 Gabriel Hoi-Huen Chan1†
Gabriel Hoi-Huen Chan1† Enoch Chan2†
Enoch Chan2† Carsten Tsun-Ka Kwok3
Carsten Tsun-Ka Kwok3 George Pak-Heng Leung4
George Pak-Heng Leung4 Simon Ming-Yuen Lee5
Simon Ming-Yuen Lee5 Sai-Wang Seto3,6,7*
Sai-Wang Seto3,6,7*- 1Division of Science, Engineering and Health Studies, College of Professional and Continuing Education, The Hong Kong Polytechnic University, Kowloon, Hong Kong SAR, China
- 2School of Clinical Medicine, The University of Hong Kong, Pokfulam, Hong Kong SAR, China
- 3Department of Applied Biology and Chemical Technology, The Hong Kong Polytechnic University, Kowloon, Hong Kong SAR, China
- 4Department of Pharmacology and Pharmacy, The University of Hong Kong, Kowloon, Hong Kong SAR, China
- 5State Key Laboratory of Quality Research in Chinese Medicine, Institute of Chinese Medical Sciences, University of Macau, Avenida da Universidade, Taipa, China
- 6Research Centre for Chinese Medicine Innovation, The Hong Kong Polytechnic University, Kowloon, Hong Kong SAR, China
- 7NICM Health Research Institute, Western Sydney University, Penrith, NSW, Australia
Ageing is a risk factor for many degenerative diseases. Cardiovascular diseases (CVDs) are usually big burdens for elderly, caregivers and the health system. During the aging process, normal functions of vascular cells and tissue progressively lost and eventually develop vascular diseases. Endothelial dysfunction, reduced bioavailability of endothelium-derived nitric oxide are usual phenomena observed in patients with cardiovascular diseases. Myriad of studies have been done to investigate to delay the vascular dysfunction or improve the vascular function to prolong the aging process. Tumor suppressor gene p53, also a transcription factor, act as a gatekeeper to regulate a number of genes to maintain normal cell function including but not limited to cell proliferation, cell apoptosis. p53 also crosstalk with other key transcription factors like hypoxia-inducible factor 1 alpha that contribute to the progression of cardiovascular diseases. Therefore, in recent three decades, p53 has drawn scientists’ attention on its effects in vascular function. Though the role of tumor suppressor gene p53 is still not clear in vascular function, it is found to play regulatory roles and may involve in vascular remodeling, atherosclerosis or pulmonary hypertension. p53 may have a divergent role in endothelial and vascular muscle cells in those conditions. In this review, we describe the different effects of p53 in cardiovascular physiology. Further studies on the effects of endothelial cell-specific p53 deficiency on atherosclerotic plaque formation in common animal models are required before the therapeutic potential can be realized.
Introduction
With an estimated 17.9 million of people died of cardiovascular diseases (CVDs) in 2019, CVDs are the leading causes of death. The diseases contributed to 32% of the total global deaths. (World Health Organization, 2021). CVDs are group of diseases affecting the cardiovascular system. The cardiovascular system consists of the blood, heart and blood vessels. Blood is in liquid form and constantly being transported inside the body. The heart is a pump that beats about 72 times per minute in human adult and keeps the blood moving throughout the body. The blood vessels are composed of different sizes of arteries, veins and capillaries as conducting duct to transport blood and body fluid. Blood vessels are essential for the transport of molecules such gases, nutrients, wastes, hormones, bioactive substances and also involve in distribution of immune cells and heat in the body. Blood pressure is the determined by the heart pumping and the blood exerted upon the walls of the blood vessels. Normal blood pressure should be less than 120 mmHg for systolic blood pressure and 80 mmg Hg for diastolic blood pressure. Blood pressure higher than 120/80 mmHg is considered to be hypertensive (or a pre-hypertensive state) and it is an indicator for vascular changes. Such vascular changes usually accompanied with altered endothelial cells, vascular smooth muscle cells migration, arterial stiffness, vascular calcification. With such alternations in the arterial walls, blood pressure usually increases (Boron and Boulpaep, 2017; Brown et al., 2018). An elevation in blood pressure lowers cardiac output by increasing afterload, the pressure that opposes the ejection of blood during ventricular systole (Klabunde, 2022). Hypertension is also an important risk factor for various kinds of cardiovascular-associated diseases like heart failure, atrial fibrillation, chronic kidney disease, heart valve diseases, aortic syndromes, and dementia (Fuchs and Whelton, 2020).
Transcription factor p53 is heavily involved in activating or suppressing various genes in response to various stress stimuli, including DNA damage, hypoxia and oxidative stress, and its activation promotes DNA repair, apoptosis, cell cycle arrest, metabolic shifts and autophagy (Vousden and Prives, 2009; Beckerman and Prives, 2010). Moreover, a recent review by Men et al. (2021) also highlighted the regulatory roles of p53 in cardiac function and dysfunction. The regulation of p53 protein is controlled by posttranslational modifications including phosphorylation, acetylation/deacetylation, glycosylation, ubiquitination, SUMOylation and more (Meek and Anderson, 2009) (Figure 1). In unstressed situation, p53 is rapidly turned over by actions of MDM2 and MDM4, which promote poly-ubiquitination, nuclear export and proteasomal degradation of p53. When stimulated by stress, phosphorylation of the amino terminus of p53 prevents the binding of MDM2, leading to stabilization of p53 protein (Meek and Anderson, 2009). Subsequently, lysine residues in the carboxyl terminal the DNA-binding domain of p53 is subject to acetylation by p300/CBP and PCAF, and this acetylation is essential for the stabilization and activation of p53 (Tang et al., 2008; Meek and Anderson, 2009). On the other hand, sirtuin 1, a histone deacetylase, is another inhibitor of p53 activity, causes deacetylation of p53 protein (Vaziri et al., 2001; Kim et al., 2007). SUMOylation also plays a complex role in regulating the turnover and transcriptional activity of p53 (Takabe et al., 2011; Santiago et al., 2013; Dehnavi et al., 2019; Celen and Sahin, 2020).
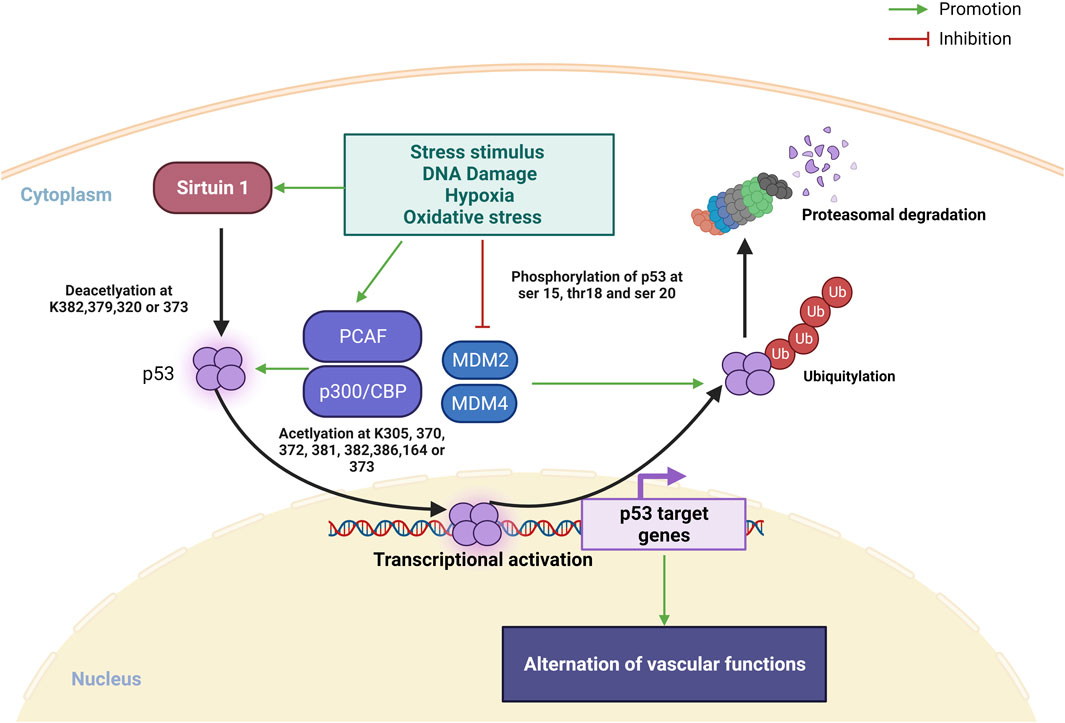
FIGURE 1. Overview on posttranslational modifications of p53 under stress stimulus. In normal condition, MDM2 and MDM4 promotes p53 ubiquitination leading to p53 degradation in proteasome. Under stress stimulus, sirtuin 1 is activated and deacetylate p53 at K382, 379, 320 or 373 for promoting p53 transcription, PACF and p300/CBP is also activated to promote p53 transcription via acetylation at k305, 370, 372, 373, 381, 282, 386 or 164. Ubiquitin proteasome of p53 degradation is inhibited via repression MDM2 and MDM4 binding to p53 by stress stimulated phosphorylation of p53 at ser 15, 20, and thr18 (Created with BioRender.com).
