 Sandra Pérez-BaosIván Prieto-PotinJorge A. Román-BlasOlga Sánchez-Pernaute
Sandra Pérez-BaosIván Prieto-PotinJorge A. Román-BlasOlga Sánchez-Pernaute Raquel Largo*
Raquel Largo* Gabriel Herrero-Beaumont
Gabriel Herrero-Beaumont- Bone and Joint Research Unit, Service of Rheumatology, IIS-Fundación Jiménez Díaz, Autonomous University of Madrid, Madrid, Spain
Besides its primary function in locomotion, skeletal muscle (SKM), which represents up to half of human's weight, also plays a fundamental homeostatic role. Through the secretion of soluble peptides, or myokines, SKM interacts with major organs involved in metabolic processes. In turn, metabolic cues from these organs are received by muscle cells, which adapt their response accordingly. This is done through an intricate intracellular signaling network characterized by the cross-talking between anabolic and catabolic pathways. A fine regulation of the network is required to protect the organism from an excessive energy expenditure. Systemic inflammation evokes a catabolic reaction in SKM known as sarcopenia. In turn this response comprises several mechanisms, which vary depending on the nature of the insult and its magnitude. In this regard, aging, chronic inflammatory systemic diseases, osteoarthritis and idiopathic inflammatory myopathies can lead to muscle loss. Interestingly, sarcopenia may persist despite remission of chronic inflammation, an issue which warrants further research. The Janus kinase/signal transducer and activator of transcription (JAK/STAT) system stands as a major participant in muscle loss during systemic inflammation, while it is also a well-recognized orchestrator of muscle cell turnover. Herein we summarize current knowledge about models of sarcopenia, their triggers and major mediators and their effect on both protein and cell growth yields. Also, the dual action of the JAK/STAT pathway in muscle mass changes is discussed. We highlight the need to unravel the precise contribution of this system to sarcopenia in order to design targeted therapeutic strategies.
Skeletal muscle (SKM) is a vast organ, which accounts for 40% of total weight in non-obese population (Janssen et al., 2000). The high metabolic activity of muscle cells, or myocytes, not only provides the necessary contraction for locomotion, but also fuels other organs' functions. Through the secretion of soluble peptides called myokines, the SKM interacts with surrounding fat, bones and skin, as well as with principal organs, including the cardiovascular system, brain, digestive tract and glands (Hartwig et al., 2014; Giudice and Taylor, 2017). In addition, some myokines exhibit autocrine/paracrine actions in the muscle, thereby helping sustain its normal growth (Pedersen and Febbraio, 2008). Considering the active interplay between SKM and other tissues, the impact of physical inactivity on general metabolism can be envisaged. In short, lack of exercise results in a lower insulin sensitivity, changes in the postprandial lipid profile, and accumulation of visceral adipose tissue (Pedersen and Febbraio, 2012). These effects are conceived to be the result of an evolutionary positive selection of pro-inflammatory pathways as well as of genes favoring gluconeogenesis, insulin resistance and fat storage. While formerly considered advantageous traits, in populations exposed to famines and epidemics, they have turned into a burden in modern ages, furthering cardiovascular diseases, because of lifestyle changes (Tuomilehto et al., 2001; Nocon et al., 2008).
Not only does SKM adjust the individual's metabolic activities, but it also suffers the consequences of perturbations in the systemic milieu. The SKM response to environmental cues is regulated by the hypothalamus, which integrates endocrine and immune signals, as well as information about physical activity and nutritional state (Clegg et al., 2013). Thus, levels of some nutrients –like glucose, fatty acids and amino acids– along with leptin and additional adiposity-related hormones (Roh et al., 2016) act as inputs for the elaboration of brain responses controlling energy expenditure, food intake, insulin secretion and glucose/fatty acid turnover in SKM (Roh et al., 2016). Muscle homeostasis requires fine-tuning of both protein turnover and cell growth in order to adapt its response to particular needs of the individual, without affecting muscle mass balance.
There is a dramatical shift in SKM homeostasis toward muscle loss during chronic inflammation. It is thought that an overacting immune system can divert energy expenditure and lead to a shortage of stored reserves affecting general metabolism (Straub, 2017). In spite of their relevance, these collateral effects are often forgotten in the assessment of prevalent conditions, while there is an increasing need of accurately measuring SKM response to stress induced by a variety of insults. However, the pathogenesis of systemic inflammation-related SKM damage, which has been termed cachexia or more accurately sarcopenia, has not until recently been understood. In this regard, sarcopenia can be observed in chronic debilitating diseases, as well as in typical inflammatory conditions, like rheumatoid arthritis, or in the proximity of injured joints, as in the case of osteoarthritis. It can also be the result of primary SKM autoimmune diseases. In the same way that the precipitating conditions are quite distinct in nature, so is the pattern of sarcopenia associated to them, while it also depends on the severity of the injury. All these factors make of sarcopenia a complex entity influencing general health which should be addressed in the therapeutic management of diseases.
This article gives an overview on triggers and mediators of SKM protein synthesis and cell turnover, especially focusing on clinical situations associated to muscle loss. The impact of systemic inflammation on muscle mass is discussed, looking into the molecular signals which disrupt muscle homeostasis in the different models of sarcopenia. In particular, the dual role of the Janus kinase/signal transducer and activator of transcription (JAK/STAT) pathway is underlined.
Muscle Protein Turnover
The major driver of SKM anabolic activity is the phosphatidylinositol 3-kinases (PIK3)/Akt signaling pathway. Along with exercise, insulin and insulin growth factor (IGF)-1 can induce phosphorylation of the insulin receptor substrate (IRS)-1 through the binding of specific receptors, subsequently activating PI3K (Sandri et al., 2013). In turn, the end product phosphatidylinositol-3, 4, 5 -trisphosphate (PI3P) facilitates membrane anchorage of Akt and its phosphorylation by 3′-phosphoinositide-dependent protein kinase-1 (PDK-1), upon which Akt enables activation of the mechanistic target of rapamycin (mTOR). The latter acts as downstream effector of the anabolic pathway through both the stimulation of ribosomal protein S6 kinase beta-1 (RS6K, or 70S6K), and the inhibition of 4E-binding protein 1 (4E-BP1) (Glass, 2003) (Figure 1). As we will discuss, Akt also shuts down the expression of forkhead O (FoxO), a transcriptional activator of muscle-specific E3 ubiquitin ligases involved in protein catabolism (Mammucari et al., 2007; Schiaffino et al., 2013). On the other hand, testosterone can induce muscle hypertrophy through the PI3K/Akt/mTOR pathway or also using androgen receptors (Basualto-Alarcón et al., 2013; Hughes et al., 2016).
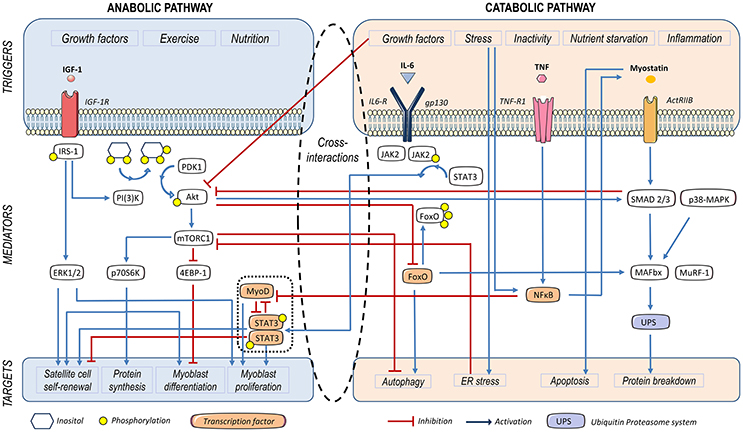
Figure 1. Triggers and mediators participating in protein and cell turnover of the skeletal muscle. Insulin growth factor (IGF)-1, leads to insulin receptor substrate-1 (IRS-1) phosphorylation. Subsequently, Akt becomes phosphorylated and activates the mammalian target of rapamycin (mTOR)-C1, thus contributing to protein synthesis and myoblast proliferation. Furthermore, Akt represses Forkhead O (FoxO), a factor that activates autophagy and the ubiquitin proteasome system (UPS) involved in protein catabolism. IRS-1 also activates the extracellular signal–regulated kinase (ERK), leading to satellite cell self-renewal and myoblast proliferation. IL-6-dependent activation of STAT-3 contributes to myogenic differentiation and SC proliferation in homeostatic conditions. Myostatin (MSTN) activates Smad2/3, which activates the ubiquitin proteasome system (UPS). Smad2/3 also inhibits the PI3K/Akt/mTOR signaling pathway, thus diminishing myoblast differentiation and protein synthesis. UPS is also activated by Forkhead O (FoxO), p38 mitogen activated protein kinases (MAPK) and nuclear factor κB (NFκB), which also induces a decay in MyoD mRNA, thus leading to a decrease in myoblast differentiation. IL-6-induced JAK2/STAT-3 signaling impairs myogenesis in a catabolic scenario.
Conversely, myostatin (MSTN) –or growth differentiation factor-8 (GDF-8)– is a member of the transforming growth factor β (TGF-β) family, and a negative regulator of muscle growth. The MSTN-Smad2/3 route inhibits Akt-dependent protein synthesis and growth of mature muscle cells (Morissette et al., 2009; Schiaffino et al., 2013; Brooks and Myburgh, 2014; Retamales et al., 2015). This myokine binds to activin type 2 receptors (ACVR2) and promotes cachexia-related catabolism, via FoxO-dependent induction of atrogenes (McFarlane et al., 2006) (Figure 1). As it has been shown both in rodents and humans, training acts as a MSTN repressor. This fact has been put in relationship with the benefit of endurance exercise on metabolism. On the other hand, both muscle and serum of obese individuals appear to be comparatively enriched in MSTN (Hittel et al., 2009).
Regulatory Networks of Protein Turnover
The concurrence of pro/anti-anabolic and pro/anti-catabolic factors, not only within the muscle milieu but also in the systemic environment, determines the yield of muscle turnover (Reed et al., 2012; Schiaffino et al., 2013). Usually, up-regulation of protein synthesis is accompanied by a reduction of protein degradation (Schiaffino et al., 2013). Likewise, under catabolic stress, a well-orchestrated network blocks new protein synthesis in order to limit energy expenditure. Nonetheless, if conditions are favorable, the amino acids released during proteolysis will boost new protein synthesis (Tran et al., 2007; Zoncu et al., 2011; Perl, 2015).
The role of microRNAs in muscle anabolic fine-tuning is noteworthy. MicroRNAs are small non-coding RNAs which act as negative regulators of gene expression. Muscle specific microRNAs, known as myomiRs, target myostatin, TGF-β and Akt-dependent pathways. In turn, these factors modify the expression rate of different myomiRs (Butz et al., 2012; Hitachi and Tsuchida, 2014; Jung and Suh, 2015). Their isolation from muscle exosomes suggests that myomiRs can be released and exert autocrine/paracrine actions in surrounding cells, in this way facilitating a synchronous muscle growth response (Demonbreun and McNally, 2017; Fry et al., 2017).
Cell Growth
Myogenesis
Although anabolic factors have commonly a role in myogenesis, processes of cell growth and protein synthesis have to be considered as independent parts of the SKM metabolism. In particular, myogenesis accounts for the capacity of cell renewal and differentiation during repair processes.
This capacity is based on the existence of a niche of quiescent myogenic cells, known as satellite cells (SC), between the outer coat of sarcolemma and the basal lamina of myofibers (Morgan and Partridge, 2003). These cells are characterized by the expression of paired box transcription factor (Pax) 7 and myogenic factor (Myf) 5 and have a crucial role in self-renewal of SKM. Upon injury, SC undergo proliferation and differentiation into myoblasts (Pax7+/Myf5+/MyoD+), which eventually lose expression of Pax7 and upregulate myogenin (Pax7-/Myf5+/MyoD+/Myogenin+). The latter determines cessation of cell proliferation and progression to terminal differentiation, maturation and fusion of cells into new myofibers (Morgan and Partridge, 2003).
