 Hui Zhang1,2†
Hui Zhang1,2† Sha Tang1†
Sha Tang1† James C. Schnable3
James C. Schnable3 Qiang He1
Qiang He1 Yuanzhu Gao1
Yuanzhu Gao1 Mingzhao Luo1
Mingzhao Luo1 Guanqing Jia1
Guanqing Jia1 Baili Feng2
Baili Feng2 Hui Zhi1
Hui Zhi1 Xianmin Diao1*
Xianmin Diao1*- 1Institute of Crop Science, Chinese Academy of Agricultural Sciences, Beijing, China
- 2State Key Laboratory of Crop Stress Biology for Arid Areas, College of Agronomy, Northwest A&F University, Yangling, China
- 3Department of Agronomy and Horticulture, University of Nebraska–Lincoln, Lincoln, NE, United States
Genome-wide DNA polymorphism analysis and molecular marker development are important for forward genetics research and DNA marker-assisted breeding. As an ideal model system for Panicoideae grasses and an important minor crop in East Asia, foxtail millet (Setaria italica) has a high-quality reference genome as well as large mutant libraries based on the “Yugu1” variety. However, there is still a lack of genetic and mutation mapping tools available for forward genetics research on S. italica. Here, we screened another S. italica genotype, “SSR41”, which is morphologically similar to, and readily cross-pollinates with, “Yugu1”. High-throughput resequencing of “SSR41” identified 1,102,064 reliable single nucleotide polymorphisms (SNPs) and 196,782 insertions/deletions (InDels) between the two genotypes, indicating that these two genotypes have high genetic diversity. Of the 8,361 high-quality InDels longer than 20 bp that were developed as molecular markers, 180 were validated with 91.5% accuracy. We used “SSR41” and these developed molecular markers to map the white leaf sheath gene SiWLS1. Further analyses showed that SiWLS1 encodes a chloroplast-localized protein that is involved in the regulation of chloroplast development in bundle sheath cells in the leaf sheath in S. italica and is related to sensitivity to heavy metals. Our study provides the methodology and an important resource for forward genetics research on Setaria.
Introduction
Foxtail millet (Setaria italica) and its wild ancestor green foxtail (Setaria viridis) are annual diploid grass species. Foxtail millet is a significant minor cereal crop and a dietary staple in a wide area of northern China and India. According to FAOSTAT reports, foxtail millet combined with other millet crops grown in an area of 9,349,969 hectares in China and India produced over 12.5 million tons of food resources in 2019.1
Setaria italica has many distinctive characteristics, including (i) C4 photosynthesis and tolerance to drought and low-quality soils; (ii) small, high-quality reference genomes (Bennetzen et al., 2012), with well-organized bioinformatic databases2,3 ; (iii) a short lifespan, the ability to self-pollinate, and prolific seed production; (iv) easy management in the laboratory (easily grown in growth chambers, no-seed shattering, and short seed dormancy). Because of these advantages, Setaria has been proposed as a molecular genetic model plant not only for C4 biology but also for crop functional genomics analyses (Diao et al., 2014).
Positional cloning of functional genes is one of the research hotspots in plant functional genomics. It has been proved to be a reliable and powerful tool for novel gene identification in well-established model systems, such as Arabidopsis and rice (Oryza sativa). In recent years, we have made significant advances in making positional cloning available for S. italica (Li et al., 2016; Liu et al., 2016; Zhang et al., 2018a), an emerging model system for Panicoideae grasses. Positional cloning starts with mutant or designed germplasm resources. “Yugu1” as its full-available genome sequences (Bennetzen et al., 2012), steady growth performance, and wide adaptability is an ideal plant material for gene functional analysis. Our team has constructed a large-scale ethyl methylsulfone (EMS)-induced mutant library based on “Yugu1”, and this library has been used in Setaria functional genomics research (Li et al., 2016; Zhang et al., 2018a). “SSR41” is another foxtail millet variety that originated from South Korea (Jia et al., 2013). We propose that “SSR41” is an ideal genotype to match to “Yugu1” mutants to construct mapping populations, because “SSR41” is morphologically similar to, but highly genetically divergent from, “Yugu1”. To data, several genes of S. italica have been cloned using “SSR41” and the corresponding “Yugu1” mutant, including DPY1 (Zhao et al., 2020), SiAGL1b (Liu et al., 2016), SiSTL1 (Tang et al., 2019), SiSTL2 (Zhang et al., 2018a), SiYGL1 (Li et al., 2016), and SiYGL2 (Zhang et al., 2018b). In addition, “SSR41” is an elite cultivar with high yields, good grain quality, resistance to diverse diseases, and wide local adaptability, so it will be useful for breeding and crop improvement.
At present, there is little genetic background and genome information available for “SSR41”. With the progress in next generation sequencing (NGS) technologies, obtaining genome variation information for various plants has become more efficient, accurate, and cost-effective (Slatko et al., 2018). Mamidi et al. (2020) used the Illumina Hiseq 2000 platform to sequence and de novo assemble 589 green foxtail accessions. Using presence-absence variation (PAV) and single nucleotide polymorphism (SNP) diversity analyses, they defined the population structure and evolutionary history of S. viridis in North America. Through a genome-wide association study (GWAS) of an agronomically important phenotype, they identified valuable loci-controlling seed shattering and a leaf angle in Setaria (Mamidi et al., 2020). Therefore, high-throughput resequencing and comparative genomics analyses of foxtail millet varieties can provide useful information for the Setaria model system.
Leaf color (including leaf blade color, leaf sheath color, and leaf ligule color) is a common and easily identifiable agronomic trait in crops. Isolation of leaf color mutants and identifying the key genes regulating this trait are of great importance for determining how chlorophyll metabolism and chloroplast development are regulated (Wu et al., 2007), and for understanding the mechanisms of photosynthesis and designing crops with high photosynthetic efficiency (Cui et al., 2019). In addition, leaf sheath color can be used as a visible morphological marker to allow breeders to identify authentic hybrids (Oshima et al., 2019). Several studies have explored the genetic control of leaf sheath color in rice and maize (Zea mays), and a few genes regulating leaf sheath color have been identified (Li et al., 2018; Hu et al., 2020). In S. italica, only one leaf sheath color gene has been fine-mapped and cloned (Bai et al., 2020), but this is insufficient to support further research on this trait.
The CLT1 gene encodes a chloroquine-resistance transporter-like transporter. This gene has been characterized in Arabidopsis and rice (Lim et al., 2011). Homozygous mutants lacking CLT1 transporter activity are sensitive to heavy metals, such as arsenic (As) and cadmium (Cd), and show disrupted glutathione (GSH) homeostasis (Yang et al., 2016). In rice, β-glucuronidase (GUS) staining of an OsCLT1 promoter-GUS transgenic line indicated that this gene is highly expressed in the leaf sheath, while subcellular localization analyses revealed that OsCLT1 localizes in chloroplasts (Yang et al., 2016). However, the role of CLT1 in regulating chloroplast or leaf sheath development is unknown.
In this study, deep whole-genome resequencing was employed to re-sequence the genome of the important S. italica genotype “SSR41”. The genomic variations and DNA polymorphisms between “SSR41” and “Yugu1” were identified. Characterization of these detected SNPs, InDels, and structure variations (SVs) shed light on the genetic basis of the desirable traits of “SSR41”. More than 8,000 genome-wide molecular markers were developed, and 180 of them were validated by PCR. Using these newly developed markers, we mapped the novel white leaf sheath gene SiWLS1 encoding a chloroquine-resistance transporter in foxtail millet by positional cloning. Our research provides useful genome resources for gene cloning and molecular breeding of foxtail millet and will be useful for the further development of Setaria as a model system.
Materials and Methods
Plant Growth and Investigation of Agronomic Traits
“Yugu1” and “SSR41” are two foxtail millet varieties deposited in the National Crop Gene Bank, Chinese Academy of Agricultural Sciences (CAAS, Beijing, China). There are no segregating variants within each cultivar so that any sequence variations identified can confidently be interpreted as between-accession polymorphisms. A white leaf sheath mutant siwls1 was isolated from the foxtail millet EMS mutant resource. The leaf sheath is white in the mutant but green in wild-type plants. An F2 mapping population of 982 individuals was generated by hybridization between “siwls1” and “SSR41”, and was used for map-based cloning. All plants were grown at the Shunyi Experimental Station at the CAAS (40°18′ N, 116°58′ E) during June 20 to October 5 in 2018. Five uniformly developed plants were collected at the seed-milking stage to investigate major agronomic traits. The assessment methods and scoring standards for agronomic traits have been described in our previous study (Jia et al., 2013).
DNA Isolation and Whole Genome Resequencing
The genomic DNAs were extracted from leaves of 4-week-old plants of “SSR41”, “Yugu1”, the siwls1 mutant, and individuals in the F2 population using a modified CTAB method (Zidani et al., 2005). Construction of DNA libraries and whole-genome resequencing were performed according to the Illumina DNA Prep workflow4 by the Beijing Berry Genomic Co. Ltd. (Beijing, China). The Illumina Hiseq 2500 platform and the 150-bp paired-end mode were employed for high-throughput sequencing. Raw sequencing reads have been submitted to the EMBL-EBI5 under the accession No. PRJEB46028. The reference genome was S. italica genome version 2.2.6 Reads filtering, reads mapping, variant calling, and variant annotation were performed as described in our previous study (He et al., 2021).
