 Zhaohui Lan1,2
Zhaohui Lan1,2 Yuhua Shi1
Yuhua Shi1 Qinggang Yin1
Qinggang Yin1 Ranran Gao1
Ranran Gao1 Chunlian Liu2
Chunlian Liu2 Wenting Wang1
Wenting Wang1 Xufang Tian2
Xufang Tian2 Jiawei Liu3Yiying Nong3
Jiawei Liu3Yiying Nong3 Li Xiang1*
Li Xiang1* Lan Wu1*
Lan Wu1*- 1Key Laboratory of Beijing for Identification and Safety Evaluation of Chinese Medicine, Artemisinin Research Center, Institute of Chinese Materia Medica, China Academy of Chinese Medical Sciences, Beijing, China
- 2College of Pharmacy, Hubei University of Chinese Medicine, Wuhan, China
- 3Department of product development, Hubei Aiaitie Health Technology Co., LTD, Huanggang, China
Artemisia Linn. is a large genus within the family Asteraceae that includes several important medicinal plants. Because of their similar morphology and chemical composition, traditional identification methods often fail to distinguish them. Therefore, developing an effective identification method for Artemisia species is an urgent requirement. In this study, we analyzed 15 chloroplast (cp) genomes, including 12 newly sequenced genomes, from 5 Artemisia species. The cp genomes from the five Artemisia species had a typical quadripartite structure and were highly conserved across species. They had varying lengths of 151,132–151,178 bp, and their gene content and codon preferences were similar. Mutation hotspot analysis identified four highly variable regions, which can potentially be used as molecular markers to identify Artemisia species. Phylogenetic analysis showed that the five Artemisia species investigated in this study were sister branches to each other, and individuals of each species formed a monophyletic clade. This study shows that the cp genome can provide distinguishing features to help identify closely related Artemisia species and has the potential to serve as a universal super barcode for plant identification.
Introduction
Artemisia Linn. is a large genus within the family Asteraceae comprising commonly used herbs that have a long history of medicinal use (Watson et al., 2002; Riggins, 2008; Kim G. B. et al., 2020). Among the various compounds present in these plants, terpenoids represent the main effective component. Modern pharmacological studies have shown that Artemisia medicinal plants exert diuretic, expectorant, antiinflammatory, hemostatic, hypotensive, and antiallergic effects (Chen et al., 2021; Hsueh et al., 2021; Hong et al., 2022). Owing to their similar morphological characteristics and chemical composition, Artemisia species are often mixed or substituted in different regions of China. According to the Chinese flora, Artemisia princeps and A. lancea are often mixed with A. argyi. It is difficult to distinguish dry herbs and raw materials using traditional identification methods. These difficulties have seriously hindered their development as medicinal plants. Some universal DNA barcodes, such as internal transcribed spacer (ITS) and ITS2, have been used to distinguish Artemisia species; however, these are inadequate for solving the classification problem because the sequences of closely related species are similar due to the hybridization of Artemisia plants (Garcia et al., 2008; Wang et al., 2016; Liu et al., 2017). Therefore, development of an accurate and effective method to identify medicinal Artemisia species is urgently needed.
The chloroplast (cp) is a multifunctional organelle with its independent genetic material. The structure of most angiosperm cp genomes is mostly conservative with a typical double-stranded, circular quadripartite structure, which includes a small single copy (SSC) region, large single copy (LSC) region, and two inverted repeat regions (IRa and IRb) (Jansen et al., 2005). With the development of sequencing technology, an increasing number of cp genomes have been published. The cp genome is usually 110–170 kb long, with 110–150 coding genes, which are highly conserved in gene type, gene number, and sequence compared with the mitochondrial or nuclear genome (Green, 2011; Zhu et al., 2016). The evolution rate of the cp genome is relatively moderate (Dong et al., 2013). Due to the lack of recombination, small genome size, and high single cell copy number, the cp genome is widely used in phylogenetic analysis and species identification (Dong et al., 2012; Twyford and Ness, 2017; Dong et al., 2017; Dong et al., 2018; Wu et al., 2018; Mader et al., 2018; Yang et al., 2018; Zhang et al., 2019). The comparison of cp genomes helps discover sequence variations, such as simple sequence repeats (SSRs), and mutation hotspots, and this has led some researchers to propose that the cp genome can be used as a super barcode for species identification (Li et al., 2015). Compared with the traditional relatively short and easily amplified DNA barcode, the cp genome has more abundant mutation site information and stronger species resolution ability, which can more accurately reflect the genetic characteristics of closely related species.
In this study, we used a second-generation sequencing platform to obtain the cp genomes from five Artemisia species. We compared their genome structure, codon usage preference, repeat sequences, and mutation hotspots. Finally, we performed a phylogenetic analysis of 29 cp genomes from 19 angiosperms. This study aimed to contribute valuable information toward the construction of the cp genome database of Artemisia species, which will aid in their identification.
Materials and methods
Sample collection, DNA extraction, and sequencing
Fifteen cp genomes from five Artemisia species were used in this study (Supplementary Table S1). Fresh leaves of 12 individuals from 5 Artemisia species were collected from Hainan, Hubei, and Beijing in China. The cp genomes of two additional individuals were downloaded from NCBI (Accession No.: MZ151340.1 and MW411453.1, A. lactiflora) and one cp genome was obtained from our previously published study (Accession No.: ON381734, A. indica) (Lan et al., 2022). The genomic DNA of each individual was extracted from fresh leaves using the plant DNA Extraction Kit (QIAGEN, Germany). The quality and concentration of genomic DNAs were evaluated using the Qubit2.0 Fluorometer (Thermo Scientific, USA) and NanoDrop 2000c spectrophotometer (Nanodrop Technologies, Wilmington, DE, USA) to ensure they met the requirements for sequencing. Based on the Illumina Nova Seq sequencing platform, 2 × 150 bp sequencing was performed with a depth of 226–578×. After quality pruning, clean reads were obtained from the original sequencing data for subsequent splicing and annotation.
Genome assembly and annotation
The NOVOPlasty software (https://github.com/ndierckx/NOVOPlasty) was used to assemble the cp genomes. We compared the clean reads with the scaffold obtained from the assembly, optimized the assembly results according to the paired-end and overlap relationships of the reads, and used the GapCloser software (v1.12, http://soap.genomics.org.cn/soapdenovo.html) to repair the inner hole of the assembly result. Finally, the reference genome was used to correct the starting position of the assembled cp sequence and determine the position and direction of four cp partitions (LSC/IRa/SSC/IRb) for obtaining the final cp genome sequence. The cp genomes were annotated using CpGAVAS (Liu et al., 2012). The genome circle maps were drawn using the online tool OGDRAWH (http://ogdraw.mpimp-golm.mpg.de/) (Lohse et al., 2007). The cp genomes and gene annotation files were uploaded to the NCBI database to obtain GenBank accession numbers.