The JAK/STAT pathway and its triggering cytokine IL-6 stand as key myogenic factors. The intracellular network exerts a relevant homeostatic role in healthy SKM. Normal proliferating myoblasts have been found to exhibit high levels of phospho-STAT-3, both in vivo (Kami and Senba, 2002) and in culture (Spangenburg and Booth, 2002; Yang et al., 2009). According to the studies performed by Hoene and co-workers in primary mouse myoblasts and in C2C12 cells, the activation of STAT-3-SOCS-3 in response to an autocrine action of IL-6 is needed for their differentiation (Hoene et al., 2013). In the same way, IL-6 stimulation of STAT-3 is required for SC proliferation following SKM overload. Moreover, not only does IL-6 stimulate myogenesis, but it also exerts some key metabolic actions (Pedersen and Febbraio, 2008; Muñoz-Cánoves et al., 2013) and blocks the production of pro-apoptotic cytokines like tumor necrosis factor alpha (TNF) (Schindler et al., 1990). Altogether, these actions place IL-6 as a pivotal myokine released from muscle cells upon exercise.
Myoblast proliferation is subject to a tight control, which prevents alterations in muscle mass volume during repair processes (Verdijk et al., 2007; McFarlane et al., 2011; Fry et al., 2015; Demonbreun and McNally, 2017). Experiments conducted in the rodent C2C12 and L6 myoblast cell lines have shown that IGF-1 plays opposite roles at distinct stages of myocyte differentiation. While the peptide initially exerts a mitogenic effect, it inhibits growth and promotes cell differentiation at later phases (Engert et al., 1996; Florini et al., 1996). In turn, these polar activities are driven by different signaling pathways. Extracellular signal-regulated kinase (ERK) appears to be involved in cell proliferation (Rosenthal and Cheng, 1995; Coolican et al., 1997), whereas PI3K/Akt/p70S6K has been shown to mediate IGF-1-induced myogenin expression (Xu and Wu, 2000) (Figure 1). Similarly, the JAK/STAT pathway may promote either proliferation or differentiation depending on the molecular isoforms involved in its activation, as we will discuss (Sun et al., 2007; Wang et al., 2008; Diao et al., 2009).
Regulatory Catabolic Processes
Metabolic disturbances and stress trigger numerous repair mechanisms in SKM aimed at restoring homeostasis. Intracellular proteolytic complexes, including calpains, the endoplasmic reticulum (ER) stress response, caspase cascades, the autophagic machinery and the ubiquitin-proteasome system (UPS) can participate in such response. Dysregulation and/or perpetuation of these reparative mechanisms may result in muscle wasting, either by increasing proteolysis or initiating muscle cell apoptosis.
The Ubiquitin-Proteasome System (UPS)
The UPS accounts for the principal mechanism of protein degradation in muscle cells (Glass, 2010; Milan et al., 2015). Two muscle-specific E3 ligases, muscle ring finger-1 (MuRF-1) and muscle atrophy F-box protein (MAFbx) –or atrogin-1– (Schiaffino et al., 2013) have been identified and their expression is commonly used as a marker of UPS activity in secondary sarcopenia models (Milan et al., 2015). UPS is regulated by FoxO (Sandri et al., 2004; Tournadre et al., 2017), p38 mitogen activated protein kinase (MAPK) (Zhang and Li, 2012), STAT-3 (Bonetto et al., 2012) and nuclear factor κB (NFκB) (Cai et al., 2004). Furthermore, STAT-3 and NFκB down-regulate MyoD expression, and, consequently, decrease myoblast differentiation (Guttridge et al., 2000) (Figure 1). While FoxO is blocked by Akt, the other mediators are activated in response to pro-inflammatory molecules (Schiaffino et al., 2013). Notably, insulin peripheral resistance, which shuts the IGF1-Akt pathway down, also raises UPS activity (de Alvaro et al., 2004; Brown et al., 2009) (Figure 1).
Autophagy
Autophagy is carried out by a cascade of proteolytic reactions which degrade and recycle malfunctioning organelles, proteins and other cytoplasmic molecules, using the lysosomal machinery and a multimolecular complex (Mizushima and Komatsu, 2011). This highly conserved mechanism of homeostasis allows cells to survive under stress conditions (Cuervo, 2008). In addition, defects in autophagy associate with muscle disease and inflammation (Levine and Kroemer, 2008). However, over-induction of autophagy is also responsible for muscle loss. Up to 35 different autophagy-related genes (ATG) encoding the autophagy machinery have been found. The inhibition of mTOR complex (mTORC)1 activates the initiation steps of autophagy, inducing autophagosome nucleation and substrate recognition. This is followed by the recruitment of different ATG products that facilitate protease digestion of the cargo (Portal-Núñez et al., 2015). Likewise, muscle mass can also be regulated by mitochondrial dysfunction, ER stress or myocyte apoptosis (Nagaraju et al., 2005; Busquets et al., 2007; Nogalska et al., 2007; Marzetti et al., 2009, 2010; Deldicque, 2013).
Endoplasmic Reticulum (ER) Stress
ER and mitochondria cooperate in SKM homeostasis. Disturbances of their balance can be induced by protein misfolding, starvation or energy deprivation, and lead to an ER stress response. Consequently, ER activates the so-called ‘unfolded protein response’, consisting in the upregulation of chaperones and other enzymes that participate in adequate protein folding. If the insult persists, the stress response can be unable to maintain cell homeostasis and give rise to the activation of NF-κB and other inflammatory pathways. Uncontrolled ER stress eventually lead to myocyte death by mechanisms which include apoptosis, autophagy and necrosis (Deldicque, 2013). Furthermore, it has been proposed that ER stress could indirectly contribute to muscle wasting through the blockage of mTORC1, thus creating a state of anabolic resistance (Deldicque, 2013).
Apoptosis
Myofiber loss accounts for the key process leading to sarcopenia during aging, skeletal muscle immobilization, muscular dystrophy and other inflammatory conditions (Dupont-Versteegden, 2006). The specific mechanisms of cell death in SKM remain largely unknown and will not be addressed in this review. Noteworthy, Dupont-Versteegden and co-workers suggested that classical apoptotic pathways could be of little relevance in muscle. Alternatively, additional molecules might exert a key role in triggering regulatory cell death processes (Dupont-Versteegden, 2006). Of note, being myofibers multinucleated syncytia, nuclear elimination does not necessarily carry cell destruction. In this regard, muscle loss can rely on loss of myonuclei or of their associated cytoplasmic domains (Allen et al., 1999).
Sarcopenia, the SKM Response to Systemic Inflammatory Stress
Systemic inflammation affects all body systems and organs, including the SKM. Muscle response can follow different patterns, namely primary sarcopenia, secondary sarcopenia, and those respectively found in osteoarthritis and in idiopathic inflammatory myopathies. These four inflammation-related clinical settings of muscle wasting are summarized in Table 1 and will be addressed below.
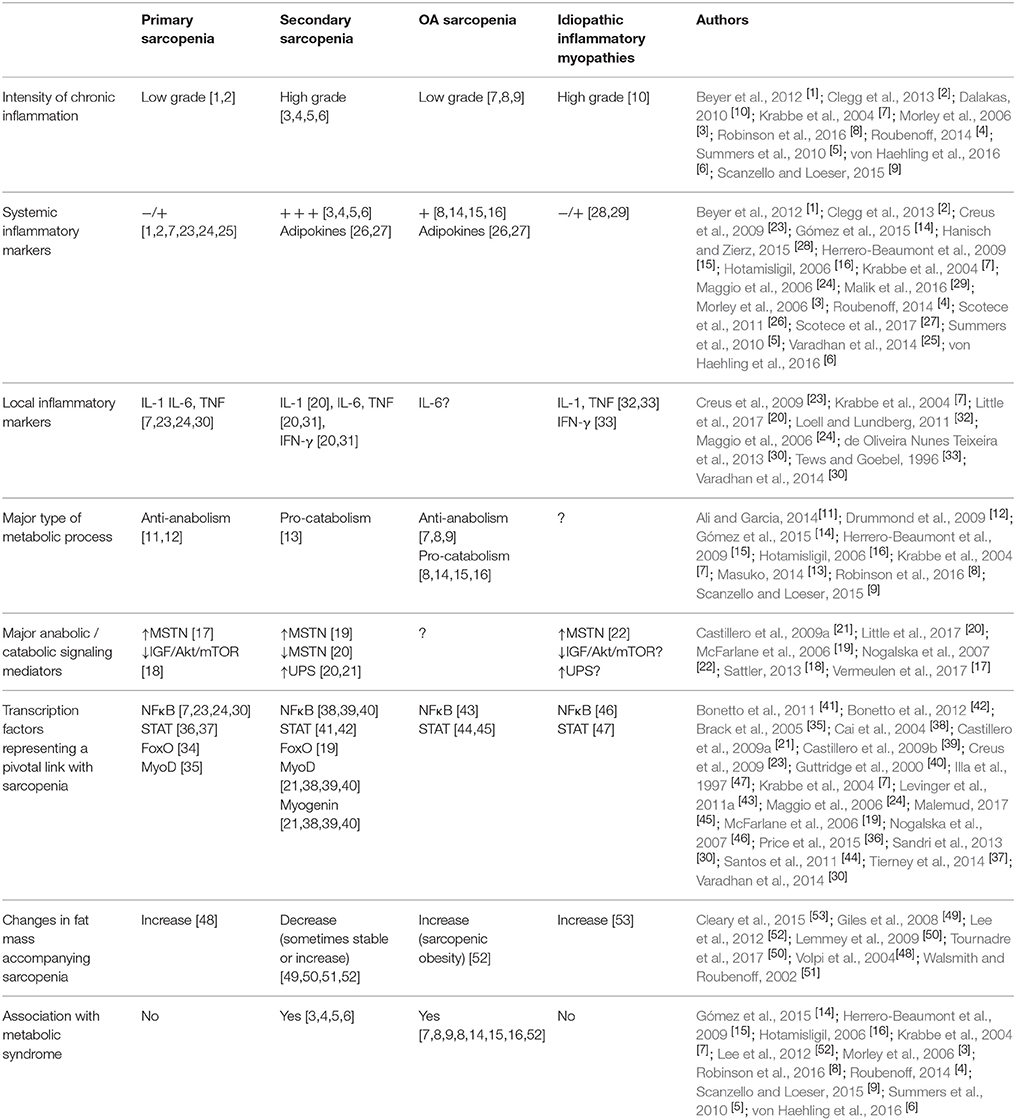
Table 1. Inflammatory description and altered molecular mediators in four clinical settings related to muscle wasting. −/+: absent / low; +: mild; +++: high.
Primary Sarcopenia
The condition known as primary sarcopenia (PS) is associated with the aging and the frailty syndrome and is defined as a progressive and generalized loss of SKM mass and/or strength leading to a significant functional impairment (Cruz-Jentoft et al., 2010). This muscle response is considered to be the consequence of aging/disease interactions at multiple systems (Cruz-Jentoft et al., 2010). Thus, PS identifies a poor health status frequently associated with disability, increasing risk of falls and fractures, and potentially dragging the elderly to dependence. On these grounds, its appearance leads to a decreased life expectancy (Cruz-Jentoft et al., 2010).
This type of sarcopenia represents a failure in muscle anabolic processes. In addition, it is likely that a mild persistent inflammatory status could play a role in its pathogenesis, promoting a catabolic scenario(Ali and Garcia, 2014).
Morphologically, atrophy of type II (fast-twitch, highly ATP-consuming) fibers can be observed, following a loss of myocyte proteins, organelles and cytoplasm size (Muscaritoli et al., 2010), and the accumulation of muscle fat. The origin of PS has been put in relationship with age-dependent functional changes in mitochondria, ER, cells and tissues. Furthermore, as suggested by studies conducted in aged mice, the niche of SC could decline with age and be insufficient for nuclear replacement (Brack et al., 2005). Additional factors are related to lack of mobility, neurodegeneration, nutritional deficiencies and hormone decrease. Progressive testosterone deficiency increases peripheral resistance to insulin and IGF-1 –both potent activators of the Akt/mTOR pathway– yielding a lower synthesis and a higher degradation of muscle proteins, through the activation of FoxO (Sandri et al., 2004). Likewise, an age-dependent impairment in the GH/IGF-1 axis has been reported. A fall in gene expression of GH receptors in the elderly inversely correlates with serum MSTN levels (Perrini et al., 2010). This fact could hamper both synthetic processes and cell renewal (Taylor et al., 2001) –and the same effects could be expected from the lowering of sex hormone levels –. Of all myokines, IGF-1 is regarded as a pivotal mediator of muscle growth because of its effect on SC proliferation, and its concentration is inversely associated with the development of PS. However, recent experiments have suggested that deficiency of GH/IGF-1 could increase longevity in animals (Sattler, 2013). Indeed, some human studies have also drawn controversial results, a matter which has diverted the attention from the GH/IGF-1 axis to MSTN up-regulation, as the cornerstone of the anti-anabolic response of PS.
