 Eva E. Vink Peter J. Blankestijn*
Eva E. Vink Peter J. Blankestijn*- Department of Nephrology, University Medical Center Utrecht, Utrecht, Netherlands
Chronic elevation of the sympathetic nervous system has been identified as a major contributor to the complex pathophysiology of hypertension, states of volume overload – such as heart failure – and progressive kidney disease. It is also a strong determinant for clinical outcome. This review focuses on the central role of the kidneys in the pathogenesis of sympathetic hyperactivity. As a consequence, renal denervation may be an attractive option to treat sympathetic hyperactivity. The review will also focus on first results and the still remaining questions of this new treatment option.
Introduction
Convincing evidence reveals chronic elevation of the sympathetic nervous system (SNS) as a major contributor to the complex pathophysiology of (essential) hypertension, states of volume overload – heart failure for example – and progressive kidney disease (Klein et al., 2003a; DiBona, 2004).
Sympathetic renal denervation offers an attractive option to treat these disease states. The concept of renal denervation is not new. Complex, non-selective, procedures were effectively used as a treatment of severe hypertension before antihypertensive drugs became generally available (Peet, 1948). These treatments were effective, but associated with a high rate of peri-operative morbidity, mortality, and long-term complications (Krum et al., 2009). Nevertheless catheter based sympathetic renal denervation is an upcoming and exciting therapeutic tool. This less invasive approach disrupts the renal efferent and afferent sympathetic nerves in the adventitia of the renal arteries, using relative low-power and precisely focused radiofrequency bursts of ≈8 W.
This short review focuses on two subjects. Firstly the central role of the kidneys in the pathogenesis of sympathetic hyperactivity will be highlighted. Secondly this review focuses on the first results of the studies on renal denervation and the still remaining questions on renal denervation.
The Central Role of the Kidney in the Pathogenesis of Chronic Elevation of the SNS
Clinical Studies on Sympathetic Activation
Kim et al. (1972) provided indirect evidence for the existence of sympathetic activation in patients with end-stage kidney disease (ESKD) and hypertension. They showed that hypertension in ESKD is caused by an increased peripheral resistance. Nephrectomy resulted in a significant reduction of blood pressure (BP) and peripheral resistance. In contrast, in normotensive ESKD patients nephrectomy had no effect on BP, peripheral resistance, and cardiac output (Kim et al., 1972).
The first direct clinical evidence pointing to a role for activation of the SNS in diseased kidneys came from Converse et al. (1992). They showed that sympathetic nerve activity assessed by muscle sympathetic nerve activity (MSNA; microneurographic technique to assess true sympathetic activity) is increased in patients with chronic kidney disease (CKD). In bilateral nephrectomized patients, MSNA was comparable to controls (Converse et al., 1992).
These clinical findings show that activation of the SNS in severe- or end-stage kidney failure is caused by diseased kidneys. A recently published study by Grassi et al. (2011) provides further evidence for a crucial involvement of the SNS in patients with a moderately impaired kidney function. In these patients and in control patients with hypertension and a normal kidney function, MSNA is significantly and inversely correlated with eGFR (Grassi et al., 2011). In patients with polycystic kidney disease, MSNA is also increased regardless of kidney function (Klein et al., 2001). The presence of cysts results in areas of kidney ischemia (Klein et al., 2001). Furthermore in young subjects with mild or borderline hypertension, MSNA is already increased. The magnitude of hypertension parallels the severity of sympathetic activation as assessed by MSNA (Grassi et al., 1998). These studies indicate that sympathetic activation is most likely an early event in the pathophysiology of chronic kidney failure.
Kidney Ischemia
Kidney ischemia is an essential step in the development of activation of the SNS and renin angiotensin system (RAS; Blankestijn et al., 2000; Blankestijn, 2004; Joles and Koomans, 2004; Koomans et al., 2004; Neumann et al., 2004; Siddiqi et al., 2009). During kidney ischemia adenosine is released as a result of decreased oxygen supply (Katholi et al., 1984). Direct infusion of adenosine into the renal artery in conscious dogs produces hypertension by activating the SNS (Katholi et al., 1984). The effects of intrarenal adenosine on BP are most likely related to increased afferent renal nerve activity and can be prevented by renal denervation (Katholi et al., 1984).
