 Sheikh Rayees
Sheikh Rayees Jagdish Chandra Joshi
Jagdish Chandra Joshi Bhagwati Joshi
Bhagwati Joshi Vigneshwaran VellingiriSomenath Banerjee
Vigneshwaran VellingiriSomenath Banerjee Dolly Mehta*
Dolly Mehta*- Department of Pharmacology and Regenerative Medicine, Centre for Lung and Vascular Biology, College of Medicine, University of Illinois, Chicago, IL, United States
Efficient phagocytosis of pathogens by the innate immune system during infectious injury is vital for restoring tissue integrity. Impaired phagocytosis, such as in the case of infection with Pseudomonas aeruginosa, a broad-spectrum antibiotic-resistant Gram-negative bacterium, can lead to a life threatening lung disorder, acute lung injury (ALI). Evidence indicates that loss of protease-activated receptor 2 (PAR2) impaired Pseudomonas aeruginosa clearance leading to non-resolvable ALI, but the mechanism remains unclear. Here, we focused on the alveolar macrophages (AMs), the predominant population of lung-resident macrophages involved in sensing bacteria, to understand their role in PAR2-mediated phagocytosis of Pseudomonas aeruginosa. We found that upon binding Pseudomonas aeruginosa, PAR2-expressing but not PAR2-null AMs had increased cAMP levels, which activated Rac1 through protein kinase A. Activated Rac1 increased actin-rich protrusions to augment the phagocytosis of Pseudomonas aeruginosa. Administration of liposomes containing constitutively active Rac1 into PAR2-null mice lungs rescued phagocytosis and enhanced the survival of PAR2-null mice from pneumonia. These studies showed that PAR2 drives the cAMP-Rac1 signaling cascade that activates Pseudomonas aeruginosa phagocytosis in AMs, thereby preventing death from bacterial pneumonia.
Introduction
Macrophages are the professional phagocytes of the innate immune system involved in host defense (Freeman and Grinstein, 2016). Phagocytosis of apoptotic cells and pathogens by the macrophages is essential for restoring the integrity of infectious tissue (Freeman and Grinstein, 2016; Han et al., 2016). Phagocytosis is a multistep process that involves the sensing of pathogens followed by their binding to the phagocytic cells for subsequent internalization (Levin et al., 2016).
The tissue-resident alveolar macrophages (AMs) are unique because they directly communicate with the environment and defend the lungs against broad-spectrum and antibiotic-resistant species of Gram-negative bacteria, including Pseudomonas aeruginosa (PA, hence forth) (Restrepo et al., 1999; Moraes et al., 2008; Nagre et al., 2019). PA, being an extracellular pathogen, prefers to associate with the surface of the host cell membrane (Lillehoj et al., 2001; Siryaporn et al., 2014). Mostly, opsonic receptors phagocytized PA, but AMs express these receptors at relatively low levels (Stokes et al., 1998; Lovewell et al., 2014). Hence, PA infections can lead to acute lung injury, a life-threatening respiratory disorder characterized by severe alveolar–capillary barrier leakage and inflammation. Also, PA can cause lethal infections in cystic fibrosis patients (Malhotra et al., 2019). Therefore, understanding mechanisms promoting PA clearance by AMs will potentially identify targets for suppressing diseases post infection.
In the present study, we focused on the protease-activated receptor 2 (PAR2) expressed on AMs as a possible mechanism inducing phagocytosis of PA. PAR2 is a G protein-coupled receptor belonging to the protease-activated receptor (PAR) family (Rayees et al., 2019). Proteases released during injury, such as thrombin, activate PAR2 (Zhao et al., 2015; Rayees et al., 2019). Interestingly, the proteases secreted by PA inactivate PAR2 (Dulon et al., 2005; Moraes et al., 2008). Mice lacking PAR2 showed severe lung inflammation and reduced PA clearance (Moraes et al., 2008). These authors also showed that PA infection increased neutrophil transmigration in PAR2-null lungs (Moraes et al., 2008). Because neutrophils require AM functions to clear PA (Mishra et al., 2012; Thanabalasuriar et al., 2017), it is unclear how the loss of PAR2 in AMs signals impaired phagocytosis of PA.
Here, we showed that upon binding PA, PAR2+ AMs increases cAMP, which in turn activates the small GTPase Rac1. Activated Rac1 forms actin-rich protrusions to phagocytize PA. Thus, AMs lacking PAR2 could not efficiently clear PA, and as a result, PAR2-null mice showed significant mortality after pneumonia. Liposomal delivery of constitutively active Rac1 into PAR2-null mice rescued phagocytosis and prevented their mortality. The studies raise the possibility of targeting the PAR2-Rac1 cascade as a tool to promote PA phagocytosis, thereby increasing bacterial clearance and hastening the resolution of pneumonia.
Material and methods
Experimental animals
All animal experiments were approved by the Institutional Animal Care and Use Committee of the University of Illinois at Chicago. C57BL/6J (WT, henceforth) and PAR2-null mice breeding pairs (Jackson Laboratory, CT, United States) were bred at the University of Illinois at Chicago. The mice colonies were maintained in a pathogen-free housing facility at the University of Illinois. For the experiments, 6–8-week-old male and female mice were used. Data represent pooled data from both female and male mice as we did not observe any effect of gender on lung injury. The wild-type strain of Pseudomonas aeruginosa (PAO1) constitutively expressing GFP (GFP-PAO1) was used in the study. The expression of GFP was plasmid-based as described previously (Hashimoto et al., 2007). The pharmacological agents NSC23766, Y-27632, and 8-bromoadenosine 3′-, 5′-cyclic monophosphate (8-Br-cAMP) were dissolved in water, whereas H-89 and ESI-09 were dissolved in DMSO.
Pseudomonas aeruginosa culture and count
GFP-tagged PA (GFP-PAO1 strain) was streaked in ampicillin-selective (HiMedia Laboratories LLC, United States) Luria–Bertani (LB) agar plates and incubated overnight at 37°C from the glycerol-preserved vial for the activation of culture. Single colonies were picked and inoculated overnight (∼17 h) in 250 ml LB broth (Amp 100 μg/ml) at 37°C. For the standard plate count method, 1 ml of the bacterial culture from the broth was serially diluted with sterile phosphate buffer up to 1 × 1010 dilutions. Each dilution plate was spread-plated in ampicillin-selective (100 μg/ml) sheep blood agar plates to get the countable bacterial colonies. The 1 × 106 CFU/25uL was calculated using the following formula: CFU/ml = (no. of colonies x dilution factor)/volume of the culture plate.
Pseudomonas aeruginosa-induced acute lung injury
Acute lung injury was induced in the WT and PAR2-null mice post PA infection and determined as described previously (Joshi et al., 2020). The mice were anesthetized by intraperitoneal injections of ketamine (100 mg/kg) and xylazine (12 mg/kg). PA (1 × 106 CFU in 50 ul PBS/mouse) was instilled into the lungs through the endotracheal route. Edema was measured in these mice at indicated times by measuring the lung wet–dry ratio. Briefly, the left lobe of the lungs was excised following PA instillation, weighed, and then dried at 55°C. The dry weight was taken after 24 h, and the wet–dry ratio was calculated, which gave a measure of edema, as described previously (Yazbeck et al., 2017).
Bronchoalveolar lavage
Bronchoalveolar lavage (BAL) and BAL-AMs were isolated as described previously (Rayees et al., 2019; Joshi et al., 2020). Briefly, after sacrificing the mice, tracheotomy was performed followed by the collection of BAL using an 18-gauge blunt needle. The BAL fluid was poured on plastic dishes, supplied with 10% serum, and the AMs allowed to adhere for 60 min. The non-adherent cells were removed by washing with PBS.
Phagocytosis assays
Phagocytosis assays in bone marrow-derived macrophages (BMDMs) or AMs were performed as described previously, with modifications (Ojielo et al., 2003; Descamps et al., 2012; Bastaert et al., 2018; Nagre et al., 2019). BMDMs were isolated as described previously (Rayees et al., 2019) and exposed to PA at MOI (multiplicity of infection) 1:10 in an antibiotic-free media containing 1% FBS for indicated times. The experiment was terminated by washing out PA with PBS. For FACS analysis, for the measurement of gene expression or immunoblotting, the cells were detached from the surface using accutase and processed further as described. For immunofluorescence, the macrophages were fixed in 2% paraformaldehyde for 15 min and stained with phalloidin (1:100) for 1 h and imaged. For AM phagocytosis, BAL-AMs were exposed to PA (MOI, 1:10) for 60 min. The AMs were fixed in 2% paraformaldehyde for 15 min after which cells were permeabilized using 0.1% triton X and stained with phalloidin (1:100) for 1 h in dark. The AMs and BMDMs were imaged on an LSM880 confocal microscope using 63x oil objective. F-actin organization in the form of pseudopods was quantified as the ratio of pseudopods/actin projections toward PA.
