 Madeline Eve
Madeline Eve Josan Gandawijaya
Josan Gandawijaya Liming Yang
Liming Yang Asami Oguro-Ando
Asami Oguro-Ando- University of Exeter Medical School, University of Exeter, Exeter, United Kingdom
Autism spectrum disorder (ASD) is a complex neurodevelopmental condition characterized by restrictive and repetitive behaviors, alongside deficits in social interaction and communication. The etiology of ASD is largely unknown but is strongly linked to genetic variants in neuronal cell adhesion molecules (CAMs), cell-surface proteins that have important roles in neurodevelopment. A combination of environmental and genetic factors are believed to contribute to ASD pathogenesis. Inflammation in ASD has been identified as one of these factors, demonstrated through the presence of proinflammatory cytokines, maternal immune activation, and activation of glial cells in ASD brains. Glial cells are the main source of cytokines within the brain and, therefore, their activity is vital in mediating inflammation in the central nervous system. However, it is unclear whether the aforementioned neuronal CAMs are involved in modulating neuroimmune signaling or glial behavior. This review aims to address the largely unexplored role that neuronal CAMs may play in mediating inflammatory cascades that underpin neuroinflammation in ASD, primarily focusing on the Notch, nuclear factor-κB (NF-κB), and mitogen-activated protein kinase (MAPK) cascades. We will also evaluate the available evidence on how neuronal CAMs may influence glial activity associated with inflammation. This is important when considering the impact of environmental factors and inflammatory responses on ASD development. In particular, neural CAM1 (NCAM1) can regulate NF-κB transcription in neurons, directly altering proinflammatory signaling. Additionally, NCAM1 and contactin-1 appear to mediate astrocyte and oligodendrocyte precursor proliferation which can alter the neuroimmune response. Importantly, although this review highlights the limited information available, there is evidence of a neuronal CAM regulatory role in inflammatory signaling. This warrants further investigation into the role other neuronal CAM family members may have in mediating inflammatory cascades and would advance our understanding of how neuroinflammation can contribute to ASD pathology.
Introduction
Autism spectrum disorder (ASD) is a complex neurodevelopmental condition characterized by restrictive and repetitive behaviors, combined with deficits in social interaction and communication (1). ASD is estimated to currently affect between 0.6 and 2% of the global population, with data suggesting prevalence is increasing over time (2–5). The etiology of ASD remains largely unknown, although it is hypothesized that a combination of environmental and genetic factors contributes to its pathogenesis (6). Monozygotic twin studies show ASD has high heritability, with estimates as great as 83%, but these figures also demonstrate the incomplete genetic concordance (7). ASD affects predominantly males compared to females for reasons yet unidentified (8).
Extensive, current research collated by the Simons Foundation Autism Research Initiative (SFARI) highlights numerous candidate genes linked to ASD development, with many of these encoding for neuronal cell adhesion molecules (CAMs) (9). Neuronal CAMs are together a diverse collection of cell-surface proteins that play roles in neurite formation, neuronal outgrowth, and axon guidance within the nervous system (10, 11). These molecules are classified into families founded on their structure, including the neurexin (NRXN) family, neuroligin (NLGN) family, and immunoglobulin superfamily CAMs (IgCAMs) (12). Amongst them, the IgCAMs represent the largest family of neuronal CAMs, encompassing neurofascins (NFASCs), neural CAMs (NCAMs), and the six-member subfamily, contactins (CNTNs) (12). Of note, variation in CNTN gene expression is linked to ASD, and various studies have demonstrated their importance in nervous system development (13). CNTNs have been reported to modulate neuronal communication and synaptic transmission, essential in the assembly of neural circuits and the formation of behavioral pathways (10, 12–15).
One key neuronal CAM, scored by the SFARI Gene database as a strong, syndromic ASD candidate gene is CNTN-associated protein-2 (CNTNAP2) (16). CNTNAP2 is localized to myelinated axons within the juxtaparanode of the nodes of Ranvier. Here, it interacts with CNTN2 and organizes axonal voltage-gated K+ channels (17). Consistent evidence reports rare variants within CNTNAP2 as a susceptibility factor for ASD and suggests that CNTNAP2 has a function in the early stages of neurodevelopment and later language development (18–21). Animal models deficient for Cntnap2 show symptoms synonymous with ASD in humans, including epileptic seizures, behavioral abnormalities, and cognitive dysfunction (22). Functional magnetic resonance imaging has illustrated atypical frontal cortex circuitry in children with ASD carrying common CNTNAP2 variants, compared to neurotypical controls (21). A case report presented by Al-Murrani and colleagues depicted a 3-year-old boy with a deletion in CNTNAP2 who displayed language delay, communication difficulties, and consequently, behavioral problems (23). However, it is important to note that two of his family members also carried the same deletion and yet exhibited no ASD phenotype. This demonstrates the presence of substantial phenotypic heterogeneity that makes it difficult to identify a solely genetic origin for many cases of ASD and, therefore, suggests an additional environmental trigger contributes to ASD symptom presentation.
Environmental factors can be categorized as prenatal, natal, and postnatal. Common prenatal risk factors include older paternal age, maternal mental and physical health, as well as familial socioeconomic status (24). A particularly promising avenue of research into the natal and postnatal environmental triggers for ASD is inflammation. The relationship between inflammatory disease, proinflammatory cytokines, and impaired immunity has been well-documented in those with ASD. Specifically, immune disorders such as asthma, gastrointestinal (GI) disorders, allergy, and maternal immune activation (MIA) have been regularly identified in studies investigating ASD comorbidities (25–30). MIA occurs when the pregnant mother acquires an infection or is exposed to immunogenic materials and has been identified to increase susceptibility to a child developing ASD (31). Consequences on the developing fetal brain can include modified expression of neuronal migration genes, increased number of microglia, aberrant dendritic morphology within the prefrontal cortex, and excessive neurogenesis (32–35). It is thought that the mother’s immune response poses a risk to fetal neurodevelopment, rather than the specific pathogen or immunogenic material (35). In murine models, ASD behaviors are observed in offspring when, throughout pregnancy, dams are exposed to immunostimulants such as lipopolysaccharide (LPS), interleukin-17 (IL-17), and polyinosinic-polycytidylic acid [poly (I:C)] (29, 36, 37). In one study, blocking IL-17A in LPS-immune activated dams resulted in the reversal of ASD behaviors in their offspring (36). These studies demonstrate that altered neuroimmune signaling adversely affects fetal neurodevelopment. Notably, MIA has also been shown to facilitate the transfer of maternal antibodies targeting fetal neural proteins, which could further impair neurodevelopment (38).
Although inflammation has been identified as an environmental risk factor for ASD, it is not entirely clear whether the neuronal CAMs discussed above may play roles in modulating neuroimmune signaling. Microglia are the main source of cytokines within the brain and, therefore, are vital in mediating inflammation in the central nervous system (CNS) (39). There is suggestion of neuronal CAMs influencing signaling cascades that then cause a proinflammatory phenotype in microglia (40).
Inflammatory processes from subsequent microglial activation indicate that neuronal CAMs may play a role in inflammatory cascades, a concept relatively unexplored in current literature. Studies of several CAMs, including CNTNAP2, have gradually revealed their functional involvement in inflammatory signaling. However, there is limited information that discusses the relation between CAMs and inflammatory systems that are risk factors in ASD. This review aims to address the association between neuronal CAMs and inflammatory systems within the scope of ASD (Figure 1). This association has been largely unexplored but is important when considering the impact of environmental factors and the inflammatory response on ASD. The following sections will cover recent research reports exploring the role of neuronal CAMs in inflammatory signaling cascades and proinflammatory cytokine production associated with neuroinflammation found in ASD. Additionally, we will focus on the dysfunction of inflammatory cascades underpinning inflammatory diseases that may link to ASD pathology. Finally, we will assess how neuronal CAMs are involved in the activation, differentiation, and proliferation of glial cells, leading to an inflammatory response and how this may impact neurodevelopmental pathways implicated in ASD.
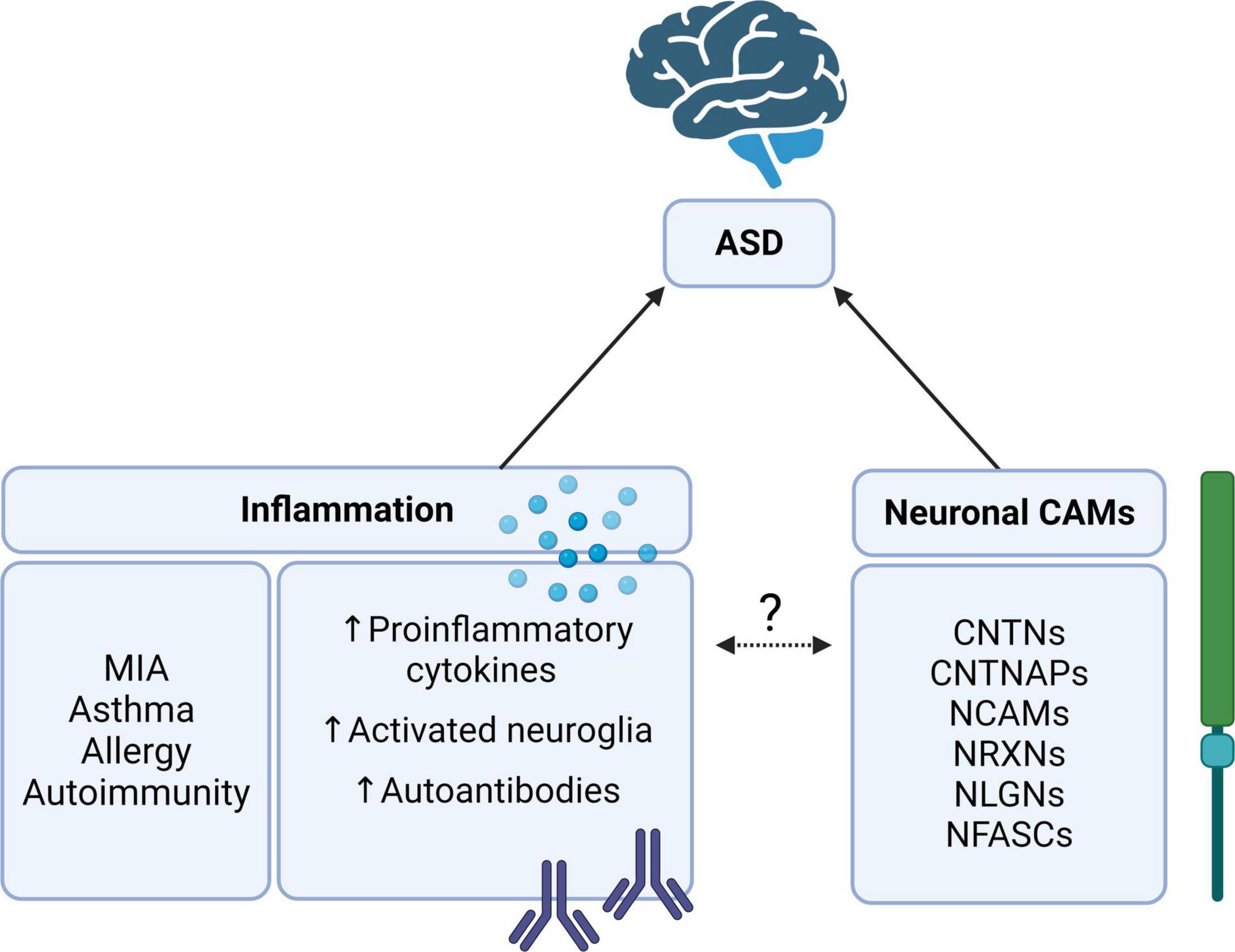
Figure 1. The relationship between autism spectrum disorder (ASD), inflammation, and cell adhesion molecules (CAMs). Neuronal CAMs such as the CNTN, CNTNAP, NCAM, NRXN, NLGN, and NFASC families of proteins are consistently implicated as risk genes for ASD (9, 16). Likewise, inflammation, particularly in MIA, has been identified as an environmental risk factor for ASD (24, 31). What is yet unclear is whether these neuronal CAMs may be involved in regulating inflammatory responses or cascades in ASD. Contactin (CNTN), CNTN-associated protein (CNTNAP), neural cell adhesion molecule (NCAM), neurexin (NRXN), neuroligin (NLGN), neurofascin (NFASC), and maternal immune activation (MIA). Created with BioRender.com.
Autism Spectrum Disorder and Inflammation
Signaling Pathways Involved in Neuroinflammation
Several signaling pathways are involved in the neuronal inflammatory response. This review will primarily focus on three of these; Notch, nuclear factor-κB (NF-κB), and mitogen-activated protein kinase (MAPK) signaling cascades, which all play key roles in inflammatory responses. Notch is a transmembrane protein that, once activated, transduces signals via direct cell-cell communication (41). There are four Notch receptors in mammals that, upon binding with a corresponding ligand, causes the intracellular domain of the Notch receptor to be cleaved and translocated to the nucleus (42). Importantly, neuronal CAMs, such as CNTN1 and CNTN6, act as ligands for Notch1 in the CNS (43, 44). In the immune system, Notch1 signaling is important for the differentiation of T cells into T-helper (Th) and regulatory T cells, whilst inhibiting the differentiation of other lymphoid lineages (45–47). Chronic inflammatory diseases associated with ASD, such as inflammatory bowel disease (IBD) and asthma, may be influenced by Notch signaling (41). Experimental alterations of the Notch signaling pathways, such as the removal of constituents responsible for Notch signal transduction, have exposed a potential role of Notch in the proinflammatory response (48, 49). Moreover, proinflammatory cytokine IL-6 is secreted upon activation of Notch2 by tumor necrosis factor-α (TNF-α) in rheumatoid arthritis models (50). Hence, inhibition of Notch signaling may be beneficial in the treatment of inflammatory diseases. However, whether neuronal CAMs acting as Notch ligands modulate the secretion of proinflammatory cytokines is unclear.
NF-κB is another signaling pathway involved in inflammation and is strongly implicated in inflammatory disease. Present in all cell types, NF-κB regulates the expression of genes encoding for proteins vital in the immune response (51). Activation of NF-κB is inducible by inflammatory cytokines, markers of infection, or stress-activated protein kinases (52). Under normal cell conditions, NF-κB is inhibited from nuclear translocation by IκB (inhibitor of κB) until activated (53). NF-κB signaling influences differentiation states of cells and regulates the production of anti-apoptotic factors (54). However, the primary role of NF-κB is to increase the production of cytokines, chemokines, and adhesion molecules involved in the immune response (55). Proinflammatory cytokines such as IL-6, IL-12, and TNF-α form common target genes for activated NF-κB (56). Although intentional activation of NF-κB is protective against pathogens and cancer, uncontrolled or dysfunctional NF-κB signaling can be causative of acute or chronic inflammatory disease (52). Within the CNS, inappropriate activation of the NF-κB signaling cascade leads to aberrant expression of proinflammatory cytokines, which can be particularly damaging. NF-κB activation in microglia, astrocytes, and oligodendrocytes has been implicated in neurodegenerative disease (57). Of note, NF-κB signaling can also be initiated downstream of the MAPK cascade (58).
The MAPK signaling cascade is activated by cytokines, mediators of stress, and inflammatory markers, such as ligands of Toll-like receptors (59). Through a series of phosphorylation cascades, MAPK proteins transduce, and propagate extracellular signals within the cell (60). During inflammation, MAPK signaling is mainly induced by Toll-like receptor activation, resulting in the phosphorylation of transcription factors (61). Once phosphorylated, these transcription factors translocate to the nucleus and transcribe genes encoding proinflammatory cytokines, such as IL-6 and TNF-α (62). Within the CNS, IL-6 signaling forms a positive feedback loop through activation of MAPK signaling, which is reported to promote neurogenesis (63). Microglial activation can also induce the MAPK signaling cascade, promoting a neuroinflammatory environment that contributes to neurodegeneration (64). Although it is uncertain, neuronal CAMs may also play roles in MAPK signaling. For example, NCAM1 was shown to activate MAPK signaling in mesenchymal stromal cells, regulating their migration to the site of inflammation where they contribute to tissue repair (65).
During an inflammatory response, Notch, NF-κB, and MAPK signaling pathways can all be activated by proinflammatory cytokines (52, 59, 66).
Cytokine and Chemokine Profiles in Autism Spectrum Disorder
In the context of the immune system, cytokines are proteins secreted by cells to coordinate, signal and recruit, normally in response to an immune insult. Generally, these signaling proteins are proinflammatory in nature and are important in orchestrating the immune response (67). Most cell types can produce cytokines, but the majority of proinflammatory cytokines are released by macrophages and lymphocytes (68). Chemokines, for example, IL-8, manage leukocyte adhesion and chemotaxis (69). Interleukins play a role in directing cell differentiation, growth, and activation (68, 70). IL-1 and IL-6 are, for the most part, produced by macrophages to stimulate the generation of proinflammatory cytokines and evoke elevation of body temperature (70). The growth and proliferation of eosinophils, T cells, natural killer cells, and B cells are mediated by a vast range of interleukins (70). Cytokines can also be anti-inflammatory and regulate or suppress an escalating immune response. For instance, IL-10 suppresses proinflammatory cytokines and modulates macrophages to dampen the immune response (71, 72).