Whether the effects of kidney injury on the activity of the SNS in the kidney are direct or mediated via angiotensin II (AngII) is unclear (Siddiqi et al., 2009). Increases in renal sympathetic nerve activity and activity of the RAS both influence renal- and vascular function. Besides the systems interact directly and indirectly (DiBona, 2000a, 2001). Direct intrarenal interactions take by specific innervation, indirect intrarenal interactions occurs via AngII. Interactions can also be extrarenal; for instance in the central nervous system (CNS) where renal sympathetic nerve activity and the baroreflex control are modulated by changes in activity of the RAS (DiBona, 2000a, 2001). For example: renovascular hypertension – a classical example of kidney ischemia – is characterized by hyperactivity of the SNS as assessed by MSNA. Treatment by angioplasty decreases MSNA, plasma renin activity, and the plasma concentration of AngII (Miyajima et al., 1991).
Even a minute injury by injection of phenol into one kidney, without affecting kidney function, stimulates central sympathetic activation (Ye et al., 1998), stimulates both afferent and efferent renal sympathetic activity, and induces hypertension in rats (Ye et al., 1997, 2002). The BP-elevating effect of phenol can be reduced or prevented by renal denervation (Ye et al., 1997, 2002). Also partial renal ablation by arterial ligation and induction of renal artery stenosis causes hypertension (Faber and Brody, 1985; Campese et al., 1995). Renal efferent and afferent innervation plays a crucial role; the effects of these abnormalities are largely prevented by renal denervation.
Kidneys as a Generator and Recipient of Sympathetic Activity
Besides being a generator of sympathetic activity, the kidney also acts as recipient of efferent signals. Renal efferent nerves are comprised of functionally specific groups of fibers which separately innervate and control the function of juxtaglomerular granular cells, tubules, and vessels. Responses of the kidney to efferent activity occur differentially in relation to graded intensity of renal nerve stimulation (DiBona, 2000b). This arrangement permits maximal flexibility.
Increased renal sympathetic activity leads to a cascade of actions: renal blood flow and glomerular filtration rate decreases by renal vasoconstriction. Secondly by stimulating the release of renin by the juxtaglomerular cells, AngII is produced. This is further amplified by direct activation of the RAS by kidney injury. Increases in renal sympathetic nerve activity also directly increase renal tubular sodium reabsorption.
Angiotensin II directly causes vasoconstriction, has trophic effects, stimulates the SNS vasomotor center in the brainstem, and plays an important role in the water and chloride reabsorption in the proximal tubule (Reid, 1992).
Angiotensin II and the renal nerves interact. An example of the interaction between AngII and renal innervation is given by Hendel and Collister (2006); a long-term low-dose AngII infusion in rats leads to a gradual increase in BP (Hendel and Collister, 2006). This effect could partially be prevented by renal denervation (Hendel and Collister, 2006). Furthermore the intravenous infusion of AngII in humans stimulates sympathetic activity as assessed by MSNA (Matsukawa et al., 1991).
Besides its peripheral actions, experimental research suggests that AngII contributes to the regulation of arterial pressure and intravascular volume through actions on several brain sites (DiBona, 1999, 2001). A hormonal-sympathetic reflex model for the long-term control of arterial pressure has been proposed (Brooks and Osborn, 1995; Brooks, 1997; Osborn, 1997). Indeed acute increases in circulating AngII concentration affect the SNS through actions on the brain, sympathetic ganglia, and sympathetic nerve endings (Reid, 1992). However the mechanism of the effect of chronic increases of AngII on sympathetic nerve activity is unclear (DiBona, 2000a, 2001). The proposed mechanism is schematically shown in Figure 1.
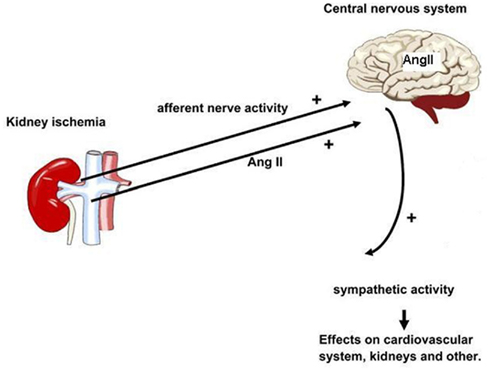
Figure 1. Schematic representation of the kidney involvement in the pathogenesis of sympathetic hyperactivity. Minimal kidney damage, not necessarily affecting kidney function, results in area(s) of ischemia. This results in increased afferent nerve activity and increased levels of AngII in plasma and CNS. Increased central sympathetic outflow affects many organs also including the cardiovascular system. In addition, AngII may enhance sympathetic activity on the peripheral level.