For measuring Rac1 activity in AMs by immunofluorescence, we quantified pseudopods, well-known indices of Rac1 activity (Sit and Manser, 2011; Flannagan et al., 2012), by measuring the spatial distribution of Rac1/F-actin fluorescent intensity peaks near cell edges. Briefly, the individual cells with protruding edges expressing fluorescent-labeled Rac1/F-actin were segmented, and the fluorescence intensity was measured using ImageJ.
Bacterial internalization assay
WT and PAR2-null BMDMs were exposed to PA for 45 min. The supernatant was centrifuged and dissolved in 50 μL sterile PBS. The BMDM monolayer was washed twice with PBS and incubated with gentamicin-containing (300 μg/ml) media for 1 h to kill non-internalized bacteria (Plotkowski et al., 1999). Following a PBS wash, the cell pellet was then lysed in 0.1% Triton X-100 (Sigma-Aldrich, United States). The dissolved supernatant and the cell lysate were spread on the blood agar plate, and PA-CFU was counted 24 h later (Almeida et al., 1999; Cavinato et al., 2020).
FACS analysis
The BMDMs exposed to PA for indicated time points were washed twice with PBS. The cells were detached from the plates using accutase, centrifuged, and fixed with fixation buffer (1:1 with PBS) (Invitrogen, United States) for 20 min. The cell suspension was washed twice with FACS buffer (0.5% BSA in PBS). All samples were then run on the BD LSR-Fortessa flow cytometer (BD Biosciences, CA, United States). The data were processed using Flow Jo software (TreeStar, Inc., CA, United States).
Rac1 activity and immunoblotting
The BMDMs were exposed to PA at MOI, 1: 10 for indicated times. Cells were lysed using RIPA buffer (Sigma-Aldrich, United States) containing 1% protease inhibitor cocktail. Lysates (120 μg protein) were incubated with PAK-GST protein beads (20 μg) (Cytoskeleton, Inc., United States) for 4 h at 4°C on gentle rotation. The samples were centrifuged, and the pellet containing the beads was washed carefully and dissolved in Laemmli buffer. The total and active Rac1 were assessed by immunoblotting, using the anti-Rac1 monoclonal antibody.
Transfection
The BMDMs were transfected as described previously (Rayees et al., 2019), using the Amaxa Nucleofactor electroporation system (Lonza). Briefly, the cells were detached from the dishes using accutase and spun down. The cell pellet was suspended in transfection reagent containing cDNA. The cells were slowly mixed with the transfection reagent to apply cDNA to the cells and electroporated. Vector plasmids were used to transfect experimental controls. The electroporated cells were processed for further experiments 48 h after transfection.
Measurement of gene expression
The RNA was extracted from AMs and BMDMs using TRIzol (Invitrogen Inc., CA, United States). The quality and integrity of RNA were verified on the NanoDrop spectrometer (Thermo Fisher Scientific, MA, United States). The cDNA was synthesized as per manufacturer’s protocol (High-Capacity cDNA Reverse Transcription Kit, Applied Biosystems), using equal quantities of RNA. Real-time q-PCR was performed for the measurement of gene expression (Applied Biosystems, CA, United States) (Rayees et al., 2015). The primer sequences used in the study were taken from our previously published study (Rayees et al., 2019).
Liposome mediated cDNA delivery to the mouse lungs
Cationic liposomes were made using a mixture of chloroform, cholesterol, and dimethyl dioctadecyl ammonium bromide, as described previously (Yazbeck et al., 2017; Rayees et al., 2019). The liposomes were filtered through a 0.45-micron filter, and the cDNA was gently added and mixed. The cDNA-loaded liposomes were injected into the mouse lungs through the endotracheal route, followed by PA administration after 24 h. The lungs were either collected for the measurement of the wet–dry ratio or BAL was performed for the bacterial colony count, immunofluorescence, or measurement of gene expression from the extracted AMs.
Statistical analysis
All values are given as mean ± SD. Statistical analysis was performed using Graph Pad Prism version 7.0 (Graph Pad Software, La Jolla, CA). Multiple groups were compared using One-way ANOVA followed by a paired Student’s t test to assess significance between two groups. The statistics details can be found in the individual figure legends.
Results
Macrophage PAR2 enables phagocytosis of PA
We exposed bone-marrow-derived Mɸ (macrophage) isolated from the WT and PAR2-null mice to the GFP-tagged PA (MOI 1:10) to determine the role of PAR2 in regulating phagocytosis of PA by flow cytometry. We found that the mean phagocytic index of WT-Mɸ was 47.2% at 15 min, which increased further to 60.4% at 45 min. In contrast, the mean phagocytic index of PAR2-deficient macrophages was only 11% and 26.5% at 15 and 45 min, respectively (Figures 1A,B). Because phagocytosis requires actin remodeling (Flannagan et al., 2012; Levin et al., 2016), we labeled F-actin using rhodamine-phalloidin to determine the effect of PAR2 deletion on the PA-induced actin reorganization. We observed that the phagocytic index of WT macrophages was ∼55% higher than that in PAR2 null-Mɸ (Figures 1C,D). The formation of membrane protrusions is an important aspect of phagocytosis, which is generated by the actin cytoskeleton in response to a phagocytic target (Flannagan et al., 2012; Levin et al., 2016). We quantified the protrusions as the ratio of pseudopods/actin projections toward PA. We found that WT-Mɸ extended pseudopods (two to four per cell) around PA within 15–45 min (Figure 1E). However, these pseudopods were barely detectable in PAR2-null Mɸ (Figure 1E), recapitulating the aforementioned findings. We next pre-treated the WT or PAR2 null Mɸ with latrunculin A, which is known to block actin polymerization (Morton et al., 2000), followed by PA exposure. We found that latrunculin A reduced phagocytosis of PA in WT-Mɸ to the level seen in PAR2-null Mɸ (Figure 1F, Supplementary Figure S1A). These findings indicate that PAR2-mediated phagocytosis of PA required actin polymerization.
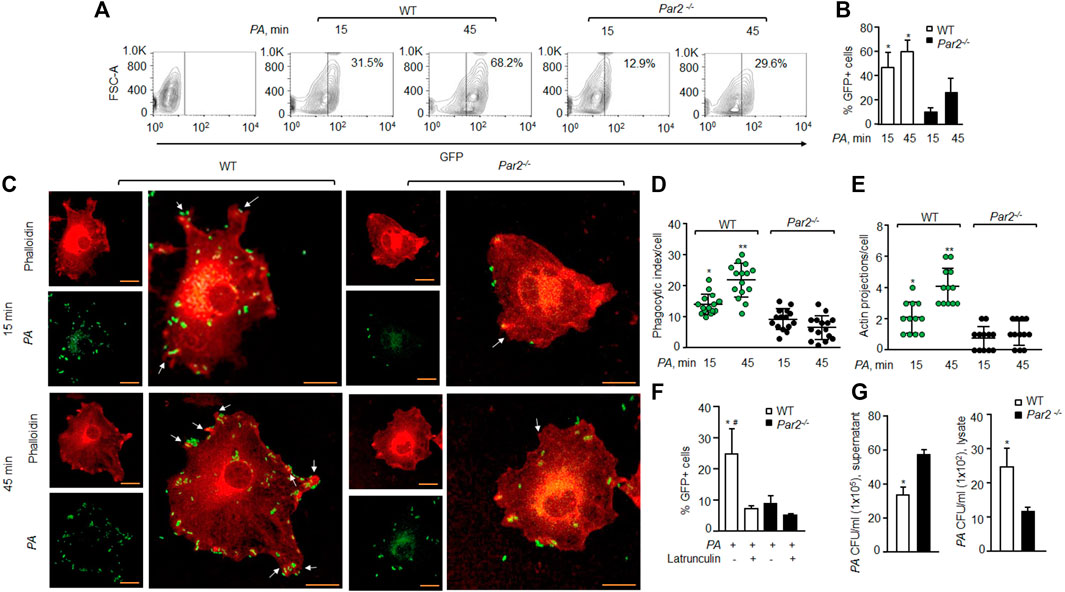
FIGURE 1. Loss of PAR2 impairs PA phagocytosis. (A) Macrophages were exposed to GFP-labeled PA (MOI, 1:10) for indicated times and then treated with accutase, spun down, and washed. The GFP+ cells were gated to assess phagocytosis. A representative contour plot is shown from experiments that were performed three times. (B) Mean ± SD of GFP+ macrophages, following PA exposure, is shown (from the aforementioned experiment-A) at indicated times from three independently performed experiments. (C) Macrophages were exposed to PA (MOI, 1:10) for indicated times and fixed with 2% paraformaldehyde. The cells were stained with rhodamine-phalloidin and imaged. Representative images are shown from the experiments that were performed three times. Bars, 5 micron meter, inset, zoomed ∼x2. (D) Phagocytic index per macrophage (of Figure 1C) was calculated by counting the total number of bacteria adhered to/engulfed by a macrophage. Individual count/phagocytic index per macrophage is shown (n = 15 cells). (E) Protrusions (of Figure 1C) were quantified per macrophage. The data are shown as individual extensions per macrophage (n = 12 cells). (F) WT and PAR2-nullpaired macrophages were pretreated with latrunculin (20 nΜ for 30 min) after which they were exposed to PA for 45 min. The GFP+ cells were gated and quantified by flow cytometric analysis. Data are shown as mean of three independent experiments. (G) WT and PAR2-null macrophages were exposed to PA for 45 min, and the supernatant and cell lysates were collected, dissolved in sterile PBS and spread on blood agar plates. CFU of PA/ml were determined. Data are shown as mean of three independent experiments. In all figures, data are presented as mean ± SD. *p < 0.05, **p < 0.001 relative to respective PAR2-null PA-exposed macrophages; #p < 0.05 relative to latrunculin-treated WT macrophages. The analysis was performed using one-way ANOVA followed by a paired t-test.