Development and Validation of Polymorphic Molecular Markers
Previous studies have developed a number of simple sequence repeat (SSR) and SNP markers that can be used for genotyping different foxtail millet accessions (Jia et al., 2013; Zhang et al., 2014). However, SSR molecular markers need to be detected by denaturing polyacrylamide gel electrophoresis, which is laborious and time-consuming. Because of their codominance, convenience, practicality, and highly polymorphic nature (Jain et al., 2019), InDel markers, especially those longer than 20 bp, are becoming a popular agar-gel-based genotyping solution. Based on the resequencing results of “SSR41”, putative polymorphic InDels with length ≥20 bp were selected to develop molecular markers for foxtail millet. To further ensure the efficiency and polymorphism of the developed markers, we compared them among 1,826 Setaria accessions (1,237 S. italica and 589 S. viridis) for which NGS data are available (Jia et al., 2013; Mamidi et al., 2020; Li et al., 2021). The polymorphism information content (PIC) value of each marker was calculated as described elsewhere (Lemos et al., 2019). Primers were designed using Primer3 software7 and were used to amplify the sequences containing the putative polymorphic InDel loci. The optimized parameters for Primer3 inputs were as follows: PCR target product size, 200–350 bp; primer size, 18–29 bp; primer melting temperature (Tm), 55–64°C; and primer GC content, 35–65%. The primers designed for all molecular markers are listed in Supplementary Table 1. To validate the polymorphic molecular markers, we selected 180 markers evenly distributed among all nine chromosomes of S. italica. The target sequences were amplified by PCR using “SSR41” and “Yugu1” genomic DNA as the templates. The PCR products were separated in 3.0% agarose gels, stained with Gelred plus (mei5bio, Beijing, China), and imaged using the Gel Doc XR system (Bio-Rad, Hercules, CA, United States).
Positional Cloning of SiWLS1 Using the Newly Developed Molecular Markers
We used the F2 mapping population of 982 individuals obtained from hybridization between “siwls1” and “SSR41” for positional cloning. Fifty-four InDel markers evenly distributed across the nine chromosomes of foxtail millet (Supplementary Tables 1, 2) were selected for primary mapping using bulked segregant analysis (BSA). The basic principles and methods of BSA have been described previously (Michelmore et al., 1991), but a brief introduction to the method is given in Supplementary Figures 1A,B. In practical terms, we selected 30 recessive individuals segregated from the “SSR41” × “siwls1” F2 population. Then, DNA was extracted from each individual and mixed in equal amounts to generate a DNA pool. We used DNA from male (♂), female (♀), and F1 plants and the DNA pool (P) as templates for PCR amplifications of all selected markers (Supplementary Figures 1C,D). For fine mapping, five new InDel markers and three SNP markers located in the candidate region were developed, and 237 recessive individuals were analyzed. Candidate genes located within the finely mapped region were amplified and sent for Sanger sequencing to determine the mutation site. The molecular markers and primer sequences used for gene mapping and sequencing are listed in Supplementary Table 2. The phylogenetic tree of the candidate gene SiWLS1 was constructed using MEGA X software with the maximum likelihood method and default parameters. The full-length protein sequences of SiWLS1 and its homologs used in this analysis are listed in Supplementary Table 3.
Treatment and Phenotyping of the siwls1 Mutant
To investigate the As and Cd sensitivity of siwls1, seeds of the mutant and wild type were germinated on moist filter paper overnight, and then exposed to solutions containing Na3AsO4 or CdSO4 at a range of concentrations (0, 10, 30, and 60 μM), respectively. Plants were grown in a growth chamber under a 10-h light (30°C)/14-h dark (26°C) photoperiod, with 40% humidity. After 7 days of treatment, five uniformly developed seedlings in each treatment were selected for measurements of plant height and root length. To investigate whether the white leaf sheath trait is related to chlorophyll (Chl) content, we measured Chl a and Chl b contents using acetone as the solvent. For these analyses, leaf sheaths of siwls1 and Yugu1 plants were collected at the heading stage (n = 15). Details of the chlorophyll measurement method have been provided in our previous study (Li et al., 2016).
Chloroplast Ultrastructure Observation and Subcellular Location Analysis
Fresh leaf sheaths of siwls1 and “Yugu1” plants were collected at the stem elongation stage. Samples were cut into 2 × 1-mm2 sections and fixed in 2.5% glutaraldehyde overnight. The plant tissues were dehydrated, embedded, and cut into sections as described previously (Zhang et al., 2018a). Ultrathin sections of embedded samples were stained with uranyl acetate, and then observed using a transmission electron microscope (TEM) (JEM 1230, JEOL, Tokyo, Japan). To investigate the subcellular localization of SiWLS1 in foxtail millet, we constructed an SiWLS1-GFP fusion protein. The primers used for vector construction were as follows: 5′-gacgatatctctagaggatccATGGCGCCGCCGTCCCCT-3′ and 5′-gcccttgctcaccatggatccGTCATTCTTGTTGTGGAAAGAATTT C-3′, where lowercase sequences are adaptors containing BamHI cleavage sites for the ClonExpress® Ultra One Step Cloning Kit (C115-02, Vazyme Biotech, Nanjing, China). The fusion vector and the control vector were then transfected into foxtail millet protoplasts (Liu et al., 2016). Then, GFP fluorescence was observed and photographed under a Zeiss LSM 700 confocal microscope (Carl Zeiss, Jena, Germany).
Results
“SSR41” Is an Ideal Material to Coordinate With “Yugu1”
Map-based cloning has become feasible for S. italica with the release of the “Yugu1” reference genome (Bennetzen et al., 2012) and the construction of a large-scale EMS-induced mutant library based on “Yugu1” (Sun et al., 2019). On that basis, we screened another S. italica germplasm “SSR41”, which readily cross-pollinates with “Yugu1”, and constructed large populations for mutation mapping. In the comparison between “Yugu1” and “SSR41” plants grown in different environments, major agronomic traits including mature plant height, leaf length and width, and panicle shape showed no significant differences (Supplementary Table 4 and Figures 1A–D). Therefore, the two varieties have similar plant stature and vegetative phenotypes, making it easy to identify mutant phenotypes in their hybrid progeny easily and clearly. Importantly, the heading dates and flowering periods of “SSR41” were almost identical to those of “Yugu1” in five different geographic regions (Figure 1E). These data indicate the two varieties are widely adapted to different environments, which is an advantage for designing cross-pollination experiments.
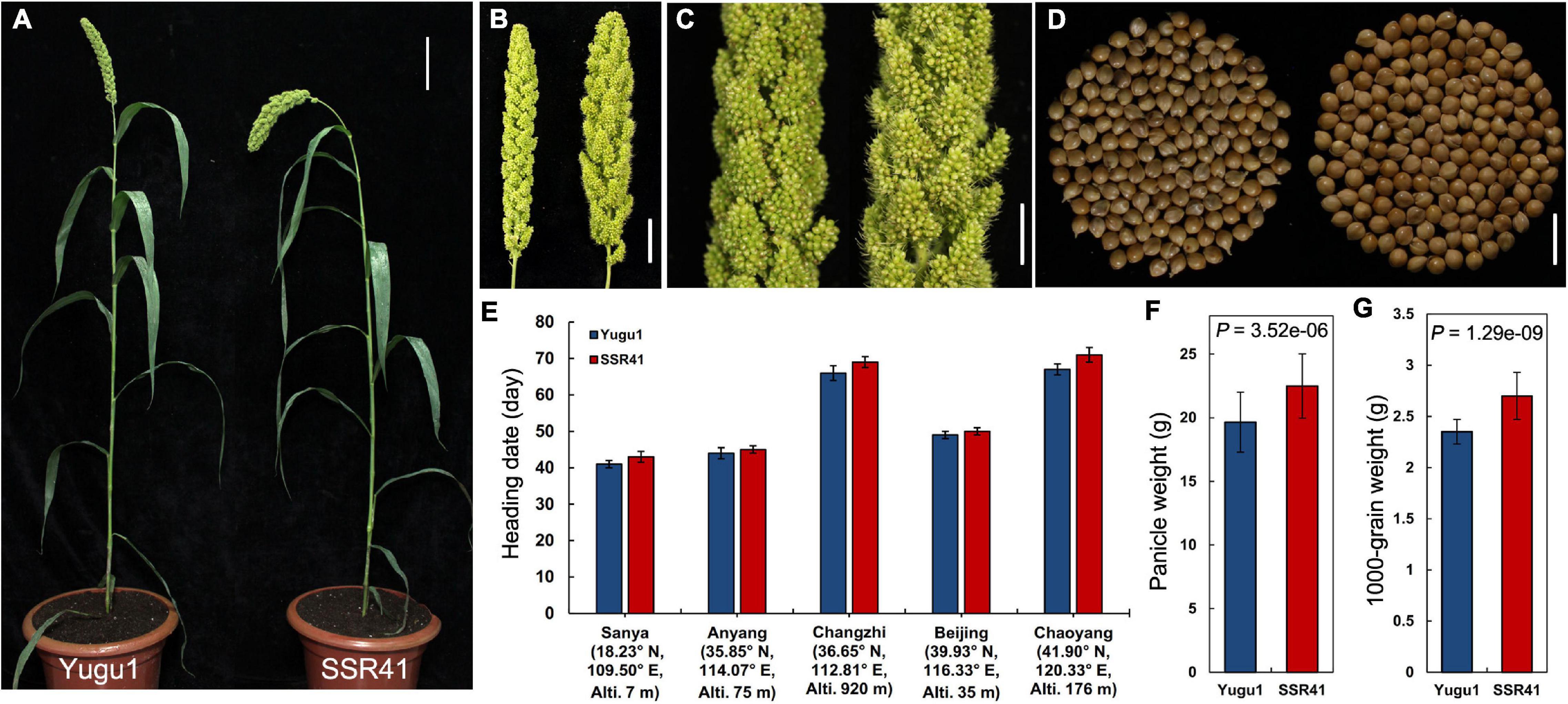
Figure 1. Phenotypes and statistical comparisons between “SSR41” and “Yugu1”. (A) Phenotype of whole plants at the heading stage. Bar = 10 cm. (B) Panicle of “Yugu1” (left) and “SSR41” (right). Bar = 3 cm. (C) An enlarged view of primary branches and spikelets of “Yugu1” (left) and “SSR41” (right). Bar = 1.5 cm. (D) Seeds of “Yugu1” (left; 150 seeds) and “SSR41” (right; 150 seeds). Bar = 5 mm. (E–G) Statistical comparisons of heading date, panicle weight, and 1,000-grain weight between “Yugu1” and “SSR41”. Values are means ± SD (n = 3 for heading date, n = 6 for panicle weight, and 1,000-grain weight). P values were determined using Student’s t-test.
