 Yu-Ying Mei
Yu-Ying Mei Dong Chuan Wu
Dong Chuan Wu Ning Zhou
Ning Zhou- 1Translational Medicine Research Center, China Medical University Hospital, Taichung, Taiwan
- 2Graduate Institute of Biomedical Sciences, China Medical University, Taichung, Taiwan
According to the glutamate hypothesis of schizophrenia, the abnormality of glutamate transmission induced by hypofunction of NMDA receptors (NMDARs) is causally associated with the positive and negative symptoms of schizophrenia. However, the underlying mechanisms responsible for the changes in glutamate transmission in schizophrenia are not fully understood. Astrocytes, the major regulatory glia in the brain, modulate not only glutamate metabolism but also glutamate transmission. Here we review the recent progress in understanding the role of astrocytes in schizophrenia. We focus on the astrocytic mechanisms of (i) glutamate synthesis via the glutamate-glutamine cycle, (ii) glutamate clearance by excitatory amino acid transporters (EAATs), (iii) D-serine release to activate NMDARs, and (iv) glutamatergic target engagement biomarkers. Abnormality in these processes is highly correlated with schizophrenia phenotypes. These findings will shed light upon further investigation of pathogenesis as well as improvement of biomarkers and therapies for schizophrenia.
Introduction
Dysregulation of glutamatergic neurotransmission is critically implicated in the pathophysiology of psychotic disorders. In the central nervous system (CNS), glutamate is the principal excitatory neurotransmitter and mediates the fast excitatory transmission by activation of ionotropic glutamate receptors, including AMPA, kainate, and NMDA receptors (NMDARs). Astrocytes affect glutamatergic transmission in several important ways, including glutamate biosynthesis, glutamate-glutamine cycle, glutamate uptake, and releasing glutamate and D-serine as gliotransmitters. This review focuses on recent advances in astrocyte-mediated dysregulation of glutamate transmission in schizophrenia.
The Glutamate Hypothesis of Schizophrenia
The “glutamate hypothesis of schizophrenia” proposes that schizophrenia symptoms and cognitive impairment are due to hypofunction of NMDARs and excessive glutamate release, especially in brain areas including prefrontal cortex and hippocampus (1). This theory was initiated from the observation that NMDAR antagonists, like phencyclidine and ketamine, could evoke negative symptoms and cognitive dysfunction resembling schizophrenic phenotypes in healthy subjects (2). In schizophrenic subjects, subanesthetic doses of ketamine could exacerbate psychotic and cognitive symptoms (3, 4). Interestingly, in encephalitis patients, the first-episode psychosis is associated with the presence of anti-NMDAR antibodies that could cause a reduction of surface expression of NMDARs (5, 6). In animal studies, suppression of NMDAR function by pharmacological or genetic approaches leads to schizophrenia-like behaviors (7, 8). Schizophrenia is also associated with dysregulation of some genes and/or proteins involved in glutamate transmission (9). Genome-wide association studies have reported that the NMDAR subunits encoding genes, GRIN2A and GRIN2B, are schizophrenia-related genes (9, 10). Two de novo mutations in GRIN2A were found in sporadic schizophrenia patients (11). The single nucleotide polymorphisms of genes related to D-serine synthesis and metabolism, such as genes encoding serine racemase (SR), D-Amino Acid Oxidase (DAAO), and the DAAO activator G72, are also associated with schizophrenia (12).
The hypofunction of NMDAR causes excessive glutamate release, hyper-glutamatergic functions, and hypermetabolism (13). However, it remains largely unknown how NMDAR hypofunction leads to elevated glutamate levels and over-activation of non-NMDA glutamate receptors. One possibility is that interneurons are more sensitive to NMDAR blockade than principal glutamatergic neurons, and interneuron inhibition leads to an overall effect of disinhibition and hyperexcitation of glutamatergic output (Figure 1) (1, 14, 15). The increased glutamate levels have been documented by brain imaging studies and the source of excessive glutamate is mainly from presynaptic release (16). This notion was strengthened by the effect of drugs that specifically reduce presynaptic glutamate release, like the mGluR2/3 agonists, which significantly reverse ketamine-evoked glutamate elevation and cerebral blood volume increase in the hippocampus (13). High extracellular glutamate levels lead to hypermetabolism, structural disorganization, and eventually hippocampal volume reduction, and these dysfunctions are correlated with disease progression from prodromal symptoms to psychosis (17). Nevertheless, many mechanisms that cause an overall increase in glutamate tone could contribute synergistically to schizophrenia pathogenesis.
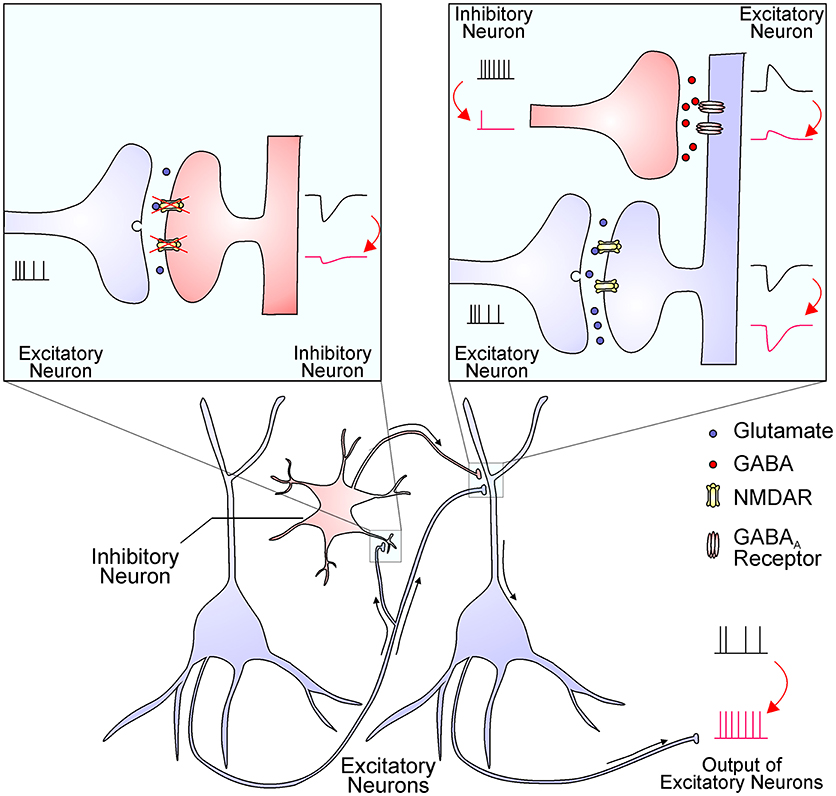
Figure 1. The NMDAR hypofunction hypothesis proposes that NMDARs in inhibitory interneurons are preferentially diminished in schizophrenia. The reduced NMDAR function in interneurons (the left inset) results in decreased excitatory postsynaptic currents, which in turn decrease interneuron output (the right inset). The reduced GABA release from interneuron terminals leads to smaller inhibitory postsynaptic responses and thereby a disinhibition of postsynaptic excitatory neurons (the right inset). This finally leads to an enhanced output of excitatory neurons, resulting in excessive glutamate release and increased activation of non-NMDA glutamate receptors. This process might be enhanced by NMDAR antagonists and ameliorated by mGluR2/3 agonists.
