 Bara A. Shraim
Bara A. Shraim Moaz O. Moursi
Moaz O. Moursi Ibrahim F. Benter
Ibrahim F. Benter Abdella M. Habib
Abdella M. Habib Saghir Akhtar
Saghir Akhtar- 1College of Medicine, QU Health, Qatar University, Doha, Qatar
- 2Biomedical and Pharmaceutical Research Unit, QU Health, Qatar University, Doha, Qatar
- 3Faculty of Medicine, Eastern Mediterranean University, Famagusta, North Cyprus
Diabetes mellitus is a major debilitating disease whose global incidence is progressively increasing with currently over 463 million adult sufferers and this figure will likely reach over 700 million by the year 2045. It is the complications of diabetes such as cardiovascular, renal, neuronal and ocular dysfunction that lead to increased patient morbidity and mortality. Of these, cardiovascular complications that can result in stroke and cardiomyopathies are 2- to 5-fold more likely in diabetes but the underlying mechanisms involved in their development are not fully understood. Emerging research suggests that members of the Epidermal Growth Factor Receptor (EGFR/ErbB/HER) family of tyrosine kinases can have a dual role in that they are beneficially required for normal development and physiological functioning of the cardiovascular system (CVS) as well as in salvage pathways following acute cardiac ischemia/reperfusion injury but their chronic dysregulation may also be intricately involved in mediating diabetes-induced cardiovascular pathologies. Here we review the evidence for EGFR/ErbB/HER receptors in mediating these dual roles in the CVS and also discuss their potential interplay with the Renin-Angiotensin-Aldosterone System heptapeptide, Angiotensin-(1-7), as well the arachidonic acid metabolite, 20-HETE (20-hydroxy-5, 8, 11, 14-eicosatetraenoic acid). A greater understanding of the multi-faceted roles of EGFR/ErbB/HER family of tyrosine kinases and their interplay with other key modulators of cardiovascular function could facilitate the development of novel therapeutic strategies for treating diabetes-induced cardiovascular complications.
Introduction
Diabetes mellites (DM) is a set of metabolic disorders arising from defective insulin secretion and/or action in which hyperglycemia (HG) is a common feature. Type 1 diabetes mellitus (T1DM) results from the autoimmune-mediated destruction of pancreatic β cells. The infiltration of T lymphocytes, release of cytokines, and generation of reactive oxygen species (ROS) ultimately results in the dysfunction and apoptosis of pancreatic β cells (Morgan et al., 2014; Crevecoeur et al., 2015; Tan et al., 2019; Toi et al., 2020). In contrast, type 2 diabetes mellitus (T2DM) is characterized by target tissue resistance to the metabolic actions of insulin as well as pancreatic β cell dysfunction. Insulin resistance is strongly associated with visceral obesity and the resulting chronic low-grade inflammation as well as increased ectopic fat deposition (Saltiel, 2021; Tsatsoulis, et al., 2013).
The International Diabetes Federation declared that approximately 463 million adults are currently living with diabetes mellitus and this figure will likely reach over 700 million by the year 2045. Half of the sufferers are unaware of their condition and hence, prone to serious hyperglycemia/diabetes-related complications (IDF Diabetes Atlas, 2019). DM results in secondary pathophysiologic alterations in several organ systems that impose an immense burden on both the individual with diabetes and the public health system. Cardiovascular (CV) complications constitute the leading cause of morbidity and mortality in individuals with DM (Forbes and Cooper, 2013; Babel and Dandekar, 2021). These include microangiopathy, abnormal vascular reactivity, atherosclerosis, hypertension, cardiomyopathy, ischemic heart disease, and myocardial infarction (Akhtar and Benter, 2013; Babel and Dandekar, 2021). Moreover, the specific underlying mechanisms leading to the development of these CV complications are not fully understood and likely involve multiple intracellular signaling networks that can be activated by hyperglycemia (HG) and/or diabetes.
Epidermal growth factor receptor (EGFR/ErbB1/HER1), one of the most versatile signaling units in biology, plays a key role in regulating many cellular functions including growth, proliferation, motility, and survival (Kumagai et al., 2021; Sharifi et al., 2021). Perturbations in EGFR expression and signalling are implicated in several pathophysiological conditions including cancer and diabetes (Matroughi, 2010; Akhtar and Benter, 2013; Xu et al., 2017; Akhtar et al., 2018; Talukdar et al., 2020; Kumagai et al., 2021; Sharifi et al., 2021). Emerging research suggests that members of the Epidermal Growth Factor Receptor (EGFR/ErbB/HER) family of tyrosine kinases can have a dual role in that they are necessary (acting as “good guys”) for the normal development and physiological functioning of the cardiovascular system (CVS) but their dysregulation may also be intricately involved in mediating cardiovascular pathologies (i.e., acting as “bad guys”) (Matrougui, 2010; Akhtar and Benter, 2013; Schreier et al., 2014). In this article, we review the evidence for EGFR/ErbB/HER receptors in mediating these dual (“good guy” verses “bad guy”) roles in the heart and then in the vascular system followed by a discussion of their potential interplay with the Renin-Angiotensin-Aldosterone System heptapeptide, Angiotensin-(1–7), as well the arachidonic acid metabolite, 20-HETE-key players in the regulation of cardiovascular function.
Epidermal Growth Factor Receptor Family of Receptor Tyrosine Kinases: An Overview
EGFR (a/k/a ErbB1 or HER1) is a 180 kDa transmembrane glycoprotein made up of 1,186 amino acids that belongs to the broader family of receptor tyrosine kinases (RTKs) (Rayego-Mateos et al., 2018; Kumagai et al., 2021). It is made up of four conserved domains: an extracellular ligand-binding domain, a transmembrane domain, a cytoplasmic tyrosine kinase-containing domain, and a regulatory domain (Zheng et al., 2014 ). Moreover, there are three other homologous members that comprise the EGFR/ErbB/HER family of RTKs: ErbB2 (EGFR2/HER2/Neu), ErbB3 (EGFR3/HER3), and ErbB4 (EGFR4/HER4). EGFR members, with the exception of ErbB2, are activated by numerous growth factors or ligands such as epidermal growth factor (EGF), betacellulin, neuregulin-1 (NRG-1) and Heparin-binding EGF-like growth factor (HB-EGF). Upon ligand interaction with its cognate receptor, either homodimerization of the receptor or heterodimerization with other members of the EGFR family occurs. ErbB2 lacks a known ligand and depends on heterodimerization with other receptors of the EGFR family for activation. ErbB3 lacks a functional kinase domain and therefore also relies on heterodimerization with other EGFR/ErbB receptors for activity. Thus, EGFR and ErbB4 receptors are the only fully functioning homodimers (Kumagai et al., 2021; Sharifi et al., 2021). Next, phosphorylation of precisely defined tyrosine residues within the cytoplasmic kinase portion of the receptor dimer occurs, eventually leading to the activation of multiple downstream signaling cascades. Such cascades include p38 Mitogen-activated protein kinase (MAPKs), Extracellular-signal regulated kinase 1/2 (ERK1/2), Phosphatidylinositol 3-kinases (PI3Ks)/Protein kinase B (AKT), phospholipase C-gamma 1 (PLCγ1), and Janus kinase (JAK) (/Signal transducers and activation of transcription (STAT) pathways (Rayego-Mateos et al., 2018). Consequently, several cellular processes, such as angiogenesis, proliferation, differentiation, motility, survival, and apoptosis can take place (Akhtar and Benter, 2013; Kumagai et al., 2021; Sharifi et al., 2021; Zheng et al., 2014).
EGFR/ErbBs can also be activated via ligand-independent pathways through a process of “transactivation” such as by peptides of the Renin-Angiotensin-Aldosterone system (RAAS) and other G-protein coupled receptors (GPCRs) (Forrester et al., 2016). For example, EGFR transactivation can occur via Angiotensin II (Ang II) Norepinephrine (NE), (leptin, thrombin, and endothelin by mechanisms involving non-ligand associated Src family kinases, and/or mediated via matrix metalloproteases (MMP), and/or a disintegrin and metalloprotease (ADAM)-dependent shedding of cell surface bound EGF-like ligands (Matrougui, 2010; Akhtar et al., 2012a; Beltowski and Jazmroz-Wisniewska, 2014; Chen et al., 2015; Forrester et al., 2016; Reichelt et al., 2017). It should be noted that all ligands of the EGFR/ErbB family of receptors exist as membrane-anchored precursors that are released by enzymatic-cleavage by sheddases such as MMPs and ADAMs (Rayego-Mateos et al., 2018). EGFR/ErbB receptors are expressed widely in epithelial, neuronal and mesenchymal tissues (Yano et al., 2003; Chen et al., 2016). Hence, EGFR/ErbBs are key regulators of many cellular homeostatic functions and their dysregulation is associated with several pathological functions including diabetes-induced cardiovascular dysfunction.
Diabetes-Induced Cardiac Dysfunction: The Dual Role of Epidermal Growth Factor Receptor Family of Receptor Tyrosine Kinases and Their Ligands in the Heart
Cardiac complications including diabetic cardiomyopathy and heart failure are 2- to 5-fold more likely in patients with diabetes (for a review see Kenny and Abel, 2019). This increased risk is dependent on blood glucose levels as higher HbA1c levels (indicating worsening hyperglycemia) are associated with increased likelihood of heart failure in patients with diabetes (Erquo et al., 2013). Even short-term episodes of hyperglycemia or “glycemic variability” such as those observed postprandially are thought to increase the risk of developing cardiovascular complications associated with diabetes (Ceriello, 2005; Brownlee and Hirsch, 2006). Interestingly, in long-term studies it has been shown that subsequent therapy-induced correction of hyperglycemia does not normalise the risk of developing cardiovascular complications back to baseline (Kenny and Abel, 2019) implying that even transient excursions into hyperglycemia may induce long-term adverse molecular or signaling changes in the cardiovascular system that can lead to cardiovascular complications. This phenomenon has been described as “glycemic” or “metabolic” memory (Chalmers and Cooper, 2008; Holman et al., 2008); though recently this concept has been challenged (Miller and Orchard, 2020). Diabetes-induced cardiac complications may arise from a direct effect of hyperglycemia-induced molecular changes in the cardiac muscle as observed in cardiac myopathy (hypertrophy and fibrosis) or in the microvasculature of the heart that manifest as atherosclerosis or coronary artery disease (Falcao-Pires and Leite-Moreira, 2012; Kenny and Abel, 2019; Babel and Dandekar, 2021). Patients with diabetes who develop cardiomyopathy initially exhibit a hidden subclinical phase of myocardial remodeling that leads to compromised diastolic function, followed by systolic dysfunction that can progress to eventual heart failure (Falcao-Pires and Leite-Moreira, 2012) The exact underlying mechanisms for the development of cardiac pathologies in diabetes are not fully understood but likely involve a wide array of cell signaling networks. These include generation of reactive oxygen species (ROS)/oxidative stress, endoplasmic reticulum (ER) stress, mitochondrial dysfunction, accumulation of advanced glycated end-products (see also section on vascular dysfunction), and hyperglycemia-induced alterations in signaling by growth factors, peptides of RAAS as well as other GPCRs that are known also to transactivate EGFR (for recent reviews see Babel and Dandekar, 2021; Kenny and Abel, 2019, Forrester et al., 2016). Emerging evidence suggests that not only do EGFR/ErbB/HER family of TK receptors play a key role in the development of diabetes-induced cardiac dysfunction but they may also represent a convergent point or “hub” and a “relay” for the multiple other signaling molecules or inputs leading to cardiac dysfunction. In fact, the current understanding is that ErbBs may play a dual role, whereby they can elicit beneficial as well as detrimental signaling in the heart. In the proceeding section, we will discuss studies highlighting the dual role of EGFR/ErbB/HER receptor signaling in the normal (physiological) and the diabetic (pathological) heart.
The Beneficial Role of Epidermal Growth Factor Receptors in the Normal and Diabetic Heart
EGFR/ErbB/HER receptors are vitally important in the physiological development of the heart irrespective of whether it is normal or diabetic. Indeed, the beneficial (good guy) role of ErbB receptors starts even before birth. These receptor tyrosine kinases (RTKs) are essential for normal cardiac morphogenesis during embryogenesis, and also in maintaining proper physiology of the adult heart (Sanchez-Soria and Camenisch, 2010; Fuller et al., 2008) see also Table 1. Observations in engineered knockout (KO) or mutant mice of ErbB1, ErbB2, ErbB3, ErbB4 highlighted the lethal phenotypes and heart defects that can arise as a result of a deficiency in the number or signaling of these receptors (for review see also Sanchez-Soria and Camenisch, 2010). ErbB1 KO and mutant mice displayed semilunar valve defects (Sibilia and Wagner, 1995; Chen et al., 2000) whilst disrupted endocardial cushion/heart valve mesenchyme formation was noted in ErbB3 KO mice that ultimately leads to death of embryos within 2 weeks (Erickson et al., 1997; Camenisch et al., 2002). ErbB2 and ErbB4 KO mice lack ventricular trabeculation-a key process in the maturation of ventricles and necessary for physical contractility and normal blood flow (Gassmann et al., 1995; Lee et al., 1995; Chan et al., 2002; Negro et al., 2004).

TABLE 1. A summary of selected studies highlighting thebeneficialrole of EGFR signaling in the heart.
ErbB2 is also important for neural development and myelination of nerve fibres (Lee et al., 1995; Garratt et al., 2000). Not only does ErbB2 signaling participate in the development of the ventricular conduction system (Negro et al., 2004) but it is also required for cardiac atrial electrical activity during development of the atrial conduction system that if disrupted leads to premature death at mid-gestation (Tenin et al., 2014). Thus, activation of ErbB signaling has been proposed as a therapeutic strategy to reduce the mortality rate of congenital heart diseases and in the prevention of cardiac damage in adults (Sanchez-Soria and Camenisch, 2010). Collectively, these studies suggest that all four EGFR/ErbB/HER receptors are necessary for the proper physiological development of the heart-the first organ to form during embryogenesis. Furthermore, all EGFR/ErbB/HER receptors and several of their ligands are now also thought to be expressed in the adult heart (either in cardiac myocytes, fibroblasts, endothelial or vascular smooth muscle cells) and are required for normal physiological functioning of the post-natal heart (Camprecios et al., 2011; Kirabo et al., 2017; Sanchez-Soria and Camenisch, 2010). Of the EGFR/ErbB receptors, ErbB2 and ErbB4 appear to be the most abundant and of the ligands, EGF, HB-EGF and NRG-1 are thought to be most important in regulating heart function (Reichelt et al., 2017).
The EGFR/ErbB1 receptor and some of its ligands, especially EGF and HB-EGF, are expressed in the adult heart and thus, both ligand-activated signaling and ligand-independent transactivation of EGFR likely contribute to specific protective responses in the adult heart (for review see Reichelt et al., 2017). EGFR/ErbB1 receptor appears to be important in cardiomyocyte survival and contractile function with its cardiac-specific deletion in the adult heart resulting in cardiac dysfunction (Rajagopalan et al., 2008). In a recent study, cardiomyocyte-specific ErbB1 downregulation in mice led to impaired contractility (Guo et al., 2021). Similarly, conditional deletion of its ligand, HB-EGF, also resulted in cardiac contractile defects (Iwamoto et al., 2003) implying EGFR/ErbB1 receptors play a critical role in maintaining contractile homeostasis in the adult heart.
Endogenous or exogenously administered EGF-like ligands (e.g., EGF, HB-EGF, betacellulin and amphiregulin) and subsequent activation of EGFR/ErbB1, is thought to play a beneficial cytoprotective role in the heart against stress induced injury (Lorita et al., 2002; Pareja et al., 2003). For example, acute, high intensity stress can cause cardiac damage via elevated catecholamine release and cardiomyocyte death by apoptosis and/or necrosis (Liaudet et al., 2014). Administration of EGF can prevent the damage caused by intense and sustained β-adrenergic stimulation and catecholamine release in the heart by interfering with β -adrenergic signaling (Lorita et al., 2002; Pareja et al., 2003). Further, it was shown that activated EGFR/ErbB1 plays a critical role in the cardiac protection against the acute, high intensity stress induced in fighting mice (Pareja et al., 2003) and may also offer protection against cardiac stressors during hibernation (Childers et al., 2019). During hibernation, animals experience a reduction in blood circulation that makes the heart prone to multiple stressors. Increased EGFR phosphorylation (Y1086) was found to play a key role in activation of MAPK signaling, inhibition of downstream p-ELK1, and reduction in both p-BAD mediated pro-apoptotic signaling and caspase-9 apoptotic signaling in the heart of hibernating ground squirrels (Childers et al., 2019). Activation of cardioprotective EGFR signaling likely explains how these mammals cope with cardiac stresses during hibernation, which otherwise can lead to serious cardiac injury (Childers et al., 2019).
To examine the physiological role of ErbB2 signaling in the adult heart, rats with a ventricular-specific deletion of ErbB2 were generated (Crone et al., 2002). After physiological analysis, they discovered that these mutant mice hearts showed wall thinning, chamber dilation and decreased contractility indicative of dilated cardiomyopathy (Crone et al., 2002). Thus, increasing expression or activation of ErbB2 might prevent dilated cardiomyopathy and heart failure accordingly. Further, in a model of type 1 diabetes, the effect of acute activation or chronic inhibition of EGFR and ErbB2 signaling on heart function was also studied (Akhtar et al., 2012a). Recovery of cardiac function following I/R injury in diabetic hearts was significantly impaired, most likely as a result of reduced dimerization and signaling by cardiac ErbB2 and EGFR receptors. Chronic administration of selective pharmacological inhibitors of either ErbB2 or EGFR/ErbB1 in diabetic animals exacerbated cardiac recovery. In contrast, acute stimulation of EGFR/ErbB2 signaling with EGF improved cardiac recovery following I/R injury (Akhtar et al., 2012a). These findings confirmed the beneficial role or EGFR and ErbB2 receptors and downstream signaling via ERK1/2, p38 MAP kinase and AKT in mediating recovery of cardiac function that is normally impaired in diabetes (Akhtar et al., 2012b).
In another study using in H9c2 rat cardiomyoblasts, pretreatment with EGF attenuated H2O2-induced oxidative stress and inhibited ROS-induced cell death and H2O2-induced apoptosis through activating Nrf2 (Ma and Jin, 2019). In animal models of myocardial I/R, in vivo administration of EGF diminished infarct size and myocardial apoptosis (Ma and Jin, 2019). Taken together, these data demonstrate that EGF attenuates oxidative stress and cardiac I/R injury by reducing myocardial infarct size and improving cardiac function possibly via activation of Nrf2 (Akhtar et al., 2012b; Ma and Jin, 2019).
During cardiac I/R injury, the myocardium becomes depleted of zinc and the capacity to mobilise labile zinc is reduced indicative of zinc dyshomeostasis. Administration of zinc pyrithione can normalize zinc levels during I/R and also prevents apoptosis by increasing ErbB1/ErbB2 levels (Bodiga et al., 2015) further confirming the cardioprotective role of these ErbB receptors. Zinc pyrithione attenuated caspase activation, decreased the proteolytic degradation of ErbB2, enhanced activation and complexation (heterodimerization) of ErbB1/ErbB2 that resulted in increased myocyte survival after hypoxia/reoxygenation injury (Bodiga et al., 2015).
EGFR/ErbB receptors also mediate the beneficial effects of cardiac preconditioning (Benter et al., 2005c; Akhtar et al., 2012b). It is well known that ischemic preconditioning, which typically involves exposure of the heart to brief episodes of ischemia-reperfusion (I/R), protects the myocardium from the greater damaging effects of subsequent more prolonged I/R. In a rat model of type 1 diabetes, chronic in vivo administration of a specific pharmacological inhibitor of EGFR abrogated the cardiac benefits of preconditioning implying a critical role of EGFR/ErbB1 in cardiac preconditioning (Akhtar et al., 2012b). In models of cardiac preconditioning induced by pharmacological agents, the cardioprotective effects of GPCR agonists such as bradykinin, A1 adenosine receptors, apelin, acetylcholine and a δ-opioid peptide were found to be also mediated via transactivation of EGFR (Krieg et al., 2004; Cohen et al., 2007; Methner et al., 2009; Williams-Pritchard et al., 2011; Folino et al., 2018). For example, apelin-induced-reduction in infarct size and myocardial contracture via its GPCR (termed APJ) were prevented by the inhibition of EGFR, Src, MMP or RISK (reperfusion injury salvage kinase) pathways (Folino et al., 2018). Recently, the cardioprotective effects of myocyte-specific hypoxia-inducible factor 2A were additionally reported to be mediated via EGFR (Lee et al., 2020) further implicating EGFR/ErbB1 receptors in a protective role during myocardial I/R injury. Moreover, the fact that a pan-ErbB inhibitor, genistein, also blocked the cardioprotective effects of I/R preconditioning (Akhtar et al., 2012b), implied that other ErbBs receptors, beyond EGFR, might also be involved in mediating the beneficial effects of cardiac preconditioning.