Irregular cytokine and chemokine profiles have been recorded in the cerebrospinal fluid (CSF), blood, and brain of those with ASD (73–76). Plasma samples from male ASD subjects revealed elevated levels of IL-1β, IL-5, IL-8, IL-12, IL-13, and IL-17 compared to controls (76). In another multiplex cytokine screen, similar differences were found in IL-1β, IL-12, and IL-17, but also IL-6, in individuals with ASD compared with age-matched typically developing children (77). A recent meta-analysis conducted with 1,393 patients with ASD found greater concentrations of IL-6 in the blood, as well as increased concentrations of proinflammatory cytokines IL-1β, TNF-α, and interferon-γ (IFN-γ) within the periphery (78). Within the CNS, localized cytokine and chemokine variances have been reported. Higher levels of IL-6, IL-8, TNF-α, and IFN-γ are observed in the frontal cerebral cortex of ASD patients compared to age-matched control cortices (79). Moreover, evidence of elevated TNF receptor I levels in the CSF of ASD children further indicates inflammation not just in the periphery, but also within the CNS (80). Although inflammatory markers may differ depending on genetic predisposition, when screening panel results are normalized to parental cytokine expression, IL-8 expression was still seen to be elevated in children with ASD (81). These cytokine profiles have been found to differ in males and females, indicating that testosterone may impinge on the inflammatory processes underlying ASD (82). Cytokine profiles may also be useful as biomarkers to predict comorbidities. Children with ASD that also suffered from epilepsy had lower peripheral IL-6 levels compared to ASD children without epilepsy (77). Distinct cytokine profiles have also been shown in ASD children who have attention deficit hyperactivity disorder, with differences in blood IL-8 levels compared to children with only ASD (74). Specific cytokine profiles have the potential to act as an ASD biomarker and aid the diagnosis of comorbidities.
For a more detailed overview of which cytokines and chemokines are most frequently found to vary in concentrations within individuals with ASD compared to typically developing controls, see Table 1. In summary, IL-1β, IL-6, IL-8, and IL-17 are the most consistently observed cytokines to be upregulated in cases of ASD, which indicates a proinflammatory response underlying neuroinflammation in ASD.
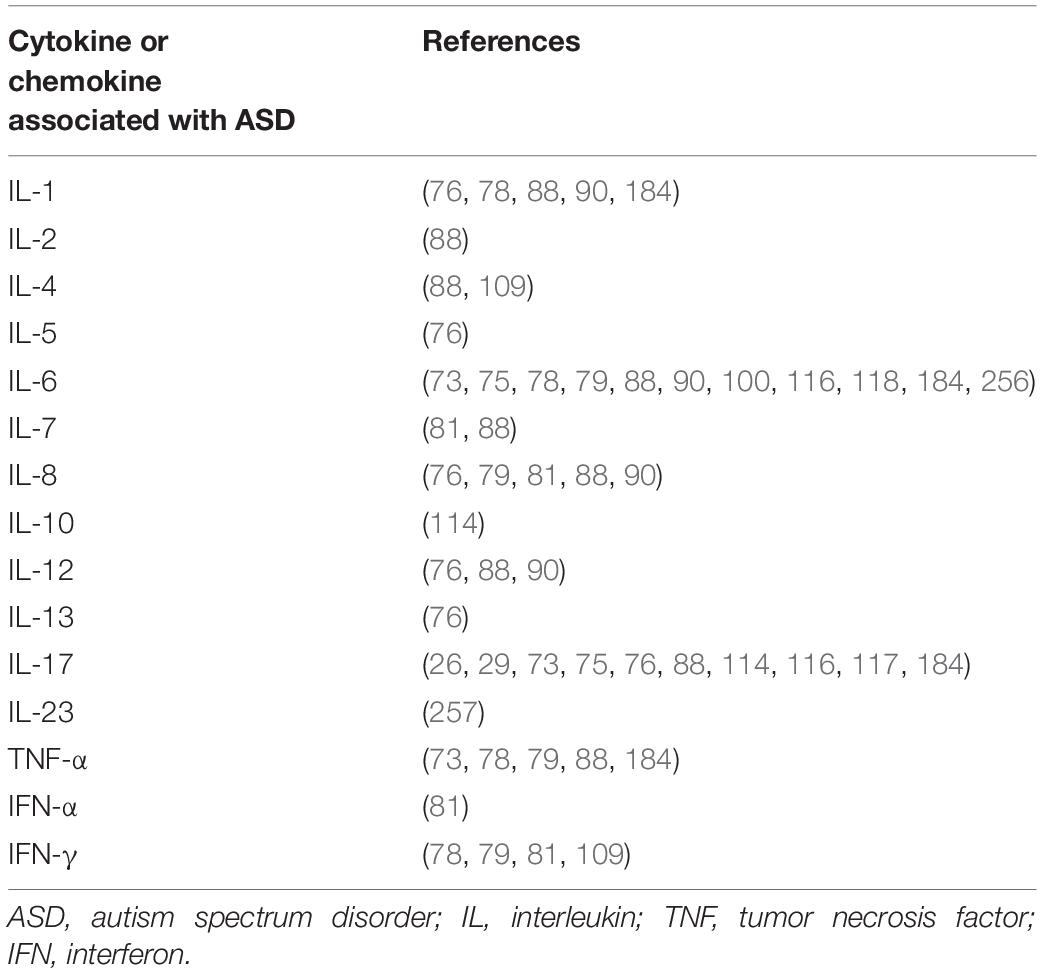
Table 1. Summary of cytokines and chemokines reported to have altered expression in autism spectrum disorder.
Interleukin-1β
Interleukin-1β is a proinflammatory cytokine found upregulated in autoinflammatory disease, chronic inflammation, and acute inflammation (83). Expression of IL-1β by monocytes and macrophages, stimulated by pathogen-associated molecular patterns and cytokines, triggers phagocytic cell activation (84). IL-1β has a role in neuropathogenesis but also neuroprotection within the CNS (85). During the development of the nervous system, IL-1β expression regulates the proliferation of neural progenitor cells (86). Hence, abnormal IL-1β levels may contribute to neurological deficits observed in ASD brains. Increased concentrations of IL-1β were shown in several studies, including a meta-analysis, in individuals with ASD compared to healthy controls (76, 78, 87, 88). Microglia can generate vast quantities of cytokines, particularly IL-1β, within the CNS (89). Extracellular vesicles isolated from the serum of children with ASD were shown, in vitro, to activate microglia to produce increased levels of IL-1β compared to neurotypical controls (40). Interestingly, IL-1β is found to be elevated in the serum and blood of both males and females with ASD, although mainly males have been included in ASD cytokine profile studies (76, 82). This increase is not as high of an increase as would be expected to be found in a person with autoimmune or inflammatory disease, suggesting that the effect of ASD on the inflammatory system is unique (76). One study showed that increased IL-1β was predominantly found in children with regressive ASD, supporting that IL-1β is influential during post-natal neurodevelopment (90). The same study also found a correlation between IL-1β concentration and aberrant behaviors (90).
Interleukin-8
Interleukin-8 production in neutrophils and mast cells is stimulated by IL-1β (91). IL-8, also known as CXCL8, is a potent chemoattractant produced by T cells and macrophages in order to recruit neutrophils and other leukocytes to a site of inflammation (92). Neutrophils can produce IL-8 to self-recruit, stimulated by IL-1 and TNF-α (92). Besides neutrophil chemotaxis, IL-8 has a role in neutrophil morphology, upregulation of adhesion molecules, migration, and exocytosis of proteolytic enzymes (92, 93). Although crucial in peripheral immunity, IL-8 has also been found to be increased within the frontal cerebral cortex of ASD brains (79). The frontal cortex is essential for cognition, emotion, and social behavior, therefore, is a brain region associated with the pathology of ASD (94). Localized inflammation due to IL-8 could affect frontal cortex processing in those with ASD. IL-8 is produced by macrophages to recruit neutrophils, eosinophils, and leukocytes (95). Increased peripheral levels of IL-8 have been found in ASD subjects compared to matched controls (76, 87, 88, 96). With IL-8 having such a key role in innate immunity, it suggests that immune dysfunction in ASD is linked to the innate immune system. Activated microglia may be able to recruit cells of the innate immune system via IL-8 secretion to exacerbate the neuroinflammatory response. One study showed that not only was serum IL-8 concentration increased in ASD children compared to healthy controls, but IL-8 was yet higher in concentration in patients with childhood ASD compared to those with Asperger syndrome (96). This data may allude to different pathophysiological mechanisms for different levels on the ASD spectrum. Once more, an increase in severity of ASD phenotype is correlated with proinflammatory cytokine concentration (90). IL-6 plasma concentrations are also demonstrated to be positively correlated with the severity of ASD traits (90).
Interleukin-6
Interleukin-6 is a pleiotropic cytokine that has both inflammatory and anti-inflammatory purposes throughout the human body (97). Functions of IL-6 during inflammation include B-cell differentiation, induction of acute-phase protein release, inhibition of regulatory T-cell differentiation, and maintenance of Th17 cell differentiation (70, 97). IL-6 activity is not limited to the immune system. It is well-documented to play roles in neurodevelopment, such as promoting neurite outgrowth, neurogenesis, and gliogenesis pathways that are implicated in ASD pathology (6, 94, 98–100). Many studies have shown increased plasma and blood IL-6 levels in individuals with ASD (73, 75, 76, 78, 88). Elevated IL-6 levels within the cerebellum are associated with impairment of neuronal cell adhesion and migration, as well as influencing synapse formation (101). CAMs are essential for the formation of behavioral pathways, synapse development, and neuronal plasticity, therefore, CAM dysfunction due to IL-6 may contribute to some ASD pathophysiologies (12, 13, 101). What is more, this data suggests IL-6 may regulate the function of neuronal CAMs (101).
There are distinguished neurological changes in the brains of those with ASD such as neuronal overgrowth in the frontal cortex and microglial activation (94, 102). In the GFAP-IL-6 mouse model of chronic neuroinflammation, where IL-6 is overexpressed in astrocytes, neurological variations like astrocytic gliosis and neurodegeneration were observed (103, 104). Increases in IL-6 serum, blood, and CSF levels have also been observed in adults with ASD, indicating IL-6 can be upregulated over a prolonged period of time, potentially contributing to neurological changes in ASD brains, similar to those observed in chronic neuroinflammation models (78, 102).
An increase of IL-6 was found in the anterior cingulate gyrus of ASD patients compared to controls (102). The anterior cingulate gyrus is responsible for emotional expression, attention allocation, and mood, the dysregulation of which are all core ASD deficits (105). Localized inflammation within this brain region via IL-6 could contribute to deficits in communication observed in those with ASD.
IL-6 appears to have a key role in MIA, one of the strongest examples of inflammation associated with ASD development. The immune response of the mother, directed against a pathogen or immunogenic molecule, adversely affects fetal neurodevelopment (31). In animal models, MIA is commonly induced in pregnant mice via administration of LPS or poly(I:C), which mimics bacterial or viral infection, respectively (106). Behavioral changes in the offspring of poly(I:C)- and LPS-induced MIA mice were observed in multiple studies (29, 36, 37, 107–109). These behaviors included enhanced marble-burying or self-grooming (represents repetitive and restricted behavior), sociability impairments, and reduced ultrasonic vocalization (which may suggest a change in social communication) (36, 108). Administration of IL-6 to pregnant dams gave rise to ASD-like behavioral traits in their offspring and further experiments utilizing poly(I:C) MIA models found offspring ASD behavior was prevented by co-administering anti-IL-6 antibodies with poly(I:C) (110). In this same study, MIA IL-6 knockout mice sired offspring without any of the behavioral deficits that MIA wild-type offspring possessed (110). Together with the upregulated IL-6 levels observed in human studies, there is substantial evidence implicating altered IL-6 signaling in the neuroimmune dysfunction underlying ASD.
Interleukin-17
Interleukin-17 is an important cytokine in the protection and clearance of bacterial and fungal infections (111). There are six members of the IL-17 family, the most studied of which is IL-17A (112). Able to act on myeloid and mesenchymal cells, IL-17 upregulates proinflammatory genes through NF-κB and MAPK signaling (113).
Th17 cells are characterized by their production of IL-17 and it has been suggested that Th17 cells are implicated in the development of inappropriate inflammatory responses in ASD (114, 115). A study conducted by Moaaz et al. found children with ASD had significantly increased Th17 cell production, alongside fewer regulatory T cells and decreased concentrations of both IL-10 and transforming growth factor-β (TGF-β), which dampen the inflammatory responses (114). This could indicate that regulation of the immune system may be malfunctioning in some cases of ASD.
Increased plasma levels of IL-17 in those with ASD have been outlined in numerous studies (73, 77, 114, 116). Additionally, increased expression of the receptor for IL-17A was found in phagocytes isolated from individuals with ASD, and IL-17 messenger RNA expression was also nearly four times higher in ASD children compared to typically developing children (114, 117, 118). One Turkish study detected reduced IL-17 expression in ASD individuals’ peripheral blood mononuclear cells (PBMC), though this could be due to demographic differences within the sample (75). The proinflammatory IL-17 signaling pathway is associated with chronic inflammatory neurological diseases (119). Activation of IL-17 receptors (IL-17R) in vitro in monocytes and neutrophils isolated from individuals with ASD led to upregulated NF-κB expression, resulting in increased expression of proinflammatory genes (117, 118). Application of anti-IL-17R antibody to ASD monocytes reversed this enhanced NF-κB expression and could be viewed as a beneficial treatment for managing the inflammation found in some cases of ASD (117).
The above data suggests that upregulation of IL-17 is associated with ASD, but the origin of IL-17 dysregulation may be maternal. In pregnant mothers, IL-17 is able to transfer from the placenta to the fetus, increasing IL-17R levels within the fetal brain (120). Additional IL-17R further increases IL-17 signaling within the fetal brain, likely initiating a neuroinflammatory response (121). This may cause a predisposed sensitivity to IL-17 and explain increased IL-17 concentrations in individuals with ASD. Abnormal IL-17 levels have featured in MIA models. IL-6 and TGF-β together facilitate Th17 cell differentiation from their CD4+ progenitors (115). Th17 cells are characterized by their production of IL-17. A rise in Th17 cell numbers is associated with autoimmune disorders and chronic inflammatory disease (122). It has been found that an increase in serum IL-6, ensuing from an immune assault, yields an elevated production of Th17 cells, increasing IL-17 levels (115). Interestingly, in two studies, the enhanced marble-burying behavior was ameliorated by IL-17A blocking, indicating that IL-17A has a role in repetitive ASD behavioral traits (36, 107). Blocking IL-17A in LPS-administered MIA dams also reversed the reduction in ultrasonic vocalization and social interaction deficits in their offspring (36). Increased levels of maternal IL-17A have been strongly associated with ASD-like behavior in rodent MIA offspring. This amplification of IL-17 has additionally been noticed in both murine MIA offspring and human ASD individuals.
In summary, specific cytokine profiles seem to highlight the presence of a proinflammatory response in individuals with ASD during early neurodevelopment, up to adulthood. Attention is drawn to abnormally elevated IL-1β, IL-6, IL-8, and IL-17 levels in children and adults with ASD (73, 74, 96). Increased IL-6 and IL-17 levels in MIA models suggest that dysregulation of inflammatory cascades begin during prenatal neurodevelopment (100). There is also some evidence that the degree of severity in ASD phenotype may be positively correlated with the concentrations of proinflammatory cytokines (90). Activation of inflammatory signaling in glial and neuronal cells by proinflammatory cytokines may alter neuronal cellular function. Significantly, IL-6 overexpression can promote astrocytic gliosis, causing neuroinflammation similar to that observed in ASD brains (104). In turn, the neuroinflammatory environment activates MAPK and NFκB signaling to enhance the neuroimmune response (52, 59). These inflammatory cascades can be regulated by NCAM1, implicating neuronal CAMs in ASD-related inflammation (65, 123). Additionally, extracellular vesicles isolated from ASD serum may activate microglia to produce increased IL-1β, further suggesting dysfunction of the neuroinflammatory response (40). IL-1β regulates the proliferation of neural progenitor cells, therefore, an increase in IL-1β in the brains of those with ASD may alter neuronal development (86). This may have implications in ASD behavioral pathways that contribute to the core deficits seen in ASD.
Involvement of Neuronal Cell Adhesion Molecules in Inflammatory and Immune Disease Associated With Autism Spectrum Disorder
Inflammatory and Immune Disease Associated With Autism Spectrum Disorder
Dysregulation of inflammatory signaling cascades and abnormal proinflammatory cytokine signaling is often present in those with ASD (77). Chronic inflammatory diseases such as asthma, IBD, and persistent neuroinflammation have frequently been reported as comorbidities to ASD, alongside immune-mediated disease (124).
Asthma is a chronic inflammatory disease of the respiratory system (125). Current literature is divided over support of a correlation between asthma and ASD. A meta-analysis of 175,406 participants found no proof of an association between asthma and ASD (126). Another study also showed no association between ASD and asthma or allergy, although allergy was linked with increased repetitive behavior (127). The same study did, however, highlight that food allergies and sensitivities were associated with ASD. Other studies report that asthma is 35% more frequently diagnosed and is more prevalent in children with ASD than in typically developing controls (26, 128). Interestingly, PBMCs isolated from children with both ASD and asthma are reported to produce higher levels of IL-17 following stimulation compared to PBMCs from children with ASD but without asthma (26). It is worth noting that upregulated IL-17, as previously discussed (previous Interleukin-17 section), can contribute to more severe ASD phenotypes.
Gastrointestinal sensitivities and general GI issues are further comorbidities to ASD (124). Children with ASD are frequently reported to have intolerances to food, abdominal pain, bloating, diarrhea, constipation, ulcerative colitis, and Crohn’s disease (129). There has been a lot of interest in the “gut-brain axis,” where gut microbiota and immune responses have a bidirectional relationship with the CNS (130). Children aged 2–18 with ASD had 67% higher odds of having Crohn’s disease and ulcerative colitis compared to a typically developing control group (131). Across four different study populations, the rates of IBD among individuals with ASD were higher than their age-matched controls (132). Impaired gut barriers from localized inflammatory cytokine production leads to increased gut permeability, allowing cytokines to access the CNS (133). These cytokines, originating from the gut, could instigate inflammatory responses within the brain and, therefore, impact cognitive function (130). Reciprocally, inflammation originating from the CNS or plasma could cross the intestinal mucosal barrier and trigger inflammatory signaling in gut-associated lymphoid tissue (133, 134).
The gut-brain axis may indirectly play a role in the neuroimmune system. Increased gut permeability from GI inflammation allows molecules that would otherwise be restricted from entering the bloodstream (30). Microglia are primarily responsible for neuroinflammation within the CNS and, once activated, produce vast quantities of proinflammatory cytokines (89). Activation of microglia and astrocytes were shown in brain tissue of patients with ASD (102). The vagus nerve interacts with the peripheral immune system, constantly surveying gut health (135). GI inflammatory markers can be sensed by the vagus nerve which transmits this information to the CNS, affecting microglial activation (136). Therefore, inflammation from the gut can trigger proinflammatory cytokine secretion via microglia activation, causing neuroinflammation (135).