Codon usage analysis
The relative synonymous codon usage (RSCU) of the 15 cp genomes from the 5 Artemisia species was determined and analyzed using the CodonW1.4.2 software (http://mobyle.pasteur.fr/cgi-bin/portal.py?form=codonw). Heat maps were constructed using the RSCU values. An RSCU value of >1 indicates that the codon is used more frequently, a value equal to 1 indicates that the codon has no usage preference, and a value of <1 indicates that the codon is used less frequently.
Repeat sequences and simple sequence repeat analysis
Four types of repeat sequences—forward, reverse, complementary, and palindromic—were identified using REPuter with a Hamming distance of 3 and a minimum repeat size of 30 bp (Kurtz et al., 2001). SSRs were detected using MISA with the following parameters: eight repeat units for mononucleotides; four for di- and trinucleotides; and three for tetra-, penta-, and hexanucleotides (Thiel et al., 2003; Lin et al., 2012).
Nucleotide diversity analysis
The nucleotide diversity of the 15 cp genomes was calculated via sliding window analysis using the DnaSP v5.10 software. The window length was set to 600 bp and the step length to 200 bp (Zhang et al., 2021).
Phylogenetic analysis
The phylogenetic tree was constructed based on the 15 whole cp genomes from 5 Artemisia species, another 13 species, and 1 outgroup Cirsium japonicum. All genomes, expect the 12 newly sequenced genomes, were downloaded from NCBI. The 29 cp genomes were compared using the MAFFT software (http://mafft.cbrc.jp/alignment/software/). Phylogenetic analysis was performed using the maximum likelihood (ML) method. Using IQtree’s default parameters, ModelFinder automatically filters the best model to build the ML tree (Bootstrap to 1000) (Nguyen et al., 2015).
Results
Chloroplast genome sequencing and features of five Artemisia species
The cp genome lengths of A. lancea, A. princeps, A. lactiflora, A. indica, and A. argyi were 151,132 bp, 151,154 bp, 151,178 bp, 151,161 bp, and 151,152 bp, respectively (Table 1 and Supplementary Table S1). These genomes have a typical quadripartite structure, including an LSC (82,870–82,911 bp), SSC (18,338–18,354 bp), and two IR (24,960–24,961 bp) regions (Table 1). The average GC content was 37.5%, and the IR regions possessed higher GC content (43.1%) than the LSC (35.5%–35.6%) and SSC (30.9%) regions. In this study, we annotated 132 genes from the 15 cp genomes, of which 7 tRNA genes, 4 rRNA genes, and 7 protein-coding genes were duplicated in the IR region. A total of 114 genes were unique, including 80 protein-coding, 30 tRNA, and 4 rRNA genes.
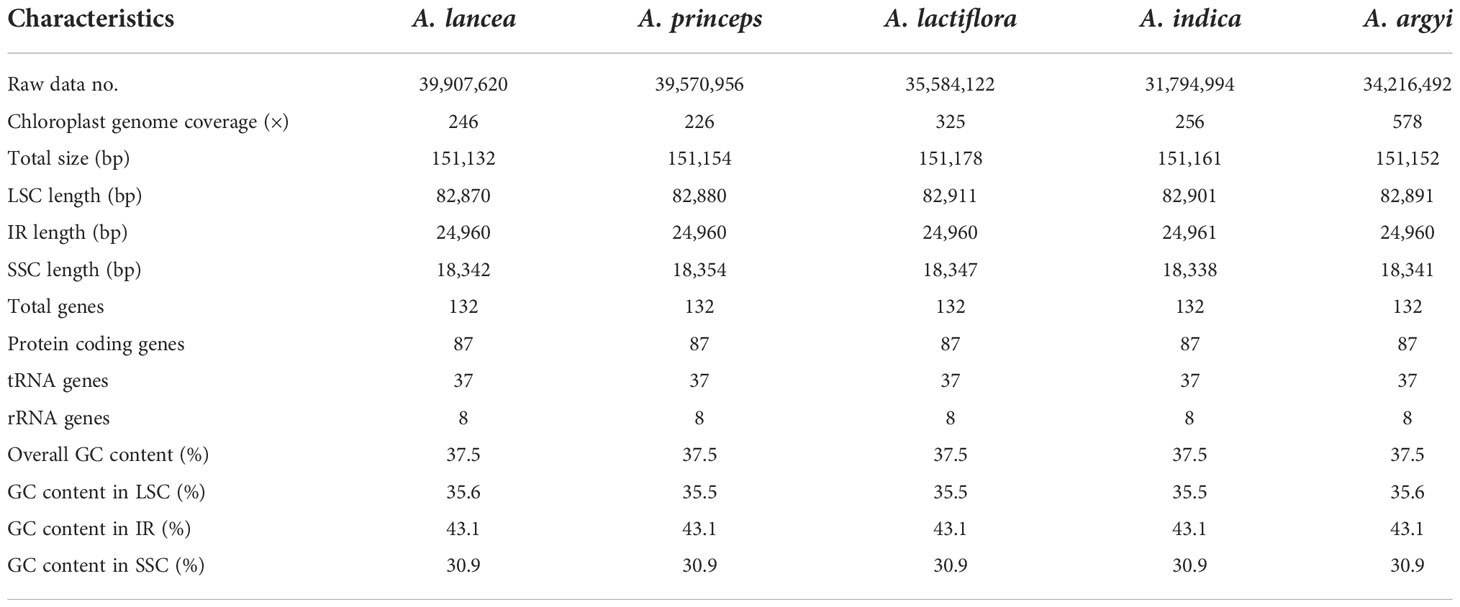
Table 1 Basic cp genome information of five Artemisia species.
Relative synonymous codons usage
The cp genomes of the 5 Artemisia species contained 64 codons. Of these, 61 codons encoded 20 proteins, and the other 3 were termination codons. The codon AUU had the highest usage frequency (1,078–1,079) with an RSCU value of 1.46–1.47. The RSCU values of the cp genomes in the five species were slightly different. Methionine (Met) and tryptophan (Trp) were encoded by a single codon, with an RSCU value of 1, indicating no preference. All other amino acids were encoded by multiple codons (Figure 1 and Supplementary Table S2). Arg, Leu, and Ser were encoded by six codons; alanine (Ala), glycine (Gly), proline (Pro), threonine (Thr), and valine (Val) by four codons; isoleucine (Ile) by three codons; and the rest were encoded by two codons.