Previously it was thought that only EGFR, ErbB2 and ErbB4 were expressed in the adult heart but subsequent studies have demonstrated that post-natal cardiomyocytes of mice express a functional ErbB3 protein as well (Camprecios et al., 2011). More recently ErbB3 gene expression has also been reported in normal human ventricular cardiac fibroblasts (Kirabo et al., 2017). Although methylation of ErbB3 gene has been reported in human dilated cardiomyopathy (Haas et al., 2013); the function of ErbB3 in the adult heart is only now being understood. For example the E3 ligase (or neuregulin receptor degradation protein-1; Nrdp1), that selectively targets ErbB3 (Diamonti et al., 2002), is upregulated following I/R injury. Mice over-expressing Nrdp1 in the heart exhibit higher infarct size as well as increased apoptosis and inflammatory cell influx following I/R (Zhang et al., 2011) implying that Nrdp1 is a pro-apoptotic signal in the heart during I/R injury and exerts its actions via degradation of ErbB3. More recently it was reported that ErbB3 is a cardioprotective/pro-survival factor against redox stress (Morano et al., 2017). ErbB3 gene expression was transiently increased in the adult heart after I/R injury (Morano et al., 2017). However, I/R reduced ErbB3 protein levels, whereas postconditioning (that was induced by brief cycles of I/R immediately after ischemia) prevented I/R-induced reduction in ErbB3 receptor protein. Furthermore, the transient over-expression of ErbB3 gene alone was able to enhance cell survival and attenuate mitochondrial dysfunction and apoptosis in H9c2 cells exposed to redox-stress (Morano et al., 2017). This study implied that ErbB3 acts beneficially as a cytoprotective and/or pro-survival factor in the heart (Morano et al., 2017).
A cardiomyocyte-specific conditional ErbB4 KO mouse model was also used to prove that ErbB4 is required for remodeling of cardiomyocytes and essential in maintaining myocardial contractile function in the post-natal heart (Garcia-Rivello et al., 2005). Despite the normal heart morphology evident at birth, conditional ErbB4 KO mice developed severe dilated cardiomyopathy and conduction abnormalities that lead to premature death (Garcia-Rivello et al., 2005).
The HB-EGF ligand binds to and triggers signaling via several ErbB receptors including EGFR/ErbB1 and ErbB4 receptors. In experiments with HB-EGF-null mice, over half of the mice lacking this ligand died within a week. The rest showed severe heart failure, enlarged cardiac valves and ventricular chambers with a resultant impairment of cardiac function. In contrast, administration of HB-EGF in WT mice increased phosphorylation of cardiac ErbB2, ErbB4, and to a lesser degree, of EGFR (Iwamoto et al., 2003) implying that HB-EGF-induced activation of multiple ErbB receptors is crucial for normal heart function.
The cardiac effects of NRG-1, another EGF-like ligand, have been extensively studied. The stimulation of ErbB signaling by NRG-1 appears crucial for cardiomyocyte survival and furthermore, is an important compensatory mechanism in the failing adult heart (De Keulenaer et al., 2019). Neuregulin ligand-mediated ErbB receptor signaling plays a key role in cellular functions such as proliferation, differentiation, migration and protection against cell death through controlling bcl-x splicing and bcl-2 family protein expression, as well as activation of the mammalian target of rapamycin (mTOR) that induces protein synthesis and hypertrophy (for reviews see Sanchez-Soria and Camenisch, 2010; De Keulenaer et al., 2019). Thus, through multiple effectors, NRG-1 induces pro-survival gene expression and enhances proliferation and survival of human cardiac fibroblasts (Fang et al., 2017; Kirabo et al., 2017; De Keulner et al., 2019). For example, NRG-1 counters the upregulation of endoplasmic reticulum (ER) stress markers like glucose-regulated protein 78 and cleaved caspase-12 in ventricular myocytes through ErbB4 receptors. Inhibition of EGFR/ErbB1 signaling or ErbB4 knockdown reversed the beneficial effects of NRG-1 in inhibiting ER stress in cultured neonatal cardiomyocytes (Fang et al., 2017). Moreover, in an in vivo rat model of cardiac I/R injury, intravenous NRG-1 administration significantly decreased ER stress and myocardial infarct size (Fang et al., 2017). Thus, NRG-1 has a protective role in I/R injury by inhibiting myocardial ER stress that is likely mediated by several ErbB receptors including ErbB4. Indeed, these pro-survival effects of NRG-1 have led to its consideration as a potential therapeutic agent for treatment of heart failure in pre-clinical and human clinical studies that have collectively shown that i. v., administration of this ligand significantly improved cardiac contractility and remodelling as well as attenuated mitochondrial dysfunction and apoptosis (Liu et al., 2006; Gao et al., 2010; Jabbour et al., 2011; Guo et al., 2012; Xiao et al., 2012; Hill et al., 2013; De Keulenaer et al., 2019). Additionally, and of relevance to the diabetic heart, is the potential glucose-lowering ability of NRG-1. Acute administration of this ligand decreased the glycaemic response to an oral glucose tolerance test in rats (Caillaud et al., 2016) implying that it might have added therapeutic value as a glucose-lowering agent in diabetic patients with heart failure.
Interestingly, monoclonal antibody and small molecule-based inhibitors of EGFR/ErbB receptors are now extensively used in the treatment of cancer (Roskoski, 2019; Kumagai et al., 2021). However, their use may lead to unwanted cardiac toxicities ranging from reduced LV ejection fraction to heart failure (Kenigsberg et al., 2017; Cuomo et al., 2019). In an early study, female (but strangely, not male) mice chronically administered either of two EGFR small molecule tyrosine kinase inhibitors, lead to physiological and pathological cardiac changes such as altered LV wall thickness and increased apoptosis (Barrick et al., 2008). Using a human-induced pluripotent stem cell-derived cardiomyocyte in vitro model, treatment with trastuzumab, a monoclonal antibody inhibitor of ErbB2, also aggravated cardiomyocyte damage, whereas activation of ErbB signaling with NRG-1 alleviated cardiomyocyte injury (Eldridge et al., 2014). In the clinic, trastuzumab is known to exhibit cardiac toxicity particularly in patients with abnormal LV function but cardiotoxicity with other ErbB inhibitors such as gefitinib (a small molecule EGFR-specific inhibitor) or even lapatinib (a dual EGFR-ErbB2 inhibitor) appears to be rare (Kenigsberg et al., 2017). Thus, studies on the cardiotoxicity of some ErbB receptor inhibitors are supportive of a cardioprotective role of ErbBs in the adult heart. However, since not all ErbB inhibitors exhibit cardiotoxicity, the possibility that the observed adverse effects on the heart might be due to other “off-target”, but drug-specific, effects remains to be elucidated.
The Detrimental Role of Epidermal Growth Factor Receptor in the Heart
The studies described in the preceding section (see also Table 1) clearly suggest a beneficial and necessary role of EGFR/ErbB/HER receptors in the developing heart. They further imply that acute or transient activation of EGFR/ErbB/HER receptor TKs and subsequent downstream activation of survival (or salvage/recovery) pathways such as PI3K/AKT/mTOR and RISK is required for mediating beneficial cardioprotective effects in I/R injury. In contrast, there is now a growing body of evidence that supports the notion that chronic or persistent overexpression/activation of EGFR/ErbB family of receptors can play a detrimental role (see Table 2) in the pathological heart including in cardiac hypertrophy, fibrosis and cardiac remodeling associated with diabetes (e.g., Kobayashi and Eguchi, 2012). As EGFR/ErbB/HER receptors are known drivers of cell growth and proliferation via mitogenic signaling through Ras/Raf/MAP kinases, AKT and ERK1/2, they play key role in mediating cardiac hypertrophy (Peng et al., 2016; Bi et al., 2020). For example, Ang II-mediated cardiac hypertrophy is reported to occur via Src-dependent EGFR transactivation through AKT and ERK pathways that can be reversed with pharmacological inhibitors of EGFR (Liang et al., 2015; Peng et al., 2016). In addition to Ang II, other members of the RAAS can also transactivate EGFR/ErbB receptors to impact cardiac function. In this regard, EGFR transactivation is known to be involved in aldosterone-induced NHE-1 (Na+/H+ Exchanger 1) stimulation that mediates oxidative stress and subsequent cardiovascular damage (De Giusti et al., 2011).
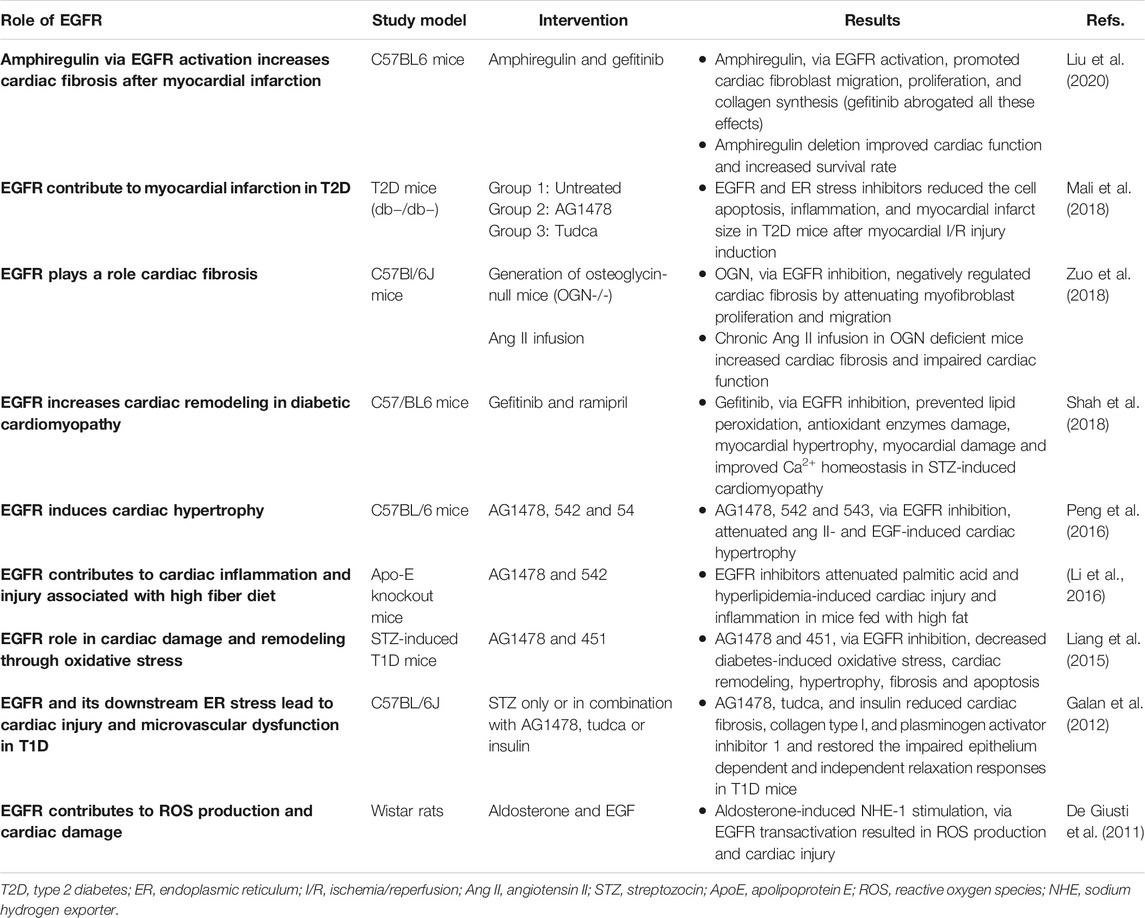
TABLE 2. A summary of selected in vivo studies highlighting the detrimental role of EGFR/ErbB signaling in the heart.
Hyperglycemia associated with diabetes is known to lead to the generation of reactive oxygen species (ROS) such as via NADPH oxidase (NOX) activity (see also The Link Between Epidermal Growth Factor Receptor & Nicotinamide Adenine Dinucleotide Phosphate Oxidase in Diabetes-Induced Vascular Dysfunction) that can lead to cardiovascular complications (Brownlee, 2005; Zhang et al., 2020; Babel and Dandekar, 2021). There is emerging evidence that EGFR inhibitors can prevent ROS formation and development of oxidative stress that leads to cardiac dysfunction (Liang et al., 2015; Shah et al., 2018). EGFR inhibition with gefitinib decreased hyperglycemia-induced cardiac remodeling by preventing oxidative stress-induced changes in the diabetic heart (Shah et al., 2018). Administration of gefitinib (a selective EGFR inhibitor) in C57/BL6 mice, significantly prevented lipid peroxidation and damage of antioxidant enzymes like glutathione and thioredoxin reductase. It also prevented myocardial hypertrophy and attenuated the diabetes-induced alterations in collagen deposition and myocardial remodeling. Administration of gefitinib also prevented depletion of SERCA2a (sarcoplasmic endoplasmic reticulum Ca2+ ATPase2a) and NCX1 (sodium-calcium exchanger-1) in streptozotocin-induced cardiomyopathy-indicative of improved Ca2+ homeostasis during myocardial contractility (Shah et al., 2018). These findings suggest that inhibition of EGFR can reduce cardiac damage in diabetic cardiomyopathy through balancing oxidant-antioxidant systems and attenuating subsequent hypertrophy and remodeling in the diabetic heart. Thus, gefitinib, and other clinically approved EGFR inhibitors that do not exhibit cardiotoxicity, may be considered for the potential treatment of diabetic cardiomyopathy.
Emerging evidence supports the view that generation of ROS and oxidative stress is strongly associated with the occurrence of endoplasmic reticulum (ER) stress (Chong et al., 2017). The ER is responsible for the correct folding, processing and trafficking of secretory and membrane-bound proteins to the Golgi apparatus, but when the capacity of this sub-cellular organelle to fold proteins becomes saturated, such as in oxidative stress, ER stress ensues that can lead to multiple pathologies (Navid and Colbert, 2017) including diabetes-induced cardiovascular complications (Xu et al., 2012; Sankrityayan et al., 2019). In a mouse model of type 1 diabetes, upregulation of EGFR tyrosine kinase led to induction of ER stress that resulted in diabetes induced cardiovascular dysfunction (Galan et al., 2012). Pharmacological inhibition of EGFR attenuated receptor phosphorylation and reduced ER stress, cardiac fibrosis, and microvascular dysfunction (Galan et al., 2012) implying a key role of EGFR in mediating ER stress and subsequent cardiac pathology. The detrimental role of EGFR in the pathogenesis of diabetes-induced heart damage was further confirmed by Liang et al. (2015). They reported that Ang II-induced cardiac hypertrophy, fibrosis and remodeling was mediated via enhanced signaling through a Src-dependent EGFR/AKT pathway. The fact that significant attenuation of these pathological changes occurred after treatment with gene silencing shRNAs or EGFR inhibitors provided strong evidence that aberrent EGFR signaling plays a crucial role in the pathogenesis of cardiac dysfunction and remodeling in diabetes (Liang et al., 2015). Additionally, EGFR signaling and ER stress are also reported to play a role in inducing myocardial infarction in type 2 diabetes (Mali et al., 2018). To investigate this role, T2D mice were treated with AG1478 (a specific EGFR inhibitor) or TUDCA (an ER stress inhibitor) for 2 weeks. After that, acute myocardial I/R injury was induced. Treatment with EGFR and ER stress inhibitors in T2D mice significantly reduced the extent of the myocardial infarct size compared to untreated T2D mice. Inhibition of EGFR and ER stress was associated with reduced myocardium p38 and ERK1/2 MAP-kinases activity, increased pro-survival signaling via AKT, and reduced inflammatory cell infiltration and apoptosis (Mali et al., 2018). By suggesting that EGFR signaling was detrimental in acute myocardial I/R injury, this study (Mali et al., 2018) was at variance with multiple other reports showing that EGFR signaling is beneficial in the recovery of cardiac function following I/R injury (Ma and Jin 2019; Akhtar et al., 2012b; Pareja et al., 2003; Lorita et al., 2002). The precise reasons for this discrepancy are not known but the use of different animal models and experimental conditions may be possible explanations. For example, Mali et al. (2018) used a T2DM (db−/db−) mouse model with regional ischemia compared to a T1DM (streptozotocin-induced) rat model with global ischemia used by Akhtar et al. (2012b). How these potential variables precisely impact on EGFR/ErbB signaling requires further study. Nonetheless, collectively these studies suggest that targeting of EGFR and ER stress may represent novel therapeutic strategies in the potential treatment of diabetes-induced cardiac pathologies.
Obesity is a leading risk factor for the development of type 2 diabetes and its cardiovascular complications (Leitner et al., 2017; Drucker, 2021). In a study aiming to determine if EGFR mediates the pathogenesis of hyperlipidemia/obesity-related cardiac diseases, EGFR inhibition significantly ameliorated myocardial fibrosis, apoptosis, and inflammation in two rat models of obesity (Li et al., 2016). Thus, consistent with reports in models of diabetes, this study also demonstrated a harmful effect of EGFR activation in the pathogenesis of obesity-induced cardiac pathology (Li et al., 2016) and thus, potential use of EGFR inhibitors for the treatment for obesity-related cardiovascular complications may be warranted.
EGFR signaling is additionally thought to be important in the cardiac actions of osteoglycin (OGN), a small leucine-rich proteoglycan that also regulates bone and glucose homeostasis (Lee et al., 2018; Zuo et al., 2018). OGN negatively regulates cardiac fibrosis by attenuating cardiac myofibroblast (CMF) proliferation and migration through inhibition of EGFR signaling. Thus, OGN deficiency promoted EGFR and ERK1/2 phosphorylation in CMFs cultured from adult and neonate mice. In contrast, restoration of OGN in OGN-null CMFs resulted in inhibition of cell migration and proliferation through decreased EGFR signaling (Zuo et al., 2018). Stimulation of EGFR/ErbB receptors by amphiregulin (AR), another member of EGF family of ligands, induced cardiac fibrosis after myocardial infarction (Liu et al., 2018; Liu et al., 2020) highlighting the detrimental role of EGFR/ErbBs in cardiac pathology. EGFR transactivation was also involved in mediating the non-canonical, cardiomyocyte hypertrophy-promoting, effects of the death receptor 5 that normally mediates apoptosis (Tanner et al., 2019). Interestingly a very recent study showed that increased EGFR signalling in the rat brain, namely within the hypothalamic paraventricular nucleus (PVN), may also play a key role in mediating myocardial-infarction-induced heart failure through enhancing ERK1/2-induced sympathetic overactivity (Yu et al., 2021). Selective gene silencing of EGFR in the PVN led to a correction of multiple central and peripheral markers associated with heart failure (Yu et al., 2021) implying that targeting brain EGFR might represent a novel approach in the treatment of MI-induced HF. Collectively, the above studies suggest a critical role for both peripheral and central EGFR/ErbB signaling in the development of cardiac pathologies, and further imply that these RTKs may serve as a “hub” or “relay” for other signaling molecules/inputs that induce cardiac complications in diabetes.
As a summary of their cardiac actions, Figure 1 highlights the overall beneficial and detrimental actions of EGFR/ErbB2/HER receptors and downstream effector signaling in the heart (see also perspectives and concluding remarks section below).
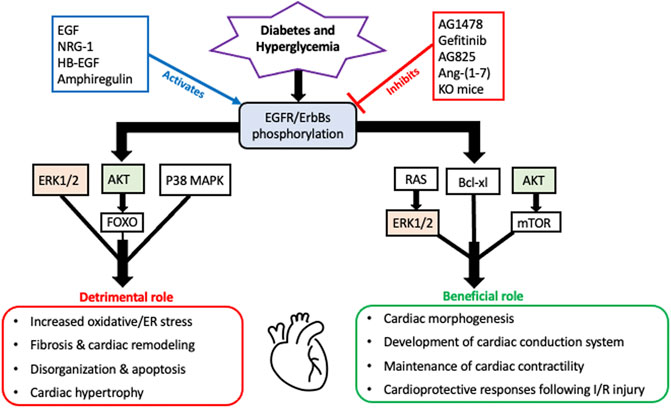
FIGURE 1. A schematic highlighting the overall beneficial and detrimental actions of EGFR/ErbB receptors and their downstream signaling effectors in the diabetic heart. It appears that all four EGFR/ErbB receptors are necessary for the development of the heart during embryogenesis and beneficial for the proper functioning of the adult heart including a key role in maintenance of cardiac contractility. EGFR/ErbB2 signaling is also essential for normal cardiomyocyte function. Acute stimulation of EGFR/ErbB receptor signaling is also essential for activation of salvage pathways and cardiac recovery following I/R injury. In addition to these beneficial actions, EGFR/ErbB receptor signaling can also have detrimental outcomes. Persistent or chronic upregulation of EGFR/ErbB receptor signaling has been involved in the development of cardiac dysfunction and pathologies including fibrosis, remodelling and hypertrophy. Several key downstream effectors are involved in mediating both actions with some notable overlaps such as AKT and ERK1/2 (highlighted)- implying that they too can have dual actions depending on the pathophysiological context akin to the role of their upstream EGFR/ErbB receptors (see main text for further details and references).