Synaptic and Extrasynaptic NMDARs
NMDARs are distributed at both synaptic and extrasynaptic sites with different subunit compositions. Their activation requires the binding of both glutamate and D-serine/glycine as a co-agonist. Synaptic NMDARs are activated by presynaptic glutamate release at low frequency, while intensive synaptic activation could lead to glutamate spillover and activation of extrasynaptic NMDARs (18). Synaptic NMDARs are important for synaptic plasticity and learning and memory, which is largely controlled by D-serine concentration within the synaptic cleft depending on astrocyte coverage and function (19, 20). Moreover, extrasynaptic NMDARs are in close proximity to synapse-enveloping astrocytes and are a preferential target for astrocyte-released glutamate, D-serine, and glycine (Figure 2). Activation of extrasynaptic NMDARs could generate two forms of responses. First, ambient glutamate could produce a tonic NMDAR current, whose amplitude is directly modulated by glial EAATs (21, 22). The tonic NMDAR currents contribute to neuronal excitability and their existence has been identified in multiple neuron types in the prefrontal cortex, hippocampus, and cerebellum (23–25). Second, activation of extrasynaptic NMDARs could produce “slow inward currents” (SICs), which are infrequent phasic events characterized by slow activation and decay kinetics, large amplitude, and insensitivity to sodium channel blockers. It is believed that SICs are due to astrocyte-originated glutamate and the co-occurrence of SICs in adjacent neurons could lead to synchronized neuronal activity (26). It is also possible that SICs are generated by the phasic release of D-serine/glycine, which potentiates extrasynaptic NMDARs that are already tonically activated by low concentrations of ambient glutamate (18). While accumulating evidence has demonstrated a critical role of deficient synaptic NMDARs in schizophrenia (27, 28), alterations in the expression and function of extrasynaptic NMDARs are less understood. It was recently reported that the tonic NMDAR conductance was significantly larger and more sensitive to NMDAR blockers in interneurons compared with pyramidal neurons in the hippocampus (29). However, it remains to be determined whether extrasynaptic NMDAR deficiency occurs under schizophrenic conditions.
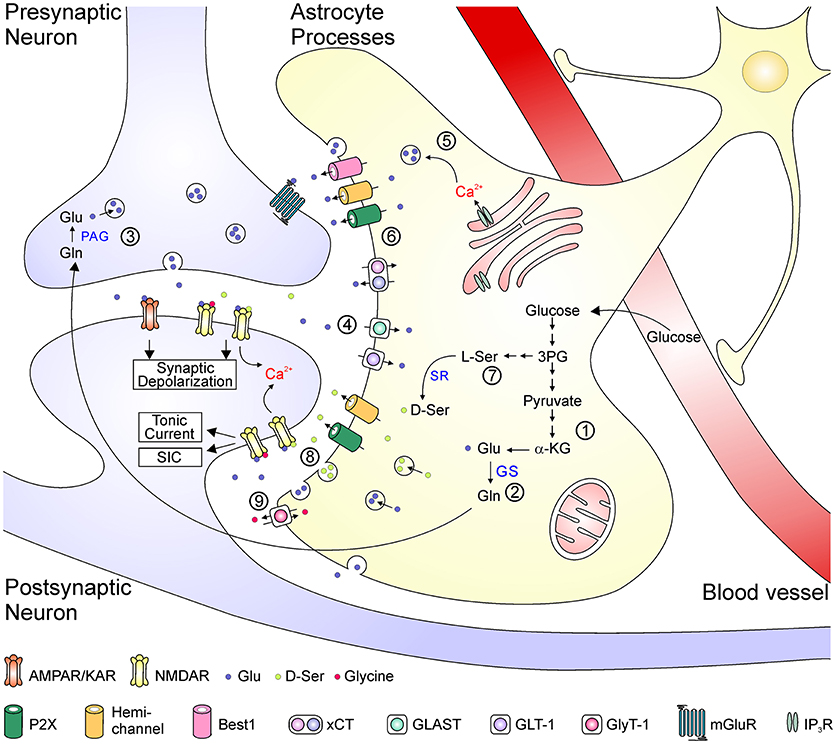
Figure 2. Schematic depiction of the astrocyte-related mechanisms in the metabolism, transport, uptake of glutamate and the regulation of glutamate transmission. Glutamate is synthesized de novo from α-KG via the TCA cycle (1). In the glutamate-glutamine cycle (2–4), glutamate is metabolized to glutamine by GS in the astrocyte and then transported to the neuron (2). Neuronal PAG catalyzes the conversion from glutamine into glutamate, which is released as the transmitter from presynaptic terminal (3). The synaptically released glutamate is rapidly uptake by EAATs back into the astrocytes (4). Glutamate can be released as a gliotransmitters by either Ca2+-dependent vesicular release (5) or efflux from transporters or large-conductance ion channels (6). D-serine is converted from L-serine, which is synthesized de novo from 3-phosphoglycerate in astrocytes (7). D-serine can be released via either vesicular or non-vesicular mechanisms and activate neuronal NMDA receptors (8). Glycine is transported across astrocytic membrane via glycine receptors (9). 3PG, 3-phosphoglycerate; α-KG, α-ketoglutarate; Glu, glutamate; Gln, glutamine; GS, glutamine synthetase; L-Ser, L-serine; D-Ser, D-serine; PAG, phosphate-activated glutaminase; SR, serine racemase; ASCT, alanine-serine-cysteine transporter; xCT, cystine-glutamate antiporter; GLAST, glutamate aspartate transporter; GLT-1, glutamate transporter 1; GlyT-1, glycine transporter 1; mGluR, metabotropic glutamate receptor; IP3R, inositol trisphosphate receptor.
Astrocytes and Glutamate Transmission
Regulation of glutamate transmission could occur at several steps, including synthesis, transportation, release, and clearance of glutamate, as well as activation of glutamate receptors by endogenous agonists and co-agonists. Figure 2 summarizes some key mechanisms underlying the impact of astrocytes in glutamate transmission in the CNS. Astrocytes are the primary locus for the biosynthesis of glutamate from glucose. Through the tricarboxylic acid cycle, the glycolysis product pyruvate is converted into α-ketoglutarate, which is then catalyzed into glutamate by aspartate aminotransferase (30). Glutamate is catalyzed by glutamine synthetase (GS), which is exclusively expressed in astrocytes in the brain, to form glutamine that is further transferred to neurons. In glutamatergic neurons, glutamate is converted from glutamine by phosphate-activated glutaminase (30). After packaged into synaptic vesicles, glutamate is released as the neurotransmitter to active postsynaptic AMPA/kainate receptors to mediate fast excitatory synaptic transmission, and to activate NMDARs to depolarize the postsynaptic membrane potential and to allow Ca2+ influx. The clearance of excessive glutamate from the synaptic cleft occurs rapidly, largely due to the function of four subtypes of EAATs (EAAT1-4), among which the EAAT1 (also termed glutamate-aspartate transporter, GLAST) and EAAT2 (also termed glutamate transporter 1, GLT-1) are mainly expressed in astrocytes (31). Once glutamate is taken up into astrocytes, it is converted into glutamine and re-enters the glutamate-glutamine cycle. Besides their key roles in the biosynthesis of glutamate and maintenance of the glutamate-glutamine cycle, astrocytes can directly release glutamate as a gliotransmitters by either Ca2+-dependent vesicular exocytosis or by non-vesicular release (32, 33). Astrocytes are also a major source of glycine and D-serine to potentiate NMDAR responses. Glycine is stored in astrocytes with high concentrations (3–6 mM) and can be released via the reversed operation of glycine transporters and other Ca2+-independent pathways (34–36). The synthesis of D-serine is initiated from the glycolytic intermediate metabolite 3-phosphoglycerate, which is synthesized into L-serine in several steps. D-serine is converted from L-serine by the action of SR and is released by Ca2+-dependent and/or -independent machineries from astrocytes (19, 37) [but see (38)]. Notably, these above processes might interact with one another; for example, the increased glutamate synthesis leads to elevated glutamate release (39), and activation of glutamate transports could evoke rapid glutamine release as a feedback mechanism (40).