In this review, we discuss the role of p53 in vascular function, and its potential as a therapeutic target for vascular dysfunction.
p53 and vascular function
Blood vessels consist of three layers: tunica externa (outermost later), tunica media (middle layer) and tunica intima (innermost later). Tunica externa is mainly made of connective tissues providing support and protection for the vessel. Tunica media comprises mainly of vascular smooth muscle cells and connective tissues. Vascular smooth muscle cells are arranged in helical or circular layers in the intima in larger vessels and as a single circular in small vessels. The contractile state of smooth muscle cells determines the diameter of blood vessels. In healthy blood vessels, when there is change in extravascular environment, local growth factors, vascular active substances, and hemodynamic stimuli initiate structural and functional adaptations of smooth muscles to maintain a normal and stable blood pressure (Renna et al., 2013).
The tunica intima consists of a single-cell layer of endothelial cells. Upon stimulation by acetylcholine, ATP, bradykinin and laminar shear stress, endothelial cells generate physiological amount of nitric oxide (NO) with an enzyme called endothelial nitric oxide synthase (eNOS), and NO stimulates the relaxation of smooth muscle (Furchgott and Vanhoutte, 1989; Fleming and Busse, 2003; Sessa, 2004). The endothelium also produces other vasorelaxant factors including prostaglandin I2 (Toda et al., 2007; Bauer and Sotníková, 2010) and endothelium-derived hyperpolarizing factor (EDHF) (Félétou and Vanhoutte, 1996) as well as vasoconstricting substances such as endothelin (Barton and Yanagisawa, 2019) and thromboxane A2 (Chen, 2018). Endothelial cells also function to keep the inner surface of blood vessel non-adhesive and non-thrombogenic, which is essential to maintain steady blood flow (O'Reilly et al., 2003).
The number of cells within tissue depends on their turnover by apoptosis and cell proliferation, which are highly regulated processes. In normal arteries, the turnover of smooth muscle cells and endothelial cells are low. eNOS inhibits the apoptosis of endothelial cells (Dimmeler et al., 1997; Polte et al., 1997), as well as the proliferation of endothelial cells and vascular smooth muscle cells (Tsihlis et al., 2011). Moreover, laminar shear stress can increase cellular level of p53 within endothelial cells. p53 is stabilized upon phosphorylation by c-Jun N-terminal kinase (JNK). This increase in p53 level leads to cell cycle arrest and the suppression of apoptosis of endothelial cells (Lin et al., 2000). Events such as healing of injuries, skeletal muscle adaptation after exercise or female reproductive cycles trigger angiogenesis (Otrock et al., 2007). Vascular endothelial growth factor (VEGF) promotes angiogenesis through promoting endothelial cell survival, proliferation and migration (Olsson et al., 2006). p53 also play a complex role in regulating VEGF expression. In the presence of an intact p21-Rb pathway, p53 represses VEGF expression. However, in the absence of p21-Rb pathway, which occurs in malignant cells, p53 promotes VEGF expression (Farhang Ghahremani et al., 2013a; Farhang Ghahremani, et al., 2013b).
p53 and endothelial dysfunction
Endothelial dysfunction is a broad term that refers to the impairment of the normal physiological functioning of the endothelium. It involves, but is not limited to, impaired endothelium-dependent vasodilatation, increased leukocyte recruitment into tunica intima, increased endothelial permeability to lipoproteins, and increased thrombotic property. A key cause of endothelial dysfunction is the reduced bioavailability of eNOS (Thomas et al., 2008).
Apart from reduction of eNOS, DNA damage, mitochondrial dysfunction, oxidative stress, telomere dysfunction and other stressors induce several tumor suppressor genes including p53 and lead to cellular senescence, the permanent state of cell arrest. (Jerafi-Vider et al., 2021; Sun and Feinberg, 2021). Cellular senescence is originally a physiological protective mechanism to prevent the development of tumor, however, when such senescent cells accumulate in number it leads to aging of cells (López-Otín et al., 2013). Senescence of vascular endothelial cells plays an important role in initiation and progression of CVDs (Hohensinner et al., 2016; Jia et al., 2019). Cellular oxidative stress, circulating IGF-1 deficiency, altered calcium signaling, may impair cell functions and contribute to endothelial damage that lead to cerebrovascular diseases and loss of cognitive functions (Tarantini et al., 2017). The senescent cells can remain alive, though accompanied by phenotypical changes, altered metabolism and gene expression. The senescent EC usually are flatter and enlarged with more polypoid nucleus and at the same time with changed structure in cytoskeleton that may lead to angiogenesis, proliferation and migration of the cells (Sun and Feinberg, 2021). Thus, endothelial senescence is an important biomarker for CVDs.
A number of biochemical pathways involving sirtuin 1, Klotho and fibroblast growth factor 21 (Figure 2) have been found to be associated with cellular senescence (Jia et al., 2019). Cellular senescence is also regulated by p53 pathway with increased inflammatory signaling in arteries from older adults (Donato et al., 2015). p53 is found to impair endothelium-dependent vasodilatation, which is important for maintaining healthy blood flow. A study found that p53 mediates angiotensin II-induced impairment of vasodilatation (Kim et al., 2008). Another study by Kumar et al. (2011) found that ex vivo adenoviral overexpression of p53 of rat aortic rings impaired endothelium-dependent vasodilatation. In vitro studies in the same paper showed that overexpression of p53 in human umbilical vein endothelial cells suppressed Kruppel-like Factor 2 (KLF2), which was known to induce eNOS expression (Kumar et al., 2011). Diabetes was also found to induce a marked increase in endothelial p53 level as well as an impairment of endothelium-dependent vasodilatation. This impairment resulted in significantly reduction of endothelial cell-specific p53 gene deletion (Yokoyama et al., 2019). In vitro studies in the same paper argued that p53 inactivates eNOS through inhibiting the phosphorylation of eNOS at its Ser1177 residue, one of the important regulatory sites for eNOS activity (Yokoyama et al., 2019; Thai et al., 2021).
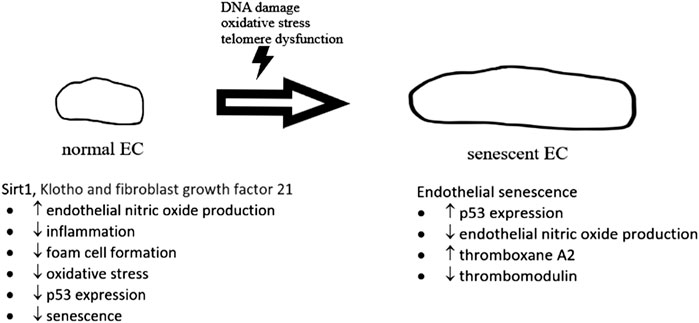
FIGURE 2. The development of normal endothelial cell (EC) to senescent EC. In normal situation, sirt 1, Klotho and fibroblast growth factor 21, etc., give protective effects on normal endothelial cells, preventing senescence. In senescent EC, expression of p53 was upregulated with a reduction of endothelial nitric oxide production.