The values of agronomic characters related to crop yield, such as panicle weight and 1,000-grain weight, were significantly higher in “SSR41” than in “Yugu1” (Figures 1F,G), suggesting that “SSR41” is an ideal germplasm resource for breeding to improve foxtail millet.
Whole Genome Resequencing of “SSR41”
For the above reasons, the S. italica genotype “SSR41” was chosen as a suitable partner with “Yugu1” for positional cloning of genes in Setaria. Hence, we used an NGS strategy for genome-wide detection of DNA polymorphism between the two genotypes. General statistics of the high throughput sequencing and reads mapping are shown in Supplementary Figure 2A. In total, 149,179,426 high-quality clean reads (>22.11 billion nucleotides) were generated. About 99.22% of the sequencing reads were successfully mapped onto the “Yugu1” reference genome. The average sequencing depth was approximately 48-fold, and the mapped sequences covered ∼95.73% of the reference genome. Analysis of the distribution of sequence coverage suggested that reads were evenly distributed throughout the available genome, indicating that the quality and randomness of our resequencing data were acceptable (Supplementary Figure 2B). The number of sequencing reads mapped to each SNP and the genomic distance between adjacent SNPs was calculated (Supplementary Figure 2C), and provided further evidence for the credibility of variation calling.
Detection and Distribution of Variations Between the Two S. italica Genotypes
After filtering, 1,102,064 reliable SNPs were detected between “Yugu1” and “SSR41”, with a low heterozygosity ratio of 21.44%. On the basis of the nucleotide substitutions, these SNPs were divided into two categories: transitions (A↔G and T↔C) (1,400,101 transitions); and transversions (C↔A, G↔C, A↔T, and T↔G) (513,280 transversions), with an overall transition (Ts)/transversion (Tv) ratio of 2.73 (Supplementary Figure 3). We also evaluated the genome-wide InDel polymorphisms. In total, 196,782 InDels were identified after aligning sequences of “SSR41” and “Yugu1”, of which 162,262 (82.46%) were homozygous. The length of InDels ranged from 1 to 298 bp. Almost half of these variations (48.07%) were single-nucleotide InDels, 44.34% were 2–20 bp in length, and a small proportion (6.70%) was longer than 20 bp.
The average densities of polymorphisms detected between the “SSR41” and “Yugu1” genomes were 2719.65 SNPs per Mb, and 485.61 InDels per Mb. The average variant rates were one SNP every 368 bases, and one InDel every 2,059 bases. The distribution of SNPs and InDels varied among chromosomes. As shown in Supplementary Figure 3 and Figure 2, Chromosome 8 had the highest density of SNPs (5440.81 Mb–1) and InDels (982.08 Mb–1), and Chromosome 9 had the lowest SNP/InDel density (1333.66/240.74 Mb–1). We also calculated the numbers of SNPs/InDels per Mb on different chromosomes and found that the distribution of SNPs/InDels was uneven within each chromosome (Figure 2). High-density regions (12.8%) had >6,000 SNPs per Mb (e.g., 10,403 SNPs distributed on Chromosome 8, 22.0–23.0 Mb region), while four low-density regions had <200 SNPs per Mb (e.g., 136 SNPs on Chromosome 2, 44.0–45.0 Mb). The trends in the variant rates and distribution of InDels were similar to those of SNPs (Figure 2).
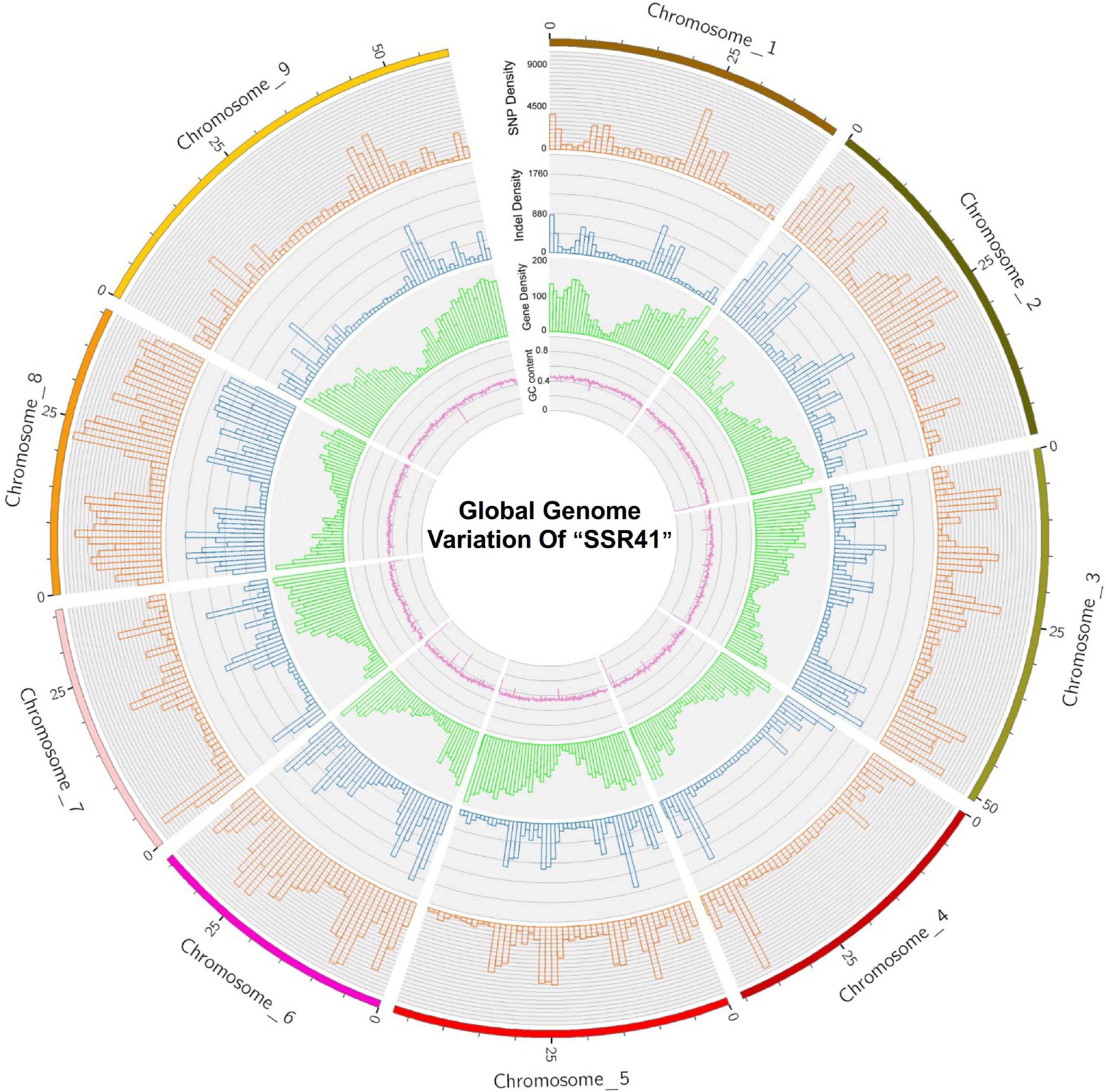
Figure 2. A circular plot showing “SSR41” genome sequence features. An orange bar shows single nucleotide polymorphism (SNP) density, a blue bar shows insertion/deletion (InDel) density, a green bar shows gene density, and a purple line shows GC content.
Characteristics of SNPs, InDels, and SVs
Among all the SNPs and InDels identified, only homozygous variations were collected for further annotation, because both “Yugu1” and “SSR41” are stable modern cultivars without phenotypic and genetic segregation. Of the 865,781 homozygous SNPs in “SSR41”, SNPs located in intergenic regions accounted for the largest proportion (37.82%), followed by those in regions 5 Kb upstream (26.44%) or downstream (24.44%) of coding regions (Figure 3A). Only 11.30% (97,833) of the SNPs were in genic regions. Of the 97,833 genic SNPs, just over half (51,514, 52.66%) were in introns (Figure 3B). Only 6.64% (3,418) of the intronic SNPs were located at splice regions, where they may change the original splicing pattern. There were 28,138 SNPs located in coding sequence (CDS) regions, 12,894 (45.82%) of which were synonymous SNPs, and 15,224 (54.18%), of which were non-synonymous variants. The ratio of non-synonymous to synonymous SNPs was about 1.18 across all gene models. We also identified that 6.81% (6,667) and 11.77% (11,515) of the SNPs occurred in the 5′ and 3′ untranslated regions. Annotation of InDel polymorphisms revealed that the general trends in the presence of 162,261 homozygous InDels in different genomic regions were similar to those of SNPs. Interestingly, there was a smaller proportion of InDels (1.59%) than SNPs (3.25%) in CDS regions, but a much higher proportion of InDels than SNPs in introns and gene upstream/downstream regions (Figure 3A).