Phagocytosis is a multi-step process initiated upon sensing pathogens, followed by phagosome formation (Flannagan et al., 2012; Levin et al., 2016). We, therefore, determined the number of colony-forming units (CFUs) in the supernatant or lysates of the WT or PAR2-null Mɸ following PA exposure to evaluate whether the impaired phagocytosis seen later was due to a defect in bacterial internalization by the Mɸ. CFUs were quantified in lysates following gentamycin treatment to remove attached bacteria. We found that the supernatant from PAR2-null Mɸ formed significantly more PA colonies than that from WT-Mɸ (Figure 1G, left), indicating a defective internalization of PA by PAR2-null macrophages as a significant factor in reducing phagocytosis. This conclusion is supported by Figures 1C–E data also. Conversely, PAR2-null Mɸ lysates formed fewer PA colonies than WT-Mɸ (Figure 1G, right).
Loss of PAR2 induces bacterial pneumonia and lethality in mice
AMs are phenotypically and genotypically different from BMDMs (Rayees et al., 2020). We thus isolated AMs from WT and PAR2-null lungs and exposed them to PA. We again found that PAR2-null AMs were defective protrusions in forming and PA phagocytosis (Figures 2A–C). Interestingly, PA markedly increased the expression of TNF-α and IL-6 in PAR2-null lung AMs relative to WT-AMs (Figure 2D). The same was true for WT and PAR2-null BMDMs exposed to PA (Supplementary Figure S2B). We also determined IL-1β and MIP-2 expression following PA infection and found a similar increase in their expression in both WT and PAR2-null BMDMs (Supplementary Figure S2B).
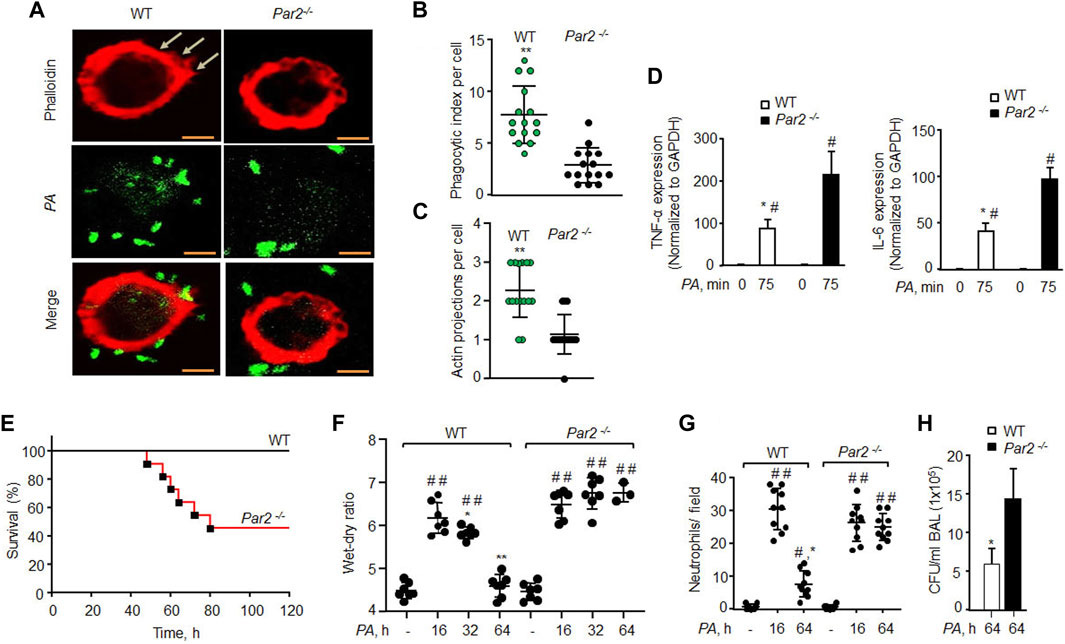
FIGURE 2. Loss of PAR2 compromises mouse recovery from bacterial pneumonia. (A) AMs isolated from BAL of mice exposed to PA (MOI,1:10) were fixed in 2% paraformaldehyde, stained with phalloidin, and imaged. Representative images are shown from three independently performed experiments. Bars, 5 micron meter. (B) Phagocytic index (of Figure 2A) was calculated by counting the total number of bacteria engulfed by a macrophage. Individual phagocytic index per macrophage is shown (n = 15 cells). (C) Protrusions (of Figure 2A) were quantified per macrophage. The data are shown as individual extensions per macrophage (n = 15 cells). (D) AMs were extracted from BAL and exposed to PA for 75 min followed by measurement of inflammatory cytokine gene expression by qPCR. The data are shown from three independent experiments. (E) Mouse survival was evaluated after administration of 1 × 106 CFU of PA in WT and PAR2-null mice (n = 10 mice/group). (F) 1 × 106 CFU of PA was injected into mice endotracheally followed by the assessment of the wet–dry ratio (n = 7 mice/group). (G) Neutrophil count (of Figure 2F) was performed from WT and PAR2-null mice post-PA infection. Individual values are shown from three independent experiments. (H) BAL was performed on PA-infected (1 × 106 CFU) mice at 64 h, and CFUs of bacteria/ml of BAL fluid were counted. Data are shown as mean of three independent experiments. Data are presented as mean ± SD. *p < 0.05, **p < 0.001 relative to respective PAR2-null PA-exposed AMs or mice; #p < 0.05 relative to corresponding control of WT or PAR2-null AMs or mice; ##p < 0.001 relative to corresponding control of WT or PAR2-null mice. The analysis was performed using one-way ANOVA followed by a paired t-test.
Bacterial pneumonia compromises patient survival in acute lung injury (Dreyfuss and Ricard, 2005; Rubenfeld et al., 2005). We instilled PA (1 × 106 CFU/mouse) into WT and PAR2-null mice, and found that 60% of PAR2-null mice died within 64 h, while all WT mice survived (Figure 2E). Upon sensing pathogens, AMs induce lung barrier leak and trigger neutrophil influx (Nagre et al., 2019; Rayees et al., 2019). We found that PA (1 × 106 CFU/mouse) induced barrier dysfunction and neutrophil accumulation in the lungs at 16 h in both WT and PAR2-null mice with the same intensity. The WT mice resolved edema formation post 32 h PA infection. However, the surviving PAR2-null mice showed massive edema and increased neutrophil influx (Figures 2F,G). We also isolated BAL from the WT and PAR2-null mice at 64 h and quantified CFUs. PAR2-null lungs failed to clear bacteria after pneumonia since BAL isolated from PAR2-null lungs showed a ∼3-fold increase in CFU relative to WT lung BAL (Figure 2H, Supplementary Figure S2A).
PAR2 mediates PA phagocytosis by activating Rac1GTPase
Small GTPases of the Rho family, such as Rho A and Rac1, are known to regulate actin organization as well as the phagocytosis of several bacterial species, including PA (Lee et al., 2000; Kazmierczak and Engel, 2002; Flannagan et al., 2012; Levin et al., 2016). We, therefore, determined if PAR2-induced PA phagocytosis occurs by activating RhoA or Rac1. We pharmacologically inhibited RhoA and Rac1 signaling using specific small molecule inhibitors, after which PA phagocytosis was determined (Routhier et al., 2010; Levay et al., 2013). We found that the inhibition of Rac1 activity using NSC23766 (Levay et al., 2013) reduced the phagocytic index to ∼50% (Figure 3A and Supplementary Figure S3A). By contrast, the inhibition of RhoA signaling using Y-27632 (Routhier et al., 2010) had no effect on PA phagocytosis in WT or PAR2-null Mɸ (Figure 3A, Supplementary Figure S3A). These findings indicate that PAR2 ligation mediates the activation of Rac1, which then induces actin polymerization to promote phagocytosis.