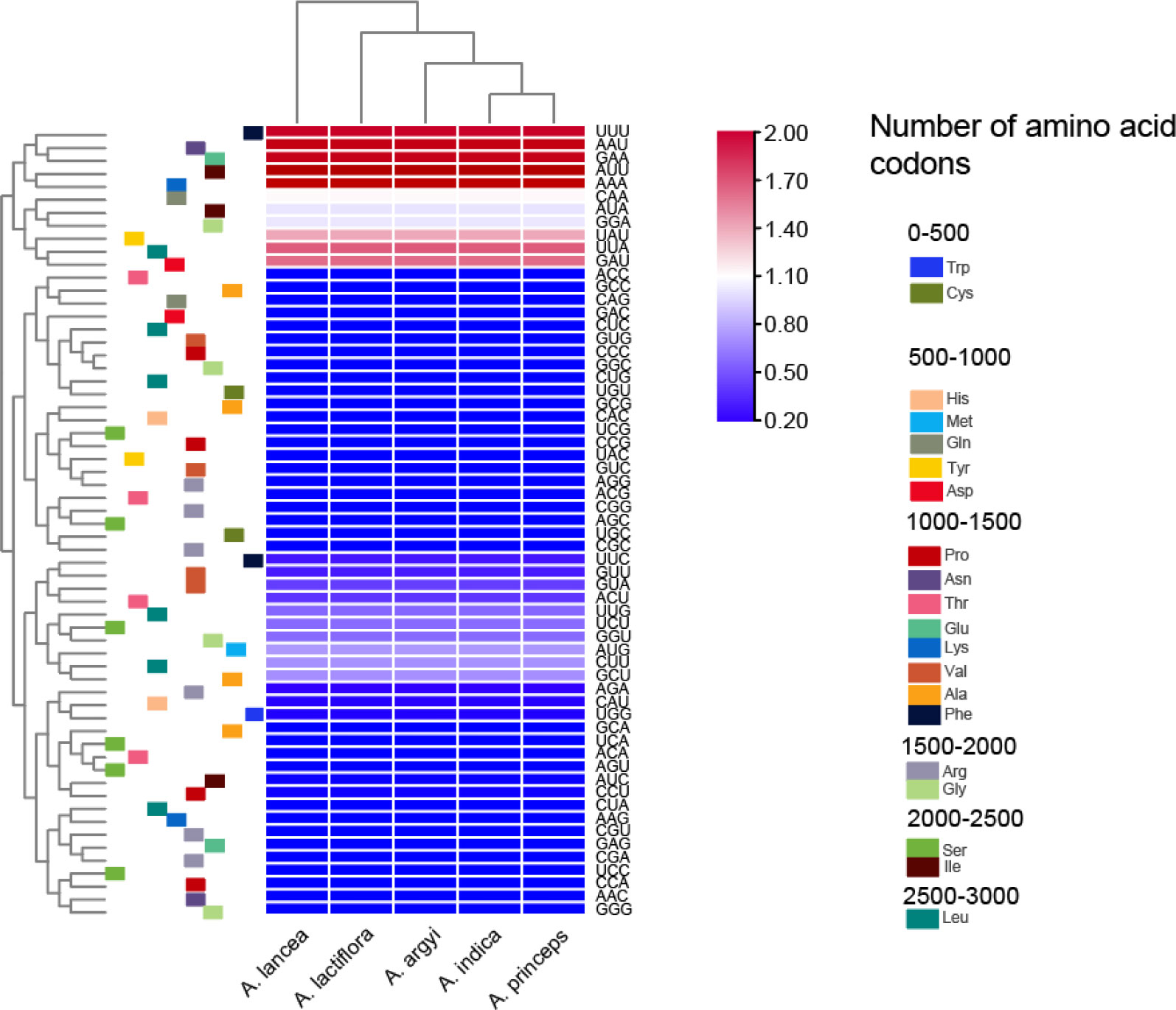
Figure 1 Heat map of the relative synonymous codons usage values of the cp genomes in the five Artemisia species.
Repeat and simple sequence repeat analyses
Some repeats with a length of ≥30 bp are known as long repeats. These are conducive to cp genome rearrangements and increase the genetic diversity of the population. In total, we found 42–50 long repeats in the 15 cp genomes of the 5 Artemisia species, including 19–22 forward, 20–22 palindromic, and 3–6 reverse repeats. Most of these repeats, which were 30–39 bp long, were located in the gene spacer and intron regions. This length of repeats was dominant in the Artemisia cp genomes, and the longest was the forward repeat. Complementary repeats were not identified in these genomes (Figure 2).
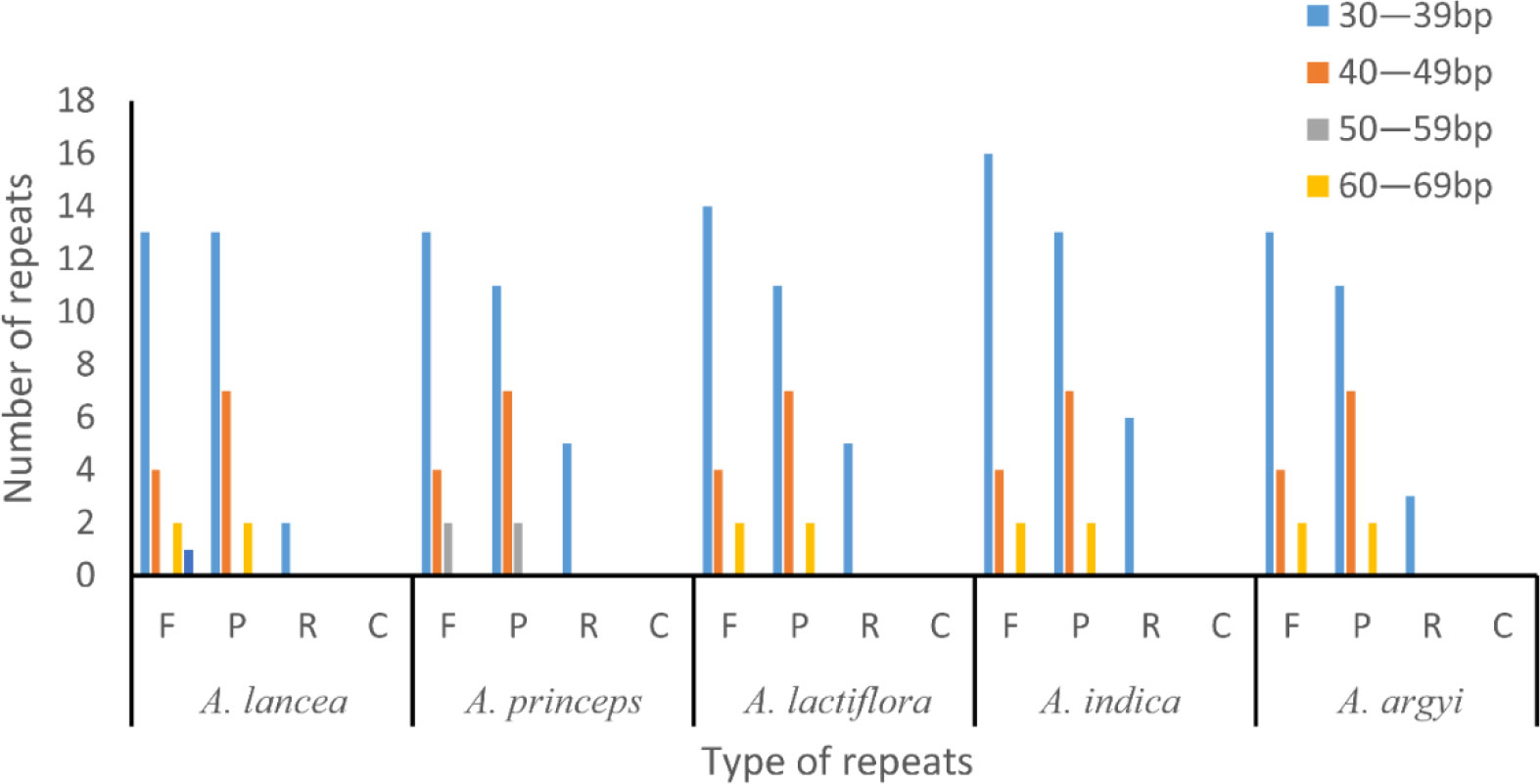
Figure 2 Long repeat sequence analysis of the genomes of five Artemisia species. F, forward repeat; P, palindromic repeat; R, reverse repeat; C, complementary repeat.