The roles of p53 in mediating vascular oxidative stress and pro-inflammatory responses in diabetes were studied. Exposure to high glucose promoted the expression of p53 in endothelial cells, and treatment with pifithrin-α (small molecule inhibitor of p53) or p53 siRNA attenuated oxidative stress and mRNA expression of adhesion molecules (VCAM-1, ICAM-1, E-selectin) and MCP-1 in high glucose-treated endothelial cells. Administration of pifithrin-α to diabetic mice in vivo significantly improved endothelium-dependent vasodilatation response of aorta (Wu et al., 2019). Interestingly, treatment with SRT2104, a sirtuin 1 activator, protects against high glucose-induced endothelial dysfunction by inhibiting p53 activity (Wu et al., 2018). In addition, SUMOylation of p53 was found to mediate disturbed flow-induced endothelial cell apoptosis (Heo et al., 2011). Disturbed flow activates PKCζ and upregulates p53 SUMOylation. SUMOylated p53 translocates from nucleus to cytoplasm, and this upregulates apoptosis (Heo et al., 2011). Subsequent studies found that increased SUMOylation of p53 promotes endothelial dysfunction and inflammation (Heo et al., 2013). The regulation and role of p53 in endothelial cell under different physiological events are summarized in Table 1.

TABLE 1. Summary of regulation of p53 in endothelial cell under different physiological or pathophysiological conditions.
p53 and atherosclerosis
Atherosclerosis is a chronic inflammatory disorder occurring in mid-sized and large arteries. It is characterized by the excessive infiltration of lipid and inflammatory cells and resultant formation of fibrous and fatty lesions within the tunica intima (Ross, 1999; Insull, 2009; Hansson and Hermansson, 2011). The progressive growth of atherosclerotic lesions leads to the impediment of blood flow. With a rupture or erosion of an unstable plaque, the contact between the blood and materials within the plaques can quickly trigger thrombosis, leading to ischemic injury or death of the tissue to which the affected artery supplies blood. The most debilitating or fatal clinical manifestations of atherosclerosis are myocardial infarction and stroke (Insull, 2009; Hansson and Hermansson, 2011).
At the initial stages of atherosclerosis, endothelial dysfunction favors the recruitment of neutrophils and monocytes into the subendothelial space of tunica media (Bevilacqua et al., 1994; Blankenberg et al., 2003). Once recruited into the subendothelial space, monocytes mature into macrophages (Negre-Salvayre et al., 2020). The endothelium then becomes more permeable to low-density lipoprotein (LDL), leading to increased accumulation of LDL within the subendothelial space (Schwenke and Carew, 1989; von Eckardstein and Rohrer, 2009). LDL is oxidized by the reactive oxygen species (ROS) produced by the activated leukocytes within the subendothelial space, and macrophages internalize oxidized LDL (oxLDL) to become cholesterol-laden foam cells (Stocker and Keaney, 2004). oxLDL also promotes inflammation of the blood vessel (Khan et al., 1995; Sawamura et al., 1997; Miller et al., 2003). Foam cells reside within the tunica media and generate various pro-inflammatory factors including cytokines, chemokines, ROS and matrix-degrading proteases (Tabas and Bornfeldt, 2016). Moreover, pro-inflammatory cytokines can stimulate the expression inducible nitric oxide synthase (iNOS) in vascular smooth muscle cells (Chan and Fiscus, 2004) and macrophages (Depre et al., 1999). Unlike eNOS, iNOS produces a large amount of NO for a prolonged period (Cho et al., 1992). Coinciding with an increased production of ROS, iNOS-drived NO is chemically converted into other reactive nitrogen species (e.g., peroxynitrite) which is a stronger oxidant, contributing to cell damage (Martínez and Andriantsitohaina, 2009; Förstermann et al., 2017).
The number of macrophages/foam cells continues to increase and lead to growth in lesion size. In more advanced lesions, this increase in lesion size is also contributed by scavenger receptor-initiated proliferation of macrophages (Robbins et al., 2013). The overall size of lesion is determined also by the ability of macrophages to migrate away from the lesion, the rate of macrophage/foam cell death (Shi et al., 2018), and the ability of lesion macrophages to clear apoptotic or necrotic materials by efferocytosis (Tabas and Bornfeldt, 2016; Brophy et al., 2017). These processes are likely impaired in advanced lesions because macrophage/foam cell migration is impaired by increased expression of iNOS and exaggerated generation of reactive nitrogen species (Huang et al., 2014), and the efferocytosis of lesion macrophages is also known to be impaired by ROS and matrix-degrading proteases (Thorp et al., 2011). Impairment of macrophage efferocytosis allows the formation of an acellular necrotic core enriched with lipid and crystalline cholesterol released by necrotic foam cells, which is a key histopathological feature of advanced atherosclerotic lesions (Tabas and Bornfeldt, 2016). Apart from the formation of necrotic core, advanced atherosclerotic lesions also feature a fibrous cap underneath the endothelium. Upon stimulation by pro-migratory signals including growth factors (e.g., PDGF, FGF), cytokines, thrombin, extracellular matrix components or high glucose level, smooth muscle cells in tunica media acquire a synthetic phenotype, resulting in their proliferation and migration to the subendothelial space. Vascular smooth muscle cells deposit collagen causing the expansion of extracellular matrix at the subendothelial space, which appears as a fibrous cap (Gerthoffer, 2007; Brophy et al., 2017). As such, advanced lesions with necrotic core and fibrous cap are called fibro-fatty atheroma (Stary et al., 1994).
In advanced atherosclerotic plaques of human, the expression and phosphorylation of p53 was found to be elevated (Iacopetta et al., 1995; Gorgoulis et al., 2005). In vivo study showed that global p53 deficiency accelerated atherosclerotic plaque formation in atherosclerosis-prone apoE−/− mice (Guevara et al., 1999; Mercer et al., 2005). Subsequently, two studies showed that LDL receptor-deficient mice and APOE*3-Leiden mice transplanted with bone marrow of p53-deficient mice also had accelerated atherosclerotic plaque formation (van Vlijmen et al., 2001; Merched et al., 2003). Based on histological evidence, two of these studies found that p53 deficiency reduced cell proliferation within atherosclerotic lesions, but did not affect apoptosis there (Guevara et al., 1999; Merched et al., 2003), while one of them found that p53 deficiency reduced apoptosis but did not affect cell proliferation (van Vlijmen et al., 2001). One of these studies also demonstrated that transplant of p53-positive bone marrow to p53−/−/apoE−/− mice reduced atherosclerotic lesion formation and cell proliferation and apoptosis within atherosclerotic lesions (Mercer et al., 2005). In vitro experiments showed that p53 differentially regulate the proliferation and apoptosis of macrophages and smooth muscle cells. For macrophages, p53 promotes their apoptosis. However, for vascular smooth muscle cells, p53 inhibits their proliferation and limits their apoptosis in vitro (Mercer et al., 2005). The regulation of p53 in vascular smooth muscle cells and macrophages are summarized in Table 2 and Table 3 respectively.