Aldosterone is another important hormone in the regulation of BP. The production of aldosterone in the adrenal glands is stimulated by AngII. In the kidneys it increases tubular sodium reabsorption, leading to an increase in renal sodium retention and a decrease in urinary sodium excretion. Aldosterone is a steroid that can easily enter the brain (Hendler and Livingston, 1978; De Nicola et al., 1981; Birmingham et al., 1984; Funder and Myles, 1996; Uhr et al., 2002). Acting via mineralocorticoid receptors, aldosterone increases sympathetic nerve activity by up regulating the brain RAS components and induction of oxidative stress in the hypothalamus (Zhang et al., 2008).
The diseased kidneys are important in the pathogenesis of sympathetic hyperactivity, however several other factors may be involved as well, including renalase (Boomsma and Tipton, 2007; Desir, 2011), NO-related mechanisms (Krukoff, 1999; Mallamaci et al., 2004), tonic chemoreflex activation (Hering et al., 2007), obesity (da Silva et al., 2009), and insulin (Lambert et al., 2010). Whether these mechanisms directly affect the CNS, exhibit their effect s via the kidneys or both is unclear.
Treatment of Sympathetic Activation
There is abundant evidence that sympathetic hyperactivity independent of BP is detrimental to health. With the pathophysiology in mind, pharmacological inhibition of the RAS is a logical step in the treatment of disease states in which sympathetic activation plays a crucial role. Treatment with ACE inhibitors (ACEi) and AngII receptor blockers (ARB) in patients with CKD resulted in a BP and MSNA lowering effect. However MSNA was not completely normalized (Klein et al., 2003b; Neumann et al., 2007). Moreover SNS as assessed by MSNA is associated with the composite of all cause mortality and non-fatal cardiovascular events in CKD patients despite treatment with ACEi or ARB (Penne et al., 2009).
The increased risk on mortality and cardiovascular events in CKD is the result of the involvement of the sympathetic activation in the pathogenesis of organ damage. Sympathetic activation has different modes of action on several pathways like structural effects on the heart and vascular system. In human studies, indexes of left ventricular hypertrophy (LVH) correlate with plasma noradrenaline values (Marcus et al., 1994) and cardiac noradrenaline spill over (Kelm et al., 1996). Also in essential hypertension, the occurrence of LVH is associated with an increase in MSNA (Greenwood et al., 2001).
The incomplete normalization of sympathetic activation in pharmacologic treatment and its associated risks, asks for new treatment options. Catheter based renal denervation may provide an important new treatment option.
The First Results and Remaining Questions
Broad Effect Range and Non-responders
Presently three studies on the effect of renal denervation on BP are available. All suggest a substantial effect (Krum et al., 2009; Esler et al., 2010; Symplicity HTN-1 Investigators, 2011). In the Simplicity HTN-2 trial (Esler et al., 2010) 106 patients with hypertension resistant to therapy were randomly allocated to treatment or control group. In patients treated with renal denervation, office-based BP declined with 32/12 mmHg 6 months after renal denervation, compared with no change in controls. However a broad range of effect was observed evidenced by a SD of 23/11 mmHg. Moreover in 10% of patients systolic BP did not decrease and were classified as non-responders. The presence of non-responders and the broad effect range has several possible explanations. Firstly it could be due to a failure of the intervention; perhaps the treatment was unsuccessful because the nerves were only partially destroyed. Another explanation is that the cardiovascular system did not respond to the decrease of the sympathetic activity. Years of hypertension made the vasculature too rigid and diseased, so that it is unable to dilate and lower vasculature resistance after the procedure. On the other hand, it is also possible that in the particular patient the renal nerves were not involved in the pathophysiology and other BP-elevating mechanisms are operational. Although, secondary forms of hypertension are among exclusion criteria, a secondary cause of hypertension was not actively ruled out in all patients. It is possible that among the group of non-responders, secondary forms of hypertension like hyperaldosteronism (a hypervolemic state with low sympathetic activity) and other forms of endocrine hypertension, are common. It seems unlikely that this procedure is effective in these patients. Therefore, it is recommended to actively exclude (treatable) secondary causes of hypertension, before performing renal denervation.
At this moment we can not distinguish between the possible explanations for treatment failure in a particular patient. As a consequence it is not possible yet to identify patients most likely to benefit from renal denervation. It is reasonable to hypothesize that patients with signs of kidney injury – for example an increased renin activity or decreased kidney perfusion/oxygenation – and patients with signs of activation of the SNS – for example a non-dipping pattern of the 24-h ambulatory BP – will probably benefit most from renal denervation.