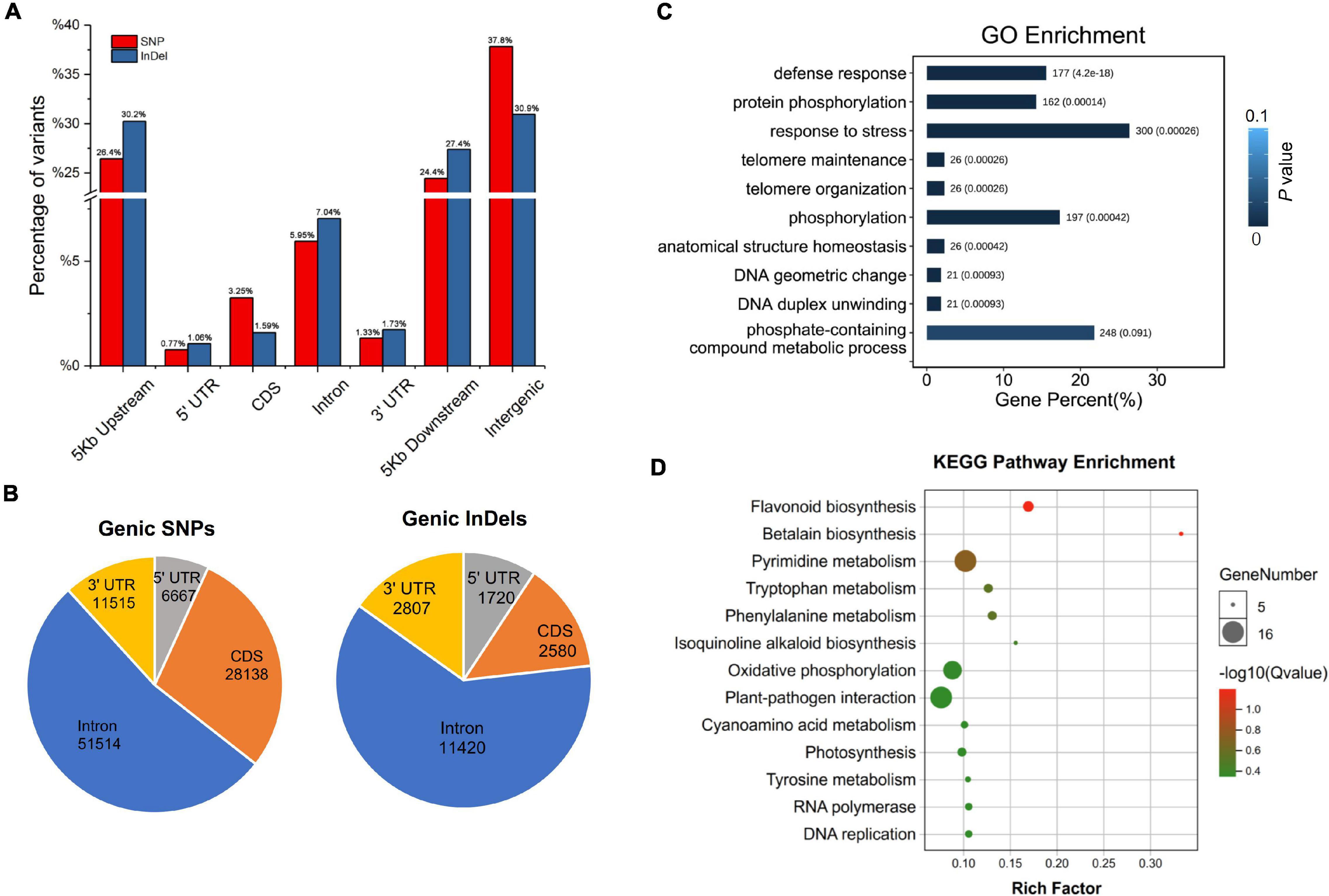
Figure 3. Annotation and functional enrichment analysis of “SSR41” genome variations. (A) Distribution of SNPs and InDels at the whole genome level. (B) Distribution of SNP and InDels in genic regions. (C) Gene ontology enrichment analysis of genes with large-effect variations. (D) Kyoto Encyclopedia of Genes and Genomes pathway enrichment analysis of genes with large-effect variations.
We also identified SVs between “SSR41” and “Yugu1”, including 388 insertions (INS), 1,197 deletions (DEL), and 18 inversions (INV) with fragment sizes larger than 100 bp. Analysis of the distribution of the fragment sizes of these SVs revealed that 65.5% of them ranged from 100 to 400 bp in length (median, 311 bp) (Supplementary Table 5).
Annotation and Effect of Genomic Variations on Functional Genes
Characterization of polymorphisms between “SSR41” and “Yugu1” revealed that approximately 89% of the variations were intergenic, while only 11% were genic. Among the genic variations, 3,979 polymorphisms were identified to have strong effects on gene function (including genomic variations leading to frame shifts, changes in splicing, loss of the start codon, and gain or loss of the stop codon) between the two accessions (Supplementary Table 6). The GO enrichment analysis showed that 10 subcategories in the “biological process” category, including defense responses (GO: 0006952), protein phosphorylation (GO: 0006468), and response to stress (GO: 0006950), were enriched with strongly affected genes (Figure 3C). The KEGG pathway enrichment analysis suggested that these genes were mainly related to 14 pathways, including secondary metabolite biosynthesis (e.g., flavonoid, tryptophan, and phenylalanine), oxidative phosphorylation, and plant-pathogen interaction (Figure 3D). Further characterization of these genes may shed light on the molecular basis of the phenotypic variations between “Yugu1” and “SSR41”.
Genome-Wide Development and Validation of Polymorphic Molecular Markers
Based on Indels and SVs identified from “SSR41” and “Yugu1”, we screened out high-quality variations that were 20 to 200 bp in length. These variations could be good candidates to design molecular markers that can be conveniently examined by PCR and DNA agarose gel electrophoresis. In total, 8,361 molecular markers evenly distributed on the nine chromosomes of foxtail millet were identified. Chromosome 8 had the largest number of InDels (1,781), followed by Chromosomes 2 (1,315) and 6 (1,287). Chromosome 1 had the fewest InDels (372). Half of these InDels had lengths of 20–32 bp (Figure 4A). To develop efficient and highly polymorphic InDel markers, the PIC values of these markers were characterized by comparison with 1,826 Setaria accessions, for which genome resequencing data are available (Jia et al., 2013; Mamidi et al., 2020; Li et al., 2021). In total, 1,551 highly polymorphic InDel markers with a PIC score higher than 0.5 were selected (Supplementary Table 1). The polymorphism levels and the number of these InDels were highest on Chromosome 8, followed by Chromosomes 3, 2, and 6. Then, 180 of these molecular markers were validated by PCR. More than 86% (156) of them generated PCR products, and 127 of them showed the expected polymorphism (Figure 4). These results confirmed that we had successfully developed polymorphic InDel markers for Setaria.
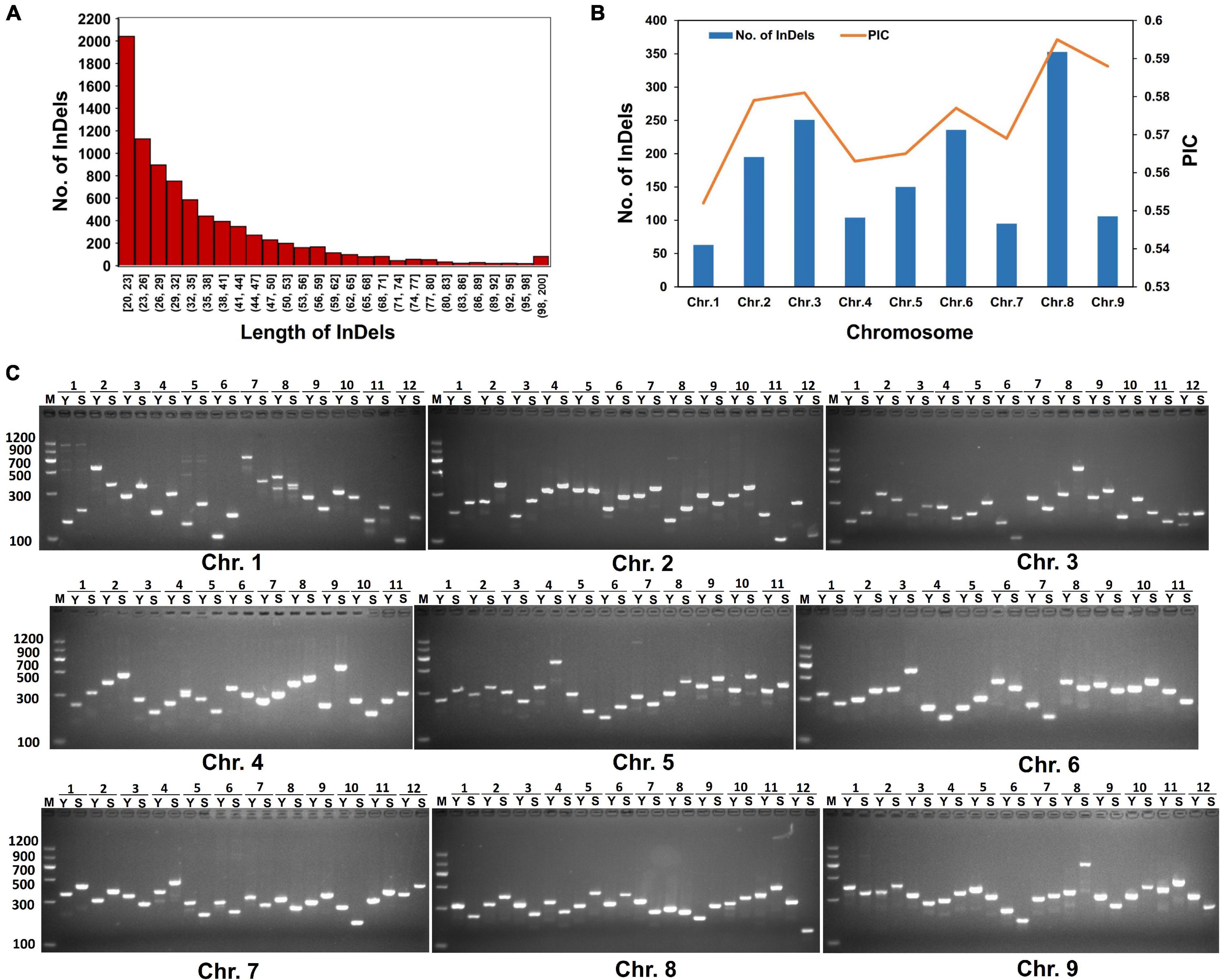
Figure 4. Characterization of polymorphic molecular markers. (A) Length distribution of InDel markers. (B) Polymorphism information content (PIC) variations of polymorphic molecular markers among different chromosomes. (C) Agarose gel electrophoresis analysis of PCR products amplified using representative molecular markers. M, DL2000 DNA marker. Y, PCR products amplified from “Yugu1” DNA as a template. S, PCR products amplified from “SSR41” DNA as a template. Numbers on top of each gel images indicate different molecular markers chosen from each chromosome.