On the other hand, although clearly regarded as an anabolic resistant process, there is also an age-related low-grade chronic inflammation that may contribute to muscle wasting in PS (Clegg et al., 2013) through the up-regulation of pro-inflammatory cytokines, like TNF, IL-1 and IL-6 (Krabbe et al., 2004; Maggio et al., 2006; Drummond et al., 2009; Ali and Garcia, 2014). This view is supported by the finding of differential levels of serum biomarkers between active and non-active elders, with an enhancement of IL-8, myeloperoxidase and TNF in the latter group (Marzetti et al., 2014).
Increased levels of IL-6 appear to be particularly associated to a higher disability in the elderly (Barbieri et al., 2003; Maggio et al., 2006). In spite of its homeostatic role in SKM, both driving myogenesis and mediating IGF-I anabolic actions (Barbieri et al., 2003; Maggio et al., 2006; Mammucari et al., 2007; Schiaffino et al., 2013), over-expression of IL-6 is known to impair myocyte functions (Roubenoff, 2014). Indeed, its levels along with those of TNF-R1 out of a group of 15 different NF-kB-dependent molecules, have been found to be the best predictor of mortality in the elderly (Varadhan et al., 2014). Altogether this suggests that the inflammatory response could account for a therapeutic target in PS.
Secondary Sarcopenia (SS)
Secondary sarcopenia (SS) occurs in the context of chronic illnesses, like cancer, renal/respiratory failure or inflammatory diseases. A paradigm of this SKM response is rheumatoid sarcopenia –or sarcopenia during rheumatoid arthritis– (RS) (Morley et al., 2006; von Haehling et al., 2016). Sarcopenia is a prominent feature of the more generalized rheumatoid cachexia syndrome, thus termed because it targets major organs and immune cells along with the SKM, leading to a profound loss of cell mass in all these tissues. In this regard, two types of cachexia have been associated to RA; the ‘classic’ type –which resembles those found in cancer and AIDS– and shows both muscle and fat mass loss, and the more typical ‘rheumatoid’ type –which results in a reduced muscle mass but an increase in fat volume– (Giles et al., 2008; Lemmey et al., 2009; Summers et al., 2010; Tournadre et al., 2017). Other forms of SS are also frequently referred to as cachexia (Roubenoff et al., 1992), due to their predominant catabolic component (Masuko, 2014). However, there are marked differences between RS and the other forms of SS (Summers et al., 2010) because of the high inflammatory status which characterizes RS, and the relevant role of the adaptive immune system in this disease.
The prevalence of RS cannot be determined with accuracy, largely due to a lack of consensus in establishing a clear cut-off in body composition for its diagnosis. Besides, the syndrome can pass unnoticed in patients with stable weight resulting from the increase in fat volume, which can mask muscle mass loss (Summers et al., 2010). It has though been estimated to be present in 38% of patients with active rheumatoid arthritis and in 10–20% of those with well-controlled disease (Elkan et al., 2009; Phillips et al., 2009; Summers et al., 2010). The appearance of RS does not carry the same poor prognosis as SS associated to AIDS or cancer. Still, its development impacts both life expectancy and quality of life, particularly due to its association with metabolic syndrome, cardiovascular disease, and weakness, in an independent fashion of disease severity (Kotler, 2000; Walsmith and Roubenoff, 2002; Fukuda et al., 2013).
Why does RS run an independent course from disease activity has been difficult to understand, since the condition is principally unchained by systemic inflammation. It appears that the concurrence of a variety of signals triggers RS, the most relevant of them being inflammation-driven increased metabolism, a reduced anabolic activity, the coexistence of malnutrition, peripheral insulin resistance and lack of exercise. Although RS has not been primarily associated with an impairment of anabolic processes, low IGF-1 has been shown to parallel muscle mass loss in experimental models (Soto et al., 2001; Castillero et al., 2009a) and also in the patients, after adjusting for age, sex, and adiposity (Lemmey et al., 2009; Baker et al., 2015). In addition, it has been proposed that rheumatoid arthritis-associated insulin resistance indirectly promotes muscle wasting, because of the physiological anti-catabolic effect of insulin (Walsmith and Roubenoff, 2002). However, further studies are needed to clarify the involvement of this pathway in RS (Lemmey et al., 2009).
Pro-inflammatory cytokines such as TNF, IL-1β and IL-6, have been proved to exert a critical contribution to the hypercatabolic state found in these patients (Roubenoff, 2014). Although RS pathogenesis is poorly understood, elevated serum levels of these cytokines are known to activate UPS. In experimental cachexia, an upregulation of muscle-specific E3 ubiquitin ligases MuRF-1 and atrogin-1 has been shown, followed by an increased myofibrillar proteolysis, through the activation of NF-κB (Li et al., 2009; Varadhan et al., 2014; Little et al., 2017). These data are consistent with the over-activation of UPS during SS, as has also been found in muscle wasting associated to experimental arthritis (Castillero et al., 2009a; Little et al., 2017). In the same way, the increased activity of FoxO resulting from MSTN-dependent PI3K/Akt inhibition enhances the expression of atrogenes, thus promoting protein catabolism during cachexia (McFarlane et al., 2006); however, the role of this transcriptional regulator in RS is not well defined.
Our group recently conducted studies to look into muscle response to chronic arthritis in a rabbit model which emulates RS. The diseased animals had a reduction in weight and muscle size, and an up-regulation of atrogin-1 both in muscle and in synovium, altogether indicating an increased protein breakdown. Strikingly, there was a paradoxical decrease in MSTN expression, along with a reduction in phospho-STAT-3 levels, which pointed to the existence of a compensatory anabolic activation (Little et al., 2017). This pattern of response suggests that the inflamed muscle could contribute to the process of SS through a mechanism of autocrine atrophy triggered by the release of muscle-derived, pro-inflammatory mediators (Little et al., 2017).
It can be envisaged how devastating the effects of atrogenes can be on the musculature of elderly RS patients (Guadalupe-Grau et al., 2015). Also interesting is that both muscle mass loss and IGF-1 content are remarkably returned to normal in patients with RS following a high intensity training program (Lemmey et al., 2009). This underlines the therapeutic goal of breaking down anabolic resistance in RS. On the other hand, standard antirheumatic therapies should not be expected to prevent RS, since changes in body composition can be observed in patients with low disease activity (Metsios et al., 2007). Previous work in our laboratory disclosed a protective role of celecoxib in the development of sarcopenia in rabbits (Romero et al., 2010). The selective inhibition of COX-2, and COX-2 derived products such as PGE2, yielded a reduction in both systemic inflammation and NF-kB activation, together with an amelioration of weight loss in arthritic rabbits. As suggested by previous studies, the inhibitory effect of NSAIDs on NF-kB signaling might be responsible for the suppression of muscle wasting induced by the activation of the ubiquitin-proteasome pathway (Wyke et al., 2004).
As regards myogenesis impairment, there are no conclusive data about its direct contribution to RS. Low MyoD and myogenin levels have been observed in a rat model of arthritis, arguing for this possibility (de Oliveira Nunes Teixeira et al., 2013). In this regard, both TNF and IFNγ can impair myogenesis, as shown in C2C12 myoblasts and in mouse muscle, where the cytokines suppressed MyoD and myogenin through an NFκB-dependent pathway (Guttridge et al., 2000). By contrast, Castillero and co-workers showed increased expression of these myogenic factors in arthritic rats (Castillero et al., 2009a,b).
OA Sarcopenia
The effect of osteoarthritis (OA) represents a third subset of muscle metabolic adaptive response to systemic inflammation. A variety of factors are involved in the pathogenesis of this complex syndrome, including biomechanical stress, senescence, hormonal status and inflammatory mediators, while the genetic background could also be relevant (Herrero-Beaumont et al., 2009). While all joint tissues are targeted by OA, the initial insult can be localized at any of them, and render different syndromes (Roman-Blas et al., 2016). As the disease progresses into advanced phases, the OA syndrome is usually more uniform, although interspersed flares in relationship with acute injuries, can occur (Castañeda et al., 2014). On the whole, the disease can be considered a protean long-course process, with many possible shifts of phenotype along time.
In the same way, inflammation in OA can take many forms. Typically, there is a low-grade inflammatory status in the OA joint, which can get temporarily higher in response to different triggers, such as capsular sprains, micro-trauma, and the presence of crystals or additional danger associated molecular patterns (DAMPs). All these factors are known to couple innate receptors, such as PI3K and the toll-like family (TLRs) (Gómez et al., 2015). Alternatively, biomechanical sensors can also contribute to these flares, since their signaling network interlinks with major inflammatory pathways.
As regards OA-related sarcopenia, it does not only target the neighboring muscles of affected joints but can also involve the whole SKM. However, the underlying mechanisms regulating joint-muscle crosstalk are not yet fully understood. A close relationship between impaired SKM functions and knee OA has been observed, along with an enhanced expression of FoxO1 reflecting lower muscle strength (Levinger et al., 2016). Since cartilage and SKM cells share some cellular pathways, paracrine communication between them remains conceivable, in addition to the influence of a close anatomical proximity (De Ceuninck et al., 2014). On the other hand, individual factors associated to OA, such as lack of physical activity and obesity can play an indirect role in the pathogenesis of OA-related sarcopenia, while they also account for an increased cardiovascular risk and a shortened life expectancy (Yoshimura et al., 2012). This places adipokines as principal actors in this model of SKM adaptive response to ‘low-grade inflammation’ (Scotece et al., 2011, 2017). Adipokines are hormone-like fat-derived factors which contribute to maintain the low-grade inflammatory status in patients with metabolic syndrome (Gómez et al., 2015). Adiponectin and leptin are by far the best characterized adipokines. They both stimulate glucose uptake and fatty acid oxidation both in muscle and in adipose tissue (Kalinkovich and Livshits, 2017). Of them leptin exerts a pro-inflammatory role and adiponectin is mainly regulatory. While serum levels of adiponectin decrease in relationship with OA, age and obesity, serum leptin is enhanced in patients with OA in parallel with the accumulation of adipose mass (Poonpet and Honsawek, 2014; Kalinkovich and Livshits, 2017). Other relevant adipokines reported to be increased in OA are resistin (Koskinen et al., 2014), which drives pro-inflammatory actions in human SKM (Carey et al., 2006) and chemerin (Ma et al., 2015), which has been shown to inhibit myogenesis and induce adipogenesis in C2C12 myoblasts (Li et al., 2015).
Both OA and sarcopenia are prevalent in the elderly (Felson et al., 1987; Fielding et al., 2011). OA is frequently considered as part of the metabolic syndrome and of senescence (Herrero-Beaumont et al., 2009). The severe peri-articular sarcopenia found in OA could be partly due to inactivity, but also due to low-grade persistent systemic inflammation, which is a feature of both syndromes (Krabbe et al., 2004; Scanzello and Loeser, 2015). In fact, sarcopenic obesity is more closely associated with knee OA than non-sarcopenic obesity, thus supporting the tight relationship between muscle metabolism and inflammation in this disease (Lee et al., 2012).
It has been suggested that sarcopenia could not only be a trigger of OA, but also a worsening factor for its progression (De Ceuninck et al., 2014). Muscle transcription factors associated to inflammation, such as STAT-3 and NFκB, correlate with the grade of joint dysfunction, disability and gait impairment in the patients (Levinger et al., 2011b). Similarly, an inverse correlation has been observed between muscular resistance of hamstrings and serum IL-6 levels in elderly women with OA (Santos et al., 2011), suggesting a role for this cytokine in OA sarcopenia. In fact, Levinger and co-workers found augmented levels of IL-6, STAT-3, SOCS-3 and NFκB, among others, in the vastus lateralis of patients with knee OA (Levinger et al., 2011a). The relevance of IL-6 in OA pathogenesis is further sustained by its association with cartilage loss and radiographic knee OA (Livshits et al., 2009; Stannus et al., 2010). The cytokine could therefore account for a pivotal link between OA and sarcopenia, and provide an attractive therapeutic target, although further research is warranted.