Diabetes-Induced Vascular Dysfunction
Both T1DM and T2DM can lead to potentially life-threatening microvascular and macrovascular complications such as retinopathy, neuropathy, nephropathy, as well as coronary heart disease and stroke (Kosiborod et al., 2018; Colom et al., 2021). Similar to that described in the heart (see above), diabetes-induced vascular complications generally result from chronic hyperglycemia (Van Wijngaarden et al., 2017) though repeated transient excursions into hyperglycemia and glycemic variability also increase the risk of developing vascular complications (Holman et al., 2008; Gray and Jandeleit-Dahm, 2014; Kenny and Abel, 2019; Babel and Dandekar, 2021). Correction of hyperglycemia in diabetic patients also does not completely restore the risk of developing vascular complications to baseline (Ceriello, 2005; Brownlee and Hirsch, 2006; Chalmers and Cooper, 2008; Kenny and Abel, 2019) implying that hyperglycemia-induced vascular changes, most likely at a molecular level, are long-lasting–a phenomenon termed “glycemic” or “metabolic memory”. Although there appears to be some differences in the pathology of micro-and macro-vascular dysfunction (e.g., the latter is most common in T2DM which comprises around 85–90% of all diabetic patients), both involve damage to the endothelium, the innermost lining of blood vessels, that is involved in many important vascular functions including maintenance of vascular tone (Shi and Vanhoutte, 2017); indeed, endothelial dysfunction represents a significant underlying cause of organ dysfunction and failure in several pathologies including diabetes and SARS-CoV2 (COVID19) infections (Shi and Vanhoutte, 2017; Jin et al., 2020; Nagele et al., 2020). In the vasculature, key dysfunctional changes of the tunica media such as increased vascular smooth muscle responsiveness to endothelium-dependent vasoconstrictors, are early features of diabetic vasculopathy (Cooper et al., 2001; Lee et al., 2012). Since diabetes-induced cardiac dysfunction also involves endothelial dysfunction, it is not surprising that endothelial dysfunction associated with micro- or macro-vascular complications most likely arises from a similar, though not necessarily identical, concoction of underlying biological mechanisms. Of note in the diabetic vasculature are increased intracellular Ca2+, diminished NO synthesis, increased polyol pathway flux, altered cellular redox state (ROS/oxidative and ER stress), prostanoid synthesis, accelerated nonenzymatic formation of advanced glycation end products (AGEs), increased formation of diacylglycerol and activation of specific protein kinase C (PKC) isoforms, inflammation and enhanced apoptosis (for review see Babel and Dandekar, 2021; Shi and Vanhoutte, 2017; Forbes and Cooper, 2013, Giacco and Brownlee, 2010; Brownlee, 2005). Since endothelial and vascular smooth muscle (VSM) cells (VSMCs) are closely linked functionally, many of the molecular pathways effecting endothelial dysfunction are also altered in VSMCs implying that a combination of endothelial and smooth muscle dysfunction is ultimately responsible for the abnormalities of vascular function in diabetes. Indeed, in patients with T2DM, VSM dysfunction was noted in both the micro- and macro-vasculature but was more pronounced in the microvasculature-i.e. blood vessels with diameter of < 150 µm (Montero et al., 2013). In terms of mechanisms, in addition to ROS generation and oxidative stress, diabetes-induced alterations in signaling by growth factors, RTKs, peptides of RAAS as well as other GPCRs that exert significant influence on intracellular calcium levels, ROS, ER stress, inflammation and apoptosis appear to be involved in mediating VSMC dysfunction (Babel and Dandekar, 2021). For example, signalling via vascular endothelial growth factor (VEGF) appears important in mediating retinopathy in diabetes (reviewed in Wong et al., 2016), and hyperglycemia-induced up-regulation of GPCR signalling by the RAAS peptide, Ang II, has been reported in VSMCs (Montezano et al., 2014; Touyz et al., 2018). Since it is now known that GPCRs can initiate transactivation of the EGFR (e.g., Akhtar et al., 2015; Forrester et al., 2016), which is expressed in both endothelial cells and VSMCs (e.g., Akhtar et al., 2012a; Schreier et al., 2021), there is strong indications that EGFR signalling may also be a key player in mediating vascular pathology in diabetes (Akhtar and Benter, 2013; Matrougui, 2010; Schreier et al., 2014; Schreier et al., 2018).
Role of Epidermal Growth Factor Receptors in the Diabetic Vasculature
EGFR/ErbB receptor signaling that typically regulates cell growth, differentiation and proliferation is essential to the normal physiologically development and functioning of the vasculature (Forrester et al., 2016). However, its chronic dysregulation appears to have a detrimental role in mediating diabetes-induced vascular dysfunction and remodelling (Benter et al., 2005; Akhtar and Benter, 2013; Matrougui, 2010; Akhtar et al., 2019; Schreier, et al., 2014; Schreier et al., 2018) see also Table 3.
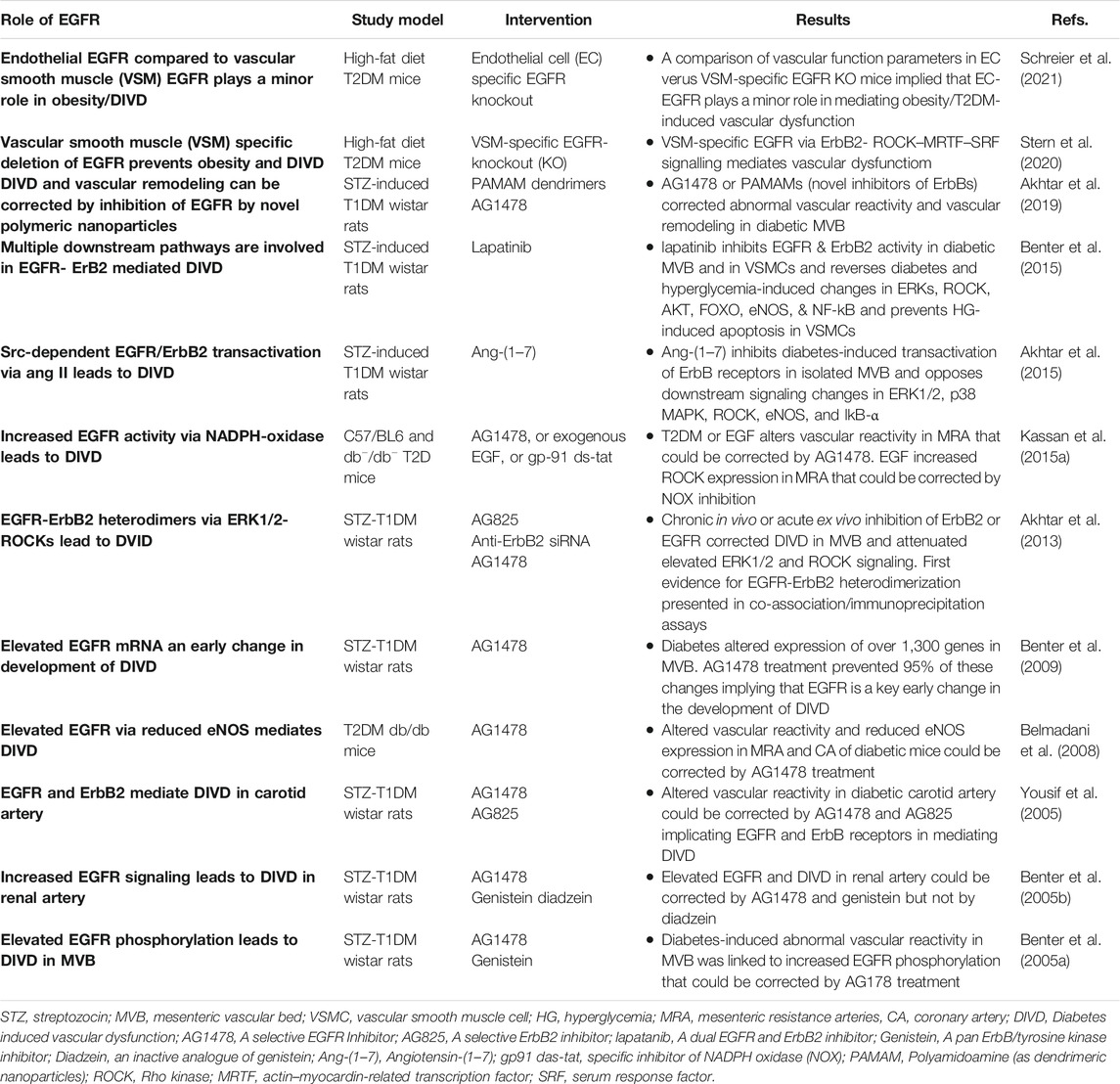
TABLE 3. A summary of selected studies implicating EGFR signaling in diabetes-induced vascular dysfunction.
Probably the early clues to the involvement of EGFR/ErbB receptors in diabetes-induced complications were the observation that excretion of EGF ligand was abnormal in many patients with diabetes (Lev-Ran et al., 1990) and that EGF gene expression was elevated in the mesenteric artery of rats bearing type 1 diabetes (Gilbert et al., 2000). Further, since vascular proliferation and remodeling (e.g., via hypertrophy, hyperplasia and fibrosis) leads to altered vascular contractility in diabetes, of note were the studies showing a pro-contractile role for EGF/EGFR signaling (Berk et al., 1985; Florian and Watts, 1999). For example, EGF via stimulation of EGFR elicited potent vasoconstriction in aortic strips from a rat model of hypertension (Florian and Watts, 1999); and EGFR activation via MMP-2 reportedly increased ROS formation and facilitated contraction in non-diabetic aortas (Prado et al., 2018). Additionally, there is also evidence in the non-diabetic vasculature that acute EGFR signaling may mediate the contractions of GPCR ligands such as Ang II and endothelin (ET-1) (Kawanabe et al., 2004; Lucchesi et al., 2004) that also play an important role in mediating diabetes-induced vascular dysfunction. A recent study also suggests that dysregulation of vascular, as opposed to endothelial, EGFR might be the dominant factor in the development of vascular pathologies associated with obesity (Schreier et al., 2021) an important risk factor for developing T2DM.
The first in vivo reports demonstrating a direct link between EGFR signaling and diabetes-induced vascular dysfunction appeared in 2005 which showed that enhanced EGFR/ErbB1 signaling was a key mediator of diabetes-induced vascular dysfunction as pharmacological inhibition of this RTK was corrective of this pathology in the mesenteric vascular bed as well as in renal and carotid arteries (Benter et al., 2005a; Benter et al., 2005b; Yousif et al., 2005). This initial research on vascular EGFR was conducted in a preclinical model of type 1 diabetes (streptozotocin-induced diabetes in rats) and its findings were subsequently replicated in the vasculature of animal models of type 2 diabetes (Belmadani et al., 2008; Palen and Matrougui, 2008; Choi et al., 2012; Galan et al., 2012; Kassan et al., 2015a; Stern et al., 2020), implying that dysregulated EGFR/ErbB1/HER1 signaling was a common mediator of vascular complications in both Type 1 and Type 2 diabetes. Further, through gene expression profiling studies of diabetic mesenteric vasculature, it was demonstrated that EGFR1/ErbB1/HER1 inhibition could normalize approximately 90% of the transcriptomic changes that occurred during the development of diabetes-induced vascular abnormalities (Benter et al., 2009). In addition, increased gene expression of EGFR1/ErbB1/HER1 appeared to be a critical early mRNA change resulting in diabetes-induced vascular dysfunction (Benter et al., 2009). Studies from the same group on the underlying mechanisms, identified that EGFR1/HER1 did not act alone, rather heterodimerization with ErbB2 (its preferred dimerization partner) and subsequently signaling via multiple pathways including PI3K/AKT were key mediators of diabetes-induced vascular complications (Akhtar et al., 2013; Akhtar et al., 2015). More recently, these authors showed that all ErbB receptors are activated by hyperglycemia (Akhtar et al., 2015; Akhtar et al., 2020b) and that pan-ErbB inhibitors can reverse these changes and correct vascular dysfunction in a rat model of type 1 diabetes (Akhtar et al., 2020b; Akhtar et al., 2019) implying a possible role for all EGFR/ErbB receptors in mediating diabetes-induced vascular dysfunction.
Src family of non-receptor tyrosine kinases appear to be key upstream effectors of EGFR/ErbB RTKs as hyperglycemia/diabetes is known to induce EGFR/ErbB signaling via a Src-dependent mechanism (Akhtar et al., 2012a; Akhtar et al., 2015; see also The Interplay of Epidermal Growth Factor Receptor and Angiotensin 1-7, a Member of the Renin-Angiotensin-Aldosterone System, in Diabetes-Induced Vascular Dysfunction below). As to the downstream effectors of EGFR/ErbB signaling in the diabetic vasculature, several studies have now implied that multiple downstream pathways are likely involved including dysregulation of NO, Rho kinases (ROCKs), ERK1/2 and PI3K/AKT pathways (Akhtar and Benter, 2013; Benter et al., 2015; Matrougui, 2010; Schreier et al., 2014; Schreier et al., 2018). For example, diabetes enhances phosphorylation of EGFR1 and ErbB2, formation of EGFR/ErbB2 heterodimers and subsequent elevation in ERK1/2 and ROCK signaling leading to dysfunction in the mesenteric vasculature of T1DM rats (Akhtar et al., 2013; Benter et al., 2015). Administration of AG1478, a selective EGFR inhibitor, or AG825, a specific ErbB2 inhibitor, or Fasudil, a ROCK inhibitor, or PD98059, an ERK1/2 signaling inhibitor, all attenuated the observed changes associated with diabetes-induced vascular dysfunction (Akhtar et al., 2013) indicating the importance of these effectors in this pathology. Similarly, in a mouse model of T2DM, EGFR inhibition rescued abnormal ROCK activity and restored vascular function (Kassan et al., 2015b). Collectively, these results implied that EGFR/ErbB2-ERK1/2-ROCK pathway is an important mediator of diabetes-induced vascular dysfunction. The role of ROCK as a downstream effector of EGFR/ErbB2 heterodimers was also confirmed recently in a mouse model of high-fat diet induced obesity/type 2 diabetes bearing vascular smooth muscle-specific deletion of EGFR (Stern et al., 2020). This study showed that EGFR expressed in vascular smooth muscle cells mediates vascular remodeling via a ROCK-dependent activation of serum response factor - a transcription factor that regulates cell growth and proliferation (Stern et al., 2020). Interestingly, administration of the ROCK inhibitor, Fasudil, in the early stages of experimental diabetes was found to markedly suppress vascular hyperreactivity, increase tissue perfusion, and prevent organ damage whereas late-stage Fasudil administration had little or no effect (Li et al., 2015). This observation is suggestive of a threshold of vascular damage beyond which recovery of function is limited with ROCK inhibitors (Li et al., 2015) as well as possibly with other types of pharmacological interventions. Also of interest here is that EGF-ligand induced vasoconstriction is dependent on ROCK signaling. In rat aortic rings, EGF induced Ca2+ sensitization of VSMCs via a Rho-kinase-dependent inactivation of myosin light chain phosphatase (MLCP) mediated by the EGFR-MEK-ERK1/2 signaling pathway (Sasahara et al., 2015). Taken together, these studies suggest an important role of hyperglycemia-induced Src-dependent transactivation of EGFR/ErbB receptors and a downstream ROCK-dependent pro-contractile action in mediating diabetes-induced vascular dysfunction.
Endothelial Nitric Oxide Synthase as an Effector of Epidermal Growth Factor Receptor or ErbB2 in Diabetes-Induced Vascular Dysfunction
Endothelial and thereby vascular dysfunction is characterised by a decreased production of nitric oxide (NO). In the vasculature, NO is mainly produced from l-arginine by endothelial nitric oxide synthase (eNOS) in a Ca2+/calmodulin-dependent protein kinase II (CaMKII) dependent reaction (Forstermann and Sessa, 2012; Zhao et al., 2015; Murthy et al., 2017). NO-mediated signaling is fundamental in maintaining vascular function as it can potentially prevent vascular inflammation, thrombosis, and cell proliferation (Bian et al., 2008; Zhao et al., 2015). eNOS activity is controlled through multi-site phosphorylation. For instance, enhanced phosphorylation of eNOS at Thr495 and decreased phosphorylation at Ser1177 site in the vascular endothelium leads to inadequate NO synthesis and impaired endothelium-dependent relaxation (Long et al., 2007). Diabetes reduces eNOS Ser1177 phosphorylation, thereby decreasing NO levels in endothelial cells and VSMCs (Tousoulis et al., 2012; Benter et al., 2015). Additionally, several studies have now determined that eNOS is a downstream effector of EGFR or ErbB2 signaling in T1DM and T2DM (Belmadani et al., 2008; Galan et al., 2012; Benter et al., 2015), and that exaggerated ErbB2-EGFR heterodimerization in the diabetic vasculature leads to diminished NO activity (Benter et al., 2015). Administration of lapatinib, which is a dual inhibitor of EGFR and ErbB2 RTKs, ameliorates vascular dysfunction by normalizing the decreased NO levels in the diabetic vasculature (Benter et al., 2015). Regarding restoration of NO activity in the diabetic vasculature by selective EGFR inhibitors, studies have shown that chronic in vivo AG1478 treatment rescued eNOS phosphorylation and expression (Belmadani et al., 2008; Galan et al., 2012) whereas acute in vitro AG1478 treatment appeared not to (Kassan et al., 2015a). It is worth mentioning that ROS generation by NADPH oxidase accounts partly for the reduced NO bioavailability in the vascular bed (Kassan et al., 2015a), consequently inducing dysfunction of the vascular endothelium (Sena et al., 2008; Akar et al., 2011; Meyrelles et al., 2011). Interestingly, a CaMKII inhibitor, KN-93, was also able to prevent the development of abnormal vascular reactivity in a rat model of diabetes and hypertension (Yousif et al., 2008) implying that NO production might also involve CaMKII-independent pathways in such pathologies.
PI3K as an Effector of Epidermal Growth Factor Receptor in Diabetes-Induced Vascular Dysfunction
PI3Ks are lipid kinases that belong to a group of enzymes involved in the regulation of multiple signaling cascades that control multiple cellular processes including cell growth, proliferation, motility, and survival. It is well known that PI3K/AKT signaling is downstream of EGFR/ErbB signalling (Roskoski 2019) and also can activate eNOS and subsequent NO generation to regulate vascular function (Kobayashi et al., 2005). Impairment in PI3K signaling pathway has been demonstrated in aortas of diabetic mice (Kobayashi et al., 2004). The activated PI3K-Akt signaling pathway can also improve insulin sensitivity and protect the vascular endothelium (Feng et al., 2018).
Activation of P13K/Akt/eNos pathway, such as by components of green tea, has been reported to have beneficial effects on diabetes-induced vascular dysfunction (Bhardwaj et al., 2014; Zhang and Zhang, 2020). For example, (-)-Epigallocatechin-3-gallate (EGCG), a highly effective component in green tea that has anti-inflammatory as well as antioxidant and free radical scavenging properties, was able to inhibit eNOS uncoupling and prevent hyperglycemia-induced endothelial dysfunction and apoptosis by activating the PI3K/AKT/eNOS pathway (Zhang and Zhang, 2020). Similarly, in a rat model of T1DM, chronic administration of catechin, another key component of green tea, markedly attenuated diabetes-induced vascular dysfunction and vascular oxidative stress via activation of endothelial PI3K signaling and the subsequent downstream eNOS signaling system that generates NO (Bhardwaj et al., 2014). Although PI3K-eNOS signaling can be a downstream consequence of EGFR activation, the relationship between EGFR activation in diabetic vasculature and administration of green tea components was not investigated in these studies. Nonetheless, these studies suggest that PI3K signalling is beneficial in maintaining vascular function as its attenuation is associated with diabetes induced vascular complications.
