surg. (1995) 5:206–10. doi: 10.1055/s-2008-1066206
36. Depaepe A, Dolk H, Lechat MF. The epidemiology of tracheo-oesophageal fistula and oesophageal atresia in Europe. EUROCAT Working Group. Arch Dis Child. (1993) 68:743–8. doi: 10.1136/adc.68.6.743
37. Robert E, Mutchinick O, Mastroiacovo P, Knudsen LB, Kjersti Daltveit A, Castilla EE, et al. An international collaborative study of the epidemiology of esophageal atresia or stenosis. Reproduct Toxicol. (1993) 7:405–21. doi: 10.1016/0890-6238(93)90085-L
38. Torfs CP, Curry CJ, Bateson TF. Population-based study of tracheoesophageal fistula and esophageal atresia. Teratology. (1995) 52:220–32. doi: 10.1002/tera.1420520408
39. Wang B, Tashiro J, Allan BJ, Sola JE, Parikh PP, Hogan AR, et al. A nationwide analysis of clinical outcomes among newborns with esophageal atresia and tracheoesophageal fistulas in the United States. J Surg Res. (2014) 190:604–12. doi: 10.1016/j.jss.2014.04.033
40. Cassina M, Ruol M, Pertile R, Midrio P, Piffer S, Vicenzi V, et al. Prevalence, characteristics, and survival of children with esophageal atresia: a 32-year population-based study including 1,417,724 consecutive newborns. Birth Defects Res A Clin Mol Teratol. (2016) 106:542–8. doi: 10.1002/bdra.23493
41. Sfeir R, Bonnard A, Khen-Dunlop N, Auber F, Gelas T, Michaud L, et al. Esophageal atresia: data from a national cohort. J Pediatr Surg. (2013) 48:1664–9. doi: 10.1016/j.jpedsurg.2013.03.075
42. Aslanabadi S, Ghabili K, Rouzrokh M, Hosseini MB, Jamshidi M, Adl FH, et al. Associated congenital anomalies between neonates with short-gap and long-gap esophageal atresia: a comparative study. Int J Gen Med. (2011) 4:487–91. doi: 10.2147/IJGM.S19301
43. Sulkowski JP, Cooper JN, Lopez JJ, Jadcherla Y, Cuenot A, Mattei P, et al. Morbidity and mortality in patients with esophageal atresia. Surgery. (2014) 156:483–91. doi: 10.1016/j.surg.2014.03.016
44. Stoll C, Alembik Y, Dott B, Roth MP. Associated malformations in patients with esophageal atresia. Eur J Med Genet. (2009) 52:287–90. doi: 10.1016/j.ejmg.2009.04.004
45. Brosens E, Ploeg M, van Bever Y, Koopmans AE, Ijsselstijn H, Rottier RJ, et al. Clinical and etiological heterogeneity in patients with tracheo-esophageal malformations and associated anomalies. Eur J Med Gene. (2014) 57:440–52. doi: 10.1016/j.ejmg.2014.05.009
46. Shaw-Smith C. Genetic factors in esophageal atresia. tracheo-esophageal fistula and the VACTERL association: roles for FOXF1 and the 16q241 FOX transcription factor gene cluster, and review of the literature. Eur J Med Gene. (2010) 53:6–13. doi: 10.1016/j.ejmg.2009.10.001
47. Li XW, Jiang YJ, Wang XQ, Yu JL, Li LQ. A scoring system to predict mortality in infants with esophageal atresia: a case-control study. Medicine (Baltimore). (2017) 96:e7755. doi: 10.1097/MD.0000000000007755
48. Rokitansky A, Kolankaya A, Bichler B, Mayr J, Menardi G. Analysis of 309 cases of esophageal atresia for associated congenital malformations. Am J Perinatol. (1994) 11:123–8. doi: 10.1055/s-2007-994571
49. Faraoni D, Zou X, DiNardo JA, Nasr VG. Integration of the intrinsic surgical risk with patient comorbidities and severity of congenital cardiac disease does not improve risk stratification in children undergoing noncardiac surgery. Anesth Analg. (2020) 131:1083–9. doi: 10.1213/ANE.0000000000004906
50. Nasr VG, Staffa SJ, Zurakowski D, DiNardo JA, Faraoni D. Pediatric risk stratification is improved by integrating both patient comorbidities and intrinsic surgical risk. Anesthesiology. (2019) 130:971–80. doi: 10.1097/ALN.0000000000002659
51. Mongerson CRL, Jaimes C, Zurakowski D, Jennings RW, Bajic D. Infant corpus callosum size after surgery and critical care for long-gap esophageal atresia: qualitative and quantitative MRI. Sci Rep. (2020) 10:6408. doi: 10.1038/s41598-020-63212-3
52. Bajic D, Rudisill SS, Jennings RW. Head circumference in infants undergoing Foker process for long-gap esophageal atresia repair: Call for attention. J Pediatr Surg. (2021) 56:1564–9. doi: 10.1016/j.jpedsurg.2021.01.030
53. Lazow SP, Ben-Ishay O, Aribindi VK, Staffa SJ, Pluchinotta FR, Schecter SC, et al. Predictors of index admission mortality and morbidity in contemporary esophageal atresia patients. J Pediatr Surg. (2020) 55:2322–8. doi: 10.1016/j.jpedsurg.2020.02.005
54. van der Zee DC, Tytgat SHA, van Herwaarden MYA. Esophageal atresia and tracheo-esophageal fistula. Semin Pediatr Surg. (2017) 26:67–71. doi: 10.1053/j.sempedsurg.2017.02.004
55. van der Zee DC, van Herwaarden MYA, Hulsker CCC, Witvliet MJ, Tytgat SHA. Esophageal atresia and upper airway pathology. Clin Perinatol. (2017) 44:753–62. doi: 10.1016/j.clp.2017.08.002
56. Petrosyan M, Estrada J, Hunter C, Woo R, Stein J, Ford HR, et al. Esophageal atresia/tracheoesophageal fistula in very low-birth-weight neonates: improved outcomes with staged repair. J Pediatr Surg. (2009) 44:2278–81. doi: 10.1016/j.jpedsurg.2009.07.047
Keywords: ASA, EA, LGEA, mortality, term, term-born infant, PRAm, premature
Citation: Evanovich DM, Wang JT, Zendejas B, Jennings RW and Bajic D (2022) From the Ground Up: Esophageal Atresia Types, Disease Severity Stratification and Survival Rates at a Single Institution. Front. Surg. 9:799052. doi: 10.3389/fsurg.2022.799052
Received: 21 October 2021; Accepted: 31 January 2022;
Published: 09 March 2022.
Edited by:
Mario Lima, University of Bologna, ItalyReviewed by:
Francesco Molinaro, University of Siena, ItalyGianluca Lista, Ospedale dei Bambini Vittore Buzzi, Italy
Copyright © 2022 Evanovich, Wang, Zendejas, Jennings and Bajic. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dusica Bajic, dusica.bajic@childrens.harvard.edu