EAATs
The efficiency of glutamate clearance directly affects extracellular resting glutamate levels and kinetics of glutamate-mediated synaptic responses. Among the total four subtypes of brain-expressed EAATs, EAAT1, and EAAT2 are mainly expressed in astrocytes, and EAAT2 is the dominant transporter in adults and is responsible for about 90% of total glutamate uptake (41, 42). The EAATs are mainly responsible for clearing synaptic glutamate and shaping postsynaptic responses. Deficits in EAAT functions cause the persistently increased glutamate levels, which represent one of the most remarkable changes in the schizophrenic brain. Accumulating evidence has reported abnormal mRNA and/or protein expression levels of EAATs in the prefrontal cortex, hippocampus, and anterior cingulate cortex in the postmortem tissue from schizophrenia patients. While many studies have revealed an overall reduction of EAAT2 levels in schizophrenia, some other studies reported unchanged or increased EAAT2 levels in certain brain regions [for review see (43)]. In a recent study using the laser-capture microdissection technique to separately harvest glutamatergic relay neurons and astrocytes from the mediodorsal nucleus of thalamus, it was reported that changes of EAAT levels could be cell-specific: mRNA expression of EAAT1 was decreased in astrocytes, whereas EAAT2 was increased in excitatory relay neurons in schizophrenia (44). The upregulation of EAATs in neurons is possibly due to a compensatory response to the reduced EAAT levels in astrocytes. Notably, the mRNA levels of EAAT2b were remarkably increased in anterior cingulate cortex pyramidal neurons and thalamus relay neurons in schizophrenia (44, 45). EAAT2b is a splicing variant of EAAT2. It contains the PDZ domain-binding motif and possibly interacts with neuronal scaffolding proteins like PSD-95 (45–47). The EAAT2b proteins are localized predominantly at the cell surface and their distribution can be regulated by intracellular Ca2+ and CaMKII (48), suggesting that EAAT2b might exhibit neuronal specific functions.
The abnormal EAAT levels are highly associated with schizophrenia phenotypes. Mice lacking EAAT1 displayed schizophrenia-like behavioral changes: they showed higher locomotor activity in the novel environment but not in the home cage, and had increased sensitivity to the locomotor hyperactivity induced by NMDA antagonists (49). The locomotor hyperactivity of EAAT1 knockout mice was reversed by either the antipsychotic haloperidol or the mGlu2/3 agonist LY379268 (49). These mice also exhibited abnormalities in behavioral tests that measure the negative and cognitive symptoms of schizophrenia, including poor nesting behaviors, lesser preference for novel social stimulus albeit normal overall social interaction, a significant reduction in acoustic startle amplitude, and impaired learning in an instrumental visual discrimination task, suggesting that EAAT1 dysfunction could generate certain behavioral changes resembling schizophrenia phenotypes (49, 50). Knockout of EAAT2 caused lethal spontaneous seizures, high susceptibility to brain injury, and short life-span in the homozygous mice (51). The heterozygous EAAT2 knockout mice had less severe abnormalities, and they exhibited increased locomotion activity in a new environment and greater freezing responses in the fear-conditioning test compared to the wildtype (52). The EAAT1/2 double knockout mice showed multiple brain defects resembling developmental defects found in schizophrenia (53). In a recent study, the inducible EAAT2 knockout mice were generated in a temporal controlled, cell-specific manner. The study demonstrated that knockout during adolescence caused 60–80% reduction of EAAT2 selectively in astrocytes, and the animals showed no lethal seizures or neuronal loss. Interestingly, these animals exhibited pathological repetitive behaviors, like excessive self-grooming and tic-like head shakes, whereas they had no obvious anxiety or social abnormality (54).
It remains unknown how EAAT abnormality leads to schizophrenia-relevant behaviors. In hippocampus, pharmacological inhibition of EAAT functions increased the peak amplitude of NMDAR-mediated EPSCs and prolonged their decay kinetics, but had no changes in AMPA receptor-mediated EPSCs, possibly due to the fast desensitization of AMPA receptor currents (55). It has been generally considered that EAATs could limit glutamate spillover to neighboring synapses and enhance synapse independence, which is related to synaptic plasticity as well as learning and memory (31). Reduced expression of glial EAATs will not only impair these important physiological functions but also increase the susceptibility of the brain to injury and cell death (51). Neuronal EAATs might have much lower efficiency than the astrocytic transports: glutamate is quickly converted into glutamine by GS, resulting in low free glutamate concentrations in astrocytes; whereas neurons are liable to accumulate intracellular glutamate concentration and thereby lead to poor operation of neuronal EAATs. Moreover, the action of EAATs will produce a non-stoichiometrically coupled anion current (56, 57), which contributes to the regulation of neuronal membrane potentials and transmitter release (58). In the zebrafish photoreceptor synapse, the EAAT2b isoform mediates a large-conductance Cl− current, potentially capable of affecting resting membrane potentials (59). Nevertheless, these mechanisms remain to be determined in schizophrenia and may vary depending on the brain region and disease progression.
The Glutamate-Glutamine Cycle
Schizophrenia is associated with the abnormal glutamate-glutamine cycle. Altered levels of brain metabolites including glutamate, GABA, and glutamine have been well-documented in human studies and these alterations can be detected in different brain regions and the cerebrospinal fluid (CSF) (60). Researchers using proton magnetic resonance spectroscopy (1H-MRS) have reported abnormal glutamine and/or glutamate levels (some studies report glutamate+glutamine levels in combination depending on the 1H-MRS approach and field strength), especially in the brain regions involving the medial temporal lobe (including hippocampus), basal ganglia, and thalamus of schizophrenia patients (61). An elevation of the glutamate+glutamine level was also observed in hippocampus of healthy humans receiving ketamine administration (62). The CSF glutamine/glutamate ratio is higher in the first-episode patients compared with normal controls (63). GS, the key enzyme involved in the glutamate-glutamine cycle, exhibits lower protein levels in spite of increased expression of mRNA in schizophrenia; but the results were less consistent among studies [for review see (43)]. Elevated glutamine levels are also directly related to psychotic symptoms and neuropsychological tests (64, 65). Although changed glutamine levels are supposedly related to functional alterations of astrocytes, it is notable that the 1H-MRS and CSF data cannot provide a precise measure of intracellular vs. extracellular substrates and further evidence is needed to establish the causality.
Disturbance of the glutamate-glutamine cycle and down-regulation of GS have also been reported in human epilepsy (66, 67). Reduced GS activity will build up intracellular glutamate in astrocytes, leading to reduced uptake capacities of EAATs. Moreover, glutamine produced by astrocytic GS is also one of the major sources for maintaining synaptic vesicle content of GABA in inhibitory interneurons (68). Pharmacological inhibition of GS reduced inhibitory postsynaptic currents in hippocampal pyramidal neurons and was reversed by exogenous application of glutamine (68). Similar dysfunction of the inhibitory synaptic response was also found in neurons near reactive astrocytes, which showed profound decreases in GS expression levels (69). These effects increased network hyperexcitability and produced spontaneous recurrent seizures (70). The loss of interneurons and deficient cortical GABA synthesis are also one of the most robust pathologies of schizophrenia (71–73). The reduced parvalbumin interneurons are associated with impaired oscillatory activity in hippocampus and imbalance of cortical excitation-inhibition (72, 74). Impaired parvalbumin interneuron activity by selective deletion of ErbB4 or dopamine D2 receptors produced schizophrenia-like behaviors in mice (75, 76). Dysfunction of neuron-astrocyte interaction has been suggested in schizophrenia models (77). However, it remains unclear whether there is any causative relationship between the dysfunction of the glutamate-glutamine cycle and interneuron deficiency.
D-Serine
D-serine plays an important role in the dysregulation of NMDAR functions in schizophrenia (78). This was supported by several lines of evidence, including the reduced serum and CSF D-serine levels (79, 80), a genetic association of SR and DAAO (the enzyme responsible for the degradation of D-serine) polymorphisms in schizophrenia patients (12, 81), the reversal effects of D-serine on schizophrenia-like behaviors in animals (82), and the beneficial effects of clinical D-serine-targeting therapies in patients (83, 84).