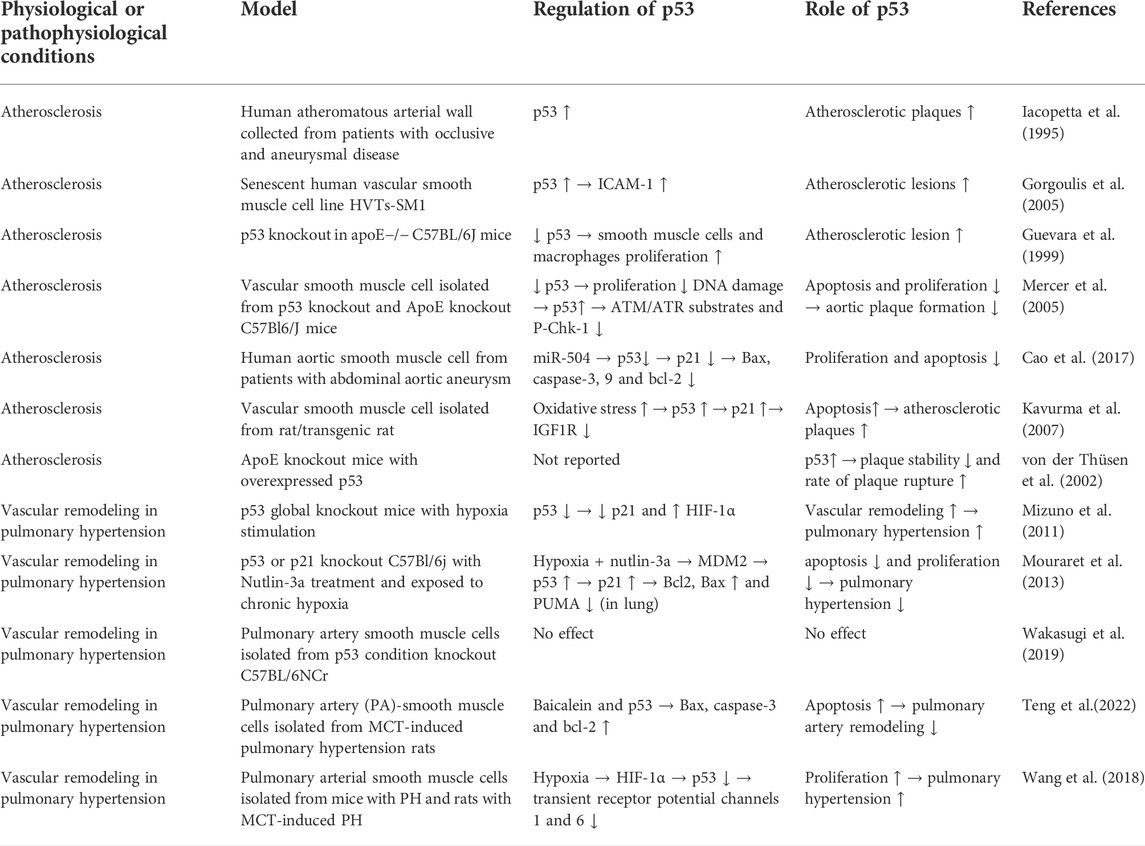
TABLE 2. Summary of regulation of p53 in smooth muscle cell under different physiological or pathophysiological conditions.
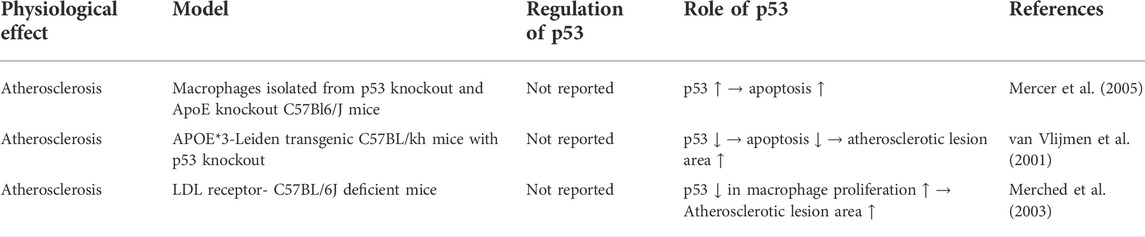
TABLE 3. Summary of regulation of p53 in macrophage during atherosclerosis.
A more recent study showed that apoE−/− mice with smooth muscle-specific p53 gene knockout had no significant difference in overall size of atherosclerotic lesions, compared to apoE−/− mice with p53 in their smooth muscle. Interestingly, this study also found that apoE−/− mice with smooth muscle-specific p53 deficiency had a significantly higher number of smooth muscle cells within the fibrous caps of atherosclerotic plaques, and the in vitro experiments of the same study showed that p53 deficiency enhanced vascular smooth muscle cell migration and invasion (Cao et al., 2017). An in vitro study showed that p53 negatively regulates the expression of IGF1 receptor, which is a key mechanism for vascular smooth muscle cell survival and proliferation (Kavurma et al., 2007). IGF1 receptor activation is also important for smooth muscle cell migration (Beneit et al., 2016). Therefore, it is plausible that p53 limited vascular smooth muscle cell migration through downregulating IGF1 receptor expression. One study looked at the effect of p53 on the stability of advanced atherosclerotic lesion through collar-induced carotid atherogenesis in apoE−/− mice, overexpression of p53 reduced plaque stability and promoted the rate of plaque rupture were observed (von der Thüsen et al., 2002). Depending on the culprit vessel, plaque rupture can lead to myocardial ischemia or cerebral ischemia. For instant, cerebral vasospasm secondary to cerebral aneurysm and hemorrhage stroke can lead to hypoperfusion resulting in delayed cerebral ischemic deficits (Sun and Feinberg, 2021), cerebral-vasospasm]. An in vivo study suggested that p53 plays an important role in the etiology of vasospasm which contributed directly to the alteration of the blood brain barrier (BBB) integrity and cerebral oedema development during the first 72 h of subarachnoid hemorrhage (SAH) (Cahill et al., 2006). Furthermore, recent study showed that angiopoietin-1 (Ang-1) inhibited the p53-mediated endoplasmic reticulum stress and apoptosis in the vascular endothelial cells in rats with SAH (Wei et al., 2022). These studies highlighted the roles of p53 in cerebral vasculature and its potential as a treatment target for SAH.
p53 and vascular remodeling in pulmonary hypertension
Pulmonary hypertension is a disease with high morbidity and mortality. After the sixth World Symposium on Pulmonary Hypertension in 2018, the diagnostic criteria of pulmonary hypertension was defined as mean pulmonary arterial pressure (mPAP) > 20 mmHg and pulmonary vascular resistance ≥3 Wood Units (Simonneau et al., 2019). Pulmonary hypertension is primarily associated with hemodynamic alterations resulting from remodeling of pulmonary arteries and veins, such as increased media/lumen ratio or increased media cross-sectional area. Pulmonary arterial smooth muscle cells and endothelial cells are the major players involved in vascular remodeling. Vascular remodeling is driven by four cellular processes: cell proliferation, cell apoptosis, cell migration, and synthesis or degradation of extracellular matrix that can be triggered by various growth factors, vasoactive substances and stimuli such as chronic hypoxia and DNA damage (Tuder, 2017).
Pulmonary arterial smooth muscle cells and endothelial cells are the major players involved in vascular remodeling. Vascular remodeling is driven by four cellular processes: cell proliferation, cell apoptosis, cell migration, and synthesis or degradation of extracellular matrix that can be triggered by various growth factors, vasoactive substances and stimuli such as chronic hypoxia and DNA damage (Tuder, 2017).
In the pathogenesis of pulmonary hypertension, conditions such as chronic hypoxia, activation of voltage-gated calcium channels, and extracellular calcium-sensing receptors contributed to vascular remodeling by promoting pulmonary arterial smooth muscle cell proliferation and retarding apoptosis of endothelial cells (Smith et al., 2016; Xiao et al., 2017; Shimoda, 2020). p53, a master regulator of cell cycle arrest, apoptosis, cell proliferation and DNA repair etc., was found to crosstalk with hypoxia-inducible transcription factors. Therefore, it is of interest to investigate whether p53 plays a role in cellular processes that drive vascular remodeling, particularly for pulmonary arterial smooth muscle cells and endothelial cells.