Similar to ASD, autoimmune disease is thought to develop through a genetic predisposition with an environmental trigger that activates the immune system (137). The connection between autoinflammatory disease and ASD is not well-defined amongst current literature. However, several previous studies have revealed an interesting relationship between ASD and autoimmune disease. Zerbo et al. discovered autoimmune disease and psoriasis were diagnosed more frequently in males and children over the age of 12 with ASD compared to controls (138). It has also been established that a familial history of autoimmune disease may increase the chance of offspring having ASD, especially when the disease is targeting the CNS, and skin or mucosal membranes (139). These results suggest that there are overlaps in the genetic predisposition to ASD and autoimmune disorders (particularly those affecting the CNS). Interestingly, an autoimmune disease in the pregnant mother of ASD children may predispose the child to an IL-17 sensitivity via MIA and as described previously, an increase in IL-17 and the Th17 cells that produce it, is linked to the development of autoimmune disorders (119, 120).
Autoantibodies against proteins found within the CNS of subjects with ASD, including myelin basic protein, have been reported in several studies (140–142). Myelin is important in nerve function and protects the axon from damage. Without myelin, nerves cannot effectively conduct electrical signals in the CNS, resulting in neuronal network dysfunction (143). Demyelination, resulting from inflammation and cytokine infiltration, is a leading cause of neurological disease, impacting sensory, motor, and cognitive function (144, 145). White matter denotes brain regions consisting mainly of myelinated axons (145). Alterations in white matter volume of ASD brains, detected through magnetic resonance imaging, suggests dysregulation of myelination in those with ASD (146). It must be acknowledged that although demyelination is not commonly reported in those with ASD, the presence of autoantibodies against myelin basic protein in those with ASD suggests the potential for demyelinating disease to affect ASD brains (140). Additionally, one case study did present a 6-year-old with ASD and demyelinating neuropathy (147).
Evidence reports that neurodegenerative disease is more prevalent in adults with ASD (148). Alzheimer’s disease (AD) is a neurodegenerative disease that has similar mechanisms supporting the pathogenesis of ASD (149). A key protein implicated in the pathology of AD is amyloid precursor protein (APP). Aberrant cleavage of APP into toxic amyloid beta (Aβ) plaques are one of the hallmarks of AD, although several studies reveal similar alterations of APP processing in those with ASD (150). A significant increase in secreted β-amyloid, a product of the pathogenic APP processing pathway, have been found in the plasma of children with severe ASD (151). Additionally, a greater intraneuronal Aβ load and increased Aβ accumulation were observed in astrocytes and some microglia in subjects with ASD and 15q11.2-13q duplication syndrome (152). Activated microglia and astrocytes clear Aβ plaques, reducing their accumulation at the synapse (153, 154). In AD models, microglia respond more readily to Aβ, producing increased proinflammatory cytokines that degrade neuronal synapses, leading to cognitive decline (155). Moreover, transgenic AD mice models showed that increased expression of APP led to elevated levels of proinflammatory cytokines, such as IL-1β and IFN-γ, in the brain (154). An increased Aβ load, as demonstrated in those with ASD, would suggest enhanced astrocyte and microglial activation, similarly, increasing proinflammatory cytokine production (152). Furthermore, in the ASD brain, astrocytes and microglia may respond more readily to Aβ, damaging synapses that could contribute to altered cognition (155).
To review, there is conflicting data for the association of asthma with ASD, but there is a strong correlation between allergy and ASD (126). The GI system is particularly susceptible to environmental triggers through ingested materials. Individuals with ASD may have a predisposition to GI disorders that cause an inflammatory immune response once activated by environmental factors. Inflammation originating from the gut may be influencing inflammatory signaling in the CNS attributable to the gut-brain axis (135). Alternatively, GI inflammation may be instigated by inflammatory signaling deriving from the CNS. There is some evidence to link autoimmunity and dysregulation of immune function to ASD, although there is a lack of support for the presence of autoantibodies in those with ASD (140). More so, there is a suggestion that myelin dysregulation may render axons vulnerable to proinflammatory cytokines that cause demyelination and cognitive impairment (144). Finally, ASD has a complex pathogenesis involving a neuroinflammatory environment comparable to some aspects found in AD.
Contactins and Contactin-Associated Proteins in Inflammatory and Immune Disease
Alongside a documented link between ASD and inflammatory disease, current literature (Table 2) also supports an association between CNTNs and CNTNAPs with inflammatory and immune-mediated disease.
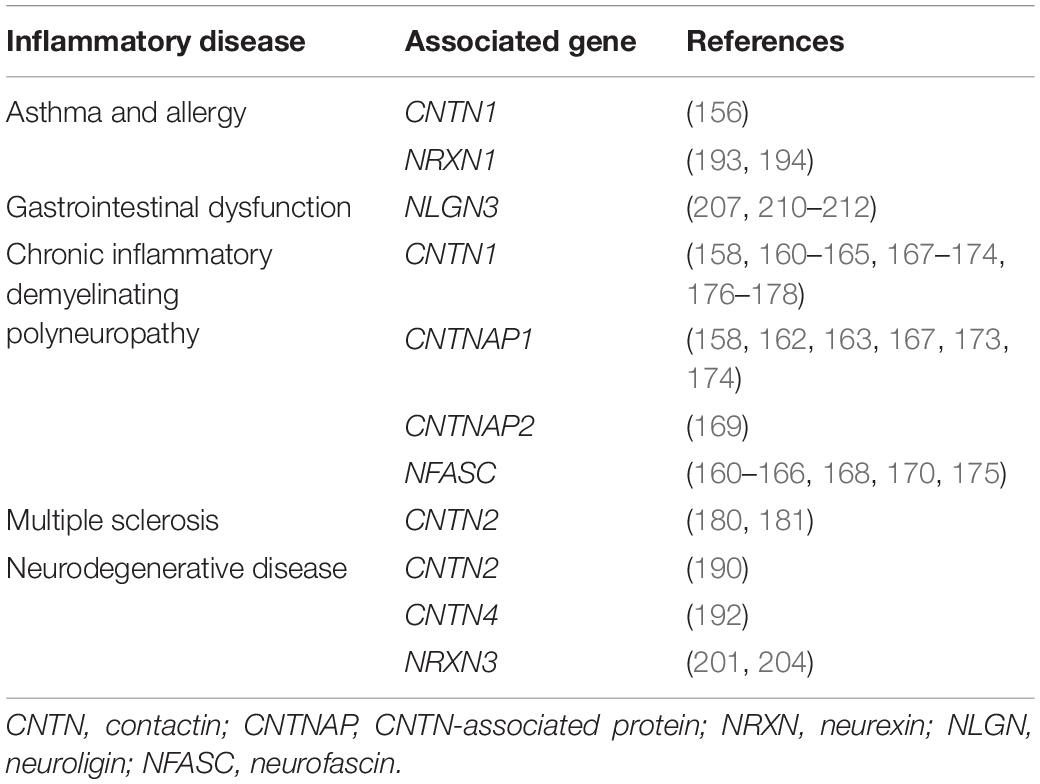
Table 2. Overview of inflammatory diseases and immune disorders that are associated with neuronal cell adhesion molecules.
Recent exosome research has identified a link between CNTN1 and asthma. CNTN1 was found to induce Notch2 signaling in asthma to activate Th17 and Th2 cells (156). CNTN1 is present on the surface of exosomes containing allergens and acts as a Notch2 ligand for monocyte-derived dendritic cells (156). These dendritic cells secrete IL-4, IL-5, IL-6, IL-13, and IL-17A to drive an enhanced inflammatory response in the airways, suggesting that CNTN1 may act as an inflammatory mediator in the pathology of asthma (156).
Likewise, chronic inflammatory demyelinating polyneuropathy (CIDP) has been strongly correlated with CNTNs alongside additional associations to CNTNAPs. CIDP is an immune-mediated neuropathy, caused by damage to the myelin sheath, with large heterogeneity (157). Antibodies against neuronal CAMs such as CNTN1, CNTNAP1, CNTNAP2, and NFASC have been found in the periphery of patients with CIDP (158–175). All these CAMs are localized to the nodes and paranodes of myelinated axons within the CNS (159). The most common autoantibody shown in seropositive CIDP patients were anti-CNTN1 IgG4 subclass antibodies (169, 176). Cytotoxic effects on cerebellar neurons were identified with chronic administration of IgG4 anti-CNTN1 serum of a patient with CIDP (177). Proteins in the CSF of seropositive CIDP patients show probable blood-brain barrier breakdown, which could increase the likelihood of anti-CNTN1 antibodies entering the brain parenchyma (163, 178). It was demonstrated that CNTN1 expression was reduced in dorsal root ganglion neurons and cerebellar granule neurons after long-term exposure to anti-CNTN1 autoantibodies (177). Notably, Vargas et al., showed that microglia and astrocytes within the cerebellum of ASD patients were activated upon the degeneration of granule cells within their vicinity (102). This raises the possibility that anti-CNTN1 antibodies targeting the cerebellar granule cells cause degeneration resulting in the activation of microglia and astrocytes. These then go on to produce proinflammatory cytokines. Like ASD, within the CSF of CIDP patients, high concentrations of proinflammatory cytokines have been observed (179). Interestingly, anti-CNTNAP2 IgG4 and anti-CNTN1 IgG1 antibodies were unable to cross the paranodal barrier, indicating these autoantibody subtypes may be less pathogenic than anti-CNTN1 IgG4 (169). However, once anti-CNTN1 IgG4 antibodies progressively deteriorate the paranode, other CAM autoantibodies may then be able to pass the paranodal barrier and accelerate demyelination.
Similar antibodies against CNTNs have been found in some cases of multiple sclerosis (MS) (180). Anti-CNTN2 antibodies were reported in a patient with MS, alongside CNTN2-specific T cells. These CNTN2-specific T cells were able to cause permeations in the blood-brain barrier and were revealed to form cortical lesions in animal models (181). Cortical lesions are the result of the inflammatory response against the myelinated sheath and can cause cognitive impairment (182). Although this data may not be directly applicable to ASD, MS demyelination pathology may provide insight into pathogenic mechanisms in ASD cases where CNS antibodies are present (140).
It has been established that the occurrence of neurodegenerative diseases is greater in adults with ASD (148). Evidence of chronic inflammation, such as activated microglia and proinflammatory cytokines, have been shown in the brains of patients with ASD, similar to that of AD (183, 184). The Notch signaling pathway regulates neurogenesis, axon guidance, and synaptic plasticity but in most cell types, can also initiate proinflammatory signaling cascades (41, 185). Therefore, dysfunction of the Notch signaling pathway has been thought to have implications in AD pathophysiology (185). By interacting with Notch1, CNTN1 may influence downstream inflammatory effects such as the expression of proinflammatory cytokines (e.g., IL-6 and IL-17) (41, 43, 186, 187). Notch signaling is found ubiquitously within the human body and is responsible for the homeostasis of many functions and for that reason, is hard to target pharmaceutically (41). However, anti-inflammatory therapies aimed at blocking Notch signaling may be able to be CNS-targeted through specifically impeding CNTN1-driven Notch signaling (188). This could be a useful approach to target neuroinflammation in ASD brains arising from aberrant Notch signaling.
As illustrated previously, Aβ has a key role in the neuropathology of AD (189). Interestingly, the CNTN family of proteins may play roles in regulating this process. Lower CNTN2 expression is observed in and around Aβ plaques that were within the hippocampus of patients with AD compared to controls (190). Additionally, CNTN2 concentrations within the CSF of those with AD were significantly reduced (190, 191). Although it is unclear what the relationship between CNTN2 and APP is, this may point toward a role for CNTN2 in the modulation of Aβ production. A similar role has been posited for CNTN4, which is known to interact with APP and promote its processing via the non-amyloidogenic (non-pathogenic) pathway (192). Altered expression of CNTN2 and CNTN4 in ASD could result in increased amyloidogenic processing of APP to Aβ, leading to plaque formation and subsequent inflammation. Furthermore, considering that CNTN2 expression in AD is correlated with increased expression of IL-1β and IFN-γ, it could be pertinent to explore how the CNTN proteins may facilitate Aβ production in ASD.
Neurexins in Inflammatory and Immune Disease
Neurexin 1 has also been implicated in asthma and ASD pathology (Table 2). In one study, 43% of patients with a 2p16.3 deletion in NRXN1 were reported to have ASD and of these, 33.5% suffered from asthma and/or allergies (193). Similarly, a de novo mutation in NRXN1α was found in a child exhibiting ASD-associated behaviors and developmental delay, alongside asthma that required recurrent hospitalization (194). NRXN1 is co-expressed with CNTN1, suggesting they may be under control of similar transcriptional regulatory programs and have similar regulatory roles in asthma to CNTN1 (195–198). Nonetheless, there is currently no evidence to support a direct link between NRXN1 and asthma, likely indicating asthma occurrence in these case studies may be unrelated to NRXN1 mutations.
In addition to the CNTNs, NRXNs may also play a role in Aβ-induced neuroinflammation. NRXNs interact with Aβ oligomers that are located between deteriorated synapses, as found in pathogenic AD brains (200, 199). NRXN3 is expressed within the hippocampus and cerebral cortex, two important regions of the brain for memory and cognition (201, 202). Variants of the NRXN3 gene have a strong association with ASD, but not much is known about its function within the scope of AD (16, 203). It was discovered that expression of NRXN3 was reduced in the hippocampus of those with AD and that this expression was inversely correlated with NLRP3 (NOD-, LRR-, and pyrin domain-containing protein 3) expression, which is a constituent of the inflammasome (204). NLRP3 inflammasome signaling leads to the production of IL-1β and IL-18, upon activation by a pathogen or cellular damage (205). This dysregulation of NRXN3 may allow deterioration of neural synapses, causing cellular damage (200). Cellular damage could activate NLRP3, resulting in the release of proinflammatory cytokines, instigating AD pathogenesis (205). Similar disruption in the brains of those with ASD could occur, equally triggering the NLRP3 inflammasome secretion of proinflammatory cytokines IL-1β and IL-18, contributing to the neuroinflammatory environment.
Neuroligins in Inflammatory and Immune Disease
Available data indicates that NLGNs play a role in GI inflammation (Table 2). The ADAMs (a disintegrin and metalloproteinase) family are enzymes that are able to cleave transmembrane neuronal CAMs including NLGNs and NRXNs, both of which are associated with ASD (16, 206). ADAMs are expressed throughout the human body but notably, ADAM10 and ADAM17 are found both in the CNS and intestines (207). ADAM17 regulates GI and neural inflammation through the cleavage of TNF-α (increased TNF-α cleavage promotes inflammation) (208). Genetic studies have revealed that whilst ADAM17 expression decreases with age in control groups, in individuals with ASD, ADAM17 expression increases with age (209). Increased ADAM17 expression in those with ASD facilitates increased TNF-α-mediated inflammation in the gut and brain. Moreover, increased cleavage of NLGN3 by ADAM10 may be causative in decreased intestinal transit seen in people with ASD. Nlgn3-deficient mice were shown to have increased colonic motility, suggesting impaired control of gut motility by the enteric nervous system (210). Like ADAM10 and ADAM17, NLGN3 is expressed in both the GI system and the CNS (16, 207). Nlgn3 mutant mice, which display ASD-associated behaviors, were found to have GI symptoms affecting the small intestine and colon function (211). Cecal weight was also decreased in Nlgn3-deficient ASD mice models, alongside increased density of enteric macrophages (212). Due to NLGN3’s role in both CNS and enteric systems, mutations affecting this gene have apparent consequences to the immune system.
Neural Cell Adhesion Molecules in Inflammatory and Immune Disease
Despite there being little evidence to support the involvement of NCAM in inflammatory and immune disease, there is some indication of NCAM1 mediating inflammatory cascades that underlie inflammatory disease. Distinct gene expression profiles were found within GI mucosal tissue in people with ASD and GI problems. However, these profiles overlapped significantly with transcriptome profiles of those with IBD, proposing a unique ASD-associated IBD variant. Genes that were exclusively differentially upregulated in ileal and colon samples from the ASD-GI group, compared to neurotypical IBD patients, included IL-2 receptor alpha (IL2RA) and IL-4-induced 1 (IL4I1) (213). IL4I1 promotes CNS remyelination and IL2RA can activate the MAPK signaling pathway (214, 215). Both remyelination processes and MAPK signaling have been associated with NCAM1 (65, 216). Not only does this link ASD with inflammatory GI disorders, but it may also implicate neuronal CAMs in their pathophysiology’s. Due to the ambiguous nature of this potential relationship, we have not included reference to NCAM1 in Table 2. This relationship needs further investigation before we can conclude its existence.
Collectively, we can conclude that NLGN3 appears to be a CAM of interest in the pathology of ASD-related GI disease owing to its expression in both the GI system and CNS, on top of its status as an ASD candidate gene (16, 210). Autoantibodies against paranodal CAMs in inflammatory autoimmune disease prove the importance of neuronal CAMs in axon myelination, which is implied to be impaired in ASD brains via white matter dysregulation, but the origin of this dysregulation is unclear (146, 164). Seropositive CIDP cases illustrate how dysfunction of neuronal CAMs, including CNTNs and CNTNAPs, leads to a neuroinflammatory response in the CNS (164). NRXN3, CNTN2, and CNTN4 stand out as key CAMs that play a role in AD-derived inflammation within the CNS (190, 192, 204). Regulation of synaptogenesis by CAMs in the development of neural pathways may also have implications in both ASD and AD. A summary of neuronal CAMs that are associated with inflammatory diseases can be found in Table 2.