Map-Based Cloning of a White Leaf Sheath Gene SiWLS1 in Setaria Using the Newly Developed Molecular Markers
A S. italica white leaf sheath mutant siwls1 was screened out from the “Yugu1” EMS mutant library. Analyses of its agronomic traits indicated that the mature plant height, panicle length, panicle diameter, length, and width of the leaf blade of siwls1 were not significantly different from those of wild-type plants (Figure 5A and Supplementary Table 7). From the heading stage to the mature stage, the siwls1 mutant exhibited a white leaf sheath, different from the green leaf sheath of “Yugu1”, while the leaf collar and leaf blade were similar to those of wild type (Figures 5B,C).
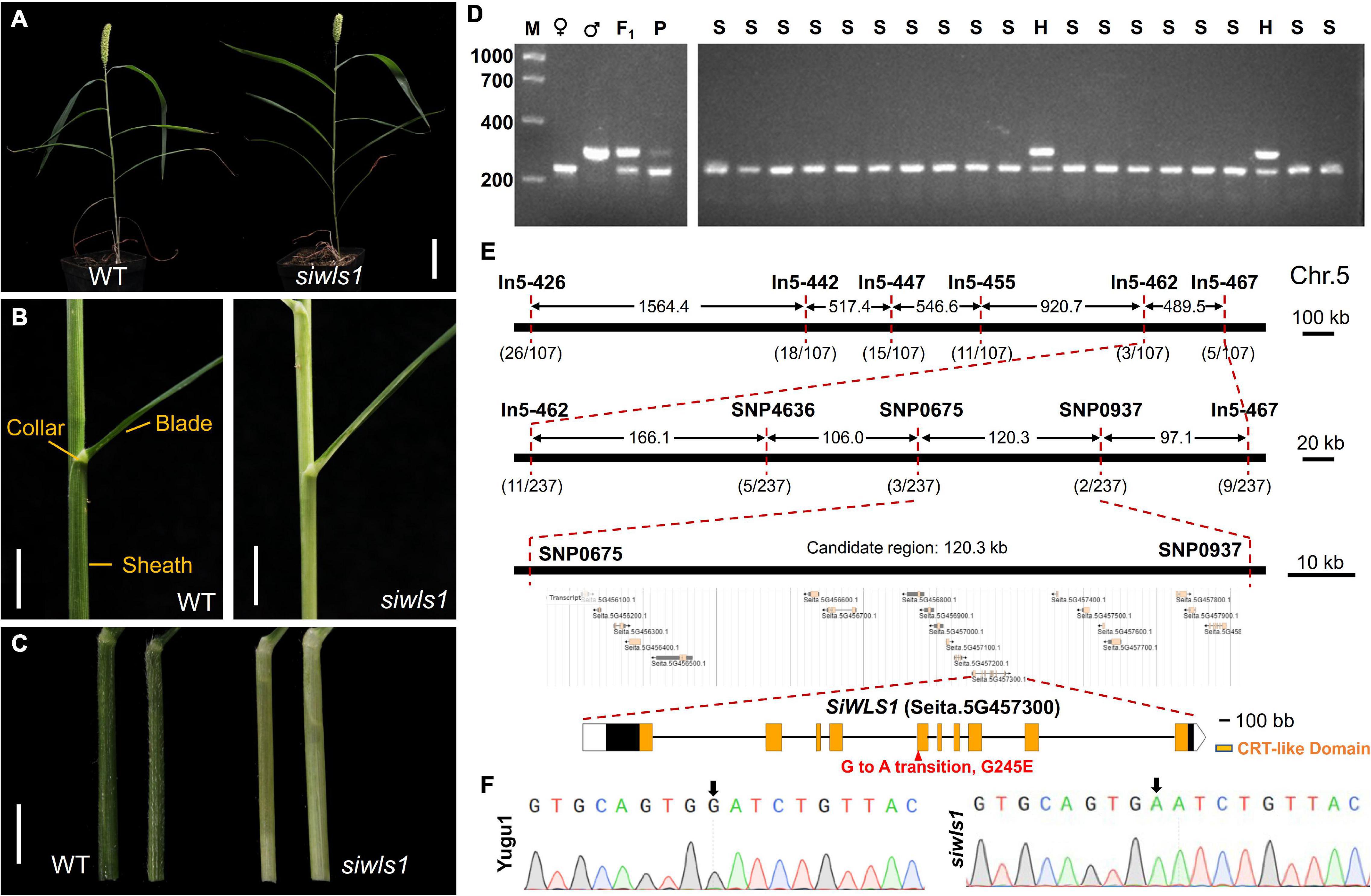
Figure 5. Characterization of foxtail millet white leaf sheath mutant siwls1 and identification of the candidate gene. (A) General statures of siwls1 mutant and wild-type plants at maturity. Bar = 5 cm. (B) Leaf blade, leaf collar, and leaf sheath morphology of “Yugu1” (WT) and siwls1. Bar = 1 cm. (C) An enlarged view of leaf sheath of wild-type and mutant plants. Bar = 1 cm. (D) An example of a molecular marker “In5-455” closely linked to the SiWLS1 locus. M, DL 2000 molecular weight marker; ♀, PCR product amplified from female parent siwls1; ♂, PCR product amplified from male parent SSR41; F1, PCR product amplified from F1 individual; P, PCR product amplified from the DNA pool of a mixture of 30 recessive homozygous individuals in F2 population; S, PCR product amplified from recessive homozygous individual in F2 population; H, PCR product amplified from heterozygous individual in F2 population. (E) Positional cloning of SiWLS1 using newly developed molecular markers. (F) Sanger sequencing validation of the causal SNP.
For the genetic analysis of the siwls1 mutant and fine mapping of its candidate gene, we constructed an F2 population from hybridization between siwls1 and “SSR41”. All individuals in the F1 generation displayed a green leaf sheath, the same as that of wild-type plants. In the F2 generation, the segregation ratio of plants with a green leaf sheath (745 individuals) and those with a white leaf sheath (237 individuals) was close to 3:1 (Supplementary Table 8). These results clearly indicated that the white leaf sheath phenotype is controlled by a single recessive nuclear gene.
Using 30 F2 homozygous recessive individuals and 54 newly developed InDel markers, we found that the target gene was located on the long arm of Chromosome 5 around the marker In5-455 using the BSA method (Supplementary Figures 1C,D and Figure 5D). For fine mapping, we further developed five InDel markers and three SNP markers, and used them to genotype all 237 recessive individuals (Figure 5E). Our gene mapping analyses eventually limited the target gene to a 120.26-kb region on Chromosome 5 between the markers SNP0675 and SNP0937 (Figure 5E). According to the foxtail millet reference genome annotation8 (S. italica v2.2), there are 19 genes within this region. To identify which one is responsible for the white leaf sheath phenotype, we performed whole genome re-sequencing of four DNA pools (from mutants, wild-type plants in the F2 population, and both parents). In a BSA sequencing (BSA-seq) analysis, only one SNP (G to A at 46,571,596 bp on Chromosome 5) met the certification standard (as described in Supplementary Table 9). Sanger sequencing and PCR analysis confirmed the presence of a G-to-A transition in the fifth exon of Seita.5G457300, leading to an amino acid change from Gly-245 to Glu in the chloroquine-resistance transporter (CRT)-like domain (Figures 5E,F). These results revealed that Seita.5G457300 corresponds to SiWLS1.
SiWSL1 Encoding a Chloroquine-Resistance Transporter Affects Chloroplast Development and Chlorophyll Accumulation in the Leaf Sheath of Foxtail Millet
Sequence analysis showed that SiWLS1 is a single-copy gene with a 1,335-bp CDS encoding a 444-amino acid protein that belongs to the CRT-like family. A phylogenetic tree was constructed to examine the relationships of the Setaria CRT-like gene family with those of Arabidopsis, rice, maize, and Sorghum bicolor. Sixteen CRT-like genes were identified: three in S. italica, four in maize, three in S. bicolor, three in rice, and three in Arabidopsis (Figure 6A and Supplementary Table 3). The genes with the strongest similarity to SiWLS1 in rice and Arabidopsis were Os01g72570 (OsCLT1) and AT5G19380 (CLT1), respectively, which are known to be key regulators of glutathione homeostasis and heavy metal tolerance (Lim et al., 2011; Yang et al., 2016). According to the results of a study on OsCLT1 in rice (Yang et al., 2016), the osclt1 mutant is hypersensitive to As and Cd. Therefore, we determined the sensitivities of siwls1 toward As and Cd treatment. As shown in Supplementary Figures 4, 5, the root growth of siwls1 was severely inhibited in response to increasing As and Cd concentrations compared with that of “Yugu1”, indicating that the siwls1 mutant is more sensitive to As and Cd. These results were consistent with those of a previous study on rice (Yang et al., 2016), supporting that SiWLS1 is the causal gene.