With the murine model of chronic hypoxia-induced pulmonary hypertension, it was found that pulmonary hypertension was promoted in p53 global knockout mice, with increased vascular remodeling, upregulated hypoxia-inducible factor 1 alpha (HIF-1α) expression and downregulated p21 expression in the pulmonary arterial smooth muscle cell (PASMC) (Mizuno et al., 2011). Mouraret et al. (2013) demonstrated the administration of Nutlin-3a to chronically hypoxic mice markedly increased p53 expression in lungs and partially reversed pulmonary hypertension and attenuated vascular remodeling. The partial reversal of pulmonary hypertension was not apparent in chronically hypoxic p53 global knockout mice and p21 global knockout mice, suggesting that p53 an p21 are required in Nutlin-3a-induced partial reversal of pulmonary hypertension (Mouraret et al., 2013). PASMC-specific gain or loss of p53 function does not affect hypoxia-induced pulmonary hypertension in mice, suggesting that the modulation of p53 signaling in other cells is required to bring about phenotypic change in the murine hypoxia-induced pulmonary hypertension (Wakasugi et al., 2019). A recent study by Teng et al. (2022) found that PASMC-targeted co-delivery of baicalein and p53 reversed pulmonary artery remodeling in monocrotaline-treated rats and promoted PASMC apoptosis via Bax/BCl2/Caspase 3 signaling pathway. However, PASMC-targeted delivery of p53 alone has no significant impact on vascular remodeling.
Recent studies provided experimental evidence delineating the complex functional role and regulation of p53 in pulmonary vessels exposed to hypoxia. Wang et al. (2018) observed that p53 expression is decreased in PASMC under hypoxia, while the opposite occurs for pulmonary arterial endothelial cells (PAEC). The proposed mechanism for this divergent regulation of p53 is that hypoxia increases the expression of HIF-1α in PASMC, while it increases the expression of hypoxia-inducible factor 2 alpha (HIF-2α) instead for PAEC. These two hypoxia-inducible factors affect p53 expression in opposite manners. While an increased expression of HIF-1α in PASMC downregulates p53 expression, an increased HIF-2α in PAEC instead upregulates p53 expression (Wang et al., 2018). Functionally, hypoxia-induced downregulation of p53 in PASMC enhanced PASMC proliferation by inactivating transient receptor potential channels 1 and 6; while the upregulation of p53 in PAEC under hypoxia promoted PAEC apoptosis. Interestingly, a more recent study found that upregulation of p53 together with Peroxisome Proliferator-Activated Receptor-gamma (PPAR-γ) in pulmonary arterial and microvascular endothelial cells can promote angiogenesis and regeneration of pulmonary microvessels (Hennigs et al., 2021).
Conclusion
Abundant evidence supports the involvement of p53 in regulating vascular function and pathogenesis of cardiovascular diseases, suggesting that p53 is of great potential as a drug target for cardiovascular diseases. However, in vivo studies with global or cell type-specific targeting of p53 produced differential effects on the pathogenesis of atherosclerosis and pulmonary hypertension in experimental models. Further in vitro studies also demonstrated diverse functional roles and regulatory mechanism of p53 in different vascular cells and macrophages. Therefore, further studies on cell-type specific function and regulation of p53 in various disease models are warranted, so that optimal p53-targetting therapy can be designed.
Author contributions
GH-HC, EC and CT-KK wrote the manuscript with input from all authors. GP-HL and SM-YL revised the manuscript. S-WS is in charge of overall direction. All authors read and approved the final manuscript.
Funding
S-WS is supported by The Hong Kong Polytechnic University—General Research Fund (Project No. P0036597) and Research Centre for Chinese Medicine Innovation, The Hong Kong Polytechnic University (Project No. P0041138).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Barton, M., and Yanagisawa, M. (2019). Endothelin: 30 years from discovery to therapy. Hypertension 74 (6), 1232–1265. doi:10.1161/HYPERTENSIONAHA.119.12105
Bauer, V., and Sotníková, R. (2010). Nitric oxide--the endothelium-derived relaxing factor and its role in endothelial functions. General Physiology Biophysics 29 (4), 319–340. doi:10.4149/gpb_2010_04_319
Beckerman, R., and Prives, C. (2010). Transcriptional regulation by p53. Cold Spring Harb. Perspect. Biol. 2 (8), a000935. doi:10.1101/cshperspect.a000935
Beneit, N., Fernández-García, C. E., Martín-Ventura, J. L., Perdomo, L., Escribano, Ó., Michel, J. B., et al. (2016). Expression of insulin receptor (IR) A and B isoforms, IGF-IR, and IR/IGF-IR hybrid receptors in vascular smooth muscle cells and their role in cell migration in atherosclerosis. Cardiovasc. Diabetol. 15, 161. doi:10.1186/s12933-016-0477-3
Bevilacqua, M. P., Nelson, R. M., Mannori, G., and Cecconi, O. (1994). Endothelial-leukocyte adhesion molecules in human disease. Annu. Rev. Med. 45, 361–378. doi:10.1146/annurev.med.45.1.361
Blankenberg, S., Barbaux, S., and Tiret, L. (2003). Adhesion molecules and atherosclerosis. Atherosclerosis 170 (2), 191–203. doi:10.1016/s0021-9150(03)00097-2
Brophy, M. L., Dong, Y., Wu, H., Rahman, H. N., Song, K., and Chen, H. (2017). Eating the dead to keep atherosclerosis at bay. Front. Cardiovasc. Med. 4, 2. doi:10.3389/fcvm.2017.00002
Brown, I., Diederich, L., Good, M. E., DeLalio, L. J., Murphy, S. A., Cortese-Krott, M. M., et al. (2018). Vascular smooth muscle remodeling in conductive and resistance arteries in hypertension. Arterioscler. Thromb. Vasc. Biol. 38 (9), 1969–1985. doi:10.1161/ATVBAHA.118.311229
Cahill, J., Calvert, J. W., Solaroglu, I., and Zhang, J. H. (2006). Vasospasm and p53-induced apoptosis in an experimental model of subarachnoid hemorrhage. Stroke 37 (7), 1868–1874. doi:10.1161/01.STR.0000226995.27230.96
Cao, X., Cai, Z., Liu, J., Zhao, Y., Wang, X., Li, X., et al. (2017). miRNA-504 inhibits p53-dependent vascular smooth muscle cell apoptosis and may prevent aneurysm formation. Mol. Med. Rep. 16 (3), 2570–2578. doi:10.3892/mmr.2017.6873
Celen, A. B., and Sahin, U. (2020). Sumoylation on its 25th anniversary: Mechanisms, pathology, and emerging concepts. FEBS J. 287 (15), 3110–3140. doi:10.1111/febs.15319
Chan, G. H., and Fiscus, R. R. (2004). Exaggerated production of nitric oxide (NO) and increases in inducible NO-synthase mRNA levels induced by the pro-inflammatory cytokine interleukin-1beta in vascular smooth muscle cells of elderly rats. Exp. Gerontol. 39 (3), 387–394. doi:10.1016/j.exger.2004.01.002