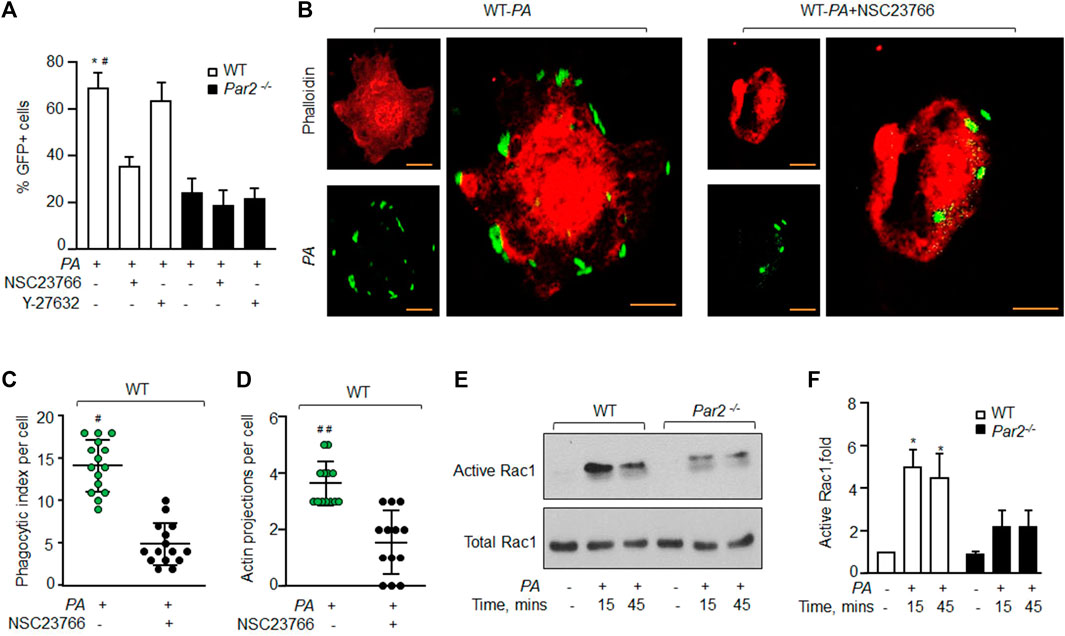
FIGURE 3. PAR2 mediates PA phagocytosis by activating Rac1. (A) WT and PAR2-null macrophages were pre-treated with 100 μM NSC23766 (Rac1 inhibitor) or 30 μM of Y-27632 (inhibitor of Rho-associated kinase) for 30 min after which they were exposed to PA for 45 min. The GFP+ cells were quantified by flow cytometric analysis. Data are shown as mean of three independent experiments. (B) WT-macrophages were pretreated with 100 μM NSC23766 for 30 min followed by PA exposure. The cells were fixed, stained with rhodamine-phalloidin, and imaged for the assessment of phagocytosis. Images shown are representative of three independently performed experiments. Bars, 5 micron meter, inset, zoomed ∼x2. (C) Phagocytic index (of Figure 3B) was calculated by counting the total number of bacteria engulfed by a macrophage. Individual count/phagocytic index per macrophage is shown (n = 15 cells). (D) Actin protrusions (of Figure 3B) were quantified per macrophage after the cells were fixed. The data are shown as individual extensions per macrophage (n = 13 cells). (E) WT and PAR2-null macrophages were exposed to PA for indicated times, and Rac1 activity was determined using GST-PAK beads. Total Rac1 was used as a loading control. A representative blot is shown from three independently performed experiments. (F) Densitometry plot (of Figure 3E) is shown. The data are shown as mean of three independently performed experiments (n = 3). In all figures, data are presented as mean ± SD. *p < 0.05 relative to PAR2-null PA-exposed macrophages; #p < 0.05, ##p < 0.001 relative to WT-macrophages treated with NSC23766. The analysis was performed using one-way ANOVA followed by a paired t-test.
To further corroborate these findings, we inhibited Rac1 activity in WT-Mɸ, which also prevented pseudopod formation and PA phagocytosis (Figures 3B–D and Supplementary Figure S3B-C). We next measured the Rac1 activity in WT and PAR2-null BMDMs following PA exposure. We observed that Rac1 activity was barely detectable in naïve WT-Mɸ. PA exposure increased Rac1 activity by ∼5-fold within 15 min, consistent with robust PA phagocytosis seen in Figure 1. Rac1 activity declined at 45 min (Figures 3E,F). Interestingly, PA only modestly increased Rac1 activity in PAR2-null Mɸ. Thus, Rac1 activity was significantly lower in PAR2-null Mɸ at 15 and 45 min than in WT-Mɸ.
We also addressed the possibility that loss of PAR2 in AMs induced their apoptosis following infection. Thus, we determined the expression of caspase 11 and PARP1 (two well-known markers of apoptosis), (Chaitanya et al., 2010; Huang et al., 2019). In WT and PAR2-null macrophages following PA infection. We failed to detect any observable differences in the expression of these proteins between the two groups (Supplementary Figure S3D). We also performed annexin V staining, another marker of cell death, (Bossy-Wetzel and Green, 2000) in WT or PAR2-null macrophages after PA infection. We again did not observe any significant apoptosis of macrophages at the indicated time (Supplementary Figure S3E).
PAR2 mediates Rac1 activity by enhancing cAMP levels
We have recently shown that the ligation of PAR2 with thrombin in macrophages induces cAMP generation (Rayees et al., 2019). cAMP is known to regulate Rac1GTPase activity (Schlegel and Waschke, 2013; Aslam et al., 2014). Thus, we tested the possibility that PAR2 stimulates cAMP generation, which activates Rac1 to mediate PA phagocytosis. Interestingly, PA increased cAMP levels by 2-fold within 15 min and 4-fold at 45 min in WT-Mɸ. PA failed to increase cAMP levels in PAR2-null Mɸ above baseline (Figure 4A).
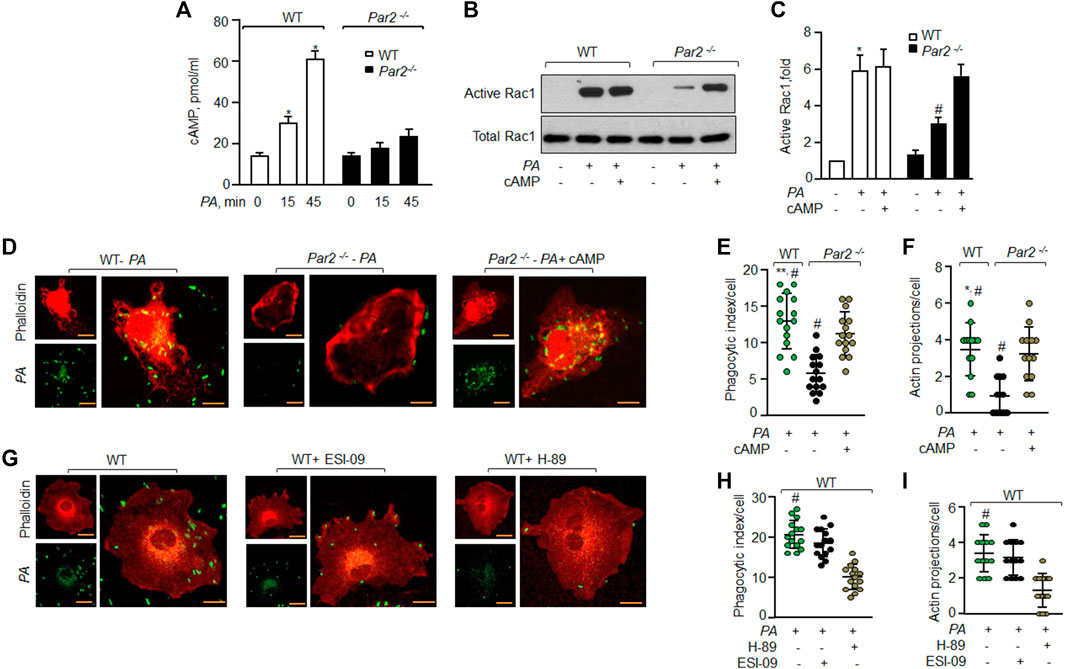
FIGURE 4. PAR2 induces cAMP that activates Rac1 to phagocytize PA. (A) WT and PAR2-null macrophages were exposed to PA for indicated times. The cells were lysed and intracellular cAMP was measured using ELISA (n = 3). (B) WT and PAR2-null macrophages were pre-treated with 10 nM of 8-Br-cAMP for 30 min followed by exposure to PA for 45 min and Rac1 activity was determined. Total Rac1 was used as the loading control. A representative blot is shown from three independently performed experiments. (C) Densitometry plot (of Figure 4B) is shown. The data are presented as mean of three independently performed experiments. (D) PAR2-null macrophages were pre-treated with 10 nM of 8-Br-cAMP for 30 min followed by exposure to PA for 45 min. The cells were fixed and stained with rhodamine-phalloidin. Images were acquired to quantify phagocytosis. The images shown are representative of results from three independently performed experiments. Bars, 5 micron meter, inset, zoomed ∼x2. (E) Phagocytic index (of Figure 4D) was calculated by counting the total number of bacteria engulfed by a macrophage. Individual count/phagocytic index per macrophage is shown (n = 15 cells). (F) Protrusions were quantified per macrophage (of Figure 4D) as described previously. Individual extensions per macrophage are shown (n = 13 cells). (G) WT macrophages were pre-treated with 20 μM of ESI-09 or 10 μM of H-89 for 30 min followed by exposure to PA for 45 min. The cells were fixed, stained with rhodamine-phalloidin and imaged for the assessment of phagocytosis. Representative images are shown from three independently performed experiments. Bars, 5 micron meter, inset, zoomed ∼x2. (H) Phagocytic index (of Figure 4G) was calculated by counting the total number of bacteria engulfed by a macrophage. The individual phagocytic index per macrophage is shown (n = 15 cells). (I) Protrusions (of Figure 4G) were quantified per macrophage. Individual extensions per macrophage are shown (n = 13 cells). Data are presented as mean ± SD. *p < 0.05, **p < 0.001 relative to respective PAR2-null PA-exposed BMDM; #p < 0.05 relative to WT BMDM or PAR2-null BMDM treated with H-89 or 8-Br-cAMP, respectively. The analysis was performed using one-way ANOVA followed by a paired t-test.