Glial Cells and Neuronal Cell Adhesion Molecules
Glia in Autism Spectrum Disorder
Glial cells encompass microglia, astrocytes, and oligodendrocytes, all found within the CNS (217). Collectively, glial cells have a major role in neuroinflammation and neurodegeneration, in addition to neuronal repair after insult (218, 219). Glial cells are sensitive to environmental cues within the CNS, such as inflammation or injury (220). Microglia act as resident macrophages of the CNS and once activated, produce proinflammatory cytokines and mediators, proliferate, migrate, and even present antigens to T cells (221). The secretion of proinflammatory cytokines, for instance, IL-1β and TNF-α, by microglia recruit immune cells to escalate the immune response and initiate the activation of astrocytes (39). Astrocytes have a similar role in inflammation as microglia. They are responsible for blood-brain barrier maintenance, immune cell activation, secretion of proinflammatory cytokines, and the induction of inflammatory-associated signaling cascades (222). After acute inflammation has been resolved through glial activation, microglia can regulate their own deactivation by the secretion of anti-inflammatory cytokines such as IL-10 and TGF-β (223). When the homeostasis of the CNS is altered, chronic activation of microglia or astrocytes can occur. This results in a prolonged inflammatory response, consequently causing damage to neuronal cells (224). Often, this is a characteristic of neurological or inflammatory disease (224). Oligodendrocytes myelinate axons within the CNS, which are important for synapse transmission and neuronal communication (225). As explained previously, dysfunction of myelination leaves neuronal cells unprotected against proinflammatory damage that may also impair cognition and sensory processing (144).
Examination of postmortem ASD brains identified microglial dysfunction as a feature of ASD pathophysiology (89, 226). Astrocytes undergo reactive gliosis and change morphology, much like microglia (227). A pivotal manuscript by Vargas et al. gave insight into the glial state within the brains of individuals with ASD. Dynamic neuroinflammation was observed within the cortex, cerebellum, and white matter of ASD subjects, as well as obvious activation of astrocytes and microglia (102). In another study, morphological changes were apparent in microglia from the prefrontal cortex of males with ASD, including decreased branching and thickening of the filopodia (226). Collectively, a neuroinflammatory state with involvement of glial cells, denoted by a change of phenotype, appear to characterize the pathophysiology of ASD. Glial reactivity could be, in part, a consequence of localized neuronal dysfunction onset by ASD and, therefore, could exacerbate synaptic and axonal aberrancy already present (226). Alternatively, glia may become activated in response to environmental cues such as LPS (89). In a rat model of LPS-induced MIA, microglia and astrocytes were activated within the fetal cortex soon after LPS administration (228). This poses an environmental source of glial activation resulting in a neuroinflammatory state. Regardless of the cause, prolonged glial activation is detrimental to neuronal health and consequently, cognitive function in ASD (224).
Microglia-derived cytokines, such as TNF-α, have been reported to regulate the pruning of neuronal synapses (229, 230). Efficient synaptic pruning is most active from the age of two and is vital for brain plasticity, which is thought to be lacking in ASD (231, 232). Researchers have found an increase in synapse number and evidence of under-pruning when examining the brains of children with ASD (233). This would agree with the onset of ASD-associated behaviors around the age of three, as well as behaviors in response to over-stimulation (3). However, there is also evidence that overproduction of TNF-α by microglia in ASD brains increases synaptic scaling, theoretically over-pruning synapses (79, 230). This may also have a detrimental effect on the formation of behavior pathways, especially during early neuronal development (234). As of yet, no consensus has been reached over these pruning hypotheses.
Neural Cell Adhesion Molecules in Glial Cell Differentiation, Proliferation, and Phenotype
Glial cell proliferation is imperative in neural development. Astrocytes originate from radial glial cells and oligodendrocytes stem from oligodendrocyte precursor cells (OPCs), whereas microglia are thought to derive from resident macrophages in the yolk sac (225, 235). OPCs, astrocytes, and microglia are incredibly sensitive to sources of deterioration, including inflammation, and respond by proliferating (218, 225, 235). Dysregulation of cell proliferation and differentiation in the prefrontal cortex may be implicated in ASD (99).
It is important to acknowledge that various neuronal CAMs implicated in ASD are also expressed by glial cells. NCAM1, for example, is expressed on the surface of astrocytes in the brain and is vital in axonal regeneration after neuronal insult (236). However, there is further evidence that NCAM1 may be an important molecule for regulating neuroimmune signaling. Studies reveal that astrocyte-derived NCAM1 can alter NF-κB activity in both bulk rat brain tissue and cerebellar granule neurons (123, 237). NF-κB-mediated transcription in neurons and astrocytes was also increased with NCAM1 homophilic binding, whereby purified NCAM1 was added in vitro (123). This indicates that NCAM1 actively moderates NF-κB activity in astrocytes and neurons, hence, altering the levels of inflammatory cytokines produced and subsequently, the neuroimmune response. The Ig domains of NCAM1 are also reported to inhibit astrocyte proliferation via inhibition of MAPK signaling (237). As astrocytes are a major source of cytokines in the CNS, their reduced proliferation presents as a mechanism by which NCAM1 can regulate inflammatory responses in the CNS. Importantly, the Ig domain of NCAM1 does share homology with other members of the IgCAM superfamily (238). Therefore, there is also the possibility that other neuronal IgCAMs implicated in ASD could also exert similar effects on astrocytes, initiating NF-κB signaling or regulating astrocyte proliferation. The introduction of purified NCAM1 to rat forebrain astrocytes inhibited astrocyte proliferation, even with the addition of growth factors (239). Likewise, the application of anti-NCAM1 IgG showed the same inhibition of astrocyte proliferation (237, 239). Genetic variation in NCAM1, as demonstrated in ASD subjects by Zhang et al., may result in the production of a structurally ineffective form of NCAM1, meaning unrestricted astrocyte proliferation may occur in ASD brains in response to an inflammatory stimulus (240). In turn, this may contribute to the increased brain volume and astrocyte density observed in human ASD brains and ASD mouse models (99, 241). A polysialylated form of NCAM (PSA-NCAM) has also been implicated in postnatal spinal cord myelination in mice (242). PSA-NCAM, a post-translational modification of NCAM, is expressed on the surface of demyelinated axons, reactive astrocytes, and OPCs (216, 242). PSA-NCAM seems to be associated with OPC migration as well as myelination, as its expression is downregulated once the production of myelin begins (243). Alterations in myelination and white matter have been associated with ASD pathology, possibly linking these findings to PSA-NCAM (146, 244). Although, it is still unclear if PSA-NCAM may regulate the way OPCs respond to inflammatory stimuli or influence their production of inflammatory cytokines. Altogether, it appears that NCAM1 may regulate astrocyte proliferation in response to inflammatory stimuli and alter NF-κB signaling in both neurons and astrocytes. Additional research is still needed to better understand this process and to ascertain if other IgCAMs may perform similar roles in modulating neuroinflammation.
Contactins and Contactin-Associated Proteins in Glial Cell Differentiation, Proliferation, and Phenotype
Another family of proteins that may alter neuroimmune responses is the CNTNs and their interacting partners, the CNTNAPs. CNTNAP1-deficient mice were found to have increased Notch signaling, which in turn promoted astrocytogenesis within the cerebral cortex (245). CNTNAP1 is expressed by the radial glial cells that differentiate into astrocytes, however, no changes in radial glial cell number were observed in CNTNAP1-deficient neonatal mice brains (245). Most radial glial cells transform into astrocytes shortly after birth, leaving differentiated astrocytes to generate new astrocytes (217, 246). During neuroinflammation, astrogliosis occurs, whereby Notch signaling promotes the proliferation of local astrocytes (247). Although CNTNAP1 deficiency may not directly alter the number of astrocytes or astrocytic function, it may alter Notch-mediated astrocyte proliferation (245). Similarly, CNTNAP2 does not appear to directly affect microglia or astrocyte number, however, it could influence astrocyte progenitor number. CNTNAP2-deficient mouse models revealed that the number of radial glial cells were decreased in the hippocampus of 5- to 6-month-old mice compared to wild-type mice (248). No differences in the number of mature astrocytes were indicated, suggesting CNTNAP2 may regulate astrocyte progenitor cell number but not the number of differentiated astrocytes. Additionally, CNTNAP2-deficient conditions alter the way astrocytes respond to stimuli. CNTNAP2-deficient mice exhibited a greater number of reactive astrocytes in the hippocampus following an induced seizure compared to wild-type mice (22). Combined, this data proposes that CNTNAP2 may moderate astrocyte activity at two neurodevelopmental stages; via astrocyte progenitor cell number at a younger age and also altering how differentiated astrocytes respond to stimuli later in life. Further experimentation on early postnatal transgenic mice would provide an understanding of alterations in the glial population as neurodevelopment progresses.
Altered OPC proliferation has been implicated in ASD pathogenesis alongside abnormalities of the white matter within the brains of those with ASD (146, 249). Analysis of differentially expressed genes in syndromic ASD models supports dysregulation of oligodendrocyte number (244). PTPRZ (receptor protein tyrosine phosphatase zeta), an interacting partner of CNTN1, is expressed in astrocytes, OPCs, and oligodendrocytes in the adult CNS (250). In the CNS, glial PTPRZ interaction with neuronal CNTN1 triggers cell signaling between glia and neuronal cells, promoting neuronal outgrowth important in neurodevelopment (251). CNTN1 is able to bind to PTPRZ at its Ig domain (252). A previous study observed that CNTN1-PTPRZ interaction on the surface of OPCs impairs OPC proliferation and induces oligodendrocyte differentiation (Figures 2A,B) (250). This indicates that CNTN1 and PTPRZ act as modulators of oligodendrogenesis. Although unexplored, alterations in CNTN1 (and other CNTN family members) expression may lead to changes in oligodendrocyte number or responses to inflammatory stimuli. Additionally, altered CNTN expression could contribute to dysfunctional myelination that exposes neurons to proinflammatory damage.
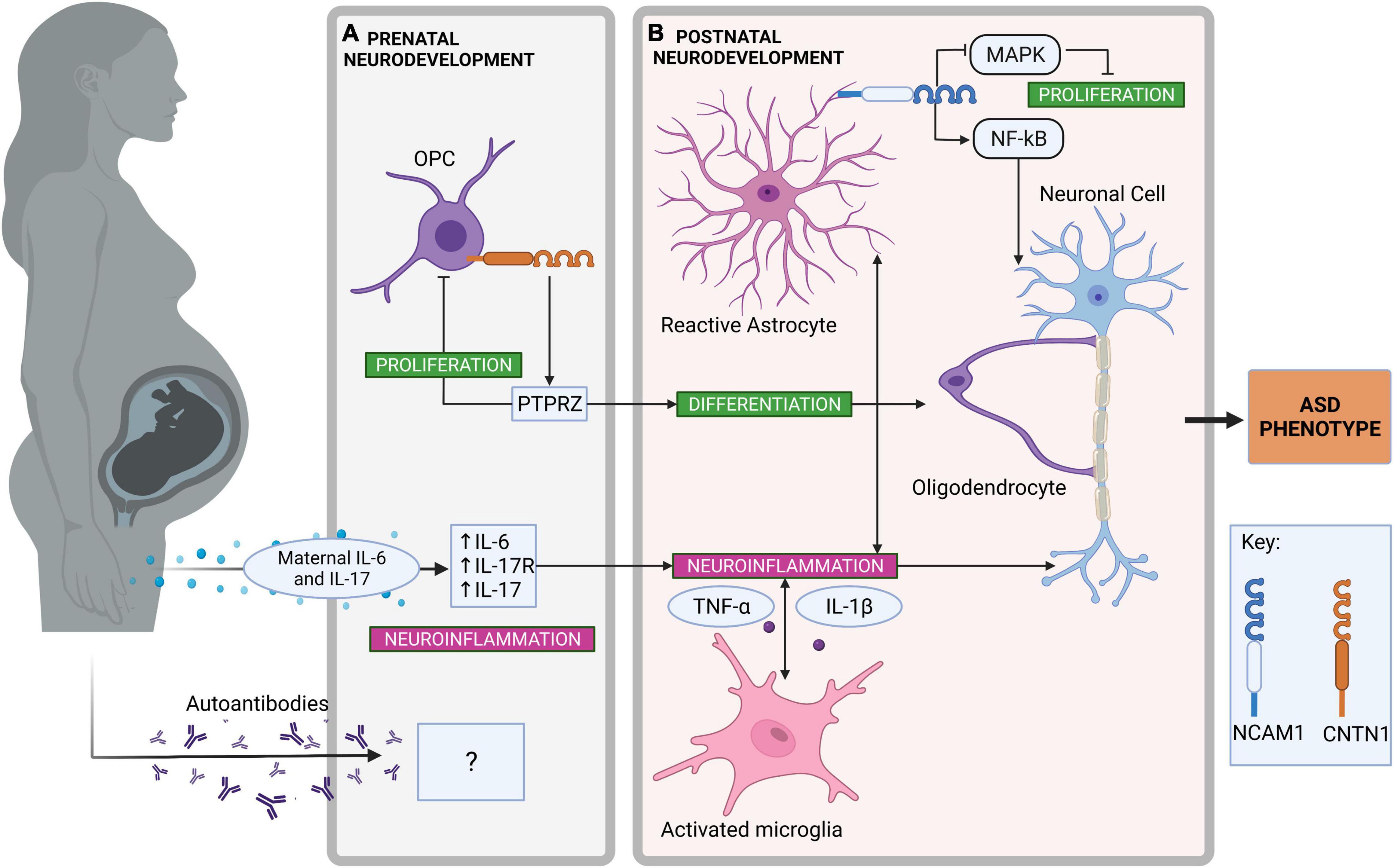
Figure 2. Neuroinflammatory pathways in prenatal and postnatal neurodevelopment contributing to autism spectrum disorder (ASD). (A) Neuroinflammatory pathways in prenatal neurodevelopment. Contactin-1 (CNTN1), expressed on the surface of oligodendrocyte precursor cells (OPC) promotes OPC differentiation to oligodendrocytes by interacting with receptor protein tyrosine phosphatase zeta (PTPRZ) (250, 258). Simultaneously, OPC proliferation is inhibited by the same PTPRZ interaction (250). The transfer of maternal proinflammatory cytokines such as interleukin (IL)-6 and IL-17 via maternal immune activation can promote neuroinflammation during prenatal neurodevelopment (115, 121). As a result, increased receptor IL-17 (IL-17R) expression is found within the fetal brain, enhancing further neuroinflammatory signaling (107). Although there is some evidence for autoimmune antibodies in association with ASD, their origin and effect on neuroinflammation in the developing ASD brain is unclear (140). (B) Neuroinflammatory pathways in postnatal neurodevelopment. Astrocytes are activated by proinflammatory cytokines and neuroinflammation in the central nervous system (227). Neural cell adhesion molecule-1 (NCAM1) expressed on the surface of astrocytes can activate nuclear factor-κB (NF-κB)-mediated transcription of proinflammatory genes (123). NCAM1 can also inhibit astrocyte proliferation through the inhibition of mitogen-activated protein kinase (MAPK) signaling (237). Changes to OPC proliferation have been observed in ASD brains, which can alter the production of myelin and possibly expose neuronal cells to inflammatory insult (146). Microglia are also activated by proinflammatory cytokines and neuroinflammation in the central nervous system (220). Activated microglia to produce proinflammatory cytokines including IL-1β and tumor necrosis factor (TNF)-α (39). Created with BioRender.com.
To summarize, glial cells are the key source of cytokines within the CNS and, therefore, are vital when considering the impact that neuroinflammation has in ASD (102). Neuronal CAMs seem to play a role, either directly or indirectly, in regulating glial activity. Importantly, neuronal CAMs may influence how glial cells respond to inflammation. One key example of this is CNTNAP2 acting as a potential moderator in astrocyte response after a stimulus (22). Over-proliferation of glial cells may contribute to ASD pathology, although there is some evidence of decreased OPC proliferation (99, 146, 226, 249). NCAM1 appears to directly regulate astrocyte proliferation via NF-κB signaling and may contribute to the increased brain volume observed in individuals with ASD (237, 239). CNTN1 may indirectly regulate OPC proliferation through the interaction of PTPRZ, which subsequently may affect neuronal myelination (250). These ideas are summarized in Figures 2A,B, depicting the role of neuronal CAMs in neuroinflammatory pathways during prenatal and postnatal neurodevelopment that may contribute to ASD. PSA-NCAM is expressed during early neurodevelopment making it a good marker to investigate the role of neuronal CAMs during behavioral pathway formation (216). Future investigations may be able to utilize PSA-NCAM to further explore the role of CAMs in inflammatory systems during neurodevelopment.
Future Directions
It is believed that both genetic and environmental factors play a role in the pathology of ASD (16, 24). Inflammation has been identified as an environmental risk factor for ASD (220). There is strong evidence highlighting the presence of immune dysfunction in those with ASD, as well as the characterization of ASD-associated behaviors in MIA models (24, 36, 80, 109, 253).
In a healthy model of inflammation, immune cells proliferate and produce proinflammatory cytokines in response to pathogens or immunogenic materials (67). This response is facilitated by signaling pathways that can be activated by cytokines including the Notch, NF-κB, and MAPK signaling cascades (41, 55, 254). Chronic inflammatory signaling can occur if there is dysfunction of the immune system, resulting in unnecessary tissue and cellular damage, instigating the pathogenesis of autoimmune or inflammatory disease (41, 224).
Abnormally elevated proinflammatory cytokines (most commonly IL-1β, IL-6, IL-8, and IL-17) are consistently observed in the CSF and blood of children and adults with ASD (73, 74, 96). Although it is not fully understood, several studies have demonstrated that chronically elevated levels of these proinflammatory cytokines impair neurodevelopmental processes and neural cell function, including impaired synaptic pruning by microglia, irregular migration of neurons, and altered synaptic plasticity (57, 101, 230). It could be of particular interest for future studies to investigate whether specific cytokine profiles can be associated with inflammation originating from different environmental triggers (e.g., upregulated IL-6 and IL-17 are often associated with MIA) (Figure 2A) (110). Additionally, it is not yet certain which cell types, either in the CNS or the periphery, are the main drivers of inflammation in ASD (and the main cell types affected by the chronic inflammation).