Chen, H. (2018). Role of thromboxane A2 signaling in endothelium-dependent contractions of arteries. Prostagl. Other Lipid Mediat. 134, 32–37. doi:10.1016/j.prostaglandins.2017.11.004
Cho, H. J., Xie, Q. W., Calaycay, J., Mumford, R. A., Swiderek, K. M., Lee, T. D., et al. (1992). Calmodulin is a subunit of nitric oxide synthase from macrophages. J. Exp. Med. 176 (2), 599–604. doi:10.1084/jem.176.2.599
Dehnavi, S., Sadeghi, M., Penson, P. E., Banach, M., Jamialahmadi, T., and Sahebkar, A. (2019). The role of protein SUMOylation in the pathogenesis of atherosclerosis. J. Clin. Med. 8 (11), 1856. doi:10.3390/jcm8111856
Depre, C., Havaux, X., Renkin, J., Vanoverschelde, J. L., and Wijns, W. (1999). Expression of inducible nitric oxide synthase in human coronary atherosclerotic plaque. Cardiovasc. Res. 41 (2), 465–472. doi:10.1016/s0008-6363(98)00304-6
Dimmeler, S., Haendeler, J., Nehls, M., and Zeiher, A. M. (1997). Suppression of apoptosis by nitric oxide via inhibition of interleukin-1beta-converting enzyme (ICE)-like and cysteine protease protein (CPP)-32-like proteases. J. Exp. Med. 185 (4), 601–607. doi:10.1084/jem.185.4.601
Donato, A. J., Morgan, R. G., Walker, A. E., and Lesniewski, L. A. (2015). Cellular and molecular biology of aging endothelial cells. J. Mol. Cell. Cardiol. 89, 122–135. doi:10.1016/j.yjmcc.2015.01.021
Farhang Ghahremani, M., Goossens, S., and Haigh, J. J. (2013a). The p53 family and VEGF regulation: "it's complicated. Cell Cycle 12 (9), 1331–1332. doi:10.4161/cc.24579
Farhang Ghahremani, M., Goossens, S., Nittner, D., Bisteau, X., Bartunkova, S., Zwolinska, A., et al. (2013b). p53 promotes VEGF expression and angiogenesis in the absence of an intact p21-Rb pathway. Cell Death Differ. 20 (7), 888–897. doi:10.1038/cdd.2013.12
Félétou, M., and Vanhoutte, P. M. (1996). Endothelium-derived hyperpolarizing factor. Clin. Exp. Pharmacol. Physiol. 23 (12), 1082–1090. doi:10.1111/j.1440-1681.1996.tb01174.x
Fleming, I., and Busse, R. (2003). Molecular mechanisms involved in the regulation of the endothelial nitric oxide synthase. Am. J. Physiol. Regul. Integr. Comp. Physiol. 284 (1), R1–R12. doi:10.1152/ajpregu.00323.2002
Förstermann, U., Xia, N., and Li, H. (2017). Roles of vascular oxidative stress and nitric oxide in the pathogenesis of atherosclerosis. Circ. Res. 120 (4), 713–735. doi:10.1161/CIRCRESAHA.116.309326
Fuchs, F. D., and Whelton, P. K. (2020). High blood pressure and cardiovascular disease. Hypertension 75 (2), 285–292. doi:10.1161/HYPERTENSIONAHA.119.14240
Furchgott, R. F., and Vanhoutte, P. M. (1989). Endothelium-derived relaxing and contracting factors. FASEB J. 3 (9), 2007–2018. doi:10.1096/fasebj.3.9.2545495
Gerthoffer, W. T. (2007). Mechanisms of vascular smooth muscle cell migration. Circ. Res. 100 (5), 607–621. doi:10.1161/01.RES.0000258492.96097.47
Gorgoulis, V. G., Pratsinis, H., Zacharatos, P., Demoliou, C., Sigala, F., Asimacopoulos, P. J., et al. (2005). p53-dependent ICAM-1 overexpression in senescent human cells identified in atherosclerotic lesions. Lab. Investig. 85 (4), 502–511. doi:10.1038/labinvest.3700241
Guevara, N. V., Kim, H. S., Antonova, E. I., and Chan, L. (1999). The absence of p53 accelerates atherosclerosis by increasing cell proliferation in vivo. Nat. Med. 5 (3), 335–339. doi:10.1038/6585
Hansson, G. K., and Hermansson, A. (2011). The immune system in atherosclerosis. Nat. Immunol. 12 (3), 204–212. doi:10.1038/ni.2001
Hennigs, J. K., Cao, A., Li, C. G., Shi, M., Mienert, J., Miyagawa, K., et al. (2021). PPARγ-p53-mediated vasculoregenerative program to reverse pulmonary hypertension. Circ. Res. 128 (3), 401–418. doi:10.1161/CIRCRESAHA.119.316339
Heo, K. S., Chang, E., Le, N. T., Cushman, H., Yeh, E. T., Fujiwara, K., et al. (2013). De-SUMOylation enzyme of sentrin/SUMO-specific protease 2 regulates disturbed flow-induced SUMOylation of ERK5 and p53 that leads to endothelial dysfunction and atherosclerosis. Circ. Res. 112 (6), 911–923. doi:10.1161/CIRCRESAHA.111.300179
Heo, K. S., Lee, H., Nigro, P., Thomas, T., Le, N. T., Chang, E., et al. (2011). PKCζ mediates disturbed flow-induced endothelial apoptosis via p53 SUMOylation. J. Cell. Biol. 193 (5), 867–884. doi:10.1083/jcb.201010051
Hohensinner, P. J., Kaun, C., Buchberger, E., Ebenbauer, B., Demyanets, S., Huk, I., et al. (2016). Age intrinsic loss of telomere protection via TRF1 reduction in endothelial cells. Biochim. Biophys. Acta 1863 (2), 360–367. doi:10.1016/j.bbamcr.2015.11.034
Huang, H., Koelle, P., Fendler, M., Schröttle, A., Czihal, M., Hoffmann, U., et al. (2014). Induction of inducible nitric oxide synthase (iNOS) expression by oxLDL inhibits macrophage derived foam cell migration. Atherosclerosis 235 (1), 213–222. doi:10.1016/j.atherosclerosis.2014.04.020
Iacopetta, B., Wysocki, S., Norman, P., and House, A. (1995). The p53 tumor-suppressor gene is overexpressed but not mutated in human atherosclerotic tissue. Int. J. Oncol. 7 (2), 399–402. doi:10.3892/ijo.7.2.399
Insull, W. (2009). The pathology of atherosclerosis: Plaque development and plaque responses to medical treatment. Am. J. Med. 122 (1), S3–S14. doi:10.1016/j.amjmed.2008.10.013
Jerafi-Vider, A., Bassi, I., Moshe, N., Tevet, Y., Hen, G., Splittstoesser, D., et al. (2021). VEGFC/FLT4-induced cell-cycle arrest mediates sprouting and differentiation of venous and lymphatic endothelial cells. Cell. Rep. 35 (11), 109255. doi:10.1016/j.celrep.2021.109255
Jia, G., Aroor, A. R., Jia, C., and Sowers, J. R. (2019). Endothelial cell senescence in aging-related vascular dysfunction. Biochim. Biophys. Acta. Mol. Basis Dis. 1865 (7), 1802–1809. doi:10.1016/j.bbadis.2018.08.008
Kavurma, M. M., Figg, N., Bennett, M. R., Mercer, J., Khachigian, L. M., and Littlewood, T. D. (2007). Oxidative stress regulates IGF1R expression in vascular smooth-muscle cells via p53 and HDAC recruitment. Biochem. J. 407 (1), 79–87. doi:10.1042/BJ20070380
Khan, B. V., Parthasarathy, S. S., Alexander, R. W., and Medford, R. M. (1995). Modified low density lipoprotein and its constituents augment cytokine-activated vascular cell adhesion molecule-1 gene expression in human vascular endothelial cells. J. Clin. Investig. 95 (3), 1262–1270. doi:10.1172/JCI117776
Kim, C. S., Jung, S. B., Naqvi, A., Hoffman, T. A., DeRicco, J., Yamamori, T., et al. (2008). p53 impairs endothelium-dependent vasomotor function through transcriptional upregulation of p66shc. Circ. Res. 103 (12), 1441–1450. doi:10.1161/CIRCRESAHA.108.181644
Kim, E. J., Kho, J. H., Kang, M. R., and Um, S. J. (2007). Active regulator of SIRT1 cooperates with SIRT1 and facilitates suppression of p53 activity. Mol. Cell. 28 (2), 277–290. doi:10.1016/j.molcel.2007.08.030
Klabunde, R. E. (2022). Cardiovascular physiology concepts. Philadelphia: Lippincott Williams and Wilkins.