The HTN-1 Investigators (Symplicity HTN-1 Investigators, 2011) only identified a higher baseline systolic BP and the use of central sympatholytic agents as significant independent predictors of response (Symplicity HTN-1 Investigators, 2011).
Theoretically, an elegant manner to identify responders is a run-in period with administration of clonidine, a centrally acting alpha agonist, which reduces central sympathetic outflow. Patients who react beneficially might be good candidates for renal denervation (Katholi et al., 2010). Of note is that in the HTN-2 trial almost halve of the participants used central acting sympathicolytic agents.
Monitoring of the Effect of Renal Denervation
Another important limitation of renal denervation is that presently there is no easy variable available to monitor the intervention. If one accepts the pathophysiologic model, MSNA would be an ideal method. It offers the opportunity to determine efferent sympathetic activity, and thus potentially makes it possible to learn more on the pathophysiology of non- and poor responders. Unfortunately MSNA is invasive and time consuming, so it is not suitable for daily practice.
In the proof-of-principle study (Krum et al., 2009) release of noradrenaline from the renal sympathetic nerves was measured in 10 patients with the isotope dilution renal noradrenaline spillover method. In these patients renal noradrenaline spillover was reduced with an average of 47% (Krum et al., 2009). This method requires the administration of radioactive isotopes and is not suitable to apply in daily patient care.
Schlaich et al. (2009) studied the effect of renal denervation, in one individual, on renal plasma flow, left ventricular mass, and MSNA. Again a pronounced BP-lowering effect was found: BP was reduced from 161/107 mmHg at baseline to 127/87 mmHg 12 months after renal denervation. Moreover, renin activity reduced by 50%; renal plasma flow increased with almost 56%; left ventricular mass decreased with almost 9%; and MSNA substantially reduced (Schlaich et al., 2009). These are intriguing results that can only be explained by accepting the pathophysiologic model presented in Figure 1. It gives strong support for a central role of the kidneys in the pathogenesis of hypertension and that renal denervation destructs the afferent renal nerves.
Physiology of the BP-lowering Effect of Renal Denervation
The BP-lowering effect of renal denervation increases in time, 6 months after treatment the effect seems to be maximal. A clear explanation for the increasing effect is not available. The effects of renal denervation occur by functional changes (rapid effects) and structural changes such as reversal of vascular hypertrophy. These structural changes may take some time to occur.
It is reasonable to hypothesize that the BP-lowering effect is due to both an altered water/sodium handling by the kidneys and a decrease in peripheral vascular resistance.
It is suggested by Liu and Cogan (1988) that for optimal water and chloride reabsorption in the proximal tubule, AngII, and renal innervation interact. After renal denervation, the effect of AngII to increase water and chloride reabsorption was decreased by ≈75% (Liu and Cogan, 1988). This indicates that the majority of the effect of AngII is dependent on intact innervation and that an important action of AngII is to facilitate the release of noradrenaline in the presynaptic site of the sympathetic nerve terminals (DiBona, 2000a).
Renal denervation also significantly increased basal renal blood flow, improved dynamic autoregulation of renal blood flow, and increased renal blood flow variability in rats with congestive heart failure and rats with spontaneous hypertension, compared with controls (DiBona and Sawin, 2004). Schlaich et al. (2009) also found an increase in renal plasma flow from 719 to 1126 mL/min after renal denervation in one single patient.
Renoprotective Effect of Renal Denervation
Conclusions from animal studies suggest that renal denervation has a renoprotective effect independent of lowering BP. In Dahl salt-sensitive hypertensive rats, renal denervation leads to local renal sympatho-inhibition in absence of a BP-lowering effect. This results in decreased urinary albumin excretion, reduced glomerulosclerosis and podocyte injury (Nagasu et al., 2010). Inflammation and oxidative stress are risk factors for progressive renal dysfunction (Modlinger et al., 2004). Veelken et al. (2008) suggested that renal denervation protects kidney function through an anti-inflammatory effect (Veelken et al., 2008). They reported that denervation significantly reduced albuminuria, mesangiolysis, formation of micro-aneurysms, deposition of glomerular collagen IV, and expression of transforming growth factor-β in experimental glomerulonephritis in rats (Veelken et al., 2008).
Also the reduction of BP alone would be expected to be beneficial to impaired kidneys and slow down kidney failure progression. An analysis of long-term clinical trials such as the Modification of Diet in Renal Disease Trial (MDRD), demonstrated that a decrease in BP results in preservation of kidney function (Peterson et al., 1995; Lazarus et al., 1997).