Although it is unclear how the neuronal CAMs commonly implicated in ASD may mediate inflammation, investigating other disorders linked to ASD with an inflammatory or immune component can advise potential molecular mechanisms. As discussed in previous sections, the gut-brain axis provides the opportunity for peripheral inflammation to influence neuroimmune signaling and glial activation within the CNS through cytokines and vagal innervation (135). Most likely, GI inflammation is triggered by an exogenous source, such as in the case of food intolerance. In cases of ASD with GI dysfunction, the treatment of GI disorders to reduce inappropriate inflammation could improve the consequences of neuroinflammation. NLGN3 is of distinct interest concerning GI inflammation and ASD, owing to its interactions with ADAM proteins which may influence the production of proinflammatory cytokines in the gut and CNS (16, 210). Further epigenetic studies to explore environmental influences on NLGN3 expression, and whether its dysregulation alters proinflammatory cytokine production, may be pertinent. There is also some evidence supporting a link to autoimmunity in dysregulation of the immune function in ASD, but literature is unclear on the origin of these autoantibodies (Figure 2A) (140, 148, 255). Additionally, there is no proof, as of yet, of the presence of autoantibodies against neuronal CAMs in those with ASD. Autoantibodies against paranodal CAMs in CIDP and MS demonstrate the importance of neuronal CAMs in the protection of neuronal cells against inflammatory damage, however, it remains uncertain how this is relevant to ASD (164). Although ASD is not typically characterized by neurodegeneration, an increase in secreted β-amyloid in the plasma of children with severe ASD identifies an association between the pathology of ASD and AD (151). CNTN4 regulates APP processing, whilst CNTN2 seems to have an association with Aβ (189, 190). The disruption of APP processing pathways may interfere with the balance of Aβ production, the clearance of which is mediated by activated astrocytes and microglia, causing a neuroinflammatory response (155). Therefore, altered expression of these CNTNs may increase Aβ production, contributing to neuroinflammation (153, 189, 192). Future research into the mechanisms by which CNTNs regulate APP processing, and whether dysregulation of this can alter glial cell activity, may reveal more about CNTNs functional role in modulating neuroinflammation.
Glial cells are the key source of cytokines within the CNS and, therefore, are vital when assessing neuroinflammation in ASD brains (102). In response to inflammation, glial cells change their morphology and proliferate (39). There is evidence that neuronal CAMs can play a direct or indirect role in the regulation of glial activity and, therefore, may influence glial responses to inflammation. CNTNAP2 may moderate astrocyte activity by influencing astrocyte progenitor cell numbers and affect how astrocytes respond to external stimuli (22, 248). Compellingly, NCAM1 has emerged as a key player in regulating neuroinflammatory cascades. NCAM1 homophilic binding can initiate NF-κB-mediated transcription in neurons and astrocytes, instigating a proinflammatory response (237). NCAM1 appears to directly regulate astrocyte proliferation (Figure 2B) and its dysregulation may, in part, account for the increased brain weight observed in individuals with ASD (237, 239). Additionally, CNTN1 may indirectly regulate OPC proliferation through interacting with PTPRZ (Figures 2A,B), which subsequently may affect neuronal myelination (250). Myelin is paramount in protecting axons from damage that may impair cognition and sensory processing (144). Further definition of neuronal CAMs regulatory role in glial activity and response to inflammation is missing from current literature. Experiments investigating CAM expression in glia at different developmental stages, and if neuroinflammation arising from this dysregulated expression can be treated in later life, would be of value for targeted anti-inflammatory therapeutics in ASD. Specifically, pre- and immediately postnatal neurodevelopment would be of interest, owing to ASD phenotypes presenting before the age of three (1).
The concept that neuronal CAMs may mediate or influence inflammatory cascades is largely unexplored in current literature. This review highlights the available evidence on the potential part neuronal CAMs play in neuroinflammation, with a particular focus on ASD. Further investigation into the role of neuronal CAMs within the context of inflammation is clearly warranted and would advance our understanding of neuroinflammation in ASD pathology.
Author Contributions
AO-A: concept, research design, editing figures and artwork, and manuscript writing and editing. ME: research, generating figures and artwork, and manuscript writing. JG: research and manuscript writing and editing. LY: research and manuscript editing. All authors read and approved the final manuscript.
Funding
This research was supported by the Northcott Devon Medical Foundation Research Grant (AO-A), ARUK Southwest Small Pump Priming Grant (JG and AO-A), and QUEX Ph.D. studentship (ME).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We acknowledge the insightful discussions with Dr. Rosie Bamford, Dr. Charli Harlow, and Miss Emily-Rose Martin (University of Exeter).
References
1. American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 5th ed. Arlington, VA: American Psychiatric Association (2013).
2. Russell G, Stapley S, Newlove-Delgado T, Salmon A, White R, Warren F, et al. Time trends in autism diagnosis over 20 years: a UK population-based cohort study. J Child Psychol Psychiatry. (2021). doi: 10.1111/jcpp.13505 [Epub ahead of print].
3. Tsai LY. Impact of DSM-5 on epidemiology of autism spectrum disorder. Res Autism Spectr Disord. (2014) 8:1454–70. doi: 10.1016/j.rasd.2014.07.016
4. Elsabbagh M, Divan G, Koh YJ, Kim YS, Kauchali S, Marcín C, et al. Global prevalence of autism and other pervasive developmental disorders. Autism Res. (2012) 5:160–79. doi: 10.1002/aur.239
5. Chiarotti F, Venerosi A. Epidemiology of autism spectrum disorders: a review of worldwide prevalence estimates since 2014. Brain Sci. (2020) 10:274. doi: 10.3390/brainsci10050274
6. Chaste P. Autism risk factors: genes, environment, and gene-environment interactions. Dialogues Clin Neurosci. (2012) 14:281–92. doi: 10.31887/DCNS.2012.14.3/pchaste
7. Sandin S, Lichtenstein P, Kuja-Halkola R, Hultman C, Larsson H, Reichenberg A. The heritability of autism spectrum disorder. JAMA. (2017) 318:1182–4. doi: 10.1001/jama.2017.12141
8. Halladay AK, Bishop S, Constantino JN, Daniels AM, Koenig K, Palmer K, et al. Sex and gender differences in autism spectrum disorder: summarizing evidence gaps and identifying emerging areas of priority. Mol Autism. (2015) 6:36. doi: 10.1186/s13229-015-0019-y
9. Gandawijaya J, Bamford RA, Burbach JPH, Oguro-Ando A. Cell adhesion molecules involved in neurodevelopmental pathways implicated in 3p-deletion syndrome and autism spectrum disorder. Front Cell Neurosci. (2021) 14:611379. doi: 10.3389/fncel.2020.611379
10. Nasser TI, Spencer GE. Neurite outgrowth. In: Reference Module in Biomedical Sciences. Amsterdam: Elsevier (2017). doi: 10.1016/B978-0-12-801238-3.99507-2
11. Binder MD, Hirokawa N, Windhorst U. Cell adhesion molecules. In: Binder MD, Hirokawa N, Windhorst U, editors. Encyclopedia of Neuroscience. Berlin: Springer (2009). p. 588. doi: 10.1007/978-3-540-29678-2_864
12. Burbach JPH. Immunoglobulin cell adhesion molecules of the Ig-FNIII type and neurodevelopment. In: Martin CR, Preedy VR, Rajendram R, editors. Factors Affecting Neurodevelopment. Cambridge, MA: Academic Press (2021). p. 105–19. doi: 10.1016/B978-0-12-817986-4.00010-9
13. Zuko A, Kleijer KTE, Oguro-Ando A, Kas MJH, van Daalen E, van der Zwaag B, et al. Contactins in the neurobiology of autism. Eur J Pharmacol. (2013) 719:63–74. doi: 10.1016/j.ejphar.2013.07.016
14. Osterhout JA, Stafford BK, Nguyen PL, Yoshihara Y, Huberman AD. Contactin-4 mediates axon-target specificity and functional development of the accessory optic system. Neuron. (2015) 86:985–99. doi: 10.1016/j.neuron.2015.04.005
15. Poot M. A candidate gene association study further corroborates involvement of contactin genes in autism. Mol Syndromol. (2014) 5:229–35. doi: 10.1159/000362891
16. Abrahams BS, Arking DE, Campbell DB, Mefford HC, Morrow EM, Weiss LA, et al. SFARI gene 2.0: a community-driven knowledgebase for the autism spectrum disorders (ASDs). Mol Autism. (2013) 4:36. doi: 10.1186/2040-2392-4-36
17. Lu Z, Reddy MVVVS, Liu J, Kalichava A, Liu J, Zhang L, et al. Molecular architecture of contactin-associated protein-like 2 (CNTNAP2) and its interaction with contactin 2 (CNTN2). J Biol Chem. (2016) 291:24133–47. doi: 10.1074/jbc.M116.748236
18. Sampath S, Bhat S, Gupta S, O’Connor A, West AB, Arking DE, et al. Defining the contribution of CNTNAP2 to autism susceptibility. PLoS One. (2013) 8:e77906. doi: 10.1371/journal.pone.0077906
19. Toma C, Hervás A, Torrico B, Balmaña N, Salgado M, Maristany M, et al. Analysis of two language-related genes in autism. Psychiatr Genet. (2013) 23:82–5. doi: 10.1097/YPG.0b013e32835d6fc6
20. Whitehouse AJO, Bishop DVM, Ang QW, Pennell CE, Fisher SE. CNTNAP2 variants affect early language development in the general population. Genes Brain Behav. (2011) 10:451–6. doi: 10.1111/j.1601-183X.2011.00684.x
21. Scott-Van Zeeland AA, Abrahams BS, Alvarez-Retuerto AI, Sonnenblick LI, Rudie JD, Ghahremani D, et al. Altered functional connectivity in frontal lobe circuits is associated with variation in the autism risk gene CNTNAP. Sci Transl Med. (2010) 2:56ra80. doi: 10.1126/scitranslmed.3001344
22. Peñagarikano O, Abrahams BS, Herman EI, Winden KD, Gdalyahu A, Dong H, et al. Absence of CNTNAP2 leads to epilepsy, neuronal migration abnormalities, and core autism-related deficits. Cell. (2011) 147:235–46. doi: 10.1016/j.cell.2011.08.040
23. Al-Murrani A, Ashton F, Aftimos S, George AM, Love DR. Amino-terminal microdeletion within the CNTNAP2 gene associated with variable expressivity of speech delay. Case Rep Genet. (2012) 2012:172408. doi: 10.1155/2012/172408
24. Karimi P, Kamali E, Mousavi SM, Karahmadi M. Environmental factors influencing the risk of autism. J Res Med Sci. (2017) 22:27. doi: 10.4103/1735-1995.200272
25. Rossignol DA, Frye RE. A systematic review and meta-analysis of immunoglobulin g abnormalities and the therapeutic use of intravenous immunoglobulins (IVIG) in autism spectrum disorder. J Pers Med. (2021) 11:488. doi: 10.3390/jpm11060488
26. Akintunde ME, Rose M, Krakowiak P, Heuer L, Ashwood P, Hansen R, et al. Increased production of IL-17 in children with autism spectrum disorders and co-morbid asthma. J Neuroimmunol. (2015) 286:33–41. doi: 10.1016/j.jneuroim.2015.07.003
27. Kordulewska NK, Kostyra E, Chwała B, Moszyñska M, Cieślińska A, Fiedorowicz E, et al. A novel concept of immunological and allergy interactions in autism spectrum disorders: molecular, anti-inflammatory effect of osthole. Int Immunopharmacol. (2019) 72:1–11. doi: 10.1016/j.intimp.2019.01.058
28. Wong H, Hoeffer C. Maternal IL-17A in autism. Exp Neurol. (2018) 299:228–40. doi: 10.1016/j.expneurol.2017.04.010
29. Gumusoglu SB, Hing BWQ, Chilukuri ASS, Dewitt JJ, Scroggins SM, Stevens HE. Chronic maternal interleukin-17 and autism-related cortical gene expression, neurobiology, and behavior. Neuropsychopharmacology. (2020) 45:1008–17. doi: 10.1038/s41386-020-0640-0
30. Pulikkan J, Mazumder A, Grace T. Role of the gut microbiome in autism spectrum disorders. In: Guest PC, editor. Reviews on Biomarker Studies in Psychiatric and Neurodegenerative Disorders. Cham: Springer International Publishing (2019). p. 253–69. doi: 10.1007/978-3-030-05542-4_13
31. Lombardo MV, Moon HM, Su J, Palmer TD, Courchesne E, Pramparo T. Maternal immune activation dysregulation of the fetal brain transcriptome and relevance to the pathophysiology of autism spectrum disorder. Mol Psychiatry. (2018) 23:1001–13. doi: 10.1038/mp.2017.15
32. le Belle JE, Sperry J, Ngo A, Ghochani Y, Laks DR, López-Aranda M, et al. Maternal inflammation contributes to brain overgrowth and autism-associated behaviors through altered redox signaling in stem and progenitor cells. Stem Cell Rep. (2014) 3:725–34. doi: 10.1016/j.stemcr.2014.09.004
33. Oskvig DB, Elkahloun AG, Johnson KR, Phillips TM, Herkenham M. Maternal immune activation by LPS selectively alters specific gene expression profiles of interneuron migration and oxidative stress in the fetus without triggering a fetal immune response. Brain Behavior Immun. (2012) 26:623–34. doi: 10.1016/j.bbi.2012.01.015
34. Smith SEP, Elliott RM, Anderson MP. Maternal immune activation increases neonatal mouse cortex thickness and cell density. J Neuroimmune Pharmacol. (2012) 7:529–32. doi: 10.1007/s11481-012-9372-1
35. Weir RK, Forghany R, Smith SEP, Patterson PH, McAllister AK, Schumann CM, et al. Preliminary evidence of neuropathology in nonhuman primates prenatally exposed to maternal immune activation. Brain Behav Immun. (2015) 48:139–46. doi: 10.1016/j.bbi.2015.03.009
36. Yasumatsu K, Nagao JI, Arita-Morioka KI, Narita Y, Tasaki S, Toyoda K, et al. Bacterial-induced maternal interleukin-17A pathway promotes autistic-like behaviors in mouse offspring. Exp Anim. (2020) 69:250–60. doi: 10.1538/expanim.19-0156
37. Mueller FS, Polesel M, Richetto J, Meyer U, Weber-Stadlbauer U. Mouse models of maternal immune activation: mind your caging system! brain. Behav Immun. (2018) 73:643–60. doi: 10.1016/j.bbi.2018.07.014
38. Turbé H, Waeckel L, Dechelotte B. Overview of prospects for inflammation pathways in autism spectrum disorders. Encephale. (2020) 46:404–7. doi: 10.1016/j.encep.2019.09.006
39. Carniglia L, Ramírez D, Durand D, Saba J, Turati J, Caruso C, et al. Neuropeptides and microglial activation in inflammation, pain, and neurodegenerative diseases. Mediators Inflamm. (2017) 2017:5048616. doi: 10.1155/2017/5048616
40. Tsilioni I, Theoharides TC. Extracellular vesicles are increased in the serum of children with autism spectrum disorder, contain mitochondrial DNA, and stimulate human microglia to secrete IL-1β. J Neuroinflamm. (2018) 15:239. doi: 10.1186/s12974-018-1275-5
41. Christopoulos PF, Gjølberg TT, Krüger S, Haraldsen G, Andersen JT, Sundlisæter E. Targeting the notch signaling pathway in chronic inflammatory diseases. Front Immunol. (2021) 12:668207. doi: 10.3389/fimmu.2021.668207
42. Fiúza UM, Arias AM. Cell and molecular biology of Notch. J Endocrinol. (2007) 194:459–74. doi: 10.1677/JOE-07-0242
43. Hu Q-D, Ang B-T, Karsak M, Hu W-P, Cui X-Y, Duka T, et al. F3/contactin acts as a functional ligand for notch during oligodendrocyte maturation. Cell. (2003) 115:163–75. doi: 10.1016/s0092-8674(03)00810-9
44. Cui XY, Hu QD, Tekaya M, Shimoda Y, Ang BT, Nie DY, et al. NB-3/Notch1 pathway via Deltex1 promotes neural progenitor cell differentiation into oligodendrocytes. J Biol Chem. (2004) 279:25858–65. doi: 10.1074/jbc.M313505200
45. Mukherjee S, Schaller MA, Neupane R, Kunkel SL, Lukacs NW. Regulation of T cell activation by notch ligand, DLL4, promotes IL-17 production and Rorc activation. J Immunol. (2009) 182:7381–8. doi: 10.4049/jimmunol.0804322
46. Samon JB, Champhekar A, Minter LM, Telfer JC, Miele L, Fauq A, et al. Notch1 and TGFbeta1 cooperatively regulate Foxp3 expression and the maintenance of peripheral regulatory T cells. Blood. (2008) 112:1813–21. doi: 10.1182/blood-2008-03-144980
47. Kawamatal S, Du C, Lavau C, Lavau C. Overexpression of the notch target genes Hes in vivo induces lymphoid and myeloid alterations. Oncogene. (2002) 21:3855–63. doi: 10.1038/sj/onc/1205487
48. Amsen D, Magarian Blander J, Lee GR, Tanigaki K, Honjo T, Flavell RA. Instruction of distinct CD4 T helper cell fates by different notch ligands on antigen-presenting cells. Cell. (2004) 117:515–26. doi: 10.1016/s0092-8674(04)00451-9
49. Bassil R, Zhu B, Lahoud Y, Riella LV, Yagita H, Elyaman W, et al. Notch ligand delta-like 4 blockade alleviates experimental autoimmune encephalomyelitis by promoting regulatory T cell development. J Immunol. (2011) 187:2322–8. doi: 10.4049/jimmunol.1100725
50. Jiao Z, Wang W, Ma J, Wang S, Su Z, Xu H. Notch signaling mediates TNF-α-Induced IL-6 production in cultured fibroblast-like synoviocytes from rheumatoid arthritis. Clin Dev Immunol. (2012) 2012:6. doi: 10.1155/2012/350209
51. Diamant G, Dikstein R. Transcriptional control by NF-κB: elongation in focus. Biochim Biophys Acta. (2013) 1829:937–45. doi: 10.1016/j.bbagrm.2013.04.007
52. Karin M, Delhase M. The IκB kinase (IKK) and NF-κB: key elements of proinflammatory signalling. Semin Immunol. (2000) 12:85–98. doi: 10.1006/smim.2000.0210
53. Yamamoto Y, Gaynor RB. IκB kinases: key regulators of the NF-κB pathway. Trends Biochem Sci. (2004) 29:72–9. doi: 10.1016/j.tibs.2003.12.003
54. Kawai T, Akira S. Signaling to NF-κB by toll-like receptors. Trends Mol Med. (2007) 13:460–9. doi: 10.1016/j.molmed.2007.09.002
55. Liu T, Zhang L, Joo D, Sun SC. NF-κB signaling in inflammation. Signal Transduct Target Ther. (2017) 2:17023. doi: 10.1038/sigtrans.2017.23
56. Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on toll-like receptors. Nat Immunol. (2010) 11:373–84. doi: 10.1038/ni.1863
57. Okun E, Griffioen KJ, Lathia JD, Tang SC, Mattson MP, Arumugam TV. Toll-like receptors in neurodegeneration. Brain Res Rev. (2009) 59:278–92. doi: 10.1016/j.brainresrev.2008.09.001
58. Schulze-Osthoff K, Ferrari D, Riehemann K, Wesselborg S. Regulation of NF-κB activation by MAP kinase cascades. Immunobiology. (1997) 198:35–49. doi: 10.1016/S0171-2985(97)80025-3
59. Kyriakis JM, Avruch J. Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol Rev. (2001) 81:807–69. doi: 10.1152/physrev.2001.81.2.807
60. Moens U, Kostenko S, Sveinbjørnsson B. The role of mitogen-activated protein kinase-activated protein kinases (MAPKAPKs) in inflammation. Genes. (2013) 4:101–33. doi: 10.3390/genes4020101
61. Kaminska B, Gozdz A, Zawadzka M, Ellert-Miklaszewska A, Lipko M. MAPK signal transduction underlying brain inflammation and gliosis as therapeutic target. Anatom Rec. (2009) 292:1902–13. doi: 10.1002/ar.21047
62. Kaminska B. MAPK signalling pathways as molecular targets for anti-inflammatory therapy—from molecular mechanisms to therapeutic benefits. Biochim Biophys Acta. (2005) 1754:253–62. doi: 10.1016/j.bbapap.2005.08.017
63. Scheller J, Chalaris A, Schmidt-Arras D, Rose-John S. The pro- and anti-inflammatory properties of the cytokine interleukin-6. Biochim Biophys Acta. (2011) 1813:878–88. doi: 10.1016/j.bbamcr.2011.01.034
64. Park SE, Sapkota K, Kim S, Kim H, Kim SJ. Kaempferol acts through mitogen-activated protein kinases and protein kinase B/AKT to elicit protection in a model of neuroinflammation in BV2 microglial cells. Br J Pharmacol. (2011) 164:1008–25. doi: 10.1111/j.1476-5381.2011.01389.x
65. Shi Y, Xia YY, Wang L, Liu R, Khoo KS, Feng ZW. Neural cell adhesion molecule modulates mesenchymal stromal cell migration via activation of MAPK/ERK signaling. Exp Cell Res. (2012) 318:2257–67. doi: 10.1016/j.yexcr.2012.05.029
66. Wang H, Tian Y, Wang J, Phillips KLE, Binch ALA, Dunn S, et al. Inflammatory cytokines induce NOTCH signaling in nucleus pulposus cells. J Biol Chem. (2013) 288:16761–74. doi: 10.1074/jbc.M112.446633
67. Zhang JM, An J. Cytokines, inflammation, and pain. Int Anesthesiol Clin. (2007) 45:27–37. doi: 10.1097/AIA.0b013e318034194e
68. Unanue ER, Calderon J, Marie Kiely J, Stadecker MJ. Regulation of immunity and inflammation by mediators from macrophages. Am J Pathol. (1976) 85:465–78.