In contrast to these studies, in a rat model of TIDM, chronic selective inhibition of PI3K with LY294002 significantly attenuated the development of diabetes-induced abnormal vascular reactivity in the carotid artery (Yousif et al., 2006) implying that that PI3K pathway might also mediate vascular dysfunction. The reasons for this discrepancy are not clear but several reports have now confirmed AKT, and by implication PI3K to which it often coupled, as a downstream player in ErbB receptor-dependent vascular dysfunction in models of diabetes (Benter et al., 2015; Kasan et al., 2015b; Amin et al., 2011). For example, in the diabetic mesenteric vascular bed, hyperglycemia-induced upregulation of Akt signaling was prevented by lapatinib, a dual inhibitor of EGFR and ErbB2 (Benter et al., 2015). With the caveat that AKT signaling does not always have to be coupled to PI3K, these studies are suggestive of a possible detrimental role of PI3K/AKT pathway in diabetes-induced vascular dysfunction. Supportive of this notion, is the finding that ANG II, that is known to be elevated in diabetes, increases aortic contractile responses via PI3-kinase pathway in a rat model of diabetes with systemic hyperinsulinemia (Kobayashi et al., 2006). Collectively, the data on the role of PI3K in mediating vascular dysfunction appears contradictory and may be indicative of a dual role for this molecule whereby depending on the context it too may behave as a good guy or a bad guy, akin to the role of EGFR/ErbB receptors in cardiovascular complications as discussed herein.
Interplay of Epidermal Growth Factor Receptor and Forkhead Transcription Factors in Diabetes-Induced Vascular Dysfunction
FOXO3 is a pro-apoptotic protein that belongs to the forkhead family of transcriptional regulators which are critical mediators of oxidative stress (Ponugoti, et al., 2012). FOXO activity, amongst others, is regulated via phosphorylation, whereby increased phosphorylation renders it inactive via mechanisms including nuclear exclusion, polyubiquitination, and degradation (Oellerich and Potente, 2012). ROS/oxidative stress can induce FOXO3 phosphorylation which results in its release from the 14–3-3 binding protein, and subsequent translocation to the nucleus where it modulates target gene expression (Ponugoti, et al., 2012). The role of FOXO3 as a downstream effector of EGFR/ErbB2 receptors was studied in the mesenteric vascular bed of T1DM rats (Benter et al., 2015). Diabetes-induced vascular dysfunction arising from enhanced EGFR/ErbB2 signaling also led to downstream activation of FOXO3 (as evidenced by reduced phosphorylation at Ser253) through an AKT-independent manner that ultimately leads to apoptosis and vascular dysfunction (Benter et al., 2015). Hyperglycemia-mediated decrease in FOXO3A phosphorylation and increased total FOXO3A levels could be reversed upon lapatinib treatment (a dual inhibitor of EGFR and ErbB2 receptors) (Benter et al., 2015). Thus, FOXO-mediated pro-apoptotic signaling is likely an important downstream contributor to EGFR/ErbB receptor-mediated vascular dysfunction in diabetes.
The Link Between Epidermal Growth Factor Receptor and NF-kB Transcription Factor in Diabetes-Induced Vascular Dysfunction
The Nuclear Factor Kappa B (NF-kB) transcription factor is a key player in the development of diabetes-induced vascular complications through regulation of many cellular processes such as inflammation, cell growth, proliferation, and apoptosis (Suryavanshi and Kulkarni., 2017; Taniguchi and Karin, 2018). In unstimulated cells, inactive NF-kB is bound in the cytoplasm to inhibitory proteins in the inhibitor of kB (IkB) family. Activation of NF-kB occurs via signal transduction pathways that stimulate the IkB kinase (IKK) complex resulting in a series of reactions that ultimately cause degradation of IkB and releasing of active NF-kB. Aberrant NF-kB activity has been implicated in many diseases including diabetes and its complications. Chronic hyperglycemia is known to activate NF-kB that triggers expression of pro-inflammatory cytokines and pro-apoptotic genes. It can also induce calcification of endothelial cells leading to endothelial and vascular dysfunction (Suryavanshi and Kulkarni., 2017). In the vasculature of T1DM rats, diabetes leads to the enhanced phosphorylation of IkB-α (Benter et al., 2015), a component of IKK complex and a universal marker of NF-kB activation. Data from this study illustrated that the increased ErbB2-EGFR hetero-dimerization in the vasculature leads to elevated NF-kB activation, which eventually induced apoptosis and vascular dysfunction (Benter et al., 2015). This was evidenced by in vivo and ex vivo lapatinib administration which attenuated the increased phosphorylation of IkB-α in VSMCs grown under high glucose conditions (Benter et al., 2015). Furthermore, administration of BAY 11–7,082, an NF-kB antagonist, corrected the altered vascular responsiveness to norephinephrine in the diabetic mesenteric vascular bed and hyperglycemia-induced apoptosis in VSMCs (Benter et al., 2015). Thus, it was determined from these results that NF-kB is downstream of EGFR-ErbB2 signaling in T1DM (Benter et al., 2015). These findings were also confirmed in coronary arteries of mice bearing T2DM (Kassan et al., 2015b). Intriguingly, it was determined that eNOS signaling was coupled with NF-kB activation in the diabetic mesenteric vasculature, where eNOS was shown to be upstream of NF-kB in mediating vascular dysfunction associated with diabetes (Benter et al., 2015).
The Link Between Epidermal Growth Factor Receptor & Nicotinamide Adenine Dinucleotide Phosphate Oxidase in Diabetes-Induced Vascular Dysfunction
Nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (NOX) enzyme complex comprises of several sub-units, namely, p22phox, NOX2; p47phox; p67phox; and p40phox; also known as NOX1, NOX2, NOX3, NOX4, and NOX5 respectively (Volpe et al., 2018). It is well established that the NOX family are major producers of reactive oxygen species that mediate cardiovascular complications associated with diabetes (Volpe et al., 2018; Iacobini et al., 2021). Several studies have now shown that EGFR inhibition leads to a reduction in NOX activity in models of diabetes-induced. cardiovascular complications (Belmadani et al., 2008; Choi et al., 2012; Galan et al., 2012; Kassan et al., 2015a)- implying that EGFR/ErbB receptor activation and subsequent NOX-mediated ROS/oxidative stress are critical in mediating vascular dysfunction associated with diabetes. In the mesenteric artery, mRNA levels of Nox2 and Nox4 and NADPH oxidase activity were upregulated in a mouse model of T1DM and reduced upon EGFR inhibition (Galan et al., 2012). Similarly, in an experimental model of TD2M, selective EGFR inhibition improved vascular function and reduced p22phox-NADPH expression (Kassan et al., 2015b). Collectively these studies provide a direct link between EGFR and NADPH oxidase in mediating oxidative stress that leads to diabetes-induced vascular dysfunction.
The Interplay of Epidermal Growth Factor Receptor and Angiotensin 1-7, a Member of the Renin-Angiotensin-Aldosterone System, in Diabetes-Induced Vascular Dysfunction
The renin-angiotensin-aldosterone system (RAAS) is a key regulator of homeostasis within the cardiovascular system (Ferrario et al., 2010; Akhtar et al., 2020a; Paz Ocaranza 2020). Its main peptide, Angiotensin II (Ang II), is known to be detrimental in the diabetic vasculature where it mediates oxidative stress, pro-inflammatory signaling and vasoconstriction. Enhanced signaling via this octapeptide in the diabetic vasculature involves transactivation and cross-talk with EGFR/ErbB/HER family of receptors (Akhtar et al., 2012a; Akhtar et al., 2015; Akhtar et al., 2016; Sur and Agrawal, 2014).
Ang II-mediated detrimental effects on the vasculature are exerted through the classical arm of the RAAS constituting angiotensin converting enzyme (ACE)/Ang II/Angiotensin II type 1 (AT1) receptors. The other arm of the RAAS that counter-regulates or opposes the actions of Ang II involves the heptapeptide, Angiotensin-(1–7) and comprises ACE2/Ang-(1–7)/Mas receptor (for recent review see Akhtar et al., 2020a). Ang-(1–7) can be derived from the catalytic metabolism of Ang II via the actions of angiotensin converting enzyme 2 (ACE2) (Ferrario et al., 2010). Ang-(1–7) induces vasodilation and attenuates Ang II-induced vasoconstriction in animal models (Benter et al., 1993). In humans, Ang-(1–7) also improves insulin-stimulated endothelium-dependent vasodilation and attenuates endothelin-1–dependent vasoconstrictor tone in obese patients (Schinzari et al., 2018). In endothelial cells Ang-(1–7) increases production of nitric oxide and prostacyclin and in vascular smooth muscle cells it inhibits pro-contractile and pro-inflammatory signalling (Benter et al., 1993; Benter et al., 1995a; Ferrario et al., 2010). It is known that Ang-(1–7) exhibits antihypertensive, antithrombotic and antiproliferative properties and can correct abnormal vascular reactivity including that observed in diabetes (Benter et al., 1995a; Benter et al., 1995b; Benter et al., 2006; Benter et al., 2007; Benter et al., 2008; Benter et al., 2011; Chappell et al., 2014; Dhaunsi et al., 2010; Patel et al., 2014; Yousif et al., 2012; Yousif et al., 2014). In animal models of diabetes, Ang-1-7 can attenuate NADPH oxidase and NF-kB activity and prevent vascular dysfunction without markedly correcting hyperglycemia (Benter et al., 2007; Benter et al., 2008; Al-Maghrebi et al., 2009; Yousif et al., 2014). Although the precise mechanistic details through which Ang-(1–7) exerts its beneficial actions are not yet fully elucidated, it appears that Ang-(1–7) prevents diabetes-induced vascular dysfunction, at least in part, via inhibiting Ang II-mediated transactivation of EGFR/ErbB family of receptor tyrosine kinases (Akhtar et al., 2012a; Akhtar et al., 2015). In addition to inhibiting EGFR phosphorylation at multiple tyrosine sites in the vasculature of diabetic animals, Ang-(1–7) inhibited ErbB2 phosphorylation at tyrosine residues Y1221/22, Y1248, Y877, with modulation of associated downstream signaling pathways involving ERK1/2, p38 MAPK, ROCK, eNOS, and lkB-α in the diabetic mesenteric vascular bed (Akhtar et al., 2015). Furthermore, high glucose- or diabetes-induced ErbB3, and Erbb4 transactivation could be ameliorated by Ang-(1–7) treatment thereby suggesting that Ang-(1–7) acts as a pan-inhibitor of the EGFR/ErbB/HER family of receptor tyrosine kinases (Akhtar et al., 2015). More recently other pan-ErbB inhibitors have been shown to be beneficial in reversing or preventing diabetes-induced vascular dysfunction (Akhtar et al., 2019; Akhtar et al., 2020b) and as such pan-ErbB inhibition might prove to be a useful strategy in the treatment of diabetes induced vascular complications.
As to the mechanism of diabetes-induced transactivation of ErbB receptors, high glucose-mediated ErbB2 transactivation occurred via a Src-dependent mechanism in VSMCs as evidenced by increased phosphorylation of Src at Y416. Ang-(1–7) acting via its MAS receptor blocked the phosphorylation of Src at this site-an upstream effector of ErbB2 transactivation (Akhtar et al., 2015). Thus, it was proposed that Ang-(1–7) inhibited diabetes or hyperglycemia induced transactivation of ErbB receptors in the diabetic vasculature by acting as an inhibitor of Src (Akhtar et al., 2012a; Akhtar et al., 2015). Subsequent studies in cardiac fibroblasts (Tao et al., 2014) and in activated macrophages (Souza and Costa-Neto, 2012) further supported the notion that Ang-(1–7) can inhibit Src phosphorylation. Recent studies in other disease models suggest that inhibition of Src-dependent EGFR transactivation by Ang-(1–7) may be a general mechanism of action of this heptapeptide (Kilarkaje et al., 2013; Yousif et al., 2014; El-Hashim et al., 2019) and may represent an important mechanism by which it may exert its beneficial effects in diabetes-induced vascular complications.
It is noteworthy that, despite its inhibitory effects on EGFR and other ErbB receptors (Akhtar et al., 2012b; Akhtar et al., 2015), Ang-(1–7) exerts considerable cardioprotective effects in models of diabetes (Dhaunsi et al., 2010; Yousif et al., 2012; Benter et al., 2014; Abwainy et al., 2016). Thus, the precise mechanisms underlying this phenomenon, especially as to how Ang-(1–7) interplays with the dual role of EGFR/ERbB signaling in the CVS, remain to be elucidated.
The Arachidonic Acid Metabolite, 20-HETE and Its Interplay With Epidermal Growth Factor Receptor in Diabetes-Induced Vascular Dysfunction
The arachidonic acid metabolite, 20-HETE (20-hydroxy-5, 8, 11, 14-eicosatetraenoic acid), plays an important role in controlling cardiovascular function (Rocic and Schwartzman, 2018; Yousif et al., 2009a; Yousif et al., 2009b). In blood vessels, it acts as a potent vasoconstrictor, regulates myogenic tone and also potentiates the vasoconstrictor response to peptides like angiotensin II and endothelin (Roman 2002; Fan et al., 2016). 20-HETE induces endothelial dysfunction and adverse vascular remodeling that leads to increased blood pressure (Fan et al., 2016). Its actions are most likely exerted through its newly discovered receptor, GPR75, an orphan G-protein (Gαq/11) coupled receptor (Garcia et al., 2017).
20-HETE is produced from arachidonic acid metabolism, not via the classical cyclooxygenase (COX) or lipoxygenase (LOX) pathways, but by a third pathway involving cytochrome P450-mediated ω-hydroxylation via CYP4A and CYP4F enzyme sub-families (Fan et al., 2016; Fan et al., 2015; Roman, 2002). Its presence has been reported in the microvasculature of the brain, lungs, kidneys, and the peripheral blood vessels (Miyata and Roman, 2005). There is accumulating evidence that CYP4A/4F and 20-HETE pathway play a major role in vascular dysfunction and changes in 20-HETE levels is implicated in many diseases like diabetes and hypertension (Fan et al., 2016; Miyata and Roman, 2005; Rocic and Schwartzman, 2018; Yousif et al., 2009a; Yousif et al., 2009b). For example, 20-HETE contributed to elevated vascular reactivity in renal and mesenteric vasculature of streptozotocin-induced diabetic mice (Yousif et al., 2009a). Moreover, 20-HETE stimulates mitogenic and angiogenic responses (in vivo and in vitro) to various growth factors such as EGF (Miyata and Roman, 2005). This was demonstrated in a study whereby inhibition of 20-HETE by HET0016 (an inhibitor of 20-HETE synthesis) almost completely abolished the angiogenic responses of EGF and other growth factors (Chen et al., 2005). Also, 20-HETE has been implicated as a key regulator of NO production and function. For instance, 20-HETE leads to eNOS uncoupling and impairs acetylcholine-induced relaxation through tyrosine kinase-, MAPK/ERK-, and IKK- dependent mechanisms that involve HSP90-eNOS dissociation and HSP90-IKKβ association (Cheng et al., 2010) leading to endothelial dysfunction. Moreover, 20-HETE stimulates NF-kB and increases levels of proinflammatory cytokines causing endothelial activation. This is partly mediated via MAPK-ERK1/2 signalling (Ishizuka et al., 2008).
Further studies on the mechanism by which 20-HETE exerts its actions in the vasculature (Chen, et al., 2012; Guo et al., 2007; Guo et al., 2009) showed that 20-HETE stimulates endothelial progenitor cell proliferation, migration and tube formation as well as enhancing release of proangiogenic factors such as VEGF. Importantly, these studies revealed the interplay with EGFR/ErbB receptors in that these vascular effects of 20-HETE were mediated through Src and EGFR with downstream activation of NADPH oxidases/reactive oxygen species (ROS) system, eNOS uncoupling and stimulation of MAPK and PI3K/Akt pathways (Chen, et al., 2012; Guo et al., 2007; Guo et al., 2009).
In VSMC, it was shown that norephinephrine (NE), Ang II, and EGF can activate the Ras/MAPK pathway through generation of 20-HETE (Muthalif et al., 1988). Since ACE/Ang II/AT1 receptor pathway is known to lead to transactivation of ErbBs in VSMC (Akhtar et al., 2015; Akhtar et al., 2016), the fact that 20-HETE is a potent activator of endothelial ACE expression and activity might represent another point of interplay between EGFR and 20-HETE signaling. Indeed, 20-HETE induces ACE expression through increased NF-κB binding to the ACE promoter (Cheng et al., 2012; Garcia et al., 2016). Endothelial ACE transcription is in turn mediated by EGFR via a MAPK/IkappaB kinase signaling cascade that ultimately results in increased ACE activity (Garcia et al., 2016). Furthermore, in parallel enhanced synthesis of 20-HETE in vivo leads to increased ACE expression in the vasculature (Garcia et al., 2015; Garcia et al., 2016; Garcia et al., 2017; Garcia and Schwartzman, 2017) as well as increased serum Ang II levels (Sodhi et al., 2010). Blocking 20-HETE synthesis prevented these changes, implicating 20-HETE in the regulation of ACE expression and AngII levels in vivo that indirectly could have an impact on ACE/AngII/AT1 receptor mediated transactivation of ErbBs. Thus, EGFR is likely involved in mediating the actions of 20-HETE in stimulating ACE expression and Ang II levels as well being involved in the downstream actions of ACE/AngII/AT1 receptor signaling possibly via Src phosphorylation. Collectively the above studies suggest that EGFR/ErbB receptor transactivation may represent an important signaling convergence point or “hub” by which many cardiovascular risk factors promote diabetes-induced vascular dysfunction and remodeling.
Perspectives and Concluding Remarks
Signaling via EGFR/ErbB family of receptor tyrosine kinases appears to be a central hub or relay in mediating vital functions in the normal development and physiology of the cardiovascular system as well as in the development of cardiovascular complications and pathologies. All four members, ErbB1-4, appear to be expressed both in the normal and diabetic heart as well as normal and diabetic vasculature where EGFR/ErbB receptors appear to have a dual (or “good guy” v “bad guy”; “Jekyll and Hyde” or “Janus-faced”) role. EGFR/ErbB receptors generally have beneficial “good-guy” actions in normal physiological function of the cardiovascular system and in mediating key “salvage” or recovery pathways following cardiac ischemia-reperfusion injury. However, several studies highlighted in this review confirm also the detrimental “bad-guy” effect of upregulated EGFR/ErbB signaling in the pathogenesis of diabetic-induced cardiac pathologies (Galan et al., 2012; Liang et al., 2015). Further highlighting the dual role of EGFR/ErbB receptors, these findings apparently contradict those showing protective effects of EGFR/ErbBs following ischemia/reperfusion injury in diabetic animals (Akhtar et al., 2012a). In an attempt to reconcile this contradiction, it has been postulated that EGFR/ErbB activation could be protective in the context of acute injury such as acute ischemia/reperfusion injury, but detrimental in chronic pathologies where persistent upregulation of EGFR/ErbB receptor signaling is observed such as diabetes-induced cardiac fibrosis and remodeling (Xu and Cai, 2015).
Similarly, upregulation of all four ErbBs has been implicated in the development of diabetes-induced vascular dysfunction through mechanisms involving multiple downstream effectors such as ERK1/2, ROCKs, p38 MAPK, FOXO, AKT, eNOS, and NF-kB [see also Figure 2] (Akhtar et al., 2015; Akhtar et al., 2020a). Both in the vasculature and in the heart, much of the evidence suggests that EGFR/ErbB receptors might function as key relays for the actions of the RAAS especially in mediating the pathological actions of its detrimental arm comprising the ACE/Ang II/AT1 receptor signaling cascade-that is known to lead to transactivation of EGFR/ErbBs with subsequent development of cardiovascular pathologies. Similarly, EGFR/ErbB receptors appear to be a key target for the actions of the beneficial counter-regulatory, ACE2/Ang-(1–7)/Mas receptor, pathway or “arm” of the RAAS. For example, the vasculo-protective effects of Ang-(1–7)/Mas receptor appear to be mediated, at least in part, via blockade of a Src-dependent EGFR/ErbB receptor phosphorylation and attenuation of downstream effectors (Akhtar et al., 2012a; Akhtar et al., 2015).