In the observational study, kidney function remained stable during the first year after renal denervation. However 2 years post-procedure data on eGFR are available of 10 patients, in them eGFR changed on average by −16.0 mL/min/1.73 m2 (Symplicity HTN-1 Investigators, 2011). The authors explained the drop in eGFR in five patients by adding spironolactone or another diuretic after the first year of follow-up. In patients without newly added spironolactone or another diuretic, eGFR changed with 7.8 mL/min/1.73 m2. Since these data are observational, without a control group, it is questionable whether the decrease in eGFR is due to renal denervation or is the effect of years of hypertension. The effect of renal denervation on kidney function is not completely clear yet.
Kidney Independent Sympathetic Activity
A substantial part of overactivity of the SNS in hypertension is kidney dependent. There is much evidence that kidney ischemia is of crucial importance in this pathway. It is unknown whether increased efferent renal nerve activity also exists as a primary abnormality. It is possible that in certain genetic forms or stress related hypertension this could be the case. However, this is difficult to determine. Since in renal denervation both the afferent, as the efferent renal nerves will be interrupted, it potentially will also be successful in these patients.
Possible indications for renal denervation
It seems worth exploring the role of renal denervation in other patient groups characterized by sympathetic activation like milder forms of hypertension, sleep apnea, heart failure, metabolic syndrome, obesity, chronic kidney failure and dialysis, hepatorenal syndrome, and cardiorenal syndrome. A pilot study shows us that renal denervation improves glucose metabolism and insulin sensitivity in addition to the BP-lowering effect. Even in the subgroup of patients with diagnosed diabetes mellitus at study entry, renal denervation significantly reduced fasting glucose and insulin sensitivity (Mahfoud et al., 2011).
Conclusion
There is convincing evidence that kidney injury can cause sympathetic overactivity. This sympathetic activation is detrimental to health and reduction may improve clinical outcome. Renal denervation offers new opportunities, not only in the treatment of patients, but also in the effort to better understand the pathophysiology in various disease conditions characterized by sympathetic overactivity.
Funding
Eva E. Vink was supported by a grant of the Dutch Kidney Foundation (IP10-15).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Birmingham, M. K., Sar, M., and Stumpf, W. E. (1984). Localization of aldosterone and corticosterone in the central nervous system, assessed by quantitative autoradiography. Neurochem. Res. 9, 333–350.
Blankestijn, P. J. (2004). Sympathetic hyperactivity in chronic kidney disease. Nephrol. Dial. Transplant. 19, 1354–1357.
Blankestijn, P. J., Ligtenberg, G., Klein, I. H., and Koomans, H. A. (2000). Sympathetic overactivity in renal failure controlled by ACE inhibition: clinical significance. Nephrol. Dial. Transplant. 15, 755–758.
Boomsma, F., and Tipton, K. F. (2007). Renalase, a catecholamine-metabolising enzyme? J. Neural Transm. 114, 775–776.
Brooks, V. L. (1997). Interactions between angiotensin II and the nervous system in the long-term control of arterial pressure. Clin. Exp. Pharmacol. Physiol. 24, 83–90.
Brooks, V. L., and Osborn, J. W. (1995). Hormonal-sympathetic interactions in long-term regulation of arterial pressure: an hypothesis. Am. J. Physiol. 268, R1343–R1358.
Campese, V. M., Kogosov, E., and Koss, M. (1995). Renal afferent denervation prevents the progression of renal disease in the renal ablation model of chronic renal failure in the rat. Am. J. Kidney Dis. 26, 861–865.
Converse, R. L. Jr., Jacobsen, T. N., Toto, R. D., Jost, C. M., Cosentino, F., Fouad-Tarazi, F., and Victor, R. G. (1992). Sympathetic overactivity in patients with chronic renal failure. N. Engl. J. Med. 327, 1912–1918.
da Silva, A. A., do Carmo, J., Dubinion, J., and Hall, J. E. (2009). The role of the sympathetic nervous system in obesity-related hypertension. Curr. Hypertens. Rep. 11, 206–211.
De Nicola, A. F., Tornello, S., Weisenberg, L., Fridman, O., and Birmingham, M. K. (1981). Uptake and binding of [3H]aldosterone by the anterior pituitary and brain regions in adrenalectomized rats. Horm. Metab. Res. 13, 103–106.
Desir, G. (2011). Novel insights into the physiology of renalase and its role in hypertension and heart disease. Pediatr. Nephrol. PMID:21424526. [Epub ahead of print].
DiBona, G. F. (1999). Central sympathoexcitatory actions of angiotensin II: role of type 1 angiotensin II receptors. J. Am. Soc. Nephrol. 10(Suppl. 11), S90–S94.