SSRs mainly comprise 1–6 types of nucleotide repeats. The cp genome exhibits characteristics of parthenogenesis, and SSRs highly vary within the same species. Therefore, SSR is widely used as a molecular marker in genetic map construction, target gene calibration, and mapping. We observed a total of 189–192 SSRs in the cp genomes of the 5 Artemisia species. Of these, 118–121 were mononucleotide SSRs, and most of them were of the A/T type (Figure 3 and Supplementary Table S3). The numbers of di-, tri-, tetra-, penta-, and hexanucleotide SSRs were 50–51, 5–6, 14–15, 1–2, and 0–1, respectively. Using comparative analysis, we found that the five Artemisia species had similar SSRs; however, the pentanucleotide repeat AAAAT/ATTTT only existed in A. argyi and the hexanucleotide repeat AAATAT/ATATTT only existed in A. indica. Overall, most SSRs comprised mono- or dinucleotide repeats. The types of oligonucleotide repeats were rich (Figure 3).
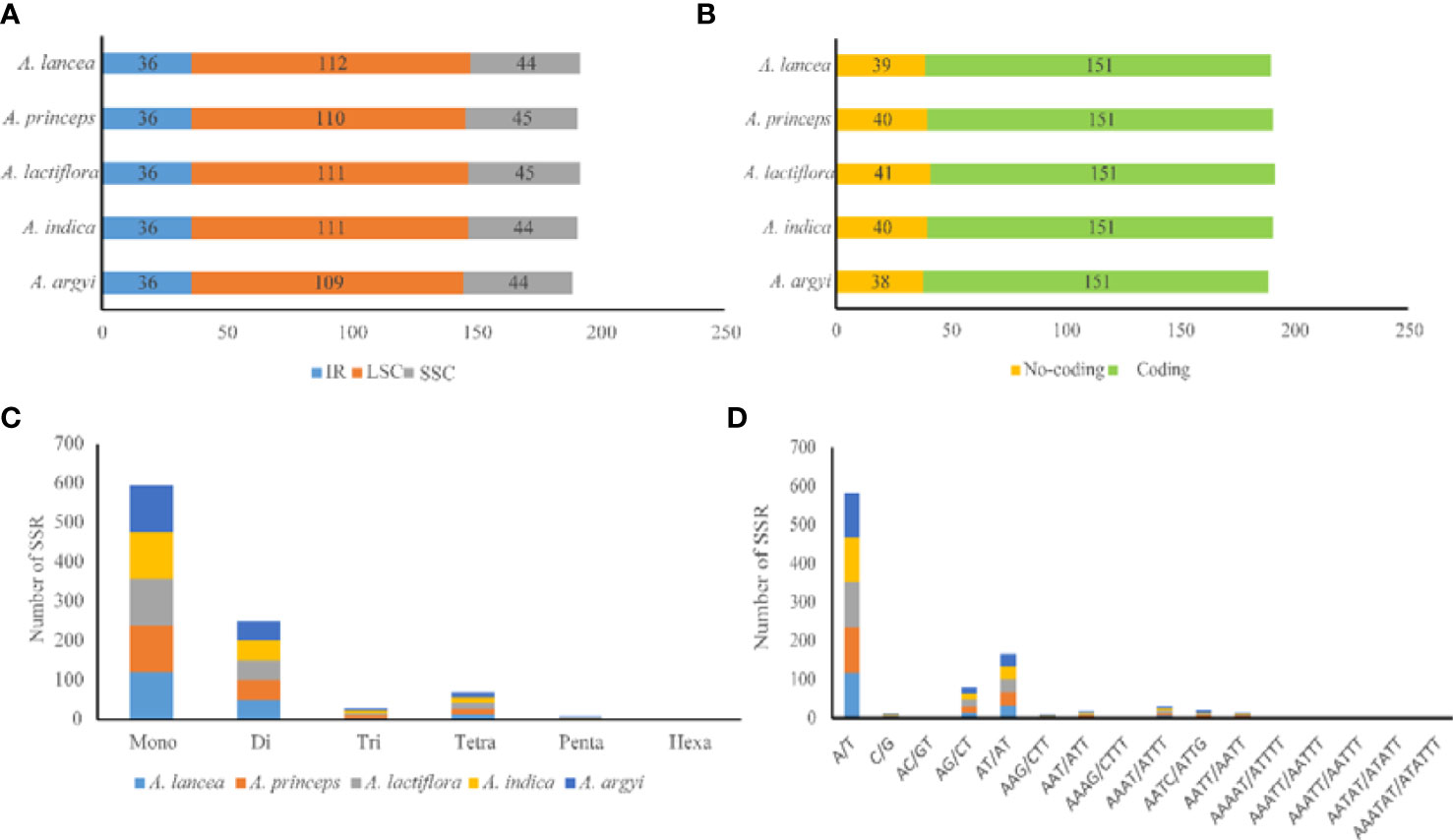
Figure 3 Type and distribution of SSRs in the five Artemisia cp genomes. (A) Frequency of SSRs in the LSC, SSC, and IR regions. (B) SSR distribution between coding and noncoding regions. (C) Number of SSR types. (D) Number of identified SSR motifs in different repeat class types. SSR, simple sequence repeat; LSC, large single copy region; SSC, small single copy region; IR, inverted repeat region.