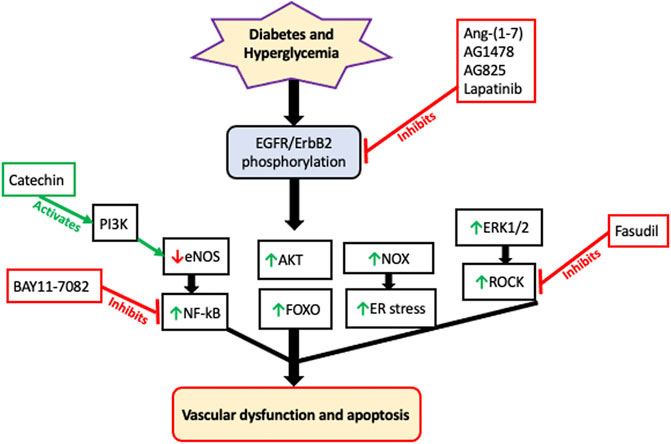
FIGURE 2. A schematic of the likely signaling effectors of EGFR/ErbB receptors in mediating diabetes-induced vascular dysfunction. Although there is now evidence that all four ErbB receptors may be activated by hyperglycemia, the majority of the data suggests that diabetes induced vascular dysfucntion proceeds via phosphorylation and subsequent hetero-dimerization EGFR and ErbB2 receptors. This is turn begins a cascade of downstream signaling pathways including attenuation of eNOS that results in decreased NO production. Activation of P13K, such as by green tea components like catechin, can rescure eNOS and reduction in NO. eNOS appears to be upstream of NF-kB activation. Additionally, ERK1/2 activation leads to downstream ROCK signaling–a key driver of pro-contractile changes leading to vascular dysfunction. Interestingly, activation of AKT and FOXO occur independently of each other implying that uncoupling of AKT-FOXO axis is a key component in developing diabetes-induced vascular complications. There is also evidence that EGFR/ErbB2 signaling via NADPH-oxidase (NOX) leads to increased ROS/oxidative and ER stress in the diabetic cardiovascular system. The precise cross-talk and interplay between these and other pathways remains to be fully elucidated. Furthermore, since multiple gene expression changes are known to corrected by EGFR inhibition in the diabetic vasculature, it is very likely that the signaling pathways discussed here will act in concert with multiple other, as yet unconfirmed, signaling cascades that are downstream of EGFR/ErbB receptors to eventually lead to diabetes-induced vascular cell apoptosis and dysfunction. Some of the reported pharmacologic interventions used in the analyses of these pathways are also shown; red dead-end arrow indicates inhibition; green arrow indicates activation (refer to the main text for more details and references).
Interestingly, EGFR/ErbB receptor signaling also appears important in mediating the vascular effects of the arachidonic acid metabolite, 20-HETE-a key regulator of cardiovascular function that also exhibits significant cross-talk with RAAS. EGFR/ErbB receptors are likely involved in mediating the actions of 20-HETE especially those leading to enhancing ACE expression and serum Ang II levels. Further, EGFR/ErbB receptors are also likely be involved in the downstream actions of ACE/AngII/AT1 receptor signaling such as in the Src-dependent transactivation of EGFR/ErbB receptor family members that ultimately leads to diabetes-induced vascular complications. Therefore, EGFR/ErbBs might represent a key point of convergence for actions of the RAAS and other molecules that are involved in cross-talk with RAAS such as 20-HETE. Thus, targeting ErbB receptor might represent a novel strategy for the treatment of cardiovascular complications.
EGFR inhibition is reported to have several benefits in diabetes including normalization of cardiovascular function, reduction of blood pressure, lowering of blood glucose levels and reversing insulin resistance (Hao et al., 2004; Nagareddy et al., 2010; Akhtar et al., 2013; Kassan et al., 2015a; Akhtar et al., 2015; Benter et al., 2015; Fountas et al., 2015). Inhibitors of EGFR/ErbB receptors (e.g., lapatinib) are already clinically available for the treatment of several cancers but could also be repurposed for treatment of diabetes-induced cardiovascular complications (Akhtar et al., 2015). In contrast, inducers or activators of EGFR/ErbB receptors (e.g., NRG or EGF) might be useful in recovering hearts from ischemia-reperfusion injury especially in diabetes where cardiac function is already compromised. Interestingly, in this regard it was found that the beneficial actions of RAAS inhibitors such as the angiotensin receptor blockers (e.g., losartan), that are clinically approved for use in myocardial infarction/ischemic heart disease, might not be optimal and cardiac function recovery could be improved by co-administering ligands or activators of EGFR/ErbB signaling (Akhtar et al., 2012b). Consistent with losartan’s known inhibition of Angiotensin II-mediated transactivation of EGFR (e.g., Akhtar et al., 2016), reduced EGFR phosphorylation was observed in losartan-treated diabetic hearts following ischemia-reperfusion injury compared to untreated diabetic hearts. Co-treatment of losartan with EGF, a ligand for EGFR, prevented the inhibitory effects of losartan on EGFR transactivation with a parallel improvement in cardiac function recovery greater than either agent alone. These findings may have important clinical implications as they suggest that rescuing the EGFR inhibitory effect of AT1 receptor antagonists by activators of the EGFR/ErbB family of receptor tyrosine kinases may represent a novel clinical approach to improve protection against end-organ damage in diabetic hearts. However, future treatments targeting EGFR/ErbBs will need to balance their beneficial versus the detrimental effects in a given cardiovascular pathology as well as the differential expression of EGFR/ErbB receptors and their ligands in different tissues. A greater understanding of the multi-faceted roles of EGFR/ErbB/HER family of tyrosine kinases, their interplay with other key modulators of cardiovascular function, and the development of smart or intelligent drug delivery systems (e.g., Mitchel et al., 2021) that can spatially and temporally control delivery of EGFR/ErbB/HER receptor modulators for a specific application, could facilitate the development of novel therapeutic strategies/formulations for treating diabetes-induced cardiovascular complications. Indeed, engineered nanoparticles, that could potentially encapsulate and site-specifically deliver EGFR/ErbB receptor modulators for a specific purpose, are now in advanced stages of development (Mitchell et al., 2021). By controlling nanoparticle surface and nanomaterial properties, their size, shape and architecture, as well as through careful selection of cell or tissue-specific targeting moieties and built-in responsiveness to environmental cues such as hyperglycemia, nanoparticle delivery systems can now be produced with intelligent designs to tailor the delivery of EGFR/ErbB receptor drugs to a specific application or condition as new-age precision medicines. Such approaches may have broader application beyond diabetes-induced cardiovascular dysfunction as dysregulated EGFR/ErbB/HER signaling also leads to diabetes-induced retinal and corneal complications (Akhtar et al., 2009; Ju et al., 2019), diabetic foot ulcers (Qi et al., 2018; Yang et al., 2020) and renal pathologies (Harskamp et al., 2016; Rayego-Mateos et al., 2018; Sheng et al., 2021) implying that aberrant signalling via EGFR/ErbB/HER receptors and their ligands might represent a common underlying mechanism in the development of diabetes complications.
Author Contributions
SA conceived the idea. SA, BS, MM, AH and IB searched, wrote and edited the manuscript.
Funding
The research in the laboratory of SA was funded by Qatar University grant QUCG-CMED-19/20-3 and by a QNRF Rapid Response call grant on COVID-19 (RRC-2-047).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Abwainy, A. a., Babiker, F., Akhtar, S., and Benter, I. F. (2016). Endogenous angiotensin-(1-7)/Mas Receptor/NO Pathway Mediates the Cardioprotective Effects of Pacing Postconditioning. Am. J. Physiology-Heart Circulatory Physiol. 310 (1), H104–H112. doi:10.1152/ajpheart.00121.2015
Akar, F., Pektas, M. B., Tufan, C., Soylemez, S., Sepici, A., Ulus, A. T., et al. (2011). Resveratrol Shows Vasoprotective Effect Reducing Oxidative Stress without Affecting Metabolic Disturbances in Insulin-dependent Diabetes of Rabbits. Cardiovasc. Drugs Ther. 25 (2), 119–131. doi:10.1007/s10557-010-6255-7
Akhtar, S., and Benter, I. F. (2013). The Role of Epidermal Growth Factor Receptor in Diabetes-Induced Cardiac Dysfunction. Bioimpacts 3 (1), 5–9. doi:10.5681/bi.2013.008
Akhtar, S., Chandrasekhar, B., Attur, S., Dhaunsi, G. S., Yousif, M. H. M., and Benter, I. F. (2015). Transactivation of ErbB Family of Receptor Tyrosine Kinases Is Inhibited by Angiotensin-(1-7) via its Mas Receptor. PLoS One 10 (11), e0141657. doi:10.1371/journal.pone.0141657
Akhtar, S., Vranic, S., Cyprian, F. S., and Al Moustafa, A. E. (20182018). Epstein-Barr Virus in Gliomas: Cause, Association, or Artifact? Front. Oncol. 8, 123. doi:10.3389/fonc.2018.00123
Akhtar, S., Almubrad, T., Bron, A. J., Yousif, M. H. M., Benter, I. F., and Akhtar, S. (20092009). Role of Epidermal Growth Factor Receptor (EGFR) in Corneal Remodelling in Diabetes. Acta Ophthalmologica 87 (8), 881–889. doi:10.1111/j.1755-3768.2008.01434.x
Akhtar, S., Benter, I. F., Danjuma, M. I., Doi, S. A. R., Hasan, S. S., and Habib, A. M. (2020a). Pharmacotherapy in COVID-19 Patients: A Review of ACE2-Raising Drugs and Their Clinical Safety. J. Drug Target. 28, 683–699. doi:10.1080/1061186X.2020.1797754
Akhtar, S., Chandrasekhar, B., and Benter, I. (2020b). 455-P: Pan-ErbB Inhibition Attenuates Diabetes-Induced Vascular Dysfunction in an Experimental Model of Diabetes. Diabetes 69 (Suppl. 1), 455–P. doi:10.2337/db20-455-P
Akhtar, S., Chandrasekhar, B., Yousif, M. H., Renno, W., Benter, I. F., and El-Hashim, A. Z. (2019). Chronic Administration of Nano-Sized PAMAM Dendrimers In Vivo Inhibits EGFR-ERK1/2-ROCK Signaling Pathway and Attenuates Diabetes-Induced Vascular Remodeling and Dysfunction. Nanomedicine: Nanotechnology, Biol. Med. 18, 78–89. doi:10.1016/j.nano.2019.02.012
Akhtar, S., El-Hashim, A. Z., Chandrasekhar, B., Attur, S., and Benter, I. F. (2016). Naked Polyamidoamine Polymers Intrinsically Inhibit Angiotensin II-Mediated EGFR and ErbB2 Transactivation in a Dendrimer Generation- and Surface Chemistry-dependent Manner. Mol. Pharmaceutics 13 (5), 1575–1586. doi:10.1021/acs.molpharmaceut.6b00045
Akhtar, S., Yousif, M. H., Dhaunsi, G. S., Chandrasekhar, B., Al-Farsi, O., and Benter, I. F. (2012a). Angiotensin-(1-7) Inhibits Epidermal Growth Factor Receptor Transactivation via a Mas Receptor-dependent Pathway. Br. J. Pharmacol. 165 (5), 1390–1400. doi:10.1111/j.1476-5381.2011.01613.x
Akhtar, S., Yousif, M. H. M., Chandrasekhar, B., and Benter, I. F. (2012b). Activation of EGFR/ERBB2 via Pathways Involving ERK1/2, P38 MAPK, AKT and FOXO Enhances Recovery of Diabetic Hearts from Ischemia-Reperfusion Injury. PLoS One 7 (6), e39066. doi:10.1371/journal.pone.0039066
Akhtar, S., Yousif, M. H. M., Dhaunsi, G. S., Sarkhouh, F., Chandrasekhar, B., Attur, S., et al. (2013). Activation of ErbB2 and Downstream Signalling via Rho Kinases and ERK1/2 Contributes to Diabetes-Induced Vascular Dysfunction. PLoS One 8 (6), e67813. doi:10.1371/journal.pone.0067813
Al-Maghrebi, M., Benter, I. F., and Diz, D. I. (2009). Endogenous Angiotensin-(1-7) Reduces Cardiac Ischemia-Induced Dysfunction in Diabetic Hypertensive Rats. Pharmacol. Res. 59 (4), 263–268. doi:10.1016/j.phrs.2008.12.008
Amin, A. H., Abd Elmageed, Z. Y., Partyka, M., and Matrougui, K. (2011). Mechanisms of Myogenic Tone of Coronary Arteriole: Role of Down Stream Signaling of the EGFR Tyrosine Kinase. Microvasc Res. 81 (1), 135–42. doi:10.1016/j.mvr.2010.11.001
Babel, R. A., and Dandekar, M. P. (2021). A Review on Cellular and Molecular Mechanisms Linked to the Development of Diabetes Complications. Cdr 17 (4), 457–473. doi:10.2174/1573399816666201103143818
Barrick, C. J., Yu, M., Chao, H.-H., and Threadgill, D. W. (2008). Chronic Pharmacologic Inhibition of EGFR Leads to Cardiac Dysfunction in C57BL/6J Mice. Toxicol. Appl. Pharmacol. 228 (3), 315–325. doi:10.1016/j.taap.2007.12.012
Belmadani, S., Palen, D. I., Gonzalez-Villalobos, R. A., Boulares, H. A., and Matrougui, K. (2008). Elevated Epidermal Growth Factor Receptor Phosphorylation Induces Resistance Artery Dysfunction in Diabetic Db/db Mice. Diabetes 57 (6), 1629–1637. doi:10.2337/db07-0739
Beltowski, J., and Jazmroz-Wisniewska, A. (2014). Transactivation of ErbB Receptors by Leptin in the Cardiovascular System: Mechanisms, Consequences and Target for Therapy. Cpd 20 (4), 616–624. doi:10.2174/138161282004140213155050
Benter, I. F., Juggi, J. S., Khan, I., Yousif, M. H., Canatan, H., and Akhtar, S. (2005c). Signal Transduction Mechanisms Involved in Cardiac Preconditioning: Role of Ras-GTPase, Ca2+/calmodulin-dependent Protein Kinase II and Epidermal Growth Factor Receptor. Mol. Cel Biochem. 268 (1-2), 175–183. doi:10.1007/s11010-005-3895-1
Benter, I. F., Yousif, M. H. M., Juggi, J., and Akhtar, S. (2014). “The Angiotensin-Converting Enzyme 2/Angiotensin-(1-7)/Mas Receptor Axis: A Potential Target for Treating Diabetic Cardiovascular Disease,” in Diabetic Cardiomyopathy: Biochemical and Molecular Mechanisms. Advances in Biocehmistry in Health and Disease. Editors B. Turan, and N. S. Dhalla (New York: Springer Science + Business Media).
Benter, I. F., Benboubetra, M., Hollins, A. J., Yousif, M. H. M., Canatan, H., and Akhtar, S. (2009). Early Inhibition of EGFR Signaling Prevents Diabetes-Induced Up-Regulation of Multiple Gene Pathways in the Mesenteric Vasculature. Vasc. Pharmacol. 51 (4), 236–245. doi:10.1016/j.vph.2009.06.008
Benter, I. F., Diz, D. I., and Ferrario, C. M. (1993). Cardiovascular Actions of Angiotensin(1-7). Peptides 14 (4), 679–684. doi:10.1016/0196-9781(93)90097-z
Benter, I. F., Diz, D. I., and Ferrario, C. M. (1995b). Pressor and Reflex Sensitivity Is Altered in Spontaneously Hypertensive Rats Treated with Angiotensin-(1-7). Hypertension 26 (6 Pt 2), 1138–1144. doi:10.1161/01.hyp.26.6.1138
Benter, I. F., Ferrario, C. M., Morris, M., and Diz, D. I. (1995a). Antihypertensive Actions of Angiotensin-(1-7) in Spontaneously Hypertensive Rats. Am. J. Physiology-Heart Circulatory Physiol. 269 (1 Pt 2), H313–H319. doi:10.1152/ajpheart.1995.269.1.h313
Benter, I. F., Juggi, J. S., Khan, I., Yousif, M. H. M., Canatan, H., and Akhtar, S. (2005). Signal Transduction Mechanisms Involved in Cardiac Preconditioning: Role of Ras-GTPase, Ca2 +/calmodulin-dependent Protein Kinase II and Epidermal Growth Factor Receptor. Mol. Cel Biochem. 268 (1-2), 175–183. doi:10.1007/s11010-005-3895-1
Benter, I. F., Sarkhou, F., Al-Khaldi, A. T., Chandrasekhar, B., Attur, S., Dhaunsi, G. S., et al. (2015). The Dual Targeting of EGFR and ErbB2 with the Inhibitor Lapatinib Corrects High Glucose-Induced Apoptosis and Vascular Dysfunction by Opposing Multiple Diabetes-Induced Signaling Changes. J. Drug Target. 23 (6), 506–518. doi:10.3109/1061186x.2015.1057150
Benter, I. F., Yousif, M. H. M., Al-Saleh, F. M., Raghupathy, R., Chappell, M. C., and Diz, D. I. (2011). Angiotensin-(1-7) Blockade Attenuates Captopril- or Hydralazine-Induced Cardiovascular protection in Spontaneously Hypertensive Rats Treated with NG-nitro-L-arginine Methyl Ester. J. Cardiovasc. Pharmacol. 57 (5), 559–567. doi:10.1097/fjc.0b013e31821324b6
Benter, I. F., Yousif, M. H. M., Anim, J. T., Cojocel, C., and Diz, D. I. (2006). Angiotensin-(1-7) Prevents Development of Severe Hypertension and End-Organ Damage in Spontaneously Hypertensive Rats Treated with L-NAME. Am. J. Physiology-Heart Circulatory Physiol. 290 (2), H684–H691. doi:10.1152/ajpheart.00632.2005
Benter, I. F., Yousif, M. H. M., Cojocel, C., Al-Maghrebi, M., and Diz, D. I. (2007). Angiotensin-(1-7) Prevents Diabetes-Induced Cardiovascular Dysfunction. Am. J. Physiology-Heart Circulatory Physiol. 292 (1), H666–H672. doi:10.1152/ajpheart.00372.2006
Benter, I. F., Yousif, M. H. M., Dhaunsi, G. S., Kaur, J., Chappell, M. C., and Diz, D. I. (2008). Angiotensin-(1-7) Prevents Activation of NADPH Oxidase and Renal Vascular Dysfunction in Diabetic Hypertensive Rats. Am. J. Nephrol. 28 (1), 25–33. doi:10.1159/000108758
Benter, I. F., Yousif, M. H. M., Griffiths, S. M., Benboubetra, M., and Akhtar, S. (2005a). Epidermal Growth Factor Receptor Tyrosine Kinase-Mediated Signalling Contributes to Diabetes-Induced Vascular Dysfunction in the Mesenteric Bed. Br. J. Pharmacol. 145 (6), 829–836. doi:10.1038/sj.bjp.0706238
Benter, I. F., Yousif, M. H. M., Hollins, A. J., Griffiths, S. M., and Akhtar, S. (2005b). Diabetes-induced Renal Vascular Dysfunction Is Normalized by Inhibition of Epidermal Growth Factor Receptor Tyrosine Kinase. J. Vasc. Res. 42 (4), 284–291. doi:10.1159/000085904
Berk, B. C., Brock, T. A., Webb, R. C., Taubman, M. B., Atkinson, W. J., Gimbrone, M. A., et al. (1985). Epidermal Growth Factor, a Vascular Smooth Muscle Mitogen, Induces Rat Aortic Contraction. J. Clin. Invest. 75, 1083–1086. doi:10.1172/jci111772
Bhardwaj, P., Khanna, D., and Balakumar, P. (2014). Catechin Averts Experimental Diabetes Mellitus-Induced Vascular Endothelial Structural and Functional Abnormalities. Cardiovasc. Toxicol. 14 (1), 41–51. doi:10.1007/s12012-013-9226-y