DiBona, G. F. (2000a). Nervous kidney. Interaction between renal sympathetic nerves and the renin-angiotensin system in the control of renal function. Hypertension 36, 1083–1088.
DiBona, G. F. (2000b). Neural control of the kidney: functionally specific renal sympathetic nerve fibers. Am. J. Physiol. Regul. Integr. Comp. Physiol. 279, R1517–R1524.
DiBona, G. F. (2001). Peripheral and central interactions between the renin-angiotensin system and the renal sympathetic nerves in control of renal function. Ann. N. Y. Acad. Sci. 940, 395–406.
DiBona, G. F. (2004). The sympathetic nervous system and hypertension: recent developments. Hypertension 43, 147–150.
DiBona, G. F., and Sawin, L. L. (2004). Effect of renal denervation on dynamic autoregulation of renal blood flow. Am. J. Physiol. Renal Physiol. 286, F1209–F1218.
Esler, M. D., Krum, H., Sobotka, P. A., Schlaich, M. P., Schmieder, R. E., and Bohm, M. (2010). Renal sympathetic denervation in patients with treatment–resistant hypertension (The Symplicity HTN-2 Trial): a randomised controlled trial. Lancet 376, 1903–1909.
Faber, J. E., and Brody, M. J. (1985). Afferent renal nerve-dependent hypertension following acute renal artery stenosis in the conscious rat. Circ. Res. 57, 676–688.
Funder, J., and Myles, K. (1996). Exclusion of corticosterone from epithelial mineralocorticoid receptors is insufficient for selectivity of aldosterone action: in vivo binding studies. Endocrinology 137, 5264–5268.
Grassi, G., Cattaneo, B. M., Seravalle, G., Lanfranchi, A., and Mancia, G. (1998). Baroreflex control of sympathetic nerve activity in essential and secondary hypertension. Hypertension 31, 68–72.
Grassi, G., Quarti-Trevano, F., Seravalle, G., Arenare, F., Volpe, M., Furiani, S., Dell’Oro, R., and Mancia, G. (2011). Early sympathetic activation in the initial clinical stages of chronic renal failure. Hypertension 57, 846–851.
Greenwood, J. P., Scott, E. M., Stoker, J. B., and Mary, D. A. (2001). Hypertensive left ventricular hypertrophy: relation to peripheral sympathetic drive. J. Am. Coll. Cardiol. 38, 1711–1717.
Hendel, M. D., and Collister, J. P. (2006). Renal denervation attenuates long-term hypertensive effects of angiotensin ii in the rat. Clin. Exp. Pharmacol. Physiol. 33, 1225–1230.
Hendler, N. H., and Livingston, A. (1978). The localization over time of exogenous aldosterone and angiotensin II in various organs. Pavlov. J. Biol. Sci. 13, 187–193.
Hering, D., Zdrojewski, Z., Krol, E., Kara, T., Kucharska, W., Somers, V. K., Rutkowski, B., and Narkiewicz, K. (2007). Tonic chemoreflex activation contributes to the elevated muscle sympathetic nerve activity in patients with chronic renal failure. J. Hypertens. 25, 157–161.
Joles, J. A., and Koomans, H. A. (2004). Causes and consequences of increased sympathetic activity in renal disease. Hypertension 43, 699–706.
Katholi, R. E., Rocha-Singh, K. J., Goswami, N. J., and Sobotka, P. A. (2010). Renal nerves in the maintenance of hypertension: a potential therapeutic target. Curr. Hypertens. Rep. 12, 196–204.
Katholi, R. E., Whitlow, P. L., Hageman, G. R., and Woods, W. T. (1984). Intrarenal adenosine produces hypertension by activating the sympathetic nervous system via the renal nerves in the dog. J. Hypertens. 2, 349–359.
Kelm, M., Schafer, S., Mingers, S., Heydthausen, M., Vogt, M., Motz, W., and Strauer, B. E. (1996). Left ventricular mass is linked to cardiac noradrenaline in normotensive and hypertensive patients. J. Hypertens. 14, 1357–1364.
Kim, K. E., Onesti, G., Schwartz, A. B., Chinitz, J. L., and Swartz, C. (1972). Hemodynamics of hypertension in chronic end-stage renal disease. Circulation 46, 456–464.
Klein, I. H., Ligtenberg, G., Neumann, J., Oey, P. L., Koomans, H. A., and Blankestijn, P. J. (2003a). Sympathetic nerve activity is inappropriately increased in chronic renal disease. J. Am. Soc. Nephrol. 14, 3239–3244.