69. Moser B, Willimann K. Chemokines: role in inflammation and immune surveillance. Ann Rheum Dis. (2004) 63:84–9. doi: 10.1136/ard.2004.028316
71. Oswald IP, Wynn TA, Sher A, James SL. Interleukin 10 inhibits macrophage microbicidal activity by blocking the endogenous production of tumor necrosis factor a required as a costimulatory factor for interferon v-induced activation. Proc Natl Acad Sci USA. (1992) 89:8676–80. doi: 10.1073/pnas.89.18.8676
72. Chadban SJ, Tesch GH, Foti R, Lan HY, Atkins RC, Nikolic-Paterson DJ. Interleukin-10 differentially modulates MHC class II expression by mesangial cells and macrophages in vitro and in vivo. Immunology. (1998) 94:72–8. doi: 10.1046/j.1365-2567.1998.00487.x
73. Eftekharian MM, Ghafouri-Fard S, Noroozi R, Omrani MD, Arsang-jang S, Ganji M, et al. Cytokine profile in autistic patients. Cytokine. (2018) 108:120–6. doi: 10.1016/j.cyto.2018.03.034
74. Han YMY, Cheung WKY, Wong CK, Sze SL, Cheng TWS, Yeung MK, et al. Distinct cytokine and chemokine profiles in autism spectrum disorders. Front Immunol. (2017) 8:11. doi: 10.3389/fimmu.2017.00011
75. Kutuk MO, Tufan E, Gokcen C, Kilicaslan F, Karadag M, Mutluer T, et al. Cytokine expression profiles in autism spectrum disorder: a multi-center study from Turkey. Cytokine. (2020) 133:155152. doi: 10.1016/j.cyto.2020.155152
76. Suzuki K, Matsuzaki H, Iwata K, Kameno Y, Shimmura C, Kawai S, et al. Plasma cytokine profiles in subjects with high-functioning autism spectrum disorders. PLoS One. (2011) 6:e20470. doi: 10.1371/journal.pone.0020470
77. Jácome MCI, Chacòn LMM, Cuesta HV, Rizo CM, Santiesteban MW, Hernandez LR, et al. Peripheral inflammatory markers contributing to comorbidities in autism. Behav Sci. (2016) 6:29. doi: 10.3390/bs6040029
78. Saghazadeh A, Ataeinia B, Keynejad K, Abdolalizadeh A, Hirbod-Mobarakeh A, Rezaei N. A meta-analysis of pro-inflammatory cytokines in autism spectrum disorders: effects of age, gender, and latitude. J Psychiatr Res. (2019) 115:90–102. doi: 10.1016/j.jpsychires.2019.05.019
79. Li X, Chauhan A, Sheikh AM, Patil S, Chauhan V, Li XM, et al. Elevated immune response in the brain of autistic patients. J Neuroimmunol. (2009) 207:111–6. doi: 10.1016/j.jneuroim.2008.12.002
80. Zimmerman AW, Jyonouchi H, Comi AM, Connors SL, Milstien S, Varsou A, et al. Cerebrospinal fluid and serum markers of inflammation in autism. Pediatr Neurol. (2005) 33:195–201. doi: 10.1016/j.pediatrneurol.2005.03.014
81. Shen Y, Li Y, Shi L, Liu M, Wu R, Xia K, et al. Autism spectrum disorder and severe social impairment associated with elevated plasma interleukin-8. Pediatr Res. (2021) 89:591–7. doi: 10.1038/s41390-020-0910-x
82. Schwarz E, Guest PC, Rahmoune H, Wang L, Levin Y, Ingudomnukul E, et al. Sex-specific serum biomarker patterns in adults with Asperger’s syndrome. Mol Psychiatry. (2011) 16:1213–20. doi: 10.1038/mp.2010.102
83. Kaneko N, Kurata M, Yamamoto T, Morikawa S, Masumoto J. The role of interleukin-1 in general pathology. Inflamm Regen. (2019) 39:12. doi: 10.1186/s41232-019-0101-5
84. Fields JK, Günther S, Sundberg EJ. Structural basis of IL-1 family cytokine signaling. Front Immunol. (2019) 10:1412. doi: 10.3389/fimmu.2019.01412
85. Rothwell NJ, Strijbos PJLM. Cytokines in neurodegeneration and repair. Int J Dev Neurosci. (1995) 13:179–85. doi: 10.1016/0736-5748(95)00018-C
86. Goines PE, Ashwood P. Cytokine dysregulation in autism spectrum disorders (ASD): Possible role of the environment. Neurotoxicol Teratol. (2013) 36:67–81. doi: 10.1016/j.ntt.2012.07.006
87. Masi A, Quintana DS, Glozier N, Lloyd AR, Hickie IB, Guastella AJ. Cytokine aberrations in autism spectrum disorder: a systematic review and meta-analysis. Mol Psychiatry. (2015) 20:440–6. doi: 10.1038/mp.2014.59
88. Zhao H, Zhang H, Liu S, Luo W, Jiang Y, Gao J. Association of peripheral blood levels of cytokines with autism spectrum disorder: a meta-analysis. Front Psychiatry. (2021) 12:670200. doi: 10.3389/fpsyt.2021.670200
89. Nayak D, Roth TL, McGavern DB. Microglia development and function. Annu Rev Immunol. (2014) 32:367–402. doi: 10.1146/annurev-immunol-032713-120240
90. Ashwood P, Krakowiak P, Hertz-Picciotto I, Hansen R, Pessah I, van de Water J. Elevated plasma cytokines in autism spectrum disorders provide evidence of immune dysfunction and are associated with impaired behavioral outcome. Brain Behav Immun. (2011) 25:40–5. doi: 10.1016/j.bbi.2010.08.003
91. Kim G-Y, Lee J-W, Ryu H-C, Wei J-D, Seong C-M, Kim J-H. Proinflammatory cytokine IL-1β stimulates IL-8 synthesis in mast cells via a leukotriene B4 receptor 2-linked pathway, contributing to angiogenesis. J Immunol. (2010) 184:3946–54. doi: 10.4049/jimmunol.0901735
92. Baggiolini M, Clark-Lewis I. Interleukin-8, a chemotactic and inflammatory cytokine. FEBS Lett. (1992) 307:97–101. doi: 10.1016/0014-5793(92)80909-Z
93. Bernhard S, Hug S, Stratmann AEP, Erber M, Vidoni L, Knapp CL, et al. Interleukin 8 elicits rapid physiological changes in neutrophils that are altered by inflammatory conditions. J Innate Immun. (2021) 13:225–41. doi: 10.1159/000514885
94. Donovan APA, Basson MA. The neuroanatomy of autism – a developmental perspective. J Anat. (2017) 230:4–15. doi: 10.1111/joa.12542
95. Brennan K, Zheng J. Interleukin 8. In: Enna SJ, Bylund DB, editors. xPharm: The Comprehensive Pharmacology Reference. New York, NY: Elsevier (2007). p. 1–4. doi: 10.1016/B978-008055232-3.61916-6
96. Bryn V, Aass HCD, Skjeldal OH, Isaksen J, Saugstad OD, Ormstad H. Cytokine profile in autism spectrum disorders in children. J Mol Neurosci. (2017) 61:1–7. doi: 10.1007/s12031-016-0847-z
97. Tanaka T, Narazaki M, Kishimoto T. Il-6 in inflammation, immunity, and disease. Cold Spring Harb Perspect Biol. (2014) 6:a016295. doi: 10.1101/cshperspect.a016295
98. Erta M, Quintana A, Hidalgo J. Interleukin-6, a major cytokine in the central nervous system. Int J Biol Sci. (2012) 8:1254–66. doi: 10.7150/ijbs.4679
99. Courchesne E, Mouton PR, Calhoun ME, Semendeferi K, Ahrens-Barbeau C, Hallet MJ, et al. Neuron number and size in prefrontal cortex of children with autism. JAMA. (2011) 306:2001. doi: 10.1001/jama.2011.1638
100. Nadeem A, Ahmad SF, Attia SM, Al-Ayadhi LY, Al-Harbi NO, Bakheet SA. Dysregulation in IL-6 receptors is associated with upregulated IL-17A related signaling in CD4+ T cells of children with autism. Prog Neuropsychopharmacol Biol Psychiatry. (2020) 97:109783. doi: 10.1016/j.pnpbp.2019.109783
101. Wei H, Zou H, Sheikh AM, Malik M, Dobkin C, Brown WT, et al. IL-6 is increased in the cerebellum of autistic brain and alters neural cell adhesion, migration and synaptic formation. J Neuroinflamm. (2011) 8:52. doi: 10.1186/1742-2094-8-52
102. Vargas DL, Nascimbene C, Krishnan C, Zimmerman AW, Pardo CA. Neuroglial activation and neuroinflammation in the brain of patients with autism. Ann Neurol. (2005) 57:67–81. doi: 10.1002/ana.20315
103. Gruol DL, Nelson E. Physiological and pathological roles of interleukin-6 in the central nervous system. Mol Neurobiol. (1997) 15:307–39. doi: 10.1007/BF02740665
104. Campbell IL, Abraham CR, Masliah E, Kemper P, Inglis JD, Oldstone MBA, et al. Neurologic disease induced in transgenic mice by cerebral overexpression of interleukin 6 (neurodegeneration/astrocytosis/angiogenesis/acute-phase response). Proc Natl Acad Sci USA. (1993) 90:10061–5. doi: 10.1073/pnas.90.21.10061
105. Stevens FL, Hurley RA, Taber KH. Anterior cingulate cortex: unique role in cognition and emotion. J Neuropsychiatry Clin Neurosci. (2011) 23:121–5. doi: 10.1176/jnp.23.2.jnp121
106. Reisinger S, Khan D, Kong E, Berger A, Pollak A, Pollak DD. The poly(I:C)-induced maternal immune activation model in preclinical neuropsychiatric drug discovery. Pharmacol Ther. (2015) 149:213–26. doi: 10.1016/j.pharmthera.2015.01.001
107. Choi GB, Yim YS, Wong H, Kim S, Kim H, Kim SV, et al. The maternal interleukin-17a pathway in mice promotes autism-like phenotypes in offspring. Science. (2016) 351:933–9. doi: 10.1126/science.aad0314
108. Malkova NV, Yu CZ, Hsiao EY, Moore MJ, Patterson PH. Maternal immune activation yields offspring displaying mouse versions of the three core symptoms of autism. Brain Behav Immun. (2012) 26:607–16. doi: 10.1016/j.bbi.2012.01.011
109. Zhang Y, Gao D, Kluetzman K, Mendoza A, Bolivar VJ, Reilly A, et al. The maternal autoimmune environment affects the social behavior of offspring. J Neuroimmunol. (2013) 258:51–60. doi: 10.1016/j.jneuroim.2013.02.019
110. Smith SEP, Li J, Garbett K, Mirnics K, Patterson PH. Maternal immune activation alters fetal brain development through interleukin-6. J Neurosci. (2007) 27:10695–702. doi: 10.1523/JNEUROSCI.2178-07.2007
111. Xu S, Cao X. Interleukin-17 and its expanding biological functions. Cell Mol Immunol. (2010) 7:164–74. doi: 10.1038/cmi.2010.21
112. Kuwabara T, Ishikawa F, Kondo M, Kakiuchi T. The role of IL-17 and related cytokines in inflammatory autoimmune diseases. Mediators Inflamm. (2017) 2017:1–11. doi: 10.1155/2017/3908061
113. Zhu S, Qian Y. IL-17/IL-17 receptor system in autoimmune disease: mechanisms and therapeutic potential. Clin Sci. (2012) 122:487–511. doi: 10.1042/CS20110496
114. Moaaz M, Youssry S, Elfatatry A, el Rahman MA. Th17/Treg cells imbalance and their related cytokines (IL-17, IL-10 and TGF-β) in children with autism spectrum disorder. J Neuroimmunol. (2019) 337:577071. doi: 10.1016/j.jneuroim.2019.577071
115. Kimura A, Kishimoto T. IL-6: regulator of Treg/Th17 balance. Eur J Immunol. (2010) 40:1830–5. doi: 10.1002/eji.201040391
116. Basheer S, Venkataswamy MM, Christopher R, van Amelsvoort T, Srinath S, Girimaji SC, et al. Immune aberrations in children with autism spectrum disorder: a case-control study from a tertiary care neuropsychiatric hospital in India. Psychoneuroendocrinology. (2018) 94:162–7. doi: 10.1016/j.psyneuen.2018.05.002
117. Nadeem A, Ahmad SF, Attia SM, Bakheet SA, Al-Harbi NO, Al-Ayadhi LY. Activation of IL-17 receptor leads to increased oxidative inflammation in peripheral monocytes of autistic children. Brain Behav Immun. (2018) 67:335–44. doi: 10.1016/j.bbi.2017.09.010
118. Nadeem A, Ahmad SF, Attia SM, Al-Ayadhi LY, Bakheet SA, Al-Harbi NO. Oxidative and inflammatory mediators are upregulated in neutrophils of autistic children: role of IL-17A receptor signaling. Prog Neuropsychopharmacol Biol Psychiatry. (2019) 90:204–11. doi: 10.1016/j.pnpbp.2018.12.002
119. Milovanovic J, Arsenijevic A, Stojanovic B, Kanjevac T, Arsenijevic D, Radosavljevic G, et al. Interleukin-17 in chronic inflammatory neurological diseases. Front Immunol. (2020) 11:947. doi: 10.3389/fimmu.2020.00947
120. Estes ML, McAllister AK. Immune mediators in the brain and peripheral tissues in autism spectrum disorder. Nat Rev Neurosci. (2015) 16:469–86. doi: 10.1038/nrn3978
121. Estes ML, McAllister AK. Maternal Th17 cells take a toll on baby’s brain. Science. (2016) 351:919–20. doi: 10.1126/science.aaf2850
122. Tesmer LA, Lundy SK, Sarkar S, Fox DA. Th17 cells in human disease. Immunol Rev. (2008) 223:87–113. doi: 10.1111/j.1600-065X.2008.00628.x
123. Krushel LA, Cunningham BA, Edelman GM, Crossin KL. NF-κB activity is induced by neural cell adhesion molecule binding to neurons and astrocytes. J Biol Chem. (1999) 274:2432–9. doi: 10.1074/jbc.274.4.2432
124. Al-Beltagi M. Autism medical comorbidities. World J Clin Pediatr. (2021) 10:15–28. doi: 10.5409/wjcp.v10.i3.15
125. Murdoch JR, Lloyd CM. Chronic inflammation and asthma. Mutat Res. (2010) 690:24–39. doi: 10.1016/j.mrfmmm.2009.09.005
126. Zheng Z, Zhang L, Zhu T, Huang J, Qu Y, Mu D. Association between asthma and autism spectrum disorder: a meta-analysis. PLoS One. (2016) 11:e0156662. doi: 10.1371/journal.pone.0156662
127. Lyall K, van de Water J, Ashwood P, Hertz-Picciotto I. Asthma and allergies in children with autism spectrum disorders: results from the CHARGE study. Autism Res. (2015) 8:567–74. doi: 10.1002/aur.1471
128. Kotey S, Ertel K, Whitcomb B. Co-occurrence of autism and asthma in a nationally-representative sample of children in the United States. J Autism Dev Disord. (2014) 44:3083–8. doi: 10.1007/s10803-014-2174-y
129. McElhanon BO, McCracken C, Karpen S, Sharp WG. Gastrointestinal symptoms in autism spectrum disorder: a meta-analysis. Pediatrics. (2014) 133:872–83. doi: 10.1542/peds.2013-3995
130. Cryan JF, Dinan TG. Mind-altering microorganisms: the impact of the gut microbiota on brain and behaviour. Nat Rev Neurosci. (2012) 13:701–12. doi: 10.1038/nrn3346
131. Lee M, Krishnamurthy J, Susi A, Sullivan C, Gorman GH, Hisle-Gorman E, et al. Association of autism spectrum disorders and inflammatory bowel disease. J Autism Dev Disord. (2018) 48:1523–9. doi: 10.1007/s10803-017-3409-5
132. Doshi-Velez F, Avillach P, Palmer N, Bousvaros A, Ge Y, Fox K, et al. Prevalence of inflammatory bowel disease among patients with autism spectrum disorders. Inflamm Bowel Dis. (2015) 21:2281–8. doi: 10.1097/MIB.0000000000000502
133. Li Q, Han Y, Dy ABC, Hagerman RJ. The gut microbiota and autism spectrum disorders. Front Cell Neurosci. (2017) 11:120. doi: 10.3389/fncel.2017.00120
134. Mörbe UM, Jørgensen PB, Fenton TM, von Burg N, Riis LB, Spencer J, et al. Human gut-associated lymphoid tissues (GALT); diversity, structure, and function. Mucosal Immunol. (2021) 14:793–802. doi: 10.1038/s41385-021-00389-4
135. Abdel Haq R, Schlachetzki JCM, Glass CK, Mazmanian SK. Microbiome–microglia connections via the gut–brain axis. J Exp Med. (2019) 216:41–59. doi: 10.1084/jem.20180794