Next, we pre-treated PAR2-null Mɸ with cell-permeable cAMP (8-Br-cAMP) to test the prediction that augmenting cAMP levels in PAR2-null Mɸ should rescue Rac1 activity and phagocytosis. We found that treatment of PAR2-null Mɸ with the cAMP analog restored the Rac1 activity (Figures 4B,C) and phagocytosis (Figures 4D–F) to the level seen in WT Mɸ. These findings demonstrate that PA activates PAR2, which induces cAMP generation to activate Rac1.
cAMP activates PKA and EPAC to signal Rac1 activation (Cheng et al., 2008; Birukova et al., 2010; Bachmann et al., 2013). Thus, we inhibited PKA and/or EPAC activity in WT-Mɸ and found that inhibition of PKA but not EPAC reduced phagocytosis significantly (Figures 4G–I).
Delivery of constitutively active Rac1 in alveolar macrophages resolves lung infection and reduces mortality
Based on our observation that constitutively active Rac1 restored PA phagocytosis in AMs lacking PAR2 expression, we surmised that rescuing constitutive Rac1 would clear infection and thereby mitigate lung injury. Indeed, we observed that constitutively active Rac1 (CA-Rac1) mutant restored Rac1 activity in PAR2-null BMDMs (Supplementary Figure S4A, B). Thus, we delivered vector or YFP-tagged CA-Rac1 in mouse lungs, using liposomes. After 48 h, we injected unlabeled PA and determined the protrusive activity of Rac1 in AMs 3 h later using Rac1 and actin antibodies. We found that CA-Rac1-receiving AMs formed numerous pseudopods upon PA infection than WT-AMs (Supplementary Figure S4D, E). Importantly, CA-Rac1-receiving PAR2-null mice resolved lung edema (Figures 5A,B). Moreover, CA-Rac1 delayed the mortality of PAR2-null mice from pneumonia such that 70% of PAR2-null mice survived at 120 h post bacterial challenge than PAR2-null mice receiving control vector (Figure 5C). Survived PAR2-null mice receiving CA-Rac1 also showed decreased CFU counts (Figure 5D, S4C). However, the restoration of Rac1 activity failed to reduce the expression of pro-inflammatory cytokines to the levels observed in WT-AMs post PA challenge (Figure 5E), indicating that the augmented expression of TNF-α and IL-6 in PA exposed PAR2-null AM (Figure 5E) is independent of Rac1 activation and hence not related to phagocytosis in the present study model.
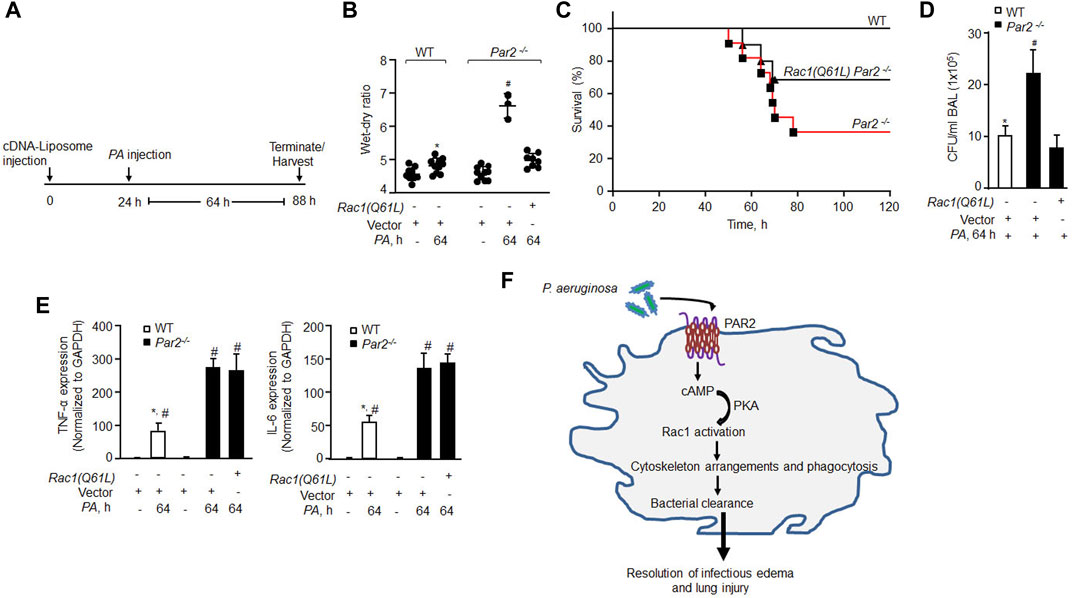
FIGURE 5. Delivery of constitutively active Rac1 rescued bacterial clearance leading to resolution of lung injury and mouse survival from bacterial pneumonia. (A) Schematics of the protocol. Liposomes complexed with constitutively active Rac1 (CA-Rac1; Q61L) or vector were administrated endotracheally and after 24 h PA was injected, and lung injury and mice survival were determined at indicated times. (B) Lung wet–dry ratio was determined to assess lung edema (n = 10 mice/group; only three mice are shown for vector Par2−/− group due to 70% mortality in this group). (C) Mouse survival after PA (1 × 106 CFU) administration (n = 10 mice/group). (D) BAL was isolated from mice (from Figure 5B) at 64 h post infection, and CFU of bacteria/ml of BAL fluid was counted. Data are shown from three independent experiments. (E) AMs were isolated from BAL of the indicated mice, and the expression of TNF-α and IL-6 was measured from the indicated groups by qPCR (n = 3). (F) Model of PAR2-mediated PA phagocytosis in AMs and resolution of acute lung injury and the resultant lethality. PA activates PAR2 in AMs stimulating cAMP generation. cAMP stimulates PKA activity which in turn activates Rac1. Activated Rac1 induces actin cytoskeleton remodeling leading to phagocytosis of PA resolving lung injury after bacterial pneumonia. Data are presented as mean ± SD. *p < 0.05 relative to respective PA-infected Par2−/− mice or AMs; #p < 0.05 relative to respective Par2−/− Rac1(Q61L) + PA-infected mice or control PAR2-null AMs. The analysis was performed using one-way ANOVA followed by a paired t-test.
Discussion
Bacterial pneumonia caused by PA remains a concern for critically ill patients suffering from conditions such as cystic fibrosis and critically ill ALI patients in the ICU despite new-generation antibiotics (Sadikot et al., 2005; Malhotra et al., 2019). AMs are the first line of defense against lung bacterial and viral infections (Hashimoto et al., 2007; Kolli et al., 2014; Dinapoli et al., 2017; He et al., 2017). How then AMs become inefficient in clearing PA? Alteration in PAR2 expression occurs in several inflammatory diseases (Yasuoka et al., 1997; Knight et al., 2001; Miotto et al., 2002). We showed that the loss of PAR2 expression in AMs reduced PA phagocytosis by half, leading to impaired bacterial clearance and irreversible lung injury, consistent with the previous study (Moraes et al., 2008). Thus, 60% of the PAR2-null mice died after lethal pneumonia. Interestingly, PAR2-null AMs generated significantly higher levels of inflammatory cytokines than the WT AMs and triggered neutrophil influx. Neutrophils recruited to the airspace are ineffective in clearing PA (Mishra et al., 2012; Thanabalasuriar et al., 2017). We showed that PAR2 expressed in AMs contributed to PA clearance and lung homeostasis. Our studies, however, did not rule out the role of PAR2 in neutrophils in co-operating with AM-PAR2 in regulating PA phagocytosis.
AMs trigger phagocytosis by mobilizing the scavenger receptors such as MARCO and Fc receptors (FcγRs) (Bonilla et al., 2013; Levin et al., 2016). We showed that about ∼60% of PA phagocytosis was PAR2-dependent. These findings also showed that the supernatant from PA-infected PAR2-null AMs formed more colonies, pointing to defective bacterial internalization. Whether AM-PAR2 regulated phagocytosis by maintaining MARCO and Fc receptor functions remains to be seen.