Comparative analysis of the Cp genome
We used the DnaSP software to compare the nucleotide variation values (Pi) between all genes and intergenic regions in the cp genomes of the five Artemisia species. The hypervariable regions were detected, and the sequence differences were analyzed. Sliding window analysis revealed that the nucleotide diversity values within 600 bp varied from 0 to 0.006. Four mutational hotspots in the LSC and SSC regions were identified, including rpl32_trnL-UAG, trnY-GUA_trnE-UUC, ndhH_rps15, and ycf1 (Figure 4). These can be used as potential sites for studying population genetics and the identification of Artemisia species.
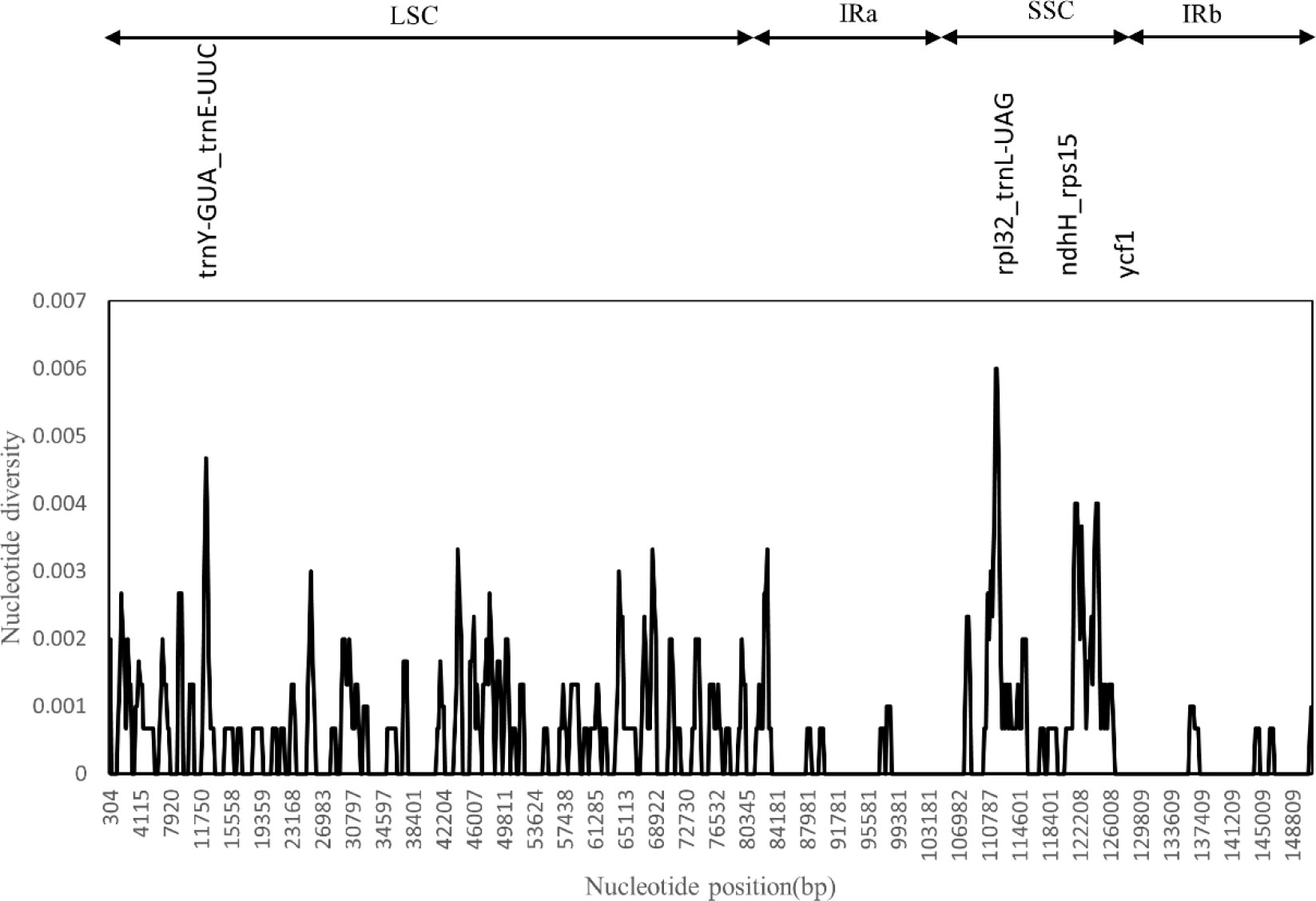
Figure 4 Sliding window test of nucleotide diversity (Pi) in the multiple alignments of the five Artemisia cp genomes. Peak regions with a Pi value of >0.004 were labeled with loci tags of the genic or intergenic region names. Pi values were calculated in the 600 bp sliding windows with steps of 200 bp. LSC, large single copy region; IRa, inverted repeat region a; SSC, small single copy region; IRb, inverted repeat region b.
Phylogenetic analysis
We constructed an ML tree using 29 cp genomes: 15 from the 5 Artemisia species used in this study and others from another 13 species and 1 outgroup. We found that all Artemisia species clustered together, and different repeat individuals in each species formed a monophyletic branch with a high branch supporting rate, indicating that the cp genome could distinguish the five Artemisia species. A. lactiflora showed the closest relationship to A. princeps, followed by A. indica and A. argyi, and was distant from A. lancea (Figure 5). A. scoparia and A. ordosica were grouped into one branch, revealing a close relationship between them.