Recent studies revealed that the abnormality in astrocyte-released D-serine plays a critical role in the DISC1-associated pathological processes. The DISC1 gene was identified from patients with familial mental disorders and has been considered a critical susceptibility gene for schizophrenia (85, 86). The density of DISC1-expressing astrocytes is largely reduced in the dentate gyrus of hippocampus in schizophrenia patients compared with healthy controls (87). Selective expression of mutant DISC1 in astrocytes decreased protein levels of SR by increasing its ubiquitination (88). Astrocytes expressing mutant DISC1 caused less elaborated dendritic arborization and decreased the density of excitatory synapses in co-cultured normal neurons (89). The downregulation of SR resulted in a significant decline in D-serine levels, an enhanced locomotor activity, and impaired responses to pre-pulse inhibition in the mutant mice (88). Mice carrying the astrocytic specific mutation of DISC1 also exhibited increased anxiety, attenuated social interaction and preference for social novelty, and impaired cognitive behaviors (90). These behavioral abnormalities in DISC1 mutant mice were reversed by treatments of D-serine (88, 90), indicating a strong correlation with NMDAR hypofunction.
The α7 subunit-containing nicotinic acetylcholine receptor (α7 nAChR) has been considered a strong genetic contribution to schizophrenia (91). It has been reported that activation of α7 nAChR contributes to dopamine release and induction of long-term potentiation (91). Interestingly, α7 nAChRs are expressed not only in neurons but also in astrocytes (92–94). ACh could induce the intracellular Ca2+ elevation in astrocytes by activation of α7 nAChRs and facilitate the synthesis and release of D-serine (94–97). Papouin et al. recently discovered that astrocytic α7 nAChRs activation could drive the vesicular release of D-serine from astrocytes to generate a wakefulness-dependent D-serine oscillation in z (98). The wakefulness and activity but not the circadian rhythms of the animal promote D-serine fluctuations over the 24 h period, during which D-serine was accumulated during the wakefulness and declined during sleep. Such an oscillation of the D-serine level is associated with NMDAR activity and the learning and memory behavior of the animal, consistent with the involvement of NMDARs in the cognitive deficits in schizophrenia. Astrocytes appear to play a central role since D-serine accumulation during the wakefulness is impaired by disruption of astrocytic vesicular exocytosis machinery or by selective deletion of α7 nAChRs from astrocytes rather than neurons. More importantly, EVP-6124, an α7 nAChR modulator tested in schizophrenia clinical trials, is able to promote D-serine release and enhance NMDAR activity (98).
Potential Biomarkers
Several types of translational biomarkers have been developed to assess prognosis and monitor disease progression and treatment in schizophrenia. First, functional magnetic imaging (fMRI) blood-oxygenation-level dependent response (BOLD) has been applied to test brain physiology following acute NMDAR antagonist administration. NMDAR antagonists evoke robust changes in relative cerebral blood volume and local metabolism, which is largely due to the energy consumption for increased glutamate release and uptake (13, 99). The second approach applies 1H-MRS to measure glutamine and/or glutamate levels in the brain (16, 100). As described above, abnormal glutamine and/or glutamate indices were found in brain regions of individuals with schizophrenia compared with healthy volunteers (61). The third method utilizes task-based fMRI to evaluate BOLD responses in hippocampus and dorsolateral prefrontal cortex (101). The most adapted task is item-specific encoding task, which is designed to assess contributions of specific encoding and retrieval processes to episodic memory (102). Schizophrenia patients with more severe negative symptoms exhibited poor BOLD responses during the encoding and retrieval of episodic memory (101, 103). And fourth, positron emission tomography (PET) technique has been used to evaluate glutamatergic, GABAergic, and monoamine neurotransmission in various brain functions and have established the association between these responses and cognitive functions in schizophrenia (104, 105). Finally, the severity of schizophrenia symptoms is associated with abnormal serum levels of several glutamate transmission related factors, including brain-derived neurotrophic factor (BDNF), vascular endothelial growth factor (VEGF), D-serine, G72, and multiple inflammatory factors (106–108). A recent clinical study by Javitt et al. compared the first three types of glutamatergic target-engagement biomarkers in healthy volunteers receiving ketamine or placebo infusions (109). The data revealed a significant increase in the NMDAR antagonist-evoked BOLD responses. The 1H-MRS results showed a smaller but significant change in glutamate+glutamine levels. However, the data from task-based fMRI failed to yield a significant difference between the ketamine and placebo group (109). These observations indicate the fMRI BOLD as a particularly relevant biomarker for NMDAR antagonist-induced responses. The potential application of one or combined biomarkers for more refined prognostic classification of schizophrenia remains to be further assessed.
Future Treatments
Based on the glutamate hypothesis, many drug targets have been proposed and some are under investigation. For example, drugs targeting the co-agonist binding site of NMDARs are of the primary interest [for review see (110)]. Direct enhancement of NMDAR functions with glycine and D-serine has been used in multiple clinical trials, among which one recent study showed improved negative symptoms in individuals at clinical high risk of schizophrenia (111). GlyT1 inhibitors, like sarcosine or Bitopertin, which are supposed to increase synaptic glycine levels, have been found effective in several trials (112). Other treatments to increase D-serine/glycine levels, like the DAAO inhibitor sodium benzoate, have demonstrated beneficial effects as an add-on drug (83, 84). The mGluR2/3 agonists have been shown to inhibit excessive glutamate release in animal studies, and the tested drug LY2140023 has produced significant improvement in patients (113). Moreover, the α7 nAChR has attracted much attention; several drugs targeting this receptor are under clinical investigation (110). Besides these, more specific regulation of certain proteins in the glutamate transmission, such as selective NMDAR subtypes or EAAT isoforms, might represent potential targets for future drug development.
Conclusions
The glutamate hypothesis has substantially advanced our understanding of the pathogenesis of schizophrenia. Although glutamate transmission is mediated by ionotropic glutamate receptors and involves other pre- and post-synaptic components at excitatory synapses, astrocytes regulate glutamate metabolism and shape glutamate transmission in several important aspects. This review has focused on some recent studies that explicitly report the important role of glutamate uptake, glutamate-glutamine cycle, and astrocyte-derived D-serine in the etiology of schizophrenia. Neuroimaging and neurochemical biomarkers based on glutamate transmission and metabolism have also demonstrated translational utility. Further understanding of neuron-astrocyte interaction during these processes will be critical to the development of diagnostic and therapeutic avenues for schizophrenia and psychotic disorders.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This study was supported by grants to NZ and DW from the Ministry of Science and Technology (MOST 107-2320-B-039-060-MY3 and MOST 107-2320-B-039-061-MY3) and China Medical University Hospital (DMR-106-113).