Kumar, A., Kim, C. S., Hoffman, T. A., Naqvi, A., DeRicco, J., Jung, S. B., et al. (2011). p53 impairs endothelial function by transcriptionally repressing kruppel-like factor 2. Arterioscler. Thromb. Vasc. Biol. 31 (1), 133–141. doi:10.1161/ATVBAHA.110.215061
Lin, K., Hsu, P. P., Chen, B. P., Yuan, S., Usami, S., Shyy, J. Y. J., et al. (2000). Molecular mechanism of endothelial growth arrest by laminar shear stress. Proc. Natl. Acad. Sci. U. S. A. 97 (17), 9385–9389. doi:10.1073/pnas.170282597
López-Otín, C., Blasco, M. A., Partridge, L., Serrano, M., and Kroemer, G. (2013). The hallmarks of aging. Cell. 153 (6), 1194–1217. doi:10.1016/j.cell.2013.05.039
Martínez, M. C., and Andriantsitohaina, R. (2009). Reactive nitrogen species: Molecular mechanisms and potential significance in health and disease. Antioxid. Redox Signal. 11 (3), 669–702. doi:10.1089/ars.2007.1993
Meek, D. W., and Anderson, C. W. (2009). Posttranslational modification of p53: Cooperative integrators of function. Cold Spring Harb. Perspect. Biol. 1 (6), a000950. doi:10.1101/cshperspect.a000950
Men, H., Cai, H., Cheng, Q., Zhou, W., Wang, X., Huang, S., et al. (2021). The regulatory roles of p53 in cardiovascular health and disease. Cell. Mol. Life Sci. 78 (5), 2001–2018. doi:10.1007/s00018-020-03694-6
Mercer, J., Figg, N., Stoneman, V., Braganza, D., and Bennett, M. R. (2005). Endogenous p53 protects vascular smooth muscle cells from apoptosis and reduces atherosclerosis in ApoE knockout mice. Circ. Res. 96 (6), 667–674. doi:10.1161/01.RES.0000161069.15577.ca
Merched, A. J., Williams, E., and Chan, L. (2003). Macrophage-specific p53 expression plays a crucial role in atherosclerosis development and plaque remodeling. Arterioscler. Thromb. Vasc. Biol. 23 (9), 1608–1614. doi:10.1161/01.ATV.0000084825.88022.53
Miller, Y. I., Viriyakosol, S., Binder, C. J., Feramisco, J. R., Kirkland, T. N., and Witztum, J. L. (2003). Minimally modified LDL binds to CD14, induces macrophage spreading via TLR4/MD-2, and inhibits phagocytosis of apoptotic cells. J. Biol. Chem. 278 (3), 1561–1568. doi:10.1074/jbc.M209634200
Mizuno, S., Bogaard, H. J., Kraskauskas, D., Alhussaini, A., Gomez-Arroyo, J., Voelkel, N. F., et al. (2011). p53 Gene deficiency promotes hypoxia-induced pulmonary hypertension and vascular remodeling in mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 300 (5), L753–L761. doi:10.1152/ajplung.00286.2010
Mouraret, N., Marcos, E., Abid, S., Gary-Bobo, G., Saker, M., Houssaini, A., et al. (2013). Activation of lung p53 by nutlin-3a prevents and reverses experimental pulmonary hypertension. Circulation 127 (16), 1664–1676. doi:10.1161/CIRCULATIONAHA.113.002434
Negre-Salvayre, A., Guerby, P., Gayral, S., Laffargue, M., and Salvayre, R. (2020). Role of reactive oxygen species in atherosclerosis: Lessons from murine genetic models. Free Radic. Biol. Med. 149, 8–22. doi:10.1016/j.freeradbiomed.2019.10.011
O'Reilly, F. M., Otto, K. B., Sexton, S. A., Swerlick, R. A., and Casper, K. A. (2003). Regulation of tissue factor in microvascular dermal endothelial cells. J. Investig. Dermatol. 120 (3), 489–494. doi:10.1046/j.1523-1747.2003.12063.x
Olsson, A. K., Dimberg, A., Kreuger, J., and Claesson-Welsh, L. (2006). VEGF receptor signalling - in control of vascular function. Nat. Rev. Mol. Cell. Biol. 7 (5), 359–371. doi:10.1038/nrm1911
Otrock, Z. K., Mahfouz, R. A., Makarem, J. A., and Shamseddine, A. I. (2007). Understanding the biology of angiogenesis: Review of the most important molecular mechanisms. Blood Cells Mol. Dis. 39 (2), 212–220. doi:10.1016/j.bcmd.2007.04.001
Polte, T., Oberle, S., and Schröder, H. (1997). Nitric oxide protects endothelial cells from tumor necrosis factor-α-mediated cytotoxicity: Possible involvement of cyclic GMP. FEBS Lett. 409 (1), 46–48. doi:10.1016/s0014-5793(97)00480-8
Renna, N. F., Heras, N., and Miatello, R. M. (2013). Pathophysiology of vascular remodeling in hypertension. Int. J. Hypertens. 2013, 808353–808357. doi:10.1155/2013/808353
Robbins, C. S., Hilgendorf, I., Weber, G. F., Theurl, I., Iwamoto, Y., Figueiredo, J. L., et al. (2013). Local proliferation dominates lesional macrophage accumulation in atherosclerosis. Nat. Med. 19 (9), 1166–1172. doi:10.1038/nm.3258
Ross, R. (1999). Atherosclerosis—An inflammatory disease. N. Engl. J. Med. 340 (2), 115–126. doi:10.1056/NEJM199901143400207
Santiago, A., Li, D., Zhao, L. Y., Godsey, A., and Liao, D. (2013). p53 SUMOylation promotes its nuclear export by facilitating its release from the nuclear export receptor CRM1. Mol. Biol. Cell. 24 (17), 2739–2752. doi:10.1091/mbc.E12-10-0771
Sawamura, T., Kume, N., Aoyama, T., Moriwaki, H., Hoshikawa, H., Aiba, Y., et al. (1997). An endothelial receptor for oxidized low-density lipoprotein. Nature 386 (6620), 73–77. doi:10.1038/386073a0
Schwenke, D. C., and Carew, T. E. (1989). Initiation of atherosclerotic lesions in cholesterol-fed rabbits. II. Selective retention of LDL vs. selective increases in LDL permeability in susceptible sites of arteries. Arteriosclerosis 9 (6), 908–918. doi:10.1161/01.atv.9.6.908
Shi, Z. W., Ge, L. S., and Li, Y. C. (2018). The role of necroptosis in cardiovascular disease. Front. Pharmacol. 9, 721. doi:10.3389/fphar.2018.00721
Shimoda, L. A. (2020). Cellular pathways promoting pulmonary vascular remodeling by hypoxia. Physiology 35 (4), 222–233. doi:10.1152/physiol.00039.2019
Simonneau, G., Montani, D., Celermajer, D. S., Denton, C. P., Gatzoulis, M. A., Krowka, M., et al. (2019). Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur. Respir. J. 53 (1), 1801913. doi:10.1183/13993003.01913-2018
Smith, K. A., Ayon, R. J., Tang, H., Makino, A., and Yuan, J. X. (2016). Calcium-sensing receptor regulates cytosolic [Ca2+] and plays a major role in the development of pulmonary hypertension. Front. Physiol. 7, 517. doi:10.3389/fphys.2016.00517
Stary, H. C., Chandler, A. B., Glagov, S., Guyton, J. R., Insull, W., Rosenfeld, M. E., et al. (1994). A definition of initial, fatty streak, and intermediate lesions of atherosclerosis. A report from the Committee on Vascular Lesions of the Council on Arteriosclerosis, American Heart Association. Circulation 89 (5), 2462–2478. doi:10.1161/01.cir.89.5.2462
Stocker, R., and Keaney, J. F. (2004). Role of oxidative modifications in atherosclerosis. Physiol. Rev. 84 (4), 1381–1478. doi:10.1152/physrev.00047.2003