Sarcopenia Associated With Idiopathic Inflammatory Myopathies (IMM)
Finally, a distinct pattern of muscle wasting can be observed in patients with idiopathic inflammatory myopathies (IMM). These systemic autoimmune diseases are characterize by weakness, muscle inflammation (Day et al., 2017) and fat mass gain (Cleary et al., 2015), without an increase in serum CRP levels (Hanisch and Zierz, 2015; Malik et al., 2016). Both the innate and the adaptive immune systems are involved in their pathogenesis. On one hand, there is a deep disturbance in the adaptive immune responses, with activation of auto-reactive cytotoxic T cells along with production of autoantibodies (Dalakas, 2010). On the other, an infiltration of inflammatory cells is responsible for cell death, and disruption of the normal muscle architecture. Local production of cytokines participates in IMM pathogenesis, both promoting cell damage and impairing muscle cell function, although some of them, such as IL-1, TNF or IL-15, appear to have a role in repair stages as well. Therefore, the complexity of muscle cytokine networks deserves especial attention at designing therapeutic strategies (Loell and Lundberg, 2011). Also to underline is the fact that muscle function restriction often persists in the patients in the long-term, despite the achievement of remission with immunosuppressive treatments (Loell et al., 2016). There appears to be therefore a permanent footprint of a previous muscle injury, which might also be present in the other three subsets of sarcopenia.
In these heterogeneous muscle disorders myocyte degeneration and necrosis occur mainly due to directed self-reactivity of CD8+ T cells. Sarcolemma disruption provokes the release of myoglobin and creatine kinase (Chargé and Rudnicki, 2004). Antigen-specific cytotoxic T lymphocytes (CTL) migrate through the endothelial wall and directly bind to muscle fibers aberrantly expressing major histocompatibility complex (MHC)-I molecules on their surface, through their T-cell receptors. Upon presentation of muscle antigens, these infiltrating CD8+ T cells undergo clonal expansion (Rayavarapu et al., 2013). Histologically, affected muscles are characterized by perivascular cell infiltration, predominantly consisting of CD8+ T cells invading and surrounding healthy-appearing muscle fibers. Direct cytotoxicity to muscle cells can then take place through the release of perforin granules. Cell infiltration is located in different regions of the muscle fascicles (i.e. interfascicular septae, periphery of the fascicle, epimysium, endomysium, etcetera) depending of the clinical subgroup (Vattemi et al., 2014). Cytokines, such as IFN-γ, IL-1, and TNF released by activated T cells, may enhance MHC class I up-regulation and T-cell cytotoxicity. There is also a shift toward Th17 CD4+ T cell differentiation, which furthers the autoimmune process (Tews and Goebel, 1996). However, as mentioned before, it has been reported that IL-1 and TNF may exert a role in muscle regeneration (Loell and Lundberg, 2011). Some of the above-mentioned cytokines signal through the NF-kB pathway and/or are controlled by this transcription factor, which indeed seems to have a key pathogenic role in IIM. It is well known that NF-kB becomes activated both in inflammatory cells and in myocytes and enhances MHC-I expression in muscle fibers. This event has been associated with ER stress, which fuels MSTN production and muscle injury (Tews and Goebel, 1996; Nagaraju et al., 2005; Nogalska et al., 2007; Creus et al., 2009). As has been previously mentioned, NF-kB is known to induce atrogene expression and loss of MyoD messenger RNA (Guttridge et al., 2000; Cai et al., 2004). On the other hand, MSTN exerts important anti-anabolic effects by blocking the PI3k/Akt/mTOR signaling pathway (Morissette et al., 2009; Schiaffino et al., 2013; Brooks and Myburgh, 2014; Retamales et al., 2015). In conclusion, these pro-catabolic and anti-anabolic mediators can elicit IIM-dependent sarcopenia.
The Dual Role of JAK/STAT and IL-6 in SKM
While the JAK/STAT pathway has been reported to play a crucial role in myogenesis, its precise contribution to muscle wasting and regeneration is yet to be defined. According to results drawn by different studies, it can be argued that the activation of JAK/STAT results in different effects in healthy and in injured muscle cells. This is likely due to the magnitude and duration of the induction.
As previously mentioned, the IL-6/STAT-3/SOCS-3 axis contributes to myogenic differentiation in physiological situations (Hoene et al., 2013). By contrast, experimental murine models of rheumatoid cachexia exhibit a profound total and muscle and total weight loss and elevated IL-6 levels are consistently found along time. IL-6 involvement could be determinant to muscle atrophy (Bonetto et al., 2012), and drive muscle wasting through the activation of UPS (Bodell et al., 2009). Furthermore, chronically elevated IL-6 levels might reflect a feedback mechanism triggered by an impairment in IL-6 dependent signaling. To our knowledge, whether this phenomenon of ‘IL-6 resistance’ takes place during muscle wasting has not been addressed. However, it is conceivable that IL-6 deregulation during disease could follow a similar pattern to those of insulin or leptin tissue resistance.
Hypothetically, during RS this phenomenon could be due to an overexpression of SOCS-3 in muscle cells. Consistently, muscle SOCS-3 overexpression is known to abrogate leptin-dependent STAT-3 phosphorylation, in this way favoring leptin resistance (Bjørbaek et al., 1998). Interestingly, increases in SOCS-3 could also inhibit the insulin receptor, thus promoting insulin resistance as well (Ueki et al., 2004). Also of interest would be the assessment of soluble gp130/sIL-6R in RS muscle, since this decoy receptor could account for an additional mechanism of IL-6 hyporesponsiveness (Jostock et al., 2001). The conflicting effects of IL-6 on muscle certainly represent a novel avenue for further research, which could be termed the ‘IL-6 paradox’.
In line with this findings, Tierney et al. and Price et al. reported that the enhancement of JAK2/STAT-3 signaling impaired myogenesis in aging mice, likely due to a loss of SC self-renewal capability (Tierney et al., 2014; Price et al., 2015). Subsequently, STAT-3 has been hold responsible for driving SS. Bonetto and co-workers found that STAT-3 and its responsive genes were up-regulated in mice with cancer-associated cachexia (Bonetto et al., 2011). In these mice, they found that constitutive activation of the transcription factor worsened their wasting status, and that JAK1/2 or STAT-3 blockade could revert this situation (Bonetto et al., 2012). As has been shown in transfected C2C12 myoblasts, overexpression of STAT-3 results in its direct interact with MyoD, thus inhibiting myogenic differentiation. Reciprocally, MyoD was shown to decrease STAT-3 activity (Kataoka et al., 2003).
Although there are scant studies looking into this signaling cascade in OA related sarcopenia, a role of the IL-6/STAT-3/SOCS-3 axis in muscle wasting following ‘chronic low-grade inflammation’ can be foreseen. Of note, in addition to experimental findings, levels of these mediators were increased in the vastus lateralis of patients with knee OA (Levinger et al., 2011a).
Globally, these data point to a role of STAT-3 in normal regeneration of healthy muscle. Nonetheless, its chronic activation during aging or chronic inflammation could carry an impairment of SC self-renewal, a lower proliferation of myoblasts and an overall abnormal muscle repair.
Among all the JAK and STAT molecular mediators, STAT-3 has received major attention in the study of healthy and damaged muscle. This transcription factor appears, nonetheless, to play distinct roles on myogenesis depending on the JAK protein kinase activated upstream. In brief, the JAK2/STAT2/STAT-3 pathway has been found to exert a pro-differentiation effect in primary muscle cells and C2C12 myoblasts (Wang et al., 2008), whereas JAK1/STAT-1/STAT-3 was shown to boost myoblast proliferation through the regulation of cell cycle-associated genes' expression. As reported, the latter could also prevent myoblast premature differentiation, thus acting as a checkpoint during myogenesis (Sun et al., 2007). In a subsequent study, the same group observed that SOCS-1 and SOCS-3 intercepted JAK1/STAT-1/STAT-3 signaling, thereby promoting myogenic differentiation (Diao et al., 2009).
Very little is known about JAK/STAT activation in IIM. According to Illa and co-workers patients with dermatomyositis could show an increased STAT-1 activation in myofibers (Illa et al., 1997). Presumably, the enhancement of JAK/STAT dependent transcription by local cytokine networks drives muscle wasting in IIM. The overall extent of muscle wasting is likely determined by activation of specific JAKs and STATs triggered by a particular group of cytokines. Whether these changes can be reverted with treatment, as well as the effects of different JAK inhibitors depending on their specificity for particular JAK/STAT cascades, deserves further research.
Conclusions and Future Perspectives
A tight regulation of protein turnover and cell growth is crucial to maintain homeostasis in SKM. Chronic systemic inflammation, however, provokes a dramatical shift in this balance, thus compromising SKM mass. Behind this fine-tuning, there is a very intricate network of catabolic and anabolic signaling pathways, which are summarized in Figure 1. In short, induction of the Akt/mTOR pathway not only enhances myogenesis and protein synthesis, but also slows down catabolic UPS and the autophagic machinery. Conversely, MSTN exerts important anti-anabolic and pro-catabolic effects, through the suppression of the Akt/mTOR axis and the induction of UPS, respectively. Pro-inflammatory cytokines mostly act as pro-catabolic and anti-anabolic factors, with IL-6 exerting a dual role in SKM turnover.
It has not been until recently that the pathogenesis of chronic inflammation-related sarcopenia has been understood. Consequently, the SKM response to the inflammatory milieu is often unadvertised in the assessment of prevalent conditions. In addition, despite the recent advances providing insight on the relationship between chronic inflammation and sarcopenia, there are still substantial gaps, which may account for the lack of treatments for sarcopenia. Intriguingly, muscle mass loss does not seem to revert despite the achievement of remission. Therefore, the mechanisms behind this permanent footprint need to be approached in future studies.
While innate and adaptive immune responses are extremely energy-consuming, metabolic inflammation does not lead to an increased energy expenditure. Notwithstanding this fact, metabolic inflammation is often said to resemble a smoldering fire, which is difficult to extinguish (Hotamisligil, 2006). Moreover, chronic low-grade inflammation and metabolic dysfunction drive the development of the most prevalent chronic diseases, particularly those targeting the musculoskeletal system such as OA (Hotamisligil, 2006; Robinson et al., 2016). Of interest, they also significantly contribute to the fragility syndrome of the elderly. As is widely acknowledged, major factors associated with accelerated atherosclerosis are the components of the metabolic cluster, namely hypertension, high blood glucose, excess in waist fat, and abnormally increased lipid levels. Obesity, which shows a higher prevalence in OA than in non-OA age-matched individuals, could be the most relevant of these traits.
Additional pathways of muscle loss are unchained by anorexia, asthenia and inactivity, all of which are typical features of chronic systemic inflammation (Phillips et al., 2009). These processes are, in fact, shared by the four previously mentioned clinical settings (Huffman et al., 2017). In this regard, as has been recently reviewed in depth by Dalle and coworkers, exercise and dietary interventions have proved beneficial against the anabolic resistance of the elderly (Dalle et al., 2017). On the other hand, considering the impact of chronic inflammation on the development of this resistance, it appears that anti-inflammatory therapies could provide an extra benefit to lifestyle and nutrition changes on restoring muscle mass in the aged population (Dalle et al., 2017). Similarly, not only suppression of disease activity but also exercise and a well-suited nutritional management have been underlined as major strategies against RS (Masuko, 2014).
An increasing attention is being currently paid to the JAK/STAT pathway as a promising target for the treatment of muscle wasting diseases. The product of this signaling cascade seems to differ, likely depending on the magnitude and duration of the input. It would be of high interest to assess therapeutic effects of JAK/STAT inhibitors not only on inflammatory parameters, but also on muscle mass and function both in clinical and preclinical studies, since they certainly stand as promising drugs in the management of sarcopenia.
On the whole, to look into muscle involvement in distinct types of systemic conditions may help identifying patterns of muscle classic inflammatory response (i.e. IMM), low-grade chronic inflammation (i.e. PS or OA) or severe chronic systemic inflammation (i.e. RS).
Author Contributions
SP-B, RL and GH-B were in charge with conception and design; SP-B, IP-P, and GH-B: were involved in drafting the article; SP-B, IP-P, JR-B, OS-P, RL, and GH-B: revise it critically for important intellectual content and approved the final version to be published.