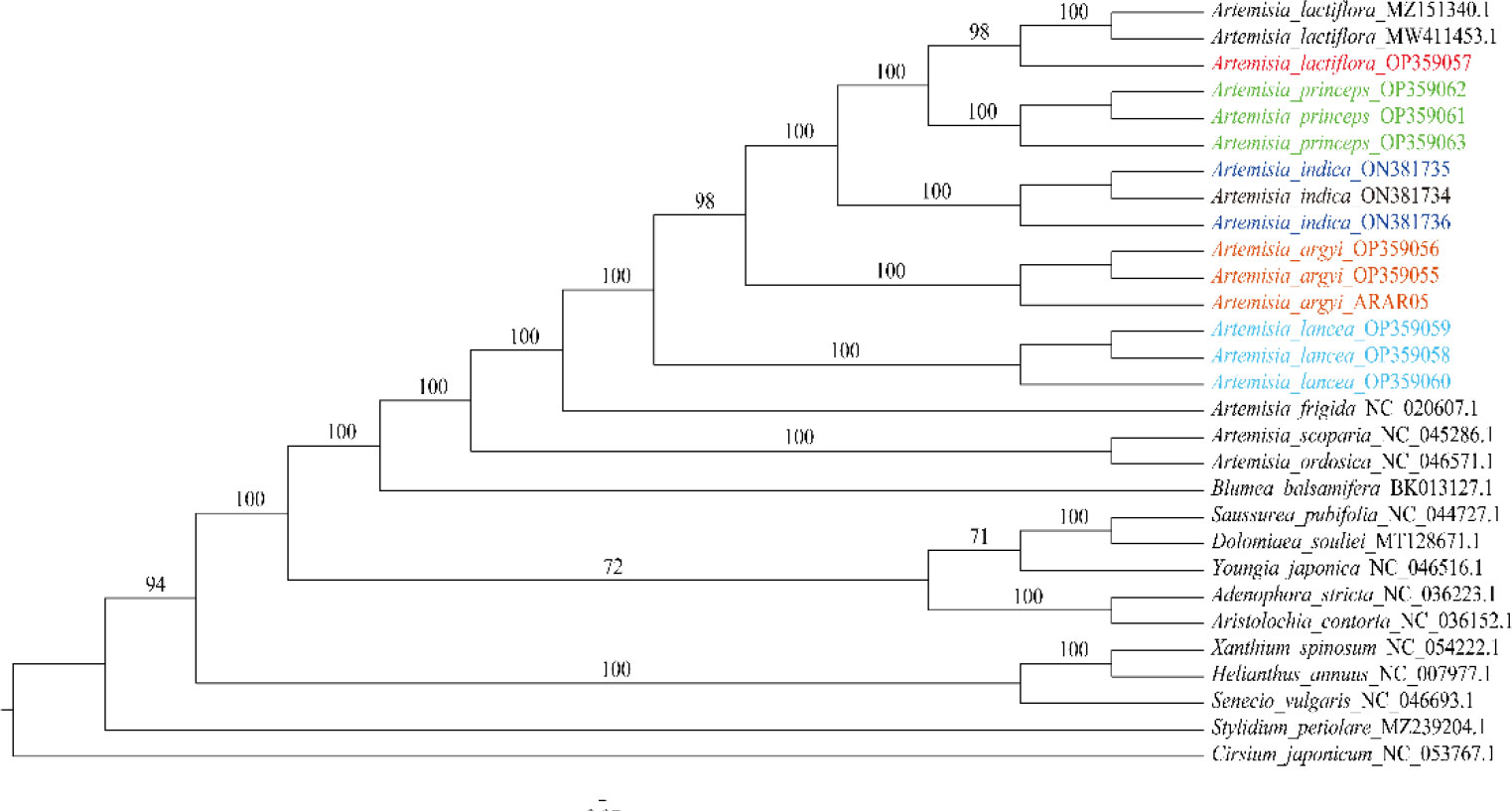
Figure 5 Phylogenetic tree constructed using the ML method based on the 29 cp genomes from 19 species. The numbers above the branches represent the ML bootstrap values. ML, maximum likelihood.
Discussion
In this study, we reported 12 newly sequenced cp genomes from 5 Artemisia medicinal species. We found that the genomes were extremely similar, with their size ranging from 151,132–151,178 bp. They belonged to medium-sized cp genomes compared to other Asteraceae species. The cp genomes of five Artemisia species contain 114 genes, which was similar to those of Artemisia annua (Shen et al., 2017). Like most other Artemisia species, ycf1 and rps19 were also detected, but the copy number and location were different, ycf1 spans the IRb/SSC boundary, this was also seen in A. scoparia and A. absinthium (Chen et al., 2022). The GC content of the IR region was significantly greater than that of the LSC and SSC regions. The AT content was higher than the GC content in all cp genomes. As observed in most plants, we found that the cp genome of Artemisia was conservative and no rearrangements were detected in the five species. Multiple codons that encode the same amino acid are known as synonymous codons. Codon usage is unequal, as some synonymous codons are used more frequently than others, a phenomenon known as codon preference. Codon preferences develop in the long-term evolution of plants, and different species have distinct preferences. In this study, we found that the amino acid Leu had the highest proportion of codons in the cp genomes of the five Artemisia species.
SSRs widely exist in the cp genome and provide important information regarding population genetics and evolution. Their types, numbers, and distribution vary in each plant. In this study, 57.29%–58.33% of the SSRs were mapped to the LSC region. An SSR-rich region may harbor mutational hotspots (George et al., 2015). The A/T type accounted for the largest proportion of SSRs. The obvious nucleotide bias may be due to the lower number of hydrogen bonds and lower energy consumption of A/T bases (Niu et al., 2017; Kim and Cheon, 2021). Previous studies have reported a higher A/T than G/C content in most plants, which may be due to the large number of A/T-type SSRs (Wang et al., 2021; Wu et al., 2021; Han et al., 2022). In this study, we analyzed the number, location, and composition of SSRs in the cp genomes of five Artemisia species and provided a new reference for further research on molecular markers, mutation hotspots, population genetics, and crop breeding.
The mutation hotspot analysis revealed a high degree of similarity among the cp genomes of the five Artemisia species, implying that the differentiation of these species was lower than that of other species. The lack of genome information has hindered the classification, identification, and protection of Artemisia species. The cp genome sequence provides a basis for the further study on genome evolution and the development of genetic resources. Mutation hotspots are often used for species identification, and these highly variable regions can serve as specific DNA barcodes. In this study, we identified four hypervariable regions—rpl32_trnL-UAG, trnY-GUA_trnE-UUC, ndhH_rps15, and ycf1—all of which have the potential to be used as DNA barcodes for subsequent studies on Artemisia species.
Phylogenetic analysis is extremely important for clarifying the genetic relationship between species and for protecting, rationally developing, and utilizing plant resources. The cp genome can solve some issues that morphological taxonomy cannot; hence, it has widely been used to explore the phylogenetic relationships between species (Kim Y. K. et al., 2020; Zhao et al., 2021). Due to the low degree of genetic differentiation and similar morphology of Artemisia species, obtaining more information on the genetic features of Artemisia is expected to improve phylogenetic resolution. In this study, using phylogenetic analysis, we showed that Artemisia was a branch of Asteraceae, the five Artemisia species are sister groups that can distinguish each other and different repeat individuals in each Artemisia species formed a monophyletic branch, indicating that the cp genome can be used as a super barcode to distinguish the five Artemisia species. The cp genome provided an effective marker for inferring the phylogenetic relationships between Artemisia species.
Conclusion
In this study, we analyzed 15 cp genomes from 5 Artemisia species, including 12 newly sequenced genomes from A. argyi, A. lactiflora, A. indica, A. princeps, and A. lancea, all of which have been used as medicinal plants for a long time. The cp genomes were similar in structure and gene content and were highly conserved. Four hotspot regions and 189–192 SSR molecular markers were identified, which can serve as potential DNA barcodes for further studies on Artemisia species. The phylogenetic analysis showed that the entire cp genome provides distinguishing features to help identify the five Artemisia species with high support rates. This study will contribute to the study of population genetics, species identification, and conservation biology of Artemisia species.