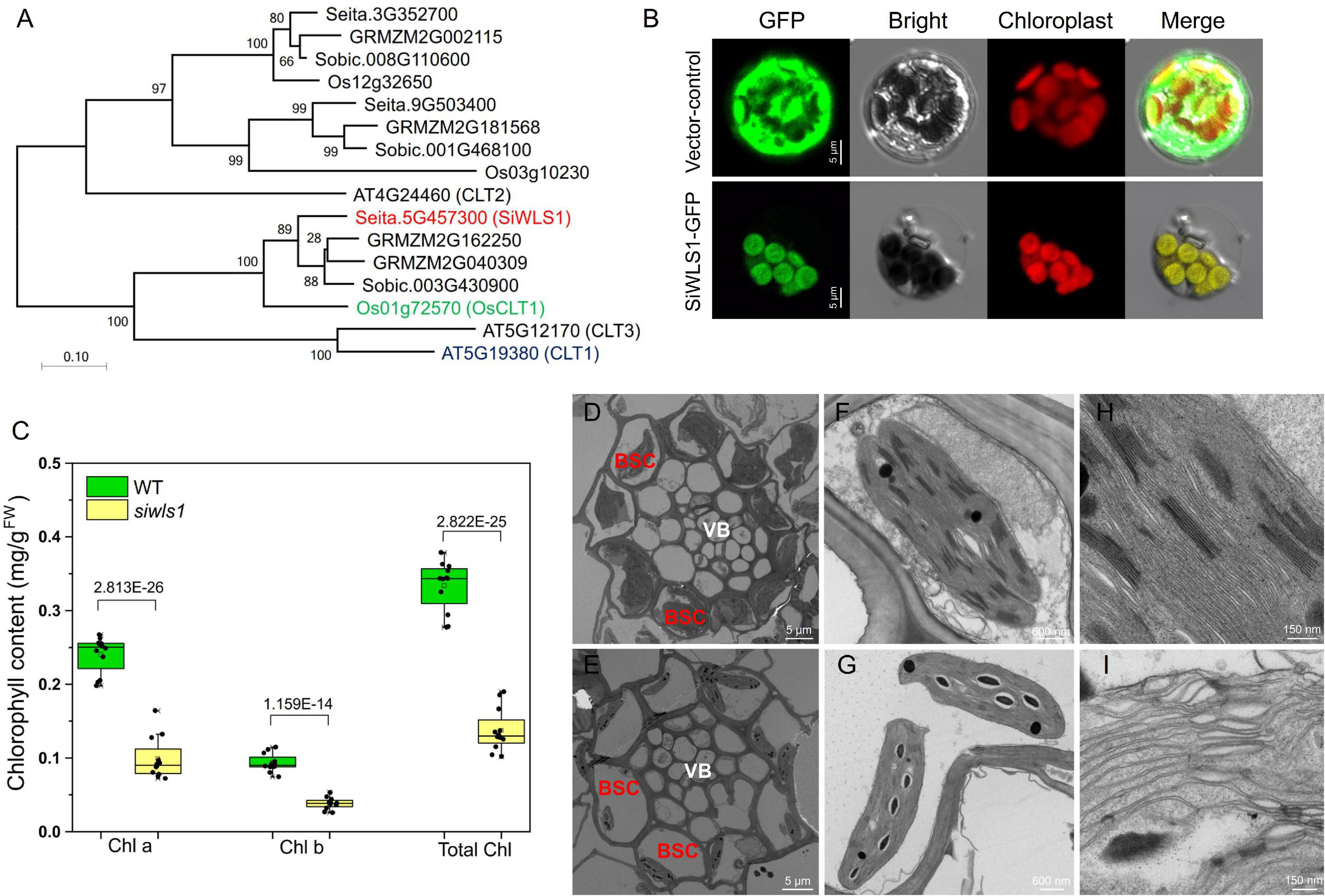
Figure 6. Functional analysis of foxtail millet white leaf sheath gene SiWLS1. (A) Phylogenetic relationship between SiWLS1 and its homologs in maize, sorghum, rice, and Arabidopsis. (B) Subcellular localization of SiWLS1 protein. (C) Comparison of chlorophyll contents between siwls1 mutant and wild-type plants. (D–I) Chloroplast ultrastructure in siwls1 mutant (the lower row) and wild-type (the upper row) plants. VB, vascular bundle; BSC, bundle sheath cells.
To determine the subcellular localization of the SiWLS1 protein, we fused the SiWLS1 CDS to GFP and transiently introduced it into foxtail millet protoplasts. The SiWLS1-GFP signal was detected in chloroplasts, as determined from the clear overlap of the GFP signal with the autofluorescence of chloroplasts (Figure 6B), suggesting that SiWLS1 functions in chloroplasts. We measured the chlorophyll contents in the leaf sheath of the siwls1 mutant and “Yugu1”, and found that the Chla, Chlb, and total Chl contents were significantly lower in siwls1 plants than in the wild type (Figure 6C). Ultrastructural observations of chloroplasts revealed dramatic reductions in the number and the size of chloroplasts in bundle sheath cells (BSC) in the siwls1 mutant compared with “Yugu1” (Figures 6D,E). The stroma lamella and grana lamella of chloroplasts were highly ordered in wild type, while the thylakoids in siwls1 chloroplasts appeared to be disorganized (Figures 6F–I). These findings, together with the white leaf sheath phenotype of siwls1, indicated that SiWLS1 plays an important role in regulating chlorophyll accumulation or chloroplast development in the leaf sheath of foxtail millet.
Discussion
“Yugu1” and “SSR41” Are Ideal Resources for Forward Genetics and Map-Based Cloning Research in S. italica
The foxtail millet variety “Yugu1” is a modern cultivar that is widely grown in northern China. Because of its outstanding agronomic performance and its wide adaptability to different growth environments, “Yugu1” was selected to generate a high-quality Setaria reference genome (Bennetzen et al., 2012) and a large-scale EMS mutant library (Sun et al., 2019). To further promote the development of the “Yugu1”-based model for gene cloning, we screened another S. italica variety, “SSR41”. As expected, “SSR41” flowered at the same time as “Yugu1” and easily hybridized with it. Multi-year and multilocation comparisons of the two varieties have shown that “Yugu1” and “SSR41” are very similar in terms of their growth, development, and general plant stature. This ensures that the hybrid offspring of “SSR41” and “Yugu1” will have a steady growth performance, which is beneficial for the selection of individuals in an F2 mapping population.
We used high-throughput sequencing to obtain detailed genome sequence information for “SSR41”. The mean sequencing depth for “SSR41” was up to 48×, deeper than that of previous sequencing of Setaria accessions (Bai et al., 2013; Mamidi et al., 2020; Li et al., 2021). High-quality sequence reads of “SSR41” were aligned to “Yugu1” (the reference genome) to discover genome-wide polymorphic molecular markers. More than 1.2 million variations were identified in “SSR41”; more than the number of DNA variations detected in another foxtail millet variety, “SLX” (Bai et al., 2013). Interestingly, although “Yugu1” and “SSR41” are morphologically similar, these two varieties have abundant DNA polymorphisms. This has allowed us to develop sufficient molecular markers to support genotyping of the genetic population generated from “Yugu1” and “SSR41”.
Due to our continuing efforts, positional cloning in Setaria is now practicable using “SSR41” and “Yugu1” EMS mutants as plant materials. To date, map-based cloning has been used to characterize at least eight S. italica genes, including SiMADS34 (Hussin et al., 2021), DPY1 (Zhao et al., 2020), SiSTL1 (Tang et al., 2019), SiSTL2 (Zhang et al., 2018a), SiYGL1 (Li et al., 2016), SiYGL2 (Zhang et al., 2018b), Loose Panicle1 (Xiang et al., 2017), and SiAGO1b (Liu et al., 2016). These studies illustrate that “SSR41” and “Yugu1” are suitable germplasm resources for forward genetics and positional cloning research on Setaria.
Development of Polymorphic InDel Markers for the Setaria Model System
With the development of high-throughput sequencing, molecular marker development has become feasible and convenient for various crops. Both small InDels and SVs have become powerful marker systems for genotyping, SV-GWAS, and map-based cloning analyses (Qin et al., 2021). Previous studies on molecular marker development in Setaria have focused on SSR markers (Jia et al., 2007). To date, more than 2,000 effective SSR markers have been developed for genetic diversity analysis and quantitative trait loci mapping (Zhang et al., 2014). However, genotyping using SSR markers is laborious and time-consuming. In comparison, InDel markers, especially those longer than 20 bp, are better for genotyping because they can easily be detected by DNA electrophoresis on agarose gels. In the present study, we identified 8,361 InDels, and all of them were compared with genome variations of 1,826 Setaria accessions to evaluate their PIC values. Among them, 1,551 were highly polymorphic with a high-average PIC value of 0.579. Of the 180 tested pairs of primers for InDel markers, 94% of them produced stable PCR amplification products, and 127 of them were polymorphic (Figure 4C). These results confirmed that we have developed a large set of highly polymorphic InDel markers for Setaria. These markers are a valuable resource for positional cloning and other genotyping analyses in Setaria.