Funding
This work was partially supported by grants from the Instituto de Salud Carlos III (PI13/00570; PI15/00340 and PI16/00065), co-funded by Fondo Europeo de Desarrollo Regional (FEDER).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
PDK-1, 3′-Phoshphoinositide-Dependent Protein Kinase-1; 4E-BP1, 4E-Binding Protein 1; PRR, Activate Pattern Recognition Receptors; ActRII, Activin Type II Receptors; ADM, Amyopathic Dermatomyositis; ATG, Autophagy-Related Genes; CRP, C-Reactive Protein; CA, Chronic Arthritis; ER, Endoplasmic Reticulum; ERK, Extracellular Signal–Regulated Kinase; FoxO, Forkhead O; GDF-8, Growth Differentiation Factor-8; GH, Growth Hormone; IMM, Idiopathic Inflammatory Myopathies; IGF, Insulin Growth Factor; IRS-1, Insulin Receptor Substrate-1; IFN, Interferon; IL, Interleukin; JAK, Janus Kinase; MHC, Major Histocompatibility Complex; mTOR, Mammalian Target of Rapamycin; MAPK, Mitogen Activated Protein Kinases; MAFbx, Muscle Atrophy F-Box Protein; MuRF-1, Muscle Ring Finger-1; MyoD, Myogenic Differentiation; Myf5, Myogenic Factor 5; MSTN, Myostatin; NFκB, Nuclear Factor KB; OA, Osteoarthritis; 70S6K, P70-S6 Kinase; Pax7, Paired Box Transcription Factor 7; PI3K, Phosphatidyilinositol 3 Kinase; PS, Primary Sarcopenia; RA, Rheumatoid Arthritis; RS, Rheumatoid Sarcopenia; RS6K, Ribosomal Protein S6 Kinase Beta-1; SC, Satellite Cells; SS, Secondary Sarcopenia; STAT, Signal Transducer and Activation Of Transcription; SKM, Skeletal Muscle; TLR, Toll-Like Receptors; TGF, Transforming Growth Factor; TNF, Tumor Necrosis Factor; UPS, Ubiquitin-Proteasome System
References
Ali, S. P., and Garcia, J. M. P. (2014). Sarcopenia, cachexia and aging: diagnosis, mechanisms and therapeutic options - a mini-review. Gerontology 60, 294–305. doi: 10.1159/000356760
Allen, D. L., Roy, R. R., and Edgerton, V. R. (1999). Myonuclear domains in muscle adaptation and disease. Muscle Nerve 22, 1350–1360. doi: 10.1002/(SICI)1097-4598(199910)22:10<1350::AID-MUS3>3.0.CO;2-8
Baker, J. F., Von Feldt, J. M., Mostoufi-Moab, S., Kim, W., Taratuta, E., and Leonard, M. B. (2015). Insulin-like growth factor 1 and adiponectin and associations with muscle deficits, disease characteristics, and treatments in rheumatoid arthritis. J. Rheumatol. 42, 2038–2045. doi: 10.3899/jrheum.150280
Barbieri, M., Ferrucci, L., Ragno, E., Corsi, A., Bandinelli, S., Bonafè, M., et al. (2003). Chronic inflammation and the effect of IGF-I on muscle strength and power in older persons. Am. J. Physiol. 284, E481–E487. doi: 10.1152/ajpendo.00319.2002
Basualto-Alarcón, C., Jorquera, G., Altamirano, F., Jaimovich, E., and Estrada, M. (2013). Testosterone signals through mTOR and androgen receptor to induce muscle hypertrophy. Med. Sci. Sport. Exerc. 45, 1712–1720. doi: 10.1249/MSS.0b013e31828cf5f3
Beyer, I., Mets, T., and Bautmans, I. (2012). Chronic low-grade inflammation and age-related sarcopenia. Curr. Opin. Clin. Nutr. Metab. Care 15, 12–22. doi: 10.1097/MCO.0b013e32834dd297
Bjørbaek, C., Elmquist, J. K., Frantz, J. D., Shoelson, S. E., and Flier, J. S. (1998). Identification of SOCS-3 as a potential mediator of central leptin resistance. Mol. Cell 1, 619–625. doi: 10.1016/S1097-2765(00)80062-3
Bodell, P. W., Kodesh, E., Haddad, F., Zaldivar, F. P., Cooper, D. M., and Adams, G. R. (2009). Skeletal muscle growth in young rats is inhibited by chronic exposure to IL-6 but preserved by concurrent voluntary endurance exercise. J. Appl. Physiol. 106, 443–453. doi: 10.1152/japplphysiol.90831.2008
Bonetto, A., Aydogdu, T., Jin, X., Zhang, Z., Zhan, R., Puzis, L., et al. (2012). JAK/STAT3 pathway inhibition blocks skeletal muscle wasting downstream of IL-6 and in experimental cancer cachexia. Am. J. Physiol. Endocrinol. Metab. 303, E410–E421. doi: 10.1152/ajpendo.00039.2012
Bonetto, A., Aydogdu, T., Kunzevitzky, N., Guttridge, D. C., Khuri, S., Koniaris, L. G., et al. (2011). STAT3 activation in skeletal muscle links muscle wasting and the acute phase response in cancer cachexia. PLoS ONE 6:e22538. doi: 10.1371/journal.pone.0022538
Brack, A. S., Bildsoe, H., and Hughes, S. M. (2005). Evidence that satellite cell decrement contributes to preferential decline in nuclear number from large fibres during murine age-related muscle atrophy. J. Cell Sci. 118, 4813–4821. doi: 10.1242/jcs.02602
Brooks, N. E., and Myburgh, K. H. (2014). Skeletal muscle wasting with disuse atrophy is multi-dimensional: the response and interaction of myonuclei, satellite cells and signaling pathways. Front. Physiol. 5:99. doi: 10.3389/fphys.2014.00099
Brown, J. P., Prince, R. L., Deal, C., Recker, R. R., Kiel, D. P., de Gregorio, L. H., et al. (2009). Comparison of the effect of denosumab and alendronate on BMD and biochemical markers of bone turnover in postmenopausal women with low bone mass: a randomized, blinded, phase 3 trial. J. Bone Miner. Res. 24, 153–161. doi: 10.1359/jbmr.0809010
Busquets, S., Deans, C., Figueras, M., Moore-Carrasco, R., López-Soriano, F. J., Fearon, K. C. H., et al. (2007). Apoptosis is present in skeletal muscle of cachectic gastro-intestinal cancer patients. Clin. Nutr. 26, 614–618. doi: 10.1016/j.clnu.2007.06.005
Butz, H., Rácz, K., Hunyady, L., and Patócs, A. (2012). Crosstalk between TGF-beta signaling and the microRNA machinery. Trends Pharmacol. Sci. 33, 382–393. doi: 10.1016/j.tips.2012.04.003
Cai, D., Frantz, J. D., Tawa, N. E., Melendez, P. A., Oh, B. C., Lidov, H. G., et al. (2004). IKKβ/NF-κB activation causes severe muscle wasting in mice. Cell 119, 285–298. doi: 10.1016/j.cell.2004.09.027
Carey, A. L., Petersen, E. W., Bruce, C. R., Southgate, R. J., Pilegaard, H., Hawley, J. A., et al. (2006). Discordant gene expression in skeletal muscle and adipose tissue of patients with type 2 diabetes: effect of interleukin-6 infusion. Diabetologia 49, 1000–1007. doi: 10.1007/s00125-006-0178-7
Castañeda, S., Roman-Blas, J. A., Largo, R., and Herrero-Beaumont, G. (2014). Osteoarthritis: a progressive disease with changing phenotypes. Rheumatology 53, 1–3. doi: 10.1093/rheumatology/ket247
Castillero, E., Martín, A. I., López-Menduiña, M., Granado, M., Villanúa, M. Á., and López-Calderón, A. (2009a). IGF-I system, atrogenes and myogenic regulatory factors in arthritis induced muscle wasting. Mol. Cell. Endocrinol. 309, 8–16. doi: 10.1016/j.mce.2009.05.017
Castillero, E., Martín, A. I., López-Menduiña, M., Villanúa, M. A., and López-Calderón, A. (2009b). Eicosapentaenoic acid attenuates arthritis-induced muscle wasting acting on atrogin-1 and on myogenic regulatory factors. Am. J. Physiol. Regul. Integr. Comp. Physiol. 297, R1322–R1331. doi: 10.1152/ajpregu.00388.2009
Chargé, S. B. P., and Rudnicki, M. A. (2004). Cellular and molecular regulation of muscle regeneration. Physiol. Rev. 84, 209–238. doi: 10.1152/physrev.00019.2003
Cleary, L. C., Crofford, L. J., Long, D., Charnigo, R., Clasey, J., Beaman, F., et al. (2015). Does computed tomography-based muscle density predict muscle function and health-related quality of life in patients with idiopathic inflammatory myopathies? Arthritis Care Res. 67, 1031–1040. doi: 10.1002/acr.22557
Clegg, A., Young, J., Iliffe, S., Rikkert, M. O., and Rockwood, K. (2013). Frailty in elderly people. Lancet 381, 752–762. doi: 10.1016/S0140-6736(12)62167-9
Coolican, S. A., Samuel, D. S., Ewton, D. Z., McWade, F. J., and Florini, J. R. (1997). The mitognic and myogenic actions of insulin-like growth factors utilize distinct signaling pathways. J. Biol. Chem. 272, 6653–6662. doi: 10.1074/jbc.272.10.6653
Creus, K. K., De Paepe, B., Werbrouck, B. F., Vervaet, V., Weis, J., and De Bleecker, J. L. (2009). Distribution of the NF-kB complex in the inflammatory exudates characterizing the idiopathic inflammatory myopathies. Ann. N. Y. Acad. Sci. 1173, 370–377. doi: 10.1111/j.1749-6632.2009.04874.x
Cruz-Jentoft, A. J., Baeyens, J. P., Bauer, J. M., Boirie, Y., Cederholm, T., Landi, F., et al. (2010). Sarcopenia: european consensus on definition and diagnosis: report of the european working group on sarcopenia in older people. Age Ageing 39, 412–423. doi: 10.1093/ageing/afq034
Cuervo, A. M. (2008). Autophagy and aging: keeping that old broom working. Trends Genet. 24, 604–612. doi: 10.1016/j.tig.2008.10.002
Dalakas, M. C. (2010). Inflammatory muscle diseases: a critical review on pathogenesis and therapies. Curr. Opin. Pharmacol. 10, 346–352. doi: 10.1016/j.coph.2010.03.001
Dalle, S., Rossmeislova, L., and Koppo, K. (2017). The role of inflammation in age-related sarcopenia. Front. Physiol. 8:1045. doi: 10.3389/fphys.2017.01045
Day, J., Otto, S., Proudman, S., Hayball, J. D., and Limaye, V. (2017). Dysregulated innate immune function in the aetiopathogenesis of idiopathic inflammatory myopathies. Autoimmun. Rev. 16, 87–95. doi: 10.1016/j.autrev.2016.09.019
de Alvaro, C., Teruel, T., Hernandez, R., and Lorenzo, M. (2004). Tumor necrosis factor alpha produces insulin resistance in skeletal muscle by activation of inhibitor kappaB kinase in a p38 MAPK-dependent manner. J. Biol. Chem. 279, 17070–17078. doi: 10.1074/jbc.M312021200
De Ceuninck, F., Fradin, A., and Pastoureau, P. (2014). Bearing arms against osteoarthritis and sarcopenia: when cartilage and skeletal muscle find common interest in talking together. Drug Discov. Tod. 19, 305–311. doi: 10.1016/j.drudis.2013.08.004
de Oliveira Nunes Teixeira, V., Filippin, L. I., Viacava, P. R., Oliveira, P. G., and Xavier, R. M. (2013). Muscle wasting in collagen-induced arthritis and disuse atrophy. Exp. Biol. Med. 238, 1421–1430. doi: 10.1177/1535370213505961
Deldicque, L. (2013). Endoplasmic reticulum stress in human skeletal muscle: any contribution to sarcopenia? Front. Physiol. 4:236. doi: 10.3389/fphys.2013.00236
Demonbreun, A. R., and McNally, E. M. (2017). Muscle cell communication in development and repair. Curr. Opin. Pharmacol. 34, 7–14. doi: 10.1016/j.coph.2017.03.008
Diao, Y., Wang, X., and Wu, Z. (2009). SOCS1, SOCS3, and PIAS1 promote myogenic differentiation by inhibiting the leukemia inhibitory factor-induced JAK1/STAT1/STAT3 pathway. Mol. Cell. Biol. 29, 5084–5093. doi: 10.1128/MCB.00267-09
Drummond, M. J., Miyazaki, M., Dreyer, H. C., Pennings, B., Dhanani, S., Volpi, E., et al. (2009). Expression of growth-related genes in young and older human skeletal muscle following an acute stimulation of protein synthesis. J. Appl. Physiol. 106, 1403–1411. doi: 10.1152/japplphysiol.90842.2008
Dupont-Versteegden, E. E. (2006). Apoptosis in skeletal muscle and its relevance to atrophy. World J. Gastroenterol. 12, 7463–7466. doi: 10.3748/wjg.v12.i46.7463