Data availability statement
The data presented in the study are deposited in the NCBI repository, accession numbers OP359055–63, and ON381735–36.
Author contributions
LW and LX designed the research study. ZL and RG performed the research. LW, QY, YS, JL, and YN collected Artemisia plant materials. ZL, CL, WW, and XT analyzed the data. ZL and LW wrote and revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by the National Natural Science Foundation of China (U1812403-1 and 81903758), the CACMS Innovation Fund (CI2021A05103 and CI2021A04112), the Fundamental Research Funds for the Central public welfare research institutes (ZZ13-YQ-106).
Acknowledgments
I would like to thank HT, GD, and TW for their valuable guidance during this research.
Conflict of interest
Authors JL and YN were employed by the company Hubei Aiaitie Health Technology Co., LTD.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2022.1049209/full#supplementary-material
Supplementary Table 1 | The cp genome information of the five Artemisia species.
Supplementary Table 2 | Relative synonymous codon usage values of the five Artemisia species.
Supplementary Table 3 | Simple sequence repeat types in the five Artemisia species.
Abbreviations
IR, inverted repeat region; LSC, large single copy region; SSC, small single copy region; ML, maximum likelihood; rRNA, ribosomal RNA; SSR, simple sequence repeats; tRNA, transfer RNA; RSCU, relative synonymous codon usage.
References
Chen, C., Miao, Y., Luo, D., Li, J., Wang, Z., Luo, M., et al. (2022). Sequence characteristics and phylogenetic analysis of the Artemisia argyi chloroplast genome. Front. Plant Sci. 13, 906725. doi: 10.3389/fpls.2022.906725
Chen, J., Xu, X., Lin, L., Guo, D., Gui, W., Lin, Y., et al. (2021). Research progress on pharmacological effects of artemisia argyi. J. Pharm. Res. 40, 5. doi: 10.13506/j.cnki.jpr.2021.12.009
Dong, W., Liu, J., Yu, J., Wang, L., Zhou, S. (2012). Highly variable chloroplast markers for evaluating plant phylogeny at low taxonomic levels and for DNA barcoding. PloS One 7, e35071. doi: 10.1371/journal.pone.0035071
Dong, W., Xu, C., Cheng, T., Lin, K., Zhou, S. (2013). Sequencing angiosperm plastid genomes made easy: A complete set of universal primers and a case study on the phylogeny of saxifragales. Genome Biol. Evol. 5, 989–997. doi: 10.1093/gbe/evt063
Dong, W., Xu, C., Li, W., Xie, X., Lu, Y., Liu, Y., et al. (2017). Phylogenetic resolution in juglans based on complete chloroplast genomes and nuclear DNA sequences. Front. Plant Sci. 8, 1148. doi: 10.3389/fpls.2017.01148
Dong, W., Xu, C., Wu, P., Cheng, T., Yu, J., Zhou, S., et al. (2018). Resolving the systematic positions of enigmatic taxa: Manipulating the chloroplast genome data of saxifragales. Mol. Phylogenet. Evol. 126, 321–330. doi: 10.1016/j.ympev.2018.04.033
Garcia, S., Canela, M. A., Garnatje, T., Mcarthur, E. D., Pellicer, J., Sanderson, S. C., et al. (2008). Evolutionary and ecological implications of genome size in the north American endemic sagebrushes and allies (Artemisia, asteraceae). Biol. J. Linn. Soc. 94, 631–649. doi: 10.1111/j.1095-8312.2008.01001.x
George, B., Bhatt, B. S., Awasthi, M., George, B., Singh, A. K. (2015). Comparative analysis of microsatellites in chloroplast genomes of lower and higher plants. Curr. Genet. 61, 665–677. doi: 10.1007/s00294-015-0495-9
Green, B. R. (2011). Chloroplast genomes of photosynthetic eukaryotes. Plant J. 66, 34–44. doi: 10.1111/j.1365-313X.2011.04541.x
Han, H., Qiu, R., Liu, Y., Zhou, X., Gao, C., Pang, Y., et al. (2022). Analysis of chloroplast genomes provides insights into the evolution of agropyron. Front. Genet. 13, 832809. doi: 10.3389/fgene.2022.832809
Hong, F., Zhao, M., Xue, L. L., Ma, X., Liu, L., Cai, X. Y., et al. (2022). The ethanolic extract of artemisia anomala exerts anti-inflammatory effects via inhibition of NLRP3 inflammasome. Phytomedicine 102, 154163. doi: 10.1016/j.phymed.2022.154163
Hsueh, T. P., Lin, W. L., Dalley, J. W., Tsai, T. H. (2021). The pharmacological effects and pharmacokinetics of active compounds of artemisia capillaris. Biomedicines 10, 1412. doi: 10.3390/biomedicines9101412
Jansen, R. K., Raubeson, L. A., Boore, J. L., Depamphilis, C. W., Chumley, T. W., Haberle, R. C., et al. (2005). Methods for obtaining and analyzing whole chloroplast genome sequences. Methods Enzymol. 395, 348–384. doi: 10.1016/S0076-6879(05)95020-9
Kim, K. A., Cheon, K. S. (2021). Complete chloroplast genome sequence of adenophora racemosa (Campanulaceae): Comparative analysis with congeneric species. PloS One 16, e0248788. doi: 10.1371/journal.pone.0248788
Kim, Y. K., Jo, S., Cheon, S. H., Joo, M. J., Kim, K. J., Hong, J. R., et al. (2020). Plastome evolution and phylogeny of orchidaceae, with 24 new sequences. Front. Plant Sci. 11, 22. doi: 10.3389/fpls.2020.00022
Kim, G. B., Lim, C. E., Kim, J. S., Kim, K., Lee, J. H., Yu, H. J., et al. (2020). Comparative chloroplast genome analysis of artemisia (Asteraceae) in East Asia: Insights into evolutionary divergence and phylogenomic implications. BMC Genomics 21, 415. doi: 10.1186/s12864-020-06812-7
Kurtz, S., Choudhuri, J. V., Ohlebusch, E., Schleiermacher, C., Stoye, J., Giegerich, R. (2001). REPuter: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 29, 4633–4642. doi: 10.1093/nar/29.22.4633
Lan, Z., Tian, X., Shi, Y., Gao, R., Yin, Q., Xiang, L., et al. (2022). Chloroplast genome structure characteristics and phylogenetic analysis of artemisia indica. China J. Chin. Materia. Med., 47, 224–231. doi: 10.19540/j.cnki.cjcmm.20220713.101