Bi, H.-L., Zhang, X.-L., Zhang, Y.-L., Xie, X., Xia, Y.-L., Du, J., et al. (2020). The Deubiquitinase UCHL1 Regulates Cardiac Hypertrophy by Stabilizing Epidermal Growth Factor Receptor. Sci. Adv. 6 (16), eaax4826. doi:10.1126/sciadv.aax4826
Bian, K., Doursout, M.-F., and Murad, F. (2008). Vascular System: Role of Nitric Oxide in Cardiovascular Diseases. J. Clin. Hypertens. 10 (4), 304–310. doi:10.1111/j.1751-7176.2008.06632.x
Blaine, S. A., Ray, K. C., Branch, K. M., Robinson, P. S., Whitehead, R. H., and Means, A. L. (2009). Epidermal Growth Factor Receptor Regulates Pancreatic Fibrosis. Am. J. Physiology-Gastrointestinal Liver Physiol. 297 (3), G434–G441. doi:10.1152/ajpgi.00152.2009
Bodiga, V. L., Thokala, S., Vemuri, P. K., and Bodiga, S. (2015). Zinc Pyrithione Inhibits Caspase-3 Activity, Promotes ErbB1-ErbB2 Heterodimerization and Suppresses ErbB2 Downregulation in Cardiomyocytes Subjected to Ischemia/reperfusion. J. Inorg. Biochem. 153, 49–59. doi:10.1016/j.jinorgbio.2015.09.010
Brownlee, M., and Hirsch, I. B. (2006). Glycemic Variability: a Hemoglobin A1c-independent Risk Factor for Diabetic Complications. JAMA 295, 1707–1708. doi:10.1001/jama.295.14.1707
Brownlee, M. (2005). The Pathobiology of Diabetic Complications: a Unifying Mechanism. Diabetes 54 (6), 1615–1625. doi:10.2337/diabetes.54.6.1615
Caillaud, K., Boisseau, N., Ennequin, G., Chavanelle, V., Etienne, M., Li, X., et al. (2016). Neuregulin 1 Improves Glucose Tolerance in Adult and Old Rats. Diabetes Metab. 42 (2), 96–104. doi:10.1016/j.diabet.2015.08.003
Camenisch, T. D., Schroeder, J. A., Bradley, J., Klewer, S. E., and McDonald, J. A. (2002). Heart-valve Mesenchyme Formation Is Dependent on Hyaluronan-Augmented Activation of ErbB2-ErbB3 Receptors. Nat. Med. 8 (8), 850–855. doi:10.1038/nm742
Camprecios, G., Lorita, J., Pardina, E., Peinado-Onsurbe, J., Soley, M., and Ramírez, I. (2011). Expression, Localization, and Regulation of the Neuregulin Receptor ErbB3 in Mouse Heart. J. Cell. Physiol. 226, 450–455. doi:10.1002/jcp.22354
Ceriello, A. (2005). Postprandial Hyperglycemia and Diabetes Complications: Is it Time to Treat?. Diabetes 54, 1–7. doi:10.2337/diabetes.54.1.1
Chalmers, J., and Cooper, M. E. (2008). UKPDS and the Legacy Effect. N. Engl. J. Med. 359, 1618–1620. doi:10.1056/nejme0807625
Chan, R., Hardy, W. R., Laing, M. A., Hardy, S. E., and Muller, W. J. (2002). The Catalytic Activity of the ErbB-2 Receptor Tyrosine Kinase Is Essential for Embryonic Development. Mol. Cel Biol 22 (4), 1073–1078. doi:10.1128/mcb.22.4.1073-1078.2002
Chappell, M. C, Marshall, A. C., Alzayadneh, E. M., Shaltout, H. A., and Diz, D. I. (2014). Update on the Angiotensin converting enzyme 2-Angiotensin (1-7)-MAS Receptor Axis: Fetal Programing, Sex Differences, and Intracellular Pathways. Front Endocrinol (Lausanne) 9 (4), 201. doi:10.3389/fendo.2013.00201
Chen, B., Bronson, R. T., Klaman, L. D., Hampton, T. G., Wang, J.-f., Green, P. J., et al. (2000). Mice Mutant for Egfr and Shp2 Have Defective Cardiac Semilunar Valvulogenesis. Nat. Genet. 24 (3), 296–299. doi:10.1038/73528
Chen, J., Chen, J.-K., and Harris, R. C. (2015). EGF Receptor Deletion in Podocytes Attenuates Diabetic Nephropathy. Jasn 26 (5), 1115–1125. doi:10.1681/asn.2014020192
Chen, J., Zeng, F., Forrester, S. J., Eguchi, S., Zhang, M.-Z., and Harris, R. C. (2016). Expression and Function of the Epidermal Growth Factor Receptor in Physiology and Disease. Physiol. Rev. 96 (3), 1025–1069. doi:10.1152/physrev.00030.2015
Chen, L., Ackerman, R., and Guo, A. M. (2012). 20-HETE in Neovascularization. Prostaglandins & Other Lipid Mediators 98, 63–68. doi:10.1016/j.prostaglandins.2011.12.005
Chen, P., Guo, M., Wygle, D., Edwards, P. A., Falck, J. R., Roman, R. J., et al. (2005). Inhibitors of Cytochrome P450 4A Suppress Angiogenic Responses. Am. J. Pathol. 166 (2), 615–624. doi:10.1016/s0002-9440(10)62282-1
Cheng, J., Garcia, V., Ding, Y., Wu, C.-C., Thakar, K., Falck, J. R., et al. (2012). Induction of Angiotensin-Converting Enzyme and Activation of the Renin-Angiotensin System Contribute to 20-Hydroxyeicosatetraenoic Acid-Mediated Endothelial Dysfunction. Atvb 32, 1917–1924. doi:10.1161/atvbaha.112.248344
Cheng, J., Wu, C.-C., Gotlinger, K. H., Zhang, F., Falck, J. R., Narsimhaswamy, D., et al. (2010). 20-Hydroxy-5,8,11,14-eicosatetraenoic Acid Mediates Endothelial Dysfunction via IκB Kinase-dependent Endothelial Nitric-Oxide Synthase Uncoupling. J. Pharmacol. Exp. Ther. 332 (1), 57–65. doi:10.1124/jpet.109.159863
Childers, C. L., Tessier, S. N., and Storey, K. B. (2019). The Heart of a Hibernator: EGFR and MAPK Signaling in Cardiac Muscle during the Hibernation of Thirteen-Lined Ground squirrels,Ictidomys Tridecemlineatus. PeerJ 7, e7587. doi:10.7717/peerj.7587
Choi, S.-K., Galán, M., Partyka, M., Trebak, M., Belmadani, S., and Matrougui, K. (2012). Chronic Inhibition of Epidermal Growth Factor Receptor Tyrosine Kinase and Extracellular Signal-Regulated Kinases 1 and 2 (ERK1/2) Augments Vascular Response to Limb Ischemia in Type 2 Diabetic Mice. Am. J. Pathol. 180 (1), 410–418. doi:10.1016/j.ajpath.2011.09.016
Chong, W., Shastri, M., and Eri, R. (2017). Endoplasmic Reticulum Stress and Oxidative Stress: A Vicious Nexus Implicated in Bowel Disease Pathophysiology. Ijms 18 (4), 771. doi:10.3390/ijms18040771
Cohen, M. V., Philipp, S., Krieg, T., Cui, L., Kuno, A., Solodushko, V., et al. (2007). Preconditioning-mimetics Bradykinin and DADLE Activate PI3-Kinase through Divergent Pathways. J. Mol. Cell. Cardiol. 42 (4), 842–851. doi:10.1016/j.yjmcc.2007.01.004
Colom, C., Rull, A., Sanchez-Quesada, J. L., and Pérez, A. (2021). Cardiovascular Disease in Type 1 Diabetes Mellitus: Epidemiology and Management of Cardiovascular Risk. Jcm 10 (8), 1798. doi:10.3390/jcm10081798
Cooper, M., Bonnet, F., Oldfield, M., and Jandeleit-Dahm, K. (2001). Mechanisms of Diabetic Vasculopathy: an Overview. Am. J. Hypertens. 14 (5 Pt 1), 475–486. doi:10.1016/s0895-7061(00)01323-6
Crèvecoeur, I., Rondas, D., Mathieu, C., and Overbergh, L. (2015). The Beta-Cell in Type 1 Diabetes: What Have We Learned from Proteomic Studies?. Prot. Clin. Appl. 9 (7-8), 755–766. doi:10.1002/prca.201400135
Crone, S. A., Zhao, Y.-Y., Fan, L., Gu, Y., Minamisawa, S., Liu, Y., et al. (2002). ErbB2 Is Essential in the Prevention of Dilated Cardiomyopathy. Nat. Med. 8 (5), 459–465. doi:10.1038/nm0502-459
Cuomo, A., Rodolico, A., Galdieri, A., Russo, M., Campi, G., Franco, R., et al. (2019). Heart Failure and Cancer: Mechanisms of Old and New Cardiotoxic Drugs in Cancer Patients. Card. Fail. Rev. 5 (2), 112–118. doi:10.15420/cfr.2018.32.2
De Giusti, V. C., Nolly, M. B., Yeves, A. M., Caldiz, C. I., Villa-Abrille, M. C., Chiappe de Cingolani, G. E., et al. (2011). Aldosterone Stimulates the Cardiac Na +/H + Exchanger via Transactivation of the Epidermal Growth Factor Receptor. Hypertension 58 (5), 912–919. doi:10.1161/hypertensionaha.111.176024
De Keulenaer, G. W., Eline, Feyen., Lindsey, Dugaucquier., Shakeri., Hadis., Anastasia, Shchendrygina., Belenkov., Yury. N., et al. (2019). Mechanisms of the Multitasking Endothelial Protein NRG-1 as a Compensatory Factor during Chronic Heart Failure Circulation. Heart Fail. 12, e006288. doi:10.1161/circheartfailure.119.006288
Dhaunsi, G. S., Yousif, M. H., Akhtar, S., Chappell, M. C., Diz, D. I., and Benter, I. F. (2010). Angiotensin-(1-7) Prevents Diabetes-Induced Attenuation in PPAR-Gamma and Catalase Activities. Eur. J. Pharmacol. 638 (1-3), 108–114. doi:10.1016/j.ejphar.2010.04.030
Diamonti, A. J., Guy, P. M., Ivanof, C., Wong, K., Sweeney, C., and Carraway, K. L. (2002). An RBCC Protein Implicated in Maintenance of Steady-State Neuregulin Receptor Levels. Proc. Natl. Acad. Sci. 99, 2866–2871. doi:10.1073/pnas.052709799
Drucker, D. J. (2021). Diabetes, Obesity, Metabolism, and SARS-CoV-2 Infection: the End of the Beginning. Cel Metab. 33 (3), 479–498. doi:10.1016/j.cmet.2021.01.016
El-Hashim, A. Z., Khajah, M. A., Babyson, R. S., Renno, W. M., Ezeamuzie, C. I., Benter, I. F., et al. (2019). Ang-(1-7)/MAS1 Receptor axis Inhibits Allergic Airway Inflammation via Blockade of Src-Mediated EGFR Transactivation in a Murine Model of Asthma. PLoS One 14 (11), e0224163. doi:10.1371/journal.pone.0224163
Eldridge, S., Guo, L., Mussio, J., Furniss, M., Hamre, J., and Davis, M. (2014). Examining the Protective Role of ErbB2 Modulation in Human-Induced Pluripotent Stem Cell-Derived Cardiomyocytes. Toxicol. Sci. 141 (2), 547–559. doi:10.1093/toxsci/kfu150
Erickson, S. L., O'Shea, K. S., Ghaboosi, N., Loverro, L., Frantz, G., Bauer, M., et al. (1997). ErbB3 Is Required for normal Cerebellar and Cardiac Development: A Comparison with ErbB2-And Heregulin-Deficient Mice. Development 124 (24), 4999–5011. doi:10.1242/dev.124.24.4999
Erqou, S., Lee, C.-T. C., Suffoletto, M., Echouffo-Tcheugui, J. B., de Boer, R. A., van Melle, J. P., et al. (2013). Association between Glycated Haemoglobin and the Risk of Congestive Heart Failure in Diabetes Mellitus: Systematic Review and Meta-Analysis. Eur. J. Heart Fail. 15, 185–193. doi:10.1093/eurjhf/hfs156
Falcão-Pires, I., and Leite-Moreira, A. F. (2012). Diabetic Cardiomyopathy: Understanding the Molecular and Cellular Basis to Progress in Diagnosis and Treatment. Heart Fail. Rev. 17 (3), 325–344. doi:10.1007/s10741-011-9257-z
Fan, F., Ge, Y., Lv, W., Elliott, M. R., Muroya, Y., Hirata, T., et al. (2016). Molecular Mechanisms and Cell Signaling of 20-hydroxyeicosatetraenoic Acid in Vascular Pathophysiology. Front. Biosci. (Landmark Ed. 21, 1427–1463. doi:10.2741/4379
Fan, F., Geurts, A. M., Murphy, S. R., Pabbidi, M. R., Jacob, H. J., and Roman, R. J. (2015). Impaired Myogenic Response and Autoregulation of Cerebral Blood Flow Is Rescued in CYP4A1 Transgenic Dahl Salt-Sensitive Rat. Am. J. Physiology-Regulatory, Integr. Comp. Physiol. 308, R379–R390. doi:10.1152/ajpregu.00256.2014
Fang, S.-J., Li, P.-Y., Wang, C.-M., Xin, Y., Lu, W.-W., Zhang, X.-X., et al. (2017). Inhibition of Endoplasmic Reticulum Stress by Neuregulin-1 Protects against Myocardial Ischemia/reperfusion Injury. Peptides 88, 196–207. doi:10.1016/j.peptides.2016.12.009
Feng, C., Jin, Z., Chi, X., Zhang, B., Wang, X., Sun, L., et al. (2018). SHBG Expression Is Correlated with PI3K/AKT Pathway Activity in a Cellular Model of Human Insulin Resistance. Gynecol. Endocrinol. 34 (7), 567–573. doi:10.1080/09513590.2017.1411474
Ferrario, C. M., Ahmad, S., Joyner, J., and Varagic, J. (2010). Advances in the Renin Angiotensin System. Adv. Pharmacol. 59, 197–233. doi:10.1016/S1054-3589(10)59007-0
Florian, J. A., and Watts, S. W. (1999). Epidermal Growth Factor: a Potent Vasoconstrictor in Experimental Hypertension. Am. J. Physiology-Heart Circulatory Physiol. 276 (3), H976–H983. doi:10.1152/ajpheart.1999.276.3.H976
Folino, A., Accomasso, L., Giachino, C., Montarolo, P. G., Losano, G., Pagliaro, P., et al. (2018). Apelin-induced Cardioprotection against Ischaemia/reperfusion Injury: Roles of Epidermal Growth Factor and Src. Acta Physiol. 222 (2), e12924. doi:10.1111/apha.12924
Forbes, J. M., and Cooper, M. E. (2013). Mechanisms of Diabetic Complications. Physiol. Rev. 93 (1), 137–188. doi:10.1152/physrev.00045.2011
Forrester, S. J., Kawai, T., O'Brien, S., Thomas, W., Harris, R. C., and Eguchi, S. (2016). Epidermal Growth Factor Receptor Transactivation: Mechanisms, Pathophysiology, and Potential Therapies in the Cardiovascular System. Annu. Rev. Pharmacol. Toxicol. 56, 627–653. doi:10.1146/annurev-pharmtox-070115-095427
Förstermann, U., and Sessa, W. C. (2012). Nitric Oxide Synthases: Regulation and Function. Eur. Heart J. 33 (7), 829–837. doi:10.1093/eurheartj/ehr304
Fountas, A., Diamantopoulos, L.-N., and Tsatsoulis, A. (2015). Tyrosine Kinase Inhibitors and Diabetes: A Novel Treatment Paradigm? Trends Endocrinol. Metab. 26 (11), 643–656. doi:10.1016/j.tem.2015.09.003
Fuller, S. J., Sivarajah, K., and Sugden, P. H. (2008). ErbB Receptors, Their Ligands, and the Consequences of Their Activation and Inhibition in the Myocardium. J. Mol. Cell Cardiol. 44 (5), 831–854. doi:10.1016/j.yjmcc.2008.02.278
Galán, M., Kassan, M., Choi, S.-K., Partyka, M., Trebak, M., Henrion, D., et al. (2012). A Novel Role for Epidermal Growth Factor Receptor Tyrosine Kinase and its Downstream Endoplasmic Reticulum Stress in Cardiac Damage and Microvascular Dysfunction in Type 1 Diabetes Mellitus. Hypertension 60 (1), 71–80. doi:10.1161/hypertensionaha.112.192500
Gao, R., Zhang, J., Cheng, L., Wu, X., Dong, W., Yang, X., et al. (2010). A Phase II, Randomized, Double-Blind, Multicenter, Based on Standard Therapy, Placebo-Controlled Study of the Efficacy and Safety of Recombinant Human Neuregulin-1 in Patients with Chronic Heart Failure. J. Am. Coll. Cardiol. 55, 1907–1914. doi:10.1016/j.jacc.2009.12.044
Garcia, V., and Schwartzman, M. L. (2017). Recent Developments on the Vascular Effects of 20-hydroxyeicosatetraenoic Acid. Curr. Opin. Nephrol. Hypertens. 26, 74–82. doi:10.1097/MNH.0000000000000302
Garcia, V., Gilani, A., Shkolnik, B., Pandey, V., Zhang, F. F., Dakarapu, R., et al. (2017). 20-HETE Signals through G-Protein-Coupled Receptor GPR75 (G Q ) to Affect Vascular Function and Trigger Hypertension. Circ. Res. 120, 1776–1788. doi:10.1161/circresaha.116.310525
Garcia, V., Joseph, G., Shkolnik, B., Ding, Y., Zhang, F. F., Gotlinger, K., et al. (2015). Angiotensin II Receptor Blockade or Deletion of Vascular Endothelial ACE Does Not Prevent Vascular Dysfunction and Remodeling in 20-HETE-dependent Hypertension. Am. J. Physiology-Regulatory, Integr. Comp. Physiol. 309, R71–R78. doi:10.1152/ajpregu.00039.2015
Garcia, V., Shkolnik, B., Milhau, L., Falck, J. R., and Schwartzman, M. L. (2016). 20-HETE Activates the Transcription of Angiotensin-Converting Enzyme via Nuclear Factor- B Translocation and Promoter Binding. J. Pharmacol. Exp. Ther. 356, 525–533. doi:10.1124/jpet.115.229377
García-Rivello, H., Taranda, J., Said, M., Cabeza-Meckert, P., Vila-Petroff, M., Scaglione, J., et al. (2005). Dilated Cardiomyopathy in Erb-B4-Deficient Ventricular Muscle. Am. J. Physiology-Heart Circulatory Physiol. 289 (3), H1153–H1160. doi:10.1152/ajpheart.00048.2005
Garratt, A. N., Voiculescu, O., Topilko, P., Charnay, P., and Birchmeier, C. (2000). A Dual Role of erbB2 in Myelination and in Expansion of the Schwann Cell Precursor Pool. J. Cel Biol. 148, 1035–1046. doi:10.1083/jcb.148.5.1035
Gassmann, M., Casagranda, F., Orioli, D., Simon, H., Lai, C., Klein, R., et al. (1995). Aberrant Neural and Cardiac Development in Mice Lacking the ErbB4 Neuregulin Receptor. Nature 378 (6555), 390–394. doi:10.1038/378390a0
Geraldes, P., Hiraoka-Yamamoto, J., Matsumoto, M., Clermont, A., Leitges, M., Marette, A., et al. (2009). Activation of PKC-δ and SHP-1 by Hyperglycemia Causes Vascular Cell Apoptosis and Diabetic Retinopathy. Nat. Med. 15 (11), 1298–1306. doi:10.1038/nm.2052
Giacco, F., and Brownlee, M. (2010). Oxidative Stress and Diabetic Complications. Circ. Res. 107 (9), 1058–1070. doi:10.1161/CIRCRESAHA.110.223545
Gilbert, R. E., Rumble, J. R., Cao, Z., Cox, A. J., van Eeden, P., Allen, T. J., et al. (2000). Endothelin Receptor Antagonism Ameliorates Mast Cell Infiltration, Vascular Hypertrophy, and Epidermal Growth Factor Expression in Experimental Diabetes. Circ. Res. 86 (2), 158–165. doi:10.1161/01.res.86.2.158
Gray, S. P., and Jandeleit-Dahm, K. (2014). The Pathobiology of Diabetic Vascular Complications-Cardiovascular and Kidney Disease. J. Mol. Med. 92 (5), 441–452. doi:10.1007/s00109-014-1146-1
Guo, A. M., Arbab, A. S., Falck, J. R., Chen, P., Edwards, P. A., Roman, R. J., et al. (2007). Activation of Vascular Endothelial Growth Factor through Reactive Oxygen Species Mediates 20-hydroxyeicosatetraenoic Acid-Induced Endothelial Cell Proliferation. J. Pharmacol. Exp. Ther. 321, 18–27. doi:10.1124/jpet.106.115360
Guo, A. M., Scicli, G., Sheng, J., Falck, J. C., Edwards, P. A., and Scicli, A. G. (2009). 20-HETE Can Act as a Nonhypoxic Regulator of HIF-1α in Human Microvascular Endothelial Cells. Am. J. Physiology-Heart Circulatory Physiol. 297, H602–H613. doi:10.1152/ajpheart.00874.2008
Guo, S., Okyere, A. D., McEachern, E., Strong, J. L., Carter, R. L., Patwa, V. C., et al. (2021). Epidermal Growth Factor Receptor-dependent Maintenance of Cardiac Contractility. Cardiovasc. Res., cvab149. doi:10.1093/cvr/cvab149
Guo, Y. F., Zhang, X. X., Liu, Y., Duan, H. Y., Jie, B. Z., and Wu, X. S. (2012). Neuregulin-1 Attenuates Mitochondrial Dysfunction in a Rat Model of Heart Failure. Chin. Med. J. (Engl) 125, 807–814.