References
1. Moghaddam B, Javitt D. From revolution to evolution: the glutamate hypothesis of schizophrenia and its implication for treatment. Neuropsychopharmacology (2012) 37:4–15. doi: 10.1038/npp.2011.181
2. Krystal JH, Karper LP, Seibyl JP, Freeman GK, Delaney R, Bremner JD, et al. Subanesthetic effects of the noncompetitive NMDA antagonist, ketamine, in humans. Psychotomimetic, perceptual, cognitive, and neuroendocrine responses. Arch Gen Psychiatry (1994) 51:199–214. doi: 10.1001/archpsyc.1994.03950030035004
3. Malhotra AK, Pinals DA, Adler CM, Elman I, Clifton A, Pickar D, et al. Ketamine-induced exacerbation of psychotic symptoms and cognitive impairment in neuroleptic-free schizophrenics. Neuropsychopharmacology (1997) 17:141–50. doi: 10.1016/S0893-133X(97)00036-5
4. Lahti AC, Koffel B, LaPorte D, Tamminga CA. Subanesthetic doses of ketamine stimulate psychosis in schizophrenia. Neuropsychopharmacology (1995) 13:9–19. doi: 10.1016/0893-133X(94)00131-I
5. Lennox BR, Palmer-Cooper EC, Pollak T, Hainsworth J, Marks J, Jacobson L, et al. Prevalence and clinical characteristics of serum neuronal cell surface antibodies in first-episode psychosis: a case-control study. Lancet Psychiatry (2017) 4:42–8. doi: 10.1016/S2215-0366(16)30375-3
6. Moscato EH, Peng X, Jain A, Parsons TD, Dalmau J, Balice-Gordon RJ. Acute mechanisms underlying antibody effects in anti-N-methyl-D-aspartate receptor encephalitis. Ann Neurol. (2014) 76:108–19. doi: 10.1002/ana.24195
7. Jones CA, Watson DJ, Fone KC. Animal models of schizophrenia. Br J Pharmacol. (2011) 164:1162–94. doi: 10.1111/j.1476-5381.2011.01386.x
8. Ramsey AJ. NR1 knockdown mice as a representative model of the glutamate hypothesis of schizophrenia. Prog Brain Res. (2009) 179:51–8. doi: 10.1016/S0079-6123(09)17906-2
9. Allen NC, Bagade S, McQueen MB, Ioannidis JP, Kavvoura FK, Khoury MJ, et al. Systematic meta-analyses and field synopsis of genetic association studies in schizophrenia: the SzGene database. Nat Genet. (2008) 40:827–34. doi: 10.1038/ng.171
10. Schizophrenia Working Group of the Psychiatric Genomics C. Biological insights from 108 schizophrenia-associated genetic loci. Nature (2014) 511:421–7. doi: 10.1038/nature13595
11. Tarabeux J, Kebir O, Gauthier J, Hamdan FF, Xiong L, Piton A, et al. Rare mutations in N-methyl-D-aspartate glutamate receptors in autism spectrum disorders and schizophrenia. Transl Psychiatry (2011) 1:e55. doi: 10.1038/tp.2011.52
12. Boks MP, Rietkerk T, van de Beek MH, Sommer IE, de Koning TJ, Kahn RS. Reviewing the role of the genes G72 and DAAO in glutamate neurotransmission in schizophrenia. Eur Neuropsychopharmacol. (2007) 17:567–72. doi: 10.1016/j.euroneuro.2006.12.003
13. Schobel SA, Chaudhury NH, Khan UA, Paniagua B, Styner MA, Asllani I, et al. Imaging patients with psychosis and a mouse model establishes a spreading pattern of hippocampal dysfunction and implicates glutamate as a driver. Neuron (2013) 78:81–93. doi: 10.1016/j.neuron.2013.02.011
14. Homayoun H, Moghaddam B. NMDA receptor hypofunction produces opposite effects on prefrontal cortex interneurons and pyramidal neurons. J Neurosci. (2007) 27:11496–500. doi: 10.1523/JNEUROSCI.2213-07.2007
15. Maccaferri G, Dingledine R. Control of feedforward dendritic inhibition by NMDA receptor-dependent spike timing in hippocampal interneurons. J Neurosci. (2002) 22:5462–72. doi: 10.1523/JNEUROSCI.22-13-05462.2002
16. Poels EM, Kegeles LS, Kantrowitz JT, Slifstein M, Javitt DC, Lieberman JA, et al. Imaging glutamate in schizophrenia: review of findings and implications for drug discovery. Mol Psychiatry (2014) 19:20–9. doi: 10.1038/mp.2013.136
17. Schobel SA, Lewandowski NM, Corcoran CM, Moore H, Brown T, Malaspina D, et al. Differential targeting of the CA1 subfield of the hippocampal formation by schizophrenia and related psychotic disorders. Arch Gen Psychiatry (2009) 66:938–46. doi: 10.1001/archgenpsychiatry.2009.115
18. Papouin T, Oliet SH. Organization, control and function of extrasynaptic NMDA receptors. Philos Trans R Soc Lond B Biol Sci. (2014) 369:20130601. doi: 10.1098/rstb.2013.0601
19. Panatier A, Theodosis DT, Mothet JP, Touquet B, Pollegioni L, Poulain DA, et al. Glia-derived D-serine controls NMDA receptor activity and synaptic memory. Cell (2006) 125:775–84. doi: 10.1016/j.cell.2006.02.051
20. Henneberger C, Papouin T, Oliet SH, Rusakov DA. Long-term potentiation depends on release of D-serine from astrocytes. Nature (2010) 463:232–6. doi: 10.1038/nature08673
21. Le Meur K, Galante M, Angulo MC, Audinat E. Tonic activation of NMDA receptors by ambient glutamate of non-synaptic origin in the rat hippocampus. J Physiol. (2007) 580(Pt 2):373–83. doi: 10.1113/jphysiol.2006.123570
22. Fleming TM, Scott V, Naskar K, Joe N, Brown CH, Stern JE. State-dependent changes in astrocyte regulation of extrasynaptic NMDA receptor signalling in neurosecretory neurons. J Physiol. (2011) 589(Pt 16):3929–41. doi: 10.1113/jphysiol.2011.207340
23. Povysheva NV, Johnson JW. Tonic NMDA receptor-mediated current in prefrontal cortical pyramidal cells and fast-spiking interneurons. J Neurophysiol. (2012) 107:2232–43. doi: 10.1152/jn.01017.2011
24. Rossi DJ, Slater NT. The developmental onset of NMDA receptor-channel activity during neuronal migration. Neuropharmacology (1993) 32:1239–48. doi: 10.1016/0028-3908(93)90018-X
25. Zhang M, Biancardi VC, Stern JE. An increased extrasynaptic NMDA tone inhibits A-type K(+) current and increases excitability of hypothalamic neurosecretory neurons in hypertensive rats. J Physiol. (2017) 595:4647–61. doi: 10.1113/JP274327
26. Fellin T, Pascual O, Gobbo S, Pozzan T, Haydon PG, Carmignoto G. Neuronal synchrony mediated by astrocytic glutamate through activation of extrasynaptic NMDA receptors. Neuron (2004) 43:729–43. doi: 10.1016/j.neuron.2004.08.011
27. Balu DT. The NMDA receptor and schizophrenia: from pathophysiology to treatment. Adv Pharmacol. (2016) 76:351–82. doi: 10.1016/bs.apha.2016.01.006
28. Banerjee A, Wang HY, Borgmann-Winter KE, MacDonald ML, Kaprielian H, Stucky A, et al. Src kinase as a mediator of convergent molecular abnormalities leading to NMDAR hypoactivity in schizophrenia. Mol Psychiatry (2015) 20:1091–100. doi: 10.1038/mp.2014.115
29. Riebe I, Seth H, Culley G, Dosa Z, Radi S, Strand K, et al. Tonically active NMDA receptors–a signalling mechanism critical for interneuronal excitability in the CA1 stratum radiatum. Eur J Neurosci. (2016) 43:169–78. doi: 10.1111/ejn.13128
30. Hertz L, Rothman DL. Glucose, lactate, beta-hydroxybutyrate, acetate, GABA, and succinate as Substrates for synthesis of glutamate and GABA in the glutamine-glutamate/GABA Cycle. Adv Neurobiol. (2016) 13:9–42. doi: 10.1007/978-3-319-45096-4_2