Elkan, A. C., Engvall, I. L., Cederholm, T., and Hafström, I. (2009). Rheumatoid cachexia, central obesity and malnutrition in patients with low-active rheumatoid arthritis: feasibility of anthropometry, Mini Nutritional Assessment and body composition techniques. Eur. J. Nutr. 48, 315–322. doi: 10.1007/s00394-009-0017-y
Engert, J., Berglund, E., and Rosenthal, N. (1996). Proliferation precedes differentiation in IGF-I stimulated myogenesis. J. Cell Biol. 135, 431–440. doi: 10.1083/jcb.135.2.431
Felson, D. T., Naimark, A., Anderson, J., Kazis, L., Castelli, W., and Meenan, R. F. (1987). The prevalence of knee osteoarthritis in the elderly. The Framingham Osteoarthritis study. Arthritis Rheum. 30, 914–918. doi: 10.1002/art.1780300811
Fielding, R. A., Vellas, B., Evans, W. J., Bhasin, S., Morley, J. E., Newman, A. B., et al. (2011). Sarcopenia: an undiagnosed condition in older adults. current consensus definition: prevalence, etiology, and consequences. international working group on sarcopenia. J. Am. Med. Dir. Assoc. 12, 249–256. doi: 10.1016/j.jamda.2011.01.003
Florini, J. R., Samuel, D. S., Ewton, D. Z., Kirk, C., and Sklar, R. M. (1996). Stimulation of myogenic differentiation by a neuregulin, glial growth factor 2. Are neuregulins the long-sought muscle trophic factors secreted by nerves? J. Biol. Chem. 271, 12699–12702. doi: 10.1074/jbc.271.22.12699
Fry, C. S., Kirby, T. J., Kosmac, K., McCarthy, J. J., and Peterson, C. A. (2017). Myogenic progenitor cells control extracellular matrix production by fibroblasts during skeletal muscle hypertrophy. Cell Stem Cell 20, 56–69. doi: 10.1016/j.stem.2016.09.010
Fry, C. S., Lee, J. D., Mula, J., Kirby, T. J., Jackson, J. R., Liu, F., et al. (2015). Inducible depletion of satellite cells in adult, sedentary mice impairs muscle regenerative capacity without affecting sarcopenia. Nat. Med. 21, 76–80. doi: 10.1038/nm.3710
Fukuda, W., Omoto, A., Ohta, T., Majima, S., Kimura, T., Tanaka, T., et al. (2013). Low body mass index is associated with impaired quality of life in patients with rheumatoid arthritis. Int. J. Rheum. Dis. 16, 297–302. doi: 10.1111/1756-185X.12079
Giles, J. T., Bartlett, S. J., Andersen, R. E., Fontaine, K. R., and Bathon, J. M. (2008). Association of body composition with disability in rheumatoid arthritis: impact of appendicular fat and lean tissue mass. Arthritis Rheum. 59, 1407–1415. doi: 10.1002/art.24109
Giudice, J., and Taylor, J. M. (2017). Muscle as a paracrine and endocrine organ. Curr. Opin. Pharmacol. 34, 49–55. doi: 10.1016/j.coph.2017.05.005
Glass, D. J. (2003). Signalling pathways that mediate skeletal muscle hypertrophy and atrophy. Nat. Cell Biol. 5, 87–90. doi: 10.1038/ncb0203-87
Glass, D. J. (2010). Signaling pathways perturbing muscle mass. Curr. Opin. Clin. Nutr. Metab. Care 13, 225–229. doi: 10.1097/MCO.0b013e32833862df
Gómez, R., Villalvilla, A., Largo, R., Gualillo, O., and Herrero-Beaumont, G. (2015). TLR4 signalling in osteoarthritis–finding targets for candidate DMOADs. Nat. Rev. Rheumatol. 11, 159–170. doi: 10.1038/nrrheum.2014.209
Guadalupe-Grau, A., Carnicero, J. A., Gómez-Cabello, A., Gutiérrez Avila, G., Humanes, S., Alegre, L. M., et al. (2015). Association of regional muscle strength with mortality and hospitalisation in older people. Age Ageing 44, 790–795. doi: 10.1093/ageing/afv080
Guttridge, D. C., Mayo, M. W., Madrid, L. V., Wang, C. Y., and Baldwin, A. (2000). NF-kappaB-induced loss of MyoD messenger RNA: possible role in muscle decay and cachexia. Science 289, 2363–2366. doi: 10.1126/science.289.5488.2363
Hanisch, F., and Zierz, S. (2015). C-reactive protein in idiopathic inflammatory myopathies. Myopain 23, 45–51. doi: 10.3109/10582452.2013.852151
Hartwig, S., Raschke, S., Knebel, B., Scheler, M., Irmler, M., Passlack, W., et al. (2014). Secretome profiling of primary human skeletal muscle cells. Biochim. Biophys. Acta 1844, 1011–1017. doi: 10.1016/j.bbapap.2013.08.004
Herrero-Beaumont, G., Roman-Blas, J. A., Castañeda, S., and Jimenez, S. A. (2009). Primary osteoarthritis no longer primary: three subsets with distinct etiological, clinical, and therapeutic characteristics. Semin. Arthritis Rheum. 39, 71–80. doi: 10.1016/j.semarthrit.2009.03.006
Hitachi, K., and Tsuchida, K. (2014). Role of microRNAs in skeletal muscle hypertrophy. Front. Physiol. 4:408. doi: 10.3389/fphys.2013.00408
Hittel, D. S., Berggren, J. R., Shearer, J., Boyle, K., and Houmard, J. A. (2009). Increased secretion and expression of myostatin in skeletal muscle from extremely obese women. Diabetes 58, 30–38. doi: 10.2337/db08-0943
Hoene, M., Runge, H., Haring, H. U., Schleicher, E. D., and Weigert, C. (2013). Interleukin-6 promotes myogenic differentiation of mouse skeletal muscle cells: role of the STAT3 pathway. Am. J. Physiol. Cell Physiol. 304, C128–C136. doi: 10.1152/ajpcell.00025.2012
Hotamisligil, G. S. (2006). Inflammation and metabolic disorders. Nature 444, 860–867. doi: 10.1038/nature05485
Huffman, K. M., Jessee, R., Andonian, B., Davis, B. N., Narowski, R., Huebner, J. L., et al. (2017). Molecular alterations in skeletal muscle in rheumatoid arthritis are related to disease activity, physical inactivity, and disability. Arthritis Res. Ther. 19:12. doi: 10.1186/s13075-016-1215-7
Hughes, D. C., Stewart, C. E., Sculthorpe, N., Dugdale, H. F., Yousefian, F., Lewis, M. P., et al. (2016). Testosterone enables growth and hypertrophy in fusion impaired myoblasts that display myotube atrophy: deciphering the role of androgen and IGF-I receptors. Biogerontology 17, 619–639. doi: 10.1007/s10522-015-9621-9
Illa, I., Gallardo, E., Gimeno, R., and Serrano, C. (1997). Signal transducer and activator of transcription 1 in human muscle implications in inflammatory myopathies. Am. J. Pathol. 151, 81–88.
Janssen, I., Heymsfield, S. B., Wang, Z. M., and Ross, R. (2000). Skeletal muscle mass and distribution in 468 men and women aged 18-88 yr. J. Appl. Physiol. 89, 81–88. doi: 10.1152/jappl.2000.89.1.81
Jostock, T., Müllberg, J., Ozbek, S., Atreya, R., Blinn, G., Voltz, N., et al. (2001). Soluble gp130 is the natural inhibitor of soluble interleukin-6 receptor transsignaling responses. Eur. J. Biochem. 167, 160–167. doi: 10.1046/j.1432-1327.2001.01867.x
Jung, H. J., and Suh, Y. (2015). Regulation of IGF−1 signaling by microRNAs. Front. Genet. 5, 1–13. doi: 10.3389/fgene.2014.00472
Kalinkovich, A., and Livshits, G. (2017). Sarcopenic obesity or obese sarcopenia: a cross talk between age-associated adipose tissue and skeletal muscle inflammation as a main mechanism of the pathogenesis. Ageing Res. Rev. 35, 200–221. doi: 10.1016/j.arr.2016.09.008
Kami, K., and Senba, E. (2002). In vivo activation of STAT3 signaling in satellite cells and myofibers in regenerating rat skeletal muscles. J. Histochem. Cytochem. 50, 1579–1589. doi: 10.1177/002215540205001202
Kataoka, Y., Matsumura, I., Ezoe, S., Nakata, S., Takigawa, E., Sato, Y., et al. (2003). Reciprocal inhibition between MyoD and STAT3 in the regulation of growth and differentiation of myoblasts. J. Biol. Chem. 278, 44178–44187. doi: 10.1074/jbc.M304884200
Koskinen, A., Vuolteenaho, K., Moilanen, T., and Moilanen, E. (2014). Resistin as a factor in osteoarthritis: synovial fluid resistin concentrations correlate positively with interleukin 6 and matrix metalloproteinases MMP-1 and MMP-3. Scand. J. Rheumatol. 43, 249–253. doi: 10.3109/03009742.2013.853096
Kotler, D. P. (2000). Cachexia. Ann. Intern. Med. 133, 622–634. doi: 10.7326/0003-4819-133-8-200010170-00015
Krabbe, K. S., Pedersen, M., and Bruunsgaard, H. (2004). Inflammatory mediators in the elderly. Exp. Gerontol. 39, 687–699. doi: 10.1016/j.exger.2004.01.009
Lee, S., Kim, T. N., and Kim, S. H. (2012). Sarcopenic obesity is more closely associated with knee osteoarthritis than is nonsarcopenic obesity: a cross-sectional study. Arthritis Rheum. 64, 3947–3954. doi: 10.1002/art.37696
Lemmey, A. B., Marcora, S. M., Chester, K., Wilson, S., Casanova, F., and Maddison, P. J. (2009). Effects of high-intensity resistance training in patients with rheumatoid arthritis: a randomized controlled trial. Arthritis Rheum. 61, 1726–1734. doi: 10.1002/art.24891
Levine, B., and Kroemer, G. (2008). Autophagy in the pathogenesis of disease. Cell 132, 27–42. doi: 10.1016/j.cell.2007.12.018
Levinger, I., Levinger, P., Trenerry, M. K., Feller, J. A., Bartlett, J. R., Bergman, N., et al. (2011a). Increased inflammatory cytokine expression in the vastus lateralis of patients with knee osteoarthritis. Arthritis Rheum. 63, 1343–1348. doi: 10.1002/art.30287
Levinger, P., Caldow, M. K., Bartlett, J. R., Peake, J. M., Smith, C., Cameron-Smith, D., et al. (2016). The level of FoxO1 and IL-15 in skeletal muscle, serum and synovial fluid in people with knee osteoarthritis: a case control study. Osteoporos. Int. 27, 2137–2143. doi: 10.1007/s00198-015-3473-7
Levinger, P., Caldow, M. K., Feller, J. A., Bartlett, J. R., Bergman, N. R., McKenna, M. J., et al. (2011b). Association between skeletal muscle inflammatory markers and walking pattern in people with knee osteoarthritis. Arthritis Care Res. 63, 1715–1721. doi: 10.1002/acr.20625
Li, H. X., Chen, K. L., Wang, H. Y., Tang, C. B., Xu, X. L., and Zhou, G. H. (2015). Chemerin inhibition of myogenesis and induction of adipogenesis in C2C12 myoblasts. Mol. Cell. Endocrinol. 414, 216–223. doi: 10.1016/j.mce.2015.07.006
Li, W., Moylan, J. S., Chambers, M. A., Smith, J., and Reid, M. B. (2009). Interleukin-1 stimulates catabolism in C2C12 myotubes. Am. J. Physiol. Cell Physiol. 297, C706–C714. doi: 10.1152/ajpcell.00626.2008
Little, R. D., Prieto-Potin, I., Pérez-Baos, S., Villalvilla, A., Gratal, P., Cicuttini, F., et al. (2017). Compensatory anabolic signaling in the sarcopenia of experimental chronic arthritis. Sci. Rep. 7:6311. doi: 10.1038/s41598-017-06581-6
Livshits, G., Zhai, G., Hart, D. J., Kato, B. S., Wang, H., Williams, F. M. K., et al. (2009). Interleukin-6 is a significant predictor of radiographic knee osteoarthritis: the Chingford Study. Arthritis Rheum. 60, 2037–2045. doi: 10.1002/art.24598
Loell, I., and Lundberg, I. E. (2011). Can muscle regeneration fail in chronic inflammation: a weakness in inflammatory myopathies? J. Intern. Med. 269, 243–257. doi: 10.1111/j.1365-2796.2010.02334.x
Loell, I., Raouf, J., Chen, Y., Shi, R., Nennesmo, I., Alexanderson, H., et al. (2016). Effects on muscle tissue remodeling and lipid metabolism in muscle tissue from adult patients with polymyositis or dermatomyositis treated with immunosuppressive agents. Arthritis Res. Ther. 18:136. doi: 10.1186/s13075-016-1033-y
Ma, J., Niu, D.-S., Wan, N.-J., Qin, Y., and Guo, C.-J. (2015). Elevated chemerin levels in synovial fluid and synovial membrane from patients with knee osteoarthritis. Int. J. Clin. Exp. Pathol. 8, 13393–13398.