Haas, J., Frese, K. S., Park, Y. J., Keller, A., Vogel, B., Lindroth, A. M., et al. (2013). Alterations in Cardiac DNA Methylation in Human Dilated Cardiomyopathy. EMBO Mol. Med. 5, 413–429. doi:10.1002/emmm.201201553
Hao, L., Du, M., Lopez-Campistrous, A., and Fernandez-Patron, C. (2004). Agonist-induced Activation of Matrix Metalloproteinase-7 Promotes Vasoconstriction through the Epidermal Growth Factor-Receptor Pathway. Circ. Res. 94 (1), 68–76. doi:10.1161/01.res.0000109413.57726.91
Harskamp, L. R., Gansevoort, R. T., van Goor, H., and Meijer, E. (2016). The Epidermal Growth Factor Receptor Pathway in Chronic Kidney Diseases. Nat. Rev. Nephrol. 12 (8), 496–506. doi:10.1038/nrneph.2016.91
Hill, M. F., Patel, A. V., Murphy, A., Smith, H. M., Galindo, C. L., Pentassuglia, L., et al. (2013). Intravenous Glial Growth Factor 2 (GGF2) Isoform of Neuregulin-1 Beta Improves Left Ventricular Function, Gene and Protein Expression in Rats after Myocardial Infarction. PLoS One 8, e55741. doi:10.1371/journal.pone.0055741
Holman, R. R., Paul, S. K., Bethel, M. A., Matthews, D. R., and Neil, H. A. W. (2008). 10-year Follow-Up of Intensive Glucose Control in Type 2 Diabetes. N. Engl. J. Med. 359, 1577–1589. doi:10.1056/nejmoa0806470
Iacobini, C., Vitale, M., Pesce, C., Pugliese, G., and Menini, S. (2021). Diabetic Complications and Oxidative Stress: A 20-Year Voyage Back in Time and Back to the Future. Antioxidants 10 (5), 727. doi:10.3390/antiox10050727
IDF Diabetes Atlas (2019). IDF Diabetes Atlas. 9th edn.. retrieved from: https://www.diabetesatlas.org/en/sections/individual-social-and-economic-impact.html (accessed on April 20, 2021).
Ishizuka, T., Cheng, J., Singh, H., Vitto, M. D., Manthati, V. L., Falck, J. R., et al. (2008). 20-Hydroxyeicosatetraenoic Acid Stimulates Nuclear Factor-κB Activation and the Production of Inflammatory Cytokines in Human Endothelial Cells. J. Pharmacol. Exp. Ther. 324 (1), 103–110. doi:10.1124/jpet.107.130336
Iwamoto, R., Yamazaki, S., Asakura, M., Takashima, S., Hasuwa, H., Miyado, K., et al. (2003). Heparin-binding EGF-like Growth Factor and ErbB Signaling Is Essential for Heart Function. Proc. Natl. Acad. Sci. 100 (6), 3221–3226. doi:10.1073/pnas.0537588100
Jabbour, A., Hayward, C. S., Keogh, A. M., Kotlyar, E., McCrohon, J. A., England, J. F., et al. (2011). Parenteral Administration of Recombinant Human Neuregulin-1 to Patients with Stable Chronic Heart Failure Produces Favourable Acute and Chronic Haemodynamic Responses. Eur. J. Heart Fail. 13, 83–92. doi:10.1093/eurjhf/hfq152
Jin, Y., Ji, W., Yang, H., Chen, S., Zhang, W., and Duan, G. (2020). Endothelial Activation and Dysfunction in COVID-19: from Basic Mechanisms to Potential Therapeutic Approaches. Sig Transduct Target. Ther. 5, 293. doi:10.1038/s41392-020-00454-7
Ju, X., Yang, X., Yang, T., Chen, H., Song, Z., Zhang, Z., et al. (2019). EGFRinhibitor,AG1478, Inhibits Inflammatory Infiltration and Angiogenesis in Mice with Diabetic Retinopathy. Clin. Exp. Pharmacol. Physiol. 46 (1), 75–85. doi:10.1111/1440-1681.13029
Kassan, M., Ait-Aissa, K., Ali, M., Trebak, M., and Matrougui, K. (2015a). Augmented EGF Receptor Tyrosine Kinase Activity Impairs Vascular Function by NADPH Oxidase-dependent Mechanism in Type 2 Diabetic Mouse. Biochim. Biophys. Acta (Bba) - Mol. Cel Res. 1853 (10 Pt A), 2404–2410. doi:10.1016/j.bbamcr.2015.05.032
Kassan, M., Choi, S.-K., Galán, M., Trebak, M., Belmadani, S., and Matrougui, K. (2015b). Nuclear Factor Kappa B Inhibition Improves Conductance Artery Function in Type 2 Diabetic Mice. Diabetes Metab. Res. Rev. 31 (1), 39–49. doi:10.1002/dmrr.2542
Kawanabe, Y., Masaki, T., and Hashimoto, N. (2004). Involvement of Epidermal Growth Factor Receptor-Protein Tyrosine Kinase Transactivation in Endothelin-1-Induced Vascular Contraction. J. Neurosurg. 100 (6), 1066–1071. doi:10.3171/jns.2004.100.6.1066
Kenigsberg, B., Jain, V., and Barac, A. (2017). Cardio-oncology Related to Heart Failure. Heart Fail. Clin. 13 (2), 297–309. doi:10.1016/j.hfc.2016.12.002
Kenny, H. C., and Abel, E. D. (2019). Heart Failure in Type 2 Diabetes Mellitus. Circ. Res. 124 (1), 121–141. doi:10.1161/CIRCRESAHA.118.311371
Kilarkaje, N., Yousif, M. H. M., El-Hashim, A. Z., Makki, B., Akhtar, S., and Benter, I. F. (2013). Role of Angiotensin II and Angiotensin-(1-7) in Diabetes-Induced Oxidative DNA Damage in the Corpus Cavernosum. Fertil. Sterility 100 (1), 226–233. doi:10.1016/j.fertnstert.2013.02.046
Kirabo, A., Ryzhov, S., Gupte, M., Sengsayadeth, S., Gumina, R. J., Sawyer, D. B., et al. (2017). Neuregulin-1β Induces Proliferation, Survival and Paracrine Signaling in normal Human Cardiac Ventricular Fibroblasts. J. Mol. Cell Cardiol. 105, 59–69. doi:10.1016/j.yjmcc.2017.03.001
Kobayashi, T., and Eguchi, S. (2012). The Epidermal Growth Factor Receptor. Hypertension 60 (1), 20–21. doi:10.1161/hypertensionaha.112.197038
Kobayashi, T., Hayashi, Y., Taguchi, K., Matsumoto, T., and Kamata, K. (2006). ANG II Enhances Contractile Responses via PI3-Kinase P110δ Pathway in Aortas from Diabetic Rats with Systemic Hyperinsulinemia. Am. J. Physiology-Heart Circulatory Physiol. 291 (2), H846–H853. doi:10.1152/ajpheart.01349.2005
Kobayashi, T., Matsumoto, T., and Kamata, K. (2005). The PI3-K/Akt Pathway: Roles Related to Alterations in Vasomotor Responses in Diabetic Models. J. Smooth Muscle Res. 41 (6), 283–302. doi:10.1540/jsmr.41.283
Kobayashi, T., Taguchi, K., Yasuhiro, T., Matsumoto, T., and Kamata, K. (2004). Impairment of PI3-K/Akt Pathway Underlies Attenuated Endothelial Function in Aorta of Type 2 Diabetic Mouse Model. Hypertension 44 (6), 956–962. doi:10.1161/01.HYP.0000147559.10261.a7
Kosiborod, M., Gomes, M. B., Gomes, M. B., Nicolucci, A., Pocock, S., Rathmann, W., et al. (2018). Vascular Complications in Patients with Type 2 Diabetes: Prevalence and Associated Factors in 38 Countries (The DISCOVER Study Program). Cardiovasc. Diabetol. 17, 150. doi:10.1186/s12933-018-0787-8
Krieg, T., Cui, L., Qin, Q., Cohen, M. V., and Downey, J. M. (2004). Mitochondrial ROS Generation Following Acetylcholine-Induced EGF Receptor Transactivation Requires Metalloproteinase Cleavage of proHB-EGF. J. Mol. Cell Cardiol. 36, 435–443. doi:10.1016/j.yjmcc.2003.12.013
Kumagai, S., Koyama, S., and Nishikawa, H. (2021). Antitumour Immunity Regulated by Aberrant ERBB Family Signalling. Nat. Rev. Cancer 21 (3), 181–197. doi:10.1038/s41568-020-00322-0
Lee, H.-Y., Youn, S.-W., Oh, B.-H., and Kim, H.-S. (2012). Krüppel-Like Factor 2 Suppression by High Glucose as a Possible Mechanism of Diabetic Vasculopathy. Korean Circ. J. 42 (4), 239–245. doi:10.4070/kcj.2012.42.4.239
Lee, J. W., Koeppen, M., Seo, S.-W., Bowser, J. L., Yuan, X., Li, J., et al. (2020). Transcription-independent Induction of ERBB1 through Hypoxia-Inducible Factor 2A Provides Cardioprotection during Ischemia and Reperfusion. Anesthesiology 132 (4), 763–780. doi:10.1097/aln.0000000000003037
Lee, K.-F., Simon, H., Chen, H., Bates, B., Hung, M.-C., and Hauser, C. (1995). Requirement for Neuregulin Receptor erbB2 in Neural and Cardiac Development. Nature 378 (6555), 394–398. doi:10.1038/378394a0
Lee, N. J., Ali, N., Zhang, L., Qi, Y., Clarke, I., Enriquez, R. F., et al. (2018). Osteoglycin, a Novel Coordinator of Bone and Glucose Homeostasis. Mol. Metab. 13, 30–44. doi:10.1016/j.molmet.2018.05.004
Leitner, D. R., Frühbeck, G., Yumuk, V., Schindler, K., Micic, D., Woodward, E., et al. (2017). Obesity and Type 2 Diabetes: Two Diseases with a Need for Combined Treatment Strategies - EASO Can Lead the Way. Obes. Facts 10 (5), 483–492. doi:10.1159/000480525
Lev-Ran, A., Hwang, D. L., Miller, J. D., and Josefsberg, Z. (1990). Excretion of Epidermal Growth Factor (EGF) in Diabetes. Clinica Chim. Acta 192 (3), 201–206. doi:10.1016/0009-8981(90)90222-e
Li, T., Yang, G.-m., Zhu, Y., Wu, Y., Chen, X.-y., Lan, D., et al. (2015). Diabetes and Hyperlipidemia Induce Dysfunction of VSMCs: Contribution of the Metabolic Inflammation/miRNA Pathway. Am. J. Physiology-Endocrinology Metab. 308 (4), E257–E269. doi:10.1152/ajpendo.00348.2014
Li, W., Fang, Q., Zhong, P., Chen, L., Wang, L., Zhang, Y., et al. (2016). EGFR Inhibition Blocks Palmitic Acid-Induced Inflammation in Cardiomyocytes and Prevents Hyperlipidemia-Induced Cardiac Injury in Mice. Sci. Rep. 6, 24580. doi:10.1038/srep24580
Liang, D., Zhong, P., Hu, J., Lin, F., Qian, Y., Xu, Z., et al. (2015). EGFR Inhibition Protects Cardiac Damage and Remodeling through Attenuating Oxidative Stress in STZ-Induced Diabetic Mouse Model. J. Mol. Cell Cardiol. 82, 63–74. doi:10.1016/j.yjmcc.2015.02.029
Liaudet, L., Calderari, B., and Pacher, P. (2014). Pathophysiological Mechanisms of Catecholamine and Cocaine-Mediated Cardiotoxicity. Heart Fail. Rev. 19 (6), 815–824. doi:10.1007/s10741-014-9418-y
Liu, L., Jin, X., Hu, C.-F., Zhang, Y.-P., Zhou, Z. e., Li, R., et al. (2018). Amphiregulin Enhances Cardiac Fibrosis and Aggravates Cardiac Dysfunction in Mice with Experimental Myocardial Infarction Partly through Activating EGFR-dependent Pathway. Basic Res. Cardiol. 113 (2), 12. doi:10.1007/s00395-018-0669-y
Liu, L., Song, S., Zhang, Y. P., Wang, D., Zhou, Z. e., Chen, Y., et al. (2020). Amphiregulin Promotes Cardiac Fibrosis post Myocardial Infarction by Inducing the Endothelial-Mesenchymal Transition via the EGFR Pathway in Endothelial Cells. Exp. Cel Res. 390 (2), 111950. doi:10.1016/j.yexcr.2020.111950
Liu, X., Gu, X., Li, Z., Li, X., Li, H., Chang, J., et al. (2006). Neuregulin-1/erbB-activation Improves Cardiac Function and Survival in Models of Ischemic, Dilated, and Viral Cardiomyopathy. J. Am. Coll. Cardiol. 48, 1438–1447. doi:10.1016/j.jacc.2006.05.057
Long, C., Cook, L. G., Hamilton, S. L., Wu, G.-Y., and Mitchell, B. M. (2007). FK506 Binding Protein 12/12.6 Depletion Increases Endothelial Nitric Oxide Synthase Threonine 495 Phosphorylation and Blood Pressure. Hypertension 49 (3), 569–576. doi:10.1161/01.hyp.0000257914.80918.72
Lorita, J., Escalona, N., Faraudo, S., Soley, M., and Ramirez, I. (2002). Effects of Epidermal Growth Factor on Epinephrine-Stimulated Heart Function in Rodents. Am. J. Physiology-Heart Circulatory Physiol. 283, H1887–H1895. doi:10.1152/ajpheart.00217.2002
Lucchesi, P. A., Sabri, A., Belmadani, S., and Matrougui, K. (2004). Involvement of Metalloproteinases 2/9 in Epidermal Growth Factor Receptor Transactivation in Pressure-Induced Myogenic Tone in Mouse Mesenteric Resistance Arteries. Circulation 110 (23), 3587–3593. doi:10.1161/01.CIR.0000148780.36121.47
Ma, J., and Jin, G. (2019). Epidermal Growth Factor Protects against Myocardial Ischaemia Reperfusion Injury through Activating Nrf2 Signalling Pathway. Free Radic. Res. 53, 313–323. doi:10.1080/10715762.2019.1584399
Mali, V., Haddox, S., Hornersmith, C., Matrougui, K., and Belmadani, S. (2018). Essential Role for EGFR Tyrosine Kinase and ER Stress in Myocardial Infarction in Type 2 Diabetes. Pflugers Arch. - Eur. J. Physiol. 470 (3), 471–480. doi:10.1007/s00424-017-2097-5
Matrougui, K. (2010). Diabetes and Microvascular Pathophysiology: Role of Epidermal Growth Factor Receptor Tyrosine Kinase. Diabetes Metab. Res. Rev. 26 (1), 13–16. doi:10.1002/dmrr.1050
Methner, C., Donat, U., Felix, S. B., and Krieg, T. (2009). Cardioprotection of Bradykinin at Reperfusion Involves Transactivation of the EGF Receptor via Metalloproteinase-8. Acta Physiol. (Oxf) 197, 265–271.
Meyrelles, S. S., Peotta, V. A., Pereira, T. M., and Vasquez, E. C. (2011). Endothelial Dysfunction in the Apolipoprotein E-Deficient Mouse: Insights into the Influence of Diet, Gender and Aging. Lipids Health Dis. 10, 211. doi:10.1186/1476-511x-10-211
Miller, R. G., and Orchard, T. J. (2020). Understanding Metabolic Memory: A Tale of Two Studies. Diabetes 69 (3), 291–299. doi:10.2337/db19-0514
Mitchell, M. J., Billingsley, M. M., Haley, R. M., Wechsler, M. E., Peppas, N. A., and Langer, R. (2021). Engineering Precision Nanoparticles for Drug Delivery. Nat. Rev. Drug Discov. 20 (2), 101–124. doi:10.1038/s41573-020-0090-8
Miyata, N., and Roman, R. J. (2005). Role of 20-hydroxyeicosatetraenoic Acid (20-HETE) in Vascular System. J. Smooth Muscle Res. 41 (4), 175–193. doi:10.1540/jsmr.41.175
Montero, D., Walther, G., Pérez-Martin, A., Vicente-Salar, N., Roche, E., and Vinet, A. (2013). Vascular Smooth Muscle Function in Type 2 Diabetes Mellitus: a Systematic Review and Meta-Analysis. Diabetologia 56 (10), 2122–2133. doi:10.1007/s00125-013-2974-1
Montezano, A. C., NguyenCat, Dinh. A., Rios, F. J., and Touyz, R. M. (2014). Angiotensin II and Vascular Injury. Curr. Hypertens. Rep. 16, 431. doi:10.1007/s11906-014-0431-2
Morano, M., Angotti, C., Tullio, F., Gambarotta, G., Penna, C., Pagliaro, P., et al. (2017). Myocardial Ischemia/reperfusion Upregulates the Transcription of the Neuregulin1 Receptor ErbB3, but Only Postconditioning Preserves Protein Translation: Role in Oxidative Stress. Int. J. Cardiol. 233, 73–79. doi:10.1016/j.ijcard.2017.01.122
Morgan, N. G., Leete, P., Foulis, A. K., and Richardson, S. J. (2014). Islet Inflammation in Human Type 1 Diabetes Mellitus. IUBMB Life 66 (11), 723–734. doi:10.1002/iub.1330
Murthy, S., Koval, O. M., Ramiro Diaz, J. M., Kumar, S., Nuno, D., Scott, J. A., et al. (2017). Endothelial CaMKII as a Regulator of eNOS Activity and NO-Mediated Vasoreactivity. PloS one 12 (10), e0186311. doi:10.1371/journal.pone.0186311
Muthalif, M. M., Benter, I. F., Karzoun, N., Fatima, S., Harper, J., Uddin, M. R., et al. (1998). 20-Hydroxyeicosatetraenoic Acid Mediates Calcium/calmodulin-dependent Protein Kinase II-Induced Mitogen-Activated Protein Kinase Activation in Vascular Smooth Muscle Cells. Proc. Natl. Acad. Sci. 95 (21), 12701–12706. doi:10.1073/pnas.95.21.12701
Nagareddy, P. R., MacLeod, K. M., and McNeill, J. H. (2010). GPCR Agonist-Induced Transactivation of the EGFR Upregulates MLC II Expression and Promotes Hypertension in Insulin-Resistant Rats. Cardiovasc. Res. 87 (1), 177–186. doi:10.1093/cvr/cvq030
Nägele, M. P., Haubner, B., Tanner, F. C., Ruschitzka, F., and Flammer, A. J. (2020). Endothelial Dysfunction in COVID-19: Current Findings and Therapeutic Implications. Atherosclerosis 314, 58–62. doi:10.1016/j.atherosclerosis.2020.10.014
Navid, F., and Colbert, R. A. (2017). Causes and Consequences of Endoplasmic Reticulum Stress in Rheumatic Disease. Nat. Rev. Rheumatol. 13, 25–40. doi:10.1038/nrrheum.2016.192
Negro, A., Brar, B. K., and Lee, K. F. (2004). Essential Roles of Her2/erbB2 in Cardiac Development and Function. Recent Prog. Horm. Res. 59 (1), 1–12. doi:10.1210/rp.59.1.1
Oellerich, M. F., and Potente, M. (2012). FOXOs and Sirtuins in Vascular Growth, Maintenance, and Aging. Circ. Res. 110 (9), 1238–1251. doi:10.1161/circresaha.111.246488
Palen, D. I., and Matrougui, K. (2008). Role of Elevated EGFR Phosphorylation in the Induction of Structural Remodelling and Altered Mechanical Properties of Resistance Artery from Type 2 Diabetic Mice. Diabetes Metab. Res. Rev. 24 (8), 651–656. doi:10.1002/dmrr.905
Pareja, M., Sánchez, O., Lorita, J., Soley, M., and Ramírez, I. (2003). Activated Epidermal Growth Factor Receptor (ErbB1) Protects the Heart against Stress-Induced Injury in Mice. Am. J. Physiology-Regulatory, Integr. Comp. Physiol. 285 (2), R455–R462. doi:10.1152/ajpregu.00588.2002
Patel, V. B., Parajuli, N., and Oudit, G. Y. (2014). Role of angiotensin-converting enzyme 2 (ACE2) in diabetic cardiovascular complications. Clin Sci (Lond) 126 (7), 471–82. doi:10.1042/CS20130344
Paz Ocaranza, M., Riquelme, J. A., García, L., Jalil, J. E., Chiong, M., Santos, R. A. S., et al. (2020). Counter-regulatory Renin-Angiotensin System in Cardiovascular Disease. Nat. Rev. Cardiol. 17 (2), 116–129. doi:10.1038/s41569-019-0244-8
Peng, K., Tian, X., Qian, Y., Skibba, M., Zou, C., Liu, Z., et al. (2016). Novel EGFR Inhibitors Attenuate Cardiac Hypertrophy Induced by Angiotensin II. J. Cell. Mol. Med. 20 (3), 482–494. doi:10.1111/jcmm.12763
Ponugoti, B., Dong, G., and Graves, D. T. (2012). Role of Forkhead Transcription Factors in Diabetes-Induced Oxidative Stress. Exp. Diabetes Res. 2012, 1–7. doi:10.1155/2012/939751