Phagosome formation aided by rapid actin remodeling is a crucial step in internalizing bacteria (Levin et al., 2016). RhoA and Rac1 regulate actin remodeling (Sit and Manser, 2011; Flannagan et al., 2012). In line with these studies, we showed that inhibiting actin polymerization or Rac1 activity blocked PA phagocytosis in WT macrophages. However, RhoA was not required. Intriguingly, PA activated Rac1 in WT-AMs, but not in PAR2-null AMs. Hence, AMs lacking PAR2 showed impaired pseudopod formation and defective phagocytosis, indicating that PAR2 induced phagocytosis by activating Rac1-mediated actin polymerization. Furthermore, delivery of constitutively active Rac1 rescued phagocytosis in PAR2-null lungs in vivo leading to 70% survival of the PAR2-null mice after PA challenge. The data presented in this study demonstrated an essential role for Rac1 but not RhoA downstream of PAR2 in phagocytizing PA. The requirement for active Rac1 in bacterial phagocytosis is consistent with its role in actin assembly for the ingestion of Gram-negative pathogens (Lee et al., 2000; Skjesol et al., 2019).
How does PAR2 induce Rac1 activity in macrophages upon PA infection? During tissue injury, PAR2 activation occurs by proteases such as trypsin, thrombin, and elastin due to the generation of tethered ligands (Zhao et al., 2015; Rayees et al., 2019; Rayees et al., 2020). However, elastase released from PA prevented PAR2 from being activated by trypsin (Dulon et al., 2003). Additionally, PA may trigger phagocytosis by generating matrix metalloproteinases, such as MMP-1, MMP-8, and MMP-13, which also cleave PAR2 (Falconer et al., 2019). Further studies are needed to explore via which of these proteases PA activated PAR2.
Controversy exists regarding the role of cAMP in regulating phagocytosis. Makranz et al. (2006) showed that cAMP promoted phagocytosis of myelin by glial macrophages. In sharp contrast, Serezani et al. (2007) showed that cAMP generation downstream of PGE2-coupled EP2/EP4 receptors suppressed the microbicidal activity of Klebsiella pneumoniae by AMs. However, mice lacking EP2 receptors showed enhanced phagocytosis of PA (Sadikot et al., 2005). In the present study, we also showed a critical role for cAMP as the second messenger in regulating the Rac1 activity and actin cytoskeletal reorganization, which induces PA phagocytosis. We showed that PA infection induced cAMP generation in macrophages in a PAR2-dependent manner. Specifically, we showed that cAMP addition to PAR2-null macrophages rescued the Rac1 activity and PA phagocytosis. cAMP activates PKA and EPAC which in turn induce Rac1 activity (Cheng et al., 2008; Birukova et al., 2010). We showed that the inhibition of PKA reduced the phagocytosis of PA in WT macrophages, indicating that PAR2 activated Rac1 via PKA.
Precisely how Rac1 was activated by PKA downstream of PAR2 to mediate phagocytosis is unknown, although it likely involves either the inhibition of guanine dissociation inhibitor (GDI) or the activation of guanine nucleotide exchange factors (GEFs) (Chahdi and Sorokin, 2008; Qiao et al., 2008). A likely scenario is the inhibition of GDI because PKA limits GDI inhibitory activity on Rac1 (Qiao et al., 2008). Another possibility is that PKA augments Rac1 activity through Vav1, a characterized GEF for Rac1 and phagocytosis (Patel et al., 2002).
Phagocytosis per se regulates pro-inflammatory gene expression (Freeman and Grinstein, 2016; Kaufmann and Dorhoi, 2016). In the present study, we established the role of PAR2 in regulating phagocytosis in association with an increase in Rac1 activity, which proved essential for the phagocytic capacity of macrophages. Thus, we showed that PAR2-null AMs have low Rac1 activity and produce cytokines at much higher levels than WT-AMs presumably because bacterial clearance becomes ineffective in these knockout mice. Accordingly, delivery of constitutively active Rac1 to PAR2-null mice rescued phagocytosis. Our findings do not rule out the involvement of other cell types, such as neutrophils, in this response. However, AMs are the most sentinel cells in the lungs and mediate phagocytosis of bacteria and viruses (Hashimoto et al., 2007; Dinapoli et al., 2017). Compared to other lung cell types, AMs predominantly expressed Rac1 (Nicholas et al., 2018). We showed that RhoA had no direct role in regulating PA phagocytosis but may mediate pro-inflammatory cytokine expression in macrophages when bacterial clearance is low (Manukyan et al., 2009; Yang et al., 2014). We concluded that the function of PAR2 in AMs is to activate Rac1, thereby facilitating PA phagocytosis, whereas RhoA may augment cytokine generation, leading to inflammatory injury (Popoff, 2014; Abedi et al., 2020). Thus, our findings show the importance of Rac1 activity in AMs in PA phagocytosis and thereby in resolving lung injury.
In summary, we demonstrated an essential role of the AM-PAR2-mediated cAMP-Rac1 pathway in triggering phagocytosis of PA (Figure 5F). We showed that loss of PAR2 expression and consequent impairment of Rac1 activity compromised PA clearance in the PAR2-null lungs, resulting in ALI and death from bacterial pneumonia. Liposomal delivery of constitutively active Rac1 in PAR2-null mice rescued phagocytosis and eliminated the lethality of PA. PAR2 in macrophages may have future therapeutic significance as a strategy to promote PA phagocytosis to treat bacterial pneumonia.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material; further inquiries can be directed to the corresponding author.
Ethics statement
The animal study was reviewed and approved by the Institutional Animal Care and Use Committee of the University of Illinois at Chicago.
Author contributions
SR and DM contributed to the conception and design of the research; SR, BJ, JC, VV, and SB performed the experiments for the study; SR and DM wrote the manuscript; and SR, BJ, JC, VV, and SB performed statistical analysis. All authors approved the manuscript.
Funding
This work was supported by NIH United States grants P01-HL151327, HL137169, and HL-155941.
Acknowledgments
The authors acknowledge Sekhar P Reddy for providing the GFP-tagged Pseudomonas aeruginosa (GFP-PAO1) strain.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2022.874197/full#supplementary-material
References
Abedi, F., Hayes, A. W., Reiter, R., and Karimi, G. (2020). Acute lung injury: The therapeutic role of Rho kinase inhibitors. Pharmacol. Res. 155, 104736. doi:10.1016/j.phrs.2020.104736
Almeida, R. A., Fang, W., and Oliver, S. P. (1999). Adherence and internalization of Streptococcus uberis to bovine mammary epithelial cells are mediated by host cell proteoglycans. FEMS Microbiol. Lett. 177, 313–317. doi:10.1111/j.1574-6968.1999.tb13748.x
Aslam, M., Tanislav, C., Troidl, C., Schulz, R., Hamm, C., and Gunduz, D. (2014). cAMP controls the restoration of endothelial barrier function after thrombin-induced hyperpermeability via Rac1 activation. Physiol. Rep. 2, e12175. doi:10.14814/phy2.12175
Bachmann, V. A., Riml, A., Huber, R. G., Baillie, G. S., Liedl, K. R., Valovka, T., et al. (2013). Reciprocal regulation of PKA and Rac signaling. Proc. Natl. Acad. Sci. U. S. A. 110, 8531–8536. doi:10.1073/pnas.1215902110
Bastaert, F., Kheir, S., Saint-Criq, V., Villeret, B., Dang, P. M., El-Benna, J., et al. (2018). Pseudomonas aeruginosa LasB subverts alveolar macrophage activity by interfering with bacterial killing through downregulation of innate immune defense, reactive oxygen species generation, and complement activation. Front. Immunol. 9, 1675. doi:10.3389/fimmu.2018.01675