31. Tzingounis AV, Wadiche JI. Glutamate transporters: confining runaway excitation by shaping synaptic transmission. Nat Rev Neurosci. (2007) 8:935–47. doi: 10.1038/nrn2274
32. Araque A, Carmignoto G, Haydon PG, Oliet SH, Robitaille R, Volterra A. Gliotransmitters travel in time and space. Neuron (2014) 81:728–39. doi: 10.1016/j.neuron.2014.02.007
33. Woo DH, Han KS, Shim JW, Yoon BE, Kim E, Bae JY, et al. TREK-1 and Best1 channels mediate fast and slow glutamate release in astrocytes upon GPCR activation. Cell (2012) 151:25–40. doi: 10.1016/j.cell.2012.09.005
34. Verleysdonk S, Martin H, Willker W, Leibfritz D, Hamprecht B. Rapid uptake and degradation of glycine by astroglial cells in culture: synthesis and release of serine and lactate. Glia (1999) 27:239–48. 35. Saransaari P, Oja SS. Characteristics of hippocampal glycine release in cell-damaging conditions in the adult and developing mouse. Neurochem Res. (2001) 26:845–52. doi: 10.1023/A:1011624421505
35. Saransaari P, Oja SS. Characteristics of hippocampal glycine release in cell-damaging conditions in the adult and developing mouse. Neurochem Res. (2001) 26:845–52. doi: 10.1023/A:1011624421505
36. Shibasaki K, Hosoi N, Kaneko R, Tominaga M, Yamada K. Glycine release from astrocytes via functional reversal of GlyT1. J Neurochem. (2017) 140:395–403. doi: 10.1111/jnc.13741
37. Pan HC, Chou YC, Sun SH. P2X7 R-mediated Ca(2+) -independent d-serine release via pannexin-1 of the P2X7 R-pannexin-1 complex in astrocytes. Glia (2015) 63:877–93. doi: 10.1002/glia.22790
38. Wolosker H, Balu DT, Coyle JT. The rise and fall of the d-serine-mediated gliotransmission hypothesis. Trends Neurosci. (2016) 39:712–21. doi: 10.1016/j.tins.2016.09.007
39. Zhu H, Wang N, Yao L, Chen Q, Zhang R, Qian J, et al. Moderate UV exposure enhances learning and memory by promoting a novel glutamate biosynthetic pathway in the brain. Cell (2018) 173:1716–27e17. doi: 10.1016/j.cell.2018.04.014
40. Uwechue NM, Marx MC, Chevy Q, Billups B. Activation of glutamate transport evokes rapid glutamine release from perisynaptic astrocytes. J Physiol. (2012) 590:2317–31. doi: 10.1113/jphysiol.2011.226605
41. Furuta A, Rothstein JD, Martin LJ. Glutamate transporter protein subtypes are expressed differentially during rat CNS development. J Neurosci. (1997) 17:8363–75. doi: 10.1523/JNEUROSCI.17-21-08363.1997
42. Haugeto O, Ullensvang K, Levy LM, Chaudhry FA, Honore T, Nielsen M, et al. Brain glutamate transporter proteins form homomultimers. J Biol Chem. (1996) 271:27715–22. doi: 10.1074/jbc.271.44.27715
43. Hu W, MacDonald ML, Elswick DE, Sweet RA. The glutamate hypothesis of schizophrenia: evidence from human brain tissue studies. Ann N Y Acad Sci. (2015) 1338:38–57. doi: 10.1111/nyas.12547
44. McCullumsmith RE, O'Donovan SM, Drummond JB, Benesh FS, Simmons M, Roberts R, et al. Cell-specific abnormalities of glutamate transporters in schizophrenia: sick astrocytes and compensating relay neurons? Mol Psychiatry (2016) 21:823–30. doi: 10.1038/mp.2015.148
45. O'Donovan SM, Hasselfeld K, Bauer D, Simmons M, Roussos P, Haroutunian V, et al. Glutamate transporter splice variant expression in an enriched pyramidal cell population in schizophrenia. Transl Psychiatry (2015) 5:e579. doi: 10.1038/tp.2015.74
46. Schmitt A, Asan E, Lesch KP, Kugler P. A splice variant of glutamate transporter GLT1/EAAT2 expressed in neurons: cloning and localization in rat nervous system. Neuroscience (2002) 109:45–61. doi: 10.1016/S0306-4522(01)00451-1
47. Gonzalez-Gonzalez IM, Garcia-Tardon N, Cubelos B, Gimenez C, Zafra F. The glutamate transporter GLT1b interacts with the scaffold protein PSD-95. J Neurochem. (2008) 105:1834–48. doi: 10.1111/j.1471-4159.2008.05281.x
48. Underhill SM, Wheeler DS, Amara SG. Differential regulation of two isoforms of the glial glutamate transporter EAAT2 by DLG1 and CaMKII. J Neurosci. (2015) 35:5260–70. doi: 10.1523/JNEUROSCI.4365-14.2015
49. Karlsson RM, Tanaka K, Heilig M, Holmes A. Loss of glial glutamate and aspartate transporter (excitatory amino acid transporter 1) causes locomotor hyperactivity and exaggerated responses to psychotomimetics: rescue by haloperidol and metabotropic glutamate 2/3 agonist. Biol Psychiatry (2008) 64:810–4. doi: 10.1016/j.biopsych.2008.05.001
50. Karlsson RM, Tanaka K, Saksida LM, Bussey TJ, Heilig M, Holmes A. Assessment of glutamate transporter GLAST (EAAT1)-deficient mice for phenotypes relevant to the negative and executive/cognitive symptoms of schizophrenia. Neuropsychopharmacology (2009) 34:1578–89. doi: 10.1038/npp.2008.215
51. Tanaka K, Watase K, Manabe T, Yamada K, Watanabe M, Takahashi K, et al. Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter GLT-1. Science (1997) 276:1699–702. doi: 10.1126/science.276.5319.1699
52. Kiryk A, Aida T, Tanaka K, Banerjee P, Wilczynski GM, Meyza K, et al. Behavioral characterization of GLT1 (+/-) mice as a model of mild glutamatergic hyperfunction. Neurotox Res. (2008) 13:19–30. doi: 10.1007/BF03033364
53. Matsugami TR, Tanemura K, Mieda M, Nakatomi R, Yamada K, Kondo T, et al. From the cover: indispensability of the glutamate transporters GLAST and GLT1 to brain development. Proc Natl Acad Sci USA. (2006) 103:12161–6. doi: 10.1073/pnas.0509144103
54. Aida T, Yoshida J, Nomura M, Tanimura A, Iino Y, Soma M, et al. Astroglial glutamate transporter deficiency increases synaptic excitability and leads to pathological repetitive behaviors in mice. Neuropsychopharmacology (2015) 40:1569–79. doi: 10.1038/npp.2015.26
55. Arnth-Jensen N, Jabaudon D, Scanziani M. Cooperation between independent hippocampal synapses is controlled by glutamate uptake. Nat Neurosci. (2002) 5:325–31. doi: 10.1038/nn825
56. Fairman WA, Vandenberg RJ, Arriza JL, Kavanaugh MP, Amara SG. An excitatory amino-acid transporter with properties of a ligand-gated chloride channel. Nature (1995) 375:599–603. doi: 10.1038/375599a0
57. Wadiche JI, Amara SG, Kavanaugh MP. Ion fluxes associated with excitatory amino acid transport. Neuron (1995) 15:721–8. doi: 10.1016/0896-6273(95)90159-0
58. Veruki ML, Morkve SH, Hartveit E. Activation of a presynaptic glutamate transporter regulates synaptic transmission through electrical signaling. Nat Neurosci. (2006) 9:1388–96. doi: 10.1038/nn1793
59. Niklaus S, Cadetti L, Vom Berg-Maurer CM, Lehnherr A, Hotz AL, Forster IC, et al. Shaping of signal transmission at the photoreceptor synapse by EAAT2 glutamate transporters. eNeuro (2017) 4:ENEURO.0339–16.2017. doi: 10.1523/ENEURO.0339-16.2017
60. Jelen LA, King S, Mullins PG, Stone JM. Beyond static measures: a review of functional magnetic resonance spectroscopy and its potential to investigate dynamic glutamatergic abnormalities in schizophrenia. J Psychopharmacol. (2018) 32:497–508. doi: 10.1177/0269881117747579