136. Goehler LE, Gaykema RPA, Nguyen KT, Lee JE, Tilders FJH, Maier SF, et al. Interleukin-1β in immune cells of the abdominal vagus nerve: a link between the immune and nervous systems? J Neurosci. (1999) 19:2799–806. doi: 10.1523/JNEUROSCI.19-07-02799.1999
137. Wang L, Wang F-S, Gershwin ME. Human autoimmune diseases: a comprehensive update. J Intern Med. (2015) 278:369–95. doi: 10.1111/joim.12395
138. Zerbo O, Leong A, Barcellos L, Bernal P, Fireman B, Croen LA. Immune mediated conditions in autism spectrum disorders. Brain Behav Immun. (2015) 46:232–6. doi: 10.1016/j.bbi.2015.02.001
139. Spann MN, Timonen-Soivio L, Suominen A, Cheslack-Postava K, Mckeague IW, Sourander A, et al. Proband and familial autoimmune diseases are associated with proband diagnosis of autism spectrum disorders. J Am Acad Child Adolesc Psychiatry. (2019) 58:496–505. doi: 10.1016/j.jaac.2018.09.444
140. Singh VK, Warren RP, Odell JD, Warren WL, Cole P. Antibodies to myelin basic protein in children with autistic behavior. Brain Behav Immun. (1993) 7:97–103. doi: 10.1006/brbi.1993.1010
141. Plioplys A, Greaves A, Yoshida W. Anti-CNS antibodies in childhood neurologic diseases. Neuropediatrics. (1989) 20:93–102. doi: 10.1055/s-2008-1071273
142. Singh VK, Warren R, Averett R, Ghaziuddin M. Brief communication circulating autoantibodies to neuronal and glial filament proteins in autism. Pediatr Neurol. (1997) 17:88–90. doi: 10.1016/s0887-8994(97)00045-3
143. Simons M, Nave KA. Oligodendrocytes: myelination and axonal support. Cold Spring Harb Perspect Biol. (2016) 8:a020479. doi: 10.1101/cshperspect.a020479
144. Schmitz T, Chew L-J. Cytokines and myelination in the central nervous system. Sci World J. (2008) 8:1119–47. doi: 10.1100/tsw.2008.140
145. Fields RD. Change in the brain’s white matter. Science. (2010) 330:768–9. doi: 10.1126/science.1199139
146. Galvez-Contreras AY, Zarate-Lopez D, Torres-Chavez AL, Gonzalez-Perez O. Role of oligodendrocytes and myelin in the pathophysiology of autism spectrum disorder. Brain Sci. (2020) 10:1–17. doi: 10.3390/brainsci10120951
147. Kamata A, Muramatsu K, Sawaura N, Makioka N, Ogata T, Kuwashima M, et al. Demyelinating neuropathy in a 6-year-old girl with autism spectrum disorder. Pediatr Int. (2017) 59:951–4. doi: 10.1111/ped.13331
148. Croen LA, Zerbo O, Qian Y, Massolo ML, Rich S, Sidney S, et al. The health status of adults on the autism spectrum. Autism. (2015) 19:814–23. doi: 10.1177/1362361315577517
149. Nadeem MS, Hosawi S, Alshehri S, Ghoneim MM, Imam SS, Murtaza BN, et al. Symptomatic, genetic, and mechanistic overlaps between autism and Alzheimer’s disease. Biomolecules. (2021) 11:1635. doi: 10.3390/biom11111635
150. Chow VW, Mattson MP, Wong PC, Gleichmann M. An overview of APP processing enzymes and products. Neuromol Med. (2010) 12:1–12. doi: 10.1007/s12017-009-8104-z
151. Sokol DK, Chen D, Farlow MR, Dunn DW, Maloney B, Zimmer JA, et al. High levels of Alzheimer beta-amyloid precursor protein (APP) in children with severely autistic behavior and aggression. J Child Neurol. (2006) 21:444–9. doi: 10.1177/08830738060210062201
152. Wegiel J, Frackowiak J, Mazur-Kolecka B, Schanen NC, Cook EH, Sigman M, et al. Abnormal intracellular accumulation and extracellular Aβ deposition in idiopathic and Dup15q11.2-q13 autism spectrum disorders. PLoS One. (2012) 7:e35414. doi: 10.1371/journal.pone.0035414
153. Fu Y, Hsiao J-HT, Paxinos G, Halliday GM, Kim WS. ABCA7 mediates phagocytic clearance of amyloid-β in the brain. J Alzheimers Dis. (2016) 54:569–84. doi: 10.3233/JAD-160456
154. Denver P, English A, McClean PL. Inflammation, insulin signaling and cognitive function in aged APP/PS1 mice. Brain Behav Immun. (2018) 70:423–34. doi: 10.1016/j.bbi.2018.03.032
155. Hong S, Beja-Glasser VF, Nfonoyim BM, Frouin A, Li S, Ramakrishnan S, et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science. (2016) 352:712–6. doi: 10.1126/science.aad8373
156. Zhang M, Yu Q, Tang W, Wu Y, Lv JJ, Sun L, et al. Epithelial exosomal contactin-1 promotes monocyte-derived dendritic cell–dominant T-cell responses in asthma. J Allergy Clin Immunol. (2021) 148:1545–58. doi: 10.1016/j.jaci.2021.04.025
157. Mathey EK, Park SB, Hughes RAC, Pollard JD, Armati PJ, Barnett MH, et al. Chronic inflammatory demyelinating polyradiculoneuropathy: from pathology to phenotype. J Neurol Neurosurg Psychiatry. (2015) 86:973–85. doi: 10.1136/jnnp-2014-309697
158. Rodríguez Y, Vatti N, Ramírez-Santana C, Chang C, Mancera-Páez O, Gershwin ME, et al. Chronic inflammatory demyelinating polyneuropathy as an autoimmune disease. J Autoimmun. (2019) 102:8–37. doi: 10.1016/j.jaut.2019.04.021
159. Fehmi J, Scherer SS, Willison HJ, Rinaldi S. Nodes, paranodes and neuropathies. J Neurol Neurosurg Psychiatry. (2018) 89:61–71. doi: 10.1136/jnnp-2016-315480
160. Kouton L, Boucraut J, Devaux J, Rajabally YA, Adams D, Antoine JC, et al. Electrophysiological features of chronic inflammatory demyelinating polyradiculoneuropathy associated with IgG4 antibodies targeting neurofascin 155 or contactin 1 glycoproteins. Clin Neurophysiol. (2020) 131:921–7. doi: 10.1016/j.clinph.2020.01.013
161. Delmont E, Manso C, Querol L, Cortese A, Berardinelli A, Lozza A, et al. Autoantibodies to nodal isoforms of neurofascin in chronic inflammatory demyelinating polyneuropathy. Brain. (2017) 140:1851–8. doi: 10.1093/brain/awx124
162. Delmont E, Brodovitch A, Kouton L, Allou T, Beltran S, Brisset M, et al. Antibodies against the node of Ranvier: a real-life evaluation of incidence, clinical features and response to treatment based on a prospective analysis of 1500 sera. J Neurol. (2020) 267:3664–72. doi: 10.1007/s00415-020-10041-z
163. Cortese A, Lombardi R, Briani C, Callegari I, Benedetti L, Manganelli F, et al. Antibodies to neurofascin, contactin-1, and contactin-associated protein 1 in CIDP: clinical relevance of IgG isotype. Neurology. (2020) 7:e639. doi: 10.1212/NXI.0000000000000639
164. Guo X, Tang L, Huang Q, Tang X. A systematic review and meta-analysis of autoantibodies for diagnosis and prognosis in patients with chronic inflammatory demyelinating polyradiculoneuropathy. Front Neurosci. (2021) 15:637336. doi: 10.3389/fnins.2021.637336
165. Mathey EK, Garg N, Park SB, Nguyen T, Baker S, Yuki N, et al. Autoantibody responses to nodal and paranodal antigens in chronic inflammatory neuropathies. J Neuroimmunol. (2017) 309:41–6. doi: 10.1016/j.jneuroim.2017.05.002
166. Querol L, Nogales-Gadea G, Rojas-Garcia R, Diaz-Manera J, Pardo J, Ortega-Moreno A, et al. Neurofascin IgG4 antibodies in CIDP associate with disabling tremor and poor response to IVIg. Neurology. (2014) 82:879–86. doi: 10.1212/WNL.0000000000000205
167. Pascual-Goñi E, Fehmi J, Lleixà C, Martín-Aguilar L, Devaux J, Höftberger R, et al. Antibodies to the Caspr1/contactin-1 complex in chronic inflammatory demyelinating polyradiculoneuropathy. Brain. (2021) 144:1183–96. doi: 10.1093/brain/awab014
168. Lin HP, Ho KWD, Chuquilin M. Presence of both anti-contactin 1 and anti-neurofascin 140 antibodies in a case of chronic inflammatory demyelinating polyneuropathy. eNeurologicalSci. (2018) 13:38–9. doi: 10.1016/j.ensci.2018.11.016
169. Manso C, Querol L, Mekaouche M, Illa I, Devaux JJ. Contactin-1 IgG4 antibodies cause paranode dismantling and conduction defects. Brain. (2016) 139:1700–12. doi: 10.1093/brain/aww062
170. Koike H, Kadoya M, Kaida KI, Ikeda S, Kawagashira Y, Iijima M, et al. Paranodal dissection in chronic inflammatory demyelinating polyneuropathy with anti-neurofascin-155 and anti-contactin-1 antibodies. J Neurol Neurosurg Psychiatry. (2017) 88:465–73. doi: 10.1136/jnnp-2016-314895
171. Carrera-García L, Natera-De Benito D, Lleixà C, Ortez C, Colomer J, Nascimento A, et al. Chronic inflammatory demyelinating polyneuropathy associated with contactin-1 antibodies in a child. Neurology. (2019) 6:e602. doi: 10.1212/NXI.0000000000000602
172. Hashimoto Y, Ogata H, Yamasaki R, Sasaguri T, Ko S, Yamashita K, et al. Chronic inflammatory demyelinating polyneuropathy with concurrent membranous nephropathy: an anti-paranode and podocyte protein antibody study and literature survey. Front Neurol. (2018) 9:997. doi: 10.3389/fneur.2018.00997
173. Labasque M, Hivert B, Nogales-Gadea G, Querol L, Illa I, Faivre-Sarrailh C. Specific contactin N-glycans are implicated in neurofascin binding and autoimmune targeting in peripheral neuropathies. J Biol Chem. (2014) 289:7907–18. doi: 10.1074/jbc.M113.528489
174. Querol L, Nogales-Gadea G, Rojas-Garcia R, Martinez-Hernandez E, Diaz-Manera J, Suárez-Calvet X, et al. Antibodies to contactin-1 in chronic inflammatory demyelinating polyneuropathy. Ann Neurol. (2013) 73:370–80. doi: 10.1002/ana.23794
175. Manso C, Querol L, Lleixà C, Poncelet M, Mekaouche M, Vallat J-M, et al. Anti–neurofascin-155 IgG4 antibodies prevent paranodal complex formation in vivo. J Clin Invest. (2019) 129:2222–36. doi: 10.1172/JCI124694
176. Miura Y, Devaux JJ, Fukami Y, Manso C, Belghazi M, Wong AHY, et al. Contactin 1 IgG4 associates to chronic inflammatory demyelinating polyneuropathy with sensory ataxia. Brain. (2015) 138:1484–91. doi: 10.1093/brain/awv054
177. Grüner J, Stengel H, Werner C, Appeltshauser L, Sommer C, Villmann C, et al. Anti-contactin-1 antibodies affect surface expression and sodium currents in dorsal root ganglia. Neurology. (2021) 8:e1056. doi: 10.1212/NXI.0000000000001056
178. Doppler K, Appeltshauser L, Wilhelmi K, Villmann C, Dib-Hajj SD, Waxman SG, et al. Destruction of paranodal architecture in inflammatory neuropathy with anti-contactin-1 autoantibodies. J Neurol Neurosurg Psychiatry. (2015) 86:720–8. doi: 10.1136/jnnp-2014-309916
179. Rentzos M, Angeli AV, Rombos A, Kyrozis A, Nikolaou C, Zouvelou V, et al. Proinflammatory cytokines in serum and cerebrospinal fluid of CIDP patients. Neurol Res. (2012) 34:842–6. doi: 10.1179/1743132812Y.0000000074
180. Boronat A, Sepúlveda M, Llufriu S, Sabater L, Blanco Y, Gabilondo I, et al. Analysis of antibodies to surface epitopes of contactin-2 in multiple sclerosis. J Neuroimmunol. (2012) 244:103–6. doi: 10.1016/j.jneuroim.2011.12.023
181. Derfuss T, Parikh K, Velhin S, Braun M, Mathey E, Krumbholz M, et al. Contactin-2/TAG-1-directed autoimmunity is identified in multiple sclerosis patients and mediates gray matter pathology in animals. Proc Natl Acad Sic USA. (2009) 106:8302–7. doi: 10.1073/pnas.0901496106
182. Mike A, Glanz BI, Hildenbrand P, Meier D, Bolden K, Liguori M, et al. Identification and clinical impact of multiple sclerosis cortical lesions as assessed by routine 3T MR imaging. Am J Neuroradiol. (2011) 32:515–21. doi: 10.3174/ajnr.A2340
183. Sudduth TL, Schmitt FA, Nelson PT, Wilcock DM. Neuroinflammatory phenotype in early Alzheimer’s disease. Neurobiol Aging. (2013) 34:1051–9. doi: 10.1016/j.neurobiolaging.2012.09.012
184. Theoharides TC, Tsilioni I, Patel AB, Doyle R. Atopic diseases and inflammation of the brain in the pathogenesis of autism spectrum disorders. Transl Psychiatry. (2016) 6:e844. doi: 10.1038/tp.2016.77
185. Ables JL, Breunig JJ, Eisch AJ, Rakic P. Not(ch) just development: notch signalling in the adult brain. Nat Rev Neurosci. (2011) 12:269–83. doi: 10.1038/nrn3024
186. Bizzoca A, Corsi P, Polizzi A, Pinto MF, Xenaki D, Furley AJW, et al. F3/contactin acts as a modulator of neurogenesis during cerebral cortex development. Dev Biol. (2012) 365:133–51. doi: 10.1016/j.ydbio.2012.02.011
187. Ma QH, Futagawa T, Yang WL, Jiang XD, Zeng L, Takeda Y, et al. A TAG1-APP signalling pathway through Fe65 negatively modulates neurogenesis. Nat Cell Biol. (2008) 10:283–94. doi: 10.1038/ncb1690
188. Gómez-Pinedo U, Galán L, Matías-Guiu JA, Pytel V, Moreno T, Guerrero-Sola A, et al. Notch signalling in the hippocampus of patients with motor neuron disease. Front Neurosci. (2019) 13:302. doi: 10.3389/fnins.2019.00302
189. Bamford RA, Widagdo J, Takamura N, Eve M, Anggono V, Oguro-Ando A. The interaction between contactin and amyloid precursor protein and its role in Alzheimer’s disease. Neuroscience. (2020) 424:184–202. doi: 10.1016/j.neuroscience.2019.10.006
190. Chatterjee M, del Campo M, Morrema THJ, de Waal M, van der Flier WM, Hoozemans JJM, et al. Contactin-2, a synaptic and axonal protein, is reduced in cerebrospinal fluid and brain tissue in Alzheimer’s disease. Alzheimers Res Ther. (2018) 10:52. doi: 10.1186/s13195-018-0383-x
191. Zoupi L, Markoullis K, Kleopa KA, Karagogeos D. Alterations of juxtaparanodal domains in two rodent models of CNS demyelination. Glia. (2013) 61:1236–49. doi: 10.1002/glia.22511
192. Zuko A, Bouyain S, van der Zwaag B, Burbach JPH. Contactins: structural aspects in relation to developmental functions in brain disease. In: Donev R, editor. Advances in Protein Chemistry and Structural Biology. Cambridge, MA: Academic Press (2011). p. 143–80. doi: 10.1016/B978-0-12-386483-3.00001-X
193. Dabell MP, Rosenfeld JA, Bader P, Escobar LF, El-Khechen D, Vallee SE, et al. Investigation of NRXN1 deletions: clinical and molecular characterization. Am J Med Genet Part A. (2013) 161:717–31. doi: 10.1002/ajmg.a.35780
194. Zahir FR, Baross A, Delaney AD, Eydoux P, Fernandes ND, Pugh T, et al. A patient with vertebral, cognitive and behavioural abnormalities and a de novo deletion of NRXN1α. J Med Genet. (2008) 45:239–43. doi: 10.1136/jmg.2007.054437
195. Kaur P, Karolina DS, Sepramaniam S, Armugam A, Jeyaseelan K. Expression profiling of RNA transcripts during neuronal maturation and ischemic injury. PLoS One. (2014) 9:e103525. doi: 10.1371/journal.pone.0103525