Characterization of Genome Variations in “SSR41” Provides Insights Into the Genetic Basis of Its Preferable Agronomic Traits
The discovery and the utilization of genome variations, including SNPs, InDels, and SVs, form the basis of gene mapping and marker-assisted breeding. “SSR41” is an elite foxtail millet variety with many desirable agronomic traits such as high yield, disease resistance, and good grain quality. In this study, we performed whole-genome sequencing of “SSR41” and identified a number of high-quality SNPs, InDels, and SVs. Among these genome variations, only a few were identified to affect gene function. Functional enrichment analysis suggested that genes related to defense response and plant-pathogen interaction were significantly enriched with sequence variations. Seita.7G239900, which is homologous to FLAGELLIN-SENSITIVE 2 (FLS2, AT5G46330), is one of the most important disease-resistance genes, and it had two insertion mutations in “SSR41”. FLS2 functions in the MAP kinase signaling pathway, which is involved in innate immunity (Gómez-Gómez and Boller, 2000). In Arabidopsis, the fls2-17 mutant exhibits a significant flg22-induced reduction in bacterial growth compared with that in wild-type plants (Zipfel et al., 2004). Thus, the variation in FLS2 in “SSR41” may contribute to its enhanced disease resistance. Interestingly, we detected mutations in some genes related to starch and sucrose metabolism, which may directly affect grain quality. Among these mutations, a C-to-A transversion leading to an early stop codon was identified at the second exon of Seita.6G036700 in “SSR41”. Seita.6G036700 is homologous to a soluble starch synthase IIIa (OsSSIIIa) in rice. The OsSSIIIa-deficient rice mutants have smaller and rounder starch granules compared with those of wild type and show reduced contents of long-chain starch in the endosperm (Ryoo et al., 2007). Thus, the mutation of this gene in foxtail millet “SSR41” may be related to its high-grain quality.
Setaria Is an Efficient Model System for Exploring Novel Gene Function in Plant Biology
For decades, Arabidopsis has been widely used as a model plant for research on many aspects of biology and technology innovation; thus, it has been at the center of basic plant basic research. However, during their long evolutionary history, plants have evolved specific characteristics and systems, such as C4 photosynthesis. Thus, more model plants with a diverse range of characters are required to detect novel gene structures and functional variations. In recent years, Setaria has been proposed as an elegant C4 model because of its distinct growth and development characteristics. Numerous research groups have made efforts to improve Setaria as a model plant, including providing high-quality Setaria genome sequence data (Yang et al., 2020), improving its Agrobacterium-mediated transformation efficiency (Van Eck, 2018) and providing new technical platforms (Fahlgren et al., 2015). Our group has focused on exploring Setaria gene functions using forward genetics approaches. In this study, we developed a sufficient number of polymorphic InDel markers that can be used to improve map-based cloning efficiency in Setaria. Leaf sheath color is an important trait that can be used not only to study the molecular mechanisms that regulate chloroplast development but also as a practical morphological marker to assist breeding. A novel white-leaf sheath mutant siwls1 was isolated. The wild-type plants have a green leaf sheath, but the mutant exhibits a white leaf sheath from the heading stage to the mature stage. The candidate gene SiWLS1 was identified by analyzing 237 F2 recessive individuals with 65 InDel markers. The laboratory work took 2 months, demonstrating that map-based cloning of a specific gene locus in Setaria is feasible and efficient.
The results of many other studies and our present study suggest that the Setaria model can reveal new functions of previously characterized genes. SiWLS1 is homologous to rice OsCLT1, which encodes a CRT-like transporter that is essential for glutathione homeostasis and As and Cd detoxification in rice (Yang et al., 2016). In the present study, we also found that the Setaria siwls1 mutant was sensitive to As and Cd treatment, indicating that CLT1 has a conserved function to regulate heavy metal accumulation and distribution in plants. However, as a chloroplast-located protein, CLT1 may also regulate chloroplast development and chlorophyll synthesis, although little is known about this role at present. Here, we identified a novel white leaf sheath Setaria mutant siwls1 harboring a homozygous recessive mutation in the CLT1 gene. Analyses by TEM revealed a defective chloroplast development phenotype in bundle sheath cells of the siwls1 leaf sheath (Figures 6D–I). Additionally, the chlorophyll content was significantly lower in siwls1 plants than in wild-type plants (Figures 6D–I), indicating that CLT1 affects chloroplast development and chlorophyll accumulation in the leaf sheath of foxtail millet. Other studies have also reported new functions of genes determined using the Setaria model, for example, Bristleless1 (Bsl1) in S. viridis. Yang et al. (2017) identified two bsl mutants in S. viridis, both of which showed defective bristle development. Then, BSAseq identified the candidate gene, BSL1, which is homolog of Dwarf11 (D11) in rice. D11 encodes a cytochrome P450 protein involved in brassinosteroid biosynthesis. Although BSL1 and D11 are syntenic orthologs, their biological function to regulate bristle and inflorescence development is unique in Setaria, and has not been reported for either Arabidopsis or rice (Yang et al., 2017). From the results of our study and other studies on gene function in Setaria, we can clearly demonstrate that both S. italica and S. viridis are ideal model systems for exploring novel gene functions in plants.
Data Availability Statement
The original contributions presented in the study are publicly available. This data can be found here: EMBL’s European Bioinformatics Institute (EMBL-EBI) BioProject database under accession number PRJEB46028.
Author Contributions
XD and ST conceived the project. HZa, ST, YG, ML, GJ, and BF conducted the experiments and collected the data. HZi constructed the mapping population. ST, JS, and QH conducted bioinformatic analysis. HZa and ST drafted the manuscript. XD and JS revised the manuscript. All the authors contributed to the article, read, and approved the manuscript.
Funding
This work was supported by National Key Research & Development Program of China (Grant Nos. 2019YFD1000700 and 2019YFD1000701), the National Natural Science Foundation of China (31871692), the China Agriculture Research System of MOF (MARACARS-06-13.5-A04), and the Agricultural Science and Technology Innovation Program of the Chinese Academy of Agricultural Sciences.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank the Beijing Berry Genome Co. and the Beijing Biomarker Co. for technical support with the high-throughput sequencing and bioinformatic analyses. We thank Jennifer Smith (1999 – Ph.D. Botany, University of Otago) from Liwen Bianji (Edanz) (www.liwenbianji.cn) for editing the English text of a draft of this manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2021.743782/full#supplementary-material
Supplementary Figure 1 | Introduction to positional cloning in foxtail millet using “SSR41”. (A) A general view of positional cloning in foxtail millet. (B) Introduction of linked and unlinked markers used in PCR tests. (C) A summary of bulked segregant analysis (BSA) for primary mapping of SiWLS1. (D) DNA agarose gel electrophoresis results for primary mapping.
Supplementary Figure 2 | General statistics of “SSR41” resequencing data. (A) Genome-wide distribution of read coverage. (B) Statistics of read coverage and sequencing depth. (C) Single nucleotide polymorphism (SNP) depth and neighboring SNP distance distributions.
Supplementary Figure 3 | Annotations of SNPs between “SSR41” and “Yugu1”.
Supplementary Figure 4 | Comparisons of plant growth between “Yugu1” and siwls1 in response to arsenic treatment. Significant differences were determined by Student’s t-test (ns = no significant difference, ∗p < 0.01, ∗∗p < 0.001).
Supplementary Figure 5 | Comparisons of plant growth between “Yugu1” and siwls1 in response to cadmium treatment. Significant differences were determined by Student’s t-test (ns = no significant difference, ∗p < 0.01, ∗∗p < 0.001).
Footnotes
- ^ http://www.fao.org
- ^ http://foxtail-millet.biocloud.net
- ^ http://www.setariamodel.cn
- ^ www.illumina.com
- ^ http://www.ebi.ac.uk/
- ^ phytozome.jgi.doe.gov/pz/portal.html#!info?alias=Org_Sitalica
- ^ https://github.com/primer3-org/primer3
- ^ http://phytozome.jgi.doe.gov/
References
Bai, H., Cao, Y., Quan, J., Dong, L., Li, Z., Zhu, Y., et al. (2013). Identifying the genome-wide sequence variations and developing new molecular markers for genetics research by re-sequencing a landrace cultivar of foxtail millet. PLoS One 8:e73514. doi: 10.1371/journal.pone.0073514
Bai, H., Song, Z., Zhang, Y., Li, Z., Wang, Y., Liu, X., et al. (2020). The bHLH transcription factor PPLS1 regulates the color of pulvinus and leaf sheath in foxtail millet (Setaria italica). Theor. Appl. Genet. 133, 1911–1926. doi: 10.1007/s00122-020-03566-4
Bennetzen, J. L., Schmutz, J., Wang, H., Percifield, R., Hawkins, J., Pontaroli, A. C., et al. (2012). Reference genome sequence of the model plant Setaria. Nat. Biotechnol. 30, 555–561. doi: 10.1038/nbt.2196