61. Merritt K, Egerton A, Kempton MJ, Taylor MJ, McGuire PK. Nature of glutamate alterations in schizophrenia: a meta-analysis of proton magnetic resonance spectroscopy studies. JAMA Psychiatry (2016) 73:665–74. doi: 10.1001/jamapsychiatry.2016.0442
62. Kraguljac NV, Frolich MA, Tran S, White DM, Nichols N, Barton-McArdle A, et al. Ketamine modulates hippocampal neurochemistry and functional connectivity: a combined magnetic resonance spectroscopy and resting-state fMRI study in healthy volunteers. Mol Psychiatry (2017) 22:562–9. doi: 10.1038/mp.2016.122
63. Hashimoto K, Engberg G, Shimizu E, Nordin C, Lindstrom LH, Iyo M. Elevated glutamine/glutamate ratio in cerebrospinal fluid of first episode and drug naive schizophrenic patients. BMC Psychiatry (2005) 5:6. doi: 10.1186/1471-244X-5-6
64. Bustillo JR, Chen H, Jones T, Lemke N, Abbott C, Qualls C, et al. Increased glutamine in patients undergoing long-term treatment for schizophrenia: a proton magnetic resonance spectroscopy study at 3 T. JAMA Psychiatry (2014) 71:265–72. doi: 10.1001/jamapsychiatry.2013.3939
65. Dempster K, Norman R, Theberge J, Densmore M, Schaefer B, Williamson P. Glutamatergic metabolite correlations with neuropsychological tests in first episode schizophrenia. Psychiatry Res. (2015) 233:180–5. doi: 10.1016/j.pscychresns.2015.06.003
66. Cavus I, Kasoff WS, Cassaday MP, Jacob R, Gueorguieva R, Sherwin RS, et al. Extracellular metabolites in the cortex and hippocampus of epileptic patients. Ann Neurol. (2005) 57:226–35. doi: 10.1002/ana.20380
67. Eid T, Thomas MJ, Spencer DD, Runden-Pran E, Lai JC, Malthankar GV, et al. Loss of glutamine synthetase in the human epileptogenic hippocampus: possible mechanism for raised extracellular glutamate in mesial temporal lobe epilepsy. Lancet (2004) 363:28–37. doi: 10.1016/S0140-6736(03)15166-5
68. Liang SL, Carlson GC, Coulter DA. Dynamic regulation of synaptic GABA release by the glutamate-glutamine cycle in hippocampal area CA1. J Neurosci. (2006) 26:8537–48. doi: 10.1523/JNEUROSCI.0329-06.2006
69. Ortinski PI, Dong J, Mungenast A, Yue C, Takano H, Watson DJ, et al. Selective induction of astrocytic gliosis generates deficits in neuronal inhibition. Nat Neurosci. (2010) 13:584–91. doi: 10.1038/nn.2535
70. Eid T, Ghosh A, Wang Y, Beckstrom H, Zaveri HP, Lee TS, et al. Recurrent seizures and brain pathology after inhibition of glutamine synthetase in the hippocampus in rats. Brain (2008) 131(Pt 8):2061–70. doi: 10.1093/brain/awn133
71. Behrens MM, Ali SS, Dao DN, Lucero J, Shekhtman G, Quick KL, et al. Ketamine-induced loss of phenotype of fast-spiking interneurons is mediated by NADPH-oxidase. Science (2007) 318:1645–7. doi: 10.1126/science.1148045
72. Lewis DA, Curley AA, Glausier JR, Volk DW. Cortical parvalbumin interneurons and cognitive dysfunction in schizophrenia. Trends Neurosci. (2012) 35:57–67. doi: 10.1016/j.tins.2011.10.004
73. Steullet P, Cabungcal JH, Coyle J, Didriksen M, Gill K, Grace AA, et al. Oxidative stress-driven parvalbumin interneuron impairment as a common mechanism in models of schizophrenia. Mol Psychiatry (2017) 22:936–43. doi: 10.1038/mp.2017.47
74. Lodge DJ, Behrens MM, Grace AA. A loss of parvalbumin-containing interneurons is associated with diminished oscillatory activity in an animal model of schizophrenia. J Neurosci. (2009) 29:2344–54. doi: 10.1523/JNEUROSCI.5419-08.2009
75. Tomasella E, Bechelli L, Ogando MB, Mininni C, Di Guilmi MN, De Fino F, et al. Deletion of dopamine D2 receptors from parvalbumin interneurons in mouse causes schizophrenia-like phenotypes. Proc Natl Acad Sci USA. (2018) 115:3476–81. doi: 10.1073/pnas.1719897115
76. Del Pino I, Garcia-Frigola C, Dehorter N, Brotons-Mas JR, Alvarez-Salvado E, Martinez de Lagran M, et al. Erbb4 deletion from fast-spiking interneurons causes schizophrenia-like phenotypes. Neuron (2013) 79:1152–68. doi: 10.1016/j.neuron.2013.07.010
77. Kondziella D, Brenner E, Eyjolfsson EM, Markinhuhta KR, Carlsson ML, Sonnewald U. Glial-neuronal interactions are impaired in the schizophrenia model of repeated MK801 exposure. Neuropsychopharmacology (2006) 31:1880–7. doi: 10.1038/sj.npp.1300993
78. Labrie V, Wong AH, Roder JC. Contributions of the D-serine pathway to schizophrenia. Neuropharmacology (2012) 62:1484–503. doi: 10.1016/j.neuropharm.2011.01.030
79. Hashimoto K, Fukushima T, Shimizu E, Komatsu N, Watanabe H, Shinoda N, et al. Decreased serum levels of D-serine in patients with schizophrenia: evidence in support of the N-methyl-D-aspartate receptor hypofunction hypothesis of schizophrenia. Arch Gen Psychiatry (2003) 60:572–6. doi: 10.1001/archpsyc.60.6.572
80. Bendikov I, Nadri C, Amar S, Panizzutti R, De Miranda J, Wolosker H, et al. A CSF and postmortem brain study of D-serine metabolic parameters in schizophrenia. Schizophr Res. (2007) 90:41–51. doi: 10.1016/j.schres.2006.10.010
81. Morita Y, Ujike H, Tanaka Y, Otani K, Kishimoto M, Morio A, et al. A genetic variant of the serine racemase gene is associated with schizophrenia. Biol Psychiatry (2007) 61:1200–3. doi: 10.1016/j.biopsych.2006.07.025
82. Labrie V, Roder JC. The involvement of the NMDA receptor D-serine/glycine site in the pathophysiology and treatment of schizophrenia. Neurosci Biobehav Rev. (2010) 34:351–72. doi: 10.1016/j.neubiorev.2009.08.002
83. Lane HY, Lin CH, Green MF, Hellemann G, Huang CC, Chen PW, et al. Add-on treatment of benzoate for schizophrenia: a randomized, double-blind, placebo-controlled trial of D-amino acid oxidase inhibitor. JAMA Psychiatry (2013) 70:1267–75. doi: 10.1001/jamapsychiatry.2013.2159
84. Lin CH, Lin CH, Chang YC, Huang YJ, Chen PW, Yang HT, et al. Sodium benzoate, a D-amino acid oxidase inhibitor, added to clozapine for the treatment of schizophrenia: a randomized, double-blind, placebo-controlled trial. Biol Psychiatry (2017) 84:422–32. doi: 10.1016/j.biopsych.2017.12.006
85. St Clair D, Blackwood D, Muir W, Carothers A, Walker M, Spowart G, et al. Association within a family of a balanced autosomal translocation with major mental illness. Lancet (1990) 336:13–6. doi: 10.1016/0140-6736(90)91520-K
86. Millar JK, Wilson-Annan JC, Anderson S, Christie S, Taylor MS, Semple CA, et al. Disruption of two novel genes by a translocation co-segregating with schizophrenia. Hum Mol Genet. (2000) 9:1415–23. doi: 10.1093/hmg/9.9.1415
87. Bernstein HG, Dobrowolny H, Keilhoff G, Bogerts B, Steiner J. Reduced density of DISC1 expressing astrocytes in the dentate gyrus but not in the subventricular zone in schizophrenia. Neuropsychopharmacology (2018) 43:457–8. doi: 10.1038/npp.2017.242