Maggio, M., Guralnik, J. M., Longo, D. L., and Ferrucci, L. (2006). Interleukin-6 in aging and chronic disease: a magnificent pathway. J. Gerontol. A. Biol. Sci. Med. Sci. 61, 575–584. doi: 10.1093/gerona/61.6.575
Malemud, C. J. (2017). Negative regulators of JAK/STAT signaling in rheumatoid arthritis and osteoarthritis. Int. J. Mol. Sci. 18:484. doi: 10.3390/ijms18030484
Malik, A., Hayat, G., Kalia, J. S., and Guzman, M. A. (2016). Idiopathic inflammatory myopathies: clinical approach and management. Front. Neurol. 7:64. doi: 10.3389/fneur.2016.00064
Mammucari, C., Milan, G., Romanello, V., Masiero, E., Rudolf, R., Del Piccolo, P., et al. (2007). FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab. 6, 458–471. doi: 10.1016/j.cmet.2007.11.001
Marzetti, E., Hwang, J. C. Y., Lees, H. A., Wohlgemuth, S. E., Dupont-Versteegden, E. E., Carter, C. S., et al. (2010). Mitochondrial death effectors: relevance to sarcopenia and disuse muscle atrophy. Biochim. Biophys. Acta 1800, 235–244. doi: 10.1016/j.bbagen.2009.05.007
Marzetti, E., Landi, F., Marini, F., Cesari, M., Buford, T. W., Manini, T. M., et al. (2014). Patterns of circulating inflammatory biomarkers in older persons with varying levels of physical performance: a partial least squares-discriminant analysis approach. Front. Med. 1:27. doi: 10.3389/fmed.2014.00027
Marzetti, E., Lees, H. A., Wohlgemuth, S. E., and Leeuwenburgh, C. (2009). Sarcopenia of aging: underlying cellular mechanisms and protection by calorie restriction. BioFactors 35, 28–35. doi: 10.1002/biof.5
Masuko, K. (2014). Rheumatoid cachexia revisited: a metabolic co-morbidity in rheumatoid arthritis. Front. Nutr. 1:20. doi: 10.3389/fnut.2014.00020
McFarlane, C., Hui, G. Z., Amanda, W. Z. W., Lau, H. Y., Lokireddy, S., Xiaojia, G., et al. (2011). Human myostatin negatively regulates human myoblast growth and differentiation. Am. J. Physiol. Cell Physiol. 301, C195–C203. doi: 10.1152/ajpcell.00012.2011
McFarlane, C., Plummer, E., Thomas, M., Hennebry, A., Ashby, M., Ling, N., et al. (2006). Myostatin induces cachexia by activating the ubiquitin proteolytic system through an NF-κB-independent, FoxO1-dependent mechanism. J. Cell. Physiol. 209, 501–514. doi: 10.1002/jcp.20757
Metsios, G. S., Stavropoulos-Kalinoglou, A., Douglas, K. M. J., Koutedakis, Y., Nevill, A. M., Panoulas, V. F., et al. (2007). Blockade of tumour necrosis factor-alpha in rheumatoid arthritis: effects on components of rheumatoid cachexia. Rheumatology 46, 1824–1827. doi: 10.1093/rheumatology/kem291
Milan, G., Romanello, V., Pescatore, F., Armani, A., Paik, J.-H., Frasson, L., et al. (2015). Regulation of autophagy and the ubiquitin-proteasome system by the FoxO transcriptional network during muscle atrophy. Nat. Commun. 6:6670. doi: 10.1038/ncomms7670
Mizushima, N., and Komatsu, M. (2011). Autophagy: renovation of cells and tissues. Cell 147, 728–741. doi: 10.1016/j.cell.2011.10.026
Morgan, J. E., and Partridge, T. A. (2003). Muscle satellite cells. Int. J. Biochem. Cell Biol. 35, 1151–1156. doi: 10.1016/S1357-2725(03)00042-6
Morissette, M. R., Cook, S. A., Buranasombati, C., Rosenberg, M. A., and Rosenzweig, A. (2009). Myostatin inhibits IGF-I-induced myotube hypertrophy through Akt. AJP Cell Physiol. 297, 1124–1132. doi: 10.1152/ajpcell.00043.2009
Morley, J. E., Thomas, D. R., and Wilson, M. M. (2006). Cachexia: pathophysiology and clinical relevance. Am. J. Clin. Nutr. 83, 735–743. doi: 10.1093/ajcn/83.4.735
Muñoz-Cánoves, P., Scheele, C., Pedersen, B. K., and Serrano, A. L. (2013). Interleukin-6 myokine signaling in skeletal muscle: a double-edged sword? FEBS J. 280, 4131–4148. doi: 10.1111/febs.12338
Muscaritoli, M., Anker, S. D., Argilés, J., Aversa, Z., Bauer, J. M., Biolo, G., et al. (2010). Consensus definition of sarcopenia, cachexia and pre-cachexia: joint document elaborated by special interest groups (SIG) “cachexia-anorexia in chronic wasting diseases” and “nutrition in geriatrics.” Clin. Nutr. 29, 154–159. doi: 10.1016/j.clnu.2009.12.004
Nagaraju, K., Casciola-Rosen, L., Lundberg, I., Rawat, R., Cutting, S., Thapliyal, R., et al. (2005). Activation of the endoplasmic reticulum stress response in autoimmune myositis: potential role in muscle fiber damage and dysfunction. Arthritis Rheum. 52, 1824–1835. doi: 10.1002/art.21103
Nocon, M., Hiemann, T., Müller-Riemenschneider, F., Thalau, F., Roll, S., and Willich, S. N. (2008). Association of physical activity with all-cause and cardiovascular mortality: a systematic review and meta-analysis. Eur. J. Cardiovasc. Prev. Rehabil. 15, 239–246. doi: 10.1097/HJR.0b013e3282f55e09
Nogalska, A., Wojcik, S., Engel, W. K., McFerrin, J., and Askanas, V. (2007). Endoplasmic reticulum stress induces myostatin precursor protein and NF-κB in cultured human muscle fibers: relevance to inclusion body myositis. Exp. Neurol. 204, 610–618. doi: 10.1016/j.expneurol.2006.12.014
Pedersen, B. K., and Febbraio, M. A. (2008). Muscle as an endocrine organ: focus on muscle-derived interleukin-6. Physiol. Rev. 88, 1379–1406. doi: 10.1152/physrev.90100.2007
Pedersen, B. K., and Febbraio, M. A. (2012). Muscles, exercise and obesity: skeletal muscle as a secretory organ. Nat. Rev. Endocrinol. 8, 457–465. doi: 10.1038/nrendo.2012.49
Perl, A. (2015). Activation of mTOR (mechanistic target of rapamycin) in rheumatic diseases. Nat. Rev. Rheumatol. 12, 169–182. doi: 10.1038/nrrheum.2015.172
Perrini, S., Laviola, L., Carreira, M. C., Cignarelli, A., Natalicchio, A., and Giorgino, F. (2010). The GH/IGF1 axis and signaling pathways in the muscle and bone: mechanisms underlying age-related skeletal muscle wasting and osteoporosis. J. Endocrinol. 205, 201–210. doi: 10.1677/JOE-09-0431
Phillips, S. M., Glover, E. I., and Rennie, M. J. (2009). Alterations of protein turnover underlying disuse atrophy in human skeletal muscle. J. Appl. Physiol. 107, 645–654. doi: 10.1152/japplphysiol.00452.2009
Poonpet, T., and Honsawek, S. (2014). Adipokines: biomarkers for osteoarthritis? World, J. Orthop. 5, 319–327. doi: 10.5312/wjo.v5.i3.319
Portal-Núñez, S., Esbrit, P., Alcaraz, M. J., and Largo, R. (2015). Oxidative stress, autophagy, epigenetic changes and regulation by miRNAs as potential therapeutic targets in osteoarthritis. Biochem. Pharmacol. 108, 1–10. doi: 10.1016/j.bcp.2015.12.012
Price, F. D., von Maltzahn, J., Bentzinger, C. F., Dumont, N. A., Yin, H., Chang, N. C., et al. (2015). Inhibition of JAK/STAT signaling stimulates adult satellite cell function. Nat. Med. 20, 1174–1181. doi: 10.1038/nm.3655
Rayavarapu, S., Coley, W., Kinder, T. B., and Nagaraju, K. (2013). Idiopathic inflammatory myopathies: pathogenic mechanisms of muscle weakness. Skelet. Muscle 3:13. doi: 10.1186/2044-5040-3-13
Reed, S. A., Sandesara, P. B., Senf, S. M., and Judge, A. R. (2012). Inhibition of FoxO transcriptional activity prevents muscle fiber atrophy during cachexia and induces hypertrophy. Faseb J. 26, 987–1000. doi: 10.1096/fj.11-189977
Retamales, A., Zuloaga, R., Valenzuela, C. A., Gallardo-Escarate, C., Molina, A., and Valdés, J. A. (2015). Insulin-like growth factor-1 suppresses the Myostatin signaling pathway during myogenic differentiation. Biochem. Biophys. Res. Commun. 464, 596–602. doi: 10.1016/j.bbrc.2015.07.018
Robinson, W. H., Lepus, C. M., Wang, Q., Raghu, H., Mao, R., Lindstrom, T. M., et al. (2016). Low-grade inflammation as a key mediator of the pathogenesis of osteoarthritis. Nat. Rev. Rheumatol. 12, 580–592. doi: 10.1038/nrrheum.2016.136
Roh, E., Song, D. K., and Kim, M.-S. (2016). Emerging role of the brain in the homeostatic regulation of energy and glucose metabolism. Exp. Mol. Med. 48:e216. doi: 10.1038/emm.2016.4
Roman-Blas, J. A., Bizzi, E., Largo, R., Migliore, A., and Herrero-Beaumont, G. (2016). An update on the up and coming therapies to treat osteoarthritis, a multifaceted disease. Expert Opin. Pharmacother. 17, 1745–1756. doi: 10.1080/14656566.2016.1201070
Romero, F. I., Martínez-Calatrava, M. J., Sánchez-Pernaute, O., Gualillo, O., Largo, R., and Herrero-Beaumont, G. (2010). Pharmacological modulation by celecoxib of cachexia associated with experimental arthritis and atherosclerosis in rabbits. Br. J. Pharmacol. 161, 1012–1022. doi: 10.1111/j.1476-5381.2010.00957.x
Rosenthal, S. M., and Cheng, Z. Q. (1995). Opposing early and late effects of insulin-like growth factor I on differentiation and the cell cycle regulatory retinoblastoma protein in skeletal myoblasts. Proc. Natl. Acad. Sci. U.S.A. 92, 10307–10311. doi: 10.1073/pnas.92.22.10307
Roubenoff, R. (2014). The “cytokine for gerontologists” has some company. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 69, 163–164. doi: 10.1093/gerona/glt184
Roubenoff, R., Roubenoff, R. A., Ward, L. M., Holland, S. M., and Hellmann, D. B. (1992). Rheumatoid cachexia: depletion of lean body mass in rheumatoid arthritis. Possible association with tumor necrosis factor. J. Rheumatol. 19, 1505–1510.