Lin, C. P., Wu, C. S., Huang, Y. Y., Chaw, S. M. (2012). The complete chloroplast genome of ginkgo biloba reveals the mechanism of inverted repeat contraction. Genome Biol. Evol. 4, 374–381. doi: 10.1093/gbe/evs021
Liu, G., Ning, H., Ayidaerhan, N., Aisa, H. A. (2017). Evaluation of DNA barcode candidates for the discrimination of artemisia l. Mitochondrial. DNA Part A. 28, 956–964. doi: 10.1080/24701394.2016.1219729
Liu, C., Shi, L., Zhu, Y., Chen, H., Zhang, J., Lin, X., et al. (2012). CpGAVAS, an integrated web server for the annotation, visualization, analysis, and GenBank submission of completely sequenced chloroplast genome sequences. BMC Genomics 13, 715. doi: 10.1186/1471-2164-13-715
Li, X., Yang, Y., Henry, R. J., Rossetto, M., Wang, Y., Chen, S. (2015). Plant DNA barcoding: from gene to genome. Biol. Rev. Camb. Philos. Soc 90, 157–166. doi: 10.1111/brv.12104
Lohse, M., Drechsel, O., Bock, R. (2007). OrganellarGenomeDRAW (OGDRAW): a tool for the easy generation of high-quality custom graphical maps of plastid and mitochondrial genomes. Curr. Genet. 52, 267–274. doi: 10.1007/s00294-007-0161-y
Mader, M., Pakull, B., Blanc-Jolivet, C., Paulini-Drewes, M., Bouda, Z. H., Degen, B., et al. (2018). Complete chloroplast genome sequences of four meliaceae species and comparative analyses. Int. J. Mol. Sci. 19, 701. doi: 10.3390/ijms19030701
Nguyen, L. T., Schmidt, H. A., von Haeseler, A., Minh, B. Q. (2015). IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274. doi: 10.1093/molbev/msu300
Niu, Z., Xue, Q., Wang, H., Xie, X., Zhu, S., Liu, W., et al. (2017). Mutational biases and GC-biased gene conversion affect GC content in the plastomes of dendrobium genus. Int. J. Mol. Sci. 18, 2307. doi: 10.3390/ijms18112307
Riggins, C. (2008). Molecular phylogenetic and biogeographic study of the genus artemisia (Asteraceae), with an emphasis on section absinthium [D] (USA:University of Illinois at Urbana-Champaign).
Shen, X., Wu, M., Liao, B., Liu, Z., Bai, R., Xiao, S., et al. (2017). Complete chloroplast fenome sequence and phylogenetic analysis of the medicinal plant artemisia annua. Molecules 22, 1330. doi: 10.3390/molecules22081330
Thiel, T., Michalek, W., Varshney, R. K., Graner, A. (2003). Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare l.). Theor. Appl. Genet. 106, 411–422. doi: 10.1007/s00122-002-1031-0
Twyford, A. D., Ness, R. W. (2017). Strategies for complete plastid genome sequencing. Mol. Ecol. Resour. 17, 858–868. doi: 10.1111/1755-0998.12626
Wang, Y., Wang, S., Liu, Y., Yuan, Q., Sun, J., Guo, L. (2021). Chloroplast genome variation and phylogenetic relationships of atractylodes species. BMC Genomics 22, 103. doi: 10.1186/s12864-021-07394-8
Wang, X., Zheng, S., Liu, Y., Han, J. (2016). ITS2, a better DNA barcode than ITS in identification of species in artemisia l. Chin. Herbal. Medicines 8, 352–358. doi: 10.1016/S1674-6384(16)60062-X
Watson, L. E., Bates, P. L., Evans, T. M., Unwin, M. M., Estes, J. R. (2002). Molecular phylogeny of subtribe artemisiinae (Asteraceae), including artemisia and its allied and segregate genera. BMC Evol. Biol. 2, 17. doi: 10.1186/1471-2148-2-17
Wu, M. L., Li, Q., Xu, J., Li, X. W. (2018). Complete chloroplast genome of the medicinal plant amomum compactum: gene organization, comparative analysis, and phylogenetic relationships within zingiberales. Chin. Med. 13, 10. doi: 10.1186/s13020-018-0164-2
Wu, L., Wu, M., Cui, N., Xiang, L., Li, Y., Li, X., et al. (2021). Plant super-barcode: a case study on genome-based identification for closely related species of fritillaria. Chin. Med. 16, 52. doi: 10.1186/s13020-021-00460-z
Yang, Z., Zhao, T., Ma, Q., Liang, L., Wang, G. (2018). Comparative genomics and phylogenetic analysis revealed the chloroplast genome variation and interspecific relationships of corylus (Betulaceae) species. Front. Plant Sci. 9, 927. doi: 10.3389/fpls.2018.00927
Zhang, L., Wang, S., Su, C., Harris, A. J., Zhao, L., Su, N., et al. (2021). Comparative chloroplast genomics and phylogenetic analysis of zygophyllum (Zygophyllaceae) of China. Front. Plant Sci. 12, 723622. doi: 10.3389/fpls.2021.723622
Zhang, T., Xing, Y., Xu, L., Bao, G., Zhan, Z., Yang, Y., et al. (2019). Comparative analysis of the complete chloroplast genome sequences of six species of pulsatilla miller, ranunculaceae. Chin. Med. 14, 53. doi: 10.1186/s13020-019-0274-5
Zhao, F., Chen, Y. P., Salmaki, Y., Drew, B. T., Wilson, T. C., Scheen, A. C., et al. (2021). An updated tribal classification of lamiaceae based on plastome phylogenomics. BMC Biol. 19, 2. doi: 10.1186/s12915-020-00931-z
Keywords: Artemisia Linn., chloroplast genome, genome comparison, species identification, phylogenetic analysis, simple sequence repeat
Citation: Lan Z, Shi Y, Yin Q, Gao R, Liu C, Wang W, Tian X, Liu J, Nong Y, Xiang L and Wu L (2022) Comparative and phylogenetic analysis of complete chloroplast genomes from five Artemisia species. Front. Plant Sci. 13:1049209. doi: 10.3389/fpls.2022.1049209
Received: 20 September 2022; Accepted: 26 October 2022;
Published: 21 November 2022.
Edited by:
Weijun Kong, Capital Medical University, ChinaReviewed by:
Long-Fei Fu, Guangxi Institute of Botany (CAS), ChinaZubaida Yousaf, Lahore College for Women University, Pakistan
Yong Gao, Qujing Normal University, China
Copyright © 2022 Lan, Shi, Yin, Gao, Liu, Wang, Tian, Liu, Nong, Xiang and Wu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lan Wu, lwu@icmm.ac.cn; Li Xiang, lxiang@icmm.ac.cn