196. Földy C, Darmanis S, Aoto J, Malenka RC, Quake SR, Südhof TC. Single-cell RNAseq reveals cell adhesion molecule profiles in electrophysiologically defined neurons. Proc Natl Acad Sci USA. (2016) 113:E5222–31. doi: 10.1073/pnas.1610155113
197. van Dam S, Võsa U, van der Graaf A, Franke L, de Magalhães JP. Gene co-expression analysis for functional classification and gene-disease predictions. Brief Bioinformatics. (2018) 19:575–92. doi: 10.1093/bib/bbw139
198. Alkhathami AG, Verma AK, Alfaifi M, Kumar L, Alshahrani MY, Hakami AR, et al. Role of miRNA-495 and NRXN-1 and CNTN-1 mRNA expression and its prognostic importance in breast cancer patients. J Oncol. (2021) 2021:1–10. doi: 10.1155/2021/9657071
199. Naito Y, Tanabe Y, Lee AK, Hamel E, Takahashi H. Amyloid-β oligomers interact with neurexin and diminish neurexin-mediated excitatory presynaptic organization. Sci Rep. (2017) 7:42548. doi: 10.1038/srep42548
200. Viola KL, Velasco PT, Klein WL. Why Alzheimer’s is a disease of memory: the attack on synapses by Aß oligomers (ADDLs). J Nutr Health Aging. (2008) 12:S51–7. doi: 10.1007/BF02982587
201. Zheng JJ, Li WX, Liu JQ, Guo YC, Wang Q, Li GH, et al. Low expression of aging-related NRXN3 is associated with Alzheimer disease: a systematic review and meta-analysis. Medicine. (2018) 97:e11343. doi: 10.1097/MD.0000000000011343
202. Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. (1991) 82:239–59. doi: 10.1007/BF00308809
203. Kasem E, Kurihara T, Tabuchi K. Neurexins and neuropsychiatric disorders. Neurosci Res. (2018) 127:53–60. doi: 10.1016/j.neures.2017.10.012
204. Hishimoto A, Pletnikova O, Lang DL, Troncoso JC, Egan JM, Liu QR. Neurexin 3 transmembrane and soluble isoform expression and splicing haplotype are associated with neuron inflammasome and Alzheimer’s disease. Alzheimers Res Ther. (2019) 11:28. doi: 10.1186/s13195-019-0475-2
205. Kelley N, Jeltema D, Duan Y, He Y. The NLRP3 inflammasome: an overview of mechanisms of activation and regulation. Int J Mol Sci. (2019) 20:3328. doi: 10.3390/ijms20133328
206. Edwards DR, Handsley MM, Pennington CJ. The ADAM metalloproteinases. Mol Aspects Med. (2009) 29:258–89. doi: 10.1016/j.mam.2008.08.001
207. Zheng Y, Verhoeff TA, Pardo PP, Garssen J, Kraneveld AD. The gut-brain axis in autism spectrum disorder: a focus on the metalloproteases adam10 and adam17. Int J Mol Sci. (2021) 22:1–30. doi: 10.3390/ijms22010118
208. Moss ML, Minond D. Recent advances in ADAM17 research: a promising target for cancer and inflammation. Mediators Inflamm. (2017) 2017:1–21. doi: 10.1155/2017/9673537
209. Ray B, Sokol DK, Maloney B, Lahiri DK. Finding novel distinctions between the sAPPα-mediated anabolic biochemical pathways in autism spectrum disorder and fragile X syndrome plasma and brain tissue. Sci Rep. (2016) 6:26052. doi: 10.1038/srep26052
210. Leembruggen AJL, Balasuriya GK, Zhang J, Schokman S, Swiderski K, Bornstein JC, et al. Colonic dilation and altered ex vivo gastrointestinal motility in the neuroligin-3 knockout mouse. Autism Res. (2020) 13:691–701. doi: 10.1002/aur.2109
211. Hosie S, Ellis M, Swaminathan M, Ramalhosa F, Seger GO, Balasuriya GK, et al. Gastrointestinal dysfunction in patients and mice expressing the autism-associated R451C mutation in neuroligin-3. Autism Res. (2019) 12:1043–56. doi: 10.1002/aur.2127
212. Sharna SS, Balasuriya GK, Hosie S, Nithianantharajah J, Franks AE, Hill-Yardin EL. Altered caecal neuroimmune interactions in the neuroligin-3R451C mouse model of autism. Front Cell Neurosci. (2020) 14:85. doi: 10.3389/fncel.2020.00085
213. Walker SJ, Fortunato J, Gonzalez LG, Krigsman A. Identification of unique gene expression profile in children with regressive autism spectrum disorder (ASD) and ileocolitis. PLoS One. (2013) 8:e58058. doi: 10.1371/journal.pone.0058058
214. Psachoulia K, Chamberlain KA, Heo D, Davis SE, Paskus JD, Nanescu SE, et al. IL4I1 augments CNS remyelination and axonal protection by modulating T cell driven inflammation. Brain. (2016) 139:3121–36. doi: 10.1093/brain/aww254
215. Ellery JM, Nicholls PJ. Alternate signalling pathways from the interleukin-2 receptor. Cytokine Growth Factor Rev. (2002) 13:27–40. doi: 10.1016/s1359-6101(01)00023-5
216. Oumesmar BN, Vignais L, Duhamel-Clerin E, Avellana-Adalid V, Rougonl G, Baron-Van Evercooren A. Expression of the highly polysialylated neural cell adhesion molecule during postnatal myelination and following chemically induced demyelination of the adult mouse spinal cord. Eur J Neurosci. (1995) 7:480–91. doi: 10.1111/j.1460-9568.1995.tb00344.x
217. Cameron RS, Rakic P. Glial cell lineage in the cerebral cortex: a review and synthesis. Glia. (1991) 4:124–37. doi: 10.1002/glia.440040204
218. Franklin RJM, Goldman SA. Glia disease and repair—remyelination. Cold Spring Harb Perspect Biol. (2015) 7:1–28. doi: 10.1101/cshperspect.a020594
219. Jha MK, Jeon S, Suk K. Glia as a link between neuroinflammation and neuropathic pain. Immune Netw. (2012) 12:41. doi: 10.4110/in.2012.12.2.41
220. Depino AM. Peripheral and central inflammation in autism spectrum disorders. Mol Cell Neurosci. (2013) 53:69–76. doi: 10.1016/j.mcn.2012.10.003
221. Harry GJ, Kraft AD. Microglia in the developing brain: a potential target with lifetime effects. Neurotoxicology. (2012) 33:191–206. doi: 10.1016/j.neuro.2012.01.012
222. Li K, Li J, Zheng J, Qin S. Reactive astrocytes in neurodegenerative diseases. Aging Dis. (2019) 10:664–75. doi: 10.14336/AD.2018.0720
223. Franco R, Fernández-Suárez D. Alternatively activated microglia and macrophages in the central nervous system. Prog Neurobiol. (2015) 131:65–86. doi: 10.1016/j.pneurobio.2015.05.003
224. Kinney JW, Bemiller SM, Murtishaw AS, Leisgang AM, Salazar AM, Lamb BT. Inflammation as a central mechanism in Alzheimer’s disease. Alzheimers Dement (N Y). (2018) 4:575–90. doi: 10.1016/j.trci.2018.06.014
225. Barateiro A, Brites D, Fernandes A. Oligodendrocyte development and myelination in neurodevelopment: molecular mechanisms in health and disease. Curr Pharm Design. (2016) 22:656–79. doi: 10.2174/1381612822666151204000636
226. Morgan JT, Chana G, Pardo CA, Achim C, Semendeferi K, Buckwalter J, et al. Microglial activation and increased microglial density observed in the dorsolateral prefrontal cortex in autism. Biol Psychiatry. (2010) 68:368–76. doi: 10.1016/j.biopsych.2010.05.024
227. Anderson MA, Ao Y, Sofroniew MV. Heterogeneity of reactive astrocytes. Neurosci Lett. (2014) 565:23–9. doi: 10.1016/j.neulet.2013.12.030
228. Ghiani CA, Mattan NS, Nobuta H, Malvar JS, Boles J, Ross MG, et al. Early effects of lipopolysaccharideinduced inflammation on foetal brain development in rat. ASN Neuro. (2011) 3:233–45. doi: 10.1042/AN20110027
229. Onore C, Careaga M, Ashwood P. The role of immune dysfunction in the pathophysiology of autism. Brain Behav Immun. (2012) 26:383–92. doi: 10.1016/j.bbi.2011.08.007
230. Stellwagen D, Malenka RC. Synaptic scaling mediated by glial TNF-α. Nature. (2006) 440:1054–9. doi: 10.1038/nature04671
231. Hansel C. Deregulation of synaptic plasticity in autism. Neurosci Lett. (2019) 688:58–61. doi: 10.1016/j.neulet.2018.02.003
232. Santos E, Noggle CA. Synaptic pruning. In: Sam G, Naglieri JA, editors. Encyclopedia of Child Behavior and Development. Boston, MA: Springer US (2011). p. 1464–5. doi: 10.1007/978-0-387-79061-9_2856
233. Tang G, Gudsnuk K, Kuo SH, Cotrina ML, Rosoklija G, Sosunov A, et al. Loss of mTOR-dependent macroautophagy causes autistic-like synaptic pruning deficits. Neuron. (2014) 83:1131–43. doi: 10.1016/j.neuron.2014.07.040
234. Thomas MSC, Davis R, Karmiloff-Smith A, Knowland VCP, Charman T. The over-pruning hypothesis of autism. Dev Sci. (2016) 19:284–305. doi: 10.1111/desc.12303
235. Ginhoux F, Lim S, Hoeffel G, Low D, Huber T. Origin and differentiation of microglia. Front Cell Neurosci. (2013) 7:45. doi: 10.3389/fncel.2013.00045
236. Fu SY, Gordon T. The cellular and molecular basis of peripheral nerve regeneration. Mol Neurobiol. (1997) 14:67–116. doi: 10.1007/BF02740621
237. Krushel LA, Tai M, Cunningham BA, Edelman GM, Crossin KL. Neural cell adhesion molecule (N-CAM) domains and intracellular signaling pathways involved in the inhibition of astrocyte proliferation. Proc Natl Acad Sci USA. (1998) 95:2592–6. doi: 10.1073/pnas.95.5.2592
238. Yoshihara Y. Immunoglobulin superfamily cell adhesion molecules. In: Binder MD, Hirokawa N, Windhorst U, editors. Encyclopedia of Neuroscience. Berlin: Springer (2009). p. 1923–6. doi: 10.1007/978-3-540-29678-2_2375
239. Sporns O, Edelman GM, Crossin KL. The neural cell adhesion molecule (N-CAM) inhibits proliferation in primary cultures of rat astrocytes (antisense/glia/regeneration/contact inhibition) contributed by. Proc Natl Acad Sci USA. (1995) 92:542–6. doi: 10.1073/pnas.92.2.542
240. Zhang J, Wang A, Li Y, Lu X, Wang F, Fang F. Association of NCAM1 polymorphisms with autism and parental age at conception in a Chinese han population. Genet Test Mol Biomark. (2014) 18:690–4. doi: 10.1089/gtmb.2014.0055
241. Kazlauskas N, Campolongo M, Lucchina L, Zappala C, Depino AM. Postnatal behavioral and inflammatory alterations in female pups prenatally exposed to valproic acid. Psychoneuroendocrinology. (2016) 72:11–21. doi: 10.1016/j.psyneuen.2016.06.001
242. Czepiel M, Leicher L, Becker K, Boddeke E, Copray S. Overexpression of polysialylated neural cell adhesion molecule improves the migration capacity of induced pluripotent stem cell-derived oligodendrocyte precursors. Stem Cells Transl Med. (2014) 3:1100–9. doi: 10.5966/sctm.2014-0041
243. Coman I, Barbin G, Charles P, Zalc B, Lubetzki C. Axonal signals in central nervous system myelination, demyelination and remyelination. J Neurol Sci. (2005) 233:67–71. doi: 10.1016/j.jns.2005.03.029
244. Phan BDN, Bohlen JF, Davis BA, Ye Z, Chen HY, Mayfield B, et al. A myelin-related transcriptomic profile is shared by Pitt–Hopkins syndrome models and human autism spectrum disorder. Nat Neurosci. (2020) 23:375–85. doi: 10.1038/s41593-019-0578-x
245. Wu ZQ, Li D, Huang Y, Chen XP, Huang W, Liu CF, et al. Caspr controls the temporal specification of neural progenitor cells through notch signaling in the developing mouse cerebral cortex. Cereb Cortex. (2017) 27:1369–85. doi: 10.1093/cercor/bhv318
246. Ge WP, Miyawaki A, Gage FH, Jan YN, Jan LY. Local generation of glia is a major astrocyte source in postnatal cortex. Nature. (2012) 484:376–80. doi: 10.1038/nature10959
247. Shimada IS, Borders A, Aronshtam A, Spees JL. Proliferating reactive astrocytes are regulated by notch-1 in the peri-infarct area after stroke. Stroke. (2011) 42:3231–7. doi: 10.1161/STROKEAHA.111.623280
248. Cope EC, Briones BA, Brockett AT, Martinez S, Vigneron PA, Opendak M, et al. Immature neurons and radial glia, but not astrocytes or microglia, are altered in adult Cntnap2 and Shank3 mice, models of autism. eNeuro. (2016) 3:ENEURO.0196-16.2016. doi: 10.1523/ENEURO.0196-16.2016
249. Noriuchi M, Kikuchi Y, Yoshiura T, Kira R, Shigeto H, Hara T, et al. Altered white matter fractional anisotropy and social impairment in children with autism spectrum disorder. Brain Res. (2010) 1362:141–9. doi: 10.1016/j.brainres.2010.09.051
250. Lamprianou S, Chatzopoulou E, Thomas JL, Bouyaing S, Harroch S. A complex between contactin-1 and the protein tyrosine phosphatase PTPRZ controls the development of oligodendrocyte precursor cells. Proc Natl Acad Sci USA. (2011) 108:17498–503. doi: 10.1073/pnas.1108774108
251. Revest JM. The interaction between F3 immunoglobulin domains and protein tyrosine phosphatases β triggers bidirectional signalling between neurons and glial cells. Eur J Neurosci. (1999) 11:1134–47. doi: 10.1046/j.1460-9568.1999.00521.x
252. Peles E, Nativ M, Campbell PL, Sakurai T, Martinez R, Lev S, et al. The carbonic anhydrase domain of receptor tyrosine phosphatase is a functional ligand for the axonal cell recognition molecule contactin. Cell. (1995) 82:251–60. doi: 10.1016/0092-8674(95)90312-7
253. Lee JH, Espinera AR, Chen D, Choi KE, Caslin AY, Won S, et al. Neonatal inflammatory pain and systemic inflammatory responses as possible environmental factors in the development of autism spectrum disorder of juvenile rats. J Neuroinflamm. (2016) 13:109. doi: 10.1186/s12974-016-0575-x
254. Manzoor Z, Koh YS. Mitogen-activated protein kinases in inflammation. J Bacteriol Virol. (2012) 42:189–95. doi: 10.4167/jbv.2012.42.3.189
255. Ashwood P, van de Water J. Is autism an autoimmune disease? Autoimmun Rev. (2004) 3:557–62. doi: 10.1016/j.autrev.2004.07.036
256. Hsiao EY, McBride SW, Chow J, Mazmanian SK, Patterson PH. Modeling an autism risk factor in mice leads to permanent immune dysregulation. Proc Natl Acad Sci USA. (2012) 109:12776–81. doi: 10.1073/pnas.1202556109
257. Fagan K, Crider A, Ahmed AO, Pillai A. Complement C3 expression is decreased in autism spectrum disorder subjects and contributes to behavioral deficits in rodents. Mol Neuropsychiatry. (2017) 3:19–27. doi: 10.1159/000465523
Keywords: autism spectrum disorder, cell adhesion molecules, neuroinflammation, inflammatory cascade, neuroinflammatory signaling, glial cells
Citation: Eve M, Gandawijaya J, Yang L and Oguro-Ando A (2022) Neuronal Cell Adhesion Molecules May Mediate Neuroinflammation in Autism Spectrum Disorder. Front. Psychiatry 13:842755. doi: 10.3389/fpsyt.2022.842755
Received: 24 December 2021; Accepted: 15 February 2022;
Published: 15 April 2022.
Edited by:
Hideo Matsuzaki, University of Fukui, JapanCopyright © 2022 Eve, Gandawijaya, Yang and Oguro-Ando. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Asami Oguro-Ando, A.Oguro-Ando@exeter.ac.uk