Sun, X., and Feinberg, M. W. (2021). Vascular endothelial senescence: Pathobiological insights, emerging long noncoding RNA targets, challenges and therapeutic opportunities. Front. Physiol. 12, 693067. doi:10.3389/fphys.2021.693067
Tabas, I., and Bornfeldt, K. E. (2016). Macrophage phenotype and function in different stages of atherosclerosis. Circ. Res. 118 (4), 653–667. doi:10.1161/CIRCRESAHA.115.306256
Takabe, W., Alberts-Grill, N., and Jo, H. (2011). Disturbed flow: p53 SUMOylation in the turnover of endothelial cells. J. Cell. Biol. 193 (5), 805–807. doi:10.1083/jcb.201104140
Tang, Y., Zhao, W., Chen, Y., Zhao, Y., and Gu, W. (2008). Acetylation is indispensable for p53 activation. Cell. 133 (4), 612–626. doi:10.1016/j.cell.2008.03.025
Tarantini, S., Tran, C., Gordon, G. R., Ungvari, Z., and Csiszar, A. (2017). Impaired neurovascular coupling in aging and alzheimer's disease: Contribution of astrocyte dysfunction and endothelial impairment to cognitive decline. Exp. Gerontol. 94, 52–58. doi:10.1016/j.exger.2016.11.004
Teng, C., Li, B., Lin, C., Xing, X., Huang, F., Yang, Y., et al. (2022). Targeted delivery of baicalein-p53 complex to smooth muscle cells reverses pulmonary hypertension. J. Control. Release 341, 591–604. doi:10.1016/j.jconrel.2021.12.006
Thai, T., Zhong, F., Dang, L., Chan, E., Ku, J., Malle, E., et al. (2021). Endothelial-transcytosed myeloperoxidase activates endothelial nitric oxide synthase via a phospholipase C-dependent calcium signaling pathway. Free Radic. Biol. Med. 166, 255–264. doi:10.1016/j.freeradbiomed.2020.12.448
Thomas, S. R., Witting, P. K., and Drummond, G. R. (2008). Redox control of endothelial function and dysfunction: Molecular mechanisms and therapeutic opportunities. Antioxid. Redox Signal. 10 (10), 1713–1765. doi:10.1089/ars.2008.2027
Thorp, E., Vaisar, T., Subramanian, M., Mautner, L., Blobel, C., and Tabas, I. (2011). Shedding of the Mer tyrosine kinase receptor is mediated by ADAM17 protein through a pathway involving reactive oxygen species, protein kinase Cδ, and p38 mitogen-activated protein kinase (MAPK). J. Biol. Chem. 286 (38), 33335–33344. doi:10.1074/jbc.M111.263020
Toda, N., Ayajiki, K., and Okamura, T. (2007). Interaction of endothelial nitric oxide and angiotensin in the circulation. Pharmacol. Rev. 59 (1), 54–87. doi:10.1124/pr.59.1.2
Tsihlis, N. D., Oustwani, C. S., Vavra, A. K., Jiang, Q., Keefer, L. K., and Kibbe, M. R. (2011). Nitric oxide inhibits vascular smooth muscle cell proliferation and neointimal hyperplasia by increasing the ubiquitination and degradation of UbcH10. Cell. biochem. Biophys. 60, 89–97. doi:10.1007/s12013-011-9179-3
Tuder, R. M. (2017). Pulmonary vascular remodeling in pulmonary hypertension. Cell Tissue Res. 367 (3), 643–649. doi:10.1007/s00441-016-2539-y
van Vlijmen, B. J., Gerritsen, G., Franken, A. L., Boesten, L. S., Kockx, M. M., Gijbels, M. J., et al. (2001). Macrophage p53 deficiency leads to enhanced atherosclerosis in APOE* 3-Leiden transgenic mice. Circ. Res. 88 (8), 780–786. doi:10.1161/hh0801.089261
Vaziri, H., Dessain, S. K., Ng, E. E., Imai, S. I., Frye, R. A., Pandita, T. K., et al. (2001). hSIR2SIRT1 functions as an NAD-dependent p53 deacetylase. Cell. 107 (2), 149–159. doi:10.1016/s0092-8674(01)00527-x
von der Thüsen, J. H., van Vlijmen, B. J., Hoeben, R. C., Kockx, M. M., Havekes, L. M., van Berkel, T. J., et al. (2002). Induction of atherosclerotic plaque rupture in apolipoprotein E-/- mice after adenovirus-mediated transfer of p53. Circulation 105 (17), 2064–2070. doi:10.1161/01.cir.0000015502.97828.93
von Eckardstein, A., and Rohrer, L. (2009). Transendothelial lipoprotein transport and regulation of endothelial permeability and integrity by lipoproteins. Curr. Opin. Lipidol. 20 (3), 197–205. doi:10.1097/MOL.0b013e32832afd63
Vousden, K. H., and Prives, C. (2009). Blinded by the light: The growing complexity of p53. Cell. 137 (3), 413–431. doi:10.1016/j.cell.2009.04.037
Wakasugi, T., Shimizu, I., Yoshida, Y., Hayashi, Y., Ikegami, R., Suda, M., et al. (2019). Role of smooth muscle cell p53 in pulmonary arterial hypertension. PLOS ONE 14 (2), e0212889. doi:10.1371/journal.pone.0212889
Wang, Z., Yang, K., Zheng, Q., Zhang, C., Tang, H., Babicheva, A., et al. (2018). Divergent changes of p53 in pulmonary arterial endothelial and smooth muscle cells involved in the development of pulmonary hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 316 (1), L216-L228–L228. doi:10.1152/ajplung.00538.2017
Wei, P., Han, Y., Chen, H., Luo, L., Liu, G., Lin, B., et al. . Ang-1 inhibited endoplasmic reticulum stress and apoptosis of VECs in rats with aSAH induced CVS through regulated PI3K/Akt pathway. Curr. Neurovasc. Res. 19, 2022. in press. doi:10.2174/1567202619666220412082145
World Health Organization, (2021). Cardiovascular diseases (CVDs). https://www.who.int/news-room/fact-sheets/detail/cardiovascular-diseases-(cvds (Accessed July 31, 2022).
Wu, H., Wu, J., Zhou, S., Huang, W., Li, Y., Zhang, H., et al. (2018). SRT2104 attenuates diabetes-induced aortic endothelial dysfunction via inhibition of p53. J. Endocrinol. 237 (1), 1–14. doi:10.1530/JOE-17-0672
Wu, J., Liang, W., Tian, Y., Ma, F., Huang, W., Jia, Y., et al. (2019). Inhibition of p53/miR-34a improves diabetic endothelial dysfunction via activation of SIRT1. J. Cell. Mol. Med. 23 (5), 3538–3548. doi:10.1111/jcmm.14253
Xiao, R., Su, Y., Feng, T., Sun, M., Liu, B., Zhang, J., et al. (2017). Monocrotaline induces endothelial injury and pulmonary hypertension by targeting the extracellular calcium-sensing receptor. J. Am. Heart Assoc. 6 (4), e004865. doi:10.1161/JAHA.116.004865
Keywords: atherosclerosis, p53, vascular smooth muscle cell, endothelial dysfunction, vascular smooth muscle proliferation, vascular smooth muscle migration
Citation: Chan GH-H, Chan E, Kwok CT-K, Leung GP-H, Lee SM-Y and Seto S-W (2022) The role of p53 in the alternation of vascular functions. Front. Pharmacol. 13:981152. doi: 10.3389/fphar.2022.981152
Received: 29 June 2022; Accepted: 17 August 2022;
Published: 06 September 2022.
Edited by:
Shailendra Pratap Singh, Central University of Rajasthan, IndiaReviewed by:
Ganapasam Sudhandiran, University of Madras, IndiaZhuoming Li, Sun Yat-sen University, China
Copyright © 2022 Chan, Chan, Kwok, Leung, Lee and Seto. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sai-Wang Seto, saiwang.seto@polyu.edu.hk
†These authors have contributed equally to this work