Klein, I. H., Ligtenberg, G., Oey, P. L., Koomans, H. A., and Blankestijn, P. J. (2003b). Enalapril and losartan reduce sympathetic hyperactivity in patients with chronic renal failure. J. Am. Soc. Nephrol. 14, 425–430.
Klein, I. H. H. T., Ligtenberg, G., Oey, P. L., Koomans, H. A., and Blankestijn, P. J. (2001). Sympathetic activity is increased in polycystic kidney disease and is associated with hypertension. J. Am. Soc. Nephrol. 12, 2427–2433.
Koomans, H. A., Blankestijn, P. J., and Joles, J. A. (2004). Sympathetic hyperactivity in chronic renal failure: a wake-up call. J. Am. Soc. Nephrol. 15, 524–537.
Krukoff, T. L. (1999). Central actions of nitric oxide in regulation of autonomic functions. Brain Res. Brain Res. Rev. 30, 52–65.
Krum, H., Schlaich, M., Whitbourn, R., Sobotka, P. A., Sadowski, J., Bartus, K., Kapelak, B., Walton, A., Sievert, H., Thambar, S., Abraham, W. T., and Esler, M. (2009). Catheter-based renal sympathetic denervation for resistant hypertension: a multicentre safety and proof-of-principle cohort study. Lancet 373, 1275–1281.
Lambert, G. W., Straznicky, N. E., Lambert, E. A., Dixon, J. B., and Schlaich, M. P. (2010). Sympathetic nervous activation in obesity and the metabolic syndrome–causes, consequences and therapeutic implications. Pharmacol. Ther. 126, 159–172.
Lazarus, J. M., Bourgoignie, J. J., Buckalew, V. M., Greene, T., Levey, A. S., Milas, N. C., Paranandi, L., Peterson, J. C., Porush, J. G., Rauch, S., Soucie, J. M., and Stollar, C. (1997). Achievement and safety of a low blood pressure goal in chronic renal disease. The Modification of Diet in Renal Disease Study Group. Hypertension 29, 641–650.
Liu, F. Y., and Cogan, M. G. (1988). Angiotensin II stimulation of hydrogen ion secretion in the rat early proximal tubule. Modes of action, mechanism, and kinetics. J. Clin. Invest. 82, 601–607.
Mahfoud, F., Schlaich, M., Kindermann, I., Ukena, C., Cremers, B., Brandt, M. C., Hoppe, U. C., Vonend, O., Rump, L. C., Sobotka, P. A., Krum, H., Esler, M., and Bohm, M. (2011). Effect of renal sympathetic denervation on glucose metabolism in patients with resistant hypertension: a pilot study. Circulation 123, 1940–1946.
Mallamaci, F., Tripepi, G., Maas, R., Malatino, L., Boger, R., and Zoccali, C. (2004). Analysis of the relationship between norepinephrine and asymmetric dimethyl arginine levels among patients with end-stage renal disease. J. Am. Soc. Nephrol. 15, 435–441.
Marcus, R., Krause, L., Weder, A. B., Meija, A. D., Schork, N. J., and Julius, S. (1994). Sex-specific determinants of increased left ventricular mass in the Tecumseh blood pressure study. Circulation 90, 928–936.
Matsukawa, T., Gotoh, E., Minamisawa, K., Kihara, M., Ueda, S., Shionoiri, H., and Ishii, M. (1991). Effects of intravenous infusions of angiotensin II on muscle sympathetic nerve activity in humans. Am. J. Physiol. 261, R690–R696.
Miyajima, E., Yamada, Y., Yoshida, Y., Matsukawa, T., Shionoiri, H., Tochikubo, O., and Ishii, M. (1991). Muscle sympathetic nerve activity in renovascular hypertension and primary aldosteronism. Hypertension 17, 1057–1062.
Modlinger, P. S., Wilcox, C. S., and Aslam, S. (2004). Nitric oxide, oxidative stress, and progression of chronic renal failure. Semin. Nephrol. 24, 354–365.
Nagasu, H., Satoh, M., Kuwabara, A., Yorimitsu, D., Sakuta, T., Tomita, N., and Kashihara, N. (2010). Renal denervation reduces glomerular injury by suppressing NAD(P)H oxidase activity in Dahl salt-sensitive rats. Nephrol. Dial. Transplant. 25, 2889–2898.