88. Ma TM, Abazyan S, Abazyan B, Nomura J, Yang C, Seshadri S, et al. Pathogenic disruption of DISC1-serine racemase binding elicits schizophrenia-like behavior via D-serine depletion. Mol Psychiatry (2013) 18:557–67. doi: 10.1038/mp.2012.97
89. Xia M, Zhu S, Shevelkin A, Ross CA, Pletnikov M. DISC1, astrocytes and neuronal maturation: a possible mechanistic link with implications for mental disorders. J Neurochem. (2016) 138:518–24. doi: 10.1111/jnc.13663
90. Terrillion CE, Abazyan B, Yang Z, Crawford J, Shevelkin AV, Jouroukhin Y, et al. DISC1 in astrocytes influences adult neurogenesis and hippocampus-dependent behaviors in mice. Neuropsychopharmacology (2017) 42:2242–51. doi: 10.1038/npp.2017.129
91. Young JW, Geyer MA. Evaluating the role of the alpha-7 nicotinic acetylcholine receptor in the pathophysiology and treatment of schizophrenia. Biochem Pharmacol. (2013) 86:1122–32. doi: 10.1016/j.bcp.2013.06.031
92. Duffy AM, Fitzgerald ML, Chan J, Robinson DC, Milner TA, Mackie K, et al. Acetylcholine alpha7 nicotinic and dopamine D2 receptors are targeted to many of the same postsynaptic dendrites and astrocytes in the rodent prefrontal cortex. Synapse (2011) 65:1350–67. doi: 10.1002/syn.20977
93. Gahring LC, Persiyanov K, Dunn D, Weiss R, Meyer EL, Rogers SW. Mouse strain-specific nicotinic acetylcholine receptor expression by inhibitory interneurons and astrocytes in the dorsal hippocampus. J Comp Neurol. (2004) 468:334–46. doi: 10.1002/cne.10943
94. Shen JX, Yakel JL. Functional alpha7 nicotinic ACh receptors on astrocytes in rat hippocampal CA1 slices. J Mol Neurosci. (2012) 48:14–21. doi: 10.1007/s12031-012-9719-3
95. Sharma G, Vijayaraghavan S. Nicotinic cholinergic signaling in hippocampal astrocytes involves calcium-induced calcium release from intracellular stores. Proc Natl Acad Sci USA. (2001) 98:4148–53. doi: 10.1073/pnas.071540198
96. Lin H, Hsu FC, Baumann BH, Coulter DA, Lynch DR. Cortical synaptic NMDA receptor deficits in alpha7 nicotinic acetylcholine receptor gene deletion models: implications for neuropsychiatric diseases. Neurobiol Dis. (2014) 63:129–40. doi: 10.1016/j.nbd.2013.11.021
97. Singh NS, Paul RK, Ramamoorthy A, Torjman MC, Moaddel R, Bernier M, et al. Nicotinic acetylcholine receptor antagonists alter the function and expression of serine racemase in PC-12 and 1321N1 cells. Cell Signal. (2013) 25:2634–45. doi: 10.1016/j.cellsig.2013.08.025
98. Papouin T, Dunphy JM, Tolman M, Dineley KT, Haydon PG. Septal cholinergic neuromodulation tunes the astrocyte-dependent gating of hippocampal NMDA receptors to wakefulness. Neuron (2017) 94:840–54.e7. doi: 10.1016/j.neuron.2017.04.021
99. Gozzi A, Large CH, Schwarz A, Bertani S, Crestan V, Bifone A. Differential effects of antipsychotic and glutamatergic agents on the phMRI response to phencyclidine. Neuropsychopharmacology (2008) 33:1690–703. doi: 10.1038/sj.npp.1301547
100. Poels EM, Kegeles LS, Kantrowitz JT, Javitt DC, Lieberman JA, Abi-Dargham A, et al. Glutamatergic abnormalities in schizophrenia: a review of proton MRS findings. Schizophr Res. (2014) 152:325–32. doi: 10.1016/j.schres.2013.12.013
101. Ragland JD, Ranganath C, Harms MP, Barch DM, Gold JM, Layher E, et al. Functional and neuroanatomic specificity of episodic memory dysfunction in schizophrenia: a functional magnetic resonance imaging study of the relational and item-specific encoding task. JAMA Psychiatry (2015) 72:909–16. doi: 10.1001/jamapsychiatry.2015.0276
102. Ragland JD, Ranganath C, Barch DM, Gold JM, Haley B, MacDonald AW III. Relational and Item-Specific Encoding (RISE): task development and psychometric characteristics. Schizophr Bull. (2012) 38:114–24. doi: 10.1093/schbul/sbr146
103. Ragland JD, Blumenfeld RS, Ramsay IS, Yonelinas A, Yoon J, Solomon M, et al. Neural correlates of relational and item-specific encoding during working and long-term memory in schizophrenia. Neuroimage (2012) 59:1719–26. doi: 10.1016/j.neuroimage.2011.08.055
104. Finnema SJ, Scheinin M, Shahid M, Lehto J, Borroni E, Bang-Andersen B, et al. Application of cross-species PET imaging to assess neurotransmitter release in brain. Psychopharmacology (2015) 232:4129–57. doi: 10.1007/s00213-015-3938-6
105. Takano H. Cognitive function and monoamine neurotransmission in schizophrenia: evidence from positron emission tomography studies. Front Psychiatry (2018) 9:228. doi: 10.3389/fpsyt.2018.00228
106. Vasic N, Connemann BJ, Wolf RC, Tumani H, Brettschneider J. Cerebrospinal fluid biomarker candidates of schizophrenia: where do we stand? Eur Arch Psychiatry Clin Neurosci. (2012) 262:375–91. doi: 10.1007/s00406-011-0280-9
107. Lin CH, Chang HT, Chen YJ, Lin CH, Huang CH, Tun R, et al. Distinctively higher plasma G72 protein levels in patients with schizophrenia than in healthy individuals. Mol Psychiatry (2014) 19:636–7. doi: 10.1038/mp.2013.80
108. Trepanier MO, Hopperton KE, Mizrahi R, Mechawar N, Bazinet RP. Postmortem evidence of cerebral inflammation in schizophrenia: a systematic review. Mol Psychiatry (2016) 21:1009–26. doi: 10.1038/mp.2016.90
109. Javitt DC, Carter CS, Krystal JH, Kantrowitz JT, Girgis RR, Kegeles LS, et al. Utility of imaging-based biomarkers for glutamate-targeted drug development in psychotic disorders: a randomized clinical trial. JAMA Psychiatry (2018) 75:11–9. doi: 10.1001/jamapsychiatry.2017.3572
110. Girgis RR, Zoghbi AW, Javitt DC, Lieberman JA. The past and future of novel, non-dopamine-2 receptor therapeutics for schizophrenia: a critical and comprehensive review. J Psychiatr Res. (2018). doi: 10.1016/j.jpsychires.2018.07.006. [Epub ahead of print].
111. Kantrowitz JT, Woods SW, Petkova E, Cornblatt B, Corcoran CM, Chen H, et al. D-serine for the treatment of negative symptoms in individuals at clinical high risk of schizophrenia: a pilot, double-blind, placebo-controlled, randomised parallel group mechanistic proof-of-concept trial. Lancet Psychiatry (2015) 2:403–12. doi: 10.1016/S2215-0366(15)00098-X
112. Harvey RJ, Yee BK. Glycine transporters as novel therapeutic targets in schizophrenia, alcohol dependence and pain. Nat Rev Drug Discov. (2013) 12:866–85. doi: 10.1038/nrd3893
Keywords: schizophrenia, glutamate, NMDA receptors, excitatory amino acid transporters, glutamate-glutamine cycle, D-serine
Citation: Mei Y-Y, Wu DC and Zhou N (2018) Astrocytic Regulation of Glutamate Transmission in Schizophrenia. Front. Psychiatry 9:544. doi: 10.3389/fpsyt.2018.00544
Received: 12 July 2018; Accepted: 12 October 2018;
Published: 06 November 2018.
Edited by:
Kenji Hashimoto, Chiba University, JapanReviewed by:
Jennifer C. Felger, Emory University, United StatesKurt Leroy Hoffman, Autonomous University of Tlaxcala, Mexico
Copyright © 2018 Mei, Wu and Zhou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ning Zhou, ningzhou.nrsc@gmail.com