Prado, A. F., Pernomian, L., Azevedo, A., Costa, R. A. P., Rizzi, E., Ramos, J., et al. (2018). Matrix Metalloproteinase-2-Induced Epidermal Growth Factor Receptor Transactivation Impairs Redox Balance in Vascular Smooth Muscle Cells and Facilitates Vascular Contraction. Redox Biol. 18, 181–190. doi:10.1016/j.redox.2018.07.005
Qi, M., Zhou, Q., Zeng, W., Wu, L., Zhao, S., Chen, W., et al. (2018). Growth Factors in the Pathogenesis of Diabetic Foot Ulcers. Front. Biosci. (Landmark Ed. 23, 310–317. doi:10.2741/4593
Rajagopalan, V., Zucker, I. H., Jones, J. A., Carlson, M., and Ma, Y. J. (2008). Cardiac ErbB-1/ErbB-2 Mutant Expression in Young Adult Mice Leads to Cardiac Dysfunction. Am. J. Physiology-Heart Circulatory Physiol. 295, H543–H554. doi:10.1152/ajpheart.91436.2007
Rayego-Mateos, S., Rodrigues-Diez, R., Morgado-Pascual, J. L., Valentijn, F., Valdivielso, J. M., Goldschmeding, R., et al. (2018). Role of Epidermal Growth Factor Receptor (EGFR) and its Ligands in Kidney Inflammation and Damage. Mediators Inflamm. 2018, 1–22. doi:10.1155/2018/8739473
Reichelt, M. E., O’Brien, S., Thomas, W. G., and Headrick, J. P. (2017). Transactivation of the Epidermal Growth Factor Receptor in Responses to Myocardial Stress and Cardioprotection. Int. J. Biochem. Cel Biol. 83, 97–110. doi:10.1016/j.biocel.2016.12.014
Rocic, P., and Schwartzman, M. L. (2018). 20-HETE in the Regulation of Vascular and Cardiac Function. Pharmacol. Ther. 192, 74–87. doi:10.1016/j.pharmthera.2018.07.004
Roman, R. J. (2002). P-450 Metabolites of Arachidonic Acid in the Control of Cardiovascular Function. Physiol. Rev. 82, 131–185. doi:10.1152/physrev.00021.2001
Roskoski, R. (2019). Small Molecule Inhibitors Targeting the EGFR/ErbB Family of Protein-Tyrosine Kinases in Human Cancers. Pharmacol. Res. 139, 395–411. doi:10.1016/j.phrs.2018.11.014
Saltiel, A. R. (2021). Insulin Signaling in Health and Disease. J. Clin. Invest. 131 (1), e142241. doi:10.1172/JCI142241
Sanchez-Soria, P., and Camenisch, T. D. (2010). ErbB Signaling in Cardiac Development and Disease. Semin. Cel Develop. Biol. 21 (9), 929–935. doi:10.1016/j.semcdb.2010.09.011
Sankrityayan, H., Oza, M. J., Kulkarni, Y. A., Mulay, S. R., and Gaikwad, A. B. (2019). ER Stress Response Mediates Diabetic Microvascular Complications. Drug Discov. Today 24 (12), 2247–2257. doi:10.1016/j.drudis.2019.08.003
Sasahara, T., Okamoto, H., Ohkura, N., Kobe, A., and Yayama, K. (2015). Epidermal Growth Factor Induces Ca2+ Sensitization through Rho-kinase-dependent Phosphorylation of Myosin Phosphatase Target Subunit 1 in Vascular Smooth Muscle. Eur. J. Pharmacol. 762, 89–95. doi:10.1016/j.ejphar.2015.05.042
Schinzari, F., Tesauro, M., Veneziani, A., Mores, N., Di Daniele, N., and Cardillo, C. (2018). Favorable Vascular Actions of Angiotensin-(1-7) in Human Obesity. Hypertension 71, 185–191. doi:10.1161/HYPERTENSIONAHA.117.10280
Schreier, B., Gekle, M., and Grossmann, C. (2014). Role of Epidermal Growth Factor Receptor in Vascular Structure and Function. Curr. Opin. Nephrol. Hypertens. 23 (2), 113–121. doi:10.1097/01.mnh.0000441152.62943.29
Schreier, B., Hünerberg, M., Mildenberger, S., Rabe, S., Bethmann, D., Wickenhauser, C., et al. (2018). Deletion of the EGF Receptor in Vascular Smooth Muscle Cells Prevents Chronic Angiotensin II-Induced Arterial wall Stiffening and media Thickening. Acta Physiol. 222 (3), e12996. doi:10.1111/apha.12996
Schreier, B., Stern, C., Dubourg, V., Nolze, A., Rabe, S., Mildenberger, S., et al. (2021). Endothelial Epidermal Growth Factor Receptor Is of Minor Importance for Vascular and Renal Function and Obesity-Induced Dysfunction in Mice. Sci. Rep. 11, 7269. doi:10.1038/s41598-021-86587-3
Sena, C. M., Nunes, E., Louro, T., Proença, T., Fernandes, R., Boarder, M. R., et al. (2008). Effects of α-lipoic Acid on Endothelial Function in Aged Diabetic and High-Fat Fed Rats. Br. J. Pharmacol. 153 (5), 894–906. doi:10.1038/sj.bjp.0707474
Shah, S., Akhtar, M. S., Hassan, M. Q., Akhtar, M., Paudel, Y. N., and Najmi, A. K. (2018). EGFR Tyrosine Kinase Inhibition Decreases Cardiac Remodeling and SERCA2a/NCX1 Depletion in Streptozotocin Induced Cardiomyopathy in C57/BL6 Mice. Life Sci. 210, 29–39. doi:10.1016/j.lfs.2018.08.018
Sharifi, J., Khirehgesh, M. R., Safari, F., and Akbari, B. (2021). EGFR and Anti-EGFR Nanobodies: Review and Update. J. Drug Target. 29 (4), 387–402. doi:10.1080/1061186X.2020.1853756
Sheng, L., Bayliss, G., and Zhuang, S. (2021). Epidermal Growth Factor Receptor: A Potential Therapeutic Target for Diabetic Kidney Disease. Front. Pharmacol. 11, 598910. doi:10.3389/fphar.2020.598910
Shi, Y., and Vanhoutte, P. M. (2017). Macro- and Microvascular Endothelial Dysfunction in Diabetes. J. Diabetes 9 (5), 434–449. doi:10.1111/1753-0407.12521
Sibilia, M., and Wagner, E. (1995). Strain-dependent Epithelial Defects in Mice Lacking the EGF Receptor. Science 269 (5221), 234–238. doi:10.1126/science.7618085
Sodhi, K., Wu, C.-C., Cheng, J., Gotlinger, K., Inoue, K., Goli, M., et al. (2010). CYP4A2-Induced Hypertension Is 20-Hydroxyeicosatetraenoic Acid- and Angiotensin II-dependent. Hypertension 56, 871–878. doi:10.1161/hypertensionaha.110.154559
Souza, L. L., and Costa-Neto, C. M. (2012). Angiotensin-(1-7) Decreases LPS-Induced Inflammatory Response in Macrophages. J. Cell. Physiol. 227 (5), 2117–2122. doi:10.1002/jcp.22940
Stern, C., Schreier, B., Nolze, A., Rabe, S., Mildenberger, S., and Gekle, M. (2020). Knockout of Vascular Smooth Muscle EGF Receptor in a Mouse Model Prevents Obesity-Induced Vascular Dysfunction and Renal Damage In Vivo. Diabetologia 63, 2218–2234. doi:10.1007/s00125-020-05187-4
Sur, S., and Agrawal, D. (2014). Transactivation of EGFR by G Protein-Coupled Receptor in the Pathophysiology of Intimal Hyperplasia. Cvp 12 (2), 190–201. doi:10.2174/1570161112666140226123745
Suryavanshi, S. V., and Kulkarni, Y. A. (2017). NF-κβ: A Potential Target in the Management of Vascular Complications of Diabetes. Front. Pharmacol. 8, 798. doi:10.3389/fphar.2017.00798
Taguchi, K., Kobayashi, T., Takenouchi, Y., Matsumoto, T., and Kamata, K. (2011). Angiotensin II Causes Endothelial Dysfunction via the GRK2/Akt/eNOS Pathway in Aortas from a Murine Type 2 Diabetic Model. Pharmacol. Res. 64 (5), 535–546. doi:10.1016/j.phrs.2011.05.001
Talukdar, S., Emdad, L., Das, S. K., and Fisher, P. B. (2020). EGFR: An Essential Receptor Tyrosine Kinase-Regulator of Cancer Stem Cells. Adv. Cancer Res. 147, 161–188. doi:10.1016/bs.acr.2020.04.003
Tan, S. Y., Mei Wong, J. L., Sim, Y. J., Wong, S. S., Mohamed Elhassan, S. A., Tan, S. H., et al. (2019). Type 1 and 2 Diabetes Mellitus: A Review on Current Treatment Approach and Gene Therapy as Potential Intervention. Diabetes Metab. Syndr. Clin. Res. Rev. 13 (Issue 1), 364–372. doi:10.1016/j.dsx.2018.10.008
Taniguchi, K., and Karin, M. (2018). NF-κB, Inflammation, Immunity and Cancer: Coming of Age. Nat. Rev. Immunol. 18, 309–324. doi:10.1038/nri.2017.142
Tanner, M. A., Thomas, T. P., and Grisanti, L. A. (2019). Death Receptor 5 Contributes to Cardiomyocyte Hypertrophy through Epidermal Growth Factor Receptor Transactivation. J. Mol. Cell Cardiol. 136, 1–14. doi:10.1016/j.yjmcc.2019.08.011
Tao, X., Fan, J., Kao, G., Zhang, X., Su, L., Yin, Y., et al. (2014). Angiotensin-(1-7) Attenuates Angiotensin II-Induced Signalling Associated with Activation of a Tyrosine Phosphatase in Sprague-Dawley Rats Cardiac Fibroblasts. Biol. Cel 106 (6), 182–192. doi:10.1111/boc.201400015
Tenin, G., Clowes, C., Wolton, K., Krejci, E., Wright, J. A., Lovell, S. C., et al. (2014). Erbb2 Is Required for Cardiac Atrial Electrical Activity during Development. PLoS One 9 (9), e107041. doi:10.1371/journal.pone.0107041
Toi, P. L., Anothaisintawee, T., Chaikledkaew, U., Briones, J. R., Reutrakul, S., and Thakkinstian, A. (2020). Preventive Role of Diet Interventions and Dietary Factors in Type 2 Diabetes Mellitus: An Umbrella Review. Nutrients 12 (9), 2722. doi:10.3390/nu12092722
Tousoulis, D., Kampoli, A.-M., Tentolouris Nikolaos Papageorgiou, C., Stefanadis, C., and Stefanadis, C. (2012). The Role of Nitric Oxide on Endothelial Function. Cvp 10 (1), 4–18. doi:10.2174/157016112798829760
Touyz, R. M., Alves-Lopes, R., Rios, F. J., Camargo, L. L., Anagnostopoulou, A., Arner, A., et al. (2018). Vascular Smooth Muscle Contraction in Hypertension. Cardiovasc. Res. 114 (4), 529–539. doi:10.1093/cvr/cvy023
Tsatsoulis, A., Mantzaris, M. D., Bellou, S., and Andrikoula, M. (2013). Insulin Resistance: an Adaptive Mechanism Becomes Maladaptive in the Current Environment - an Evolutionary Perspective. Metabolism 62 (5), 622–633. doi:10.1016/j.metabol.2012.11.004
van Wijngaarden, R. P. T., Overbeek, J. A., Heintjes, E. M., Schubert, A., Diels, J., Straatman, H., et al. (2017). Relation between Different Measures of Glycemic Exposure and Microvascular and Macrovascular Complications in Patients with Type 2 Diabetes Mellitus: an Observational Cohort Study. Diabetes Ther. 8, 1097–1109. doi:10.1007/s13300-017-0301-4
Volpe, C. M. O., Villar-Delfino, P. H., dos Anjos, P. M. F., and Nogueira-Machado, J. A. (2018). Cellular Death, Reactive Oxygen Species (ROS) and Diabetic Complications. Cell Death Dis 9, 119. doi:10.1038/s41419-017-0135-z
Williams-Pritchard, G., Knight, M., Hoe, L. S., Headrick, J. P., and Peart, J. N. (2011). Essential Role of EGFR in Cardioprotection and Signaling Responses to A1 Adenosine Receptors and Ischemic Preconditioning. Am. J. Physiology-Heart Circulatory Physiol. 300 (6), H2161–H2168. doi:10.1152/ajpheart.00639.2010
Wong, T. Y., Cheung, C. M., Larsen, M., Sharma, S., and Simó, R. (2016). Diabetic Retinopathy. Nat. Rev. Dis. Primers 2, 16012. doi:10.1038/nrdp.2016.12
Xiao, J., Li, B., Zheng, Z., Wang, M., Peng, J., Li, Y., et al. (2012). Therapeutic Effects of Neuregulin-1 Gene Transduction in Rats with Myocardial Infarction. Coron. Artery Dis. 23, 460–468. doi:10.1097/mca.0b013e32835877da
Xu, J., Zhou, Q., Xu, W., and Cai, L. (2012). Endoplasmic Reticulum Stress and Diabetic Cardiomyopathy. Exp. Diabetes Res. 2012, 1–12. doi:10.1155/2012/827971
Xu, Z., and Cai, L. (2015). Diabetic Cardiomyopathy: Role of Epidermal Growth Factor Receptor Tyrosine Kinase. J. Mol. Cell Cardiol. 84, 10–12. doi:10.1016/j.yjmcc.2015.04.002
Xu, Z., Zhao, Y., Zhong, P., Wang, J., Weng, Q., Qian, Y., et al. (2017). EGFR Inhibition Attenuates Diabetic Nephropathy through Decreasing ROS and Endoplasmic Reticulum Stress. Oncotarget 8 (20), 32655–32667. doi:10.18632/oncotarget.15948
Yang, Q., Zhang, Y., Yin, H., and Lu, Y. (2020). Topical Recombinant Human Epidermal Growth Factor for Diabetic Foot Ulcers: A Meta-Analysis of Randomized Controlled Clinical Trials. Ann. Vasc. Surg. 62, 442–451. doi:10.1016/j.avsg.2019.05.041
Yano, S., Kondo, K., Yamaguchi, M., Richmond, G., Hutchison, M., Wakeling, A., et al. (2003). Distribution and Function of EGFR in Human Tissue and the Effect of EGFR Tyrosine Kinase Inhibition. Anticancer Res. 23 (5A), 3639–3650.
Yousif, M. H., Benter, I. F., and Roman, R. J. (2009a). Cytochrome P450 Metabolites of Arachidonic Acid Play a Role in the Enhanced Cardiac Dysfunction in Diabetic Rats Following Ischaemic Reperfusion Injury. Auton. Autacoid Pharmacol. 29 (1-2), 33–41. doi:10.1111/j.1474-8673.2009.00429.x
Yousif, M. H. M., Makki, B., El-Hashim, A. Z., Akhtar, S., and Benter, I. F. (2014). Chronic Treatment with Ang-(1-7) Reverses Abnormal Reactivity in the Corpus Cavernosum and Normalizes Diabetes-Induced Changes in the Protein Levels of ACE, ACE2, ROCK1, ROCK2 and omega-hydroxylase in a Rat Model of Type 1 Diabetes. J. Diabetes Res. 2014, 142154. doi:10.1155/2014/142154
Yousif, M. H. M., Akhtar, S., Walther, T., and Benter, I. F. (2008). Role of Ca2+/calmodulin-dependent Protein Kinase II in Development of Vascular Dysfunction in Diabetic Rats with Hypertension. Cell Biochem. Funct. 26 (2), 256–263. doi:10.1002/cbf.1446
Yousif, M. H. M., Benter, I. F., and Akhtar, S. (2005). The Role of Tyrosine Kinase-Mediated Pathways in Diabetes-Induced Alterations in Responsiveness of Rat Carotid Artery. Autonom Auta Pharm. 25 (2), 69–78. doi:10.1111/j.1474-8673.2004.00333.x
Yousif, M. H. M., Benter, I. F., Dunn, K. M. J., Dahly-Vernon, A. J., Akhtar, S., and Roman, R. J. (2009b). Role of 20-hydroxyeicosatetraenoic Acid in Altering Vascular Reactivity in Diabetes. Auton. Autacoid Pharmacol. 29 (1-2), 1–12. doi:10.1111/j.1474-8673.2009.00426.x
Yousif, M. H. M., Benter, I. F., Hares, N., Canatan, H., and Akhtar, S. (2006). Phosphoinositide 3-kinase Mediated Signalling Contributes to Development of Diabetes-Induced Abnormal Vascular Reactivity of Rat Carotid Artery. Cel Biochem. Funct. 24 (1), 13–22. doi:10.1002/cbf.1278
Yousif, M. H. M., Dhaunsi, G. S., Makki, B. M., Qabazard, B. A., Akhtar, S., and Benter, I. F. (2012). Characterization of Angiotensin-(1-7) Effects on the Cardiovascular System in an Experimental Model of Type-1 Diabetes. Pharmacol. Res. 66 (3), 269–275. doi:10.1016/j.phrs.2012.05.001
Yu, Y., Wei, S.-G., Weiss, R. M., and Felder, R. B. (2021). Silencing Epidermal Growth Factor Receptor in Hypothalamic Paraventricular Nucleus Reduces Extracellular Signal-Regulated Kinase 1 and 2 Signaling and Sympathetic Excitation in Heart Failure Rats. Neuroscience 463, 227–237. doi:10.1016/j.neuroscience.2021.01.025
Zhang, C., Booz, G. W., Yu, Q., He, X., Wang, S., and Fan, F. (2018). Conflicting Roles of 20-HETE in Hypertension and Renal End Organ Damage. Eur. J. Pharmacol. 833, 190–200. doi:10.1016/j.ejphar.2018.06.010
Zhang, Y., Murugesan, P., Huang, K., and Cai, H. (2020). NADPH Oxidases and Oxidase Crosstalk in Cardiovascular Diseases: Novel Therapeutic Targets. Nat. Rev. Cardiol. 17 (3), 170–194. doi:10.1038/s41569-019-0260-8
Zhang, Y., Zeng, Y., Wang, M., Tian, C., Ma, X., Chen, H., et al. (2011). Cardiacspecific Overexpression of E3 Ligase Nrdp1 Increases Ischemia and Reperfusioninduced Cardiac Injury. Basic Res. Cardiol. 106, 371–383. doi:10.1007/s00395-011-0157-0
Zhang, Z., and Zhang, D. (2020). (-)-Epigallocatechin-3-Gallate Inhibits eNOS Uncoupling and Alleviates High Glucose-Induced Dysfunction and Apoptosis of Human Umbilical Vein Endothelial Cells by PI3K/AKT/eNOS Pathway. Dmso 13, 2495–2504. doi:10.2147/DMSO.S260901
Zhao, Y., Vanhoutte, P. M., and Leung, S. W. S. (2015). Vascular Nitric Oxide: Beyond eNOS. J. Pharmacol. Sci. 129 (2), 83–94. doi:10.1016/j.jphs.2015.09.002
Zheng, K., Kitazato, K., and Wang, Y. (2014). Viruses Exploit the Function of Epidermal Growth Factor Receptor. Rev Med Virol. 24 (4), 274–86. doi:10.1002/rmv.1796
Zhou, J., Du, T., Li, B., Rong, Y., Verkhratsky, A., and Peng, L. (2015). Crosstalk between MAPK/ERK and PI3K/AKT Signal Pathways during Brain Ischemia/Reperfusion. ASN Neuro 7 (5), 175909141560246. doi:10.1177/1759091415602463
Zuo, C., Li, X., Huang, J., Chen, D., Ji, K., Yang, Y., et al. (2018). Osteoglycin Attenuates Cardiac Fibrosis by Suppressing Cardiac Myofibroblast Proliferation and Migration through Antagonizing Lysophosphatidic Acid 3/matrix Metalloproteinase 2/epidermal Growth Factor Receptor Signalling. Cardiovasc. Res. 114 (5), 703–712. doi:10.1093/cvr/cvy035
Keywords: epidermal growth factor receptor, ErbB2, ErbB3, ErbB4, diabetes, heart, cardiac dysfunction, vascular dysfunction
Citation: Shraim BA, Moursi MO, Benter IF, Habib AM and Akhtar S (2021) The Role of Epidermal Growth Factor Receptor Family of Receptor Tyrosine Kinases in Mediating Diabetes-Induced Cardiovascular Complications. Front. Pharmacol. 12:701390. doi: 10.3389/fphar.2021.701390
Received: 27 April 2021; Accepted: 14 July 2021;
Published: 02 August 2021.
Edited by:
Venkatesh Sundararajan, The State University of New Jersey, United StatesReviewed by:
Raminderjit Kaur, Cleveland Clinic, United StatesShougang Zhuang, Brown University, United States
Copyright © 2021 Shraim, Moursi, Benter, Habib and Akhtar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Saghir Akhtar, s.akhtar@qu.edu.qa
†These authors have contributed equally to this work and share first authorship