Birukova, A. A., Burdette, D., Moldobaeva, N., Xing, J., Fu, P., and Birukov, K. G. (2010). Rac GTPase is a hub for protein kinase A and Epac signaling in endothelial barrier protection by cAMP. Microvasc. Res. 79, 128–138. doi:10.1016/j.mvr.2009.11.007
Bonilla, D. L., Bhattacharya, A., Sha, Y., Xu, Y., Xiang, Q., Kan, A., et al. (2013). Autophagy regulates phagocytosis by modulating the expression of scavenger receptors. Immunity 39, 537–547. doi:10.1016/j.immuni.2013.08.026
Bossy-Wetzel, E., and Green, D. R. (2000). Detection of apoptosis by annexin V labeling. Methods Enzymol. 322, 15–8. doi:10.1016/s0076-6879(00)22004-1
Cavinato, L., Genise, E., Luly, F. R., Di Domenico, E. G., Del Porto, P., and Ascenzioni, F. (2020). Escaping the phagocytic oxidative burst: The role of SODB in the survival of Pseudomonas aeruginosa within macrophages. Front. Microbiol. 11, 326. doi:10.3389/fmicb.2020.00326
Chahdi, A., and Sorokin, A. (2008). Protein kinase A-dependent phosphorylation modulates beta1Pix guanine nucleotide exchange factor activity through 14-3-3beta binding. Mol. Cell. Biol. 28, 1679–1687. doi:10.1128/MCB.00898-07
Chaitanya, G. V., Steven, A. J., and Babu, P. P. (2010). PARP-1 cleavage fragments: signatures of cell-death proteases in neurodegeneration. Cell Commun. Signal. 8, 31. doi:10.1186/1478-811X-8-31
Cheng, X., Ji, Z., Tsalkova, T., and Mei, F. (2008). Epac and PKA: a tale of two intracellular cAMP receptors. Acta Biochim. Biophys. Sin. 40, 651–662. doi:10.1111/j.1745-7270.2008.00438.x
Descamps, D., Le Gars, M., Balloy, V., Barbier, D., Maschalidi, S., Tohme, M., et al. (2012). Toll-like receptor 5 (TLR5), IL-1β secretion, and asparagine endopeptidase are critical factors for alveolar macrophage phagocytosis and bacterial killing. Proc. Natl. Acad. Sci. U. S. A. 109, 1619–1624. doi:10.1073/pnas.1108464109
Dinapoli, S., Ortiz, A., Wu, F., Matsuda, K., Twigg, H., Hirsch, V., et al. (2017). Tissue-resident macrophages can contain replication-competent virus in antiretroviral-naive, SIV-infected Asian macaques. JCI Insight 2, e91214. doi:10.1172/jci.insight.91214
Dreyfuss, D., and Ricard, J. D. (2005). Acute lung injury and bacterial infection. Clin. Chest Med. 26, 105–112. doi:10.1016/j.ccm.2004.10.014
Dulon, S., Cande, C., Bunnett, N. W., Hollenberg, M. D., Chignard, M., and Pidard, D. (2003). Proteinase-activated receptor-2 and human lung epithelial cells: disarming by neutrophil serine proteinases. Am. J. Respir. Cell Mol. Biol. 28, 339–346. doi:10.1165/rcmb.4908
Dulon, S., Leduc, D., Cottrell, G. S., D'alayer, J., Hansen, K. K., Bunnett, N. W., et al. (2005). Pseudomonas aeruginosa elastase disables proteinase-activated receptor 2 in respiratory epithelial cells. Am. J. Respir. Cell Mol. Biol. 32, 411–419. doi:10.1165/rcmb.2004-0274OC
Falconer, A. M. D., Chan, C. M., Gray, J., Nagashima, I., Holland, R. A., Shimizu, H., et al. (2019). Collagenolytic matrix metalloproteinases antagonize proteinase-activated receptor-2 activation, providing insights into extracellular matrix turnover. J. Biol. Chem. 294, 10266–10277. doi:10.1074/jbc.RA119.006974
Flannagan, R. S., Jaumouille, V., and Grinstein, S. (2012). The cell biology of phagocytosis. Annu. Rev. Pathol. 7, 61–98. doi:10.1146/annurev-pathol-011811-132445
Freeman, S. A., and Grinstein, S. (2016). Phagocytosis: How macrophages tune their non-professional counterparts. Curr. Biol. 26, R1279–R1282. doi:10.1016/j.cub.2016.10.059
Han, C. Z., Juncadella, I. J., Kinchen, J. M., Buckley, M. W., Klibanov, A. L., Dryden, K., et al. (2016). Macrophages redirect phagocytosis by non-professional phagocytes and influence inflammation. Nature 539, 570–574. doi:10.1038/nature20141
Hashimoto, Y., Moki, T., Takizawa, T., Shiratsuchi, A., and Nakanishi, Y. (2007). Evidence for phagocytosis of influenza virus-infected, apoptotic cells by neutrophils and macrophages in mice. J. Immunol. 178, 2448–2457. doi:10.4049/jimmunol.178.4.2448
He, W., Chen, C. J., Mullarkey, C. E., Hamilton, J. R., Wong, C. K., Leon, P. E., et al. (2017). Alveolar macrophages are critical for broadly-reactive antibody-mediated protection against influenza A virus in mice. Nat. Commun. 8, 846. doi:10.1038/s41467-017-00928-3
Huang, X., Feng, Y., Xiong, G., Whyte, S., Duan, J., Yang, Y., et al. (2019). Caspase-11, a specific sensor for intracellular lipopolysaccharide recognition, mediates the non-canonical inflammatory pathway of pyroptosis. Cell Biosci. 9, 31. doi:10.1186/s13578-019-0292-0
Joshi, J. C., Joshi, B., Rochford, I., Rayees, S., Akhter, M. Z., Baweja, S., et al. (2020). SPHK2-Generated S1P in CD11b(+) macrophages blocks STING to suppress the inflammatory function of alveolar macrophages. Cell Rep. 30, 4096–4109. doi:10.1016/j.celrep.2020.02.112
Kaufmann, S. H. E., and Dorhoi, A. (2016). Molecular determinants in phagocyte-bacteria interactions. Immunity 44, 476–491. doi:10.1016/j.immuni.2016.02.014
Kazmierczak, B. I., and Engel, J. N. (2002). Pseudomonas aeruginosa ExoT acts in vivo as a GTPase-activating protein for RhoA, Rac1, and Cdc42. Infect. Immun. 70, 2198–2205. doi:10.1128/iai.70.4.2198-2205.2002
Knight, D. A., Lim, S., Scaffidi, A. K., Roche, N., Chung, K. F., Stewart, G. A., et al. (2001). Protease-activated receptors in human airways: upregulation of PAR-2 in respiratory epithelium from patients with asthma. J. Allergy Clin. Immunol. 108, 797–803. doi:10.1067/mai.2001.119025
Kolli, D., Gupta, M. R., Sbrana, E., Velayutham, T. S., Chao, H., Casola, A., et al. (2014). Alveolar macrophages contribute to the pathogenesis of human metapneumovirus infection while protecting against respiratory syncytial virus infection. Am. J. Respir. Cell Mol. Biol. 51, 502–515. doi:10.1165/rcmb.2013-0414OC
Lee, D. J., Cox, D., Li, J., and Greenberg, S. (2000). Rac1 and Cdc42 are required for phagocytosis, but not NF-kappaB-dependent gene expression, in macrophages challenged with Pseudomonas aeruginosa. J. Biol. Chem. 275, 141–146. doi:10.1074/jbc.275.1.141
Levay, M., Krobert, K. A., Wittig, K., Voigt, N., Bermudez, M., Wolber, G., et al. (2013). NSC23766, a widely used inhibitor of Rac1 activation, additionally acts as a competitive antagonist at muscarinic acetylcholine receptors. J. Pharmacol. Exp. Ther. 347, 69–79. doi:10.1124/jpet.113.207266
Levin, R., Grinstein, S., and Canton, J. (2016). The life cycle of phagosomes: formation, maturation, and resolution. Immunol. Rev. 273, 156–179. doi:10.1111/imr.12439
Lillehoj, E. P., Hyun, S. W., Kim, B. T., Zhang, X. G., Lee, D. I., Rowland, S., et al. (2001). Muc1 mucins on the cell surface are adhesion sites for Pseudomonas aeruginosa. Am. J. Physiol. Lung Cell. Mol. Physiol. 280, L181–L187. doi:10.1152/ajplung.2001.280.1.L181
Lovewell, R. R., Patankar, Y. R., and Berwin, B. (2014). Mechanisms of phagocytosis and host clearance of Pseudomonas aeruginosa. Am. J. Physiol. Lung Cell. Mol. Physiol. 306, L591–L603. doi:10.1152/ajplung.00335.2013
Makranz, C., Cohen, G., Reichert, F., Kodama, T., and Rotshenker, S. (2006). cAMP cascade (PKA, Epac, adenylyl cyclase, Gi, and phosphodiesterases) regulates myelin phagocytosis mediated by complement receptor-3 and scavenger receptor-AI/II in microglia and macrophages. Glia 53, 441–448. doi:10.1002/glia.20303
Malhotra, S., Hayes, D., and Wozniak, D. J. (2019). Cystic fibrosis and Pseudomonas aeruginosa: the host-microbe interface. Clin. Microbiol. Rev. 32, e00138–18. doi:10.1128/CMR.00138-18
Manukyan, M., Nalbant, P., Luxen, S., Hahn, K. M., and Knaus, U. G. (2009). RhoA GTPase activation by TLR2 and TLR3 ligands: connecting via src to NF-kappa B. J. Immunol. 182, 3522–3529. doi:10.4049/jimmunol.0802280