Cui, T., Luo, S., Du, Y., Yu, L., Yang, J., Li, W., et al. (2019). Research of photosynthesis and genomewide resequencing on a yellow-leaf Lotus japonicus mutant induced by carbon ion beam irradiation. Grassl. Sci. 65, 41–48. doi: 10.1111/grs.12216
Diao, X., Schnable, J., Bennetzen, J. L., and Li, J. (2014). Initiation of Setaria as a model plant. Front. Agr. Sci. Eng. 1:16. doi: 10.15302/j-fase-2014011
Fahlgren, N., Feldman, M., Gehan, M. A., Wilson, M. S., Shyu, C., Bryant, D. W., et al. (2015). A versatile phenotyping system and analytics platform reveals diverse temporal responses to water availability in Setaria. Mol. Plant 8, 1520–1535. doi: 10.1016/j.molp.2015.06.005
Gómez-Gómez, L., and Boller, T. (2000). FLS2: an LRR receptor-like kinase involved in the perception of the bacterial elicitor flagellin in Arabidopsis. Mol. Cell 5, 1003–1011. doi: 10.1016/s1097-2765(00)80265-8
He, Q., Zhi, H., Tang, S., Xing, L., Wang, S., Wang, H., et al. (2021). QTL mapping for foxtail millet plant height in multi-environment using an ultra-high density bin map. Theor. Appl. Genet. 134, 557–572. doi: 10.1007/s00122-020-03714-w
Hu, W., Zhou, T., Han, Z., Tan, C., and Xing, Y. (2020). Dominant complementary interaction between OsC1 and two tightly linked genes, Rb1 and Rb2, controls the purple leaf sheath in rice. Theor. Appl. Genet. 133, 2555–2566. doi: 10.1007/s00122-020-03617-w
Hussin, S. H., Wang, H., Tang, S., Zhi, H., Tang, C., Zhang, W., et al. (2021). SiMADS34, an E-class MADS-box transcription factor, regulates inflorescence architecture and grain yield in Setaria italica. Plant Mol. Biol. 105, 419–434. doi: 10.1007/s11103-020-01097-6
Jain, A., Roorkiwal, M., Kale, S., Garg, V., Yadala, R., and Varshney, R. K. (2019). InDel markers: an extended marker resource for molecular breeding in chickpea. PLoS One 14:e0213999. doi: 10.1371/journal.pone.0213999
Jia, G., Huang, X., Zhi, H., Zhao, Y., Zhao, Q., Li, W., et al. (2013). A haplotype map of genomic variations and genome-wide association studies of agronomic traits in foxtail millet (Setaria italica). Nat. Genet. 45, 957–961. doi: 10.1038/ng.2673
Jia, X. P., Shi, Y. S., Song, Y. C., Wang, G. Y., Wang, T. Y., and Yu, L. (2007). Development of EST-SSR in foxtail millet (Setaria italica). Genet. Resour. Crop Evol. 54, 233–236. doi: 10.1007/s10722-006-9139-8
Lemos, S., Silveira, R., Buuron, S. K., Santos, R., and Moro, S. C. (2019). Determining the polymorphism information content of a molecular marker. Gene 726:144175. doi: 10.1016/j.gene.2019.144175
Li, C., Wang, G., Li, H., Wang, G., Ma, J., Zhao, X., et al. (2021). High-depth resequencing of 312 accessions reveals the local adaptation of foxtail millet. Theor. Appl. Genet. 134, 1303–1317. doi: 10.1007/s00122-020-03760-4
Li, P., Du, C., Zhang, Y., Yin, S., Zhang, E., Fang, H., et al. (2018). Combined bulked segregant sequencing and traditional linkage analysis for identification of candidate gene for purple leaf sheath in maize. PLoS One 13:e0196296. doi: 10.1371/journal.pone.0196296
Li, W., Tang, S., Zhang, S., Shan, J., and Diao, X. (2016). Gene mapping and functional analysis of the novel leaf color gene SiYGL1 in foxtail millet [Setaria italica (L.) P. Beauv]. Physiol. Plant 157, 24–37. doi: 10.1111/ppl.12405
Lim, B., Meyer, A. J., and Cobbett, C. S. (2011). Development of glutathione-deficient embryos in Arabidopsis is influenced by the maternal level of glutathione. Plant Biol. 13, 693–697. doi: 10.1111/j.1438-8677.2011.00464.x
Liu, X., Tang, S., Jia, G., Schnable, J. C., Su, H., Tang, C., et al. (2016). The C-terminal motif of SiAGO1b is required for the regulation of growth, development and stress responses in foxtail millet (Setaria italica (L.) P. Beauv). J. Exp. Bot. 67, 3237–3249. doi: 10.1093/jxb/erw135
Mamidi, S., Healey, A., Huang, P., Grimwood, J., Jenkins, J., Barry, K., et al. (2020). A genome resource for green millet Setaria viridis enables discovery of agronomically valuable loci. Nat. Biotechnol. 38, 1203–1210. doi: 10.1038/s41587-020-0681-2
Michelmore, R. W., Paran, I., and Kesseli, R. V. (1991). Identification of markers linked to disease-resistance genes by bulked segregant analysis: a rapid method to detect markers in specific genomic regions by using segregating populations. Proc. Natl. Acad. Sci. U.S.A. 88, 9828–9832. doi: 10.1073/pnas.88.21.9828
Oshima, M., Taniguchi, Y., Akasaka, M., Abe, K., Ichikawa, H., Tabei, Y., et al. (2019). Development of a visible marker trait based on leaf sheath-specific anthocyanin pigmentation applicable to various genotypes in rice. Breed. Sci. 69, 244–254. doi: 10.1270/jsbbs.18151
Qin, P., Lu, H., Du, H., Wang, H., Chen, W., Chen, Z., et al. (2021). Pan-genome analysis of 33 genetically diverse rice accessions reveals hidden genomic variations. Cell 184, 3542–3558. doi: 10.1016/j.cell.2021.04.046
Ryoo, N., Yu, C., Park, C.-S., Baik, M.-Y., Park, I. M., Cho, M.-H., et al. (2007). Knockout of a starch synthase gene OsSSIIIa/Flo5 causes white-core floury endosperm in rice (Oryza sativa L.). Plant Cell Rep. 26, 1083–1095. doi: 10.1007/s00299-007-0309-8
Slatko, B. E., Gardner, A. F., and Ausubel, F. M. (2018). Overview of next-generation sequencing technologies. Curr. Protoc. Mol. Biol. 122:e59. doi: 10.1002/cpmb.59
Sun, J., Luu, N. S., Chen, Z., Chen, B., Cui, X., Wu, J., et al. (2019). Generation and characterization of a foxtail millet (Setaria italica) mutant library. Front. Plant Sci. 10:369. doi: 10.3389/fpls.2019.00369
Tang, C., Tang, S., Zhang, S., Luo, M., Jia, G., Zhi, H., et al. (2019). SiSTL1, encoding a large subunit of ribonucleotide reductase, is crucial for plant growth, chloroplast biogenesis, and cell cycle progression in Setaria italica. J. Exp. Bot. 70, 1167–1182. doi: 10.1093/jxb/ery429
Van Eck, J. (2018). The status of Setaria viridis transformation: Agrobacterium-mediated to floral dip. Front. Plant Sci. 9:652. doi: 10.3389/fpls.2018.00652
Wu, Z., Zhang, X., He, B., Diao, L., Sheng, S., Wang, J., et al. (2007). A chlorophyll-deficient rice mutant with impaired chlorophyllide esterification in chlorophyll biosynthesis. Plant Physiol. 145, 29–40. doi: 10.1104/pp.107.100321
Xiang, J., Tang, S., Zhi, H., Jia, G., Wang, H., and Diao, X. (2017). Loose panicle1 encoding a novel WRKY transcription factor, regulates panicle development, stem elongation, and seed size in foxtail millet [Setaria italica (L.) P. Beauv.]. PLoS One 12:e0178730. doi: 10.1371/journal.pone.0178730
Yang, J., Gao, M. X., Hu, H., Ding, X. M., Lin, H. W., Wang, L., et al. (2016). OsCLT1, a CRT-like transporter 1, is required for glutathione homeostasis and arsenic tolerance in rice. New Phytol. 211, 658–670. doi: 10.1111/nph.13908
Yang, J., Thames, S., Best, N. B., Jiang, H., Huang, P., Dilkes, B. P., et al. (2017). Brassinosteroids modulate meristem fate and differentiation of unique inflorescence morphology in Setaria viridis. The Plant Cell 30, 48–66. doi: 10.1105/tpc.17.00816
Yang, Z., Zhang, H., Li, X., Shen, H., Gao, J., Hou, S., et al. (2020). A mini foxtail millet with an Arabidopsis-like life cycle as a C4 model system. Nat. Plants 6, 1167–1178. doi: 10.1038/s41477-020-0747-7
Zhang, S., Tang, C., Zhao, Q., Li, J., Yang, L., Qie, L., et al. (2014). Development of highly polymorphic simple sequence repeat markers using genome-wide microsatellite variant analysis in foxtail millet [Setaria italica (L.) P. Beauv.]. BMC Genomics 15:78. doi: 10.1186/1471-2164-15-78
Zhang, S., Tang, S., Tang, C., Luo, M., Jia, G., Hui, Z., et al. (2018a). SiSTL2 is required for cell cycle, leaf organ development, chloroplast biogenesis, and has effects on C4 photosynthesis in Setaria italica (L.) P. Beauv. Front. Plant Sci. 9:1103. doi: 10.3389/fpls.2018.01103
Zhang, S., Zhi, H., Li, W., Shan, J., Tang, C., Jia, G., et al. (2018b). SiYGL2 is involved in the regulation of leaf senescence and photosystem II efficiency in Setaria italica (L.) P. Beauv. Front. Plant Sci. 9:1308. doi: 10.3389/fpls.2018.01308
Zhao, M., Tang, S., Zhang, H., He, M., Liu, J., Zhi, H., et al. (2020). DROOPY LEAF1 controls leaf architecture by orchestrating early brassinosteroid signaling. Proc. Natl. Acad. Sci. U.S.A. 117, 21766–21774. doi: 10.1073/pnas.2002278117
Zidani, S., Ferchichi, A., and Chaieb, M. (2005). Genomic DNA extraction method from pearl millet (Pennisetum glaucum) leaves. Afr. J. Biotechnol. 4, 862–866. doi: 10.4314/ajb.v4i8.15198
Keywords: Setaria italica, molecular marker, positional cloning, white leaf sheath, mutant
Citation: Zhang H, Tang S, Schnable JC, He Q, Gao Y, Luo M, Jia G, Feng B, Zhi H and Diao X (2021) Genome-Wide DNA Polymorphism Analysis and Molecular Marker Development for the Setaria italica Variety “SSR41” and Positional Cloning of the Setaria White Leaf Sheath Gene SiWLS1. Front. Plant Sci. 12:743782. doi: 10.3389/fpls.2021.743782
Received: 19 July 2021; Accepted: 13 October 2021;
Published: 11 November 2021.
Edited by:
Andrew Doust, Oklahoma State University, United StatesReviewed by:
Mingsheng Chen, Institute of Genetics and Developmental Biology, Chinese Academy of Sciences (CAS), ChinaChengjun Zhang, Kunming Institute of Botany, Chinese Academy of Sciences (CAS), China
Harriet Hunt, University of Cambridge, United Kingdom
Copyright © 2021 Zhang, Tang, Schnable, He, Gao, Luo, Jia, Feng, Zhi and Diao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xianmin Diao, diaoxianmin@caas.cn
†These authors have contributed equally to this work