Sandri, M., Barberi, L., Bijlsma, A. Y., Blaauw, B., Dyar, K. A., Milan, G., et al. (2013). Signalling pathways regulating muscle mass in ageing skeletal muscle: the role of the IGF1-Akt-mTOR-FoxO pathway. Biogerontology 14, 303–323. doi: 10.1007/s10522-013-9432-9
Sandri, M., Sandri, C., Gilbert, A., Skurk, C., Calabria, E., Picard, A., et al. (2004). Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell 117, 399–412. doi: 10.1016/S0092-8674(04)00400-3
Santos, M. L., Gomes, W. F., Pereira, D. S., Oliveira, D. M., Dias, J. M., Ferrioli, E., et al. (2011). Muscle strength, muscle balance, physical function and plasma interleukin-6 (IL-6) levels in elderly women with knee osteoarthritis (OA). Arch. Gerontol. Geriatr. 52, 322–326. doi: 10.1016/j.archger.2010.05.009
Sattler, F. R. (2013). Best practice & research clinical endocrinology & metabolism growth hormone in the aging male. Best Pract. Res. Clin. Endocrinol. Metab. 27, 541–555. doi: 10.1016/j.beem.2013.05.003
Scanzello, C. R., and Loeser, R. F. (2015). Editorial: inflammatory activity in symptomatic knee osteoarthritis: not all inflammation is local. Arthritis Rheumatol. 67, 2797–2800. doi: 10.1002/art.39304
Schiaffino, S., Dyar, K. A., Ciciliot, S., Blaauw, B., and Sandri, M. (2013). Mechanisms regulating skeletal muscle growth and atrophy. FEBS J. 280, 4294–4314. doi: 10.1111/febs.12253
Schindler, R., Mancilla, J., Endres, S., Ghorbani, R., Clark, S. C., and Dinarello, C. A. (1990). Correlations and interactions in the production of interleukin-6 (IL- 6), IL-1, and tumor necrosis factor (TNF) in human blood mononuclear cells: IL-6 suppresses IL-1 and TNF. Blood 75, 40–47.
Scotece, M., Conde, J., Gómez, R., López, V., Lago, F., Gómez-Reino, J. J., et al. (2011). Beyond fat mass: exploring the role of adipokines in rheumatic diseases. Sci. World J. 11, 1932–1947. doi: 10.1100/2011/290142
Scotece, M., Pérez, T., Conde, J., Abella, V., López, V., Pino, J., et al. (2017). Adipokines induce pro-inflammatory factors in activated Cd4+ T cells from osteoarthritis patient. J. Orthop. Res. 35, 1299–1303. doi: 10.1002/jor.23377
Soto, L., Martin, A. I., Vara, E., and López-Calderón, A. (2001). Cyclosporin A treatment is able to revert the decrease in circulating GH and IGF-I and the increase in IGFBPs induced by adjuvant arthritis. Horm. Metab. Res. 33, 590–595. doi: 10.1055/s-2001-17905
Spangenburg, E. E., and Booth, F. W. (2002). Multiple signaling pathways mediate LIF-induced skeletal muscle satellite cell proliferation. Am. J. Physiol. Cell Physiol. 283, 204–211. doi: 10.1152/ajpcell.00574.2001
Stannus, O., Jones, G., Cicuttini, F., Parameswaran, V., Quinn, S., Burgess, J., et al. (2010). Circulating levels of IL-6 and TNF-alpha are associated with knee radiographic osteoarthritis and knee cartilage loss in older adults. Osteoarthr. Cartil. 18, 1441–1447. doi: 10.1016/j.joca.2010.08.016
Straub, R. H. (2017). The brain and immune system prompt energy shortage in chronic inflammation and ageing. Nat. Rev. Rheumatol. 13, 743–751. doi: 10.1038/nrrheum.2017.172
Summers, G. D., Metsios, G. S., Stavropoulos-Kalinoglou, A., and Kitas, G. D. (2010). Rheumatoid cachexia and cardiovascular disease. Nat. Rev. Rheumatol. 6, 445–451. doi: 10.1038/nrrheum.2010.105
Sun, L., Ma, K., Wang, H., Xiao, F., Gao, Y., Zhang, W., et al. (2007). JAK1-STAT1-STAT3, a key pathway promoting proliferation and preventing premature differentiation of myoblasts. J. Cell Biol. 179, 129–138. doi: 10.1083/jcb.200703184
Taylor, W. E., Bhasin, S., Artaza, J., Byhower, F., Azam, M., Willard, D. H., et al. (2001). Myostatin inhibits cell proliferation and protein synthesis in C2C12 muscle cells. Am. J. Physiol. Endocrinol. Metab. 280, E221–E228. doi: 10.1152/ajpendo.2001.280.2.E221
Tews, D. S., and Goebel, H. H. (1996). Cytokine expression profile in idiopathic inflammatory myopathies. J. Neuropathol. Exp. Neurol. 55, 342–347. doi: 10.1097/00005072-199603000-00009
Tierney, M. T., Aydogdu, T., Sala, D., Malecova, B., Gatto, S., Puri, P. L., et al. (2014). STAT3 signaling controls satellite cell expansion and skeletal muscle repair. Nat. Med. 20, 1182–1186. doi: 10.1038/nm.3656
Tournadre, A., Pereira, B., Dutheil, F., Giraud, C., Courteix, D., Sapin, V., et al. (2017). Changes in body composition and metabolic profile during interleukin 6 inhibition in rheumatoid arthritis. J. Cachexia. Sarcopenia Muscle 8, 639–646. doi: 10.1002/jcsm.12189
Tran, P. K., Agardh, H. E., Tran-Lundmark, K., Ekstrand, J., Roy, J., Henderson, B., et al. (2007). Reduced perlecan expression and accumulation in human carotid atherosclerotic lesions. Atherosclerosis 190, 264–270. doi: 10.1016/j.atherosclerosis.2006.03.010
Tuomilehto, J., Lindström, J., Eriksson, J. G., Valle, T. T., Hämäläinen, H., Ilanne-Parikka, P., et al. (2001). Prevention of type 2 diabetes mellitus by changes in lifestyle among subjects with impaired glucose tolerance. N. Engl. J. Med. 344, 1343–1350. doi: 10.1056/NEJM200105033441801
Ueki, K., Kondo, T., and Kahn, C. R. (2004). Suppressor of cytokine signaling 1 (SOCS-1) and SOCS-3 cause insulin resistance through inhibition of tyrosine phosphorylation of insulin receptor substrate proteins by discrete mechanisms. Mol. Cell. Biol. 24, 5434–5446. doi: 10.1128/MCB.24.12.5434-5446.2004
Varadhan, R., Yao, W., Matteini, A., Beamer, B. A., Xue, Q. L., Yang, H., et al. (2014). Simple biologically informed infammatory index of two serum cytokines predicts 10 year all-cause mortality in older adults. J. Gerontol. A Biol. Sci. Med. Sci. 69, 165–173. doi: 10.1093/gerona/glt023
Vattemi, G., Mirabella, M., Guglielmi, V., Lucchini, M., Tomelleri, G., Ghirardello, A., et al. (2014). Muscle biopsy features of idiopathic inflammatory myopathies and differential diagnosis. Autoimmun. Highlights 5, 77–85. doi: 10.1007/s13317-014-0062-2
Verdijk, L. B., Koopman, R., Schaart, G., Meijer, K., Savelberg, H. H. C. M., and van Loon, L. J. C. (2007). Satellite cell content is specifically reduced in type II skeletal muscle fibers in the elderly. Am. J. Physiol. Endocrinol. Metab. 292, E151–E157. doi: 10.1152/ajpendo.00278.2006
Vermeulen, C., Grade, C., Mantovani, C. S., Alves, M., Faisal, F., Beate, Y., et al. (2017). CREB, NF-Y and MEIS1 conserved binding sites are essential to balance Myostatin promoter/enhancer activity during early myogenesis. Mol. Biol. Rep. 44, 419–427. doi: 10.1007/s11033-017-4126-z
Volpi, E., Nazemi, R., and Fujita, S. (2004). Muscle tissue changes with aging. Curr. Opin. Clin. Nutr. Metab. Care 7, 405–410. doi: 10.1097/01.mco.0000134362.76653.b2
von Haehling, S., Anker, M. S., and Anker, S. D. (2016). Prevalence and clinical impact of cachexia in chronic illness in Europe, USA, and Japan: facts and numbers update 2016. J. Cachexia. Sarcopenia Muscle 7, 507–509. doi: 10.1002/jcsm.12167
Walsmith, J., and Roubenoff, R. (2002). Cachexia in rheumatoid arthritis. Int. J. Cardiol. 85, 89–99. doi: 10.1016/S0167-5273(02)00237-1
Wang, K., Wang, C., Xiao, F., Wang, H., and Wu, Z. (2008). JAK2/STAT2/STAT3 are required for myogenic differentiation. J. Biol. Chem. 283, 34029–34036. doi: 10.1074/jbc.M803012200
Wyke, S. M., Russell, S. T., and Tisdale, M. J. (2004). Induction of proteasome expression in skeletal muscle is attenuated by inhibitors of NF-kappaB activation. Br. J. Cancer 91, 1742–1750. doi: 10.1038/sj.bjc.6602165
Xu, Q., and Wu, Z. (2000). The insulin-like growth factor-phosphatidylinositol 3-kinase-Akt signaling pathway regulates myogenin expression in normal myogenic cells but not in rhabdomyosarcoma-derived RD cells. J. Biol. Chem. 275, 36750–36757. doi: 10.1074/jbc.M005030200
Yang, Y., Xu, Y., Li, W., Wang, G., Song, Y., Yang, G., et al. (2009). STAT3 induces muscle stem cell differentiation by interaction with myoD. Cytokine 46, 137–141. doi: 10.1016/j.cyto.2008.12.015
Yoshimura, N., Muraki, S., Oka, H., Tanaka, S., Kawaguchi, H., Nakamura, K., et al. (2012). Accumulation of metabolic risk factors such as overweight, hypertension, dyslipidaemia, and impaired glucose tolerance raises the risk of occurrence and progression of knee osteoarthritis: a 3-year follow-up of the ROAD study. Osteoarthritis Cartilage 20, 1217–1226. doi: 10.1016/j.joca.2012.06.006
Zhang, G., and Li, Y.-P. (2012). p38β MAPK upregulates atrogin1/MAFbx by specific phosphorylation of C/EBPβ. Skelet. Muscle 2:20. doi: 10.1186/2044-5040-2-20
Keywords: skeletal muscle, turnover, anabolism, catabolism, sarcopenia, myokines, inflammation
Citation: Pérez-Baos S, Prieto-Potin I, Román-Blas JA, Sánchez-Pernaute O, Largo R and Herrero-Beaumont G (2018) Mediators and Patterns of Muscle Loss in Chronic Systemic Inflammation. Front. Physiol. 9:409. doi: 10.3389/fphys.2018.00409
Received: 20 December 2017; Accepted: 04 April 2018;
Published: 24 April 2018.
Edited by:
Alexandrina Ferreira Mendes, University of Coimbra, PortugalReviewed by:
Luca De Toni, Università degli Studi di Padova, ItalyAnna Picca, Università Cattolica del Sacro Cuore, Italy
Massimo Negro, Centro di Medicina dello Sport Voghera, Università degli Studi di Pavia, Italy
Copyright © 2018 Pérez-Baos, Prieto-Potin, Román-Blas, Sánchez-Pernaute, Largo and Herrero-Beaumont. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Raquel Largo, rlargo@fjd.es