Miotto, D., Hollenberg, M. D., Bunnett, N. W., Papi, A., Braccioni, F., Boschetto, P., et al. (2002). Expression of protease activated receptor-2 (PAR-2) in central airways of smokers and non-smokers. Thorax 57, 146–151. doi:10.1136/thorax.57.2.146
Mishra, M., Byrd, M. S., Sergeant, S., Azad, A. K., Parsek, M. R., Mcphail, L., et al. (2012). Pseudomonas aeruginosa Psl polysaccharide reduces neutrophil phagocytosis and the oxidative response by limiting complement-mediated opsonization. Cell. Microbiol. 14, 95–106. doi:10.1111/j.1462-5822.2011.01704.x
Moraes, T. J., Martin, R., Plumb, J. D., Vachon, E., Cameron, C. M., Danesh, A., et al. (2008). Role of PAR2 in murine pulmonary pseudomonal infection. Am. J. Physiol. Lung Cell. Mol. Physiol. 294, L368–L377. doi:10.1152/ajplung.00036.2007
Morton, W. M., Ayscough, K. R., and Mclaughlin, P. J. (2000). Latrunculin alters the actin-monomer subunit interface to prevent polymerization. Nat. Cell Biol. 2, 376–378. doi:10.1038/35014075
Nagre, N., Cong, X., Pearson, A. C., and Zhao, X. (2019). Alveolar macrophage phagocytosis and bacteria clearance in mice. J. Vis. Exp. 145, 59088. doi:10.3791/59088
Nicholas, S., Jim Karkanias, , Norma, F. N., Al, E., et al. (2018). Single-cell transcriptomics of 20 mouse organs creates a Tabula Muris. Nature 562, 367–372. doi:10.1038/s41586-018-0590-4
Ojielo, C. I., Cooke, K., Mancuso, P., Standiford, T. J., Olkiewicz, K. M., Clouthier, S., et al. (2003). Defective phagocytosis and clearance of Pseudomonas aeruginosa in the lung following bone marrow transplantation. J. Immunol. 171, 4416–4424. doi:10.4049/jimmunol.171.8.4416
Patel, J. C., Hall, A., and Caron, E. (2002). Vav regulates activation of Rac but not Cdc42 during FcgammaR-mediated phagocytosis. Mol. Biol. Cell 13, 1215–1226. doi:10.1091/mbc.02-01-0002
Plotkowski, M. C., De Bentzmann, S., Pereira, S. H., Zahm, J. M., Bajolet-Laudinat, O., Roger, P., et al. (1999). Pseudomonas aeruginosa internalization by human epithelial respiratory cells depends on cell differentiation, polarity, and junctional complex integrity. Am. J. Respir. Cell Mol. Biol. 20, 880–890. doi:10.1165/ajrcmb.20.5.3408
Popoff, M. R. (2014). Bacterial factors exploit eukaryotic Rho GTPase signaling cascades to promote invasion and proliferation within their host. Small GTPases 5, e28209. doi:10.4161/sgtp.28209
Qiao, J., Holian, O., Lee, B. S., Huang, F., Zhang, J., and Lum, H. (2008). Phosphorylation of GTP dissociation inhibitor by PKA negatively regulates RhoA. Am. J. Physiol. Cell Physiol. 295, C1161–C1168. doi:10.1152/ajpcell.00139.2008
Rayees, S., Joshi, J. C., Tauseef, M., Anwar, M., Baweja, S., Rochford, I., et al. (2019). PAR2-Mediated cAMP generation suppresses TRPV4-dependent Ca(2+) signaling in alveolar macrophages to resolve TLR4-induced inflammation. Cell Rep. 27, 793–805. doi:10.1016/j.celrep.2019.03.053
Rayees, S., Mabalirajan, U., Bhat, W. W., Rasool, S., Rather, R. A., Panda, L., et al. (2015). Therapeutic effects of R8, a semi-synthetic analogue of Vasicine, on murine model of allergic airway inflammation via STAT6 inhibition. Int. Immunopharmacol. 26, 246–256. doi:10.1016/j.intimp.2015.03.035
Rayees, S., Rochford, I., Joshi, J. C., Joshi, B., Banerjee, S., and Mehta, D. (2020). Macrophage TLR4 and PAR2 signaling: Role in regulating vascular inflammatory injury and repair. Front. Immunol. 11, 2091. doi:10.3389/fimmu.2020.02091
Restrepo, C. I., Dong, Q., Savov, J., Mariencheck, W. I., and Wright, J. R. (1999). Surfactant protein D stimulates phagocytosis of Pseudomonas aeruginosa by alveolar macrophages. Am. J. Respir. Cell Mol. Biol. 21, 576–585. doi:10.1165/ajrcmb.21.5.3334
Routhier, A., Astuccio, M., Lahey, D., Monfredo, N., Johnson, A., Callahan, W., et al. (2010). Pharmacological inhibition of Rho-kinase signaling with Y-27632 blocks melanoma tumor growth. Oncol. Rep. 23, 861–867. doi:10.3892/or_00000708
Rubenfeld, G. D., Caldwell, E., Peabody, E., Weaver, J., Martin, D. P., Neff, M., et al. (2005). Incidence and outcomes of acute lung injury. N. Engl. J. Med. 353, 1685–1693. doi:10.1056/NEJMoa050333
Sadikot, R. T., Blackwell, T. S., Christman, J. W., and Prince, A. S. (2005). Pathogen-host interactions in Pseudomonas aeruginosa pneumonia. Am. J. Respir. Crit. Care Med. 171, 1209–1223. doi:10.1164/rccm.200408-1044SO
Schlegel, N., and Waschke, J. (2013). cAMP with other signaling cues converges on Rac1 to stabilize the endothelial barrier- a signaling pathway compromised in inflammation. Cell Tissue Res. 355, 587–596. doi:10.1007/s00441-013-1755-y
Serezani, C. H., Chung, J., Ballinger, M. N., Moore, B. B., Aronoff, D. M., and Peters-Golden, M. (2007). Prostaglandin E2 suppresses bacterial killing in alveolar macrophages by inhibiting NADPH oxidase. Am. J. Respir. Cell Mol. Biol. 37, 562–570. doi:10.1165/rcmb.2007-0153OC
Siryaporn, A., Kuchma, S. L., O'toole, G. A., and Gitai, Z. (2014). Surface attachment induces Pseudomonas aeruginosa virulence. Proc. Natl. Acad. Sci. U. S. A. 111, 16860–16865. doi:10.1073/pnas.1415712111
Sit, S. T., and Manser, E. (2011). Rho GTPases and their role in organizing the actin cytoskeleton. J. Cell Sci. 124, 679–683. doi:10.1242/jcs.064964
Skjesol, A., Yurchenko, M., Bosl, K., Gravastrand, C., Nilsen, K. E., Grovdal, L. M., et al. (2019). The TLR4 adaptor TRAM controls the phagocytosis of Gram-negative bacteria by interacting with the Rab11-family interacting protein 2. PLoS Pathog. 15, e1007684. doi:10.1371/journal.ppat.1007684
Stokes, R. W., Thorson, L. M., and Speert, D. P. (1998). Nonopsonic and opsonic association of Mycobacterium tuberculosis with resident alveolar macrophages is inefficient. J. Immunol. 160, 5514–5521.
Thanabalasuriar, A., Surewaard, B. G., Willson, M. E., Neupane, A. S., Stover, C. K., Warrener, P., et al. (2017). Bispecific antibody targets multiple Pseudomonas aeruginosa evasion mechanisms in the lung vasculature. J. Clin. Invest. 127, 2249–2261. doi:10.1172/JCI89652
Yang, Z., Sun, D., Yan, Z., Reynolds, A. B., Christman, J. W., Minshall, R. D., et al. (2014). Differential role for p120-catenin in regulation of TLR4 signaling in macrophages. J. Immunol. 193, 1931–1941. doi:10.4049/jimmunol.1302863
Yasuoka, S., Ohnishi, T., Kawano, S., Tsuchihashi, S., Ogawara, M., Masuda, K., et al. (1997). Purification, characterization, and localization of a novel trypsin-like protease found in the human airway. Am. J. Respir. Cell Mol. Biol. 16, 300–308. doi:10.1165/ajrcmb.16.3.9070615
Yazbeck, P., Tauseef, M., Kruse, K., Amin, M. R., Sheikh, R., Feske, S., et al. (2017). STIM1 phosphorylation at Y361 recruits Orai1 to STIM1 puncta and induces Ca(2+) entry. Sci. Rep. 7, 42758. doi:10.1038/srep42758
Zhao, P., Lieu, T., Barlow, N., Sostegni, S., Haerteis, S., Korbmacher, C., et al. (2015). Neutrophil elastase activates protease-activated receptor-2 (PAR2) and transient receptor potential vanilloid 4 (TRPV4) to cause inflammation and pain. J. Biol. Chem. 290, 13875–13887. doi:10.1074/jbc.M115.642736
Keywords: acute lung injury, cAMP, Pseudomonas aeruginosa, alveolar macrophage, PAR2, Rac1
Citation: Rayees S, Joshi JC, Joshi B, Vellingiri V, Banerjee S and Mehta D (2022) Protease-activated receptor 2 promotes clearance of Pseudomonas aeruginosa infection by inducing cAMP-Rac1 signaling in alveolar macrophages. Front. Pharmacol. 13:874197. doi: 10.3389/fphar.2022.874197
Received: 17 February 2022; Accepted: 22 August 2022;
Published: 20 September 2022.
Edited by:
Heike Wulff, University of California, Davis, United StatesReviewed by:
Silvia C. Finnemann, Fordham University, United StatesSisi He, Zunyi Medical University, China
Copyright © 2022 Rayees, Joshi, Joshi, Vellingiri, Banerjee and Mehta. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dolly Mehta, dmehta@uic.edu