Neumann, J., Ligtenberg, G., Klein, I. H., Boer, P., Oey, P. L., Koomans, H. A., and Blankestijn, P. J. (2007). Sympathetic hyperactivity in hypertensive chronic kidney disease patients is reduced during standard treatment. Hypertension 49, 506–510.
Neumann, J., Ligtenberg, G., Klein, I. I., Koomans, H. A., and Blankestijn, P. J. (2004). Sympathetic hyperactivity in chronic kidney disease: pathogenesis, clinical relevance, and treatment. Kidney Int. 65, 1568–1576.
Osborn, J. W. (1997). Hormones as long-term error signals for the sympathetic nervous system: importance of a new perspective. Clin. Exp. Pharmacol. Physiol. 24, 109–115.
Peet, M. M. (1948). Hypertension and its surgical treatment by bilateral supradiaphragmatic splanchnicectomy. Am. J. Surg. 75, 48–68.
Penne, E. L., Neumann, J., Klein, I. H., Oey, P. L., Bots, M. L., and Blankestijn, P. J. (2009). Sympathetic hyperactivity and clinical outcome in chronic kidney disease patients during standard treatment. J. Nephrol. 22, 208–215.
Peterson, J. C., Adler, S., Burkart, J. M., Greene, T., Hebert, L. A., Hunsicker, L. G., King, A. J., Klahr, S., Massry, S. G., and Seifter, J. L. (1995). Blood pressure control, proteinuria, and the progression of renal disease. The Modification of Diet in Renal Disease Study. Ann. Intern. Med. 123, 754–762.
Reid, I. A. (1992). Interactions between ANG II, sympathetic nervous system, and baroreceptor reflexes in regulation of blood pressure. Am. J. Physiol. 262, E763–E778.
Schlaich, M. P., Sobotka, P. A., Krum, H., Lambert, E., and Esler, M. D. (2009). Renal sympathetic-nerve ablation for uncontrolled hypertension. N. Engl. J. Med. 361, 932–934.
Siddiqi, L., Joles, J. A., Grassi, G., and Blankestijn, P. J. (2009). Is kidney ischemia the central mechanism in parallel activation of the renin and sympathetic system? J. Hypertens. 27, 1341–1349.
Symplicity HTN-1 Investigators. (2011). Catheter-based renal sympathetic denervation for resistant hypertension: durability of blood pressure reduction out to 24 months. Hypertension 57, 911–917.
Uhr, M., Holsboer, F., and Muller, M. B. (2002). Penetration of endogenous steroid hormones corticosterone, cortisol, aldosterone and progesterone into the brain is enhanced in mice deficient for both mdr1a and mdr1b P-glycoproteins. J. Neuroendocrinol. 14, 753–759.
Veelken, R., Vogel, E. M., Hilgers, K., Amann, K., Hartner, A., Sass, G., Neuhuber, W., and Tiegs, G. (2008). Autonomic renal denervation ameliorates experimental glomerulonephritis. J. Am. Soc. Nephrol. 19, 1371–1378.
Ye, S., Gamburd, M., Mozayeni, P., Koss, M., and Campese, V. M. (1998). A limited renal injury may cause a permanent form of neurogenic hypertension. Am. J. Hypertens. 11, 723–728.
Ye, S., Ozgur, B., and Campese, V. M. (1997). Renal afferent impulses, the posterior hypothalamus, and hypertension in rats with chronic renal failure. Kidney Int. 51, 722–727.
Ye, S., Zhong, H., Yanamadala, V., and Campese, V. M. (2002). Renal injury caused by intrarenal injection of phenol increases afferent and efferent renal sympathetic nerve activity. Am. J. Hypertens. 15, 717–724.
Keywords: renal denervation, sympathetic activity, kidney disease, hypertension
Citation: Vink EE and Blankestijn PJ (2012) Evidence and consequences of the central role of the kidneys in the pathophysiology of sympathetic hyperactivity. Front. Physio. 3:29. doi: 10.3389/fphys.2012.00029
Received: 04 November 2011; Accepted: 06 February 2012;
Published online: 20 February 2012.
Edited by:
Jacqueline Kathleen Phillips, Macquarie University, AustraliaReviewed by:
Ulla Kopp, University of Iowa Carver College of Medicine, USARohit Ramchandra, Florey Neuroscience Institute, Australia
Copyright: © 2012 Vink and Blankestijn. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Peter J. Blankestijn, Department of Nephrology, University Medical Center Utrecht, Room F03.220, Heidelberglaan 100, PO Box 85500, 3508 GA Utrecht, Netherlands. e-mail: p.j.blankestijn@umcutrecht.nl